
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 07 May 2015
Sec. Cellular Endocrinology
Volume 6 - 2015 | https://doi.org/10.3389/fendo.2015.00058
This article is part of the Research TopicEndocrine Disrupters: Defining the new insights!View all 7 articles
 Virginie Rouiller-Fabre1,2,3*
Virginie Rouiller-Fabre1,2,3* Marie Justine Guerquin1,2,3
Marie Justine Guerquin1,2,3 Thierry N’Tumba-Byn1,2,3
Thierry N’Tumba-Byn1,2,3 Vincent Muczynski1,2,3Delphine Moison1,2,3
Vincent Muczynski1,2,3Delphine Moison1,2,3 Sophie Tourpin1,2,3Sébastien Messiaen1,2,3René Habert1,2,3Gabriel Livera1,2,3
Sophie Tourpin1,2,3Sébastien Messiaen1,2,3René Habert1,2,3Gabriel Livera1,2,3
During the last decades, many studies reported that male reproductive disorders are increasing among humans. It is currently acknowledged that these abnormalities can result from fetal exposure to environmental chemicals that are progressively becoming more concentrated and widespread in our environment. Among the chemicals present in the environment (air, water, food, and many consumer products), several can act as endocrine disrupting compounds (EDCs), thus interfering with the endocrine system. Phthalates, bisphenol A (BPA), and diethylstilbestrol (DES) have been largely incriminated, particularly during the fetal and neonatal period, due to their estrogenic and/or anti-androgenic properties. Indeed, many epidemiological and experimental studies have highlighted their deleterious impact on fetal and neonatal testis development. As EDCs can affect many different genomic and non-genomic pathways, the mechanisms underlying the adverse effects of EDC exposure are difficult to elucidate. Using literature data and results from our laboratory, in the present review, we discuss the role of classical nuclear receptors (genomic pathway) in the fetal and neonatal testis response to EDC exposure, particularly to phthalates, BPA, and DES. Among the nuclear receptors, we focused on some of the most likely candidates, such as peroxisome-proliferator activated receptor (PPAR), androgen receptor (AR), estrogen receptors (ERα and β), liver X receptors (LXR), and small heterodimer partner (SHP). First, we describe the expression and potential functions (based on data from studies using receptor agonists and mouse knockout models) of these nuclear receptors in the developing testis. Then, for each EDC studied, we summarize the main evidences indicating that the reprotoxic effect of each EDC under study is mediated through a specific nuclear receptor(s). We also point-out the involvement of other receptors and nuclear receptor-independent pathways.
During the last 50 years, the frequency of various male reproductive disorders, such as cryptorchidism, hypospadias, testicular cancer, and low sperm count, has progressively increased. Many clinical, epidemiological, and experimental studies suggest that these disorders arise during fetal development (1, 2).
The fetal period is critical for proper testis development. Indeed, gametogenesis and steroidogenesis, the two major functions of the testis, are set up during this period and their proper onset is fundamental for the adult reproductive function. For instance, the number of germ cells formed during fetal life will strongly affect adult fertility. In mutant mice with pronounced germ cell loss during fetal life, adult fertility is altered (3, 4). Similarly, exposure of mouse perinatal testes to gamma rays decreases the sperm counts at sexual maturity (5). Concerning steroidogenesis, fetal Leydig cells produce testosterone that is responsible for the masculinization of the male genital tract and external genitalia (6, 7). Importantly, androgens must act during a specific period, called the «masculinization programing window»that predates genital masculinization. Genital development in the rat is programed between 15.5 and 18.5 day post-coitum (dpc). This corresponds to the 13.5–17.5 dpc period in the mouse and to the 6.5–14 gestational week (GW) time in humans (8). Fetal androgens are also required for the proper development of adult Leydig stem cells (9) and for testis descent, which depends also on Insulin-like 3 (INSL3), another hormone produced by fetal Leydig cells (10, 11).
It has been hypothesized that male reproductive system abnormalities could be related to the massively increased presence in the environment of natural and synthetic chemicals during the last decades (1, 12, 13). Among the chemicals present in air, water, food, and in a variety of consumer products, many can interfere with the endocrine system and therefore are called endocrine disrupting compounds (EDCs). EDCs can affect the production, release, transport, metabolism, binding or elimination of natural hormones (2). Among the EDCs present in the environment, phthalates, bisphenol A (BPA), and diethylstilbestrol (DES) have been chiefly incriminated, particularly during the fetal and neonatal period due to their estrogenic and/or anti-androgenic properties (14–17).
A large amount of data shows the effects of EDCs on testis development. EDCs can act by affecting different tissue-specific genomic and non-genomic pathways. It is now crucial to identify which nuclear receptors and downstream signaling pathways are altered by exposure to DES, BPA, or phthalates in human and rodent fetal gonads.
In the present study, we highlight the involvement of some classical nuclear receptors in the response of fetal and neonatal testes to DES, phthalates, and BPA exposure. We focused particularly on peroxisome-proliferator activated receptor (PPAR), androgen receptor (AR), estrogen receptors (ER1 and 2), liver X receptor (LXR), and small heterodimer partner (SHP) due to their affinity or involvement in mediating some EDC effects (Table 1).
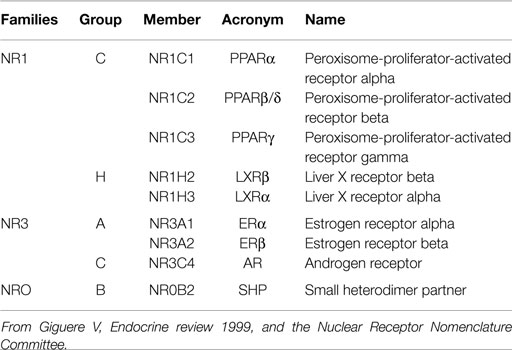
Table 1. Nomenclature and classification of the chosen receptors.
Here, we describe the expression profile of these nuclear receptors in the male gonad based on literature and personal data (Figure 1).
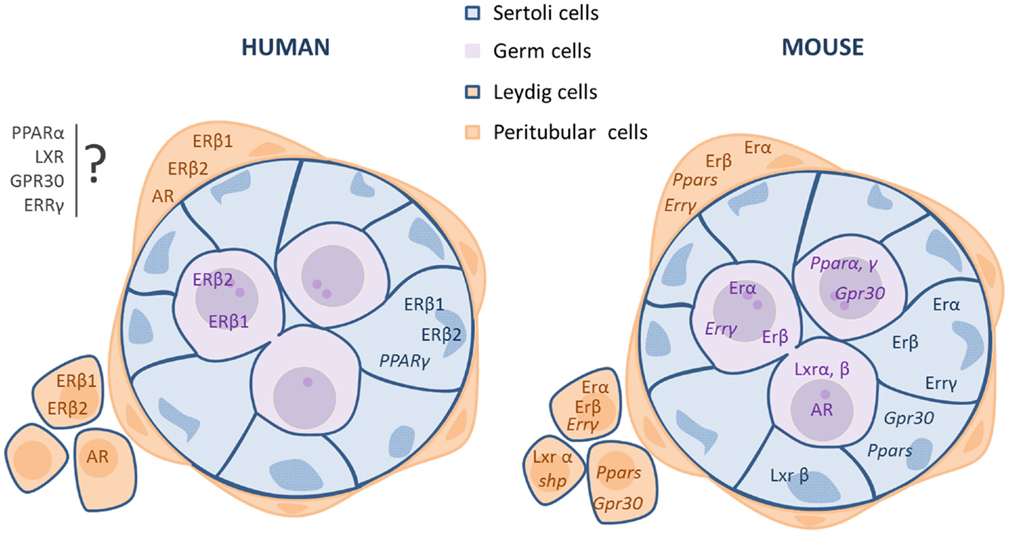
Figure 1. Localization of the receptors involved in EDCs effects in fetal or neonatal mammalian testes (human and rodents). The mRNA (italic type) or protein (roman type) expression of the receptors is indicated in each cell type: germ, Sertoli, and interstitial (Leydig and peritubular cells) cells. *Potential localization in somatic cells.
Estrogen action is mainly mediated through the activation of two specific estrogen receptors (ER), ERα (ESR1/NR3A1) and ERβ (ESR2/NR3A2), in target cells (18–21). The expression of these two receptors in the testis varies according to the species and developmental stage (Figure 1).
In rat fetal testes, ERβ protein is present in all cell types, whereas ERα is expressed only in Leydig cells (22). In the mouse, immunohistochemical studies showed that ERα is expressed in undifferentiated gonads as early as 10.5 dpc and then is localized in fetal Leydig cells until birth, while it is absent in germ cells during the fetal period (23, 24). ERα signaling is functional in the fetal testis, as ERα knockout leads to an increase in testosterone production by mouse fetal testes as soon as 13.5 dpc (25). ERβ is expressed in rat and mouse testis germ cells (Figure 1) from 14.5 dpc (26). In the rat, it is also expressed in Sertoli and Leydig cells from 16.5 dpc (26–28). In sorted cells from mouse fetal testes, we detected that ERα mRNA level is higher than that of ERß. Interestingly, ERα transcripts are mainly detected in germ cells, but also in interstitial and Sertoli cells (Figure 1). ERβ is mainly expressed in interstitial cells and to a lower extent in Sertoli and germ cells (Figure 1).
In humans, ER α and β expression in the testis have been assessed mainly during the second quarter of pregnancy (Figure 1). These studies highlighted the presence of two ERß variants (ERß1 and ERß2) (29, 30) that are expressed (mRNA) in Sertoli and germ cells and also in some interstitial cells (29, 31, 32). Conversely, ERβ1 protein is specifically detected in Sertoli and interstitial cells, whereas ERβ2 protein is only detected in germ cells (32). Concerning ERα expression, some studies did not detect neither mRNA nor protein expression in human fetal testis (31–33), while others detected both (29, 34).
Estrogen effects in the testis have been elucidated using genetically modified mice that lack ERα (ERαKO), ERβ (ERβKO) (35), or both (ERαβKO), or the CYP19/aromatase gene (ArKO) (36, 37). ERαKO, ERβKO, ERαβKO, and ArKO male mice show reproductive disorders. ERαKO and ArKO mice are sterile due to epididymal fluid reabsorption deficiency and spermatogenesis disorders, respectively (14, 38). Endogenous estrogens also play an important role during fetal and neonatal testis development (25, 39). ERβ knock out induces a 50% increase in the number of gonocytes observed after birth, due to increased proliferation and decreased apoptosis (39), but no changes during fetal life. Conversely, ERα gene deletion does not modify the number of gonocytes, but increases testosterone production during fetal life from 13.5 dpc (25). Thus, ERβ is involved in the control of neonatal gametogenesis, whereas ERα regulates fetal and neonatal steroidogenesis.
