 Joseph A. M. J. L. Janssen
Joseph A. M. J. L. Janssen Aimee J. Varewijck
Aimee J. Varewijck
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 23 December 2014
Sec. Cancer Endocrinology
Volume 5 - 2014 | https://doi.org/10.3389/fendo.2014.00224
This article is part of the Research Topic The role of the Insulin/IGFs system in cancer: critical points in dysregulation, targeting problems and new blocking strategies View all 17 articles
The IGF-I receptor (IGF-IR) has been studied as an anti-cancer target. However, monotherapy trials with IGF-IR targeted antibodies or with IGF-IR specific tyrosine kinase inhibitors have, overall, been very disappointing in the clinical setting. This review discusses potential reasons why IGF-I R targeted therapy fails to inhibit growth of human cancers. It has become clear that intracellular signaling pathways are highly interconnected and complex instead of being linear and simple. One of the most potent candidates for failure of IGF-IR targeted therapy is the insulin receptor isoform A (IR-A). Activation of the IR-A by insulin-like growth factor-II (IGF-II) bypasses the IGF-IR and its inhibition. Another factor may be that anti-cancer treatment may reduce IGF-IR expression. IGF-IR blocking drugs may also induce hyperglycemia and hyperinsulinemia, which may further stimulate cell growth. In addition, circulating IGF-IRs may reduce therapeutic effects of IGF-IR targeted therapy. Nevertheless, it is still possible that the IGF-IR may be a useful adjuvant or secondary target for the treatment of human cancers. Development of functional inhibitors that affect the IGF-IR and IR-A may be necessary to overcome resistance and to make IGF-IR targeted therapy successful. Drugs that modify alternative downstream effects of the IGF-IR, so called “biasing agonists,” should also be considered.
Activation of the insulin-like growth factor-I receptor (IGF-IR) pathway has been found to be essential for initiation and growth of cancers (1). Many types of tumor cells often overexpress IGF-IRs (1). In addition, surrounding stromal tissue of tumor cells produces IGF-I and IGF-II (2) and activation of the IGF-IRs of tumor cells may be mediated by IGFs in a paracrine and autocrine way (3). Prospective studies suggest that individuals with circulating IGF-I concentrations at the high end of the normal range have an increased risk for several common cancers (1, 4). In many tumors, binding of IGF-I and IGF-II to the IGF-IR, inhibits apoptosis and promotes cell proliferation (5). In the last years, insulin has been also associated with the development of cancer in subjects with type 2 diabetes. Although all the underlying mechanisms need to be clarified, it has been suggested that hyperinsulinemia upregulates growth hormone receptor (GHR) expression and thereby upregulates hepatic IGF-I production (6). In addition, hyperinsulinemia also suppresses the levels of IGFBP1 and IGFBP2, thereby increasing IGF-I bioavailability (7).
The insulin-IGF family comprises a complex molecular signaling pathway (8). Depending on which regions are being compared, the IR and IGF-IR have sequence similarities varying from 41 to 84% (9). Both the IR and IGF-IR belong to the family of ligand-activated receptor kinases. The gene coding for the IR is localized on chromosome 19 and is composed of 22 exons (10, 11). Alternative mRNA splicing results in the expression of two isoforms: one containing exon 11 (the “classical” IR-B) and one missing exon 11 (the IR-A) (12). Exon 11 encodes for 12 amino acids localized at the C-terminal part of the alpha chain of the IR. Deletion of this exon has important functional consequences: IR-A has high affinity for insulin and IGF-II but binds IGF-I with low affinity. IR-A binds insulin with 1.5-fold higher affinity than IR-B and possesses a higher dissociation and internalization rate (13). Therefore, in cells with increased IR-A: IR-B ratios, insulin mainly signals through IR-A (14). IR-B is responsible for the classic metabolic responses induced by insulin, and also binds IGF-I and IGF-II with low and intermediate affinity, respectively (15). The expression profile of both IR isoforms is tissue-specific (12, 16). Interestingly, an increase in the IR-A to IR-B ratio have been reported in type 2 diabetes (17, 18).
All this information is mainly obtained by measurements of mRNA levels of IR-A and IR-B in different tissues. However, at present there is no good information about the exact expression of protein levels of IR-A and IR-B at the cell surface due to a lack of antibodies which can distinguish both isoforms of the IR.
In contrast to insulin, most of the IGFs in the circulation and in other extracellular fluids, form complexes to six high-affinity IGF-binding proteins (IGFBPs) (5). IGFBPs regulate the half-life and bioavailability of IGF-I and IGF-II and modulate their accessibility to the receptor (19). Under physiological circumstances but also in cancers partial proteolysis of IGFBP-3 and other IGFBPs may be an important mechanism for regulating IGF-I bioavailability (20).
