 Sandra Merscher
Sandra Merscher Christopher E. Pedigo
Christopher E. Pedigo Armando J. Mendez
Armando J. Mendez
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 14 October 2014
Sec. Cellular Endocrinology
Volume 5 - 2014 | https://doi.org/10.3389/fendo.2014.00169
This article is part of the Research TopicMetabolism, energetics, and lipid biology in the podocyteView all 4 articles
Chronic kidney disease (CKD) is associated with a high risk of death. Dyslipidemia is commonly observed in patients with CKD and is accompanied by a decrease in plasma high-density lipoprotein, and an increase in plasma triglyceride-rich lipoproteins and oxidized lipids. The observation that statins may decrease albuminuria but do not stop the progression of CKD indicates that pathways other than the cholesterol synthesis contribute to cholesterol accumulation in the kidneys of patients with CKD. Recently, it has become clear that increased lipid influx and impaired reverse cholesterol transport can promote glomerulosclerosis, and tubulointerstitial damage. Lipid-rafts are cholesterol-rich membrane domains with important functions in regulating membrane fluidity, membrane protein trafficking, and in the assembly of signaling molecules. In podocytes, which are specialized cells of the glomerulus, they contribute to the spatial organization of the slit diaphragm (SD) under physiological and pathological conditions. The discovery that podocyte-specific proteins such as podocin can bind and recruit cholesterol contributing to the formation of the SD underlines the importance of cholesterol homeostasis in podocytes and suggests cholesterol as an important regulator in the development of proteinuric kidney disease. Cellular cholesterol accumulation due to increased synthesis, influx, or decreased efflux is an emerging concept in podocyte biology. This review will focus on the role of cellular cholesterol accumulation in the pathogenesis of kidney diseases with a focus on glomerular diseases.
The glomerulus is a highly specialized structure that ensures that essential proteins are retained in the blood through the selective ultrafiltration of plasma (1). Intensive research has outlined the crucial role of podocytes and the slit diaphragm (SD) in the size-selective filtration of proteins from plasma. Podocytes are visceral glomerular epithelial cells consisting of a cell body, major and minor foot processes. Numerous foot processes from neighboring podocytes form a unique interdigitating pattern leaving filtration slits between them, which are bridged by a 40-nm wide SD (2, 3). The integrity of this filtration barrier is important to prevent the loss of protein into the urine (proteinuria). Research of the past two decades has revealed that the SD is a lipid raft-like structure that contains multi-protein complexes, including ion channels, receptors (transcient receptor potential cation channel 6, TRPC6), transmembrane proteins (nephrin, NPHS1), integral membrane proteins (podocin, NPHS2), structural proteins (alpha-actinin-4, ACTN4), signaling adaptors (CD2 associated protein, CD2AP), and other proteins involved in cell signaling, and that proper signaling at the SD is essential for proper glomerular filtration. Thus, mutations in genes coding for SD proteins were shown to cause proteinuria-associated nephropathies (4–8). The observation that the SD represents a lipid-raft like structure suggested that cholesterol might play an important role in granting proper localization of SD proteins and initiated recent research to investigate the role of cellular cholesterol metabolism in proteinuric kidney disease. It was shown that binding of SD proteins such as podocin to cholesterol is necessary for the podocin-dependent activation of TRPC6 (9, 10), further demonstrating the importance of cholesterol in the proper function and localization of SD proteins. This review will focus on the role of cellular cholesterol imbalance in the pathogenesis of proteinuric glomerular diseases.
Cellular cholesterol homeostasis is regulated by de novo synthesis, cholesterol influx, and efflux (11) and is crucial for proper cell function (Figure 1). Cholesterol synthesis is tightly regulated through the action of the sterol regulatory element binding proteins (SREBPs) that play a critical role in the regulation of genes involved in cholesterol and fatty acid synthesis and of the low-density lipoprotein receptor (LDLR) gene. Of the three known isoforms of SREBP (SREBP-1a, SREBP-1c, and SREBP-2), SREBP-2 preferentially activates genes involved in cholesterol metabolism, whereas SREB-1c primarily acts on genes important in fatty acid and triglyceride metabolism (12, 13). SREBPs reside in the endoplasmatic reticulum (ER) membrane bound in complexes with SREBP cleavage-activating protein (SCAP) (14–16). When cellular cholesterol levels are low in the ER membrane, a conformational change occurs in SCAP, allowing SREBPs to be transported to the Golgi apparatus where two sequential proteolytic cleavages occur allowing for the release of the active SREBP fragment that can transcriptionally regulate several sterol response element genes including 3-hydroxy-3-methyl-glutaryl-CoA reductase (HMG-CoA reductase, HMGCR), the rate-limiting enzyme of cholesterol biosynthesis. When ER membrane cholesterol levels are elevated, SCAP remains associated with SREBP and prevents its transport and activation in the Golgi. This pathway provides a feedback mechanism to prevent the over accumulation of cellular cholesterol (Figure 1).
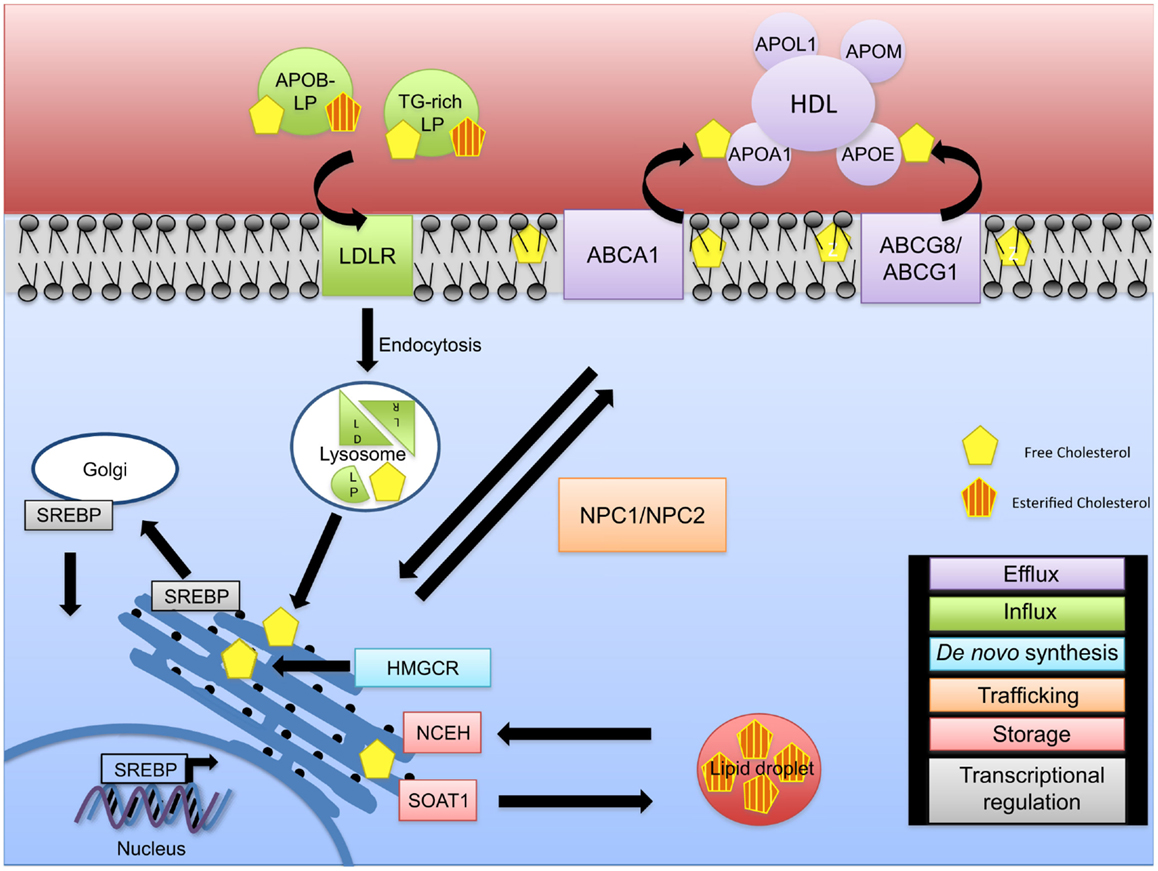
Figure 1. Intracellular cholesterol trafficking. The fine regulation of cholesterol homeostasis is maintained via different mechanisms. De novo synthesis (blue) of free cholesterol occurs via the rate-limiting enzyme HMG-CoA reductase (HMGCR) at the endoplasmic reticulum (ER). In the event of a cellular cholesterol deficit, cholesterol influx (green) mediated through APOB-rich lipoproteins and triglyceride-rich (TG-rich) lipoproteins occurs. LDLR/lipoprotein complexes are internalized via endocytosis and transported to the lysosome for degradation resulting in LDL and VLDL remnants, thus releasing free cholesterol. As excess free cholesterol is toxic, it is transported to the plasma membrane via NPC1/2 for efflux by an ABCA1-ApoAI/L1- or ABCG1/8-HDL-mediated mechanism (purple), or it is converted to cholesteryl esters via SOAT1 leading to the formation of cholesterol enriched lipid droplets (red). Cholesteryl esters can be converted back to unesterified (free) cholesterol via NCEH. Cholesterol pathways are regulated on a transcriptional level (gray). During cholesterol deficits, SREBP is transported to the Golgi apparatus and cleaved, allowing its translocation to the nucleus to regulate expression of cholesterol genes.
In the blood, cholesterol is transported by two major lipoproteins, low-density lipoprotein (LDL) and high-density lipoprotein (HDL). Circulating LDL is the major source for cholesterol uptake into cells in a LDLR dependent manner (17). Binding of cholesterol-containing LDL to its receptor is followed by endocytosis and fusion of the LDL-containing vesicle with a lysosome. Lysosomal enzymes digest the LDL, breaking down the proteins to amino acids and liberating the free cholesterol. A concerted action between Niemann–Pick C1 and C2 proteins (NPC1 and NPC2) allows for the transport of unesterified (free) cholesterol from the endosome/lysosome complex to the ER and then to the plasma membrane (18, 19). The increase in cellular cholesterol induces cellular cholesteryl ester formation through activation of sterol-O-acyltransferase 1 (SOAT1 or acyl-Coenzyme A: cholesterol acyltransferase, ACAT1) and reduces cholesterol synthesis through inhibition of HMGCR and decreases LDLR synthesis (17). Abnormal accumulation of free cholesterol leads to cell toxicity underlining the importance of cholesterol esterification in maintaining proper levels of free cholesterol needed for optimal cell and cell membrane function (20–22). Enzymes involved in conversion of free cholesterol to cholesteryl esters and vice versa at the ER are SOAT1 and neutral cholesterol ester hydrolase 1 (NCEH) (23). Cellular levels of cholesterol are also modulated through the reverse cholesterol transport pathways that promote cholesterol efflux to HDL (Figure 1).
