
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 09 October 2014
Sec. Neuroendocrine Science
Volume 5 - 2014 | https://doi.org/10.3389/fendo.2014.00161
 Enrique Blázquez1,2,3*
Enrique Blázquez1,2,3* Esther Velázquez1,2,3Verónica Hurtado-Carneiro1,2,3Juan Miguel Ruiz-Albusac1,2,3
Esther Velázquez1,2,3Verónica Hurtado-Carneiro1,2,3Juan Miguel Ruiz-Albusac1,2,3
Although the brain has been considered an insulin-insensitive organ, recent reports on the location of insulin and its receptors in the brain have introduced new ways of considering this hormone responsible for several functions. The origin of insulin in the brain has been explained from peripheral or central sources, or both. Regardless of whether insulin is of peripheral origin or produced in the brain, this hormone may act through its own receptors present in the brain. The molecular events through which insulin functions in the brain are the same as those operating in the periphery. However, certain insulin actions are different in the central nervous system, such as hormone-induced glucose uptake due to a low insulin-sensitive GLUT-4 activity, and because of the predominant presence of GLUT-1 and GLUT-3. In addition, insulin in the brain contributes to the control of nutrient homeostasis, reproduction, cognition, and memory, as well as to neurotrophic, neuromodulatory, and neuroprotective effects. Alterations of these functional activities may contribute to the manifestation of several clinical entities, such as central insulin resistance, type 2 diabetes mellitus (T2DM), and Alzheimer’s disease (AD). A close association between T2DM and AD has been reported, to the extent that AD is twice more frequent in diabetic patients, and some authors have proposed the name “type 3 diabetes” for this association. There are links between AD and T2DM through mitochondrial alterations and oxidative stress, altered energy and glucose metabolism, cholesterol modifications, dysfunctional protein O-GlcNAcylation, formation of amyloid plaques, altered Aβ metabolism, and tau hyperphosphorylation. Advances in the knowledge of preclinical AD and T2DM may be a major stimulus for the development of treatment for preventing the pathogenic events of these disorders, mainly those focused on reducing brain insulin resistance, which is seems to be a common ground for both pathological entities.
Although the control of peripheral glucose homeostasis is one of the main functions of insulin, its action on the brain is now also being studied carefully, as it is considered an insulin-sensitive organ because insulin receptors (IR) and their signal transduction pathways have been identified in several regions of the brain that mediate important physiological effects on this organ, such as neuronal development, glucoregulation, feeding behavior, and body weight, as well as cognitive processes, including attention, executive functioning, learning, and memory (1).
In the late 1970s, the central nervous system (CNS) was not considered to be an insulin-dependent tissue, but it is now well known that insulin plays a major physiologic role in this tissue and its disturbances, being involved in certain neurodegenerative states, such as Alzheimer’s disease (AD). The presence of insulin in the brain was first detected by Havrankova et al. (2), who used radioimmunoassay to determine high levels of insulin in brain extracts. Likewise, they reported that insulin content in the brain was independent of the peripheral insulin, since circulating insulin levels had no effect on the brain’s insulin concentration (3). In addition, high insulin concentrations had also been reported not only in the human brain but also in several experimental animals (4).
The detection of insulin in cerebrospinal fluid (CSF) should not be interpreted as a robust indication of blood–brain barrier (BBB) transport. There is a blood:CSF barrier located at the choroid plexus, and insulin entering the brain from the blood cannot be expected rapidly be excreted in CSF (5, 6).
The presence of high concentrations of insulin in brain samples has raised the question of its origin. This question continues to be one of the most debated aspects of the research into cerebral insulin. Previous findings support the hypothesis that, at least in part, “brain insulin” is produced in the CNS. However, as posited by Havrankova et al. (2), other sources for cerebral insulin should be considered, such as its peripheral origin and then crossing the BBB, versus a central origin, or both.
The notion that insulin could cross the BBB was first suggested by Margolis and Altszuler (7), who showed that insulin levels in the CSF of rats increased slightly after peripheral infusions of this hormone, suggesting that insulin crossed the BBB possibly by means of a saturable transport system. These results were later confirmed in dogs after the iv administration of insulin (8). They found a large, rapid increase in blood insulin, but a relatively small increase in the hormone in the CSF. These findings confirmed a non-linear correlation between plasma and CSF levels of insulin, providing the first evidence for a saturable transport system for insulin from blood to the brain. Although there is no direct evidence on whether the insulin transport system and the IR are the same protein, this seems to be widely assumed (9), as they have similar physicochemical properties (saturability, specificity, affinity, immunoneutralization, cooperative interactions, and kinetics of dissociation) to IRs (10) in typical target tissues (11). On the other hand, differences in the activity of the BBB transporter system may be responding to regional differences in insulin permeability, and also to the hormone concentration, recording the highest values in the pons, medulla, and hypothalamus, and the lowest in the occipital cortex and thalamus (12). This insulin transport may be regulated by multiple factors, such as glucocorticoids (13), or in several pathophysiological situations, such as fasting and re-feeding (14), obesity (15), and hibernation (16), as well as during aging and in patients with diabetes mellitus (DM), and AD (17, 18).
The local production of insulin in the CNS has also been widely studied, and suggestions of possible insulin biosynthesis in the brain have been based on different experimental evidence, as cited below.
Immunoreactive insulin and C-peptide were found in the brain from human cadavers (19), recording concentrations that were much higher in the brain than in the blood, with the highest content in the hypothalamus. In addition, C-peptide concentration decreased after 72 h of fasting, and increased after oral glucose administration in both plasma and hypothalamus (20). On the other hand, C-peptide was found in the post-mortem brain cortex of elderly and AD subjects, showing for the first time a direct correlation between C-peptide concentrations and a decrease in the number of brain IRs (21). These findings led the authors to conclude that, at least in part, cerebral insulin was a product of the brain itself.
Evidence of the presence of insulin mRNA was found in the periventricular nucleus of the rat hypothalamus by in situ hybridization (22). Furthermore, the use of RNase-protection and sensitive reverse transcription-polymerase chain reaction (RT-PCR) assays led to the detection of insulin II mRNA in the brains of fetal, neonatal, and adult rats, which suggests that the ancestral insulin II gene expression in rat brains belongs to the pre-pancreatic stage of embryonic development (23). Likewise, insulin mRNA was located in the CA1 and CA3 regions of the hippocampus, in the dentate gyrus, and in the granule cell layer of the olfactory bulbs of the neonatal rabbit brain by in situ hybridization experiments (24).
Evidence supporting the synthesis of insulin in the CNS has also been obtained from brain cell cultures. Thus, incubation with [3H]valine resulted in the incorporation of radioactivity into immunoprecipitable insulin, and with cycloheximide caused an 80% decrease in the number of insulin-like immunoreactive neurons from primary cultures of rat brain (25). In addition, two main forms of the immunoreactive insulin (IRI) were detected in cultures from fetal mouse brain. The major component resembling pro-insulin was converted by trypsin into the minor form, which was similar to real pancreatic insulin (26). Likewise, using immunohistochemical and in situ hybridization techniques it has been showed the ability of fetal neuron cell cultures to produce and secrete an insulin-like mRNA and an insulin-like substance (ILS) that was indistinguishable from real insulin (27). On the other hand, the novo synthesis and secretion of insulin by central mammalian neurons in both neuron-enriched and glial-enriched postnatal rabbit brain cell cultures was studied (24), indicating that specific neurons, but not astrocytes, produced an extracellular secretion of immunoprecipitable insulin.
The molecular mechanisms involved in the production and secretion of insulin in the CNS reveal similarities between beta cells and neurons, particularly in relation to ATP-sensitive K+ channel depolarization (28). Both beta cells and neurons are electrically excitable and respond to hormonal stimuli and glucose by depolarization and exocytosis. The depolarization by potassium ions (in the presence of calcium) of primary cultures of neuronal cells caused a threefold stimulation of insulin release. This depolarization-induced release of insulin was inhibited by cycloheximide, and was specific for neurons, but not for astrocytes (29). Similarly, insulin was also released from adult rat brain synaptosomes under depolarizing conditions, and depending on calcium influx, which suggested that insulin was stored in the adult rat brain in synaptic vesicles within nerve endings, from which it can be mobilized by exocytosis related to neural activity (30). In synaptosomes, it has been shown that insulin secretion was increased by glucose, and that the addition of the glycolytic inhibitor, iodoacetic acid (IAA), produced a 50% decrease in the glucose-induced release of IRI, suggesting that, as occurs in the pancreas, glucose metabolism is also involved in brain insulin release (31). These results imply that the brain itself might synthesize some portion of the insulin detected locally, which is not an unusual occurrence (32).
The BBB is formed by a type of brain endothelial cell (33) that is unique, since the cell membranes are exposed both to the blood stream and to the CNS, whereby these cells receive signals from both the periphery and the CNS (17). BECs have insulin-binding sites that appear to have two distinct functions: as transporters of insulin across the BBB (Figure 1) and as classic receptors (34), both affecting the function of the barrier cell by activating intracellular machinery and mediating the effects of insulin on these cells, such as the increase in the transport of tyrosine and tryptophan (35), azidothymidine (36), and leptin (37) from blood to brain. In addition, insulin modifies the expression and/or activity of certain efflux transporters. Thus, insulin induces P-glycoprotein expression (the 170-kDa protein product of the multidrug resistance one gene), which plays an important role in the integrity of the BBB, protects the brain from many exogenous toxins (38), and suppresses the expression and function of the breast cancer resistance protein (39). Likewise, insulin induces neurochemical modifications in brain microvessels by inhibiting the activity of alkaline phosphatase (40), and increasing the expression and activity of the glutamate–cysteine ligase catalytic subunit by activating the antioxidant response element-4 (41). In addition, insulin inhibits the activity of the serotonin receptor 5-HT2c in choroid plexus, showing that this G-protein-coupled receptor (GPCR) is modulated by the tyrosine-kinase receptor-MAP kinase pathway (42).
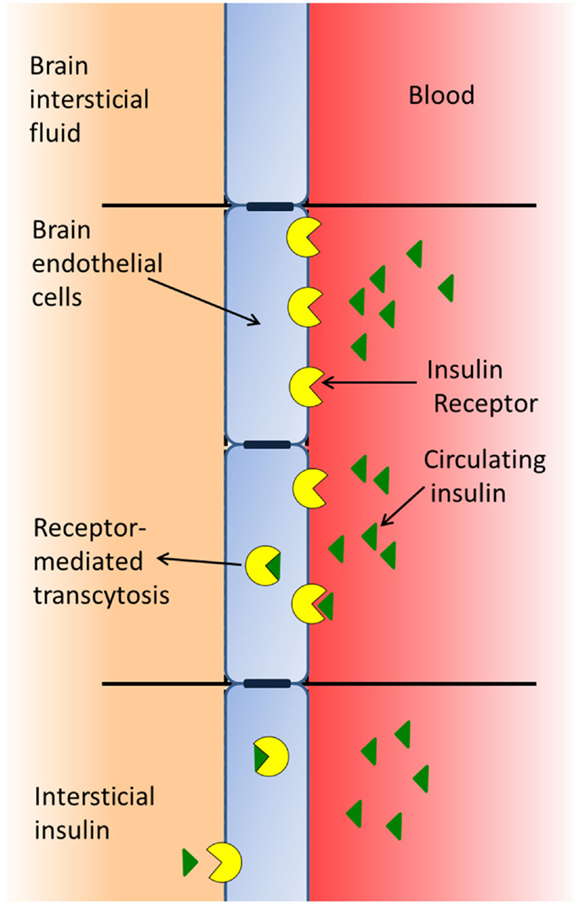
Figure 1. Insulin of peripheral origin may pass through the blood–brain barrier using a receptor-mediated transport system.
On the other hand, the insulin-degrading enzyme (IDE) is present in several brain regions, including multiple cortical areas, hippocampus, cerebellum, and brain stem. At cellular level, IDE was confined mainly to neurons, but it was also present in oligodendrocytes, choroid plexus, and some blood vessel endothelial cells (43). IDE is upregulated by exposure to low levels of amyloid-beta peptide (Abeta), which may be an important therapeutic target because of its role in the degradation of Abeta and other substances (44).
