 Lauren M. Rota
Lauren M. Rota Teresa L. Wood
Teresa L. Wood
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol., 09 June 2015
Sec. Cancer Endocrinology
Volume 6 - 2015 | https://doi.org/10.3389/fendo.2015.00092
This article is part of the Research TopicThe role of the Insulin/IGFs system in cancer: critical points in dysregulation, targeting problems and new blocking strategiesView all 17 articles
The insulin-like growth factor system has long been considered a pathway that promotes cell proliferation, survival, and transformation, and is thus a promoter of tumorigenesis. However, recent failure of clinical trials for IGF-1R inhibitors reveals the need for a better understanding of how this pathway functions in specific tumor subtypes. Ongoing studies are designed to uncover biomarkers and downstream targets to enhance therapeutic strategies. Other approaches in specific tumor models reveal complex interactions between IGF signaling and other tumor initiating pathways. Here, we review relevant background and recent studies suggesting that inhibiting the IGF-1R can amplify Wnt and Notch signaling pathways in a model of triple negative breast cancer.
The insulin/IGF system has very potent mitogenic, survival, and pro-migratory properties for breast cancer cells in vitro and in animal models [for reviews, see Ref. (1–3)]. Moreover, high serum levels of IGF-I are correlated with an increased risk for breast cancer, particularly in premenopausal women (4, 5). Collectively, these findings led to development of multiple monoclonal antibodies designed to target the IGF-1R or the IGF ligands, and tyrosine kinase inhibitors to target both the IGF-1R and insulin receptor (IR) [for reviews, see Ref. (6, 7)]. Despite the preclinical findings, the usefulness of disrupting IGF-1R/IR signaling in clinical trials has been less than promising and in some cases has led to worse outcomes (6, 7).
The failure of these trials illuminates the need to better understand patient cohorts that will best be served by disrupting the IGF signaling pathway. Recent studies have demonstrated that an “IGF gene signature” correlating to a set of genes that are up and down regulated by IGF-I is present in human breast cancers, specifically luminal B and triple negative breast cancer (TNBC) (8). Another approach to establish the function of IGF-1R in different types of breast cancers is to disrupt IGF-1R in mouse tumor models with distinct phenotypes. Early studies reported that expression of the IGF-1R predicted a favorable phenotype and a correlation with estrogen receptor (ER) expression (9, 10). Numerous studies have further confirmed crosstalk between the ER and IGF-1R [for reviews, see Ref. (11, 12)]. Consistent with these data, loss of IGF-1R has been associated with breast tumor progression into a more undifferentiated phenotype (13). The studies establishing the IGF-1R as growth promoting for breast cancers suggests some complexity concerning IGF-1R function in breast cancers. One question that has not been well addressed is whether IGF-1R has distinct functions in breast tumors depending on other active signaling pathways and/or the specific mutation(s) or oncogene driving the tumor. We have recently begun to examine this question using a mouse model of TNBC, the MMTV-Wnt1 mouse.
A variety of studies support the conclusion that Wnt pathway hyperactivation contributes to mammary/breast cancers in rodents and humans [for reviews, see Ref. (14–16)]. The Wnt pathway in mammals was first investigated by Nusse and Varmus in 1982, with the observation that overexpression of Wnt-1 in the mammary gland from the mouse mammary tumor virus (MMTV) promoter resulted in mammary hyperplasias by early puberty and mammary tumors between 3 and 8 months (17, 18). The MMTV-Wnt1 tumor model has been well-characterized as a basal tumor model (19–22). Using flow cytometry markers to label the mammary epithelial lineages in preneoplastic epithelium, Shackelton and colleagues observed a significant increase in the mammary stem cell (MaSC)/myoepithelial (CD24+CD29hi) population in MMTV-Wnt-1 epithelium compared to wild type epithelium (23). Other studies suggested that the overexpression of Wnt-1 via the MMTV promoter led to the expansion of mammary progenitor populations, based on the increase in the side population and Sca1+ population (20, 21). The overexpression of Wnt-1 in mammary progenitor cells appears to confer radioresistance (24). More recently, isolation of luminal progenitors from MMTV-Wnt-1 mice were found capable of reconstituting a mammary gland upon transplantation into a cleared fat pad (25). These studies led investigators to raise the interesting possibility that Wnt-1 regulates the MaSCs, which then change their cell surface phenotype during or after oncogenic transformation (25). However, other studies now support the hypothesis that it is the expanded luminal progenitor population that gives rise to Wnt1 tumors in this mouse model (26). Similarly, recent studies have defined the luminal progenitor as the cell of origin for BRCA tumors in both mouse and human (27–29). These studies strongly support the hypothesis that the basal phenotype of both the Wnt1-driven and the BRCA mutated tumors is acquired subsequent to transformation of a luminal progenitor cell.