Since most mammalian cells express both the IGF-IR and the IR, although at different expression levels, functional heterodimers can form between the IGF-IR and IR isoforms (15). A significant fraction of both IRs and IGF-IRs is present as hybrids in most mammalian tissues, including those that are considered to be classic targets of insulin (21). The IR-A/IGF-IR hybrids bind insulin and both IGFs with similar affinity, whereas IR-B/IGF-IR hybrids mainly behave like an IGF-IR, with high affinity for IGF-I, low affinity for IGF-II, and insignificant affinity for insulin (22). Slaaby et al. postulated that a possible explanation for this latter phenomenon may be that to achieve high-affinity binding, IGF-I requires a receptor monomer, while insulin requires a homodimeric IR (23). Thus, IGF-I may bind to the IGF-IR half of a hybrid receptor irrespective of what the molecular structure of the other half receptor is, while insulin requires a homodimeric IR to achieve this (23). Both IGFs and insulin stimulated cell proliferation and migration more effectively in cells containing IGF-IR/IR-A hybrids than in cells containing IGF-IR/IR-B hybrids (22). When considering all possible combinations of homodimer and hybrid receptors of the insulin/IGF signaling pathway, there are at least six tyrosine kinase receptors potentially involved in signal transduction (Figure 1).
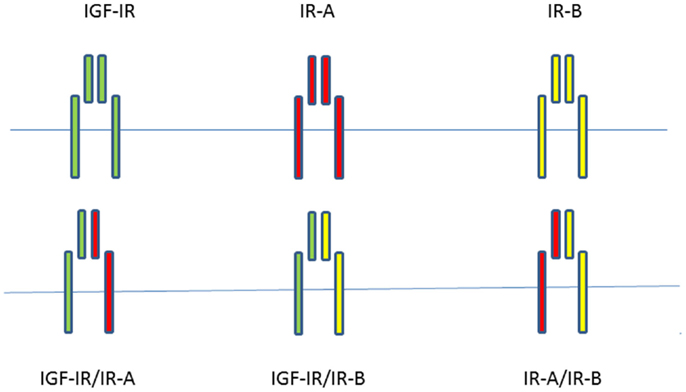
Figure 1. When considering all possible combinations of homodimer and hybrid receptors of the insulin/IGF signaling pathway, there are at least six potentially tyrosine kinase receptors involved in signal transduction.
The IR/IGF-IR signaling pathway also has extensive cross-talk with the GHR, estrogen receptor (ER), androgen receptor (AR), epidermal growth factor receptor (EGFR), and human epidermal growth factor receptor 2 (HER-2) signaling pathways (24–27) (see below). On the other hand EGFR mediated growth requires the presence of functional IGF-IRs for its mitogenic activities (28).
While IGF-IR can heterodimerize with the IRs, as discussed above, it can also form heterodimers with receptors from other families (8). For example, the IGF-IR may form a heterodimer with the EGFR (29). Inhibition of one receptor in these hybrids may shift the signaling pathway in favor of the other available counterpart receptor (8, 30, 31).
Specific signaling pathway (like IGF-IR pathway) dependence of certain cancers creates, at least theoretically, an Achilles heel for tumor maintenance, which can be exploited therapeutically (32). As an anti-cancer target, the IGF-IR has been studied in many clinical trials over the past years (33). Three major strategies have been used: (1) monoclonal antibodies against the IGF-IR, (2) monoclonal antibodies against IGF-I or IGF-II, and (3) tyrosine kinase inhibitors of the IGF-IR (33). Monoclonal IGF-IR antibodies block ligand binding and induce receptor internalization and degradation (34). As a class effect, IGF-IR blocking drugs cause insulin resistance, hyperinsulinemia, and (often mild and reversible) hyperglycemia (35). Besides through decreased IGF-I action, insulin resistance induced by IGF-IR blocking drugs is also thought to be caused by an increased secretion of GH and some IGFBPs (36). The increased GH secretion is the result of a direct blockade of the pituitary IGF-IRs by these agents (36). Monoclonal antibodies against IGF-I or IGF-II act by neutralizing IGF-I and IGF-II, preventing receptor activation without affecting glucose tolerance (37). The majority of IGF-IR tyrosine kinase inhibitors act by competing with ATP for binding in the IGF-IR kinase domain, although also non-competitive agents are being developed (37). They may induce hyperglycemia by direct inhibition of IR-kinase (37).
Although, initially promising effects have been reported in phase 2 trials, more extensive experience has learned that a potential clinical benefit for monotherapies blocking IGF-I or the IGF-IR is only limited to a small subset of patients with specific cancers (sarcoma, Ewing sarcoma non-small cell lung cancer and some other chemotherapy-refractory solid tumors) (38–40), while the majority of studies have been disappointing and have failed to show any clinical benefit in the treatment of cancers (33). In the next sections, we will discuss some background and possible reasons for the failure of these therapies.
Recent DNA and RNA sequencing projects have revealed recurrent somatic alterations in several genes that are drivers of oncogenesis. Most cancers contain many mutations and are thus not dependent on one single oncogenic mutation (32). They activate RAS, BRAF, EGFR, and many others. These driver alterations can give rise to a tumor dependency on a particular signaling pathway (32). However, in clinical settings no cancer specific mutations of the IGF-IR or its ligands have been described to date (41). Moreover, the IGF-IRs of cancer cells also do not contain intrinsic (post) receptor abnormalities (42).