The single gene of the human IR, located on chromosome 19p13.2–19p13.3, has 22 exons (11 each for coding the α and β subunits). Two isoforms of the precursor protein are generated by the alternative splicing of +/− exon 11 (IR-B/IR-A, respectively) in a tissue-specific manner. This exon encodes a small amino acid sequence that is located at the C-terminal of the extracellular α-subunit (45). In humans, IR-B (the longer isoform) is the most prominent isoform in classical insulin-sensitive tissues, skeletal muscle, adipose tissue, and liver, as opposed to IR-A in the brain (46–48).
The heterotetrameric IR is composed of two ligand-binding sites, disulfide-linked extracellular α subunits, which are linked by disulfide bonds to two membrane-spanning β subunits. The α subunit is predominantly hydrophilic in nature, lacks membrane anchor regions, and contains 15 potential N-glycosylation sites and 37 cysteine residues. The β subunit contains a portion that is extracellular, a portion that comprises the transmembrane region of the receptor, and a portion that is intracellular, and which possess inherent tyrosine-protein-kinase activity (49).
Although the presence of IRs in many tissues in the periphery, and their main function of mediating glucose transport into cells, was well known, the existence of IRs within the brain was poorly understood, and their function sometimes seemed to be something of an enigma because brain cells are not fully reliant upon insulin for glucose supply inasmuch as they have insulin-independent means of obtaining glucose (49). However, we now know that insulin signaling in the brain affects several important functions.
Studies on the presence of IRs in the CNS began in the early 1970s with the observation that systemic glucose concentration decreased after the injection of 500 μU of insulin into the carotid artery of rats (50), and through the report of specific binding of radiolabeled 125I-insulin in a crude membrane preparation of several tissues from monkeys, rats, and pigeons (51). The hepatic carbohydrate metabolism was thus reported to be under cholinergic influence through efferent neural pathways, and not due to a modification of pancreatic hormone secretion. IR was located and quantified in the CNS for the first time in 1978 (52), being present in membrane preparations from the brain at all stages of the development studied (53). Since then, a wide but uneven distribution of IR in the CNS has been reported. Accordingly, it was shown that membrane preparations from the hypothalami specifically bound greater [125I]insulin than membranes from the cortex and thalamus, and that this binding was higher for preparations from the anterior rather than the posterior portions of the hypothalamus (54). Likewise, the binding of [125I]insulin was high not only in all olfactory areas and in closely related limbic regions, but also in the neocortex and accessory motor areas of the basal ganglia, hippocampus, cerebellum, and choroid plexus, which suggested a neuromodulatory function for insulin in the brain (55). When IRs were quantified by autoradiography and computerized densitometry, the highest concentrations were detected in regions concerned with olfaction, appetite, and autonomic functions, all of which contain dendritic fields receiving rich synaptic input (56). In situ, hybridization showed that IR mRNA was the most abundant in the granule cell layers of the olfactory bulb, cerebellum, dentate gyrus, in the pyramidal cell body layers of the piriform cortex, hippocampus, in the choroid plexus, and in the arcuate nucleus of the hypothalamus; these findings were consistent with the distribution of IR binding (57). Interestingly, the expression of IR mRNA seems to be higher in the brain from obese (fa/fa) Zucker rats as compared with lean (Fa/−) age-matched controls (58). However, brain homogenates from normal and streptozotocin-induced diabetic rats showed similar specific insulin-binding, which indicated the absence of the upregulation of these receptors (59).
As compared with IRs, IGF1 receptors (IGF1R) are also widespread throughout the rat brain, but they have a distinct distribution, with a high concentration in regions concerned with olfaction, autonomy, and sensory processing, as well as in the pituitary gland, where they are involved in the regulation of growth hormone release (60). What’s more, the existence has been reported of a differential expression of both IGF-1R and IR in the left–right of male–female developing rat hippocampus, which may be responsible for the etiology of several mental health disorders, as well as sex differences in hippocampal-associated behaviors such as spatial learning strategies and stress response (61).
Insulin receptors are also widely distributed in the human brain, with the highest specific binding of [125I]labeled human insulin in homogenates prepared from hypothalamus, cerebral cortex, and cerebellum obtained post-mortem from non-diabetic subjects (62). Iodinated insulin-binding to synaptosomal membranes within the human cortex was found to be a function of age. Binding to IR was observed as early as week 14 of gestation, with a slight decrease around week 30, and a marked decrease after birth (63).
Brain IRs have similar kinetics and pharmacological properties to those described in peripheral tissues (64), although they differ in molecular size (as indicated, the α subunits of brain IR, named IR-A, are smaller than the α subunits of peripheral ones, called IR-B), degree of glycosylation (being higher in peripheral than in brain IR), and antigenicity. In addition, regulation by insulin also occurs in a different way, thus, while peripheral IRs are downregulated in response to insulin excess, their counterparts in the brain do not record such downregulation (65). Receptor heterogeneity is a powerful principle that allows the independent and specific regulation of cellular functions via identical hormones or second messengers. Furthermore, the presence of different receptor isoforms allows an independent regulation of their expression by different mechanisms (66). Some regions show a marked difference in IR density between the embryonic and adult brain, which may play a developmental role. Thus, high concentrations of IR are found in the thalamus, caudate–putamen, and some mesencephalic and brainstem nuclei during neurogenesis, but these same areas have a low IR density in adult rat brains (67).
Insulin-binding to α subunits of the IRs triggers the activation of the β subunit tyrosine-kinase activity by stimulating the phosphorylation of its own receptor in both neuronal and glial cells (68). In most higher animals, the mechanism of insulin signal transduction (Figure 2) is modulated through the tyrosine phosphorylation of cellular substrates, including several insulin receptor substrates (IRSs) (69), as well as other scaffold proteins (70), which initiate divergent signal transduction pathways (71). Likewise, following the binding of insulin, aggregated IRs are rapidly internalized into the cell by a process that at least in part involves coated pits and vesicles (72). It has been suggested that aggregation or internalization could be essential for insulin signaling (73). The internalized IRs can then be degraded or recycled back to the cell membrane.
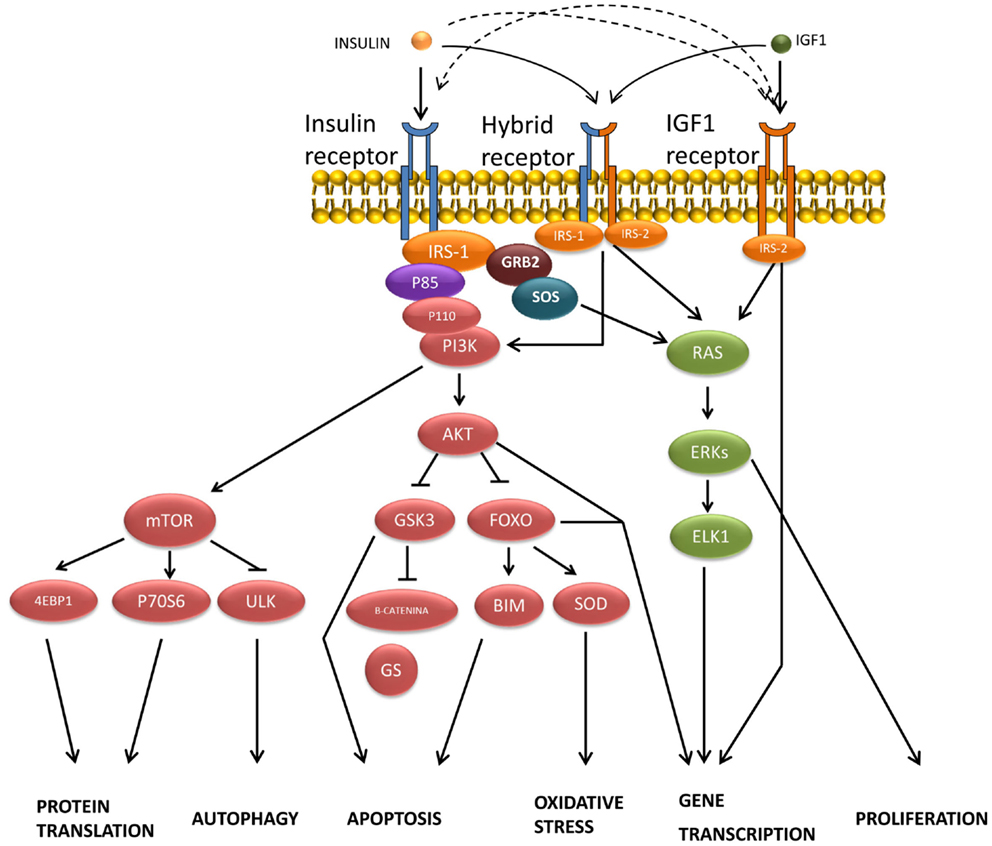
Figure 2. Transduction of signals and biological actions induced by insulin or IGF-1 through their receptor or through their hybrid receptors.
Most insulin responses are mediated by IRS-1 and IRS-2. IRS-1 controls body growth and peripheral insulin action, while IRS-2 regulates brain growth, body weight control, glucose homeostasis, and female fertility (74). IRS proteins are composed of an NH2-terminal pleckstrin homology (PH) domain adjacent to a phosphotyrosine-binding (PTB) domain, and followed by a tail containing numerous tyrosine and Ser/Thr phosphorylation sites (75). The Tyr phosphorylation sites coordinate downstream signaling cascades by binding the SH2 domains present in common effector proteins, including enzymes (the phosphoinositide 3-kinase, PI3K; the phosphatase SHP2; or the tyrosine-kinase Fyn) or adapters (SOCS1, SOCS-3, GRB2, and others) (70, 74). By contrast, the specific serine phosphorylation of the IRS-1/2 by the c-Jun N-terminal kinase (JNK1) and other protein kinases inhibits insulin-stimulated tyrosine phosphorylation, which correlates closely with insulin resistance (76). Likewise, the ubiquitin-mediated degradation of IRS-1/2 also promotes insulin resistance (77). However, the agonists that increase IRS-2 expression through cAMP production and CREB activation improve insulin signaling (78).
The synapse is the primary locus of cell–cell communication in the nervous system. It has been reported that IR was co-expressed with the insulin receptor tyrosine-kinase substrate p58/53 (IRSp53) in the synapse-rich molecular layer and in the granule cell layer of the cerebellum, as well as in the synapses of the cultured hippocampal neuron, which suggested that these molecules could be part of an insulin-dependent signaling pathway at the post-synaptic apparatus (79). IRSp53, which is phosphorylated upon stimulation with insulin (80, 81), is a key factor in cytoskeleton reorganization that mediates neurite outgrowth (82), being also involved in several neurodegenerative disorders (83), because IRSp53-deficient animals record cognitive deficits in the contextual fear-conditioning paradigm (84).
The association of IRS proteins and PI3K triggers the activation of this enzyme, which phosphorylates an inositol phospholipid in the plasma membrane, named PI (4,5)P2, to PI (3,4,5)P3, which recruits both the Ser/Thr kinase PDK (3-phosphatidylinositol-dependent protein kinase) and protein kinase B (PKB or Akt) to the plasma membrane, where Akt is activated by PDK1- and PDK2-mediated phosphorylation (85). This signaling pathway is antagonized by the action of the phospholipid phosphatases PTEN or SHIP2. Akt phosphorylates several substrates, including TSC2 (tuberous sclerosis complex, tuberin), which finally activates the mammalian target of rapamycin (mTOR) and provides a direct link between insulin signaling and nutrient sensing (70, 86). In addition to the IRS/PI3K/Akt, a second signaling pathway has been reported in peripheral tissues for the translocation of the glucose transporter GLUT-4 by insulin, involving other substrates of IR such as Cbl and APS. Following the recruitment of several proteins, including TC10, into the lipid raft, the trafficking of GLUT-4 vesicles is stimulated until their fusion with the plasma membrane (71, 85).