In breast cancer, Wnt signaling is activated in the absence of downstream mutations and can occur via autocrine or paracrine mechanisms (30, 31). A multitude of Wnt ligands and Fzd receptors are overexpressed in breast cancer cell lines and primary human breast tumors (32–34). Secreted frizzled-related protein 1, a negative regulator of the Wnt pathway, is lost in 46–80% of breast cancers and is associated with a poor prognosis (35). Up to 50% of breast tumors have hypermethylation of the adenomatous polyposis coli (APC) promoter, and transcript loss leads to hyperactivation of the Wnt pathway (36). APC is a negative regulator of β-catenin (see Figure 1). Thus, hyperactivation of the Wnt pathway is common in breast carcinomas. More recently, TNBCs have been classified into six subtypes based on gene expression profiles; Wnt pathway activation was identified in several of the TNBC subtypes (37).
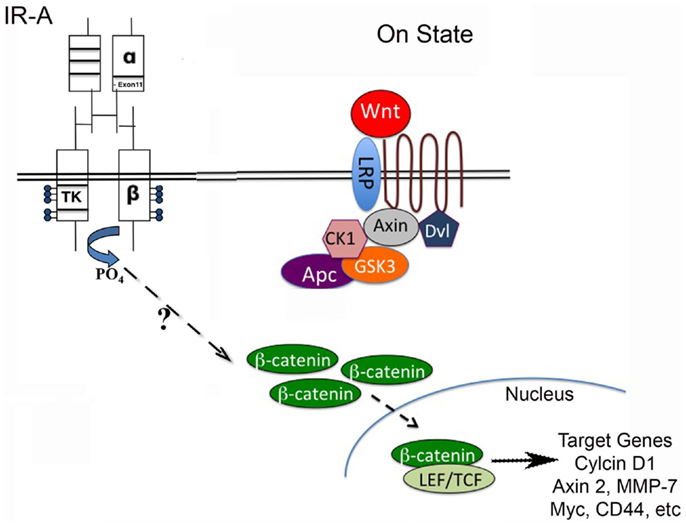
Figure 1. Wnt/β-catenin pathway regulates stem cell pluripotency and cell fate decisions during development. The Wnt pathway is activated when Wnt ligand binds to a Frizzled receptor, which then is brought into complex with the co-receptor LRP5/6. The activation of the Wnt pathway leads to stabilization of β-catenin through inactivation of the destruction complex (containing Axin, CK1, Gsk3β, and Apc). β-catenin can then translocate to the nucleus and interact with LEF/TCF to regulate Wnt target genes. The stimulation of the insulin receptor isoform A (IR-A) in IGF-1R null (R-/IR-A) fibroblasts by insulin was shown to increase levels of β-catenin through a mechanism that is currently unknown.
The Wnt signaling pathway is complex and there are still many questions about how to interfere with this pathway in breast and other cancers. Several therapeutic treatments are being explored, such as monoclonal antibodies that target Wnt ligand–receptor interaction and small molecule inhibitors designed to down-regulate Wnt secretion; for review, see Ref. (38, 39). Understanding pathways that interact with and potentially inactivate Wnt signaling, however, is of vital importance to develop more effective therapeutic treatments.