Owing to the presence of an intracellular tyrosine kinase domain, the IGF-IR is usually classified as a tyrosine kinase receptor. Accordingly, phosphorylation was until very recently, considered to be the central process governing IGF-IR signaling (42). As a consequence, to date most therapies targeting the IGF-IR have mainly been designed aiming to block phosphorylation mediated signaling by preventing receptor–ligand interaction or by limiting kinase activation (42).
The common mechanisms of action of IGF-IR targeted antibodies are that they block the IGF-IR from ligand binding and induce internalization/degradation of the IGF-IR (43). Most developed antibodies directed against the IGF-IR are highly specific for the IGF-IR and do not bind to IRs. Nevertheless, it has been found that antibodies directed against the IGF-IR may not only downregulate IGF-IR homodimers but also the IGF-IR/IR hybrids (44). Interestingly, in a model of prostate cancer, following IGF-IR inhibition, the IR was able to compensate for and mediate IGF-I induced mitogenic signaling (45). Moreover, in breast cancer cells, downregulation of IGF-IRs by siRNA sensitized these cells to insulin-mediated activation of intracellular downstream signaling pathways (46).
The following reasons for the failure of IGF-IR targeted therapy to inhibit the growth of human cancers have been suggested (47):
(1) Mutations in PI3K (or PTEN deletions) constitutively activate Akt (and subsequent cell cycle progression genes) and render cells resistant to IGF-IR targeted therapy. This has not been proven so far.
(2) Targeting the IGF-IR in some cells (e.g., hematopoietic precursors and neuronal cells) could actually inhibit a tendency to differentiation.
(3) An increased IR-A cell surface expression upon downregulation of IGF-IR signaling. The Vignieri–Belfiori group has convincingly demonstrated that stimulation of the IR-A – especially by IGF-II – may induce mitogenic signals and replace IGF-IR signaling in human tumors (13, 47). Thus activation of the IR-A by IGF-II bypasses IGF-IR signaling and its inhibition.
(4) Anti-cancer treatment may cause a reduction of IGF-IR expression at the cellular membrane. For example, treatment of human breast tumors with the anti-estrogen tamoxifen causes reduction in membranous IGF-IR expression (48). A reduction in membranous IGF-IR expression may occur as an adaptive resistance mechanism and this may play an important role in the inability to sustain IGF-IR blockade (8). In addition, absence of N-linked glycosylation has been reported to change insertion of the IGF-IR into the cell membrane and induces resistance to IGF-IR directed antibodies (49). Nuclear translocation of the IGF-IR may be another factor, which limits accessibility and actions of IGF-IR directed antibodies. On the other hand, exclusive nuclear localization of IGF-IR has been reported to be associated with a better response when patients were treated with IGF-IR targeted antibodies (50).
(5) Most important is the complexity of regulatory mechanisms in cancer cells, including the existence of subpopulations in each cancer with different mutations and sensitivities. As discussed in the previous paragraph, the IGF system is not a unique tumor driver and each of these subpopulations can be regulated by a variety of genes and large and small non-coding RNAs (the “dark matter” of cellular function) (47). Moreover, signaling pathways in cells are interconnected and these interactions are dynamic in time (51). It has become clear that all pathways previously thought to be linear are in fact highly interconnected into complex signaling pathways (32). Receptor cross-talk can occur through ligand interactions or via common downstream molecular pathways and modulation (8). This cross-talk gives tumors additional signaling opportunities and the potential to evade regulatory checkpoints (8). For example, the inhibition of the IGF-1R pathway by cixutumumab results in stimulation of the Akt/mTOR pathway by increasing synthesis of EGFR, Akt1, and antiapoptotic surviving proteins (52).
As discussed above, IGF-IR blocking drugs may also induce hyperglycemia, hyperinsulinemia, and an increased GH secretion. This may paradoxically contribute to proliferation of tumor cells by several mechanisms:
(1) Although phosphorylation of IGF-IR tyrosine residues is generally considered to be the initial activation step within the intracellular IGF-IR signaling pathway, it has been found that cells undergo a signaling switch under hyperglycemic conditions (53). After this switch, a completely different mechanism is utilized to activate mitogenic pathways downstream of the IGF-IR and this activation is independent of IGF-IR tyrosine phosphorylation (54).
(2) Secretion of insulin is mainly regulated by glucose levels. IGF-IR blocking drugs increase glucose levels, which may induce hyperinsulinemia and thereby tumor growth by directly stimulating IR-A and indirectly by inducing resistance to chemotherapy (55).
(3) An increased expression of the GHR is a characteristic of a variety of human cancers while GH may induce tumor growth, independent of IGF-I action (56).
Recently circulating IGF-IR levels were reported (57). We hypothesize that circulating (soluble) IGF-IRs in cancer patients may be another important factor responsible for the observed discrepancy between in vitro and in vivo effects of IGF-IR targeted therapy: antibodies against a tumor-target largely remain in the blood and usually no more than 20% of the administered antibody dose typically interacts with the tumor (58). Formation of complexes between the soluble IGF-IRs and antibodies directed against the IGF-IR may further reduce the amount of IGF-IR antibodies leaving the circulation to interact with IGF-IRs expressed on the surface of cancer cells (Figure 2A). In addition, these complexes displace IGF-I and IGF-II from the circulating soluble IGF-IRs thereby (paradoxically) increasing the amount of free IGF-I and IGF-II that can leave the circulation to stimulate IGF-IRs displayed on the surface of cancer cells (Figures 2B,C).