Mitogen-activated protein kinase is another signaling pathway activated by insulin through tyrosine phosphorylation of certain prototypical signaling adaptors such as Gab-1/Shp2, Shc/Grb2, and SOS/Grb2, which activate the small G-protein Ras by stimulating GDP:GTP exchange. Raf activation then takes place through a multi-step process (87), initiating an activation cascade of several protein kinases that include MAPK/ERK kinase (MEK) and extracellular signal-regulated kinase (88). ERK phosphorylates and activates several cytosolic proteins including p90rsk (89) cytoskeletal proteins, phospholipase A2 (PLA2), and signaling proteins, such as tyrosine-kinase receptors, estrogen receptors, SOS, and STATs (signal transducer and activator of transcription proteins). ERK also enters the nucleus, where it controls gene expression by phosphorylating transcription factors such as Elk-1 and other Ets-family proteins (18, 70).
Some brain dysfunction might result not only from an aberrant IR expression or function that occurs either during development or later, but also from single-point mutations, such as F382V (delayed transport of IR components to cell surface); R735S (insulin resistance due to the inhibition of precursor processing); L1018A (absence of tyrosine-kinase activity); and Y960F (multiple functional defects) (49).
Although the brain uses ketone bodies during starvation, glucose is its main fuel, which is needed in a continuous and permanent supply (90). Besides being an energy substrate, glucose is a signaling molecule involved in glucoregulatory mechanisms of primary functional concern to provide an uninterrupted glucose supply to the CNS and meet the metabolic needs of peripheral tissues. Given the vital importance of the continuous supply of glucose to the brain and the high prevalence of DM, the possible lack of insulin-dependent glucose uptake may be considered as an advantage.
The brain has two groups of glucose-sensitive neurons named glucose-excited (GE) and glucose-inhibited (GI) by rises and falls in glucose concentrations, respectively. These neurons are involved in the control of feeding, energy expenditure, and glucose homeostasis (49) and in addition the glucokinase acts as a glucose sensor in those neurons, facilitating the control of food intake (91–94). These various glucoregulatory functions are usually secondary to glucose uptake, a step that in most tissues is controlled by the level of glucose transporter (Table 1) and glucose sensor expression (95). GLUT-1, the more abundant glucose transporter in the brain, is expressed as two isoforms that differ in their degree of glycosylation. The 45-kD isoform expressed in astrocytes is resistant to both hypoglycemia and hyperglycemia, while the expression of the 55-kD isoform, originally located in the capillary endothelial cells, is increased under conditions of hypoglycemia, but remains unchanged during hyperglycemia. GLUT-1 has a widespread distribution in the brain (96), where it seems to have tissue-specialized functions, and some isoforms could be sensitive to acute insulin regulation (49). GLUT-2 is expressed in several neuronal populations, including specific neurons in the hypothalamus such as the paraventricular nucleus, the arcuate nucleus, and the lateral region (97, 98), where GLUT-2 is co-expressed with glucokinase (49, 93) and sulfonylurea receptor-1 (SUR1) (99). GLUT-3, the major glucose transporter in the neurons of the cerebellum, striatum, cortex, and hippocampus (100), has also been detected in brain glial and endothelial cells (101) operating at lower glucose levels, which is important given that the glucose concentration in the brain interstitium is relatively low as compared to in the blood.
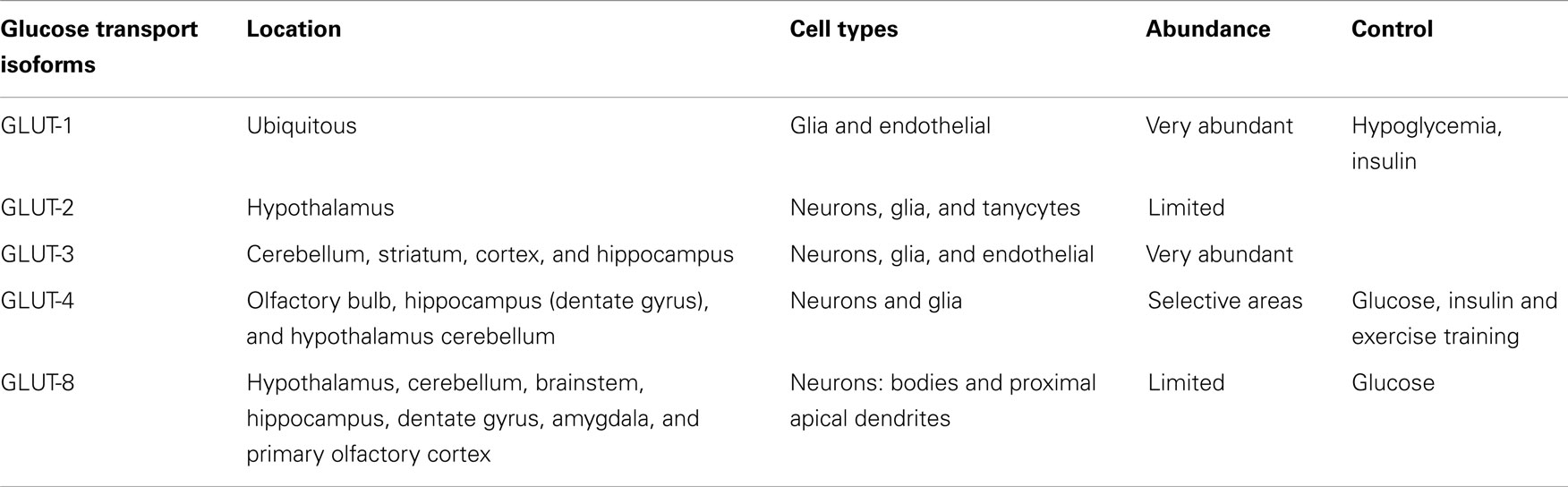
Table 1. Main glucose transport (GLUT) isoforms in the brain.
In contrast with peripheral tissues, the brain is considered an insulin-insensitive organ because GLUT-4 is present at low level and it does not seem to be significantly regulated by insulin. Thus, GLUT-4 was located in selective areas of the brain, including the olfactory bulb, dentate gyrus of the hippocampus, hypothalamus, and cortex, but at low amounts compared to the other isoforms, GLUT-1 and GLUT-3. As in those tissues, GLUT-4 was also located in both the plasma membrane and cytoplasm, which could suggest that a readily mobilizable pool was available for translocation to the plasma membrane (102). Surprisingly, in cerebellar membranes, GLUT-4 was present in significant amounts and its expression was insulin-dependent (103). In addition, the trafficking of GLUT-4 to the plasma membrane was modulated in the cerebellum, cortex, and hippocampus under conditions that increased plasma insulin levels (104), such as after peripheral glucose administration. Also, as GLUT-4, GK, and IR were co-expressed in both GE and GI hypothalamic neurons, these findings could suggest that this brain region, may experience stimulation of glucose uptake in response to insulin (105). However, the observation that GE and GI neurons respond to alterations of ambient glucose levels in the complete absence of insulin (97, 98, 106), and that insulin fails to induce neuronal glucose uptake in hippocampal formation, and that IR activation with insulin in humans has no effect on AS160-dependent GLUT-4 translocation (104), it seems possible to conclude that insulin-mediated glucose transport is at least not required by glucosensing neurons.
The neuron-specific glucose transporter GLUT-8, which has limited association with the plasma membrane in the CNS under physiological settings or in experimental models of type 1 diabetes (107), is expressed in bodies and in the most proximal apical dendrites of several brain areas (108), including both excitatory and inhibitory neurons in the hippocampus (109). Its functional role is still little known, but being an insulin-responsive isoform, it may play a role in augmenting substrate delivery under conditions of increased demand (110). As GLUT-8 is present in the rough endoplasmic reticulum and cytosol, a new role for this glucose transport has been proposed. Thus, since glucose is released from oligosaccharides during protein glycosylation events that occur in the rough endoplasmic reticulum, GLUT-8 may transport glucose out of the rough endoplasmic reticulum and into the cytosol, and thereby contribute to glucose homeostasis in hippocampal neurons, which is impaired under hyperglycemic/insulinopenic conditions (111, 112). As with GLUT-4, glucose administration also stimulates GLUT-8 trafficking from the cytosol to the rough endoplasmic reticulum, but does not result in GLUT-8 association with the plasma membrane.
Although insulin does not induce a significant glucose uptake in the brain as compared with peripheral tissues, it may play other important roles in glucose homeostasis. Thus, the experimental inhibition of insulin action in the hypothalamus, or the direct stimulus of the arcuate nucleus with this hormone, induces a reduction in insulin’s ability to block the production of liver glucose (113). To produce this effect, insulin acts through its own receptors in the liver and hypothalamus. Accordingly, a reduction in insulin sensitivity in the hypothalamus might lead to the diminished efficiency of this hormone in blocking glucose formation (114), which might contribute to the hyperglycemia of diabetic patients (115, 116). The effect of insulin on hypothalamic glucose-sensitive neurons might be to induce an opening of the ATP-sensitive K+-channels (117), causing a cell-hyperpolarization that ameliorates the functional capacity to modify the glucose response of these glucose-sensitive cells (118). The signals generated in this process are transmitted to the motor nucleus of the vagus nerve that carries this information to the liver, which produces the appropriate response. In fact, the resection of the liver branch of the vagus nerve induces a decrease in the insulin inhibitory effect on glucose hepatic production (116). All these findings lend support to the report by Claude Bernard, who in 1855 (119) showed that puncturing the fourth cerebral ventricle produced glycosuria in mice, which gives rise to the assumption that the brain is involved in glucose homeostasis.
We now consider the existence of specific populations of neurons involved in energy homeostasis, located on the so-called satiety and hunger networks rather than centers. They contain orexigenic and anorexigenic molecules, and their receptors are closely interrelated with metabolic sensors such as the glucose transporter isoform GLUT-2, glucokinase (92–94), AMPK, PASK, and others. They generate integrated responses to afferent stimuli related to modifications in metabolites or in fuel storage. It is accepted that the cells of several hypothalamic nuclei detect circulating satiety signals and transmit this information to other brain areas. The interactions among orexigenic and anorexigenic molecules and transmitters located in different hypothalamic nuclei may induce a characteristic feeding behavior. The activation of the PI3K pathway is common to insulin, leptin, and serotonin, and it is believed that the dialog between these biomolecules is potentially a major event in the pathophysiology of the brain from a general perspective, and in the control of food intake in particular. Insulin is a powerful anabolic hormone in the periphery, and behaves as a catabolic hormone in the brain because of its anorexigenic properties (120). Insulin also strengthens the signals induced by leptin through JAK2 and SHT3 in the hypothalamus (121). Leptin and insulin resistance has recently been reported in the hypothalamus of diabetic mice, thereby opening new ways for a better understanding of insulin resistance and type 2 diabetes (33).
Fertility is closely dependent on energy reserves, since negative alterations of energy homeostasis produce changes in the control of reproduction (122) through the hypothalamic–pituitary–gonadal axis (123). By contrast, an abundance of nutrients enables the reproductive process and survival of offspring. To control the relationship between the reproductive function and metabolic activities, several hormones such as insulin are involved. This hormone acts at several levels in the interplay between the hypothalamus, pituitary gland, and gonads. Thus, when hypothalamic pieces were perfused with low concentrations of insulin, a stimulatory effect on LHRH secretion was observed, which was dependent on the availability of glucose (124). However, high levels of glucose alone did not modify the release of LHRH (124). In the same way, the intracerebral infusion of insulin also increased the luteinizing hormone (LH) pulse frequency, but glucose infusion did not modify gonadotropin secretion (125). In diabetic animals, low levels of circulating insulin were accompanied by a reduced LH release (124), while the central or peripheral insulin administration restored LH pulse frequency (126). Other authors have found that low levels of circulating insulin in diabetic rats decrease both gonadotropin-releasing hormone (GnRH) release from the hypothalamus and the response of pituitary LH-releasing cells to GnRH (126). These results led Tanaka et al. (127) to propose that intracerebral insulin is a key regulator of pulsatile GnRH secretion in diabetic sheep. However, other authors have not ruled out the effects of glucose in the process, indicating that LH secretion is not wholly dependent on insulin activity because specialized glucodetectors in the hypothalamus can also modulate GnRH secretion, depending or not on insulin (128).