In recent studies, we attenuated IGF-1R signaling in the MMTV-Wnt1 tumor model and demonstrated decreased tumor latency, increased tumor incidence, and development of a metastatic tumor phenotype (40). These results were surprising given that the IGF-1R has been considered predominantly a positive-mediator of breast cancer growth. Moreover, transgenic mice overexpressing or with constitutive activation of the IGF-1R develop mammary tumors as early as 8 weeks of age (41–43). A recent study further demonstrated that the ability of a Wnt pathway inhibitor to reduce tumor growth in the MMTV-Wnt1 model was due to upregulation of the IGF-binding protein-5 (44). The authors of this study further suggested that part of the mechanism for IGFBP-5 in reducing tumor volume after acute treatment with the Wnt pathway inhibitor was due to down-regulation of IGF-1R signaling. However, our data suggest an opposing role for the IGF-1R in the context of active Wnt signaling in a chronic model and support the conclusion that the IGF-1R is protective against Wnt-mediated oncogenesis. In the remainder of this review, we focus on the possible mechanisms for and implications of our data on the function of the IGF-1R in the MMTV-Wnt1 tumor model and in TNBCs.
One explanation for our findings is that the loss of the IGF-1R alters mammary epithelial lineages leading to amplification of the luminal progenitor population responsible for Wnt-mediated tumors. Support for the IGF-1R in maintaining normal MEC lineage distribution is our observation that the attenuation of the IGF-1R in the normal mammary epithelium leads to an accumulation of luminal progenitors that are enriched in the CD61+CD29lo flow cytometry profile (40). The mechanism for this shift in lineage is likely due to the dysregulation of Notch signaling with down-regulation of the IGF-1R in both normal mammary epithelium and the MMTV-Wnt1 hyperplasias (40). In addition, we showed a decrease in Elf-5 expression in bigenic tumors compared to MMTV-Wnt1 tumors. Loss of Elf-5 leads to an increase in the CD61+ luminal progenitors and hyperactive Notch signaling (45, 46). Finally, previous data demonstrated that Wnt-mediated oncogenic conversion of breast epithelial cells is Notch-dependent (33). Taken together, these data support the hypothesis that dysregulation of Notch signaling with down-regulation of IGF-1R signaling leads to accelerated tumor development in the presence of active Wnt signaling. How the attenuation of IGF-1R signaling leads to dysregulation in Notch signaling is not entirely clear. Previous studies have demonstrated that the IGF-1R is a substrate of, and upregulated by, Notch1 signaling in T-ALL cells (47) and in lung adenocarcinomas (48). However, no studies to date have shown regulation of Notch signaling by IGF-1R activation.
A second potential mechanism for enhanced tumorigenesis of MMTV-Wnt1 tumors with decreased IGF-1R signaling is through enhanced IR signaling, in particular through the IR-A isoform [for review of IGF-1R/IR isoform signaling, see Ref. (49, 50)] (see Figure 1). Prior studies from the Yee laboratory demonstrated that down-regulation of IGF-1R signaling in a variety of breast cancer cell lines increases insulin sensitivity by increasing cell surface expression of holo-IR due to a reduction in hybrid IR/IGF-1R cell surface expression (51). Previous studies have shown that the overexpression of IR-A is increased in breast tumor samples (52). Furthermore, IR-A has been suspected as one of the possible mechanisms responsible for resistance to IGF-1R targeted therapies (53, 54).