Efflux of cellular cholesterol occurs through several distinct pathways (24). Cholesterol efflux by aqueous diffusion is a bi-directional, energy independent process and involves equilibrium of cholesterol molecules between cellular membranes and any acceptor, including HDL. Scavenger receptor B-I (SR-BI) mediates the selective uptake of HDL cholesteryl esters into cells, and also facilitates the passive efflux of cholesterol from cells to HDL. Third, efflux of phospholipids and cholesterol mediated by the ATP-binding cassette transporter A1 (ABCA1) transporter is an active, energy-requiring process that requires the presence of extracellular lipid-poor apolipoproteins (APO proteins) including APOAI and APOE. Another ATP-binding cassette transporter, ABCG1, enhances efflux by increasing desorption of plasma membrane cholesterol and enhancing removal by HDL (Figure 1). The contribution of the various efflux pathways to reverse cholesterol transport, especially in vivo, remains unknown and it is likely that all pathways contribute to the overall extent of reverse cholesterol transport. While the first step in reverse cholesterol transport is removal of cellular cholesterol to HDL, several other key steps in reverse cholesterol transport involve cholesterol esterification by lecithin-cholesterol acyltransferase (LCAT) and transfer of HDL-associated cholesteryl esters to APOB containing lipoproteins through the action of cholesteryl ester transferase protein (CETP) and subsequent uptake of cholesterol by the liver for excretion or redistribution.
Dyslipidemia is a common disorder observed in subjects with chronic kidney disease (CKD). The most common lipid abnormalities observed are elevated fasting triglyceride levels and reduced HDL cholesterol, while total cholesterol and LDL cholesterol can be elevated or in the normal range and is often below normal in subjects with renal failure (25–27). In addition to the quantitative changes in lipoprotein levels, there are a multitude of qualitative changes in the lipoproteins of CKD patients that include reduced clearance rates of triglyceride-rich lipoproteins resulting in increased blood levels of atherogenic remnant particles (28), increased levels of small dense LDL particles (29), increased levels of oxidized LDL (30), altered HDL subfraction distribution with reductions in larger HDL particles consistent with delayed maturation of HDL (31), reduced LCAT activity (32), and decreased HDL anti-oxidant and anti-inflammatory activity (27). Each of these lipoprotein abnormalities can contribute to the increased risk of cardiovascular disease observed in patients with CKD and likely contribute to altered cellular cholesterol homeostasis in kidney cells.
Defective cellular cholesterol trafficking was described in diseases of genetic and non-genetic origin with and without renal involvement (33–36). Hypercholesterolemia due to impaired reverse cholesterol transport and abnormalities of lipid metabolism are common features observed in patients with end-stage renal disease (ESRD), including patients on hemodialysis, patients with diabetic kidney disease (DKD), nephrotic syndrome, and uremia (37–40). In addition to hypercholesterolemia in the blood, lipid accumulation in glomeruli of patients with kidney disease was described. Although hyperlipidemia is a known risk factor for the development of cardiovascular disease and atherosclerosis, the role of hyperlipidemia as a cause for the development of renal diseases and the mechanisms leading to glomerular lipid accumulation remain less understood (41–44).
Within the kidney, the glomerulus and proximal tubules express various apolipoproteins and proteins important in lipid and cholesterol metabolism. More than 30 years ago, apoA1 and apoB were the first apolipoproteins shown to be synthesized in kidneys from chickens suggesting that the kidney might be a major source for the synthesis of plasma lipoproteins (45, 46). Later, it was shown that both are secreted as constituents of lipoprotein particles from kidney cells (47). APOM, a lipoprotein associated with HDL (48), APOE (49, 50), APOB (50), and APOL1 (51), was consequently shown to be expressed in human kidneys, and we recently demonstrated that human glomerular podocytes express ABCA1, HMGCR, and LDL receptor (52). These observations indicate the possibility of apolipoprotein-mediated cholesterol homeostasis in kidney cells and may suggest that dysregulated cholesterol homeostasis may represent a pathogenic mechanism in kidney disease.
Within the glomerulus, podocytes are able to uptake LDL, APOB, and APOE containing lipoproteins. Their capacity to uptake APOB and APOE containing lipoproteins is increased when compared to LDL, and this process is associated with the suppression of cellular sterol synthesis and cholesteryl ester formation (50).
Lipoprotein glomerulopathy [LPG, Online Mendelian Inheritance in Man (OMIM) #611771] is a rare genetic disorder mainly affecting people of Japanese and Chinese origin that is caused by a heterozygous mutation in the APOE gene on chromosome 19q13. Several mutations in APOE were identified resulting in the expression of a dysfunctional APOE protein with impaired LDL receptor binding (34, 53, 54). Patients with LPG have proteinuria and nephrotic syndrome with elevated serum APOE levels, abnormal lipoprotein deposition in glomerular capillaries in form of laminated thrombi that contain APOB and APOE, a variable degree of mesangial proliferation, and dysbetalipoproteinemia (55, 56). Occasionally, a similar renal phenotype can be observed in type III hyperlipoproteinemia, a disease that is characterized by homozygous mutation in the ε2 allele of the APOE2 gene. The kidney phenotype includes proteinuria, glomerulosclerosis with mesangial and interstitial foam-cell accumulation, and the presence of intraglomerular thrombi (57, 58). Genetic polymorphisms in APOE were shown to differentially affect LDLR binding. Thus, the APOE2 variant binds poorly to LDLR, whereas the APOE4 variant binds with high affinity (59). The APOE2 variant was associated with persistent proteinuria and renal failure in patients with non-insulin-dependent diabetes mellitus (NIDDM) (60) and may contribute to the severity of the renal phenotype in IgA nephropathy (61). Conversely, the polymorphism in APOE4 is thought to constitute a protective factor in patients with NIDDM (62).
Constitutive ApoE−/− mice generated through partial replacement of exon 3 and part of intron 3 of the ApoE gene by a neomycin cassette (63) are a model for experimental atherosclerosis and develop a renal phenotype similar to that observed in patients with type III hyperlipoproteinemia and LPG (64–66). Kidneys from ApoE−/− mice showed glomerular macrophage infiltration with foam-cell formation, deposition of extracellular matrix, glomerular hyperplasia, mesangiolysis, and lipid deposits in glomerular capillaries at 24 weeks, which further progressed to a LPG-like phenotype at 36 weeks (66). In a different study, in which the APOE-Sendai mutation (Arg145Pro mutation found in patients with LPG) was introduced into ApoE−/− mice showed that, although similar, the LPG-like lesions observed in the APOE-Sendai mice were different from the lesions in aged ApoE−/− mice (65, 67) indicating that APOE variants may contribute to a differential phenotype in LPG. ApoE−/− mice, which were fed a high fat diet developed a renal phenotype at 16 weeks of age with lipid droplet and cholesterol crystal accumulation within the glomerulus. This phenotype was associated with decreased basal plasma renin concentrations and low mean arterial blood pressure, indicating impaired renin-angiotensin system (RAS) function. After renal artery constriction, these mice were unable to increase renin secretion from the juxtaglomerular cells, demonstrating a lack of a normal response to renal hypoperfusion (68). Similar glomerular changes were observed in another study when male mice were fed a high cholesterol diet for 20 weeks (69) indicating that high fat diet accelerates the glomerular injury phenotype. Likewise, streptozotocin (STZ) injected into ApoE−/− mice accelerated renal injury when compared to non-diabetic mice and was attenuated when advanced glycation end product (AGE) accumulation was prevented by the use of an AGE formation inhibitor. Renal changes observed in diabetic ApoE−/− mice included increased albuminuria and structural changes in glomeruli and tubulointerstitium (70). Taken together, these studies suggest that ApoE deficiency and mutations that affect ApoE binding to LDLR and the resulting hyperlipidemia play a role in rendering renal cells more susceptible to glomerular injury.
In further support of this hypothesis, mice with a combined deficiency of inhibitor of differentiation 3 (Id3) and ApoE−/− spontaneously develop glomerulonephritis characterized by lipid deposition specifically in glomeruli, the presence of enlarged hypercellular glomeruli, mesangial expansion, and increased extracellular matrix deposition (71). ApoE deficiency also accelerates the renal phenotype in MRL-Faslpr mice, a mouse model of human lupus (72). Further, decreased ApoE expression in glomerular podocytes is observed in Tg26 (human immunodeficiency virus, HIV) and Nef (negative regulatory factor) transgenic mice, two models of HIV-associated nephropathy (HIVAN) (73), whereas increased ApoE and ApoB expression was found in association with accumulation of oil red O positive lipid droplets in glomerular visceral epithelial cells and mesangial cells in rats with puromycin aminonucleoside or adriamycin induced nephrosis (74). Increased glomerular APOE expression was observed in patients with idiopathic nephrotic syndrome but it is rather a rare occurrence in focal segmental glomerulosclerosis (FSGS) (75).
APOM is a 26-kDa apolipoprotein and is a member of the lipocalin family expressed in the liver and in the kidney. In the plasma, APOM is associated with HDL particles (48); in kidney proximal tubular cells, APOM binds to megalin, thus preventing its excretion in the urine by megalin-mediated endocytosis (76). Mutations, polymorphisms, or allelic variants of the APOM gene have not been associated with any glomerular phenotype. Although APOM is strongly expressed in kidney tubular epithelial cells, it is not found in the urine of mice or human beings. However, megalin knockout mice were shown to be characterized by urinary loss of low molecular weight proteins underlining an important role of proximal tubules in resorption of proteins (77). Megalin is expressed in glomerular podocytes where it can bind to α-galactosidase A, suggesting a potential role in Fabry disease, and it may act as a pathogenic antigen in membranous glomerulonephritis (78, 79).
APOL1 is a secreted HDL-associated protein that binds to APOA1 and promotes cholesterol efflux. It is coded by a gene on human chromosome 22q12, and APOL1 mutations are associated with increased susceptibility to FSGS (OMIM #612551), HIVAN, and hypertensive nephropathy in patients of African ancestry (80–82). Using immunofluorescence staining of normal human kidneys, APOL1 was localized to podocytes of the glomerulus, the promixal tubules, and the extraglomerular arterial endothelium. In kidney biopsies from patients with FSGS and HIVAN, APOL1 expression levels in podocytes were found decreased and de novo appearance of APOL1 within cells of the arterial medial wall was observed (51). The observation that normal human podocytes express APOL1 opens up the possibility that APOL1 may be contributing to cellular cholesterol homeostasis under physiological conditions. Likewise, it is possible that decreased podocyte APOL1 expression under disease conditions leads to decreased cholesterol efflux from podocytes and cellular cholesterol accumulation, thus contributing to the pathogenesis of glomerular diseases such as FSGS and HIVAN. Future studies to investigate the role of the different APOL1 variants and their role in podocyte cholesterol homeostasis under physiological and disease conditions are needed to shed light on their contribution to the pathogenesis of glomerular diseases.
Familial LCAT deficiency (OMIM #245900) is a rare genetic disorder caused by a mutation in the LCAT gene. LCAT is a HDL-associated enzyme that converts cholesterol to cholesteryl esters by transfer of the SN2 fatty acid from a phosphatidylcholines (lecithins) with the formation of lyso-phosphatidylcholine on the surface of HDLs. It, therefore, plays an important role in reverse cholesterol transport from peripheral tissues to the liver. LCAT deficiency leads to cholesterol accumulation in many tissues (83, 84). Patients with familial LCAT deficiency are characterized by diffuse corneal opacities, target cell hemolytic anemia, and proteinuria with chronic progressive glomerulopathy resulting in renal failure. Massive lipid deposits in the glomerular basement membrane (GBM) and in the mesangial region can be detected (85, 86). The appearance of abnormal choleastic lipoprotein, LpX, in the plasma of the patients was described (84, 87, 88).
Lecithin-cholesterol acyltransferase was originally shown to be expressed only in liver (89) but later also in brain, testis, ileum, kidney, spleen, and adrenal tissue (84, 90). More detailed analysis of LCAT expression in kidneys from Mongolian gerbils by immunohistochemistry showed expression mainly in the proximal and distal convoluted tubules and in the collecting duct epithelial cells (91).
Lcat deficient mice are characterized by reduced total cholesterol, HDL cholesterol, and APOA1 plasma levels (92, 93). When fed a regular chow diet, Lcat deficient mice show no glomerular phenotype. However, when fed a high fat diet, Lcat deficient mice developed glomerular lesions including reduction of vascular space, mesangial expansion and sclerosis, and increased extracellular matrix with accumulation of lipid droplets and macrophages. Lipid droplets in glomeruli were composed of free cholesterol and polar lipids as shown by filipin and oil red O staining. Interestingly, the glomerular phenotype was only observed in mice that simultaneously accumulated LpX (94). In support of the observation that elevated plasma LpX levels are necessary to mediate the glomerular phenotype, double mutant mice with Lcat deficiency that constitutively overexpress a NH2-terminus segment of SREBP-1a, which leads to accumulation of predominantly LpX in plasma were shown to spontaneously develop glomerular lesions with glomerular and tubulointerstitial lipid deposits at 6 months (95).
Niemann–Pick disease type C (OMIM#607616) is a genetic, neurogenerative disorder caused by mutations in the genes NPC1 on chromosome 18q11 in the vast majority of the cases, and NPC2 on chromosome 14q24. Patients present with ataxia, vertical supranuclear gaze palsy (VSGP), and dementia. Mutations in this gene(s) lead to the inability to transport free cholesterol between intracellular compartments. As a consequence, free cholesterol and other lipids accumulate in late endosomes/lysosomes in all organs including the kidney simultaneously causing a delayed response in sterol-responsive pathways to exogenous cholesterol (96, 97). NPC1 and NPC2 are expressed in the kidney (98, 99). NPC2 expression was further localized to the distal and proximal convoluted tubules of the kidney (99). Although rather rare, Niemann–Pick disease-associated renal pathology was described in biopsies from patients and included foamy podocytes, vacuolated tubular epithelial cells, and collections of foam cells in the interstitium (100). Association with a phenotype resembling membranoproliferative glomerulonephritis type II was described as well (101). These observations indicate that cholesterol accumulation in glomerular or tubular cells contributes to the renal pathogenesis observed in Niemann–Pick disease. In Npc1 deficient mice, a single injection with hydroxy-propyl-beta cyclodextrin, a cholesterol-depleting agent, was shown to increase the life span and weekly injections normalized cholesterol metabolism in nearly every organ (102–104).
Tangier disease (OMIM #205400) is an autosomal recessive and familial hypoalphalipoproteinemia (FHA, OMIM #604091), an autosomal dominant disorder caused by a mutation in the ABCA1 gene on chromosome 9q31 leading to accumulation of esterified cholesterol in tissues due to impaired reverse cholesterol transport resulting in reduced levels of plasma HDL (105–108). Patients present with heterogeneous clinical symptoms including liver, spleen, lymph node, and tonsil enlargement and peripheral neuropathy in children and adolescents. Occasionally, cardiovascular disease is observed in adults but the presence of a renal phenotype in Tangier patients is extremely rare (109, 110). FHA is the more common disease and is, like Tangier disease, characterized by low plasma HDL but without the clinical manifestations of Tangier disease. Abca1 deficient mice as a model for Tangier disease were generated by partial (111) or complete (112) replacement of the exons encoding the first ATP-binding cassette of Abca1. When fed on a high fat diet, Abca1 deficient mice were characterized by significantly decreased plasma HDL levels and lipid accumulation in several tissues including the kidney. Phenotypical differences were observed within the two models of Abca1 deficiency, possibly due to differences in the high fat diet composition or to slight differences in the targeting strategies (111, 112). In the study by Christiansen-Weber, a glomerular phenotype was observed, which included the presence of a thickened and “split” GBM and mesangial cell proliferation. Furthermore, immunoglobulin and C3 complement complexes characteristic of menbranoproliferative glomerulonephritis were detected (111), whereas no renal phenotype was described in the other study. The observation that Abca1 deficient mice develop a glomerular phenotype on a high fat diet deficiency may indicate that Abca1 deficiency leads to increased susceptibility for the development of renal disease.
Lipid droplets in kidney biopsies from patients with DKD were first identified by Kimmelstiel and Wilson (42) and were more recently localized within podocyte foot processes of patients with DKD (41). We recently demonstrated that glomerular ABCA1 expression is decreased in patients with type 2 diabetes (T2D) and early DKD and in human podocytes treated with the sera from patients with type 1 diabetes (T1D) and DKD in the absence of changes in LDLR and HMGCR expression (52). These observations indicate that lipid accumulation in podocytes due to defective cellular cholesterol homeostasis may play an important role in glomerular injury in DKD.
In support of these observations, lipid droplets in various experimental models of DKD have been reported. In two models for DKD with type 1 diabetes, Akita, and OVE26 mice, renal triglycerides and cholesterol accumulation was described and was associated with increased renal cholesterol synthesis and decreased efflux. This coincided with increased expression of SREBP-2 and HMGCR and decreased expression of liver X receptor (LXR)-α, LXR-β, and ABCA1 in kidneys of these mice (113). In diabetic STZ-injected DBA2/J mice, the DKD phenotype, which includes albuminuria, mesangial expansion, fibrosis and lipid accumulation in glomeruli, and tubulointerstitium is associated with increased HMGCR expression in kidneys. Aliskiren (a renin inhibitor) and valsartan (an angiotensin II receptor antagonist) reduced albuminuria and glomerulosclerosis in these animals and was associated with decreased HMGCR expression and decreased lipid accumulation in glomeruli (114), suggesting a potential crosstalk between the RAS and lipid metabolism in the kidney. FvB-Leprdb/db mice, a model for DKD with type 2 diabetes, spontaneously develop a renal DKD-like phenotype that includes glomerulosclerosis, tubulointerstitial fibrosis, GBM thickening, and proteinuria. In addition, renal triglyceride and cholesterol accumulation in glomeruli and tubules associated with increased activity of SREBP-1 and -2 was observed (115). Likewise, renal cholesterol accumulation in high fat/sucrose-fed (116) (type 2 diabetes model for DKD) and STZ-induced rats (type 1 diabetes model for DKD) was associated with increased expression of SCAP, SREBP-2, HMGCR, and LDLR. Atorvastatin, a lipid-lowering agent with antioxidative and anti-inflammatory effects reduced cholesterol synthesis in the kidneys improving renal function and kidney morphology in these models (116).
It should be noted that STZ has also been shown to impair renal function during the first week after injection in rodents affecting not only proteinuria but also renal microcirculation and oxidative pathways (117, 118). Studies to elucidate the contribution of hyperglycemia after STZ treatment or STZ-induced renal toxicity demonstrated that a majority of the renal damage was a result of the diabetic condition and only early proteinuria was caused by the nephrotoxic effects of STZ (119).
In the non-obese diabetic (NOD) mouse model of T1D, ABCA1 protein expression was decreased in kidneys and circulating macrophages, and associated with increased cholesterol content indicating that impaired reverse cholesterol transport may contribute to kidney phenotype observed in diabetic NOD mice (120). Diabetes-induced reduction of kidney ABCA1 expression in association with cholesterol accumulation was observed in cyclophosphamide-induced diabetic NOD and in LDLR-deficient-GP (glycoprotein) lymphocytic choriomeningitis virus (LCMV)-induced diabetic mice (121). In BTBR ob/ob mice, a type 2 diabetes model of DKD, cholesterol accumulation occurs in kidneys, a phenotype that can be prevented by cholesterol depletion with cyclodextrin, underlining the causative role of cholesterol accumulation in glomerular injury (52). Together these studies indicate that cholesterol accumulation due to impaired reverse cholesterol transport occurs in glomerular podocytes and contribute to the pathogenesis of DKD. As statins, which decrease cholesterol synthesis do not substantially affect the progression of kidney disease (122–124), other specific strategies that facilitate transport of sequestered cholesterol such as targeting reverse cholesterol transport are needed and may prove beneficial in treatment of DKD.