Although the role of insulin as a neurotrophic agent in the adult brain is little known, the trophic function of insulin referred to proliferation, differentiation, and neurite growth has been reported in developing nervous systems. In fact, the systemic administration of insulin increases the ornithine decarboxylase activity in the brain of neonatal rats, which is an indicator of growth stimulation, indicating in this case that insulin was involved in the regulation of brain development (129). In addition, with the observation that the number of IRs increases during cell differentiation in the developing brain (130), IR signaling was suggested to play an important role in neuronal proliferation during development, which was confirmed when it was shown that IRS-2 mediated the effects of insulin on brain growth (131), as well as on the outgrowth, maturation, and regeneration of axons (132), as well as on neurite growth (18).
Nevertheless, the most abundant evidence on the neurotrophic effects of insulin has been obtained by in vitro studies using different neural cell cultures. Thus, it was determined that insulin-stimulated nucleotide incorporation in rat brain (133) and induced the growth and differentiation of a fraction of neurons isolated from the chick forebrain (134). The effects of insulin on growth and development mediated by IRs have been reported in both neurons and glial cells (135, 136), where both the number and the activity of the IRs may be regulated oppositely depending on cell type. On the other hand, insulin and IGF2 are necessary for NGF to stimulate neurite formation (137), while this effect is not observed in their absence. It has been also reported that these actions were dependent on the presence of astrocytes (138). In fact, astrocytes are known to modulate neuronal functions, and they may contribute to the cerebral actions of insulin, including cell growth. Thus, it has been reported that insulin induces the proliferation of both cultured rat (139) and human (140) astrocytes, in which the expression of several key proteins of insulin signaling was shown to increase.
In cultured fetal neurons, insulin increased both ribosomal protein S6 phosphorylation (136) and PKC-epsilon activity via a mechanism that does not involve the translocation of the enzyme from cytosol to the membrane (141), which could be closely related to neurite outgrowth (142, 143). This hormone also modulates the growth of neuronal cells by activating other protein kinases, such as phosphatidylinositol 3-kinase (PI3K) (144). Likewise, insulin increased the protein expression of the dendritic scaffolding protein post-synaptic density-95 (PSD-95) in hippocampal area CA1 through the activation of the PI3K/mTOR pathway, providing a molecular mechanism that could explain the effect of insulin on synaptogenesis and on the modulation of the synaptic function in area CA1 (145), as well as on the regulation of dendritic spine formation and excitatory synapse development in hippocampal neurons (146). Other proposed mechanisms for explaining the effect of insulin on neurite formation could be the upregulation of tau protein, the main microtubule-associated protein in the CNS that participates in the axon/neurite growth, which also involves the activation of the PI3K/mTOR pathway, or the upregulation or stabilization of tubulin mRNA followed by an increase in protein levels (147). It has also been reported that endogenous insulin synthesized by neurons was capable of promoting neurofilament distribution, which was abolished in the presence of IR inhibitors or antibodies against insulin (148).
On the other hand, both the proliferation and differentiation of multipotent neural stem cells are regulated by insulin, while the withdrawal of this hormone causes non-apoptotic autophagic cell death (130). Likewise, a reduction in the activation of the PI3K/Akt pathway has proven to be critical to the cell survival signaling of differentiated human neurons and human-derived neural stem cells (hNSC), which unlike the NSCs of rodent origin are extremely sensitive to insulin, and growth healthily in a narrow range of relatively low insulin concentrations (149). It is now accepted that IR pathways function as an integrating factor that correlate neuronal differentiation with nutritional information, to the extent that the degree of differentiation adapts to modifications of the nutritional state (150).
Insulin is a potent neuroprotective agent that acts mainly against apoptosis, beta amyloid toxicity, oxidative stress, and ischemia. It has been reported that the antiapoptotic effect of insulin is dependent on the PI3K pathway, but not on the mitogen-activated protein kinase (MAPK) route, because the inhibition of mTOR activity by rapamycin avoids the antiapoptotic effects of insulin, suggesting that the protein p70SK, one of the downstream targets of the PI3K/Akt/mTOR pathway, may be one of the mechanisms through which insulin prevents apoptosis (151). Several authors have reported that insulin protects against beta amyloid-induced cell death. They have shown that the formation of Aβ fibrils is prevented by insulin, and suggest that Aβ damage may be caused through the regulation of Aβ fibrillation (152).
Insulin antagonizes the deleterious effects of oxidative stress in the CNS. Lipid and protein oxidation occur as a consequence of oxidative stress, which may alter proteins such as GLUT-3, modifying glucose uptake and then lactate accumulation, acidosis, and mitochondrial dysfunction (153). By stimulating glucose uptake and pyruvate formation, insulin restores intracellular ATP formation as well as reduces oxidative stress (154). Likewise, GABA and glutamate uptake are decreased under oxidative stress, which induces the accumulation of these neurotransmitters in the extrasynaptosomal space, while the addition of insulin reverses these changes (155). On the other hand, under situations of severe oxidative stress, the elevation of uric acid by insulin might provide some antioxidant benefit because uric acid, glutathione (GSH), and vitamins C and E are important as components of a neuronal antioxidant pool (156).
Two mechanisms have been proposed as being involved in insulin protection against ischemia; one by the direct effect of insulin on the brain tissue and the other one by an indirect mechanism in which insulin reduces peripheral glucose levels (157). As regards the former, it has been reported that insulin treatment increases the extracellular GABA during transient ischemia, independent of hypoglycemia, which can inhibit pyramidal neurons and protect them against ischemia (158). Another explanation is based on alterations of glucose metabolism and decreased lactic acidosis (159). Thus, insulin has a stimulatory effect on the Na+/K+ ATP pump, reducing both extracellular K+ and intracellular Na+, which may change the neuronal firing rate and its metabolic demands, while preventing water accumulation and the subsequent post-ischemic edema (160). On the other hand, as cerebral ischemia-reperfusion induces JNK1/2 phosphorylation, Bcl-2 expression, and caspase-3 cleavage in the rat hippocampus, and insulin reverses all the changes mentioned, it should be concluded that a cross-talk exists between Akt and JNK1/2 that could play a role in the anti-ischemic effects of insulin.
It is now widely recognized that insulin has neuromodulatory effects on mammalian CNS by acting both at electrophysiological levels, as well as on the concentration and function of certain neurotransmitters. Thus, it has been shown that insulin has direct and reversible electrophysiological effects on all of the recorded neurons in vivo, and these effects are highly dependent with respect to GABA pretreatment, being blocked by the co-administration of IR inhibitors (161). In addition, as the GABA receptor is a substrate of Akt phosphorylation, insulin may play an important role in the control of GABA receptor density in the post-synaptic domain (162). Insulin also affects intracellular ion concentrations by modulating the activity of certain ion channels. Thus, insulin in hypothalamic neurons activates K+ ATP channels producing membrane hyperpolarization, which has an inhibiting effect (163). In addition, insulin has stimulatory effects on Na+/K+ ATPase, producing an acute rise in intracellular Ca2+ concentration that triggers the release of neuropeptides (164).
Insulin may modulate neurotransmitter concentration through different mechanisms. Thus, insulin induces both the inhibition of norepinephrine and the stimulation of serotonin reuptake in neuronal cells (165, 166), which may increase glucose homeostasis through the interrelationship between brain IRs and the neurotransmitter function (130). Insulin also has modulatory effects on neurotransmitter receptor density. Thus, insulin reduced the increased number of dopamine receptors in striatal membranes from rats that were rendered diabetic with alloxan or streptozotocin (167), and in rats treated with haloperidol (168), but it had no effect on binding in normal rats. However, systemic insulin administration produced an increase in dopamine and serotonin levels in the CSF, while this hormone downregulated the α2-adrenergic receptors in hypothalamic neurons (169). On the other hand, insulin has a stimulatory effect on the uptake of amino acids by the neurons required for neurotransmitter synthesis (170).
It has been widely reported that the peripheral or central administration of insulin by icv or intrahippocampal routes to experimental animals has positive effects on memory and learning processes (171). The improvement in these activities is related with an increase in both the IR expression and its signal transduction pathways in the hippocampus (172), and the loss of memory due to ischemic lesions in this structure can be avoided by insulin administration (173). Streptozotocin has been used to develop experimental models of type 1 diabetes. However, when injected directly at low doses into the brain, it produced a central resistance to insulin by interfering with the binding of the hormone to its own receptor and blocking insulin actions, which was related to deficits in memory and altered behavior (174). Besides, epidemiological studies have shown that both type 1 and type 2 diabetic patients have cognitive impairment and an increased risk of AD, mainly in older patients, and also that insulin administered to AD patients to keep glycemic levels constant can improve memory formation (175). Likewise, the systemic administration to healthy humans of insulin under euglycemic hyperinsulinemic conditions yields a significant improvement in verbal memory and selective attention.
It is believed that an event is fixed in the memory by modifications of the neuron networks based on the processes called long-term potentiation (LTP) and long-term depression (LTD) (176). The LTP process occurs when the presynaptic neuron excites the post-synaptic ones in a repetitive and prolonged manner, and then the depolarization of the post-synaptic neuron is reinforced and maintained for a long time. Accordingly, there is a significant increase in Ca2+ input, and the metabolic activities for this cation are prolonged, and as a result the memory is consolidated. The LTD is a compensatory process that in the post-synaptic neuron, still under the effect of a LTP, facilitates a decrease in transmission efficiency, permitting cell activity to return to the previous excitatory level, and then get ready to store new information. Memory and learning functions need LTP and LTD processes, but also the remodeling of the dendritic spine morphology and modifications in the cytoskeleton produced during synaptic transmission. These processes, involve the glutamate, as well as two of its receptors, AMPA and NMDA, in which neurotransmission is regulated by changing the amount of receptors present in the membrane, or by covalent modification of their subunit components. Thus, whereas LTP increases the post-synaptic density of the AMPA receptors, LTD is associated to a decrease. Likewise, the phosphorylation of these receptors increases the efficiency of the ionic channel during LTP, while dephosphorylation during LTD decreases it (177).
Insulin modulates glutamatergic neurotransmission at the synapses. This hormone induces the LTD process by decreasing the amount of AMPA receptors in the post-synaptic membrane. Besides, this process depends on the phosphorylation of the hormone receptor, PI3-kinase activation, and on a process of protein synthesis (178). Other authors reported that insulin also induces the phosphorylation of the GluR2 subunit in the AMPA receptors of hippocampal neurons, producing endocytosis and a decrease in the post-synaptic excitatory ability (179). There is also experimental evidence that insulin affects learning and memory through GABA receptors by stimulating the translocation of these receptors to the plasma membrane. This effect is abolished by the action of a PI3K inhibitor. Insulin also increases the functional GABA receptor expression on the post-synaptic and dendritic membranes of the CNS neurons (18). Likewise, NO has also been reported to be involved in insulin-induced memory improvement, since the administration of the NOS inhibitor L-NAME avoids insulin-induced memory improvement (180).
IGF1 increases the synaptic transmission in the rat hippocampus through a mechanism in which AMPA receptors and PI3K activity are involved (181). Furthermore, GH increases the expression of the subunits of the NMDA in the hippocampus, thereby facilitating LTP induction and improving the memory (182).
There is experimental evidence to indicate that inflammatory responses are closely associated with the development of insulin resistance in peripheral and central tissues, as well as to show that these processes are present in obesity and in type 2 diabetes, which may increase the risk or incidence of AD (183). Thus, high concentrations of interleukin IL-6 have been determined in the CSF of patients with AD (184), while studies in animals suggest that inflammation interacts with the processing and deposit of β-amyloid peptide (Aβ) (185), with low amounts of insulin producing anti-inflammatory effects (186). The administration of lipopolysaccharide increases in plasma concentrations of the C-reactive protein and pro-inflammatory cytokines IL-1β, IL-6, and TNF (187). TNFα and IL-6 also induce the activation on NFkβ and subsequent transcription of the pro-inflammatory genes TNFα, IL-6, and IL-1b (188). Increased levels of inflammatory cytokines alter hippocampal synaptic plasticity and the components of spatial learning (189). Furthermore, obesity induces a peripheral insulin resistance that is related with a marked elevation of pro-inflammatory cytokines and of free fatty acids. Chronic inflammation may contribute to the appearance of insulin resistance and type 2 diabetes, as well as to the association of AD and type 2 diabetes mellitus (T2DM) (190). In addition, the increased concentrations of TNFα and Aβ within the brain of obese hyperinsulinemic persons facilitates the formation of amyloid plaques (191). Obese and AD patients have CSF insulin concentrations lower than in control subjects, suggesting a reduction in both insulin transport across the BBB and hormone sensitivity (192).