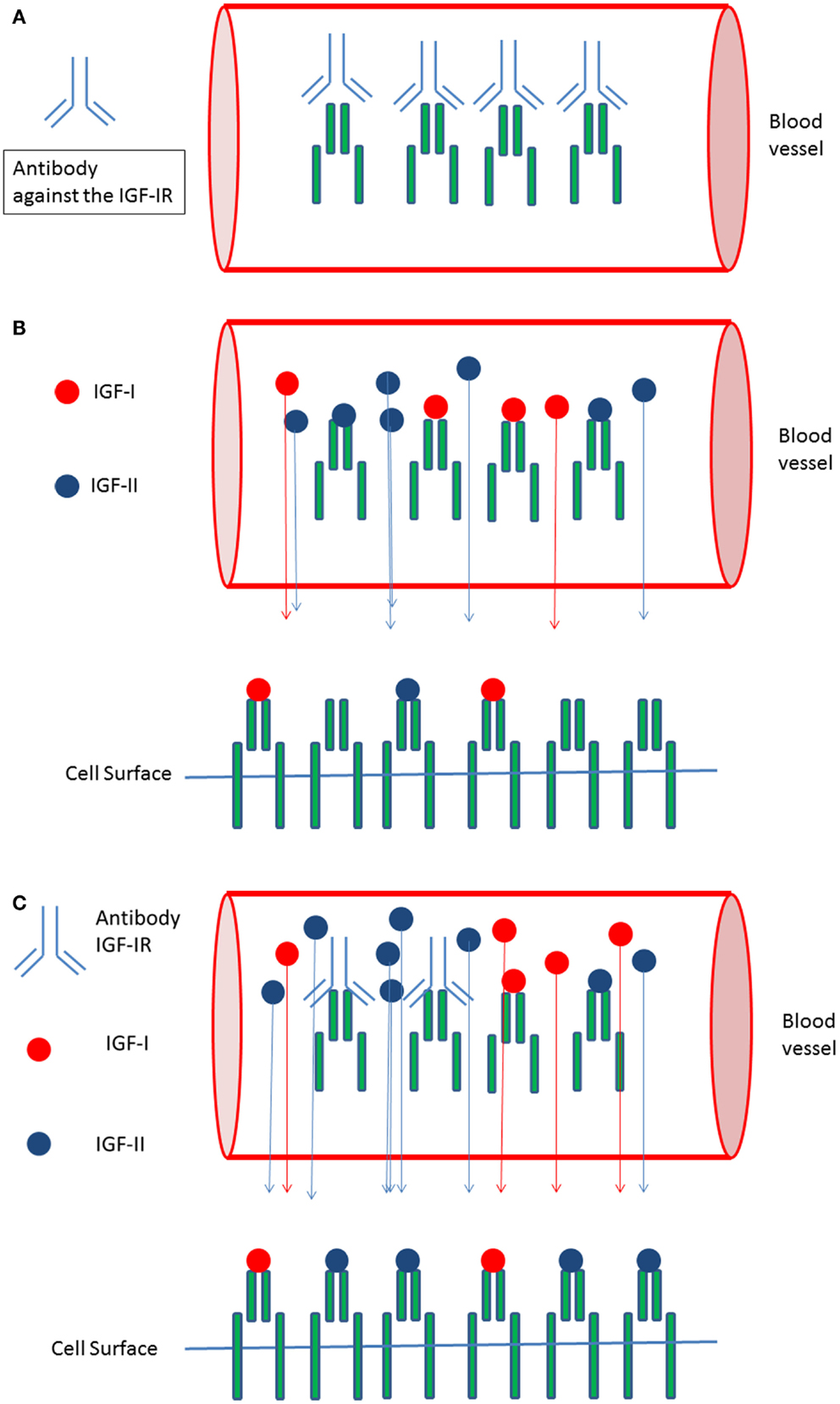
Figure 2. Circulating IGF-I receptors in cancer patients may form complexes with IGF-IR directed antibodies and this may reduce therapeutic effects of IGF-I receptor antibodies in vivo: (A) In the circulation soluble IGF-I receptors and antibodies directed against the IGF-IR may form complexes. This may limit the availability of IGF-IR antibodies leaving the circulation and reduce the amount of IGF-IR antibodies leaving the circulation to inhibit IGF-I receptors present at the cell surface of cancer cells. (B) Only “free” IGF-I and IGF-II may leave the circulation to bind to IGF-I receptors present at the cell surface of cancer cells. Complexes formed between the IGFs and the soluble IGF-Rs are to big to pass the vessel wall and to leave the circulation. (C) Formation of complexes between IGF-IR directed antibodies and soluble IGF-IR displace IGF-I and IGF-II from the soluble IGF-I receptors thereby (paradoxically) increasing the total amount of free IGF-I and IGF-II that can leave the circulation to stimulate IGF receptors displayed at the cell surface of cancer cells.
Given the complex signaling pathways that converge on those activated by the IGF-IR, it is not surprising that for most cancers IGF-IR targeted monotherapy in vivo has been disappointing (8, 59). Although IGF-IR targeted monotherapy has essentially been abandoned, it is still possible that targeting the IGF-IR may have an important role as adjuvant treatment of human cancers (47) (see below).
Since the IGF-IR has extensive cross-talk with other receptor tyrosine kinases and their downstream effectors, inhibition of the IGF-IR by a specific antibody may be compensated by other pathways (37). The most obvious candidates responsible for this compensation are the IR-A and IGF-II. In vitro, we recently showed that circulating IGF-II may contribute substantially to IR-A and IR-B signaling (60). It has been also suggested that therapeutic interventions of cancers may trigger a phenotype switch in surviving cells to a more primitive and immature cell state (61). In favor of this latter possibility, as discussed above, the IR-A and IGF-II are frequently highly expressed by human tumors. In addition, there is further evidence that resistance to IGF-IR directed therapy is the direct result from formation of IGF-I/IR-A hybrids and IGF-II signaling via the IR-A isoform (37). Thus the development of functional inhibitors that affect IGF-IR and IR-A may be necessary to overcome resistance to IGF-IR directed therapy.
On the other hand, IGF-IR blocking therapy is expected to be most effective in tumors with increased IGF-IR and poor IR-activation. Thus, identification of a subset of patients most likely to benefit from IGF signaling interference should be pursued.
Combining the diagnostic and therapeutic potential of an antibody, thereby selecting those patients who are most likely to benefit from antibody treatment, could be an important step to improve IGF-IR targeted therapy (62). In contrast to immunohistochemical imaging of the IGF-IR, molecular IGF-IR imaging (for example 111In-R1507 SPECT) in vivo might be able to predict and to identify who will benefit from IGF-IR targeted therapy (63).
Combining IGF-IR targeted therapy to chemotherapy may be another potential successful strategy since IGF-I may protect tumor cells from being killed by cytotoxic drugs (37, 64). In addition, this may help to suppress chemotherapy induced IGF-IR activation and DNA repair mechanisms (37). Furthermore, the feasibility and timing of combining multiple targeted therapies (IGF-IR and IR-A) and conventional cytotoxic drugs need to be explored. Recently it was suggested that the IGF-IR also behaves like a functional receptor tyrosine kinase/G-protein related coupled receptor (GPCR) hybrid “borrowing” components of GPCR signaling (42). As a consequence, the IGF-IR (and IR) can activate signaling as a GPCR, using different G-proteins (42). IGF-I activity and its biological effects are further controlled by a variety of adaptor proteins/signaling proteins through IGF-IR posttranslational modifications including tyrosine and serine phosphorylation, dephosphorylation, ubiquitination, and sumoylation (42). Therefore potential drugs that modify alternative downstream effects of the IGF-IR, the “biasing agonists,” should also be considered (42).
Both Joseph A. M. J. L. Janssen and Aimee J. Varewijck researched data, wrote manuscript, and reviewed/edited the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Clemmons DR. Targeting the insulin-like growth factor-I receptor in cancer therapy. In: Leroith D, editor. Insulin-Like Growth Factors and Cancer: From Basic Biology to Therapeutics. New York, NY: Springer (2012). p. 193–213.
2. Moschos SJ, Mantzoros CS. The role of the IGF system in cancer: from basic to clinical studies and clinical applications. Oncology (2002) 63:317–32. doi: 10.1159/000066230
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Yee D, Paik S, Lebovic GS, Marcus RR, Favoni RE, Cullen KJ, et al. Analysis of insulin-like growth factor I gene expression in malignancy: evidence for a paracrine role in human breast cancer. Mol Endocrinol (1989) 3:509–17. doi:10.1210/mend-3-3-509
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
4. Juul A. Serum levels of insulin-like growth factor I and its binding proteins in health and disease. Growth Horm IGF Res (2003) 13:113–70. doi:10.1016/S1096-6374(03)00038-8
5. Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev (1995) 16:3–34. doi:10.1210/edrv-16-1-3
6. Baxter RC, Bryson JM, Turtle JR. Somatogenic receptors of rat liver: regulation by insulin. Endocrinology (1980) 107:1176–81. doi:10.1210/endo-107-4-1176
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
7. Wang HS, Wang TH. Polycystic ovary syndrome (PCOS), insulin resistance and insulin-like growth factors (IGfs)/IGF-binding proteins (IGFBPs). Chang Gung Med J (2003) 26:540–53.
8. King ER, Wong KK. Insulin-like growth factor: current concepts and new developments in cancer therapy. Recent Pat Anticancer Drug Discov (2012) 7:14–30. doi:10.2174/157489212798357930
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Varewijck AJ, Janssen JA. Insulin and its analogues and their affinities for the IGF1 receptor. Endocr Relat Cancer (2012) 19:F63–75. doi:10.1530/ERC-12-0026
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Ebina Y, Ellis L, Jarnagin K, Edery M, Graf L, Clauser E, et al. The human insulin receptor cDNA: the structural basis for hormone-activated transmembrane signalling. Cell (1985) 40:747–58. doi:10.1016/0092-8674(85)90334-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Seino S, Seino M, Nishi S, Bell GI. Structure of the human insulin receptor gene and characterization of its promoter. Proc Natl Acad Sci U S A (1989) 86:114–8. doi:10.1073/pnas.86.1.114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
12. Moller DE, Yokota A, Caro JF, Flier JS. Tissue-specific expression of two alternatively spliced insulin receptor mRNAs in man. Mol Endocrinol (1989) 3:1263–9. doi:10.1210/mend-3-8-1263
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Frasca F, Pandini G, Scalia P, Sciacca L, Mineo R, Costantino A, et al. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol Cell Biol (1999) 19:3278–88.
14. Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev (2009) 30:586–623. doi:10.1210/er.2008-0047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. Chao W, D’Amore PA. IGF2: epigenetic regulation and role in development and disease. Cytokine Growth Factor Rev (2008) 19:111–20. doi:10.1016/j.cytogfr.2008.01.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Mosthaf L, Grako K, Dull TJ, Coussens L, Ullrich A, Mcclain DA. Functionally distinct insulin receptors generated by tissue-specific alternative splicing. EMBO J (1990) 9:2409–13.
17. Mosthaf L, Vogt B, Haring HU, Ullrich A. Altered expression of insulin receptor types A and B in the skeletal muscle of non-insulin-dependent diabetes mellitus patients. Proc Natl Acad Sci U S A (1991) 88:4728–30. doi:10.1073/pnas.88.11.4728
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
18. Sesti G, Marini MA, Tullio AN, Montemurro A, Borboni P, Fusco A, et al. Altered expression of the two naturally occurring human insulin receptor variants in isolated adipocytes of non-insulin-dependent diabetes mellitus patients. Biochem Biophys Res Commun (1991) 181:1419–24. doi:10.1016/0006-291X(91)92097-4
19. Baxter RC. Insulin-like growth factor (IGF)-binding proteins: interactions with IGFs and intrinsic bioactivities. Am J Physiol Endocrinol Metab (2000) 278:E967–76.
20. Rechler MM, Clemmons DR. Regulatory actions of insulin-like growth factor-binding proteins. Trends Endocrinol Metab (1998) 9:176–83. doi:10.1016/S1043-2760(98)00047-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Bailyes EM, Nave BT, Soos MA, Orr SR, Hayward AC, Siddle K. Insulin receptor/IGF-I receptor hybrids are widely distributed in mammalian tissues: quantification of individual receptor species by selective immunoprecipitation and immunoblotting. Biochem J (1997) 327(Pt 1):209–15.
22. Pandini G, Frasca F, Mineo R, Sciacca L, Vigneri R, Belfiore A. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J Biol Chem (2002) 277:39684–95. doi:10.1074/jbc.M202766200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Slaaby R, Schaffer L, Lautrup-Larsen I, Andersen AS, Shaw AC, Mathiasen IS, et al. Hybrid receptors formed by insulin receptor (IR) and insulin-like growth factor I receptor (IGF-IR) have low insulin and high IGF-1 affinity irrespective of the IR splice variant. J Biol Chem (2006) 281:25869–74. doi:10.1074/jbc.M605189200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Gee JM, Robertson JF, Gutteridge E, Ellis IO, Pinder SE, Rubini M, et al. Epidermal growth factor receptor/HER2/insulin-like growth factor receptor signalling and oestrogen receptor activity in clinical breast cancer. Endocr Relat Cancer (2005) 12(Suppl 1):S99–111. doi:10.1677/erc.1.01005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
25. Pandini G, Mineo R, Frasca F, Roberts CT Jr, Marcelli M, Vigneri R, et al. Androgens up-regulate the insulin-like growth factor-I receptor in prostate cancer cells. Cancer Res (2005) 65:1849–57. doi:10.1158/0008-5472.CAN-04-1837
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
26. Xu J, Messina JL. Crosstalk between growth hormone and insulin signaling. Vitam Horm (2009) 80:125–53. doi:10.1016/S0083-6729(08)00606-7
27. Malaguarnera R, Sacco A, Morcavallo A, Squatrito S, Migliaccio A, Morrione A, et al. Metformin inhibits androgen-induced IGF-IR up-regulation in prostate cancer cells by disrupting membrane-initiated androgen signaling. Endocrinology (2014) 155:1207–21. doi:10.1210/en.2013-1925
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Coppola D, Ferber A, Miura M, Sell C, D’Ambrosio C, Rubin R, et al. A functional insulin-like growth factor I receptor is required for the mitogenic and transforming activities of the epidermal growth factor receptor. Mol Cell Biol (1994) 14:4588–95.
29. Morgillo F, Woo JK, Kim ES, Hong WK, Lee HY. Heterodimerization of insulin-like growth factor receptor/epidermal growth factor receptor and induction of survivin expression counteract the antitumor action of erlotinib. Cancer Res (2006) 66:10100–11. doi:10.1158/0008-5472.CAN-06-1684
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Guix M, Faber AC, Wang SE, Olivares MG, Song Y, Qu S, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J Clin Invest (2008) 118:2609–19. doi:10.1172/JCI34588
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
31. van der Veeken J, Oliveira S, Schiffelers RM, Storm G, Van Bergen En Henegouwen PM, Roovers RC. Crosstalk between epidermal growth factor receptor- and insulin-like growth factor-1 receptor signaling: implications for cancer therapy. Curr Cancer Drug Targets (2009) 9:748–60. doi:10.2174/156800909789271495
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
32. Groenendijk FH, Bernards R. Drug resistance to targeted therapies: Deja vu all over again. Mol Oncol (2014) 8(6):1067–83. doi:10.1016/j.molonc.2014.05.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Chen HX, Sharon E. IGF-1R as an anti-cancer target – trials and tribulations. Chin J Cancer (2013) 32:242–52. doi:10.5732/cjc.012.10263
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Sachdev D, Li SL, Hartell JS, Fujita-Yamaguchi Y, Miller JS, Yee D. A chimeric humanized single-chain antibody against the type I insulin-like growth factor (IGF) receptor renders breast cancer cells refractory to the mitogenic effects of IGF-I. Cancer Res (2003) 63:627–35.
35. Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer (2012) 12:159–69. doi:10.1038/nrc3215
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Gualberto A, Pollak M. Emerging role of insulin-like growth factor receptor inhibitors in oncology: early clinical trial results and future directions. Oncogene (2009) 28:3009–21. doi:10.1038/onc.2009.172
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. King H, Aleksic T, Haluska P, Macaulay VM. Can we unlock the potential of IGF-1R inhibition in cancer therapy? Cancer Treat Rev (2014) 40(9):1096–105. doi:10.1016/j.ctrv.2014.07.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
38. Karp DD, Paz-Ares LG, Novello S, Haluska P, Garland L, Cardenal F, et al. Phase II study of the anti-insulin-like growth factor type 1 receptor antibody CP-751,871 in combination with paclitaxel and carboplatin in previously untreated, locally advanced, or metastatic non-small-cell lung cancer. J Clin Oncol (2009) 27:2516–22. doi:10.1200/JCO.2008.19.9331
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Olmos D, Postel-Vinay S, Molife LR, Okuno SH, Schuetze SM, Paccagnella ML, et al. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: a phase 1 expansion cohort study. Lancet Oncol (2010) 11:129–35. doi:10.1016/S1470-2045(09)70354-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
40. Atzori F, Tabernero J, Cervantes A, Prudkin L, Andreu J, Rodriguez-Braun E, et al. A phase I pharmacokinetic and pharmacodynamic study of dalotuzumab (MK-0646), an anti-insulin-like growth factor-1 receptor monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res (2011) 17:6304–12. doi:10.1158/1078-0432.CCR-10-3336
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
41. Arcaro A. Targeting the insulin-like growth factor-1 receptor in human cancer. Front Pharmacol (2013) 4:30. doi:10.3389/fphar.2013.00030
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Girnita L, Worrall C, Takahashi S, Seregard S, Girnita A. Something old, something new and something borrowed: emerging paradigm of insulin-like growth factor type 1 receptor (IGF-1R) signaling regulation. Cell Mol Life Sci (2014) 71:2403–27. doi:10.1007/s00018-013-1514-y
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
43. Burtrum D, Zhu Z, Lu D, Anderson DM, Prewett M, Pereira DS, et al. A fully human monoclonal antibody to the insulin-like growth factor I receptor blocks ligand-dependent signaling and inhibits human tumor growth in vivo. Cancer Res (2003) 63:8912–21.
44. Cohen BD, Baker DA, Soderstrom C, Tkalcevic G, Rossi AM, Miller PE, et al. Combination therapy enhances the inhibition of tumor growth with the fully human anti-type 1 insulin-like growth factor receptor monoclonal antibody CP-751,871. Clin Cancer Res (2005) 11:2063–73. doi:10.1158/1078-0432.CCR-04-1070
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
45. Weinstein D, Sarfstein R, Laron Z, Werner H. Insulin receptor compensates for IGF1R inhibition and directly induces mitogenic activity in prostate cancer cells. Endocr Connect (2014) 3:24–35. doi:10.1530/EC-13-0086
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
46. Zhang H, Pelzer AM, Kiang DT, Yee D. Down-regulation of type I insulin-like growth factor receptor increases sensitivity of breast cancer cells to insulin. Cancer Res (2007) 67:391–7. doi:10.1158/0008-5472.CAN-06-1712
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
47. Baserga R. The decline and fall of the IGF-I receptor. J Cell Physiol (2013) 228:675–9. doi:10.1002/jcp.24217
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
48. Heskamp S, Boerman OC, Molkenboer-Kuenen JD, Koornstra RH, Linn SC, Oyen WJ, et al. Dynamics of IGF-1R expression during endocrine breast cancer treatment. Mol Imaging Biol (2014) 16:529–37. doi:10.1007/s11307-014-0723-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Kim JG, Kang MJ, Yoon YK, Kim HP, Park J, Song SH, et al. Heterodimerization of glycosylated insulin-like growth factor-1 receptors and insulin receptors in cancer cells sensitive to anti-IGF1R antibody. PLoS One (2012) 7:e33322. doi:10.1371/journal.pone.0033322
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Asmane I, Watkin E, Alberti L, Duc A, Marec-Berard P, Ray-Coquard I, et al. Insulin-like growth factor type 1 receptor (IGF-1R) exclusive nuclear staining: a predictive biomarker for IGF-1R monoclonal antibody (Ab) therapy in sarcomas. Eur J Cancer (2012) 48:3027–35. doi:10.1016/j.ejca.2012.05.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell (2010) 141:1117–34. doi:10.1016/j.cell.2010.06.011
52. Shin DH, Min HY, El-Naggar AK, Lippman SM, Glisson B, Lee HY. Akt/mTOR counteract the antitumor activities of cixutumumab, an anti-insulin-like growth factor I receptor monoclonal antibody. Mol Cancer Ther (2011) 10:2437–48. doi:10.1158/1535-7163.MCT-11-0235
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Clemmons D, Maile L, Xi G, Shen X, Radhakrishnan Y. Igf-I signaling in response to hyperglycemia and the development of diabetic complications. Curr Diabetes Rev (2011) 7:235–45. doi:10.2174/157339911796397848
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Janssen JA, Varewijck AJ. Insulin analogs and cancer: a note of caution. Front Endocrinol (2014) 5:79. doi:10.3389/fendo.2014.00079
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Baricevic I, Roberts DL, Renehan AG. Chronic insulin exposure does not cause insulin resistance but is associated with chemo-resistance in colon cancer cells. Horm Metab Res (2014) 46:85–93. doi:10.1055/s-0033-1354414
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Perry JK, Liu DX, Wu ZS, Zhu T, Lobie PE. Growth hormone and cancer: an update on progress. Curr Opin Endocrinol Diabetes Obes (2013) 20:307–13. doi:10.1097/MED.0b013e328363183a
57. Xu JW, Wang TX, You L, Zheng LF, Shu H, Zhang TP, et al. Insulin-like growth factor 1 receptor (IGF-1R) as a target of MiR-497 and plasma IGF-1R levels associated with TNM stage of pancreatic cancer. PLoS One (2014) 9:e92847. doi:10.1371/journal.pone.0092847
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
58. Beckman RA, Weiner LM, Davis HM. Antibody constructs in cancer therapy: protein engineering strategies to improve exposure in solid tumors. Cancer (2007) 109:170–9. doi:10.1002/cncr.22402
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Chitnis MM, Yuen JS, Protheroe AS, Pollak M, Macaulay VM. The type 1 insulin-like growth factor receptor pathway. Clin Cancer Res (2008) 14:6364–70. doi:10.1158/1078-0432.CCR-07-4879
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Varewijck AJ, Brugts MP, Frystyk J, Goudzwaard JA, Uitterlinden P, Waaijers AM, et al. Circulating insulin-like growth factors may contribute substantially to insulin receptor isoform A and insulin receptor isoform B signalling. Mol Cell Endocrinol (2013) 365:17–24. doi:10.1016/j.mce.2012.08.021
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Davies PC, Lineweaver CH. Cancer tumors as Metazoa 1.0: tapping genes of ancient ancestors. Phys Biol (2011) 8:015001. doi:10.1088/1478-3975/8/1/015001
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Fleuren ED, Versleijen-Jonkers YM, Heskamp S, Van Herpen CM, Oyen WJ, Van Der Graaf WT, et al. Theranostic applications of antibodies in oncology. Mol Oncol (2014) 8:799–812. doi:10.1016/j.molonc.2014.03.010
63. Fleuren ED, Versleijen-Jonkers YM, Van De Luijtgaarden AC, Molkenboer-Kuenen JD, Heskamp S, Roeffen MH, et al. Predicting IGF-1R therapy response in bone sarcomas: immuno-SPECT imaging with radiolabeled R1507. Clin Cancer Res (2011) 17:7693–703. doi:10.1158/1078-0432.CCR-11-1488
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Awasthi N, Zhang C, Ruan W, Schwarz MA, Schwarz RE. BMS-754807, a small-molecule inhibitor of insulin-like growth factor-1 receptor/insulin receptor, enhances gemcitabine response in pancreatic cancer. Mol Cancer Ther (2012) 11:2644–53. doi:10.1158/1535-7163.MCT-12-0447
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: IGF-IR targeted therapy, IGF-IR antibodies, IGF-I receptor, insulin receptor-A, insulin receptor-B, cancer, hyperglycemia
Citation: Janssen JAMJL and Varewijck AJ (2014) IGF-IR targeted therapy: past, present and future. Front. Endocrinol. 5:224. doi: 10.3389/fendo.2014.00224
Received: 24 October 2014; Accepted: 08 December 2014;
Published online: 23 December 2014.
Edited by:
Antonino Belfiore, University “Magna Graecia” of Catanzaro, ItalyReviewed by:
Roberta Malaguarnera, University “Magna Graecia” of Catanzaro, ItalyCopyright: © 2014 Janssen and Varewijck. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joseph A. M. J. L. Janssen, Department of Internal Medicine, Erasmus MC, Room D-443, ‘s-Gravendijkwal 230, Rotterdam 3015 CE, Netherlands e-mail:ai5hLm0uai5sLmphbnNzZW5AZXJhc211c21jLm5s
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.