Estrogens play a vital role in the control of human reproductive functions as well. Studies in patients with ERα or CYP19 inactivating mutations suggest a role of estrogens in human male fertility (40).
Androgen actions are mediated through the AR (Figure 1). Androgen and AR roles in the masculinization of male genitals are well known. AR mutations cause androgen insensitivity syndrome (AIS), possibly the most frequently described hormone insensitivity syndrome. The most serious AIS phenotype is testicular feminization (Tfm) where genetic males are females in appearance. In adult life, androgen action on seminiferous tubules is essential for normal spermatogenesis and fertility and most evidences suggest that this effect is mediated by Sertoli cells (41). In adult testis, AR is expressed in Sertoli cells in almost all species tested, but also in peritubular cells, Leydig cells, and spermatids (42).
In the mouse, AR mRNA and protein are expressed in germ cells during fetal life (43), when gonocytes are the main testis cell type physiologically controlled by endogenous androgens. Leydig cells are largely independent of endogenous androgens during fetal development. On the other hand, peritubular myoid and Sertoli cells seem to become androgen-dependent mainly in the latest part of fetal development (44). However, during late fetal life, androgen positive effect on Sertoli cell proliferation is probably indirect because AR is expressed in Sertoli cells only after birth (45). In human fetal testis, AR is expressed in peritubular and Leydig cells, but not in germ or Sertoli cells (46) (Figure 1).
Few studies exist on androgen and AR functions in fetal testis. Nevertheless, alterations of testis functions resulting from exposure to anti-androgenic EDCs during fetal testis development suggest a key role for androgens and AR during this period. AR knock out (and thus decreased fetal androgen signaling) in mice leads to a reduction of intratesticular testosterone level and of the number of adult Leydig stem cell by 40% at birth to adulthood (9). Similarly, the analysis of a mouse model in which AR was selectively invalidated in Leydig cells from fetal life onward and of patients with complete AIS showed that androgen autocrine action is essential for Leydig cell maturation and function (47). Finally, increased prenatal exposure to androgens alters the development and function of Leydig cells at a time when androgen production is paramount for male development (48). Similarly, in male lambs exposed prenatally to an excess of testosterone, the number of Sertoli cells is increased and this effect could explain the testicular dysfunction observed in adult rams (49).
Concerning androgen role in germ cells, AR deficiency results in increased gonocyte numbers during fetal life due to higher proliferative activity in Tfm mice. Conversely, gonocyte proliferation is decreased by the addition of DES in fetal testis organotypic cultures (43).
The PPAR family includes three members: PPARα (NR1C1), PPARβ/δ (NR1C2), and PPARγ (NR1C3). They are mostly involved in lipid and cholesterol metabolism and in fatty acid synthesis (50). They are also present in the female and male reproductive organs (51).
In adult rat testis, the three PPAR members are located in the interstitial space and in the seminiferous cords (52) (Figure 1). PPARβ/δ is the most expressed isoform in the testis, followed by PPARα. Although PPARγ protein is barely detectable (52), PPARγ mRNA expression was detected mostly in Sertoli cells (53), but also in spermatocytes (54) and in fetal germ cells (55). In mouse fetal testes, PPARα is mainly expressed in interstitial cells, but also in Sertoli and germ cells. PPARγ is mostly expressed in Sertoli cells and then in interstitial and germ cells (personal data Figure 1). PPARγ expression is higher in testicular cancer cells than in normal cells (56).
For human fetal testes, we developed a novel flow cytometric cell sorting approach based on the staining of M2A, a trans-membrane antigen that is expressed in 90–95% of germ cells during the first trimester of pregnancy. The M2A-positive cell fraction expresses only germ cell markers, whereas the M2A-negative fraction expresses exclusively somatic cell markers. We have shown that PPARγ mRNA is expressed only in M2A-positive cells (55) (Figure 1).
Concerning PPAR implication in the reproductive function, PPARα deletion in mice did not have any effect on their viability and fertility (57). Furthermore, gestational exposure to a PPARα agonist did not reduce fetal testicular testosterone production (58). However, PPARα and PPARβ/δ have been recently identified as regulators of Sertoli cell metabolism (59). As PPARγ knockout mice die at 10 dpc (60), its effect on the reproductive functions could not be tested. The results of microarray analyses suggest that PPARγ could play an important role in regulating the expression of key lipid metabolic genes in Sertoli cells during postnatal development (61).
The liver X receptors LXRα (NR1H3) and LXRβ (NR1H2) belong to a subclass of nuclear receptors that form obligate heterodimers with retinoid X receptors (RXRs) and that are activated upon binding of their ligands (oxysterols) (62). They are mainly involved in the regulation of cholesterol and fatty acid homeostasis, but also in glucose homeostasis, immunity, skin development, and brain functions (63).
In the mouse, quantitative PCR analysis has shown that LXRα is expressed in Leydig cells, LXRβ in Sertoli cells, and both in germ cells (64) (Figure 1). In human fetal testes, we demonstrated, using our cell sorting approach, that LXRα mRNA is expressed both in M2A-negative and in M2A-positive cells (55) (Figure 1).
Recently, their physiological role in the regulation of male reproductive functions has been elucidated using LXRα and LXRβ single and double knockout mouse models (64). Mice in which LXRα or LRXβ has been invalidated show reduced fertility throughout life, whereas double knockout mice show reduced fertility at 5 months which progresses to complete sterility by the age of 9 months (65). Moreover, LXRs seem to have distinct roles in sustaining spermatogenesis (66, 67). Indeed, germ cell apoptosis is increased in 3.5-month-old LXRα-deficient mice, whereas the number of proliferating germ cells is reduced in LXRβ-deficient mice compared to wild type animals. Therefore, deregulation of one of the LXR pathways can be easily related to the disruption of the fine equilibrium between germ cell proliferation and apoptosis, leading to alterations of the reproductive function. For instance, a recent study has reported a positive correlation between reduced number of germ cells and decreased LXR mRNA level in testis biopsies of patients with various degrees of azoospermia (68). Furthermore, in 2.5-month-old LRXα-deficient mice, testosterone production is significantly lower than in wild type controls and the level of 3β-hydroxysteroid dehydrogenase isomerase mRNA (an enzyme that plays a crucial role in the biosynthesis of hormonal steroids) is significantly reduced in these mice (65, 66).
Small heterodimer partner (SHP/NR0B2) is a member of the nuclear receptor superfamily and is classified as an “orphan” receptor, because its ligand has not been identified yet. This receptor is mainly known for its role in liver and in the control of bile acid homeostasis (69, 70).
Small heterodimer partner expression in the testis is very low (62). In a study performed with purified cells (using laser microdissection), SHP mRNA was mostly expressed in interstitial cells of adult mouse testis. SHP is also transiently expressed in the tubular cells of seminiferous tubules during early postnatal development and its expression declines as the mice reach sexual maturity (71, 72) (Figure 1). Interestingly, SHP is not detectable in human fetal testes (55).
Small heterodimer partner deletion in mice results in higher testosterone production due to enhanced expression of steroidogenic genes, such as steroidogenic acute regulatory protein (StAR) and CYP11A1 (71). SHP knockout mice also show earlier differentiation of germ cells compared with control littermates, as suggested by the number of tubules with elongated spermatids.
The retinoic acid (RA) metabolic pathway is affected by SHP knock out as indicated by the altered expression of several RA receptor (RAR) target genes in these mice. Among them, Stra8, a key gene in meiotic initiation (73, 74), is up-regulated, possibly as a result of the increased RA expression in the testes of NR0B2−/− mice (71).
The so-called “estrogen hypothesis,” which was first proposed 20 years ago, suggests that the growing incidence of male reproductive abnormalities in humans could be related to increased estrogen exposure (75). Over the years, this hypothesis has been supported by a large number of epidemiological and experimental studies.
Diethylstilbestrol is a synthetic estrogen and a recognized EDC. Between 1945 and 1971, DES was administered to pregnant women to prevent miscarriage or premature delivery. This was associated with an increased incidence of reproductive tract abnormalities in their male and female offspring (75). Specifically, several studies have reported alterations in sperm quality and higher incidence of genital malformations, cryptorchidism, and testicular cancer than in untreated populations (76, 77). DES seems to have a negative effect on sperm count when administered at high dose during the first semester of pregnancy (78).
Similarly, experimental studies support the estrogen hypothesis (14), in utero, rodents exposed to DES during development have abnormal testicular histology and altered adult male fertility (14, 79). Also, in utero exposure to DES of 9 and 10 dpc rat embryos can advance testicular development, alter the differentiation of gonocytes and Sertoli cells, and cause fetal Leydig cell hyperplasia from 16 dpc onward (33, 80–82). In the same way, in vitro incubation of rat fetal testes with DES reduces testosterone production and the number of gonocytes by inhibiting proliferation and increasing apoptosis (83, 84). Likewise, in a mouse fetal testis organ culture system, DES exposure reduces testosterone production (25). In utero exposure to DES also alters mouse testis development by decreasing testosterone levels in testes and reducing the expression of StAR (85) (Table 2).
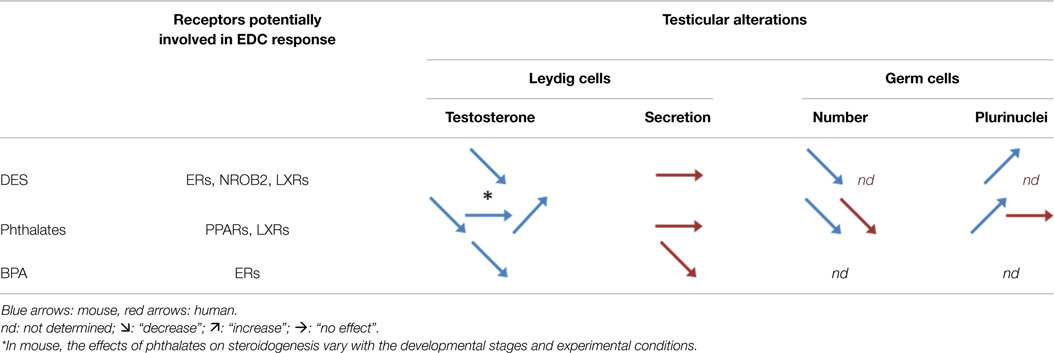
Table 2. Involvement of nuclear receptors in EDCs effects in human and mouse testes.
Surprisingly (relative to the epidemiological data), testosterone secretion by cultured human fetal testes is barely affected by DES exposure, while a strong effect is observed in mouse testes (33, 55) (Table 2).
DES and ERs. Diethylstilbestrol exerts its anti-androgen effects mainly through classical ER signaling, particularly via ERα (25, 86). In an organ culture system of mouse fetal testes, the reduction in testosterone production observed following DES exposure in wild type testes does not occur in ERα-deficient mice (25). Similarly, INSL3 gene expression and testis descent are not affected by in utero exposure to DES in ERαKO mice, whereas ERβ invalidation does not protect from DES effect (86).