Chronic renal failure induced by 5/6 nephrectomy in rats is associated with neutral lipid accumulation in the remnant renal tissue, glomerulosclerosis and tubulointerstitial injury, and proteinuria. In remnant kidneys, increased expression of LXR, ABCA1, ABCG1, ACAT1, SR-B1, scavenger receptor A1 (SR-A1), lectin-type oxidized LDL receptor (LOX-1), carbohydrate-responsive element binding protein (ChREBP), fatty acid synthase (FAS), and acyl-CoA carboxylase (ACC) was detected, whereas expression of SREBP-1, SREBP-2, HMGCR, peroxisome proliferator-activated receptor alpha (PPAR-α), fatty acid binding protein (L-FABP), and carnitine palmitoyltransferase 1A (CPT1A) was decreased (125, 126). Cholesterol accumulation in proximal tubules is associated with HMGCR activation in acute kidney injury (AKI) (127, 128). The observation that cholesterol accumulation can result from glomerular injury suggested that renal cholesterol accumulation may represent a mechanism of stress response (129). In a model of experimental glomerulonephritis with concomitant proteinuria, cholesterol accumulation occurred in the renal cortex, i.e., the glomerulus and proximal tubular cells. While esterified cholesterol accumulation was present in glomerular and tubular cells, free cholesterol accumulation occurred in proximal tubular cells and was associated with increased ABCA1 and decreased SR-B1 expression (130).
Hypoxia-induced dyslipidemia is a phenomenon that was first described decades ago (131–133) and shown to play an important role in foam-cell formation and cytokine secretion in atherosclerosis and in non-alcoholic steatohepatitis (NASH) (134–137). Hypoxia is also a pathological feature observed in kidney disease and hypoxia-inducible factors (HIFs), such as HIF-1 and HIF-2 are thought to play an important role in AKI. HIFs initiate the transcription of a variety of genes involved in the regulation of tissue oxygen levels, glucose metabolism, apoptosis, lipid metabolism, and immune responses (138–143). HIF target genes include hemeoxygenase-1, vascular endothelial growth factor, plasminogen activator inhibitor-1, tissue-inhibitor of metalloproteinase-1, and connective tissue growth factor (139, 142, 144), all of which have been linked to the pathogenesis of a variety of glomerular diseases including DKD, FSGS, and HIVAN (145, 146). HIF-1 was shown to contribute to lipid accumulation by increasing lipid influx and synthesis in hepatocytes through increased LDL and very low-density lipoprotein (VLDL) uptake and increased levels and activity of HMGCR, respectively (147). More recently, it was demonstrated that activation of HIF-2 can also induce changes in hepatic lipid metabolism leading to the development of severe fatty liver disease in mice (148). Additionally, hypoxia-inducible protein 2 (HIG2) was identified as a novel lipid droplet protein and as a specific target gene of HIF-1 (149), further underlining a role for hypoxia in altered lipid metabolism in disease. In the kidney, HIF-1 was localized to the tubular epithelia, whereas HIF-2 was found predominantly in glomerular (podocytes), endothelial, and interstitial cells (150, 151). The induction of HIFs or inhibition of HIF degradation through hypoxia seems to have protective effects in AKI (152–154). Chronic hypoxia may also contribute to the pathogenesis of DKD (155) and genes induced by hypoxia can promote tubulointerstitial injury and renal fibrosis. Thus, novel therapeutic approaches targeting mechanisms of hypoxia-induced transcription may prove beneficial in targeting dyslipidemia in a variety of kidney diseases.
Inflammation, foam-cell formation, and lipid accumulation are characteristic occurrences associated with glomerulosclerosis. In non-alcoholic fatty liver disease (NAFLD), a two hit hypothesis proposed that a first hit such as obesity, T2D, or the initial lipid accumulation in the liver sensitizes it to a second hit such oxidative stress and proinflammatory cytokines ultimately causing hepatocellular injury and liver inflammation (156). In hepatocytes, tumor necrosis factor alpha (TNFα) or interleukin-1 beta (IL-1β) mediated inflammatory stress significantly reduced intracellular cholesterol efflux by inhibiting PPAR, LXR, and ABCA1 expression and increased LDLR and SREBP-2 expression and suggesting that inflammatory stress may exacerbates progression of fatty liver in NAFLD (157). We recently described an important role of circulating factors in the pathogenesis of DKD and showed that treatment of human podocytes with the sera from patients with DKD leads to cholesterol accumulation (52). Therefore, it seems possible that circulating inflammatory factors, such as TNFα or IL-1β, contribute to the progression of DKD. In support of this observation, TNFα is a major predictor of DKD progression in T1D and T2D (158–160) and inflammatory cytokines, such as TNFα or IL-1β, were shown to modify cholesterol-mediated LDL receptor regulation in mesangial cells. These studies suggest that inflammatory cytokines contribute to lipid-mediated renal damage (161, 162). Furthermore, IL-1β was shown to promote intracellular cholesterol accumulation in human mesangial cells via downregulation of ABCA1 (163). Interestingly, IL-1β treatment of hepatic and mesangial cells also interrupted LDLR feedback regulation, thus causing statin resistance (164), which might explain why statins are not effective in preventing the progression of DKD. More recently, Nod-like receptor protein 3 (Nlrp3) knockout mice were shown to be protected against obesity-induced renal fibrosis and microalbuminuria which, in wildtype mice, was associated with cholesterol and lipid accumulation in kidneys and increased SREBP-2, LDLR, and SREBP-1c expression (165). Whether cytokines contribute to lipid accumulation observed in podocytes and the exact mechanisms that lead to cytokine-induced cholesterol accumulation remain to be investigated.
Emerging data strongly suggest a role of impaired cholesterol homeostasis within glomerular cells as an important mediator of CKD. This is supported by the observation that a variety of glomerular diseases of genetic and non-genetic origin in humans and targeted mutations in mice that affect lipid and lipoprotein metabolism are associated with impaired glomerular structure.
Podocytes express genes and proteins that modulate cellular cholesterol homeostasis, such as LDLR, ABC transporters, and apolipoproteins involved in maintaining cellular cholesterol levels. Recent studies demonstrated that the dysregulation of a single gene involved in cholesterol homeostasis is usually not sufficient to cause a glomerular phenotype per se but renders renal cells more susceptible to glomerular injury. Based on these observations, we propose that in the kidney, similar to what was proposed in NAFLD, a first insult such as obesity, T2D, or an initial lipid accumulation occurs, which then sensitizes the kidney to a second insult such as oxidative stress, proinflammatory cytokines, or other stimuli ultimately leading to renal damage. Further research is needed to determine what are the exact stimuli and mechanisms that lead to glomerular cholesterol accumulation and ultimately to impaired glomerular function. In addition, significant advances in recent years have led to the identification of new regulators of cellular cholesterol homeostasis. The observation that circulating factors such as cytokines and hypoxia-induced transcription factors, such as HIFs, can directly regulate genes important for cholesterol homeostasis in hepatocytes opens new avenues to investigate if they also contribute to lipid dysregulation observed within glomerular cells. Such research will not only increase our insights into the physiology of cellular cholesterol homeostasis and trafficking in glomerular cells but it will also help to identify new drug targets and strategies to treat a variety of glomerular diseases, which are characterized by cellular dyslipidemia.
Sandra Merscher is the inventor on pending or issued patents aimed to diagnose or treat proteinuric renal diseases. She stands to gain royalties from their future commercialization. The other co-authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Sandra Merscher is supported by the US National Institutes of Health (NIH, http://www.nih.gov/) (DK090316), by the 2013 Stanley J. Glaser Foundation Research Award, and by the Peggy and Harold Katz Family Foundation. Christopher E. Pedigo is supported by an American Heart Association Greater Southeast Affiliate Pre-Doctoral fellowship 14PRE20380743.
1. Somlo S, Mundel P. Getting a foothold in nephrotic syndrome. Nat Genet (2000) 24:333–5. doi:10.1038/71724
2. Ruotsalainen V, Ljungberg P, Wartiovaara J, Lenkkeri U, Kestilä M, Jalanko H, et al. Nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc Natl Acad Sci U S A (1999) 96:7962–7. doi:10.1073/pnas.96.14.7962
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
3. Tryggvason K. Unraveling the mechanisms of glomerular ultrafiltration: nephrin, a key component of the slit diaphragm. J Am Soc Nephrol (1999) 10:2440–5.
4. Kestilä M, Lenkkeri U, Männikkö M, Lamerdin J, McCready P, Putaala H, et al. Positionally cloned gene for a novel glomerular protein – nephrin – is mutated in congenital nephrotic syndrome. Mol Cell (1998) 1:575–82. doi:10.1016/S1097-2765(00)80057-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
5. Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet (2000) 24:349–54. doi:10.1038/74166
6. Li C, Ruotsalainen V, Tryggvason K, Shaw AS, Miner JH. CD2AP is expressed with nephrin in developing podocytes and is found widely in mature kidney and elsewhere. Am J Physiol Renal Physiol (2000) 279:F785–92.
7. Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science (2005) 308:1801–4. doi:10.1126/science.1106215
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
8. Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet (2000) 24:251–6. doi:10.1038/73456
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
9. Huber TB, Schermer B, Müller RU, Höhne M, Bartram M, Calixto A, et al. Podocin and MEC-2 bind cholesterol to regulate the activity of associated ion channels. Proc Natl Acad Sci U S A (2006) 103:17079–86. doi:10.1073/pnas.0607465103
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
10. Schermer B, Benzing T. Lipid-protein interactions along the slit diaphragm of podocytes. J Am Soc Nephrol (2009) 20:473–8. doi:10.1681/ASN.2008070694
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
11. Tabas I. Consequences of cellular cholesterol accumulation: basic concepts and physiological implications. J Clin Invest (2002) 110:905–11. doi:10.1172/JCI0216452
12. Hua X, Yokoyama C, Wu J, Briggs MR, Brown MS, Goldstein JL, et al. SREBP-2, a second basic-helix-loop-helix-leucine zipper protein that stimulates transcription by binding to a sterol regulatory element. Proc Natl Acad Sci U S A (1993) 90:11603–7. doi:10.1073/pnas.90.24.11603
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
13. Yokoyama C, Wang X, Briggs MR, Admon A, Wu J, Hua X, et al. SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell (1993) 75:187–97. doi:10.1016/S0092-8674(05)80095-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
14. Brown MS, Goldstein JL. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc Natl Acad Sci U S A (1999) 96:11041–8. doi:10.1073/pnas.96.20.11041
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
15. McPherson R, Gauthier A. Molecular regulation of SREBP function: the Insig-SCAP connection and isoform-specific modulation of lipid synthesis. Biochem Cell Biol (2004) 82:201–11. doi:10.1139/o03-090
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
16. Ye J, DeBose-Boyd RA. Regulation of cholesterol and fatty acid synthesis. Cold Spring Harb Perspect Biol (2011) 3:a004754. doi:10.1101/cshperspect.a004754
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
17. Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol (2009) 29:431–8. doi:10.1161/ATVBAHA.108.179564
18. Abi-Mosleh L, Infante RE, Radhakrishnan A, Goldstein JL, Brown MS. Cyclodextrin overcomes deficient lysosome-to-endoplasmic reticulum transport of cholesterol in Niemann-Pick type C cells. Proc Natl Acad Sci U S A (2009) 106:19316–21. doi:10.1073/pnas.0910916106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
19. Maxfield FR, Wustner D. Intracellular cholesterol transport. J Clin Invest (2002) 110:891–8. doi:10.1172/JCI200216500
20. Kellner-Weibel G, Geng YJ, Rothblat GH. Cytotoxic cholesterol is generated by the hydrolysis of cytoplasmic cholesteryl ester and transported to the plasma membrane. Atherosclerosis (1999) 146:309–19. doi:10.1016/S0021-9150(99)00155-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
21. Warner GJ, Stoudt G, Bamberger M, Johnson WJ, Rothblat GH. Cell toxicity induced by inhibition of acyl coenzyme A:cholesterol acyltransferase and accumulation of unesterified cholesterol. J Biol Chem (1995) 270:5772–8. doi:10.1074/jbc.270.11.5772
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
22. Yao PM, Tabas I. Free cholesterol loading of macrophages is associated with widespread mitochondrial dysfunction and activation of the mitochondrial apoptosis pathway. J Biol Chem (2001) 276:42468–76. doi:10.1074/jbc.M101419200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
23. Ghosh S. Macrophage cholesterol homeostasis and metabolic diseases: critical role of cholesteryl ester mobilization. Expert Rev Cardiovasc Ther (2011) 9:329–40. doi:10.1586/erc.11.16
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
24. Rosenson RS, Brewer HB Jr, Davidson WS, Fayad ZA, Fuster V, Goldstein J, et al. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation (2012) 125:1905–19. doi:10.1161/CIRCULATIONAHA.111.066589
25. Keane WF, Tomassini JE, Neff DR. Lipid abnormalities in patients with chronic kidney disease. Contrib Nephrol (2011) 171:135–42. doi:10.1159/000327317
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
27. Vaziri ND, Norris K. Lipid disorders and their relevance to outcomes in chronic kidney disease. Blood Purif (2011) 31:189–96. doi:10.1159/000321845
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
28. Nestel PJ, Fidge NH, Tan MH. Increased lipoprotein-remnant formation in chronic renal failure. N Engl J Med (1982) 307:329–33. doi:10.1056/NEJM198208053070601
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
29. Al-Shahrouri HZ, Ramirez P, Fanti P, Abboud H, Lorenzo C, Haffner S. NMR identifies atherogenic lipoprotein abnormalities in early diabetic nephropathy that are unrecognized by conventional analysis. Clin Nephrol (2010) 73:180–9. doi:10.5414/CNP73180
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
30. Kuchta A, Pacanis A, Kortas-Stempak B, Cwiklinska A, Zietkiewicz M, Renke M, et al. Estimation of oxidative stress markers in chronic kidney disease. Kidney Blood Press Res (2011) 34:12–9. doi:10.1159/000321508
31. de Boer IH, Brunzell JD. HDL in CKD: how good is the “good cholesterol?”. J Am Soc Nephrol (2014) 25:871–4. doi:10.1681/ASN.2014010062
32. Calabresi L, Simonelli S, Conca P, Busnach G, Cabibbe M, Gesualdo L, et al. Acquired lcat deficiency as a major factor in lowering plasma HDL levels in chronic kidney disease. J Intern Med (2014). doi:10.1111/joim.12290
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
33. Peake KB, Vance JE. Defective cholesterol trafficking in Niemann-Pick C-deficient cells. FEBS Lett (2010) 584:2731–9. doi:10.1016/j.febslet.2010.04.047
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
34. Oikawa S, Matsunaga A, Saito T, Sato H, Seki T, Hoshi K, et al. Apolipoprotein E sendai (arginine 145 – >proline): a new variant associated with lipoprotein glomerulopathy. J Am Soc Nephrol (1997) 8:820–3.
35. Khera AV, Cuchel M, de laLlera-Moya M, Rodrigues A, Burke MF, Jafri K, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med (2011) 364:127–35. doi:10.1056/NEJMoa1001689
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
36. Le Lay S, Krief S, Farnier C, Lefrère I, Le Liepvre X, Bazin R, et al. Cholesterol, a cell size-dependent signal that regulates glucose metabolism and gene expression in adipocytes. J Biol Chem (2001) 276:16904–10. doi:10.1074/jbc.M010955200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
37. Avram MM, Fein PA, Antignani A, Mittman N, Mushnick RA, Lustig AR, et al. Cholesterol and lipid disturbances in renal disease: the natural history of uremic dyslipidemia and the impact of hemodialysis and continuous ambulatory peritoneal dialysis. Am J Med (1989) 87:55N–60N.
38. Joven J, Villabona C, Vilella E, Masana L, Albertí R, Vallés M. Abnormalities of lipoprotein metabolism in patients with the nephrotic syndrome. N Engl J Med (1990) 323:579–84. doi:10.1056/NEJM199008303230905
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
39. Wheeler DC. Lipid abnormalities in the nephrotic syndrome: the therapeutic role of statins. J Nephrol (2001) 14(Suppl 4):S70–5.
40. Attman PO, Samuelsson O, Alaupovic P. Lipoprotein metabolism and renal failure. Am J Kidney Dis (1993) 21:573–92. doi:10.1016/S0272-6386(12)80030-8
41. Herman-Edelstein M, Scherzer P, Tobar A, Levi M, Gafter U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J Lipid Res (2013) 55:561–72. doi:10.1194/jlr.P040501
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
42. Kimmelstiel P, Wilson C. Intercapillary lesions in the glomeruli of the kidney. Am J Pathol (1936) 12(83–98):87.
43. Moorhead JF, Chan MK, El-Nahas M, Varghese Z. Lipid nephrotoxicity in chronic progressive glomerular and tubulo-interstitial disease. Lancet (1982) 2:1309–11. doi:10.1016/S0140-6736(82)91513-6
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
44. Keane WF, O’Donnell MP, Kasiske BL, Schmitz PG. Lipids and the progression of renal disease. J Am Soc Nephrol (1990) 1:S69–74.
45. Blue ML, Protter AA, Williams DL. Biosynthesis of apolipoprotein B in rooster kidney, intestine, and liver. J Biol Chem (1980) 255:10048–51.
46. Blue ML, Ostapchuk P, Gordon JS, Williams DL. Synthesis of apolipoprotein AI by peripheral tissues of the rooster. A possible mechanism of cellular cholesterol efflux. J Biol Chem (1982) 257:11151–9.
47. Tarugi P, Ballarini G, Pinotti B, Franchini A, Ottaviani E, Calandra S. Secretion of apoB- and apoA-I-containing lipoproteins by chick kidney. J Lipid Res (1998) 39:731–43.
48. Xu N, Dahlback B. A novel human apolipoprotein (apoM). J Biol Chem (1999) 274:31286–90. doi:10.1074/jbc.274.44.31286
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
49. Blue ML, Williams DL, Zucker S, Khan SA, Blum CB. Apolipoprotein E synthesis in human kidney, adrenal gland, and liver. Proc Natl Acad Sci U S A (1983) 80:283–7. doi:10.1073/pnas.80.1.283
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
50. Gröne HJ, Walli AK, Gröne E, Krämer A, Clemens MR, Seidel D. Receptor mediated uptake of apo B and apo E rich lipoproteins by human glomerular epithelial cells. Kidney Int (1990) 37:1449–59. doi:10.1038/ki.1990.135
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
51. Madhavan SM, O’Toole JF, Konieczkowski M, Ganesan S, Bruggeman LA, Sedor JR. APOL1 localization in normal kidney and nondiabetic kidney disease. J Am Soc Nephrol (2011) 22:2119–28. doi:10.1681/ASN.2011010069
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
52. Merscher-Gomez S, Guzman J, Pedigo CE, Lehto M, Aguillon-Prada R, Mendez A, et al. Cyclodextrin protects podocytes in diabetic kidney disease. Diabetes (2013) 62(11):3817–27. doi:10.2337/db13-0399
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
53. Toyota K, Hashimoto T, Ogino D, Matsunaga A, Ito M, Masakane I, et al. A founder haplotype of APOE-Sendai mutation associated with lipoprotein glomerulopathy. J Hum Genet (2013) 58:254–8. doi:10.1038/jhg.2013.8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
54. Matsunaga A, Sasaki J, Komatsu T, Kanatsu K, Tsuji E, Moriyama K, et al. A novel apolipoprotein E mutation, E2 (Arg25Cys), in lipoprotein glomerulopathy. Kidney Int (1999) 56:421–7. doi:10.1046/j.1523-1755.1999.00572.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
55. Saito T, Ishigaki Y, Oikawa S, Yamamoto TT. Etiological significance of apolipoprotein E mutations in lipoprotein glomerulopathy. Trends Cardiovasc Med (2002) 12:67–70. doi:10.1016/S1050-1738(01)00148-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
56. Zhang P, Matalon R, Kaplan L, Kumar A, Gallo G. Lipoprotein glomerulopathy: first report in a Chinese male. Am J Kidney Dis (1994) 24:942–50. doi:10.1016/S0272-6386(12)81066-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
57. Amatruda JM, Margolis S, Hutchins GM. Type 3 hyperlipoproteinemia with mesangial foam cells in renal glomeruli. Arch Pathol (1974) 98:51–4.
58. Sakatsume M, Kadomura M, Sakata I, Imai N, Kondo D, Osawa Y, et al. Novel glomerular lipoprotein deposits associated with apolipoprotein E2 homozygosity. Kidney Int (2001) 59:1911–8. doi:10.1046/j.1523-1755.2001.0590051911.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
59. Ruiz J, Kouiavskaia D, Migliorini M, Robinson S, Saenko EL, Gorlatova N, et al. The apoE isoform binding properties of the VLDL receptor reveal marked differences from LRP and the LDL receptor. J Lipid Res (2005) 46:1721–31. doi:10.1194/jlr.M500114-JLR200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
60. Horita K, Eto M, Makino I. Apolipoprotein E2, renal failure and lipid abnormalities in non-insulin-dependent diabetes mellitus. Atherosclerosis (1994) 107:203–11. doi:10.1016/0021-9150(94)90021-3
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
61. Yorioka N, Nishida Y, Oda H, Watanabe T, Yamakido M. Apolipoprotein E polymorphism in IgA nephropathy. Nephron (1999) 83:246–9. doi:10.1159/000045517
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
62. Kimura H, Suzuki Y, Gejyo F, Karasawa R, Miyazaki R, Suzuki S, et al. Apolipoprotein E4 reduces risk of diabetic nephropathy in patients with NIDDM. Am J Kidney Dis (1998) 31:666–73. doi:10.1053/ajkd.1998.v31.pm9531184
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
63. Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc Natl Acad Sci U S A (1992) 89:4471–5. doi:10.1073/pnas.89.10.4471
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
64. Feussner G, Dobmeyer J, Grone HJ, Lohmer S, Wohlfeil S. A 10-bp deletion in the apolipoprotein epsilon gene causing apolipoprotein E deficiency and severe type III hyperlipoproteinemia. Am J Hum Genet (1996) 58:281–91.
65. Ishigaki Y, Oikawa S, Suzuki T, Usui S, Magoori K, Kim DH, et al. Virus-mediated transduction of apolipoprotein E (ApoE)-sendai develops lipoprotein glomerulopathy in ApoE-deficient mice. J Biol Chem (2000) 275:31269–73. doi:10.1074/jbc.M005906200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
66. Wen M, Segerer S, Dantas M, Brown PA, Hudkins KL, Goodpaster T, et al. Renal injury in apolipoprotein E-deficient mice. Lab Invest (2002) 82:999–1006. doi:10.1097/01.LAB.0000022222.03120.D4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
67. Ishimura A, Watanabe M, Nakashima H, Ito K, Miyake K, Mochizuki S, et al. Lipoprotein glomerulopathy induced by ApoE-Sendai is different from glomerular lesions in aged apoE-deficient mice. Clin Exp Nephrol (2009) 13:430–7. doi:10.1007/s10157-009-0195-1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
68. Mazzolai L, Korber M, Bouzourene K, Aubert JF, Nussberger J, Stamenkovic I, et al. Severe hyperlipidemia causes impaired renin-angiotensin system function in apolipoprotein E deficient mice. Atherosclerosis (2006) 186:86–91. doi:10.1016/j.atherosclerosis.2005.07.017
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
69. Bruneval P, Bariéty J, Bélair MF, Mandet C, Heudes D, Nicoletti A. Mesangial expansion associated with glomerular endothelial cell activation and macrophage recruitment is developing in hyperlipidaemic apoE null mice. Nephrol Dial Transplant (2002) 17:2099–107. doi:10.1093/ndt/17.12.2099
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
70. Lassila M, Seah KK, Allen TJ, Thallas V, Thomas MC, Candido R, et al. Accelerated nephropathy in diabetic apolipoprotein e-knockout mouse: role of advanced glycation end products. J Am Soc Nephrol (2004) 15:2125–38. doi:10.1097/01.ASN.0000133025.23732.46
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
71. Bagavant H, Scindia Y, Nackiewicz D, Nandula SR, Doran A, Cutchins A, et al. Deficiency of a transcriptional regulator, inhibitor of differentiation 3, induces glomerulonephritis in apolipoprotein E-deficient mice: a model linking hyperlipidemia and renal disease. Am J Pathol (2011) 179:651–60. doi:10.1016/j.ajpath.2011.04.029
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
72. Feng X, Li H, Rumbin AA, Wang X, La Cava A, Brechtelsbauer K, et al. ApoE-/-Fas-/- C57BL/6 mice: a novel murine model simultaneously exhibits lupus nephritis, atherosclerosis, and osteopenia. J Lipid Res (2007) 48:794–805. doi:10.1194/jlr.M600512-JLR200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
73. Arora S, Husain M, Kumar D, Patni H, Pathak S, Mehrotra D, et al. Human immunodeficiency virus downregulates podocyte apoE expression. Am J Physiol Renal Physiol (2009) 297:F653–61. doi:10.1152/ajprenal.90668.2008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
74. van Goor H, van derHorst ML, Atmosoerodjo J, Joles JA, van Tol A, Grond J. Renal apolipoproteins in nephrotic rats. Am J Pathol (1993) 142:1804–12.
75. Bruschi M, Catarsi P, Candiano G, Rastaldi MP, Musante L, Scolari F, et al. Apolipoprotein E in idiopathic nephrotic syndrome and focal segmental glomerulosclerosis. Kidney Int (2003) 63:686–95. doi:10.1046/j.1523-1755.2003.00777.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
76. Faber K, Hvidberg V, Moestrup SK, Dahlback B, Nielsen LB. Megalin is a receptor for apolipoprotein M, and kidney-specific megalin-deficiency confers urinary excretion of apolipoprotein M. Mol Endocrinol (2006) 20:212–8. doi:10.1210/me.2005-0209
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
77. Leheste JR, Rolinski B, Vorum H, Hilpert J, Nykjaer A, Jacobsen C, et al. Megalin knockout mice as an animal model of low molecular weight proteinuria. Am J Pathol (1999) 155:1361–70. doi:10.1016/S0002-9440(10)65238-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
78. Prabakaran T, Nielsen R, Larsen JV, Sørensen SS, Feldt-Rasmussen U, Saleem MA, et al. Receptor-mediated endocytosis of alpha-galactosidase A in human podocytes in Fabry disease. PLoS One (2011) 6:e25065. doi:10.1371/journal.pone.0025065
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
79. Kerjaschki D, Exner M, Ullrich R, Susani M, Curtiss LK, Witztum JL, et al. Pathogenic antibodies inhibit the binding of apolipoproteins to megalin/gp330 in passive Heymann nephritis. J Clin Invest (1997) 100:2303–9. doi:10.1172/JCI119768
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
80. Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol (2011) 22:2129–37. doi:10.1681/ASN.2011040388
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
81. Tzur S, Rosset S, Shemer R, Yudkovsky G, Selig S, Tarekegn A, et al. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet (2010) 128:345–50. doi:10.1007/s00439-010-0861-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
82. Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science (2010) 329:841–5. doi:10.1126/science.1193032
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
83. Norum KR, Gjone E. Familial serum-cholesterol esterification failure. A new inborn error of metabolism. Biochim Biophys Acta (1967) 144:698–700. doi:10.1016/0005-2760(67)90064-1
84. Glomset JA, Nichols AV, Norum KR, King W, Forte T. Plasma lipoproteins in familial lecithin: cholesterol acyltransferase deficiency. Further studies of very low and low density lipoprotein abnormalities. J Clin Invest (1973) 52:1078–92. doi:10.1172/JCI107273
85. Ohta Y, Yamamoto S, Tsuchida H, Murano S, Saitoh Y, Tohjo S, et al. Nephropathy of familial lecithin-cholesterol acyltransferase deficiency: report of a case. Am J Kidney Dis (1986) 7:41–6. doi:10.1016/S0272-6386(86)80055-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
86. Borysiewicz LK, Soutar AK, Evans DJ, Thompson GR, Rees AJ. Renal failure in familial lecithin: cholesterol acyltransferase deficiency. Q J Med (1982) 51:411–26.
87. Seidel D, Agostini B, Muller P. Structure of an abnormal plasma lipoprotein (LP-X) characterizing obstructive jaundice. Biochim Biophys Acta (1972) 260:146–52. doi:10.1016/0005-2760(72)90082-3
88. Torsvik H, Berg K, Magnani HN, McConathy WJ, Alaupovic P, Gjone E. Identification of the abnormal cholestatic lipoprotein (LP-X) in familial lecithin:cholesterol acyltransferase deficiency. FEBS Lett (1972) 24:165–8. doi:10.1016/0014-5793(72)80758-0
89. McLean J, Wion K, Drayna D, Fielding C, Lawn R. Human lecithin-cholesterol acyltransferase gene: complete gene sequence and sites of expression. Nucleic Acids Res (1986) 14:9397–406. doi:10.1093/nar/14.23.9397
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
90. Hixson JE, Driscoll DM, Birnbaum S, Britten ML. Baboon lecithin cholesterol acyltransferase (LCAT): cDNA sequences of two alleles, evolution, and gene expression. Gene (1993) 128:295–9. doi:10.1016/0378-1119(93)90578-Q
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
91. Liu YH, Wu JS, Wang ZY, Yu CH, Ying HZ, Xu NY. Characteristic, polymorphism and expression distribution of LCAT gene in a Mongolian gerbil model for hyperlipidemia. Exp Mol Pathol (2014). doi:10.1016/j.yexmp.2014.07.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
92. Sakai N, Vaisman BL, Koch CA, Hoyt RF Jr, Meyn SM, Talley GD, et al. Targeted disruption of the mouse lecithin:cholesterol acyltransferase (LCAT) gene. Generation of a new animal model for human LCAT deficiency. J Biol Chem (1997) 272:7506–10. doi:10.1074/jbc.272.11.7506
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
93. Ng DS, Francone OL, Forte TM, Zhang J, Haghpassand M, Rubin EM. Disruption of the murine lecithin:cholesterol acyltransferase gene causes impairment of adrenal lipid delivery and up-regulation of scavenger receptor class B type I. J Biol Chem (1997) 272:15777–81. doi:10.1074/jbc.272.25.15777
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
94. Lambert G, Sakai N, Vaisman BL, Neufeld EB, Marteyn B, Chan CC, et al. Analysis of glomerulosclerosis and atherosclerosis in lecithin cholesterol acyltransferase-deficient mice. J Biol Chem (2001) 276:15090–8. doi:10.1074/jbc.M008466200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
95. Zhu X, Herzenberg AM, Eskandarian M, Maguire GF, Scholey JW, Connelly PW, et al. A novel in vivo lecithin-cholesterol acyltransferase (LCAT)-deficient mouse expressing predominantly LpX is associated with spontaneous glomerulopathy. Am J Pathol (2004) 165:1269–78. doi:10.1016/S0002-9440(10)63386-X
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
96. Pentchev PG, Blanchette-Mackie EJ, Dawidowicz EA. The NP-C gene: a key to pathways of intracellular cholesterol transport. Trends Cell Biol (1994) 4:365–9. doi:10.1016/0962-8924(94)90086-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
97. Pentchev PG, Comly ME, Kruth HS, Vanier MT, Wenger DA, Patel S, et al. A defect in cholesterol esterification in Niemann-Pick disease (type C) patients. Proc Natl Acad Sci U S A (1985) 82:8247–51. doi:10.1073/pnas.82.23.8247
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
98. Loftus SK, Morris JA, Carstea ED, Gu JZ, Cummings C, Brown A, et al. Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science (1997) 277:232–5. doi:10.1126/science.277.5323.232
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
99. Liao YJ, Lin MW, Yen CH, Lin YT, Wang CK, Huang SF, et al. Characterization of Niemann-Pick Type C2 protein expression in multiple cancers using a novel NPC2 monoclonal antibody. PLoS One (2013) 8:e77586. doi:10.1371/journal.pone.0077586
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
100. Grafft CA, Fervenza FC, Semret MH, Orloff S, Sethi S. Renal involvement in Neimann-pick disease. NDT Plus (2009) 2:448–51. doi:10.1093/ndtplus/sfp101
101. Philit JB, Queffeulou G, Walker F, Gubler MC, Dupuis E, Vrtovsnik F, et al. Membranoproliferative glomerulonephritis type II and Niemann-Pick disease type C. Nephrol Dial Transplant (2002) 17:1829–31. doi:10.1093/ndt/17.10.1829
102. Liu B, Turley SD, Burns DK, Miller AM, Repa JJ, Dietschy JM. Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1-/- mouse. Proc Natl Acad Sci U S A (2009) 106:2377–82. doi:10.1073/pnas.0810895106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
103. Ramirez CM, Liu B, Taylor AM, Repa JJ, Burns DK, Weinberg AG, et al. Weekly cyclodextrin administration normalizes cholesterol metabolism in nearly every organ of the Niemann-Pick type C1 mouse and markedly prolongs life. Pediatr Res (2010) 68:309–15. doi:10.1203/00006450-201011001-00604
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
104. Davidson CD, Ali NF, Micsenyi MC, Stephney G, Renault S, Dobrenis K, et al. Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS One (2009) 4:e6951. doi:10.1371/journal.pone.0006951
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
105. Mott S, Yu L, Marcil M, Boucher B, Rondeau C, Genest J Jr. Decreased cellular cholesterol efflux is a common cause of familial hypoalphalipoproteinemia: role of the ABCA1 gene mutations. Atherosclerosis (2000) 152:457–68. doi:10.1016/S0021-9150(99)00498-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
106. Bodzioch M, Orsó E, Klucken J, Langmann T, Böttcher A, Diederich W, et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat Genet (1999) 22:347–51. doi:10.1038/11914
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
107. Brooks-Wilson A, Marcil M, Clee SM, Zhang LH, Roomp K, van Dam M, et al. Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency. Nat Genet (1999) 22:336–45. doi:10.1038/11905
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
108. Rust S, Rosier M, Funke H, Real J, Amoura Z, Piette JC, et al. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat Genet (1999) 22:352–5. doi:10.1038/11921
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
109. Ferrans VJ, Fredrickson DS. The pathology of Tangier disease. A light and electron microscopic study. Am J Pathol (1975) 78:101–58.
110. Mautner SL, Sanchez JA, Rader DJ, Mautner GC, Ferrans VJ, Fredrickson DS, et al. The heart in Tangier disease. Severe coronary atherosclerosis with near absence of high-density lipoprotein cholesterol. Am J Clin Pathol (1992) 98:191–8.
111. Christiansen-Weber TA, Voland JR, Wu Y, Ngo K, Roland BL, Nguyen S, et al. Functional loss of ABCA1 in mice causes severe placental malformation, aberrant lipid distribution, and kidney glomerulonephritis as well as high-density lipoprotein cholesterol deficiency. Am J Pathol (2000) 157:1017–29. doi:10.1016/S0002-9440(10)64614-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
112. Orsó E, Broccardo C, Kaminski WE, Böttcher A, Liebisch G, Drobnik W, et al. Transport of lipids from golgi to plasma membrane is defective in tangier disease patients and Abc1-deficient mice. Nat Genet (2000) 24:192–6. doi:10.1038/72869
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
113. Proctor G, Jiang T, Iwahashi M, Wang Z, Li J, Levi M. Regulation of renal fatty acid and cholesterol metabolism, inflammation, and fibrosis in Akita and OVE26 mice with type 1 diabetes. Diabetes (2006) 55:2502–9. doi:10.2337/db05-0603
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
114. Wang W, Qiu L, Howard A, Solis N, Li C, Wang X, et al. Protective effects of aliskiren and valsartan in mice with diabetic nephropathy. J Renin Angiotensin Aldosterone Syst (2014). doi:10.1177/1470320313507123
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
115. Wang Z, Jiang T, Li J, Proctor G, McManaman JL, Lucia S, et al. Regulation of renal lipid metabolism, lipid accumulation, and glomerulosclerosis in FVBdb/db mice with type 2 diabetes. Diabetes (2005) 54:2328–35. doi:10.2337/diabetes.54.8.2328
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
116. Sun H, Yuan Y, Sun ZL. Cholesterol contributes to diabetic nephropathy through SCAP-SREBP-2 pathway. Int J Endocrinol (2013) 2013:592576. doi:10.1155/2013/592576
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
117. Inada A, Kanamori H, Arai H, Akashi T, Araki M, Weir GC, et al. A model for diabetic nephropathy: advantages of the inducible cAMP early repressor transgenic mouse over the streptozotocin-induced diabetic mouse. J Cell Physiol (2008) 215:383–91. doi:10.1002/jcp.21316
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
118. Tesch GH, Allen TJ. Rodent models of streptozotocin-induced diabetic nephropathy (methods in renal research). Nephrology (2007) 12:261–6. doi:10.1111/j.1440-1797.2007.00796.x
119. Palm F, Ortsäter H, Hansell P, Liss P, Carlsson P-O. Differentiating between effects of streptozotocin per se and subsequent hyperglycemia on renal function and metabolism in the streptozotocin-diabetic rat model. Diabetes Metab Res Rev (2004) 20:452–9. doi:10.1002/dmrr.472
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
120. Tang C, Kanter JE, Bornfeldt KE, Leboeuf RC, Oram JF. Diabetes reduces the cholesterol exporter ABCA1 in mouse macrophages and kidneys. J Lipid Res (2009) 51:1719–28. doi:10.1194/jlr.M003525
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
121. Renard CB, Kramer F, Johansson F, Lamharzi N, Tannock LR, von Herrath MG, et al. Diabetes and diabetes-associated lipid abnormalities have distinct effects on initiation and progression of atherosclerotic lesions. J Clin Invest (2004) 114:659–68. doi:10.1172/JCI17867
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
122. Agarwal R. Anti-inflammatory effects of short-term pioglitazone therapy in men with advanced diabetic nephropathy. Am J Physiol Renal Physiol (2006) 290:F600–5. doi:10.1152/ajprenal.00289.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
123. Colhoun HM, Betteridge DJ, Durrington PN, Hitman GA, Neil HA, Livingstone SJ, et al. Effects of atorvastatin on kidney outcomes and cardiovascular disease in patients with diabetes: an analysis from the collaborative atorvastatin diabetes study (CARDS). Am J Kidney Dis (2009) 54:810–9. doi:10.1053/j.ajkd.2009.03.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
124. Haynes R, Lewis D, Emberson J, Reith C, Agodoa L, Cass A, et al. Effects of lowering LDL cholesterol on progression of kidney disease. J Am Soc Nephrol (2014) 25(8):1825–33. doi:10.1681/ASN.2013090965
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
125. Cho KH, Kim HJ, Kamanna VS, Vaziri ND. Niacin improves renal lipid metabolism and slows progression in chronic kidney disease. Biochim Biophys Acta (2009) 1800:6–15. doi:10.1016/j.bbagen.2009.10.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
126. Moradi H, Yuan J, Ni Z, Norris K, Vaziri ND. Reverse cholesterol transport pathway in experimental chronic renal failure. Am J Nephrol (2009) 30:147–54. doi:10.1159/000210020
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
127. Johnson AC, Ware LB, Himmelfarb J, Zager RA. HMG-CoA reductase activation and urinary pellet cholesterol elevations in acute kidney injury. Clin J Am Soc Nephrol (2011) 6:2108–13. doi:10.2215/CJN.02440311
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
128. Zager RA, Johnson A. Renal cortical cholesterol accumulation is an integral component of the systemic stress response. Kidney Int (2001) 60:2299–310. doi:10.1046/j.1523-1755.2001.00071.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
129. Zager RA, Andoh T, Bennett WM. Renal cholesterol accumulation: a durable response after acute and subacute renal insults. Am J Pathol (2001) 159:743–52. doi:10.1016/S0002-9440(10)61745-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
130. Johnson AC, Yabu JM, Hanson S, Shah VO, Zager RA. Experimental glomerulopathy alters renal cortical cholesterol, SR-B1, ABCA1, and HMG CoA reductase expression. Am J Pathol (2003) 162:283–91. doi:10.1016/S0002-9440(10)63819-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
131. Gordon GB. Saturated free fatty acid toxicity. II. lipid accumulation, ultrastructural alterations, and toxicity in mammalian cells in culture. Exp Mol Pathol (1977) 27:262–76. doi:10.1016/0014-4800(77)90035-1
132. Gordon GB, Barcza MA, Bush ME. Lipid accumulation of hypoxic tissue culture cells. Am J Pathol (1977) 88:663–78.
133. Whitmer JT, Idell-Wenger JA, Rovetto MJ, Neely JR. Control of fatty acid metabolism in ischemic and hypoxic hearts. J Biol Chem (1978) 253:4305–9.
134. Aron-Wisnewsky J, Minville C, Tordjman J, Lévy P, Bouillot JL, Basdevant A, et al. Chronic intermittent hypoxia is a major trigger for non-alcoholic fatty liver disease in morbid obese. J Hepatol (2011) 56:225–33. doi:10.1016/j.jhep.2011.04.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
135. Boström P, Magnusson B, Svensson PA, Wiklund O, Borén J, Carlsson LM, et al. Hypoxia converts human macrophages into triglyceride-loaded foam cells. Arterioscler Thromb Vasc Biol (2006) 26:1871–6. doi:10.1161/01.ATV.0000229665.78997.0b
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
136. Byrne CD. Hypoxia and non-alcoholic fatty liver disease. Clin Sci (Lond) (2009) 118:397–400. doi:10.1042/CS20090565
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
137. Sluimer JC, Gasc JM, van Wanroij JL, Kisters N, Groeneweg M, Sollewijn Gelpke MD, et al. Hypoxia, hypoxia-inducible transcription factor, and macrophages in human atherosclerotic plaques are correlated with intraplaque angiogenesis. J Am Coll Cardiol (2008) 51:1258–65. doi:10.1016/j.jacc.2007.12.025
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
138. Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell (2003) 112:645–57. doi:10.1016/S0092-8674(03)00154-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
139. Greijer AE, van derGroep P, Kemming D, Shvarts A, Semenza GL, Meijer GA, et al. Up-regulation of gene expression by hypoxia is mediated predominantly by hypoxia-inducible factor 1 (HIF-1). J Pathol (2005) 206:291–304. doi:10.1002/path.1778
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
140. Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, et al. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood (2005) 105:659–69. doi:10.1182/blood-2004-07-2958
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
141. Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature (2006) 441:437–43. doi:10.1038/nature04871
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
142. Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med (2003) 9:677–84. doi:10.1038/nm0603-677
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
143. Vengellur A, Woods BG, Ryan HE, Johnson RS, LaPres JJ. Gene expression profiling of the hypoxia signaling pathway in hypoxia-inducible factor 1alpha null mouse embryonic fibroblasts. Gene Expr (2003) 11:181–97. doi:10.3727/000000003108749062
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
144. Wenger RH. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J (2002) 16:1151–62. doi:10.1096/fj.01-0944rev
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
145. Reis VO, Silva JC, Souza GT, Semedo P, Buscariollo B, Pereira RL, et al. The polysaccharide fraction of Propionibacterium acnes modulates the development of experimental focal segmental glomerulosclerosis. Immunobiology (2011) 217:831–41. doi:10.1016/j.imbio.2011.12.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
146. Pereira RL, Buscariollo BN, Corrêa-Costa M, Semedo P, Oliveira CD, Reis VO, et al. Bradykinin receptor 1 activation exacerbates experimental focal and segmental glomerulosclerosis. Kidney Int (2011) 79:1217–27. doi:10.1038/ki.2011.14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
147. Pallottini V, Guantario B, Martini C, Totta P, Filippi I, Carraro F, et al. Regulation of HMG-CoA reductase expression by hypoxia. J Cell Biochem (2008) 104:701–9. doi:10.1002/jcb.21757
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
148. Rankin EB, Rha J, Selak MA, Unger TL, Keith B, Liu Q, et al. Hypoxia-inducible factor 2 regulates hepatic lipid metabolism. Mol Cell Biol (2009) 29:4527–38. doi:10.1128/MCB.00200-09
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
149. Gimm T, Wiese M, Teschemacher B, Deggerich A, Schödel J, Knaup KX, et al. Hypoxia-inducible protein 2 is a novel lipid droplet protein and a specific target gene of hypoxia-inducible factor-1. FASEB J (2010) 24:4443–58. doi:10.1096/fj.10-159806
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
150. Haase VH. Hypoxia-inducible factors in the kidney. Am J Physiol Renal Physiol (2006) 291:F271–81. doi:10.1152/ajprenal.00071.2006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
151. Freeburg PB, Robert B, St John PL, Abrahamson DR. Podocyte expression of hypoxia-inducible factor (HIF)-1 and HIF-2 during glomerular development. J Am Soc Nephrol (2003) 14:927–38. doi:10.1097/01.ASN.0000059308.82322.4F
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
152. Kojima I, Tanaka T, Inagi R, Kato H, Yamashita T, Sakiyama A, et al. Protective role of hypoxia-inducible factor-2alpha against ischemic damage and oxidative stress in the kidney. J Am Soc Nephrol (2007) 18:1218–26. doi:10.1681/ASN.2006060639
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
153. Matsumoto M, Makino Y, Tanaka T, Tanaka H, Ishizaka N, Noiri E, et al. Induction of renoprotective gene expression by cobalt ameliorates ischemic injury of the kidney in rats. J Am Soc Nephrol (2003) 14:1825–32. doi:10.1097/01.ASN.0000074239.22357.06
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
154. Weidemann A, Bernhardt WM, Klanke B, Daniel C, Buchholz B, Câmpean V, et al. HIF activation protects from acute kidney injury. J Am Soc Nephrol (2008) 19:486–94. doi:10.1681/ASN.2007040419
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
155. Singh DK, Winocour P, Farrington K. Mechanisms of disease: the hypoxic tubular hypothesis of diabetic nephropathy. Nat Clin Pract Nephrol (2008) 4:216–26. doi:10.1038/ncpneph0757
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
156. Musso G, Gambino R, Cassader M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog Lipid Res (2012) 52:175–91. doi:10.1016/j.plipres.2012.11.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
157. Ma KL, Ruan XZ, Powis SH, Chen Y, Moorhead JF, Varghese Z. Inflammatory stress exacerbates lipid accumulation in hepatic cells and fatty livers of apolipoprotein E knockout mice. Hepatology (2008) 48:770–81. doi:10.1002/hep.22423
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
158. Gohda T, Niewczas MA, Ficociello LH, Walker WH, Skupien J, Rosetti F, et al. Circulating TNF receptors 1 and 2 predict stage 3 CKD in type 1 diabetes. J Am Soc Nephrol (2012) 23:516–24. doi:10.1681/ASN.2011060628
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
159. Niewczas MA, Ficociello LH, Johnson AC, Walker W, Rosolowsky ET, Roshan B, et al. Serum concentrations of markers of TNFalpha and Fas-mediated pathways and renal function in nonproteinuric patients with type 1 diabetes. Clin J Am Soc Nephrol (2009) 4:62–70. doi:10.2215/CJN.03010608
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
160. Niewczas MA, Gohda T, Skupien J, Smiles AM, Walker WH, Rosetti F, et al. Circulating TNF receptors 1 and 2 predict ESRD in type 2 diabetes. J Am Soc Nephrol (2012) 23:507–15. doi:10.1681/ASN.2011060627
161. Zhang G, Li Q, Wang L, Chen Y, Zhang W. Interleukin-1beta enhances the intracellular accumulation of cholesterol by up-regulating the expression of low-density lipoprotein receptor and 3-hydroxy-3-methylglutaryl coenzyme A reductase in podocytes. Mol Cell Biochem (2011) 346:197–204. doi:10.1007/s11010-010-0605-4
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
162. Ruan XZ, Varghese Z, Powis SH, Moorhead JF. Dysregulation of LDL receptor under the influence of inflammatory cytokines: a new pathway for foam cell formation. Kidney Int (2001) 60:1716–25. doi:10.1046/j.1523-1755.2001.00025.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
163. Ruan XZ, Moorhead JF, Fernando R, Wheeler DC, Powis SH, Varghese Z. PPAR agonists protect mesangial cells from interleukin 1beta-induced intracellular lipid accumulation by activating the ABCA1 cholesterol efflux pathway. J Am Soc Nephrol (2003) 14:593–600. doi:10.1097/01.ASN.0000050414.52908.DA
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
164. Chen Y, Ruan XZ, Li Q, Huang A, Moorhead JF, Powis SH, et al. Inflammatory cytokines disrupt LDL-receptor feedback regulation and cause statin resistance: a comparative study in human hepatic cells and mesangial cells. Am J Physiol Renal Physiol (2007) 293:F680–7. doi:10.1152/ajprenal.00209.2007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
165. Bakker PJ, Butter LM, Kors L, Teske GJ, Aten J, Sutterwala FS, et al. Nlrp3 is a key modulator of diet-induced nephropathy and renal cholesterol accumulation. Kidney Int (2014) 85:1112–22. doi:10.1038/ki.2013.503
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: cholesterol, podocyte, renal disease, kidney disease, glomerular disease, ABCA1, reverse cholesterol transport, apolipoprotein
Citation: Merscher S, Pedigo CE and Mendez AJ (2014) Metabolism, energetics, and lipid biology in the podocyte – cellular cholesterol-mediated glomerular injury. Front. Endocrinol. 5:169. doi: 10.3389/fendo.2014.00169
Received: 14 August 2014; Paper pending published: 08 September 2014;
Accepted: 28 September 2014; Published online: 14 October 2014.
Edited by:
Sonia Q. Doi, Uniformed Services University, USAReviewed by:
Rudy M. Ortiz, University of California Merced, USACopyright: © 2014 Merscher, Pedigo and Mendez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandra Merscher, Peggy and Harold Katz Family Drug Discovery Center and Division of Nephrology and Hypertension, Department of Medicine, University of Miami,1580 NW 10th Ave, Batchelor Bldg, Room 628, Miami, FL 33136, USA e-mail:c21lcnNjaGVyQG1lZC5taWFtaS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.