Consistent with these findings, we found that the MMTV-Wnt1 tumors with decreased IGF-1R signaling have an increased IR-A:IR-B ratio, as well as increased expression of IGF-II, a high-affinity ligand for the IR-A. To determine if IGF-II signaling through the IR-A can activate canonical Wnt signaling, we stimulated IGF-1R null/IR-A overexpressing fibroblasts with IGF-II and analyzed protein levels of β-catenin, that is stabilized by canonical Wnt signaling (Figure 1). We found that levels of β-catenin are increased in a dose-dependent manner by IGF-II stimulation of IR-A expressing fibroblasts (40). The Wnt/β-catenin as well as IR-A signaling pathways have been implicated in stem cell and cancer stem cell renewal [for reviews, see Ref. (31, 55, 56)]. Although both pathways are expressed in similar tissues and seem to have overlapping actions, the interaction between these two pathways remains unclear. Our data suggest a novel role for IR-A in Wnt-mediated oncogenesis in the absence of IGF-1R signaling, and further support the hypothesis that inhibiting the IGF-1R may amplify this signaling cascade.
The Wnt pathway is commonly altered in basal-like or TNBC subtypes (37, 57). Our data suggest a complex interaction between IGF-1R/IR and Wnt pathway signaling and support the need for screening breast cancer patients for expression of Wnt pathway components in combination with IGF-1R as well as the IR-A:IR-B ratio and potentially IGF-II to better understand how inhibitors of these pathways might be best employed in different tumor subtypes. It is possible that inhibiting the IGF-1R or downstream signaling will lead to amplification of more undifferentiated tumors containing increased stem/progenitor populations with higher self-renewal potential through induction of Wnt and/or Notch pathway signaling. Dual inhibition of the IGF-1R/IR receptors or downstream signaling targets in the IR-A pathway along with Wnt pathway inhibitors may be beneficial in specific TNBCs. Understanding the mechanism(s) responsible for IR-A stimulation of the canonical Wnt pathway could prove helpful in designing therapeutic targets in subclasses of TNBCs.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Belfiore A, Frasca F. IGF and insulin receptor signaling in breast cancer. J Mammary Gland Biol Neoplasia (2008) 13:381–406. doi: 10.1007/s10911-008-9099-z
2. Lann D, Leroith D. The role of endocrine insulin-like growth factor-I and insulin in breast cancer. J Mammary Gland Biol Neoplasia (2008) 13:371–9. doi:10.1007/s10911-008-9100-x
3. Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer (2008) 8:915–28. doi:10.1038/nrc2536
4. Byrne C, Colditz GA, Willett WC, Speizer FE, Pollak M, Hankinson SE. Plasma insulin-like growth factor (IGF) I, IGF-binding protein 3, and mammographic density. Cancer Res (2000) 60:3744–8.
5. Stoll BA. Biological mechanisms in breast cancer invasiveness: relevance to preventive interventions. Eur J Cancer Prev (2000) 9:73–9. doi:10.1097/00008469-200004000-00002
6. Boone DN, Lee AV. Targeting the insulin-like growth factor receptor: developing biomarkers from gene expression profiling. Crit Rev Oncog (2012) 17:161–73. doi:10.1615/CritRevOncog.v17.i2.30
7. Yang Y, Yee D. Targeting insulin and insulin-like growth factor signaling in breast cancer. J Mammary Gland Biol Neoplasia (2012) 17:251–61. doi:10.1007/s10911-012-9268-y
8. Creighton CJ, Casa A, Lazard Z, Huang S, Tsimelzon A, Hilsenbeck SG, et al. Insulin-like growth factor-I activates gene transcription programs strongly associated with poor breast cancer prognosis. J Clin Oncol (2008) 26:4078–85. doi:10.1200/JCO.2007.13.4429
9. Bonneterre J, Peyrat JP, Beuscart R, Demaille A. Prognostic significance of insulin-like growth factor 1 receptors in human breast cancer. Cancer Res (1990) 50:6931–5.
10. Papa V, Gliozzo B, Clark GM, Mcguire WL, Moore D, Fujita-Yamaguchi Y, et al. Insulin-like growth factor-I receptors are overexpressed and predict a low risk in human breast cancer. Cancer Res (1993) 53:3736–40.
11. Fagan DH, Yee D. Crosstalk between IGF1R and estrogen receptor signaling in breast cancer. J Mammary Gland Biol Neoplasia (2008) 13:423–9. doi:10.1007/s10911-008-9098-0
12. Bartella V, De Marco P, Malaguarnera R, Belfiore A, Maggiolini M. New advances on the functional cross-talk between insulin-like growth factor-I and estrogen signaling in cancer. Cell Signal (2012) 24:1515–21. doi:10.1016/j.cellsig.2012.03.012
13. Schnarr B, Strunz K, Ohsam J, Benner A, Wacker J, Mayer D. Down-regulation of insulin-like growth factor-I receptor and insulin receptor substrate-1 expression in advanced human breast cancer. Int J Cancer (2000) 89:506–13. doi:10.1002/1097-0215(20001120)89:6<506::AID-IJC7>3.0.CO;2-F
14. Incassati A, Chandramouli A, Eelkema R, Cowin P. Key signaling nodes in mammary gland development and cancer: beta-catenin. Breast Cancer Res (2010) 12:213. doi:10.1186/bcr2723
15. Roarty K, Rosen JM. Wnt and mammary stem cells: hormones cannot fly wingless. Curr Opin Pharmacol (2010) 10:643–9. doi:10.1016/j.coph.2010.07.004
16. King TD, Suto MJ, Li Y. The Wnt/beta-catenin signaling pathway: a potential therapeutic target in the treatment of triple negative breast cancer. J Cell Biochem (2012) 113:13–8. doi:10.1002/jcb.23350
17. Nusse R, Varmus HE. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell (1982) 31:99–109. doi:10.1016/0092-8674(82)90409-3
18. Tsukamoto A, Grosschedl R, Guzman R, Parslow T, Varmus H. Expression of the int-1 gene in transgenic mice is associated with mammary gland hyperplasia and adenocarcinomas in male and female mice. Cell (1988) 55:619–25. doi:10.1016/0092-8674(88)90220-6
19. Li Y, Hively WP, Varmus HE. Use of MMTV-Wnt-1 transgenic mice for studying the genetic basis of breast cancer. Oncogene (2000) 19:1002–9. doi:10.1038/sj.onc.1203273
20. Li Y, Welm B, Podsypanina K, Huang S, Chamorro M, Zhang X, et al. Evidence that transgenes encoding components of the Wnt signaling pathway preferentially induce mammary cancers from progenitor cells. Proc Natl Acad Sci U S A (2003) 100:15853–8. doi:10.1073/pnas.2136825100
21. Liu BY, Mcdermott SP, Khwaja SS, Alexander CM. The transforming activity of Wnt effectors correlates with their ability to induce the accumulation of mammary progenitor cells. Proc Natl Acad Sci U S A (2004) 101:4158–63. doi:10.1073/pnas.0400699101
22. Lindvall C, Evans NC, Zylstra CR, Li Y, Alexander CM, Williams BO. The Wnt signaling receptor Lrp5 is required for mammary ductal stem cell activity and Wnt1-induced tumorigenesis. J Biol Chem (2006) 281:35081–7. doi:10.1074/jbc.M607571200
23. Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML, et al. Generation of a functional mammary gland from a single stem cell. Nature (2006) 439:84–8. doi:10.1038/nature04372
24. Woodward WA, Chen MS, Behbod F, Alfaro MP, Buchholz TA, Rosen JM. WNT/beta-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc Natl Acad Sci U S A (2007) 104:618–23. doi:10.1073/pnas.0606599104
25. Vaillant F, Asselin-Labat ML, Shackleton M, Forrest NC, Lindeman GJ, Visvader JE. The mammary progenitor marker CD61/beta3 integrin identifies cancer stem cells in mouse models of mammary tumorigenesis. Cancer Res (2008) 68:7711–7. doi:10.1158/0008-5472.CAN-08-1949
26. Kim S, Goel S, Alexander CM. Differentiation generates paracrine cell pairs that maintain basaloid mouse mammary tumors: proof of concept. PLoS One (2011) 6:e19310. doi:10.1371/journal.pone.0019310
27. Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med (2009) 15:907–13. doi:10.1038/nm.2000
28. Molyneux G, Geyer FC, Magnay FA, Mccarthy A, Kendrick H, Natrajan R, et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell (2010) 7:403–17. doi:10.1016/j.stem.2010.07.010
29. Proia TA, Keller PJ, Gupta PB, Klebba I, Jones AD, Sedic M, et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell (2011) 8:149–63. doi:10.1016/j.stem.2010.12.007
30. Howe LR, Brown AM. Wnt signaling and breast cancer. Cancer Biol Ther (2004) 3:36–41. doi:10.4161/cbt.3.1.561
31. Alexander CM, Goel S, Fakhraldeen SA, Kim S. Wnt signaling in mammary glands: plastic cell fates and combinatorial signaling. Cold Spring Harb Perspect Biol (2012) 4. doi:10.1101/cshperspect.a008037
32. Milovanovic T, Planutis K, Nguyen A, Marsh JL, Lin F, Hope C, et al. Expression of Wnt genes and frizzled 1 and 2 receptors in normal breast epithelium and infiltrating breast carcinoma. Int J Oncol (2004) 25:1337–42. doi:10.3892/ijo.25.5.1337
33. Ayyanan A, Civenni G, Ciarloni L, Morel C, Mueller N, Lefort K, et al. Increased Wnt signaling triggers oncogenic conversion of human breast epithelial cells by a Notch-dependent mechanism. Proc Natl Acad Sci U S A (2006) 103:3799–804. doi:10.1073/pnas.0600065103
34. Schlange T, Matsuda Y, Lienhard S, Huber A, Hynes NE. Autocrine WNT signaling contributes to breast cancer cell proliferation via the canonical WNT pathway and EGFR transactivation. Breast Cancer Res (2007) 9:R63. doi:10.1186/bcr1769
35. Klopocki E, Kristiansen G, Wild PJ, Klaman I, Castanos-Velez E, Singer G, et al. Loss of SFRP1 is associated with breast cancer progression and poor prognosis in early stage tumors. Int J Oncol (2004) 25:641–9. doi:10.3892/ijo.25.3.641
36. Virmani AK, Rathi A, Sathyanarayana UG, Padar A, Huang CX, Cunnigham HT, et al. Aberrant methylation of the adenomatous polyposis coli (APC) gene promoter 1A in breast and lung carcinomas. Clin Cancer Res (2001) 7:1998–2004.
37. Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest (2011) 121:2750–67. doi:10.1172/JCI45014
38. Curtin JC, Lorenzi MV. Drug discovery approaches to target Wnt signaling in cancer stem cells. Oncotarget (2010) 1:552–66.
39. Takebe N, Harris PJ, Warren RQ, Ivy SP. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat Rev Clin Oncol (2011) 8:97–106. doi:10.1038/nrclinonc.2010.196
40. Rota LM, Albanito L, Shin ME, Goyeneche CL, Shushanov S, Gallagher EJ, et al. IGF1R Inhibition in mammary epithelia promotes canonical Wnt signaling and Wnt1-driven tumors. Cancer Res (2014) 74:1–12. doi:10.1158/0008-5472.CAN-14-0970
41. Bonnette SG, Hadsell DL. Targeted disruption of the IGF-I receptor gene decreases cellular proliferation in mammary terminal end buds. Endocrinology (2001) 142:4937–45. doi:10.1210/endo.142.11.8500
42. Carboni JM, Lee AV, Hadsell DL, Rowley BR, Lee FY, Bol DK, et al. Tumor development by transgenic expression of a constitutively active insulin-like growth factor I receptor. Cancer Res (2005) 65:3781–7. doi:10.1158/0008-5472.CAN-04-4602
43. Jones RA, Moorehead RA. The impact of transgenic IGF-IR overexpression on mammary development and tumorigenesis. J Mammary Gland Biol Neoplasia (2008) 13:407–13. doi:10.1007/s10911-008-9097-1
44. Liu BY, Soloviev I, Huang X, Chang P, Ernst JA, Polakis P, et al. Mammary tumor regression elicited by Wnt signaling inhibitor requires IGFBP5. Cancer Res (2012) 72:1568–78. doi:10.1158/0008-5472.CAN-11-3668
45. Chakrabarti R, Hwang J, Andres Blanco M, Wei Y, Lukacisin M, Romano RA, et al. Elf5 inhibits the epithelial-mesenchymal transition in mammary gland development and breast cancer metastasis by transcriptionally repressing Snail2. Nat Cell Biol (2012) 14:1212–22. doi:10.1038/ncb2607
46. Chakrabarti R, Wei Y, Romano RA, Decoste C, Kang Y, Sinha S. Elf5 regulates mammary gland stem/progenitor cell fate by influencing notch signaling. Stem Cells (2012) 30:1496–508. doi:10.1002/stem.1112
47. Medyouf H, Gusscott S, Wang H, Tseng JC, Wai C, Nemirovsky O, et al. High-level IGF1R expression is required for leukemia-initiating cell activity in T-ALL and is supported by Notch signaling. J Exp Med (2011) 208:1809–22. doi:10.1084/jem.20110121
48. Eliasz S, Liang S, Chen Y, De Marco MA, Machek O, Skucha S, et al. Notch-1 stimulates survival of lung adenocarcinoma cells during hypoxia by activating the IGF-1R pathway. Oncogene (2010) 29:2488–98. doi:10.1038/onc.2010.7
49. Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev (2009) 30:586–623. doi:10.1210/er.2008-0047
50. Janssen JA, Varewijck AJ. IGF-IR targeted therapy: past, present and future. Front Endocrinol (2014) 5:224. doi:10.3389/fendo.2014.00224
51. Zhang H, Pelzer AM, Kiang DT, Yee D. Down-regulation of type I insulin-like growth factor receptor increases sensitivity of breast cancer cells to insulin. Cancer Res (2007) 67:391–7. doi:10.1158/0008-5472.CAN-06-1712
52. Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A, et al. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol (1999) 19: 3278–88.
53. Haluska P, Shaw HM, Batzel GN, Yin D, Molina JR, Molife LR, et al. Phase I dose escalation study of the anti insulin-like growth factor-I receptor monoclonal antibody CP-751,871 in patients with refractory solid tumors. Clin Cancer Res (2007) 13:5834–40. doi:10.1158/1078-0432.CCR-07-1118
54. Avnet S, Sciacca L, Salerno M, Gancitano G, Cassarino MF, Longhi A, et al. Insulin receptor isoform A and insulin-like growth factor II as additional treatment targets in human osteosarcoma. Cancer Res (2009) 69:2443–52. doi:10.1158/0008-5472.CAN-08-2645
56. Malaguarnera R, Belfiore A. The emerging role of insulin and insulin-like growth factor signaling in cancer stem cells. Front Endocrinol (2014) 5:10. doi:10.3389/fendo.2014.00010
Keywords: IGF, Wnt, beta-catenin, insulin receptor, mouse models, IGF-1R, IGF-II, TNBC
Citation: Rota LM and Wood TL (2015) Crosstalk of the insulin-like growth factor receptor with the Wnt signaling pathway in breast cancer. Front. Endocrinol. 6:92. doi: 10.3389/fendo.2015.00092
Received: 15 March 2015; Accepted: 17 May 2015;
Published: 09 June 2015
Edited by:
Antonino Belfiore, University Magna Graecia of Catanzaro, ItalyReviewed by:
Marcello Maggiolini, University of Calabria, ItalyCopyright: © 2015 Rota and Wood. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Teresa L. Wood, Department of Pharmacology, Physiology and Neuroscience, New Jersey Medical School and Cancer Center, Rutgers University, 205 South Orange Avenue, Newark, NJ 07101, USA,dGVycmkud29vZEBydXRnZXJzLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.