DES and ARs and PPARs. To our knowledge, there is no data showing the direct involvement of AR or PPARs in DES testis effects. However, an indirect action of DES cannot be excluded in relation with the reduction of testosterone secretion observed in vitro in rodent testes incubated with DES (14).
DES and LXRs. Some studies have linked estrogens and LXRs in breast and in mouse adipose tissue (87, 88). In the testis, LXRs could partially interfere with DES effects. Daily treatment with DES from day 1 to day 5 after birth induces an important increase in cell apoptosis in LXR-deficient mice at day 10 compared to wild type animals. Likewise, LXRs modify the neonatal effects of DES on the expression of Leydig and Sertoli cell markers (67). However, whether LXRs have a protective effect against or contribute to DES effects remains unclear.
DES and SHP. Treatment with DES promotes SHP mRNA accumulation in the testes of wild type SHP male mice (NR0B2+/+) at postnatal day 10 (P10). Moreover, neonatal DES exposure induces apoptosis, in P10 NR0B2+/+ mice, without any effect on cell proliferation. Conversely, DES does not have any effect on apoptosis in the testes of NR0B2L−/L− males, suggesting that SHP inactivation protects against DES effects. This seems to be germ cell-specific because DES treatment drastically decreases intratesticular testosterone in both NR0B2L−/L− and NR0B2L−/L− males (89). Interestingly, SHP, mediating the deleterious effects of DES in mice, is not detectable in human fetal testes, and incubation with DES does not modify testosterone production by human fetal testes in culture (90). SHP absence in human fetal testes could be an additional explanation for their lack of sensitivity to DES.
Phthalates (phthalic acid esters) are industrial chemicals commonly found in many consumer products, such as soaps, shampoos, cosmetics, and hairsprays. They are also used in flexible plastics, such as food and beverage packaging, children’s toys, and biomedical equipment (91). Phthalates are not covalently bound to plastic products and therefore may leak out. Di-2-ethylhexyl phthalate (DEHP) is the most produced phthalate. In the gut, liver, and blood, DEHP is rapidly hydrolyzed by esterases into its monoester metabolite mono-2-ethylhexyl phthalate (MEHP), considered to be the active metabolite and a recognized active testicular toxicant (55, 92–95), although DEHP, the parental compound, is also suspected to affect steroidogenesis (93).
Epidemiological studies in humans did not clearly prove the association between masculinization defects and phthalate exposure during fetal life (96, 97). Conversely, experimental studies in rats have shown that phthalates (di-butyl phthalate, DBP) administered during fetal life in vivo reduce testosterone production (98) (Table 2). This effect could be due to a decrease in Leydig stem/progenitor cells (9).
Surprisingly, MEHP does not affect in vitro testosterone production in human fetal testis explants (94). This was surprising because phthalates are considered to be anti-androgenic compounds based on their in vivo inhibitory action on testosterone production in the rat (7, 16, 99, 100). The recent observations that DBP decreases the steroidogenic activity of rat fetal testes, but not of human fetal testes, grafted into a mouse or rat host (101–103) definitively confirm that phthalates are not anti-androgenic compounds for human fetal testis (Table 2).
Phthalates also impair gonocyte development in the rat (104, 105) and increase gonocyte apoptosis in vitro in rat (93) and mouse fetal testes (95). Using an organotypic culture system, we demonstrated that MEHP also induces germ cell apoptosis in human fetal testes (94) after only 3 days of culture and at doses (10−5M) in the range of human exposure. In vitro, MEHP induces the appearance of multinucleated gonocytes (MNGs) in mouse and rat, but not in human fetal testis explants. However, exposure of human fetal testis xenografts to 100–500 mg/kg of DBP significant increases MNG content compared with untreated control (101).
Phthalates and PPARs. Peroxisome-proliferator activated receptor activation has been widely linked to the adverse effects of exposure to phthalates in adult mice (106). PPAR role in phthalate toxicity was first demonstrated in the liver (107).
Several studies using reporter genes for the different PPARs showed that phthalates can directly activate PPARs, particularly PPARα and PPARγ (53, 108).
In vitro transactivation assays demonstrated that MEHP can activate human and rodent PPARα and PPARγ (109). However, in PPARα knockout mice, the deleterious effects of DEHP exposure on male genitals were only partially blocked, suggesting that some of the toxicological effects of phthalate esters are mediated also by other nuclear receptors (57, 110).
A large risk assessment study demonstrated PPAR involvement in di-isobutyl phthalate (DiBP) toxic effects in the testis (111). Particularly, DEHP or DiBP treatment between 7 and 21 dpc induces an increase in PPARα gene expression and PPARγ protein expression in Leydig cells of fetal rat testes (112, 113). Moreover, DEHP induces apoptosis in adult rat testes via PPARγ and the ERK1/2 pathway (114). We demonstrated that MEHP (55, 94, 95) and PPARα or PPARγ agonists (unpublished data) have similar apoptotic effects in mouse and human fetal germ cells.
Phthalates and ERs. Phthalate actions in the developing testis seem independent of ER signaling. A study performed in our laboratory showed that the effects of phthalates on steroidogenesis vary with the developmental stage. Conversely, the strong deleterious effect of phthalates on germ cells is present during the active phases of gonocyte development and thus is unaffected by the steroidogenic status. Moreover, phthalate effects were comparable in testes from ERαKO or ERβKO mice and wild type controls (95). Interestingly, it has been suggested that the onset of dysgenesis in fetal rat testes exposed to phthalates may be mediated by estradiol (115).
Phthalates and ARs. Studies performed in our laboratory have shown that after MEHP treatment, (MNGs are similarly increased in testes from AR-deficient mice (Tfm) and wild type controls. Furthermore, incubation with MEHP for 3 days reduces the number of gonocytes by ∼40–50% in cultured 18.5 dpc testes from both wild type and Tfm mice. These results demonstrate that MEHP reduces the number of germ cells independently of the AR pathways (95).
Phthalates and LXRs. LXRs have also been related to the deleterious effect of exposure to environmental pollutants, particularly phthalates, during fetal life. Exposure of human fetal testis to MEHP during the first trimester of pregnancy leads to up-regulation of LXRα mRNA expression specifically in testis somatic cells, but not in germ cells (55). This effect is associated with increased transcription of Sterol Response Element Binding Protein 1c (SREBP1c) and downstream effectors involved in cholesterol and lipid synthesis and lipid accumulation in somatic cells. Somatic cells support the testis architecture that is essential for maintaining germ cell development. Therefore, the MEHP-induced modulation of LXRα expression specifically in these cells could lead to a deregulation of cell interactions, ultimately increasing germ cell apoptosis and reducing their number (94, 116).
Phthalates and SHP. To our knowledge, there is no data on SHP involvement in phthalate testis effects. However, LXRs can be modulated by SHP (89), thus suggesting a potential complex mechanism of EDC effect through several nuclear receptors.
Bisphenol A [BPA, 2,2-Bis (4-hydroxyphenol) propane] is one of the most studied EDCs. BPA was first synthesized by Dianin in 1891 and its estrogenic activity was discovered in 1936 (117). It is, therefore, one of the oldest synthetic compounds known for its endocrine activity, although DES was preferred because of its stronger estrogenic activity.
Bisphenol A is widely used as a monomer for the industrial production by polymerization of polycarbonate plastics (72%) that are used in a variety of common products (optical, media, automotive, electrical and electronics, housewares and appliances, construction, medical, packaging…). BPA is also used (21%) as an essential component of epoxy resins that are mainly employed to coat the inner surface of food and beverage metallic cans (118). Lastly, BPA is used as anti-oxidant or inhibitor of polymerization in some plasticizers, polyvinyl chloride, and in thermal cash register paper (119).
BisphenolA can leach into the content of food containers made of polycarbonate plastic or coated with epoxy resins and then be ingested. This is the main source of human contamination, although its ubiquitous distribution leads also to contamination from dermal exposure through the skin, especially from thermal paper, and from inhalation of household dusts.
Growing evidence from research on laboratory animals, wildlife, and humans supports the view that BPA has an endocrine disrupting effect and adversely affects male reproductive function (100, 120).
In humans, most studies reported the association between masculinization defects and BPA exposure during fetal life. In China, sons of workers who were occupationally exposed to BPA during pregnancy showed shorter anogenital distance (AGD) (121). In the general Korean population, BPA level in the plasma of newborn boys with hypospadias was seven times higher than in newborns without hypospadias, and the difference was statistically significant (122). Lastly, although no BPA increase was first detected in newborn boys with undescended testes in France (123), recent data from this group showed a negative correlation between the cord blood concentrations of BPA and Insulin like 3 (INSL3) (124).
The in vivo effects of BPA on fetal Leydig cell function in rodents are not clear. Some studies reported an inhibitory effect of BPA on plasma testosterone at birth (125) or on the AGD in male pups (126), but others did not (127–130). Recently, using an in vitro organotypic culture system, we analyzed the testosterone production of mouse, rat, and human fetal testes in response to various doses of BPA. At high doses, BPA reduces testosterone production in all three species. However, only human fetal testes are sensitive to lower doses (10−8M). Furthermore, BPA treatment reduces INSL3 mRNA level in human testes (by more than 20%), but not in rat and mouse testes (17, 90) (Table 2). This might explain the difficulty to observe a masculinization defect associated with BPA exposure in rodents (above).
Very few studies have investigated in detail BPA effects on fetal germ cells. Exposure in utero to low BPA concentrations by daily oral administration to pregnant female rats induces a decrease in litter size and in sperm number and motility in the adult progeny. It is important to note that these effects are maintained in the subsequent generations (F2, F3), suggesting a reprograming of germ cells by (genetic/epigenetic) mechanisms that persists well beyond the initial exposure (131). A potential explanation is that BPA exposure could affect DNA methylation of imprinted genes in fetal mouse germ cells (132).
BPA and ERs. Using in vitro bioassays based on competitive binding to nuclear receptors, reporter gene expression, and cell proliferation assessment, it has been shown that BPA efficiently activates both ERs (133).
Bisphenol A binds to ERα and ERß, but its affinity for these receptors is weak (∼1000-fold lower than estradiol) (134). This low affinity and the non-detectable expression of ERα in human fetal testes (32, 33, 135) suggest that ERα is not involved in BPA effect in humans. This is probably true also for the mouse species because the negative effect of BPA on mouse testis steroidogenesis is maintained after ERα invalidation (90).
BPA and ARs. In reporter cell lines, BPA or its halogenated derivatives inhibit ARs (136). Using in vitro bioassays, BPA has been described as a full human AR antagonist and a weak human AR agonist (133).