TNF-α has both neurotoxic and neuroprotective effects, depending on the receptor subtypes TNF-R1 and TNF-R2. Thus, TNF-R1 is involved in pro-apoptotic actions, while TNF-R2 promotes cell survival. High levels of TNF-R1 and decreased concentrations of TNF-R2 have been described in the brain of AD patients (193). Altered levels of TNF-R1 and TNF-R2 have been reported in persons with diabetes and impaired glucose tolerance (194), which was normalized after a 3-week low calorie diet (195). The accumulation of glycation end products, oxidative stress, and the resulting brain cellular and molecular damage may contribute to diabetes-induced brain aging (196).
Alterations of some of the insulin signaling pathways such as PI3K/Akt and GSK-3 are recorded in central inflammation and insulin resistance (197). It is known that the PI3K pathway has a negative effect on IL-12 formation by dendritic cells, while GSK-3 is a tau kinase involved in hyperphosphorylation and in the modulation of Aβ metabolism (198). There are some connections between the insulin signaling pathway and this protein, as IR activation phosphorylates and inhibits GSK-3β (199), while in AD, GSK-3β activity is increased as well as in T2DM, and this process enables it to phosphorylate the IR and IRS-1, which decrease the phosphorylation of tyrosine residues of the IR and IRS-1 (200). In addition, the activation of STAT-3 in astrocytes and microglia depends on GSK-3β activity (201), while the inhibition of GSK-3 stimulates the production of anti-inflammatory cytokines such as IL-10, and decreases pro-inflammatory cytokines such as IL-1β, IL-6, and IFN-γ in response to toil receptors (202). These findings suggest that PI3K/Akt/GSK-3 play an important role in controlling inflammation, and that the inhibitory effect of insulin on GSK-3 activity shows how insulin controls inflammatory responses.
It is well accepted that neuroinflammation occurs in AD, with infiltrates of T-lymphocytes and monocytes that are responsible, together with microglia cells, for the increase in a number cytokines in the CNS (203, 204). Inflammation is needed for the expression of insulin resistance, as demonstrated by the inhibition of inflammatory pathways, which avoid diet-induced insulin resistance in experimental animals (205). In fact, the initial part of high-fat diet-induced insulin resistance is independent of inflammation (206), while the chronic states are dependent on inflammatory processes (207). Accordingly, TNF-α reduces the tyrosine-kinase of the IR, resulting in insulin resistance, and the reduction in other mediators such as SOCS-3 inhibits IRS-1 (208). In addition, the activation of JNK by cytokines, inflammatory mediators, and fatty acids phosphorylate IRS-1, modifying insulin signaling. Thus, animals lacking JNK are protected against the high-fat diet (209). On the other hand, JNK inhibitors improve the neuroinflammatory response, while JNK activation promotes neuroinflammation and facilitates insulin resistance (210). Sartorius et al. (211) suggest that the activation of neuroinflammatory reactions may be responsible for the high-fat diet-induced insulin resistance in the brain, since the prevention of a neuroinflammatory response blocks this resistance (211, 212). In the same way, the icv administration of TNF-α produces hypothalamic inflammation, as well as the expression of the phenotype present in type 2 diabetic patients with high insulin levels and altered insulin signaling in peripheral tissues. It is accepted that central insulin resistance can favor an adaptive increase in food intake that facilitates the peripheral alteration of glucose homeostasis. Alternatively, the elevation of free fatty acids is a signal to increase the release of pro-inflammatory cytokines, which then activates the inflammatory signaling pathways responsible for insulin inhibition signaling and the promotion of insulin resistance (213).
Impaired insulin signal transduction with reduced tyrosine-kinase activity of the IR has been reported in the brain cells of AD patients. Moreover, the expression of insulin and IGF1 mRNA and protein levels, their own receptors, and the downstream signaling elements are decreased in the brain of AD patients (214). Insulin resistance is thus associated with reduced responses to insulin signaling in the IR/IRS-1/PI3K and greatly with IGF1 in the IGF1R/IRS-2/PI3K signaling pathways. Reduced insulin responses peaked at the level of IRS-1, and were consistently associated with basal elevations of IRS-1 phosphorylated at serine 616 (IRS-1 pS616) and IRS-1 pS636/639. These potential biomarkers of insulin resistance increased significantly from normal cases, through mild cognitively impaired cases, to frank AD cases, regardless of APOE-4 status (104). Related with this insulin insensitivity, the protective role of this hormone against Aβ accumulation is reduced, at the same time as the expression and function of insulin are downregulated by Aβ deposits (215). Thus, Aβ peptides inhibit the binding of insulin to its receptors (216), reduce receptor autophosphorylation, and impair insulin-induced signaling pathways (217). The alterations of these effects in AD and T2DM patients interfere with the neuroprotective actions of insulin, facilitating the brain’s susceptibility to neurodegeneration (218). Both brain insulin and IGF1 resistance are considered an early and common feature of AD, which seem to be closely associated with the IRS-I dysfunction triggered by Aβ oligomers that promote cognitive decline (104).
There is sound evidence about the role of insulin in brain functions, as well as of the close relationships between AD and T2DM (219), two highly prevalent nosological entities (Figure 3). AD is a neurological disorder that causes profound memory loss and progressive dementia, with histological manifestations of amyloid plaques, neurofibrillary tangles, and amyloid angiopathy, accompanied by widespread loss of neurons and synapses (220). There are more than 30 million people suffering from AD, with this number increasing very quickly and expected to exceed 120 million by 2040 (197). T2DM is characterized by impaired insulin secretion and by a resistance to the action of this hormone. It has been estimated that there were 250 million diabetic patients worldwide in 2010, with 90% of the patients having T2DM (221). Aging is a high-risk factor for both AD and T2DM. Accordingly, T2DM is a disease that seriously affects the quality of life and longevity of elderly people, although in recent years it also affects obese young people.
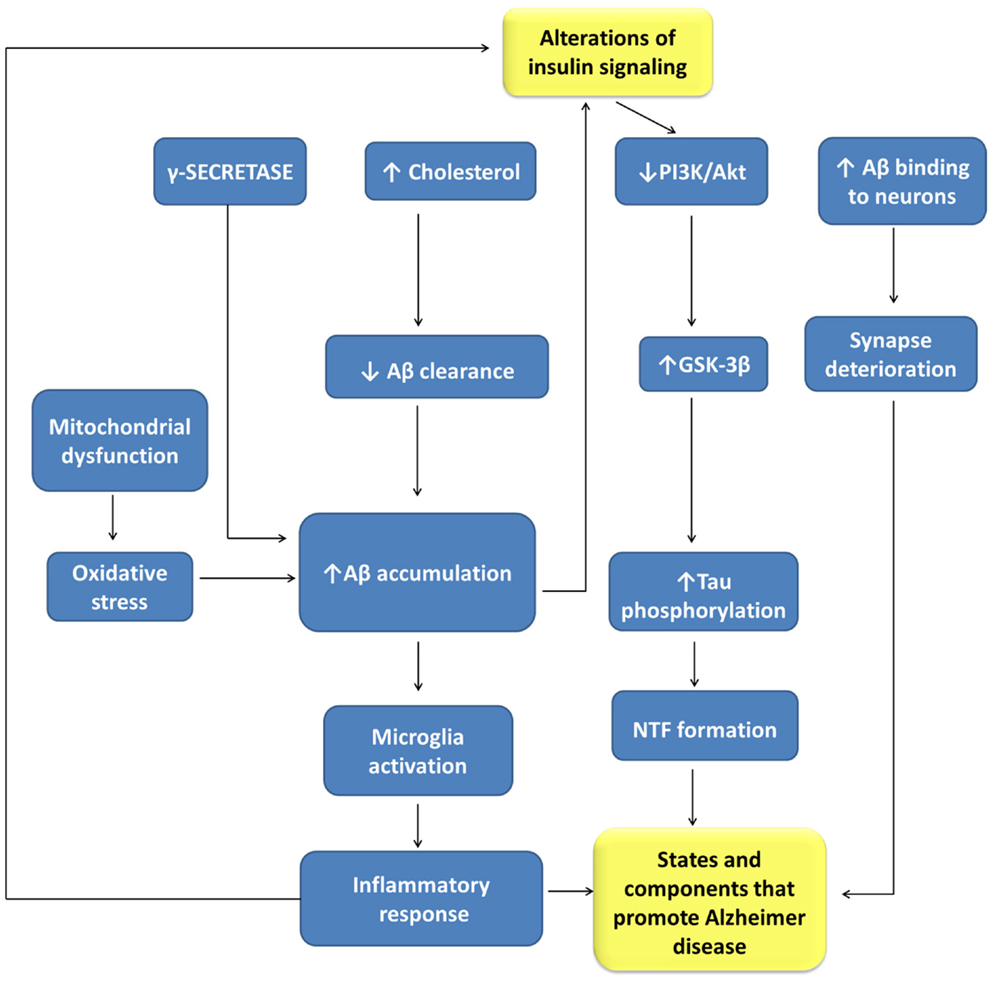
Figure 3. Relationships between alterations of insulin signaling and Alzheimer disease pathogenesis. Aβ, beta amyloid-peptide; GLUT-3, glucose transporter isoform-3; GSK-3β, glycogen synthase kinase 3-β; NFT, neurofibrillary tangles; PI3K, phosphatidyl inositol 3-kinases.
Both disturbances share many pathophysiological features, such as insulin resistance, amyloid aggregation, inflammatory stress, and cognitive disturbances that suggest common or related pathogenic processes. Insulin resistance is a risk factor for AD, being a common feature of AD patients with or without T2DM. Besides peripheral insulin resistance, this alteration may be present in the brain accompanied by IGF1 resistance, and IRS-1 and IRS-2 dysfunction, potentially triggered by Aβ oligomers and cognitive decline.
Interestingly, peripheral insulin resistance begins a long period of time before the appearance of a frank T2DM, which might permit a preventive therapeutic treatment during this period (222). To know whether peripheral or central insulin resistance are components of the same disease or are expressed independently of one another is a matter of potential interest. What’s more, the existence of AD in patients without T2DM could be related to a lack of time to develop the diabetic process, or be the consequence of different nosological entities. AD is a progressive neurodegenerative disease, and when the symptoms and signs of cognitive dysfunction appear, the disease has already been present for many years. Accordingly, four stages for AD (223) have been proposed: a pre-disease stage without detectable pathophysiological alterations, a preclinical stage with pathophysiological alterations but without cognitive alterations, a stage of pre-dementia or mild cognitive impairment (224), and the dementia stage. It would be of great interest to know whether these stages coincide in time with the predicted stage in the peripheral tissues during the long period of insulin resistance. This period of time during prediabetes or the four stages of AD may constitute an opening for the development of preventive programs or new therapeutic approaches.
Many authors have suggested that insulin alterations and changes in glucose metabolism may condition the risk of developing dementia (225, 226). A recent study indicates that the altered expression of genes related to T2DM and AD brains (227) is a result of AD pathology, which may be exacerbated by insulin resistance or DM (227). This statement can be stressed because cognitive deficits are associated with insulin signaling abnormalities. The close relationship between these two pathological disturbances, because of the common presence of insulin resistance, has led to the use of the term type 3 diabetes, which means it is considered a neuropathogenic expression of AD. However, this entity cannot be included (228) within the classic concept of diabetes, because AD patients are not hyperglycemic, as happens with both T1DM and T2DM, nor does insulin stimulate glucose uptake in the brain, by contrast with the strong stimulatory effect observed in muscle, fat, and the liver. Nevertheless, an insulin-resistant brain state (229) exists as a variant of the hormone manifestations in several peripheral tissues, with different properties as corresponds to the brain functions. Insulin resistance in the peripheral tissues could facilitate insulin resistance in the brain by reducing brain insulin uptake and by increasing levels of Aβ (230). It seems that insulin resistance is a prior and common manifestation of these diseases, which over a long period of time deteriorate central or peripheral tissues before the appearance of diabetes and AD. This period, which lasts around 10–15 years, and is referred to as prediabetes, is more frequent in older people at a time when the incidence of AD is increasing significantly. The prevalence of prediabetes and diabetes in AD is of 81% in the USA (226), in which peripheral insulin resistance was accompanied with or without T2DM. The long duration of prediabetes might explain why brain insulin resistance in some cases of AD may be found without diabetes and/or the high prevalence of both prediabetes and AD in the elderly.
Both experimental and epidemiological studies provide evidence to show that the risk of AD dementia or vascular dementia is increased in diabetic patients (231). Thus, large epidemiological series have reported the association between insulin resistance or DM and the risk of AD, albeit independent of the APOE-4 phenotype (232). Furthermore, hyperinsulinemia and hyperglycemia caused by insulin resistance accelerate the formation of neurite plaques (233). The results from a nationwide case-control study in Finland (234) show that individuals with AD are more likely to have a history of medically treated diabetes than the older population in general, and that the association is independent of vascular diseases.
Most studies have focused on the role of type 2 diabetes, while the association between type 1 diabetes and alterations of CNS has received less attention (235). Then, T2DM is clearly associated with AD, while T1DM is more related to other brain alterations (235–240). However, learning and memory impairments and deficits in mental flexibility and problem-solving are more frequent in patients with type 1 diabetes than in the general population (241, 242). Interestingly, these alterations improve after the establishment of insulin therapy, when the glycemia is controlled (243, 244). These alterations may have a morphological explanation, since patients with type 1 diabetes have a reduced number of dendrites in the gray matter (245). Accordingly, intranasal insulin administration in diabetic mice reverses morphological and cognitive alterations (246). In addition, type 1 diabetes and type 2 diabetes are associated with brain atrophy and cognitive impairments, which are prevented by insulin and IGF1 (247).
Besides the studies cited above establishing a risk of AD development in diabetic patients, there are other reports with opposite findings in which they have stated that although diabetes produces alterations in cognitive performance or increases the risk of dementia, there is no association between DM and AD (248, 249).
In AD patients, the CSF concentrations of insulin are decreased, while the blood plasma concentrations are raised, which is more evident in advanced stages of AD and in patients without APOE-4 allele (197). High levels of circulating insulin may be the consequence of hormone resistance, while the reduction in CSF insulin may be related to a decrease in insulin clearance and/or to a reduction in insulin uptake from a peripheral source through the BBB.
Several experimental evidences support the link between AD and DM through mitochondrial alterations and oxidative stress, altered energy and glucose metabolism, cholesterol modifications, and dysfunctional protein O-GlcNAcylation (197). Since insulin avoids the reduction in mitochondrial oxidative phosphorylation and any increase in oxidative stress that protects against Aβ-protein toxicity, we can infer that diabetes is a risk factor for AD development. In fact, diabetes induces alterations in the mitochondrial antioxidant defense, facilitating brain susceptibility to Aβ toxicity (250).
Impairments in energy and glucose metabolism, manifested as reduced cerebral glucose and oxygen utilization, are present both in non-diabetic and diabetic patients with AD, indicating another link between these two nosological entities (223). PET technology has been used to find a depressed glucose metabolism in the temporal–parietal cortex, posterior cingulate cortex, and the frontal areas of AD patients. The administration of glucose to these patients therefore improves memory alterations (251, 252), although this effect seems to be due more to insulin than to hexose. At this point, it is important to remember that glucose metabolism dysfunction increases the risk of cognitive disturbances in the elderly.
Contributions to cerebral glucose hypometabolism may be related to glucose transportation abnormalities, intracellular glucose metabolic disturbances, and the altered functional status of thiamine metabolism. AD patients have decreased GLUT-1 and GLUT-3 expressions (253), especially in the cerebral cortex. The dentate gyrus of the hippocampus also has reduced GLUT-3 expression. Liu et al. (254) have explained that glucose hypometabolism results in both reduced glucose transporter expression and the decreased O-GlcNAcylation of tau. They proposed that hypometabolism in the brain reduces the O-GlcNAcylation of tau that conversely increases its phosphorylation, which induces the NFTs that underlie the cognitive deficits of AD subjects. Three key enzymes in the Krebs cycle and pentose phosphate pathway (PPP)-pyruvate dehydrogenase complex, α-ketoglutarate dehydrogenase complex, and transketolase, and their common coenzyme thiamine diphosphate (255), recorded altered levels in the brain of AD patients. These findings suggest the relevant roles of mitochondrial dysfunction and impaired thiamine-dependent processes in the cerebral glucose hypometabolism of AD.
Impaired cerebral glucose metabolism is a pathophysiological feature that may even precede pathological alterations by decades (222). Accordingly, Chen and Zhong proposed the hypothesis that impaired cerebral glucose metabolism, mainly thiamine metabolism and insulin resistance, could promote Aβ accumulation and tau hyperphosphorylation, as well as many other pathogenic factors that might contribute to the pathological dysfunction of the brain in AD. These pathophysiological cascades include inflammatory factors, mitochondrial dysfunction, and oxidative stress, Advances Glycation End products (AGEs), apoptosis, excitotoxicity, and the hyper-activation of protein kinases. All these factors are involved in cognitive dysfunction (223).
In addition, changes in brain cholesterol metabolism facilitate the interactions between DM and AD pathogenesis. Thus, cholesterol accumulation alters beta cell functions and insulin secretion (256), as well as insulin-stimulated cholesterol biosynthesis, and controls its circulating levels (257), which form a strong bond between hypercholesterolemia and T2DM. In addition, elevated levels of circulating cholesterol increase the risk of AD (88) because it modulates Aβ synthesis, inhibits the clearance of Aβ, and potentiates the interaction of Aβ with neuronal membranes (10). Furthermore, Aβ binds to cholesterol to catalyze the formation of oxysterols, products with highly neurotoxic properties, which alter the insulin signaling pathway by inhibiting the phosphorylation of the ERK/Akt route (258, 259). It has been reported that hyperglycemia and hyperinsulinemia activate the O-GlcNAcylation of proteins included in the insulin signaling pathway, facilitating insulin resistance (260, 261). Studies on autopsied frontal cortices have found that the protein content and activity of the brain insulin/PI3K/Akt signaling pathway are decreased in T2DM and AD patients, and more so in AD-T2DM patients (262). The reduced brain insulin/PI3K/Akt pathway leads to the overactivation of GSK-3β calpain-1 and downregulation of O-GlcNAcylation, which promoted altered tau hyperphosphorylation and neurodegeneration (262). These data suggest that the alteration of GlcNAcylation may be another mechanism by which the predisposition for AD is increased by DM.
Other situations, such as the formation of amyloid plaques, altered Aβ metabolism, and tau hyperphosphorylation, are favored by the interactions of AD and DM. Aβ are the components of the amyloid deposits in the AD brain, while the protein deposited in the islets of Langerhans is the islet amyloid polypeptide IAPP. Both proteins are able to form amyloid aggregates (263), with the Aβ protein being toxic for neurons, and IAPP toxic for pancreatic islet cells (224). These two proteins have a high sequence similarity, where the chaperone protein pathway preventing IAPP and Aβ aggregation may be common and act on both of them. It has been suggested that the decreased capacity of this shared chaperone protein is responsible for the development of AD and T2DM (226). This means that islet amylogenesis is increased in patients with AD, and that the density of neurite plaques and their diffusion are positively related to the duration of diabetes (226).
Aβ is degraded and cleared by several proteases that avoid the accumulation of Aβ. IDE is a metalloendopeptidase that inactivates peptides such as insulin, IGFs, glucagon, and others. The alterations in insulin signaling also affect the Aβ metabolism, with Aβ and phosphorylated tau accumulation associated to neuronal loss and neurite degeneration. These effects are more significant in T2DM (264). Some data show that the activation of insulin signaling in CNS can upregulate IDE activity, and may correct the IDE defects present in AD (265, 266). Insulin also stimulates the internalization of Aβ oligomers and inhibits their binding to neurons, and thereby protects synapses against Aβ oligomers.
Neurofibrillary tangles are mainly composed of hyperphosphorylated tau molecules (267), which may be cleavaged by several proteases such as caspases and calpains, which play an important role in AD pathology (268). Alterations of insulin signaling in types 1 or 2 diabetes increase tau phosphorylation and tau cleavage, promoting AD pathology (235). Insulin resistance in T2DM, and the corresponding peripheral hyperinsulinemia, reduces insulin transport through the BBB, and the systemic insulin deficiency in T1DM increases tau phosphorylation (235).
Besides in AD, there are several mental diseases such as Huntington’s disease, depression, and schizophrenia in which insulin disturbances play a pathogenic role (197).
We should propose that resistance to the central action of insulin may be a meeting point between T2DM and AD and the slow pathological progression of both nosological entities should facilitates the deterioration of neurons mainly in the hippocampus and the cerebral cortex. Further studies are needed to accept or refuse this hypothesis, as well as with the possibility that AD may be caused though a slow deteriorating effect by microbial infection (269) by some viruses and bacteria; recently has been also proposed that fungal infections may represent a risk factor or possibly the cause of AD (270, 271). All the entities cited above have in common a slow deteriorating effect on CNS and represent an interesting matter to be further investigated. It should be also interesting to know whether patients with AD and cerebral infection with some viruses, bacteria, or fungus have central resistance to the action of insulin.
Scientific advances in cognitive functions, brain metabolic, and energy control, have provided new openings for trials on insulin resistance and on therapeutic approaches associated to nosological entities such as AD and DM. Therapeutics for AD treatment based on amyloid hypothesis (272) and tau hyperphosphorylation hypothesis (273) have been developed, but they perform poorly. Likewise, antioxidants, anti-inflammatory, and neuroprotective agents have been used with negative results (274, 275).
As thiamine deficiency is frequent in AD patients, drugs targeting altered thiamine metabolism have been used with conflicting results. Based on the hypothesis of multiple pathogenic cascades induced by glucose metabolism dysfunction, “cocktail therapies,” or drugs acting at multiple pathogenic cascades have been developed for AD (276).
Advances in the knowledge of preclinical AD and T2DM have provided a major stimulus for the development of treatments for preventing the pathogenic events of these disorders, focusing mainly on reducing brain insulin resistance.
At present, the main therapeutics available are insulin-sensitizing agents, metformin and the peroxisome proliferator-activated receptor gamma (PPARγ) agonists, and the incretin insulin mimetics molecules, glucagon-like peptide-1 (GLP-1), and gastric inhibitory peptide (GIP), which are insulin secretagogues. In addition, the beneficial effects of intranasal insulin or GLP-1 administration to patients with mild cognitive impairment (224) or T2DM have to be considered.
The biguanide metformin is one of the more used agents in T2DM that improves fasting insulin levels and the control of insulin on hepatic glucose production. Considering that its major action is on the liver, it has been reported in recent years that it can also cross the BBB (277), which given its insulin-sensitizing properties suggests that it may play a role in insulin resistance and dementia. In fact, an epidemiological study reported that treatment with metformin reduces the incidence of dementia in diabetic patients (278).
In addition, PPAR gamma agonists have been used in the treatment of T2DM because they improve insulin sensitivity (279), increasing the function of adipose tissue, and moving triglycerides and fatty acids away from the liver and muscle. Furthermore, they reduce both Aβ accumulation and neuroinflammation (280, 281), which may improve pathologies related with AD and T2DM, such as MCI (Mild Cognitive Impairment) associated with insulin resistance. Treatment with the PPAR gamma agonist rosiglitazone improved attention and memory, reducing fasting insulin levels in patients in the first stages of AD (282). However, the increased incidence of heart failure in patients treated with PPARγ agonists has reduced their use in diabetic patients, which has forced the development of new procedures and drugs.
Glucagon-like peptide-1 is an incretin that works as an insulin secretagogue in a glucose-dependent manner. Gliptins that delay the degradation of GLP-1 or exenatide and liraglutide or GLP-1 mimetics, with a more stable structure, reduce the degradation of GLP-1, and are therefore very useful in diabetes therapy. As metformin, GLP-1 mimetics readily cross the BBB and they induce several actions through GLP-1 brain receptors. Both exenatide and liraglutide were found to antagonize processes related to neurodegeneration and AD progression in mouse models (283). This may be because these agents are neuroprotectants (284, 285) that decrease oligomeric Aβ, neuritic plaque load, and microglial activation (283, 286). Furthermore, they stimulate neurogenesis and improve object recognition and spatial memory (283, 287). These GLP-1 mimetics reduce insulin resistance in MCI and AD, which suggests they could be used in the treatment of dementia with or without diabetes.
Intranasal insulin administration has a beneficial effect on patients with MCI or T2DM. Excellent scientific contributions have been done in human patients and others in animal models (240, 288–290). This procedure has the advantage of avoiding certain shortcomings in insulin transport through the BBB, added to the fact it does not produce hypoglycemia, as sometimes happens after peripheral insulin administration. This kind of treatment should be of interest for attenuating central insulin resistance, but it was abandoned because of secondary complications. In addition, GLP-1 analogs through intranasal administration could be potential therapeutics for AD (291).
Certain molecules involved in the transduction of signals induced by insulin might be targets for new therapeutic approaches in the future. The desensitization of insulin signaling by IRS-1 Ser/Tht phosphorylation should play an important role in insulin resistance. As only phospho-Ser/Thr on IRS-1 can have negative or positive effects on the insulin signaling of healthy and diseased tissues, those molecules might play an important pathophysiological role (292). Whether or not IRSs constitute a drug target for the treatment of insulin resistance and gaining a comprehensive understanding of Ser/Thr phosphorylation will help to explain the pathophysiological processes that facilitate insulin resistance.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors are indebted to Dr. Manuel Serrano Ríos for the critical review of the manuscript and for his interesting comments and suggestions. This work was supported by Mutua Madrileña Medical Research Foundation, the CIBER for Diabetes and Associated Metabolic Disorders (CIBERDEM) of the Carlos III Health Institute (ISCIII, Ministry of Science and Innovation), and Instituto de Investigación Sanitaria, Hospital Clínico San Carlos (IdISSC), Madrid, Spain.
1. Derakhshan F, Toth C. Insulin and the brain. Curr Diabetes Rev (2013) 9(2):102–16. doi: 10.2174/1573399811309020002
2. Havrankova J, Schmechel D, Roth J, Brownstein M. Identification of insulin in rat brain. Proc Natl Acad Sci U S A (1978) 75(11):5737–41. doi:10.1073/pnas.75.11.5737
3. Havrankova J, Roth J, Brownstein MJ. Concentrations of insulin and insulin receptors in the brain are independent of peripheral insulin levels. Studies of obese and streptozotocin-treated rodents. J Clin Invest (1979) 64(2):636–42. doi:10.1172/JCI109504
4. Dorn A, Bernstein HG, Rinne A, Ziegler M, Hahn HJ, Ansorge S. Insulin and glucagonlike peptides in the brain. Anat Rec (1983) 207(1):69–77. doi:10.1002/ar.1092070108
5. Pardridge WM. Blood-brain barrier biology and methodology. J Neurovirol (1999) 5(6):556–69. doi:10.3109/13550289909021285
6. Banks WA, Jaspan JB, Huang W, Kastin AJ. Transport of insulin across the blood-brain barrier: saturability at euglycemic doses of insulin. Peptides (1997) 18(9):1423–9. doi:10.1016/S0196-9781(97)00231-3
7. Margolis RU, Altszuler N. Insulin in the cerebrospinal fluid. Nature (1967) 215(5108):1375–6. doi:10.1038/2151375a0
8. Woods SC, Porte D Jr. Relationship between plasma and cerebrospinal fluid insulin levels of dogs. Am J Physiol (1977) 233(4):E331–4.
9. Duffy KR, Pardridge WM. Blood-brain barrier transcytosis of insulin in developing rabbits. Brain Res (1987) 420(1):32–8. doi:10.1016/0006-8993(87)90236-8
10. Eckert GP, Kirsch C, Leutz S, Wood WG, Muller WE. Cholesterol modulates amyloid beta-peptide’s membrane interactions. Pharmacopsychiatry (2003) 36(Suppl 2):S136–43. doi:10.1055/s-2003-43059
11. Baura GD, Foster DM, Porte D Jr, Kahn SE, Bergman RN, Cobelli C, et al. Saturable transport of insulin from plasma into the central nervous system of dogs in vivo. A mechanism for regulated insulin delivery to the brain. J Clin Invest (1993) 92(4):1824–30. doi:10.1172/JCI116773
12. Banks WA, Kastin AJ. Differential permeability of the blood-brain barrier to two pancreatic peptides: insulin and amylin. Peptides (1998) 19(5):883–9. doi:10.1016/S0196-9781(98)00018-7
13. Baura GD, Foster DM, Kaiyala K, Porte D Jr, Kahn SE, Schwartz MW. Insulin transport from plasma into the central nervous system is inhibited by dexamethasone in dogs. Diabetes (1996) 45(1):86–90. doi:10.2337/diab.45.1.86
14. Strubbe JH, Porte D Jr, Woods SC. Insulin responses and glucose levels in plasma and cerebrospinal fluid during fasting and refeeding in the rat. Physiol Behav (1988) 44(2):205–8. doi:10.1016/0031-9384(88)90139-4
15. Kaiyala KJ, Prigeon RL, Kahn SE, Woods SC, Schwartz MW. Obesity induced by a high-fat diet is associated with reduced brain insulin transport in dogs. Diabetes (2000) 49(9):1525–33. doi:10.2337/diabetes.49.9.1525
16. Florant GL, Richardson RD, Mahan S, Singer L, Woods SC. Seasonal changes in CSF insulin levels in marmots: insulin may not be a satiety signal for fasting in winter. Am J Physiol (1991) 260(4 Pt 2):R712–6.
17. Banks WA, Owen JB, Erickson MA. Insulin in the brain: there and back again. Pharmacol Ther (2012) 136(1):82–93. doi:10.1016/j.pharmthera.2012.07.006
18. Ghasemi R, Haeri A, Dargahi L, Mohamed Z, Ahmadiani A. Insulin in the brain: sources, localization and functions. Mol Neurobiol (2013) 47(1):145–71. doi:10.1007/s12035-012-8339-9
19. Dorn A, Rinne A, Bernstein HG, Hahn HJ, Ziegler M. Insulin and C-peptide in human brain neurons (insulin/C-peptide/brain peptides/immunohistochemistry/radioimmunoassay). J Hirnforsch (1983) 24(5):495–9.
20. Jezova D, Vigas M, Sadlon J. C-peptide-like material in rat brain: response to fasting and glucose ingestion. Endocrinol Exp (1985) 19(4):261–6.
21. Frolich L, Blum-Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S, et al. Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J Neural Transm (1998) 105(4–5):423–38. doi:10.1007/s007020050068
22. Young WS III. Periventricular hypothalamic cells in the rat brain contain insulin mRNA. Neuropeptides (1986) 8(2):93–7. doi:10.1016/0143-4179(86)90035-1
23. Devaskar SU, Singh BS, Carnaghi LR, Rajakumar PA, Giddings SJ. Insulin II gene expression in rat central nervous system. Regul Pept (1993) 48(1–2):55–63. doi:10.1016/0167-0115(93)90335-6
24. Devaskar SU, Giddings SJ, Rajakumar PA, Carnaghi LR, Menon RK, Zahm DS. Insulin gene expression and insulin synthesis in mammalian neuronal cells. J Biol Chem (1994) 269(11):8445–54.
25. Raizada MK. Localization of insulin-like immunoreactivity in the neurons from primary cultures of rat brain. Exp Cell Res (1983) 143(2):351–7. doi:10.1016/0014-4827(83)90061-7
26. Birch NP, Christie DL, Renwick AG. Proinsulin-like material in mouse foetal brain cell cultures. FEBS Lett (1984) 168(2):299–302. doi:10.1016/0014-5793(84)80266-5
27. Schechter R, Whitmire J, Wheet GS, Beju D, Jackson KW, Harlow R, et al. Immunohistochemical and in situ hybridization study of an insulin-like substance in fetal neuron cell cultures. Brain Res (1994) 636(1):9–27. doi:10.1016/0006-8993(94)90170-8
28. Gerozissis K. Brain insulin: regulation, mechanisms of action and functions. Cell Mol Neurobiol (2003) 23(1):1–25. doi:10.1023/A:1025021529347
29. Clarke DW, Mudd L, Boyd FT Jr, Fields M, Raizada MK. Insulin is released from rat brain neuronal cells in culture. J Neurochem (1986) 47(3):831–6. doi:10.1111/j.1471-4159.1986.tb00686.x
30. Wei LT, Matsumoto H, Rhoads DE. Release of immunoreactive insulin from rat brain synaptosomes under depolarizing conditions. J Neurochem (1990) 54(5):1661–5. doi:10.1111/j.1471-4159.1990.tb01219.x
31. Santos MS, Pereira EM, Carvaho AP. Stimulation of immunoreactive insulin release by glucose in rat brain synaptosomes. Neurochem Res (1999) 24(1):33–6. doi:10.1023/A:1020971812098
32. Gerozissis K. Brain insulin, energy and glucose homeostasis; genes, environment and metabolic pathologies. Eur J Pharmacol (2008) 585(1):38–49. doi:10.1016/j.ejphar.2008.01.050
33. Burgos-Ramos E, Gonzalez-Rodriguez A, Canelles S, Baquedano E, Frago LM, Revuelta-Cervantes J, et al. Differential insulin receptor substrate-1 (IRS1)-related modulation of neuropeptide Y and proopiomelanocortin expression in nondiabetic and diabetic IRS2-/- mice. Endocrinology (2012) 153(3):1129–40. doi:10.1210/en.2011-1278
34. Miller DW, Keller BT, Borchardt RT. Identification and distribution of insulin receptors on cultured bovine brain microvessel endothelial cells: possible function in insulin processing in the blood-brain barrier. J Cell Physiol (1994) 161(2):333–41. doi:10.1002/jcp.1041610218
35. Tagliamonte A, DeMontis MG, Olianas M, Onali PL, Gessa GL. Possible role of insulin in the transport of tyrosine and tryptophan from blood to brain. Adv Exp Med Biol (1976) 69:89–94. doi:10.1007/978-1-4684-3264-0_7
36. Ayre SG, Skaletski B, Mosnaim AD. Blood-brain barrier passage of azidothymidine in rats: effect of insulin. Res Commun Chem Pathol Pharmacol (1989) 63(1):45–52.
37. Kastin AJ, Akerstrom V. Glucose and insulin increase the transport of leptin through the blood-brain barrier in normal mice but not in streptozotocin-diabetic mice. Neuroendocrinology (2001) 73(4):237–42. doi:10.1159/000054640
38. Liu H, Yang H, Wang D, Liu Y, Liu X, Li Y, et al. Insulin regulates P-glycoprotein in rat brain microvessel endothelial cells via an insulin receptor-mediated PKC/NF-kappaB pathway but not a PI3K/Akt pathway. Eur J Pharmacol (2009) 602(2–3):277–82. doi:10.1016/j.ejphar.2008.11.026
39. Liu X, Jing XY, Jin S, Li Y, Liu L, Yu YL, et al. Insulin suppresses the expression and function of breast cancer resistance protein in primary cultures of rat brain microvessel endothelial cells. Pharmacol Rep (2011) 63(2):487–93. doi:10.1016/S1734-1140(11)70515-1
40. Catalan RE, Martinez AM, Aragones MD, Miguel BG, Robles A. Insulin action on brain microvessels; effect on alkaline phosphatase. Biochem Biophys Res Commun (1988) 150(2):583–90. doi:10.1016/0006-291X(88)90433-0
41. Langston JW, Li W, Harrison L, Aw TY. Activation of promoter activity of the catalytic subunit of gamma-glutamylcysteine ligase (GCL) in brain endothelial cells by insulin requires antioxidant response element 4 and altered glycemic status: implication for GCL expression and GSH synthesis. Free Radic Biol Med (2011) 51(9):1749–57. doi:10.1016/j.freeradbiomed.2011.08.004
42. Hurley JH, Zhang S, Bye LS, Marshall MS, DePaoli-Roach AA, Guan K, et al. Insulin signaling inhibits the 5-HT2C receptor in choroid plexus via MAP kinase. BMC Neurosci (2003) 4:10. doi:10.1186/1471-2202-4-10
43. Bernstein HG, Lendeckel U, Bukowska A, Ansorge S, Ernst T, Stauch R, et al. Regional and cellular distribution patterns of insulin-degrading enzyme in the adult human brain and pituitary. J Chem Neuroanat (2008) 35(2):216–24. doi:10.1016/j.jchemneu.2007.12.001
44. Lynch JA, George AM, Eisenhauer PB, Conn K, Gao W, Carreras I, et al. Insulin degrading enzyme is localized predominantly at the cell surface of polarized and unpolarized human cerebrovascular endothelial cell cultures. J Neurosci Res (2006) 83(7):1262–70. doi:10.1002/jnr.20809
45. Seino S, Seino M, Nishi S, Bell GI. Structure of the human insulin receptor gene and characterization of its promoter. Proc Natl Acad Sci U S A (1989) 86(1):114–8. doi:10.1073/pnas.86.1.114
46. Moller DE, Yokota A, Caro JF, Flier JS. Tissue-specific expression of two alternatively spliced insulin receptor mRNAs in man. Mol Endocrinol (1989) 3(8):1263–9. doi:10.1210/mend-3-8-1263
47. Seino S, Bell GI. Alternative splicing of human insulin receptor messenger RNA. Biochem Biophys Res Commun (1989) 159(1):312–6. doi:10.1016/0006-291X(89)92439-X
48. Vienberg SG, Bouman SD, Sorensen H, Stidsen CE, Kjeldsen T, Glendorf T, et al. Receptor-isoform-selective insulin analogues give tissue-preferential effects. Biochem J (2011) 440(3):301–8. doi:10.1042/BJ20110880
49. Schulingkamp RJ, Pagano TC, Hung D, Raffa RB. Insulin receptors and insulin action in the brain: review and clinical implications. Neurosci Biobehav Rev (2000) 24(8):855–72. doi:10.1016/S0149-7634(00)00040-3
50. Szabo O, Szabo AJ. Evidence for an insulin-sensitive receptor in the central nervous system. Am J Physiol (1972) 223(6):1349–53.
51. Posner BI, Kelly PA, Shiu RP, Friesen HG. Studies of insulin, growth hormone and prolactin binding: tissue distribution, species variation and characterization. Endocrinology (1974) 95(2):521–31. doi:10.1210/endo-95-2-521
52. Havrankova J, Roth J, Brownstein M. Insulin receptors are widely distributed in the central nervous system of the rat. Nature (1978) 272(5656):827–9. doi:10.1038/272827a0
53. Lowe WL Jr, Boyd FT, Clarke DW, Raizada MK, Hart C, LeRoith D. Development of brain insulin receptors: structural and functional studies of insulin receptors from whole brain and primary cell cultures. Endocrinology (1986) 119(1):25–35. doi:10.1210/endo-119-1-25
54. Landau BR, Takaoka Y, Abrams MA, Genuth SM, van Houten M, Posner BI, et al. Binding of insulin by monkey and pig hypothalamus. Diabetes (1983) 32(3):284–91. doi:10.2337/diabetes.32.3.284
55. Hill JM, Lesniak MA, Pert CB, Roth J. Autoradiographic localization of insulin receptors in rat brain: prominence in olfactory and limbic areas. Neuroscience (1986) 17(4):1127–38. doi:10.1016/0306-4522(86)90082-5
56. Werther GA, Hogg A, Oldfield BJ, McKinley MJ, Figdor R, Allen AM, et al. Localization and characterization of insulin receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry. Endocrinology (1987) 121(4):1562–70. doi:10.1210/endo-121-4-1562
57. Marks JL, Porte D Jr, Stahl WL, Baskin DG. Localization of insulin receptor mRNA in rat brain by in situ hybridization. Endocrinology (1990) 127(6):3234–6. doi:10.1210/endo-127-6-3234
58. Amessou M, Tahiri K, Chauvet G, Desbuquois B. Age-related changes in insulin receptor mRNA and protein expression in genetically obese Zucker rats. Diabetes Metab (2010) 36(2):120–8. doi:10.1016/j.diabet.2009.09.004
59. Pacold ST, Blackard WG. Central nervous system insulin receptors in normal and diabetic rats. Endocrinology (1979) 105(6):1452–7.
60. Werther GA, Hogg A, Oldfield BJ, McKinley MJ, Figdor R, Mendelsohn FA. Localization and characterization of insulin-like growth factor-I receptors in rat brain and pituitary gland using in vitro autoradiography and computerized densitometry* A distinct distribution from insulin receptors. J Neuroendocrinol (1989) 1(5):369–77. doi:10.1111/j.1365-2826.1989.tb00131.x
61. Hami J, Sadr-Nabavi A, Sankian M, Haghir H. Sex differences and left-right asymmetries in expression of insulin and insulin-like growth factor-1 receptors in developing rat hippocampus. Brain Struct Funct (2012) 217(2):293–302. doi:10.1007/s00429-011-0358-1
62. Hopkins DF, Williams G. Insulin receptors are widely distributed in human brain and bind human and porcine insulin with equal affinity. Diabet Med (1997) 14(12):1044–50. doi:10.1002/(SICI)1096-9136(199712)14:12<1044::AID-DIA508>3.3.CO;2-6
63. Potau N, Escofet MA, Martinez MC. Ontogenesis of insulin receptors in human cerebral cortex. J Endocrinol Invest (1991) 14(1):53–8. doi:10.1007/BF03350263
64. Zahniser NR, Goens MB, Hanaway PJ, Vinych JV. Characterization and regulation of insulin receptors in rat brain. J Neurochem (1984) 42(5):1354–62. doi:10.1111/j.1471-4159.1984.tb02795.x
65. Heidenreich KA, Zahniser NR, Berhanu P, Brandenburg D, Olefsky JM. Structural differences between insulin receptors in the brain and peripheral target tissues. J Biol Chem (1983) 258(14):8527–30.
66. Joost HG. Structural and functional heterogeneity of insulin receptors. Cell Signal (1995) 7(2):85–91. doi:10.1016/0898-6568(94)00071-I
67. Kar S, Chabot JG, Quirion R. Quantitative autoradiographic localization of [125I]insulin-like growth factor I, [125I]insulin-like growth factor II, and [125I]insulin receptor binding sites in developing and adult rat brain. J Comp Neurol (1993) 333(3):375–97. doi:10.1002/cne.903330306
68. Shemer J, Adamo M, Raizada MK, Heffez D, Zick Y, LeRoith D. Insulin and IGF-I stimulate phosphorylation of their respective receptors in intact neuronal and glial cells in primary culture. J Mol Neurosci (1989) 1(1):3–8. doi:10.1007/BF02918899
69. Brummer T, Schmitz-Peiffer C, Daly RJ. Docking proteins. FEBS J (2010) 277(21):4356–69. doi:10.1111/j.1742-4658.2010.07865.x
70. Taguchi A, White MF. Insulin-like signaling, nutrient homeostasis, and life span. Annu Rev Physiol (2008) 70:191–212. doi:10.1146/annurev.physiol.70.113006.100533
71. Saltiel AR, Pessin JE. Insulin signaling pathways in time and space. Trends Cell Biol (2002) 12(2):65–71. doi:10.1016/S0962-8924(01)02207-3
72. Pilch PF, Shia MA, Benson RJ, Fine RE. Coated vesicles participate in the receptor-mediated endocytosis of insulin. J Cell Biol (1983) 96(1):133–8. doi:10.1083/jcb.96.1.133
73. Heffetz D, Zick Y. Receptor aggregation is necessary for activation of the soluble insulin receptor kinase. J Biol Chem (1986) 261(2):889–94.
74. White MF. Insulin signaling in health and disease. Science (2003) 302(5651):1710–1. doi:10.1126/science.1092952
75. Myers MG Jr, Sun XJ, White MF. The IRS-1 signaling system. Trends Biochem Sci (1994) 19(7):289–93. doi:10.1016/0968-0004(94)90007-8
76. Aguirre V, Werner ED, Giraud J, Lee YH, Shoelson SE, White MF. Phosphorylation of Ser307 in insulin receptor substrate-1 blocks interactions with the insulin receptor and inhibits insulin action. J Biol Chem (2002) 277(2):1531–7. doi:10.1074/jbc.M101521200
77. Krebs DL, Hilton DJ. A new role for SOCS in insulin action. Suppressor of cytokine signaling. Sci STKE (2003) 2003(169):e6. doi:10.1126/stke.2003.169.pe6
78. Jhala US, Canettieri G, Screaton RA, Kulkarni RN, Krajewski S, Reed J, et al. cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes Dev (2003) 17(13):1575–80. doi:10.1101/gad.1097103
79. Abbott MA, Wells DG, Fallon JR. The insulin receptor tyrosine kinase substrate p58/53 and the insulin receptor are components of CNS synapses. J Neurosci (1999) 19(17):7300–8.
80. Okamura-Oho Y, Miyashita T, Yamada M. Distinctive tissue distribution and phosphorylation of IRSp53 isoforms. Biochem Biophys Res Commun (2001) 289(5):957–60. doi:10.1006/bbrc.2001.6102
81. Miyahara A, Okamura-Oho Y, Miyashita T, Hoshika A, Yamada M. Genomic structure and alternative splicing of the insulin receptor tyrosine kinase substrate of 53-kDa protein. J Hum Genet (2003) 48(8):410–4. doi:10.1007/s10038-003-0047-x
82. Choi J, Ko J, Racz B, Burette A, Lee JR, Kim S, et al. Regulation of dendritic spine morphogenesis by insulin receptor substrate 53, a downstream effector of Rac1 and Cdc42 small GTPases. J Neurosci (2005) 25(4):869–79. doi:10.1523/JNEUROSCI.3212-04.2005
83. Mackie S, Aitken A. Novel brain 14-3-3 interacting proteins involved in neurodegenerative disease. FEBS J (2005) 272(16):4202–10. doi:10.1111/j.1742-4658.2005.04832.x
84. Sawallisch C, Berhorster K, Disanza A, Mantoani S, Kintscher M, Stoenica L, et al. The insulin receptor substrate of 53 kDa (IRSp53) limits hippocampal synaptic plasticity. J Biol Chem (2009) 284(14):9225–36. doi:10.1074/jbc.M808425200
85. Lizcano JM, Alessi DR. The insulin signalling pathway. Curr Biol (2002) 12(7):R236–8. doi:10.1016/S0960-9822(02)00777-7
86. Jewell JL, Guan KL. Nutrient signaling to mTOR and cell growth. Trends Biochem Sci (2013) 38(5):233–42. doi:10.1016/j.tibs.2013.01.004
87. Kolch W, Kotwaliwale A, Vass K, Janosch P. The role of Raf kinases in malignant transformation. Expert Rev Mol Med (2002) 4(8):1–18. doi:10.1017/S1462399402004386
88. Evans RM, Hui S, Perkins A, Lahiri DK, Poirier J, Farlow MR. Cholesterol and APOE genotype interact to influence Alzheimer disease progression. Neurology (2004) 62(10):1869–71. doi:10.1212/01.WNL.0000125323.15458.3F
89. Frodin M, Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol (1999) 151(1–2):65–77. doi:10.1016/S0303-7207(99)00061-1
90. White H, