 Agnieszka Bierzynska
Agnieszka Bierzynska Katrina Soderquest
Katrina Soderquest Ania Koziell
Ania Koziell
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol., 23 January 2015
Sec. Clinical Diabetes
Volume 5 - 2014 | https://doi.org/10.3389/fendo.2014.00226
This article is part of the Research TopicPodocyte Pathology and NephropathyView all 11 articles
After decades of primarily morphological study, positional cloning of the NPHS1 gene was the landmark event that established aberrant podocyte genetics as a pivotal cause of malfunction of the glomerular filter. This ended any uncertainty whether genetic mutation plays a significant role in hereditary nephrotic syndromes (NS) and confirmed podocytes as critical players in regulating glomerular protein filtration. Although subsequent sequencing of candidate genes chosen on the basis of podocyte biology had less success, unbiased analysis of genetically informative kindreds and syndromic disease has led to further gene discovery. However, the 45 genes currently associated with human NS explain not more than 20–30% of hereditary and only 10–20% of sporadic cases. It is becoming increasingly clear both from genetic analysis and phenotypic data – including occasional response to immunosuppressive agents and post-transplant disease recurrence in Mendelian disease – that monogenic inheritance of abnormalities in podocyte-specific genes disrupting filter function is only part of the story. Recent advances in genetic screening technology combined with increasingly robust bioinformatics are set to allow identification and characterization of novel disease causing variants and more importantly, disease modifying genes. Emerging data also support a significant but incompletely characterized immunoregulatory component.
Nephrotic syndrome (NS) is one of the commonest kidney conditions to affect children and adults. It manifests as excessive leak of protein into the urine, with interstitial edema occurring through albumin loss, aggravated by salt and water retention. Moreover, secondary knock-on effects on lipid metabolism, hemostasis, and the endocrine system through loss of key binding proteins serve to augment morbidity and mortality. NS represents a heterogeneous group of conditions, some idiopathic, others with a clear genetic basis. It may be highly kidney specific, associated with extra-renal developmental malformations or occur as a complication of systemic disease. The mainstay of treatment is immunosuppression, also effective in about 8–10% of genetic cases. Non-responders generally progress to renal failure with kidney transplantation, the only life-saving treatment.
Despite the apparent heterogeneity, the unifying feature in NS is malfunction of the glomerular filtration barrier (GFB). This highly sophisticated macromolecular sieve with size and charge restricting characteristics is the primary ultra-filter of plasma by the kidney. It allows free flow of water and small solutes but restricts the passage of molecules >15 kDa; proteinuria occurs through loss of these normal permselective properties. The GFB comprises three layers: fenestrated endothelium, glomerular basement membrane (GBM) and podocytes, specialized terminally differentiated epithelial cells connected by slit diaphragms (SD), unique intercellular junctions interposed between interdigitating foot processes (1). Endothelial cells and podocytes have a negatively charged surface glycocalyx, which together with GBM sialoproteins and heparan sulfate gives the GFB an overall negative charge at physiological pH. Although size is the primary determinant of molecular filterability, recent detection of mutations in the main component of the podocyte glycocalyx, podocalyxin, in familial NS (2) supports additional charge selection through electrostatic repulsion.
Although damage to any of the three layers can result in significant proteinuria and kidney disease (3), podocytes are considered pivotal for maintaining barrier integrity. They encircle the glomerular capillaries creating a compact interdigitating network on the urinary side of the GFB. The connecting SDs integrate structural components of tight, adhesion, gap, and neuronal junctions to meet the diverse functional requirements of macromolecular filtering under high pressure while subjected to rapid changes in mechanical shear stress (4). SDs connect with the actin cytoskeleton to initiate signaling pathways that regulate podocyte function, namely plasticity of foot processes, mechanosensation, calcium flux, endocytosis, cell polarity, and cell survival. It is perhaps not surprising that podocyte gene mutations link to these key cellular functions. Podocytes also react very stereotypically to injury irrespective of whether this is acquired or resulting from an intrinsic developmental defect with reorganization of the actin cytoskeleton, foot process effacement, molecular re-characterization of SDs, apoptosis, and detachment from the GBM (5). These dramatic morphological changes correlate with dysregulation of specific markers of podocyte differentiation including WT1, PAX2, and nephrin again signifying an underlying molecular basis. Additionally, de-differentiated podocytes can attempt healing sometimes by excessive proliferation, with the eventual outcome of repair or cell death attributable to complex interplay of poorly defined genetic and epigenetic mechanisms (6–8).
Clinical data support the hypothesis that the predominant cellular lesion targets podocytes; GBM gene defects usually result in insidious plasma protein leak whereas defects in podocyte/slit-diaphragm genes cause precipitous leak and NS. Moreover, there is evidence for a potential role for immune-regulation, particularly in childhood. Podocytes express cytokine and chemokine receptors as well as Toll Like receptors (TLRs) (9) and can respond to immune stimuli both in vivo and in vitro. Moreover, emerging evidence indicates that podocyte injury in NS may sometimes result from an unknown circulating factor, cytokine imbalance, or immune complex injury, with rare genomic variants potentially dictating susceptibility or resistance to immune triggers and the degree of subsequent response.
Initial evidence supporting a molecular basis for NS came from positional cloning in familial cases and experimental animal models. Inherited diseases are frequently caused by mutations in genes with restricted expression patterns that generally do not cause early embryonic lethality but instead manifest at the time when gene function becomes critical for a specific tissue and subsequent survival (10). This is certainly the case for NPHS1 and NPHS2 (11, 12), the first podocyte genes cloned in NS encoding nephrin, an immunoglobulin superfamily member, and podocin an integral membrane protein both exclusively expressed at the podocyte SD supporting a key role in protein filtration. Mutations are associated with autosomal recessive NS manifesting at birth or early life during post-natal glomerular maturation and resultant increase in glomerular filtration.
Detection of WT1 mutations in human syndromic NS (13), together with murine transgenic approaches using WT1, CD2AP (14), and NEPH1 (15) underscored the contribution of developmentally regulated podocyte genes. Subsequent identification of mutations in ACTN4 (16) and INF2 (17) emphasized the central role of the actin cytoskeleton whereas gain of function mutations in TRCP6 linked podocyte disease to abnormalities of cellular calcium flux and associated signaling pathways (18). Inherited defects of mitochondrial (19) and lysosomal components (20) have also been shown to lead to profound podocyte dysfunction, suggesting that they have high energy requirements and turn over. This also lead to the discovery that lysosomes participate in autophagy, increasingly recognized as protecting podocytes against injury (21, 22) Other genes such as CD2AP that participate in endocytosis and/or actin assembly have also been detected in glomerular disease providing further important clues about podocyte biology.
Familial and candidate gene studies have provided important information about the molecular basis of fundamental podocyte functions such as slit-diaphragm signaling, regulation of actin cytoskeleton dynamics, and cell–matrix interactions. However, until recently, only limited information about the overall genetic landscape was available, which posed a challenge to correct interpretation of genetic findings. Previously, podocytes could be extracted with high purity, but low cell yields during isolation hampered complete characterization of the transcriptome and proteome. Primary cells are difficult to grow in culture, so immortalized podocytes provided a useful tool to study gene function but incompletely mirrored their in vivo counterparts at molecular level. However, recent development of a double fluorescent reporter mouse model and optimized bead perfusion protocol, combined with FACS sorting, microarray, proteomics, and unbiased mathematical biology has yielded >5223 differentially regulated genes within the podocyte transcriptome and >1280 translated proteins (23). These studies have revealed that the podocyte proteome is enriched with plasma membrane, cytoskeleton, and neuronal-type proteins. Subsequent functional analysis of transcriptome and proteome data resulted in seven gene clusters: endoplasmatic reticulum, ubiquitination, cytoskeleton, nuclear elements, mitochondria, peroxisome and protein transport, and cell junction. Interestingly, nuclear and translational proteins were depleted, reflecting the post-mitotic nature of podocytes and alternative splicing was reduced, perhaps to ensure discreet cellular regulation. This has also enabled more meaningful RNA sequencing, which has identified eight miRNA’s preferentially expressed in podocytes with significantly reduction fold change of miRNA target genes supporting miRNA-mediated mRNA degradation.
Improved description of the podocyte genome has also resulted in more effective interpretation of data from next generation sequencing (NGS) and to detect new variants or genes that cause or contribute to NS. NGS allows rapid sequencing of the whole exome or genome, and is becoming an increasingly powerful and cost effective method of analyzing the genetic contribution to any disorder. An unbiased approach can be taken to highlight new genes, new pathways, and new disease mechanisms through rigorous statistical genetics followed by functional experiments. This is especially relevant to NS where there is already known association with complex genetic determinants and this is likely to prove important mechanistically. However, as with any rare diseases, finding causal variants in NS suffers from twin problems of moderately low sample size and disease heterogeneity. In presumed Mendelian NS, the approach is still to search for genetic variants that segregate with disease within a family. In sporadic NS, we hypothesize that the condition will be caused by a rare variant in a small number of cases (limiting sample size and statistical power) and that the variant and, indeed the gene, may vary between cases. Added to this, although it is often assumed that there will be a heterozygous, homozygous, or compound heterozygous change that is causal, in common with related glomerular disorders, there is evidence for modifier effects in NS, at the genetic (24) and environmental level (25). This introduces the concept that NS is more complex than the Mendelian one podocyte gene, one disease situation as previously thought as variants are not always completely penetrant (26), synergistic activation between genes may occur and in non-heritable disease, an environmental trigger is needed to cause disease. There is now clear evidence that in at least one podocyte gene, the pathogenicity of a particular allele depends on the trans-associated mutation (27). These concepts are supported by increasing evidence that expected phenotype–genotype correlations are not always adhered to and pathological changes may develop focally (not in all glomeruli) and segmentally (only in parts of a glomerulus), a pattern suggestive of an initial insult precipitated by an environmental stressor, perhaps a viral infection, that leads to localized cell injury (28).
Attempts to analyze NGS data in rare yet moderately complex diseases such as NS initially tend to focus on three main issues, namely how to best divide the genome for maximum signal, which statistical test to use and how to computationally assess the function of any variants found. Unlike common complex conditions, variants in conditions such as NS are too rare to provide meaningful sample sizes for statistical testing on their own and must be collapsed or aggregated to the level of gene, network, or other biologically meaningful unit. That is, testing for enrichment of variants in NS versus a control population will likely involve counting the number of rare protein altering variants in each gene or other biological unit in cases and controls and finding genes with an excess of these rare variants, rather than searching for an enrichment of any one variant as would be the case in a common complex Genome Wide Association Study (GWAS) approach. In addition to deciding on the biological unit by which to combine variants, decisions also need to be made about the statistical techniques used to combine and then analyze variants (29, 30). There are then several databases and algorithms by which the functional deleteriousness of potential variants can be assessed [(31): CADD; (32): ClinVar; (33): HGMD; (34): SIFT; (35): PolyPhen]. These might be used throughout the analysis to remove samples with known causal variants before statistical testing, or choose likely deleterious variants during testing. They can also be used to examine the likelihood that new variants might be causal prior to investing resources in replication studies or functional testing in vitro or in vivo. Nonetheless, understanding genotype/phenotype relationships without powerful systems biology tools in place has become increasingly complicated as emerging NGS data reveal not only considerable genetic heterogeneity but also evidence showing that factors such as epigenetic modifications, imprinting, non-coding RNAs, and RNA editing may play an important role in determining phenotype (36).
Mutations in 45 genes have been associated with familial and sporadic NS to date (Figure 1). These disrupt function either through SD disassembly, damaging cell architecture or metabolism, disturbing cell–matrix interactions, and/or impeding signaling pathways. All are expressed in podocytes, but there is increasing evidence to suggest that other systems, including immunoregulatory, play a role. Moreover, mutations in podocyte genes currently only explain 20–30% of familial and 10–20% of sporadic NS. From a clinical perspective, the renal phenotype rarely correlates absolutely with genotype as histology overlaps, suggesting a final common pathway to glomerular damage. Nonetheless, mode of inheritance and age of onset can give important clues. Gene mutations detected in early life are biased toward developmental genes, podocyte/SD/GBM malformation, and autosomal recessive inheritance whereas in later life, mutations are more frequently autosomal dominant (AD), preferentially affecting genes that participate directly or indirectly in regulation of the actin cytoskeleton. Moreover, combined gene defects in more than one podocyte gene may play a role in the development of NS, for example, mutations in both NPHS1 and NPHS2 can cause a tri-allelic hit modifying phenotype (37), and bi-allelic trans-heterozygosity has been described for CD2AP and NPHS2 in sporadic NS (38). Additionally, ADCK4 appears to modify CoQ10 (39) Furthermore, R229Q, a non-neutral NPHS2 polymorphism that may predispose to NS in adults appears pathogenic only when associated with 3′ NPHS2 mutations in trans- (27), or deleterious mutation in another podocyte gene (37). Another emerging layer of complexity is miRNAs, for example, miR-193a downregulates the expression of WT1 in transgenic mice resulting in rapid and progressive NS (40). Other genes regulate podocyte differentiation and SD function by interacting synergistically on common enhancers or repressors, e.g., LMX1B, which combinatorially regulates NPHS2 with FoxC (41). This underlines the likely diversity of genetic interaction within podocytes, supporting a requirement for additional environmental factors and/or modifier genes for full phenotypic expression.
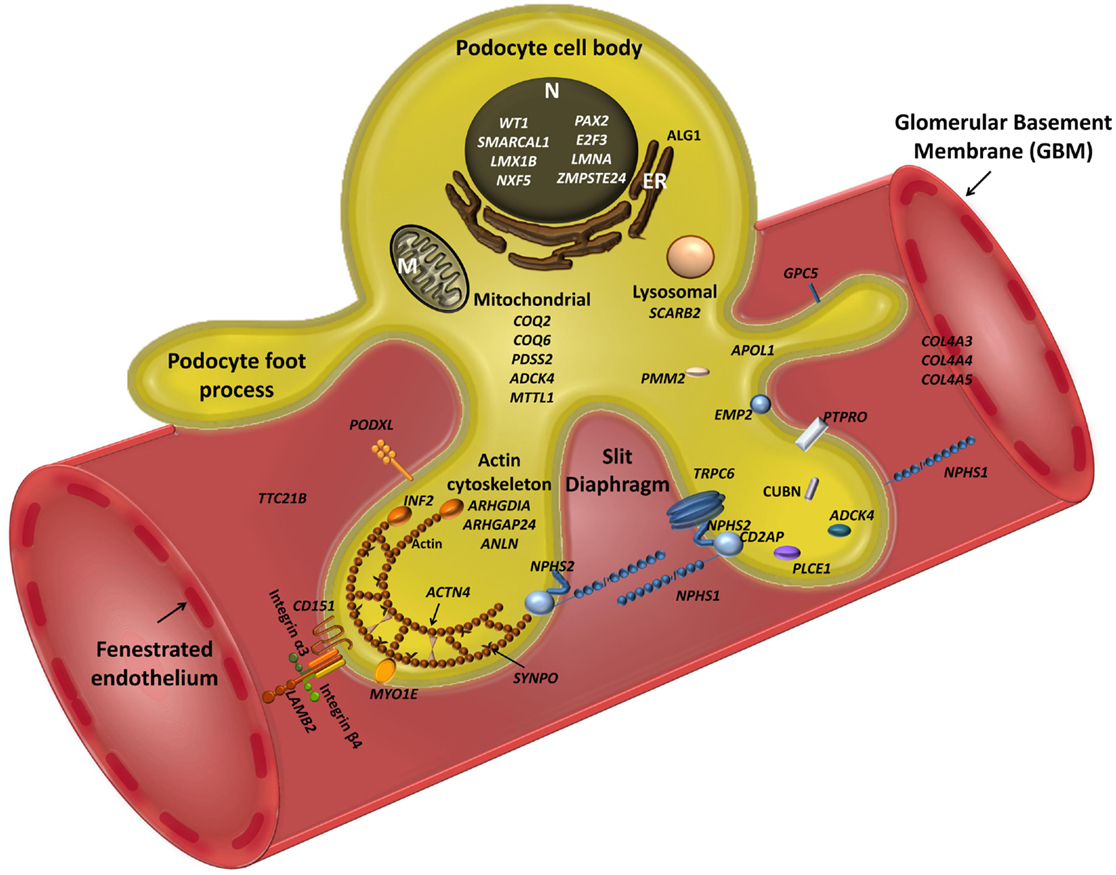
Figure 1. Schematic view of podocyte genes associated with steroid resistant nephrotic syndrome (with reference to Table 1). Podocyte cell with foot processes, slit diaphragm, the glomerular basement membrane (GBM), and fenestrated endothelium are shown. N, podocyte nucleus; ER, endoplasmic reticulum; M, mitochondrion. Slit diaphragm associated and adaptor proteins: nephrin (NPHS1), podocin (NPHS2), CD2-associated protein (CD2AP), PLCε1 (PLCE1), transient receptor potential channel 6 (TRPC6), and protein tyrosine phosphatase receptor type O (PTPRO). Nuclear proteins and transcription factors: Wilm’s tumor protein (WT1), SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A-like protein 1 (SMARCAL1), LIM Homeobox Transcription Factor 1β (LMX1B), nuclear RNA export factor 5 (NXF5), paired box protein (PAX2) and transcription factor E2F3 (E2F3), lamin A/C (LMNA), zinc metallopeptidase STE24 (ZMPSTE24). Actin cytoskeleton and signalling: podocalyxin (PODXL), inverted formin 2 (INF2), α-actinin-4 (ACTN4), synaptopodin (SYNPO), myosin 1E (MYO1E), Rho-GDP-dissociation inhibitor 1 (ARHGDIA), and Rho-GTPase-activating protein 24 (ARHGAP24), and actin-binding protein anillin (ANLN), cubilin (CUBN), tetratricopeptide repeat protein 21B (IFT139, TTC21B); epithelial membrane protein 2 (EMP2). Mitochondria-associated proteins: parahydroxybenzoate-polyprenyl transferase (COQ2), ubiquinone biosynthesis monooxygenase (COQ6), decaprenyl-diphosphate synthase subunit 2 (PDSS2), AarF domain-containing kinase 4 (ADCK4), and mitochondrially encoded tRNA leucine 1 (MTTL1). Metabolic and lysosomal proteins: chitobiosyldiphosphodolichol β-mannosyltransferase (ALG1), phosphomannomutase 2 (PMM2), and scavenger receptor class B, member 2 (SCARB2). GBM associated proteins: collagen 4 – COL4A3, 4, 5; integrin-α3 (ITGA3), integrin-β4 (ITGB4), laminin-β2 (LAMB2); CD151 antigen (CD151).
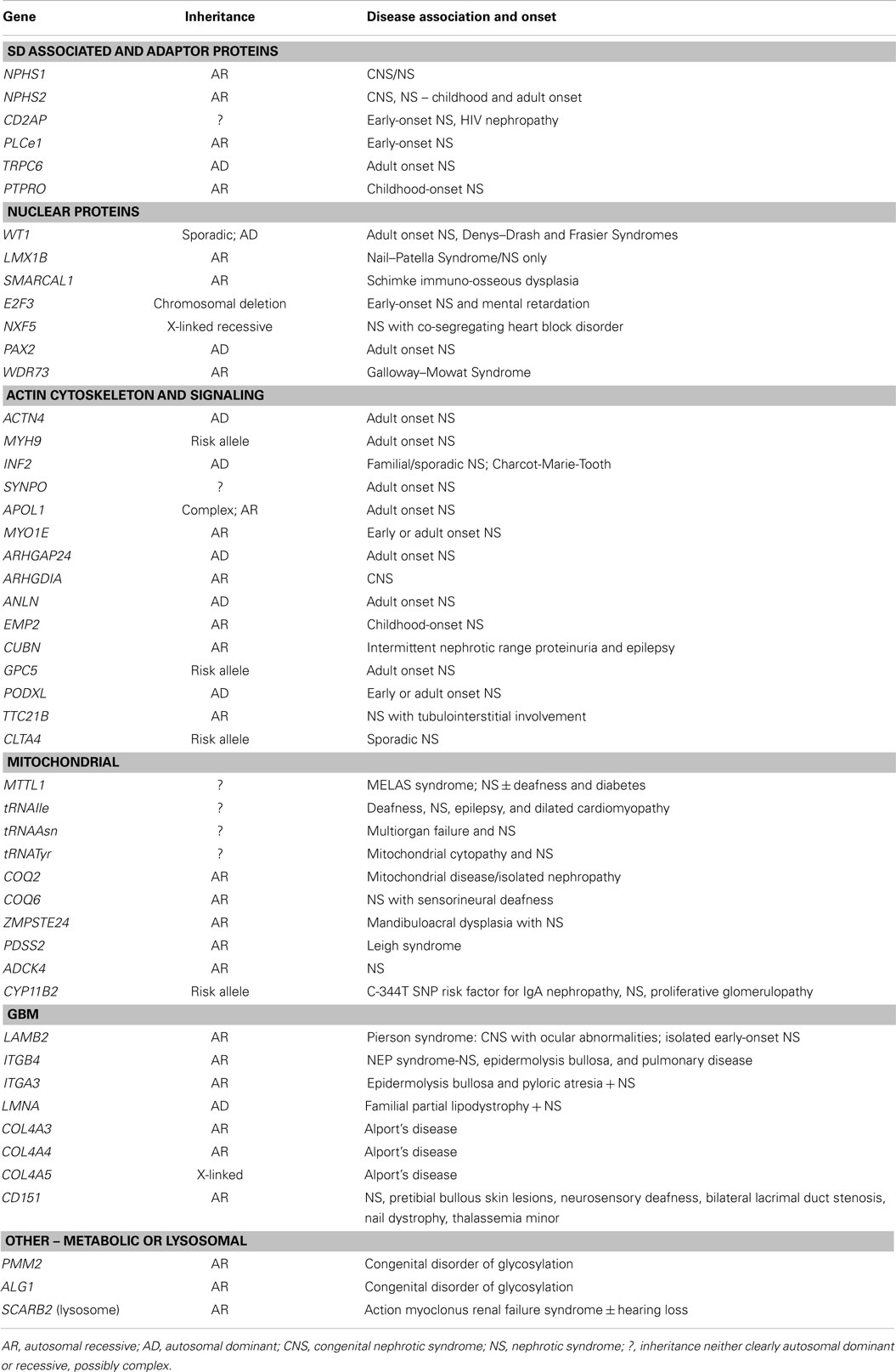
Table 1. Genes directly associated nephrotic syndrome.
Table 1 summarizes all genes currently associated with human NS. Broadly speaking, these can be categorized on the basis of function substantiated by the predicted effect of mutations. SD associated and adaptor proteins normally communicate between the SD and the podocyte cytoskeleton, forming multimeric signaling complexes associated with lipid raft micro-domains coordinating actin remodeling, cell survival, and endocytosis. The prototypes are nephrin (NPHS1), and podocin (NPHS2), which interact with proteins such as CD2AP (42) and TRPC6 to adapt between SD and cytoskeleton through a number of cytoskeletal linkers including F-actin and synaptopodin (43). More than 236 mutations in NPHS1 have been described (HGMD® Professional 2014.2), and 171 mutations in NPHS2 (http://www.lovd.nl/NPHS2, HGMD® Professional 2014.2). CD2AP haploinsufficiency causes NS, and is associated with trans-heterozygosity (38). Gain of function TRPC6 mutations cause AD NS mainly but not exclusively in adult life, by increasing calcium influx and dysregulating the actin cytoskeleton via aberrant signaling (18). Nuclear proteins regulate transcription, podocyte differentiation and homeostasis; mutations result in dysregulation of downstream targets often initiating catastrophic collapse of the entire podocyte-stabilizing system. WT1 mutations cause Denys–Drash (intersex, Wilm’s tumor, NS) and Frasier syndromes (intersex, NS, gonadoblastoma) and AD adult onset NS (44). LMX1B, a homeobox transcription factor, regulates SDs and is essential for the maintenance of the actin cytoskeleton. Haploinsufficiency causes Nail–Patella syndrome, a rare AD NS that can occur as just kidney specific disease (45). The most recent gene to be cloned is WDR73, its exact function in the podocyte is as yet unknown but mutations result in Galloway–Mowat syndrome, a rare association between microcephaly, diaphragmatic hernia, and SRNS (46). Proteins regulating the actin cytoskeleton are also targeted. ACTN4 (α-actinin-4) causes AD adult onset NS with incomplete penetrance. Here, mutant proteins show stronger affinity for F-actin and impair cytoskeletal function. Another actin regulating gene, INF2 belongs to the formin family and normally homodimerizes to inhibit actin depolymerization. Mutations are detected in a significant proportion of familial but not sporadic NS. Mutations in PLCe1 (phospholipase C epsilon) mostly cause early-onset NS by dysregulating the cytoskeleton and signaling, and may be variably penetrant with some missense mutations resulting in later onset disease responding to immunosuppression (47). PLCe1 mutations may also co-exist with trans-heterozygote mutations in other podocyte genes. PTPRO encodes a protein tyrosine phosphatase receptor located on the apical side of podocyte foot processes that controls the glomerular pressure/filtration rate (48). Only two splice-site mutations have been detected in PTPRO and cases partially responded to immunosuppression (49). Recently, mutations in ANLN (anillin), another F-Actin binding protein, were detected in familial NS (50), and other mutation screens of inherited disease have identified ARHGDIA (51) and ARHGAP24 (52), underpinning a key role for RHO GTPases and Rho–Rac signaling in regulating the podocyte cytoskeleton. MYO1E (53) and MYH9 (24), mutated in autosomal recessive NS regulate actin cytoskeletal function and cell shape. APOL1 mutations also result in increased NS in African Americans but the mechanism remains unclear (54). Mutations affecting protein expressed in the GBM may also disrupt GFB functions sufficiently to cause NS. LAMB2 mutations, encoding laminin β2, cause Pierson’s syndrome characterized by early-onset NS with or without microcoria, depending on the severity of the mutation (55). ITGA3 and ITGB4 encode integrins thought to be mainly passive GBM stabilizers, with mutations causing hyperglycosylation that prevents formation of functional integrins manifesting as NS and skin defects (56). COL4A3, A4, and A5 are key GBM proteins that cause Alport’s syndrome; recent data suggest caution in interpreting COL4A variants detected in apparent familial NS (57). Mitochondrial and other rare metabolic syndromes also present with NS, by disrupting podocyte metabolism.
Initial identification of monogenic defects correlating with morphological findings in podocytes in health and disease provided important mechanistic insights. However, this is now too simplistic, a model; podocyte genetic mechanisms are becoming increasingly complex, perhaps not surprising considering their super-specialist role within the GFB. Multiple levels of control involving modifier genes and di-genic and multi-genic inheritance are undoubtedly necessary to deal with epigenetic events and environmental effects (58). It is also likely that some gene variants are not directly causative, but rather modifiers of phenotype that may even arise from primary immune injury or presence of a circulating permeability factor. The advent of NGS and increasingly sophisticated bioinformatics approaches pave the way for exciting exploration of these alternative mechanisms, which undoubtedly play a key role in podocyte pathobiology.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Kriz W, Lemley KV. The role of the podocyte in glomerulosclerosis. Curr Opin Nephrol Hypertens (1999) 8:489–97. doi:10.1097/00041552-199907000-00014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
2. Barua M, Shieh E, Schlondorff J, Genovese G, Kaplan BS, Pollak MR. Exome sequencing and in vitro studies identified podocalyxin as a candidate gene for focal and segmental glomerulosclerosis. Kidney Int (2014) 85:124–33. doi:10.1038/ki.2013.354
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Menon MC, Chuang PY, He CJ. The glomerular filtration barrier: components and crosstalk. Int J Nephrol (2012) 2012:749010. doi:10.1155/2012/749010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Grahammer F, Schell C, Huber TB. The podocyte slit diaphragm – from a thin grey line to a complex signalling hub. Nat Rev Nephrol (2013) 9:587–98. doi:10.1038/nrneph.2013.169
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Mundel P, Shankland SJ. Podocyte biology and response to injury. J Am Soc Nephrol (2002) 13:3005–15. doi:10.1097/01.ASN.0000039661.06947.FD
6. Lasagni L, Lazzeri E, Shankland SJ, Anders HJ, Romagnani P. Podocyte mitosis – a catastrophe. Curr Mol Med (2013) 13:13–23. doi:10.2174/15665240130103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Hagemann JH, Haegele H, Muller S, Anders HJ. Danger control programs cause tissue injury and remodeling. Int J Mol Sci (2013) 14(2013):11319–. doi:10.3390/ijms140611319
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Hayashi K, Sasamura H, Nakamura M, Azegami T, Oguchi H, Sakamaki Y, et al. KLF4-dependent epigenetic remodeling modulates podocyte phenotypes and attenuates proteinuria. J Clin Invest (2014) 124:2523–37. doi:10.1172/JCI69557
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Ma J, Chadban SJ, Zhao CY, Chen X, Kwan T, Panchapakesan U, et al. TLR4 activation promotes podocyte injury and interstitial fibrosis in diabetic nephropathy. PLoS One (2014) 9:e97985. doi:10.1371/journal.pone.0097985
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Cai JJ, Petrov DA. Relaxed purifying selection and possibly high rate of adaptation in primate lineage-specific genes. Genome Biol Evol (2010) 2:393–409. doi:10.1093/gbe/evq019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Kestila M, Lenkkeri U, Mannikko M, Lamerdin J, McCready P, Putaala H, et al. Positionally cloned gene for a novel glomerular protein – nephrin – is mutated in congenital nephrotic syndrome. Mol Cell (1998) 1:575–82. doi:10.1016/S1097-2765(00)80057-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet (2000) 24:349–54. doi:10.1038/74166
13. Pelletier J, Bruening W, Kashtan CE, Mauer SM, Manivel JC, Striegel JE, et al. Germline mutations in the Wilms’ tumor suppressor gene are associated with abnormal urogenital development in Denys-Drash syndrome. Cell (1991) 67:437–47. doi:10.1016/0092-8674(91)90194-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Kim JM, Wu H, Green G, Winkler CA, Kopp JB, Miner JH, et al. CD2-associated protein haploinsufficiency is linked to glomerular disease susceptibility. Science (2003) 300:1298–300. doi:10.1126/science.1081068
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Donoviel DB, Freed DD, Vogel H, Potter DG, Hawkins E, Barrish JP, et al. Proteinuria and perinatal lethality in mice lacking NEPH1, a novel protein with homology to NEPHRIN. Mol Cell Biol (2001) 21:4829–36. doi:10.1128/MCB.21.14.4829-4836.2001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet (2000) 24:251–6. doi:10.1038/73456
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Brown EJ, Schlondorff JS, Becker DJ, Tsukaguchi H, Tonna SJ, Uscinski AL, et al. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat Genet (2010) 42:72–6. doi:10.1038/ng.505
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science (2005) 308:1801–4. doi:10.1126/science.1106215
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Diomedi-Camassei F, Di GS, Santorelli FM, Caridi G, Piemonte F, Montini G, et al. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol (2007) 18:2773–80. doi:10.1681/ASN.2006080833
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
20. Berkovic SF, Dibbens LM, Oshlack A, Silver JD, Katerelos M, Vears DF, et al. Array-based gene discovery with three unrelated subjects shows SCARB2/LIMP-2 deficiency causes myoclonus epilepsy and glomerulosclerosis. Am J Hum Genet (2008) 82:673–84. doi:10.1016/j.ajhg.2007.12.019
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Hartleben B, Godel M, Meyer-Schwesinger C, Liu S, Ulrich T, Kobler S, et al. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest (2010) 120:1084–96. doi:10.1172/JCI39492
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Zeng C, Fan Y, Wu J, Shi S, Chen Z, Zhong Y, et al. Podocyte autophagic activity plays a protective role in renal injury and delays the progression of podocytopathies. J Pathol (2014) 234:203–13. doi:10.1002/path.4382
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Boerries M, Grahammer F, Eiselein S, Buck M, Meyer C, Goedel M, et al. Molecular fingerprinting of the podocyte reveals novel gene and protein regulatory networks. Kidney Int (2013) 83:1052–64. doi:10.1038/ki.2012.487
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Kopp JB, Smith MW, Nelson GW, Johnson RC, Freedman BI, Bowden DW, et al. MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat Genet (2008) 40:1175–84. doi:10.1038/ng.226
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Kriz W, Shirato I, Nagata M, LeHir M, Lemley KV. The podocyte’s response to stress: the enigma of foot process effacement. Am J Physiol Renal Physiol (2013) 304:F333–47. doi:10.1152/ajprenal.00478.2012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Cooper DN, Krawczak M, Polychronakos C, Tyler-Smith C, Kehrer-Sawatzki H. Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease. Hum Genet (2013) 132:1077–130. doi:10.1007/s00439-013-1331-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Tory K, Menyhard DK, Woerner S, Nevo F, Gribouval O, Kerti A, et al. Mutation-dependent recessive inheritance of NPHS2-associated steroid-resistant nephrotic syndrome. Nat Genet (2014) 46:299–304. doi:10.1038/ng.2898
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Yamashita M, Millward CA, Inoshita H, Saikia P, Chattopadhyay S, Sen GC, et al. Antiviral innate immunity disturbs podocyte cell function. J Innate Immun (2013) 5:231–41. doi:10.1159/000345255
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Dering C, Hemmelmann C, Pugh E, Ziegler A. Statistical analysis of rare sequence variants: an overview of collapsing methods. Genet Epidemiol (2011) 35(Suppl 1):S12–7. doi:10.1002/gepi.20643
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Stitziel NO, Kiezun A, Sunyaev S. Computational and statistical approaches to analyzing variants identified by exome sequencing. Genome Biol (2011) 12(9):227. doi:10.1186/gb-2011-12-9-227
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet (2014) 46:310–5. doi:10.1038/ng.2892
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res (2014) 42:D980–5. doi:10.1093/nar/gkt1113
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet (2014) 133:1–9. doi:10.1007/s00439-013-1358-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res (2001) 11:863–74. doi:10.1101/gr.176601
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
35. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet (2013) 7:7. doi:10.1002/0471142905.hg0720s76
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Gottlieb B, Beitel LK, Trifiro M. Changing genetic paradigms: creating next-generation genetic databases as tools to understand the emerging complexities of genotype/phenotype relationships. Hum Genomics (2014) 8:9. doi:10.1186/1479-7364-8-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Koziell A, Grech V, Hussain S, Lee G, Lenkkeri U, Tryggvason K, et al. Genotype/phenotype correlations of NPHS1 and NPHS2 mutations in nephrotic syndrome advocate a functional inter-relationship in glomerular filtration. Hum Mol Genet (2002) 11:379–88. doi:10.1093/hmg/11.4.379
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Lowik M, Levtchenko E, Westra D, Groenen P, Steenbergen E, Weening J, et al. Bigenic heterozygosity and the development of steroid-resistant focal segmental glomerulosclerosis. Nephrol Dial Transplant (2008) 23:3146–51. doi:10.1093/ndt/gfn208
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Ashraf S, Gee HY, Woerner S, Xie LX, Vega-Warner V, Lovric S, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest (2013) 123:5179–89. doi:10.1172/JCI69000
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Gebeshuber CA, Kornauth C, Dong L, Sierig R, Seibler J, Reiss M, et al. Focal segmental glomerulosclerosis is induced by microRNA-193a and its downregulation of WT1. Nat Med (2013) 19:481–7. doi:10.1038/nm.3142
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. He B, Ebarasi L, Zhao Z, Guo J, Ojala JR, Hultenby K, et al. Lmx1b and FoxC combinatorially regulate podocin expression in podocytes. J Am Soc Nephrol (2014) 25(12):2764–77. doi:10.1681/ASN.2012080823
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Shih NY, Li J, Karpitskii V, Nguyen A, Dustin ML, Kanagawa O, et al. Congenital nephrotic syndrome in mice lacking CD2-associated protein. Science (1999) 286:312–5. doi:10.1126/science.286.5438.312
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Tang VW, Brieher WM. FSGS3/CD2AP is a barbed-end capping protein that stabilizes actin and strengthens adherens junctions. J Cell Biol (2013) 203:815–33. doi:10.1083/jcb.201304143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Hall G, Gbadegesin RA, Lavin P, Wu G, Liu Y, Oh EC, et al. A novel missense mutation of Wilms’ tumor 1 causes autosomal dominant FSGS. J Am Soc Nephrol (2014). doi:10.1681/ASN.2013101053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Boyer O, Woerner S, Yang F, Oakeley EJ, Linghu B, Gribouval O, et al. LMX1B mutations cause hereditary FSGS without extrarenal involvement. J Am Soc Nephrol (2013) 24:1216–22. doi:10.1681/ASN.2013020171
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Colin E, Huynh Cong E, Mollet G, Guichet A, Gribouval O, Arrondel C, et al. Loss-of-function mutations in WDR73 are responsible for microcephaly and steroid-resistant nephrotic syndrome: Galloway-Mowat syndrome. Am J Hum Genet (2014) 95(6):637–48. doi:10.1016/j.ajhg.2014.10.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Hinkes B, Wiggins RC, Gbadegesin R, Vlangos CN, Seelow D, Nurnberg G, et al. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet (2006) 38:1397–405. doi:10.1038/ng1918
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Wharram BL, Goyal M, Gillespie PJ, Wiggins JE, Kershaw DB, Holzman LB, et al. Altered podocyte structure in GLEPP1 (Ptpro)-deficient mice associated with hypertension and low glomerular filtration rate. J Clin Invest (2000) 106:1281–90. doi:10.1172/JCI7236
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Ozaltin F, Ibsirlioglu T, Taskiran EZ, Baydar DE, Kaymaz F, Buyukcelik M, et al. Disruption of PTPRO causes childhood-onset nephrotic syndrome. Am J Hum Genet (2011) 89:139–47. doi:10.1016/j.ajhg.2011.05.026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Gbadegesin RA, Hall G, Adeyemo A, Hanke N, Tossidou I, Burchette J, et al. Mutations in the gene that encodes the F-actin binding protein anillin cause FSGS. J Am Soc Nephrol (2014) 25:1991–2002. doi:10.1681/ASN.2013090976
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Gupta IR, Baldwin C, Auguste D, Ha KC, El AJ, Fahiminiya S, et al. ARHGDIA: a novel gene implicated in nephrotic syndrome. J Med Genet (2013) 50:330–8. doi:10.1136/jmedgenet-2012-101442
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Akilesh S, Suleiman H, Yu H, Stander MC, Lavin P, Gbadegesin R, et al. Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J Clin Invest (2011) 121:4127–37. doi:10.1172/JCI46458
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Mele C, Iatropoulos P, Donadelli R, Calabria A, Maranta R, Cassis P, et al. MYO1E mutations and childhood familial focal segmental glomerulosclerosis. N Engl J Med (2011) 365:295–306. doi:10.1056/NEJMoa1101273
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science (2010) 329:841–5. doi:10.1126/science.1193032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Matejas V, Hinkes B, Alkandari F, Al-Gazali L, Annexstad E, Aytac MB, et al. Mutations in the human laminin beta2 (LAMB2) gene and the associated phenotypic spectrum. Hum Mutat (2010) 31:992–1002. doi:10.1002/humu.21304
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Kambham N, Tanji N, Seigle RL, Markowitz GS, Pulkkinen L, Uitto J, et al. Congenital focal segmental glomerulosclerosis associated with beta4 integrin mutation and epidermolysis bullosa. Am J Kidney Dis (2000) 36:190–6. doi:10.1053/ajkd.2000.8293
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Malone AF, Phelan PJ, Hall G, Cetincelik U, Homstad A, Alonso AS, et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int (2014) 86(6):1253–9. doi:10.1038/ki.2014.305
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Stieger N, Worthmann K, Teng B, Engeli S, Das AM, Haller H, et al. Impact of high glucose and transforming growth factor-β on bioenergetic profiles in podocytes. Metabolism (2012) 61(8):1073–86. doi:10.1016/j.metabol.2011.12.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: podocyte nephropathy, podocytes, genetic predisposition to disease, gene mutation, nephrotic syndrome, nephrotic genes
Citation: Bierzynska A, Soderquest K and Koziell A (2015) Genes and podocytes – new insights into mechanisms of podocytopathy. Front. Endocrinol. 5:226. doi: 10.3389/fendo.2014.00226
Received: 28 September 2014; Paper pending published: 06 November 2014;
Accepted: 09 December 2014; Published online: 23 January 2015.
Edited by:
Barbara Lewko, Medical University of Gdansk, PolandReviewed by:
Kazunori Sango, Tokyo Metropolitan Institute of Medical Science, JapanCopyright: © 2015 Bierzynska, Soderquest and Koziell. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ania Koziell, Division of Transplantation Immunology and Mucosal Biology, Department of Experimental Immunobiology, Faculty of Life Sciences and Medicine, 5th Floor Tower Wing, Guy’s Hospital, Great Maze Pond, London SE1 9RT, UK e-mail:YW5pYS5rb3ppZWxsQGtjbC5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.