BPA and PPARs. In in vitro bioassays, BPA, and the halogenated forms used in flame retardants, such as tetrabromobisphenol A (TBBPA) and tetrachlorobisphenol A (TCBPA), bind to PPARγ (137, 138). The toxicity of those pollutants in the testis and the direct implication of PPARs still need to be demonstrated.
To our knowledge, there are no data showing PPAR involvement in BPA testicular effects. Conversely, PPARγ activation has been associated with BPA-induced adipogenesis (139).
BPA and LXRs. LXRs have never been incriminated in the mediation of the toxicological effects of BPA exposure in the testis. However, in mouse 3T3-L1 adipocyte cells, very low BPA doses (1 pM) increase lipid accumulation. This effect is correlated with up-regulation of SREBP1c mRNA, a gene that is directly regulated by LXRs, and also of downstream effectors involved in lipid synthesis and homeostasis (140). In vivo, rat exposed to BPA during fetal and neonatal life display adipocyte hypertrophy in the perigonadal adipose tissue at 21 dpp and increased SREBP1c expression (141). Moreover, analysis of the hepatic transcriptome of adult male mice exposed to various doses of BPA (5–5000 μg/kg/day in the food) for 28 days showed that low doses of BPA directly increase LXRs and SREBP1c expression and alter fatty acid biosynthesis (142). As LXRs appear to be involved in the regulation of both steroidogenesis and spermatogenesis, it is crucial to further investigate their potential role in the effects on male reproductive functions of BPA exposure during fetal and neonatal life.
BPA and SHP. To our knowledge, no data is available on SHP involvement in BPA testicular effects.
Based on their reprotoxic effects, most EDCs were thought to have pro-estrogenic or anti-androgenic effects via ERs and ARs (1, 143–145). However, evidences based on transgenic models, genomic analyses, and in vitro binding bioassays highlighted the involvement of additional or alternative pathways. This may account for the effects observed at much lower concentrations and for the non-monotonic dose-response often observed with EDCs (146, 147).
Some studies highlighted the involvement of the glucocorticoid and mineralocorticoid receptors (MRs) in the mechanisms of action of phthalates. Indeed, prepubertal exposure to DBP inhibits testosterone production through a glucocorticoid-mediated pathway (148). Moreover, in utero exposure to DEHP results in decreased MR mRNA and protein expression in adult interstitial Leydig cells and reduced mRNA expression of MR-regulated genes (149).
The molecular basis of BPA deleterious effects is poorly known, and currently the effects of low BPA doses are the focus of major discussions. Although ERα and ERβ are considered to be the main BPA targets, several other cellular targets have been proposed. Specifically, BPA could be a ligand of estrogen-related receptor gamma (ERRγ) (150) and of G protein-coupled estrogen receptor (GPER) (151). Moreover, low BPA concentrations trigger effects via G-protein coupled receptor 30 (GPR30) (152, 153).
Bisphenol A binds to the orphan nuclear receptor ERRγ at nanomolar concentrations with high specificity (154), and BPA positively regulates the transcriptional activity of human ERRγ. Moreover, these in vitro effects of BPA on ERRγ have been observed also in vivo during zebrafish development (154). Interestingly, ERRγ has higher affinity for BPA than for DES. The mechanism of action of BPA may be complex and it could also antagonize the binding of an unknown ERRγ ligand, thus maintaining the receptor in a constitutively active form (155).
Both GPR30 and ERRγ are expressed in mouse and human fetal testes. Using sorted cells from mouse 13.5 dpc testes, we observed that ERRγ is mainly expressed in interstitial cells and GPR30 in germ cells at the same level as ERβ (data not shown, Figure 1).
Other endocrine receptors, such as glucocorticoid receptor, thyroid receptor, and aryl hydro-carbon receptor (AhR), are also possible BPA targets, although their in vivo relevance is still debated (156).
Diethylstilbestrol has been considered for years as the paradigm of environmental EDCs with estrogen-like activity. Indeed, DES acts through ERs. However, recent studies suggest that LXRα and LXRβ not only are important for testis physiology, but could also exert a protective effect against estrogen-like endocrine disruptors (67).
Historically, the concept of EDCs arose from observations in wild fauna. During the last decades, it has become obvious, based on the analysis of the effects of a limited number of compounds, that some chemicals, to which the general population might be exposed, can negatively affect also human health, particularly by targeting the reproductive function and testis development. We suggest that the huge number of different signaling pathways required for gametogenesis and steroidogenesis also might explain/contribute to the high vulnerability of this system to exogenous interferences. One consequence of the wealth of studies on the effects of EDCs on human male reproduction is the growing interest of the general population and of policy makers. This has been translated rapidly into the legal ban of some highly mediatized compounds. While this seemed the proper response at first, in reality, it did not change much the way of consuming, and many less studied or less well-known compounds, which could be as deleterious as those that have been banned, remain in the environment. Even more worrying, “new” molecules for which very few or no data on their potential toxicity exist are put on the market to palliate for the withdrawal of some “old” ones. This increases the diversity and the number of compounds we may be concomitantly exposed to, another risk that is difficult to assess as these compound mixtures change over time. On the other hand, the action of a single EDC may involve directly or indirectly several nuclear receptors, while multiple EDCs may act through a single nuclear receptor. Despite all these difficulties, it is surprising that few studies have investigated the activation of nuclear receptors upon EDC exposure simply by measuring the target gene expression. Indeed, nuclear receptors constitute a large family of transcription factors that regulate developmental and physiological processes by directly controlling gene expression. Moreover, nuclear receptor-mediated transcription is often modulated through tissue-specific coregulators (coactivators or corepressors) (157); however, very few studies have investigated EDC effects on the expression of these coregulators. These issues will have to be addressed in future studies to provide reliable predictions about chemicals suspected to threaten human reproductive health. The formidable explosion of cre-lox-based approaches and transcriptomic analyses has rendered this goal achievable.
Another important point to address is whether it is worth to continue working on specific compounds for which a wealth of scientific data and experimental models are available, even though these compounds are going to be legally banned soon. The answer is unexpectedly yes! For many EDCs, we close to understand the mechanism of reprotoxicity. Indeed, the finding that reprotoxicity is not obligatorily mediated through estrogen- or androgen-disrupting activities allows now focusing on structure-based approach. Additionally, the identification of the mechanisms of action of a given compound is a mandatory step that will allow screening at medium throughput the thousands of remaining pollutants, which may use the same mechanism to target reproductive functions, and help predicting their potential reprotoxic effect. However, this will only be possible if the mechanism underlying their deleterious effect within the reproductive organs, and not only in a reporter cell line, is deciphered. On the other hand, major advances from in silico, in vitro, and in vivo (notably model organisms) studies are now helping to define some unsuspected challenges. For instance, it is now possible to propose that two substances together may activate an unexpected signaling cascade. Such evidences are expected to multiply in the near future and raise a major question: how are we expected to investigate the potential effect of combinations of hundreds of compounds? For instance, if among the 900 existing EDCs, only 10 compounds are sufficiently abundant, this would already lead to billions (9 × 1022) of possible different exposure combinations. Is this a lost battle? Possibly not. Indeed scientists, and particularly toxicologists, always aim at performing experiments in specifically defined conditions and by taking into account similar experiments for reasoning. This might be a narrow view and one may wish to test now more realistic exposure conditions with less defined mixtures but that reflect a real life situation. For instance, it has already been proposed to start using sewage sludge and from there move to airborne mixtures, compounds extracted from serum…etc.
In the end, every effort needs to keep in mind human health. From this point of view, the potential reprotoxicity of chemicals is worrying and alarming. The potential transgenerational effect of some substances is an additional issue. Indeed, we may need to determine not only the current exposure but also what our ancestors have been exposed to. This alarming scenery is definitively a factor of stress and stress is an endocrine based/endocrine disrupting situation that affects negatively the reproductive function as well. This is something we all need to consider when communicating to broad audiences. On an optimistic note, we hope that as EDC deleterious effects are under the spotlight, this will help gathering efforts to understand the mechanism of action of some of them. However, this is insufficient and a sustained research effort on the fundamental mechanisms that govern the development of the reproductive system, especially in humans, is obviously needed. Indeed, unraveling the key signaling cascades underlying the development and maintenance of the reproductive function is a complementary approach to identify altered signaling pathways using model compounds. Only, the synergy of both approaches will ensure the efficient prediction of EDC reprotoxicity and preserve the health of the future human generations.
Despite the wealth of data showing the deleterious effects of phthalates, BPA, and DES on testis development, their mechanisms of action remain poorly understood. In a general way, EDCs can act by modulating many genomic and non-genomic pathways. While non-genomic mechanisms are already seriously investigated, the deregulation of genomic signaling through nuclear receptors has not been fully elucidated yet. Two main reasons could explain this: first, the embryonic lethality of some mutants (e.g., PPARγ) may preclude later investigations; second, the complexity of the system. We believe that it is still essential to identify the mechanism of action of compounds, the harmful effects of which have been already demonstrated in the developing testis. This knowledge might help us predicting the reprotoxic potential of other substances that, alone or in combination with other molecules, use the same mechanism to target reproduction. Such prediction tools are crucially needed to supervise efficiently the replacement of compounds, such as BPA or phthalates, by other molecules that are currently poorly known.
All the authors have nothing to declare. All the authors or institutions never received any personal payment or service or funding for research studies from any chemical industrial company.
We are grateful to Aurélie Gouret for skillful secretarial assistance, Véronique Neuville for animal care, and the staff of the Department of Obstetrics and Gynecology of the Antoine Béclère Hospital (Clamart, France) for collecting human fetuses. We also thank Elisabetta Andermarcher for english-proofing the manuscript. This work was supported by the University of Paris Diderot-Paris 7, the Atomic Energy Committee (CEA), the National Institute for Health and Medical Research (INSERM), the French Ministry of Ecology, Sustainable Development, Transports and Housing. (Program ANTIOPES – Storm), and by the French National Agency for Safety in Food, Environment, and Work (ANSES) (Environment and Health Program). VM is supported by a fellowship from the Ministère de l’Education Nationale de la Recherche et de la Technologie and a grant from the Association pour la Recherche sur le Cancer. TT-B received a studentship from the Ile de France region for his Ph.D.
1. Toppari J, Larsen JC, Christiansen P, Giwercman A, Grandjean P, Guillette LJ Jr, et al. Male reproductive health and environmental xenoestrogens. Environ Health Perspect (1996) 104(Suppl 4):741–803. doi: 10.2307/3432709
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
2. Sharpe RM, Irvine DS. How strong is the evidence of a link between environmental chemicals and adverse effects on human reproductive health? BMJ (2004) 328:447–51. doi:10.1136/bmj.328.7437.447
3. Rucker EB III, Dierisseau P, Wagner KU, Garrett L, Wynshaw-Boris A, Flaws JA, et al. Bcl-x and Bax regulate mouse primordial germ cell survival and apoptosis during embryogenesis. Mol Endocrinol (2000) 14:1038–52. doi:10.1210/mend.14.7.0465
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
4. Lu B, Bishop CE. Late onset of spermatogenesis and gain of fertility in POG-deficient mice indicate that POG is not necessary for the proliferation of spermatogonia. Biol Reprod (2003) 69:161–8. doi:10.1095/biolreprod.102.014654
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
5. Forand A, Messiaen S, Habert R, Bernardino-Sgherri J. Exposure of the mouse perinatal testis to radiation leads to hypospermia at sexual maturity. Reproduction (2009) 137:487–95. doi:10.1530/REP-08-0358
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
6. Habert R, Lejeune H, Saez JM. Origin, differentiation and regulation of fetal and adult Leydig cells. Mol Cell Endocrinol (2001) 179:47–74. doi:10.1016/S0303-7207(01)00461-0
7. Scott HM, Mason JI, Sharpe RM. Steroidogenesis in the fetal testis and its susceptibility to disruption by exogenous compounds. Endocr Rev (2009) 30:883–925. doi:10.1210/er.2009-0016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
8. Welsh M, Suzuki H, Yamada G. The masculinization programming window. Endocr Dev (2014) 27:17–27. doi:10.1159/000363609
9. Kilcoyne KR, Smith LB, Atanassova N, Macpherson S, Mckinnell C, Van Den Driesche S, et al. Fetal programming of adult leydig cell function by androgenic effects on stem/progenitor cells. Proc Natl Acad Sci U S A (2014) 111:E1924–32. doi:10.1073/pnas.1320735111
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
10. Bay K, Main KM, Toppari J, Skakkebaek NE. Testicular descent: INSL3, testosterone, genes and the intrauterine milieu. Nat Rev Urol (2011) 8:187–96. doi:10.1038/nrurol.2011.23
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
11. Ivell R, Heng K, Anand-Ivell R. Insulin-like factor 3 and the HPG axis in the male. Front Endocrinol (2014) 5:6.
12. Sharpe RM, Skakkebaek NE. Testicular dysgenesis syndrome: mechanistic insights and potential new downstream effects. Fertil Steril (2008) 89:e33–8. doi:10.1016/j.fertnstert.2007.12.026
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
13. Main KM, Skakkebaek NE, Virtanen HE, Toppari J. Genital anomalies in boys and the environment. Best Pract Res Clin Endocrinol Metab (2010) 24:279–89. doi:10.1016/j.beem.2009.10.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
14. Delbes G, Levacher C, Habert R. Estrogen effects on fetal and neonatal testicular development. Reproduction (2006) 132:527–38. doi:10.1530/rep.1.01231
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
15. Livera G, Delbes G, Pairault C, Rouiller-Fabre V, Habert R. Organotypic culture, a powerful model for studying rat and mouse fetal testis development. Cell Tissue Res (2006) 324:507–21. doi:10.1007/s00441-006-0167-7
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
16. Habert R, Muczynski V, Lehraiki A, Lambrot R, Lecureuil C, Levacher C, et al. Adverse effects of endocrine disruptors on the foetal testis development: focus on the phthalates. Folia Histochem Cytobiol (2009) 47:S67–74. doi:10.2478/v10042-009-0056-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
17. Eladak S, Grisin T, Moison D, Guerquin MJ, N’tumba-Byn T, Pozzi-Gaudin S, et al. A new chapter in the bisphenol A story: bisphenol S and bisphenol F are not safe alternatives to this compound. Fertil Steril (2015) 103:11–21. doi:10.1016/j.fertnstert.2014.11.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
18. Green S, Walter P, Kumar V, Krust A, Bornert JM, Argos P, et al. Human oestrogen receptor cDNA: sequence, expression and homology to v-erb-A. Nature (1986) 320:134–9. doi:10.1038/320134a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
19. Koike S, Sakai M, Muramatsu M. Molecular cloning and characterization of rat estrogen receptor cDNA. Nucleic Acids Res (1987) 15:2499–513. doi:10.1093/nar/15.6.2499
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
20. Kuiper GG, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson JA. Cloning of a novel receptor expressed in rat prostate and ovary. Proc Natl Acad Sci U S A (1996) 93:5925–30. doi:10.1073/pnas.93.12.5925
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
21. Nuclear Receptors Nomenclature C. A unified nomenclature system for the nuclear receptor superfamily. Cell (1999) 97:161–3. doi:10.1016/S0092-8674(00)80726-6
22. Williams K, Mckinnell C, Saunders PT, Walker M, Fisher JS, Turner KJ, et al. Neonatal exposure to potent and environmental oestrogens and abnormalities of the male reproductive system in the rat: evidence for importance of the androgen-oestrogen balance and assessment of the relevance to man. Hum Reprod Update (2001) 7:236–47. doi:10.1093/humupd/7.3.236
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
23. Greco TL, Furlow JD, Duello TM, Gorski J. Immunodetection of estrogen receptors in fetal and neonatal male mouse reproductive tracts. Endocrinology (1992) 130:421–9. doi:10.1210/en.130.1.421
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
24. Nielsen M, Bjornsdottir S, Hoyer PE, Byskov AG. Ontogeny of oestrogen receptor alpha in gonads and sex ducts of fetal and newborn mice. J Reprod Fertil (2000) 118:195–204. doi:10.1530/jrf.0.1180195
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
25. Delbes G, Levacher C, Duquenne C, Racine C, Pakarinen P, Habert R. Endogenous estrogens inhibit mouse fetal leydig cell development via estrogen receptor alpha. Endocrinology (2005) 146:2454–61. doi:10.1210/en.2004-1540
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
26. Jefferson WN, Couse JF, Banks EP, Korach KS, Newbold RR. Expression of estrogen receptor beta is developmentally regulated in reproductive tissues of male and female mice. Biol Reprod (2000) 62:310–7. doi:10.1095/biolreprod62.2.310
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
27. Saunders PT, Fisher JS, Sharpe RM, Millar MR. Expression of oestrogen receptor beta (ER beta) occurs in multiple cell types, including some germ cells, in the rat testis. J Endocrinol (1998) 156:R13–7. doi:10.1677/joe.0.156R013
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
28. van Pelt AM, De Rooij DG, Van Der Burg B, Van Der Saag PT, Gustafsson JA, Kuiper GG. Ontogeny of estrogen receptor-beta expression in rat testis. Endocrinology (1999) 140:478–83. doi:10.1210/en.140.1.478
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
29. Brandenberger AW, Tee MK, Lee JY, Chao V, Jaffe RB. Tissue distribution of estrogen receptors alpha (ER-alpha) and beta (ER-beta) mRNA in the midgestational human fetus. J Clin Endocrinol Metab (1997) 82:3509–12. doi:10.1210/jcem.82.10.4400
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
30. Saunders PT, Millar MR, Macpherson S, Irvine DS, Groome NP, Evans LR, et al. ERbeta1 and the ERbeta2 splice variant (ERbetacx/beta2) are expressed in distinct cell populations in the adult human testis. J Clin Endocrinol Metab (2002) 87:2706–15. doi:10.1210/jcem.87.6.8619
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
31. Takeyama J, Suzuki T, Inoue S, Kaneko C, Nagura H, Harada N, et al. Expression and cellular localization of estrogen receptors alpha and beta in the human fetus. J Clin Endocrinol Metab (2001) 86:2258–62. doi:10.1210/jcem.86.5.7447
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
32. Gaskell TL, Robinson LL, Groome NP, Anderson RA, Saunders PT. Differential expression of two estrogen receptor beta isoforms in the human fetal testis during the second trimester of pregnancy. J Clin Endocrinol Metab (2003) 88:424–32. doi:10.1210/jc.2002-020811
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
33. Mitchell RT, Sharpe RM, Anderson RA, Mckinnell C, Macpherson S, Smith LB, et al. Diethylstilboestrol exposure does not reduce testosterone production in human fetal testis xenografts. PLoS One (2013) 8:e61726. doi:10.1371/journal.pone.0061726
34. Pelletier G, El-Alfy M. Immunocytochemical localization of estrogen receptors alpha and beta in the human reproductive organs. J Clin Endocrinol Metab (2000) 85:4835–40. doi:10.1210/jcem.85.12.7029
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
35. Korach KS. Insights from the study of animals lacking functional estrogen receptor. Science (1994) 266:1524–7. doi:10.1126/science.7985022
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
36. Fisher CR, Graves KH, Parlow AF, Simpson ER. Characterization of mice deficient in aromatase (ArKO) because of targeted disruption of the cyp19 gene. Proc Natl Acad Sci U S A (1998) 95:6965–70. doi:10.1073/pnas.95.12.6965
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
37. Robertson KM, O’donnell L, Jones ME, Meachem SJ, Boon WC, Fisher CR, et al. Impairment of spermatogenesis in mice lacking a functional aromatase (cyp 19) gene. Proc Natl Acad Sci U S A (1999) 96:7986–91. doi:10.1073/pnas.96.14.7986
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
38. O’Donnell L, Robertson KM, Jones ME, Simpson ER. Estrogen and spermatogenesis. Endocr Rev (2001) 22:289–318. doi:10.1210/edrv.22.3.0431
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
39. Delbes G, Levacher C, Pairault C, Racine C, Duquenne C, Krust A, et al. Estrogen receptor beta-mediated inhibition of male germ cell line development in mice by endogenous estrogens during perinatal life. Endocrinology (2004) 145:3395–403. doi:10.1210/en.2003-1479
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
40. Carreau S, Wolczynski S, Galeraud-Denis I. Aromatase, oestrogens and human male reproduction. Philos Trans R Soc Lond B Biol Sci (2010) 365:1571–9. doi:10.1098/rstb.2009.0113
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
41. Smith LB, Walker WH. The regulation of spermatogenesis by androgens. Semin Cell Dev Biol (2014) 30:2–13. doi:10.1016/j.semcdb.2014.02.012
42. Zhou Q, Nie R, Prins GS, Saunders PT, Katzenellenbogen BS, Hess RA. Localization of androgen and estrogen receptors in adult male mouse reproductive tract. J Androl (2002) 23:870–81.
43. Merlet J, Racine C, Moreau E, Moreno SG, Habert R. Male fetal germ cells are targets for androgens that physiologically inhibit their proliferation. Proc Natl Acad Sci U S A (2007) 104:3615–20. doi:10.1073/pnas.0611421104
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
44. Merlet J, Moreau E, Habert R, Racine C. Development of fetal testicular cells in androgen receptor deficient mice. Cell Cycle (2007) 6:2258–62. doi:10.4161/cc.6.18.4654
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
45. Sharpe RM, Mckinnell C, Kivlin C, Fisher JS. Proliferation and functional maturation of Sertoli cells, and their relevance to disorders of testis function in adulthood. Reproduction (2003) 125:769–84. doi:10.1530/rep.0.1250769
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
46. Boukari K, Meduri G, Brailly-Tabard S, Guibourdenche J, Ciampi ML, Massin N, et al. Lack of androgen receptor expression in sertoli cells accounts for the absence of anti-mullerian hormone repression during early human testis development. J Clin Endocrinol Metab (2009) 94:1818–25. doi:10.1210/jc.2008-1909
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
47. O’Hara L, Mcinnes K, Simitsidellis I, Morgan S, Atanassova N, Slowikowska-Hilczer J, et al. Autocrine androgen action is essential for Leydig cell maturation and function, and protects against late-onset leydig cell apoptosis in both mice and men. FASEB J (2015) 29:894–910. doi:10.1096/fj.14-255729
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
48. Connolly F, Rae MT, Bittner L, Hogg K, Mcneilly AS, Duncan WC. Excess androgens in utero alters fetal testis development. Endocrinology (2013) 154:1921–33. doi:10.1210/en.2012-2153
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
49. Rojas-Garcia PP, Recabarren MP, Sir-Petermann T, Rey R, Palma S, Carrasco A, et al. Altered testicular development as a consequence of increase number of sertoli cell in male lambs exposed prenatally to excess testosterone. Endocrine (2013) 43:705–13. doi:10.1007/s12020-012-9818-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
50. Berger JP, Akiyama TE, Meinke PT. PPARs: therapeutic targets for metabolic disease. Trends Pharmacol Sci (2005) 26:244–51. doi:10.1016/j.tips.2005.03.003
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
51. Froment P, Gizard F, Defever D, Staels B, Dupont J, Monget P. Peroxisome proliferator-activated receptors in reproductive tissues: from gametogenesis to parturition. J Endocrinol (2006) 189:199–209. doi:10.1677/joe.1.06667
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
52. Braissant O, Foufelle F, Scotto C, Dauca M, Wahli W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-alpha, -beta, and -gamma in the adult rat. Endocrinology (1996) 137:354–66. doi:10.1210/endo.137.1.8536636
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
53. Lapinskas PJ, Brown S, Leesnitzer LM, Blanchard S, Swanson C, Cattley RC, et al. Role of PPARalpha in mediating the effects of phthalates and metabolites in the liver. Toxicology (2005) 207:149–63. doi:10.1016/j.tox.2004.09.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
54. Marugo M, Aste H, Bernasconi D, Fazzuoli L, Conio M, Cuva A, et al. Cytosolic and nuclear androgen receptors in colorectal adenomas. Anticancer Res (1992) 12:705–8.
55. Muczynski V, Lecureuil C, Messiaen S, Guerquin M, N’tumba-Byn T, Moison D, et al. Cellular and molecular effect of MEHP Involving LXRalpha in human fetal testis and ovary. PLoS One (2012) 7:e48266. doi:10.1371/journal.pone.0048266
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
56. Hase T, Yoshimura R, Mitsuhashi M, Segawa Y, Kawahito Y, Wada S, et al. Expression of peroxisome proliferator-activated receptors in human testicular cancer and growth inhibition by its agonists. Urology (2002) 60:542–7. doi:10.1016/S0090-4295(02)01747-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
57. Ward JM, Peters JM, Perella CM, Gonzalez FJ. Receptor and nonreceptor-mediated organ-specific toxicity of di(2-ethylhexyl)phthalate (DEHP) in peroxisome proliferator-activated receptor alpha-null mice. Toxicol Pathol (1998) 26:240–6. doi:10.1177/019262339802600208
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
58. Hannas BR, Lambright CS, Furr J, Evans N, Foster PM, Gray EL, et al. Genomic biomarkers of phthalate-induced male reproductive developmental toxicity: a targeted RT-PCR array approach for defining relative potency. Toxicol Sci (2012) 125:544–57. doi:10.1093/toxsci/kfr315
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
59. Regueira M, Riera MF, Galardo MN, Pellizzari EH, Cigorraga SB, Meroni SB. Activation of PPAR alpha and PPAR beta/delta regulates sertoli cell metabolism. Mol Cell Endocrinol (2014) 382:271–81. doi:10.1016/j.mce.2013.10.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
60. Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, et al. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell (1999) 4:585–95. doi:10.1016/S1097-2765(00)80209-9
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
61. Thomas K, Sung DY, Chen X, Thompson W, Chen YE, Mccarrey J, et al. Developmental patterns of PPAR and RXR gene expression during spermatogenesis. Front Biosci (Elite Ed) (2011) 3:1209–20.
62. Lu TT, Repa JJ, Mangelsdorf DJ. Orphan nuclear receptors as eLiXiRs and FiXeRs of sterol metabolism. J Biol Chem (2001) 276:37735–8. doi:10.1074/jbc.R100035200
63. Cao G, Liang Y, Jiang XC, Eacho PI. Liver X receptors as potential therapeutic targets for multiple diseases. Drug News Perspect (2004) 17:35–41. doi:10.1358/dnp.2004.17.1.829024
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
64. El-Hajjaji FZ, Oumeddour A, Pommier AJ, Ouvrier A, Viennois E, Dufour J, et al. Liver X receptors, lipids and their reproductive secrets in the male. Biochim Biophys Acta (2011) 1812:974–81. doi:10.1016/j.bbadis.2011.02.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
65. Volle DH, Mouzat K, Duggavathi R, Siddeek B, Dechelotte P, Sion B, et al. Multiple roles of the nuclear receptors for oxysterols liver X receptor to maintain male fertility. Mol Endocrinol (2007) 21:1014–27. doi:10.1210/me.2006-0277
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
66. Volle DH, Lobaccaro JM. Role of the nuclear receptors for oxysterols LXRs in steroidogenic tissues: beyond the “foie gras”, the steroids and sex? Mol Cell Endocrinol (2007) 26(5–266):183–9. doi:10.1016/j.mce.2006.12.018
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
67. Oumeddour A, Viennois E, Caira F, Decourbey C, Maqdasy S, Tahraoui A, et al. Liver X receptors interfere with the deleterious effect of diethylstilbestrol on testicular physiology. Biochem Biophys Res Commun (2014) 446:656–62. doi:10.1016/j.bbrc.2013.12.005
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
68. Rondanino C, Ouchchane L, Chauffour C, Marceau G, Dechelotte P, Sion B, et al. Levels of liver X receptors in testicular biopsies of patients with azoospermia. Fertil Steril (2014) 102(361–371):e365. doi:10.1016/j.fertnstert.2014.04.033
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
69. Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, et al. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell (2000) 6:507–15. doi:10.1016/S1097-2765(00)00050-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
70. Kerr TA, Saeki S, Schneider M, Schaefer K, Berdy S, Redder T, et al. Loss of nuclear receptor SHP impairs but does not eliminate negative feedback regulation of bile acid synthesis. Dev Cell (2002) 2:713–20. doi:10.1016/S1534-5807(02)00154-5
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
71. Volle DH, Duggavathi R, Magnier BC, Houten SM, Cummins CL, Lobaccaro JM, et al. The small heterodimer partner is a gonadal gatekeeper of sexual maturation in male mice. Genes Dev (2007) 21:303–15. doi:10.1101/gad.409307
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
72. Maqdasy S, Baptissart M, Vega A, Baron S, Lobaccaro JM, Volle DH. Cholesterol and male fertility: what about orphans and adopted? Mol Cell Endocrinol (2013) 368:30–46. doi:10.1016/j.mce.2012.06.011
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
73. Bowles J, Knight D, Smith C, Wilhelm D, Richman J, Mamiya S, et al. Retinoid signaling determines germ cell fate in mice. Science (2006) 312:596–600. doi:10.1126/science.1125691
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
74. Koubova J, Menke DB, Zhou Q, Capel B, Griswold MD, Page DC. Retinoic acid regulates sex-specific timing of meiotic initiation in mice. Proc Natl Acad Sci U S A (2006) 103:2474–9. doi:10.1073/pnas.0510813103
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
75. Sharpe RM, Skakkebaek NE. Are oestrogens involved in falling sperm counts and disorders of the male reproductive tract? Lancet (1993) 341:1392–5. doi:10.1016/0140-6736(93)90953-E
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
76. Glaze GM. Diethylstilbestrol exposure in utero: review of literature. J Am Osteopath Assoc (1984) 83:435–8.
77. Strohsnitter WC, Noller KL, Hoover RN, Robboy SJ, Palmer JR, Titus-Ernstoff L, et al. Cancer risk in men exposed in utero to diethylstilbestrol. J Natl Cancer Inst (2001) 93:545–51. doi:10.1093/jnci/93.7.545
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
78. Storgaard L, Bonde JP, Olsen J. Male reproductive disorders in humans and prenatal indicators of estrogen exposure. A review of published epidemiological studies. Reprod Toxicol (2006) 21:4–15. doi:10.1016/j.reprotox.2005.05.006
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
79. Sharpe RM. The ‘oestrogen hypothesis’ – where do we stand now? Int J Androl (2003) 26:2–15. doi:10.1046/j.1365-2605.2003.00367.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
80. Yasuda Y, Kihara T, Tanimura T. Effect of ethinyl estradiol on the differentiation of mouse fetal testis. Teratology (1985) 32:113–8. doi:10.1002/tera.1420320115
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
81. Yasuda Y, Kihara T, Tanimura T, Nishimura H. Gonadal dysgenesis induced by prenatal exposure to ethinyl estradiol in mice. Teratology (1985) 32:219–27. doi:10.1002/tera.1420320210
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
82. Perez-Martinez C, Garcia-Iglesias MJ, Ferreras-Estrada MC, Bravo-Moral AM, Espinosa-Alvarez J, Escudero-Diez A. Effects of in-utero exposure to zeranol or diethylstilboestrol on morphological development of the fetal testis in mice. J Comp Pathol (1996) 114:407–18. doi:10.1016/S0021-9975(96)80016-8
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
83. Lassurguere J, Livera G, Habert R, Jegou B. Time- and dose-related effects of estradiol and diethylstilbestrol on the morphology and function of the fetal rat testis in culture. Toxicol Sci (2003) 73:160–9. doi:10.1093/toxsci/kfg065
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
84. Delbes G, Duquenne C, Szenker J, Taccoen J, Habert R, Levacher C. Developmental changes in testicular sensitivity to estrogens throughout fetal and neonatal life. Toxicol Sci (2007) 99:234–43. doi:10.1093/toxsci/kfm160
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
85. Guyot R, Odet F, Leduque P, Forest MG, Le Magueresse-Battistoni B. Diethylstilbestrol inhibits the expression of the steroidogenic acute regulatory protein in mouse fetal testis. Mol Cell Endocrinol (2004) 220:67–75. doi:10.1016/j.mce.2004.03.008
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
86. Cederroth CR, Schaad O, Descombes P, Chambon P, Vassalli JD, Nef S. Estrogen receptor alpha is a major contributor to estrogen-mediated fetal testis dysgenesis and cryptorchidism. Endocrinology (2007) 148:5507–19. doi:10.1210/en.2007-0689
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
87. Lundholm L, Moverare S, Steffensen KR, Nilsson M, Otsuki M, Ohlsson C, et al. Gene expression profiling identifies liver X receptor alpha as an estrogen-regulated gene in mouse adipose tissue. J Mol Endocrinol (2004) 32:879–92. doi:10.1677/jme.0.0320879
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
88. Gong H, Guo P, Zhai Y, Zhou J, Uppal H, Jarzynka MJ, et al. Estrogen deprivation and inhibition of breast cancer growth in vivo through activation of the orphan nuclear receptor liver X receptor. Mol Endocrinol (2007) 21:1781–90. doi:10.1210/me.2007-0187
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
89. Volle DH, Decourteix M, Garo E, Mcneilly J, Fenichel P, Auwerx J, et al. The orphan nuclear receptor small heterodimer partner mediates male infertility induced by diethylstilbestrol in mice. J Clin Invest (2009) 119:3752–64. doi:10.1172/JCI38521
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
90. N’Tumba-Byn T, Moison D, Lacroix M, Lecureuil C, Lesage L, Prud’homme S, et al. Differential effects of bisphenol A and diethylstilbestrol on human, rat and mouse fetal leydig cell function. PLoS One (2012) 7:e51579. doi:10.1371/journal.pone.0051579
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
91. Latini G, Del Vecchio A, Massaro M, Verrotti A, DE Felice C. In utero exposure to phthalates and fetal development. Curr Med Chem (2006) 13:2527–34. doi:10.2174/092986706778201666
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
92. Fisher JS. Environmental anti-androgens and male reproductive health: focus on phthalates and testicular dysgenesis syndrome. Reproduction (2004) 127:305–15. doi:10.1530/rep.1.00025
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
93. Chauvigne F, Menuet A, Lesne L, Chagnon MC, Chevrier C, Regnier JF, et al. Time- and dose-related effects of di-(2-ethylhexyl) phthalate and its main metabolites on the function of the rat fetal testis in vitro. Environ Health Perspect (2009) 117:515–21. doi:10.1289/ehp.11870
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
94. Lambrot R, Muczynski V, Lecureuil C, Angenard G, Coffigny H, Pairault C, et al. Phthalates impair germ cell development in the human fetal testis in vitro without change in testosterone production. Environ Health Perspect (2009) 117:32–7. doi:10.1289/ehp.11146
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
95. Lehraiki A, Racine C, Krust A, Habert R, Levacher C. Phthalates impair germ cell number in the mouse fetal testis by an androgen- and estrogen-independent mechanism. Toxicol Sci (2009) 111:372–82. doi:10.1093/toxsci/kfp153
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
96. Swan SH, Main KM, Liu F, Stewart SL, Kruse RL, Calafat AM, et al. Decrease in anogenital distance among male infants with prenatal phthalate exposure. Environ Health Perspect (2005) 113:1056–61. doi:10.1289/ehp.8100
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
97. Huang PC, Kuo PL, Chou YY, Lin SJ, Lee CC. Association between prenatal exposure to phthalates and the health of newborns. Environ Int (2009) 35:14–20. doi:10.1016/j.envint.2008.05.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
98. Fisher JS, Macpherson S, Marchetti N, Sharpe RM. Human ‘testicular dysgenesis syndrome’: a possible model using in-utero exposure of the rat to dibutyl phthalate. Hum Reprod (2003) 18:1383–94. doi:10.1093/humrep/deg273
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
99. Habert R, Livera G, Rouiller-Fabre V. Man is not a big rat: concerns with traditional human risk assessment of phthalates based on their anti-androgenic effects observed in the rat foetus. Basic Clin Androl (2014) 24:14. doi:10.1186/2051-4190-24-14
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
100. Habert R, Muczynski V, Grisin T, Moison D, Messiaen S, Frydman R, et al. Concerns about the widespread use of rodent models for human risk assessments of endocrine disruptors. Reproduction (2014) 147:R119–29. doi:10.1530/REP-13-0497
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
101. Heger NE, Hall SJ, Sandrof MA, Mcdonnell EV, Hensley JB, Mcdowell EN, et al. Human fetal testis xenografts are resistant to phthalate-induced endocrine disruption. Environ Health Perspect (2012) 120:1137–43. doi:10.1289/ehp.1104711
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
102. Mitchell RT, Childs AJ, Anderson RA, Van Den Driesche S, Saunders PT, Mckinnell C, et al. Do phthalates affect steroidogenesis by the human fetal testis? Exposure of human fetal testis xenografts to di-n-butyl phthalate. J Clin Endocrinol Metab (2012) 97:E341–8. doi:10.1210/jc.2011-2411
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
103. Spade DJ, Hall SJ, Saffarini CM, Huse SM, Mcdonnell EV, Boekelheide K. Differential response to abiraterone acetate and di-n-butyl phthalate in an androgen-sensitive human fetal testis xenograft bioassay. Toxicol Sci (2014) 138:148–60. doi:10.1093/toxsci/kft266
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
104. Barlow NJ, Phillips SL, Wallace DG, Sar M, Gaido KW, Foster PM. Quantitative changes in gene expression in fetal rat testes following exposure to di(n-butyl) phthalate. Toxicol Sci (2003) 73:431–41. doi:10.1093/toxsci/kfg087
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
105. Ferrara D, Hallmark N, Scott H, Brown R, Mckinnell C, Mahood IK, et al. Acute and long-term effects of in utero exposure of rats to di(n-butyl) phthalate on testicular germ cell development and proliferation. Endocrinology (2006) 147:5352–62. doi:10.1210/en.2006-0527
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
106. Latini G, Scoditti E, Verrotti A, De Felice C, Massaro M. Peroxisome proliferator-activated receptors as mediators of phthalate-induced effects in the male and female reproductive tract: epidemiological and experimental evidence. PPAR Res (2008) 2008:359267. doi:10.1155/2008/359267
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
107. Rusyn I, Peters JM, Cunningham ML. Modes of action and species-specific effects of di-(2-ethylhexyl)phthalate in the liver. Crit Rev Toxicol (2006) 36:459–79. doi:10.1080/10408440600779065
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
108. Hurst CH, Waxman DJ. Activation of PPARalpha and PPARgamma by environmental phthalate monoesters. Toxicol Sci (2003) 74:297–308. doi:10.1093/toxsci/kfg145
109. Maloney EK, Waxman DJ. Trans-activation of PPARalpha and PPARgamma by structurally diverse environmental chemicals. Toxicol Appl Pharmacol (1999) 161:209–18. doi:10.1006/taap.1999.8809
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
110. Hurst CH, Waxman DJ. Environmental phthalate monoesters activate pregnane X receptor-mediated transcription. Toxicol Appl Pharmacol (2004) 199:266–74. doi:10.1016/j.taap.2003.11.028
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
111. Euling SY, White LD, Kim AS, Sen B, Wilson VS, Keshava C, et al. Use of genomic data in risk assessment case study: II. Evaluation of the dibutyl phthalate toxicogenomic data set. Toxicol Appl Pharmacol (2013) 271:349–62. doi:10.1016/j.taap.2011.06.014
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
112. Borch J, Metzdorff SB, Vinggaard AM, Brokken L, Dalgaard M. Mechanisms underlying the anti-androgenic effects of diethylhexyl phthalate in fetal rat testis. Toxicology (2006) 223:144–55. doi:10.1016/j.tox.2006.03.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
113. Boberg J, Metzdorff S, Wortziger R, Axelstad M, Brokken L, Vinggaard AM, et al. Impact of diisobutyl phthalate and other PPAR agonists on steroidogenesis and plasma insulin and leptin levels in fetal rats. Toxicology (2008) 250:75–81. doi:10.1016/j.tox.2008.05.020
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
114. Ryu JY, Whang J, Park H, Im JY, Kim J, Ahn MY, et al. Di(2-ethylhexyl) phthalate induces apoptosis through peroxisome proliferators-activated receptor-gamma and ERK 1/2 activation in testis of Sprague-Dawley rats. J Toxicol Environ Health A (2007) 70:1296–303. doi:10.1080/15287390701432160
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
115. Klinefelter GR, Laskey JW, Winnik WM, Suarez JD, Roberts NL, Strader LF, et al. Novel molecular targets associated with testicular dysgenesis induced by gestational exposure to diethylhexyl phthalate in the rat: a role for estradiol. Reproduction (2012) 144:747–61. doi:10.1530/REP-12-0266
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
116. Muczynski V, Cravedi JP, Lehraiki A, Levacher C, Moison D, Lecureuil C, et al. Effect of mono-(2-ethylhexyl) phthalate on human and mouse fetal testis: in vitro and in vivo approaches. Toxicol Appl Pharmacol (2012) 261:97–104. doi:10.1016/j.taap.2012.03.016
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
117. Dodds EC, Lawson W. Synthetic oestrogenic agents without the phenanthrene nucleus. Nature (1936) 137:996. doi:10.1038/137996a0
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
118. Vandenberg LN, Maffini MV, Wadia PR, Sonnenschein C, Rubin BS, Soto AM. Exposure to environmentally relevant doses of the xenoestrogen bisphenol-A alters development of the fetal mouse mammary gland. Endocrinology (2007) 148:116–27. doi:10.1210/en.2006-0561
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
119. Zalko D, Jacques C, Duplan H, Bruel S, Perdu E. Viable skin efficiently absorbs and metabolizes bisphenol A. Chemosphere (2011) 82:424–30. doi:10.1016/j.chemosphere.2010.09.058
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
120. Rochester JR. Bisphenol A and human health: a review of the literature. Reprod Toxicol (2013) 42:132–55. doi:10.1016/j.reprotox.2013.08.008
121. Miao M, Yuan W, He Y, Zhou Z, Wang J, Gao E, et al. In utero exposure to bisphenol-A and anogenital distance of male offspring. Birth Defects Res A Clin Mol Teratol (2011) 91:867–72. doi:10.1002/bdra.22845
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
122. Choi H, Kim J, Im Y, Lee S, Kim Y. The association between some endocrine disruptors and hypospadias in biological samples. J Environ Sci Health A Tox Hazard Subst Environ Eng (2012) 47:2173–9. doi:10.1080/10934529.2012.680387
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
123. Fenichel P, Dechaux H, Harthe C, Gal J, Ferrari P, Pacini P, et al. Unconjugated bisphenol A cord blood levels in boys with descended or undescended testes. Hum Reprod (2012) 27:983–90. doi:10.1093/humrep/der451
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
124. Fenichel P, Chevalier N, Brucker-Davis F. Bisphenol A: an endocrine and metabolic disruptor. Ann Endocrinol (Paris) (2013) 74:211–20. doi:10.1016/j.ando.2013.04.002
125. Tanaka M, Nakaya S, Katayama M, Leffers H, Nozawa S, Nakazawa R, et al. Effect of prenatal exposure to bisphenol A on the serum testosterone concentration of rats at birth. Hum Exp Toxicol (2006) 25:369–73. doi:10.1191/0960327106ht638oa
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
126. Murray TJ, Maffini MV, Ucci AA, Sonnenschein C, Soto AM. Induction of mammary gland ductal hyperplasias and carcinoma in situ following fetal bisphenol A exposure. Reprod Toxicol (2007) 23:383–90. doi:10.1016/j.reprotox.2006.10.002
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
127. Kobayashi K, Miyagawa M, Wang RS, Sekiguchi S, Suda M, Honma T. Effects of in utero and lactational exposure to bisphenol A on somatic growth and anogenital distance in F1 rat offspring. Ind Health (2002) 40:375–81. doi:10.2486/indhealth.40.375
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
128. Tyl RW, Myers CB, Marr MC, Thomas BF, Keimowitz AR, Brine DR, et al. Three-generation reproductive toxicity study of dietary bisphenol A in CD Sprague-Dawley rats. Toxicol Sci (2002) 68:121–46. doi:10.1093/toxsci/68.1.121
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
129. Howdeshell KL, Furr J, Lambright CR, Wilson VS, Ryan BC, Gray LE Jr. Gestational and lactational exposure to ethinyl estradiol, but not bisphenol A, decreases androgen-dependent reproductive organ weights and epididymal sperm abundance in the male long evans hooded rat. Toxicol Sci (2008) 102:371–82. doi:10.1093/toxsci/kfm306
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
130. LaRocca J, Boyajian A, Brown C, Smith SD, Hixon M. Effects of in utero exposure to bisphenol A or diethylstilbestrol on the adult male reproductive system. Birth Defects Res B Dev Reprod Toxicol (2011) 92:526–33. doi:10.1002/bdrb.20336
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
131. Salian S, Doshi T, Vanage G. Perinatal exposure of rats to bisphenol A affects the fertility of male offspring. Life Sci (2009) 85:742–52. doi:10.1016/j.lfs.2009.10.004
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
132. Zhang HQ, Zhang XF, Zhang LJ, Chao HH, Pan B, Feng YM, et al. Fetal exposure to bisphenol A affects the primordial follicle formation by inhibiting the meiotic progression of oocytes. Mol Biol Rep (2012) 39:5651–7. doi:10.1007/s11033-011-1372-3
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
133. Molina-Molina JM, Amaya E, Grimaldi M, Saenz JM, Real M, Fernandez MF, et al. In vitro study on the agonistic and antagonistic activities of bisphenol-S and other bisphenol-A congeners and derivatives via nuclear receptors. Toxicol Appl Pharmacol (2013) 272:127–36. doi:10.1016/j.taap.2013.05.015
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
134. Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, Van Der Saag PT, et al. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology (1998) 139:4252–63. doi:10.1210/endo.139.10.6216
135. Boukari K, Ciampi ML, Guiochon-Mantel A, Young J, Lombes M, Meduri G. Human fetal testis: source of estrogen and target of estrogen action. Hum Reprod (2007) 22:1885–92. doi:10.1093/humrep/dem091
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
136. Paris F, Balaguer P, Terouanne B, Servant N, Lacoste C, Cravedi JP, et al. Phenylphenols, biphenols, bisphenol-A and 4-tert-octylphenol exhibit alpha and beta estrogen activities and antiandrogen activity in reporter cell lines. Mol Cell Endocrinol (2002) 193:43–9. doi:10.1016/S0303-7207(02)00094-1
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
137. Riu A, Grimaldi M, Le Maire A, Bey G, Phillips K, Boulahtouf A, et al. Peroxisome proliferator-activated receptor gamma is a target for halogenated analogs of bisphenol A. Environ Health Perspect (2011) 119:1227–32. doi:10.1289/ehp.1003328
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
138. Riu A, Le Maire A, Grimaldi M, Audebert M, Hillenweck A, Bourguet W, et al. Characterization of novel ligands of ERalpha, Erbeta, and PPARgamma: the case of halogenated bisphenol A and their conjugated metabolites. Toxicol Sci (2011) 122:372–82. doi:10.1093/toxsci/kfr132
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
139. Pereira-Fernandes A, Demaegdt H, Vandermeiren K, Hectors TL, Jorens PG, Blust R, et al. Evaluation of a screening system for obesogenic compounds: screening of endocrine disrupting compounds and evaluation of the PPAR dependency of the effect. PLoS One (2013) 8:e77481. doi:10.1371/journal.pone.0077481
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
140. Helies-Toussaint C, Peyre L, Costanzo C, Chagnon MC, Rahmani R. Is bisphenol S a safe substitute for bisphenol A in terms of metabolic function? An in vitro study. Toxicol Appl Pharmacol (2014) 280:224–35. doi:10.1016/j.taap.2014.07.025
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
141. Somm E, Schwitzgebel VM, Toulotte A, Cederroth CR, Combescure C, Nef S, et al. Perinatal exposure to bisphenol a alters early adipogenesis in the rat. Environ Health Perspect (2009) 117:1549–55. doi:10.1289/ehp.11342
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
142. Marmugi A, Ducheix S, Lasserre F, Polizzi A, Paris A, Priymenko N, et al. Low doses of bisphenol A induce gene expression related to lipid synthesis and trigger triglyceride accumulation in adult mouse liver. Hepatology (2012) 55:395–407. doi:10.1002/hep.24685
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
143. Colborn T, Vom Saal FS, Soto AM. Developmental effects of endocrine-disrupting chemicals in wildlife and humans. Environ Health Perspect (1993) 101:378–84. doi:10.1289/ehp.93101378
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
144. Sonnenschein C, Soto AM. An updated review of environmental estrogen and androgen mimics and antagonists. J Steroid Biochem Mol Biol (1998) 65:143–50. doi:10.1016/S0960-0760(98)00027-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
145. Edwards TM, Moore BC, Guillette LJ Jr. Reproductive dysgenesis in wildlife: a comparative view. Int J Androl (2006) 29:109–21. doi:10.1111/j.1365-2605.2005.00631.x
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
146. Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, Jacobs DR Jr, Lee DH, et al. Hormones and endocrine-disrupting chemicals: low-dose effects and nonmonotonic dose responses. Endocr Rev (2012) 33:378–455. doi:10.1210/er.2011-1050
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
147. Vandenberg LN. Non-monotonic dose responses in studies of endocrine disrupting chemicals: bisphenol a as a case study. Dose Response (2014) 12:259–76. doi:10.2203/dose-response.13-020.Vandenberg
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
148. Xiao-feng Z, Nai-Qiang Q, Jing Z, Zi L, Yang Z. Di (n-butyl) phthalate inhibits testosterone synthesis through a glucocorticoid-mediated pathway in rats. Int J Toxicol (2009) 28:448–56. doi:10.1177/1091581809342596
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
149. Martinez-Arguelles DB, Culty M, Zirkin BR, Papadopoulos V. In utero exposure to di-(2-ethylhexyl) phthalate decreases mineralocorticoid receptor expression in the adult testis. Endocrinology (2009) 150:5575–85. doi:10.1210/en.2009-0847
150. Okada H, Tokunaga T, Liu X, Takayanagi S, Matsushima A, Shimohigashi Y. Direct evidence revealing structural elements essential for the high binding ability of bisphenol A to human estrogen-related receptor-gamma. Environ Health Perspect (2008) 116:32–8. doi:10.1289/ehp.10587
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
151. Levin ER. Minireview: extranuclear steroid receptors: roles in modulation of cell functions. Mol Endocrinol (2011) 25:377–84. doi:10.1210/me.2010-0284
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
152. Bouskine A, Nebout M, Brucker-Davis F, Benahmed M, Fenichel P. Low doses of bisphenol A promote human seminoma cell proliferation by activating PKA and PKG via a membrane G-protein-coupled estrogen receptor. Environ Health Perspect (2009) 117:1053–8. doi:10.1289/ehp.0800367
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
153. Chevalier N, Vega A, Bouskine A, Siddeek B, Michiels JF, Chevallier D, et al. GPR30, the non-classical membrane G protein related estrogen receptor, is overexpressed in human seminoma and promotes seminoma cell proliferation. PLoS One (2012) 7:e34672. doi:10.1371/journal.pone.0034672
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
154. Tohme M, Prud’homme SM, Boulahtouf A, Samarut E, Brunet F, Bernard L, et al. Estrogen-related receptor gamma is an in vivo receptor of bisphenol A. FASEB J (2014) 28:3124–33. doi:10.1096/fj.13-240465
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
155. Takayanagi S, Tokunaga T, Liu X, Okada H, Matsushima A, Shimohigashi Y. Endocrine disruptor bisphenol A strongly binds to human estrogen-related receptor gamma (ERRgamma) with high constitutive activity. Toxicol Lett (2006) 167:95–105. doi:10.1016/j.toxlet.2006.08.012
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
156. Delfosse V, Grimaldi M, Le Maire A, Bourguet W, Balaguer P. Nuclear receptor profiling of bisphenol-A and its halogenated analogues. Vitam Horm (2014) 94:229–51. doi:10.1016/B978-0-12-800095-3.00009-2
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
157. Lonard DM, O’Malley BW. The expanding cosmos of nuclear receptor coactivators. Cell (2006) 125:411–4. doi:10.1016/j.cell.2006.04.021
PubMed Abstract | Full Text | CrossRef Full Text | Google Scholar
Keywords: DES, BPA, phthalates, nuclear receptors, human fetal testis
Citation: Rouiller-Fabre V, Guerquin MJ, N’Tumba-Byn T, Muczynski V, Moison D, Tourpin S, Messiaen S, Habert R and Livera G (2015) Nuclear receptors and endocrine disruptors in fetal and neonatal testes: a gapped landscape. Front. Endocrinol. 6:58. doi: 10.3389/fendo.2015.00058
Received: 16 December 2014; Accepted: 07 April 2015;
Published: 07 May 2015
Edited by:
David H. Volle, INSERM, FranceReviewed by:
Pascale Crepieux, Centre National de la Recherche Scientifique, FranceCopyright: © 2015 Rouiller-Fabre, Guerquin, N’Tumba-Byn, Muczynski, Moison, Tourpin, Messiaen, Habert and Livera. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Virginie Rouiller-Fabre, Unit of Genetic Stability, Stem Cells and Radiation, Laboratory of Development of the Gonads, CEA, DSV, iRCM, SCSR, LDG, BP6, Fontenay aux Roses F-92265, France,dmlyZ2luaWUucm91aWxsZXItZmFicmVAY2VhLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.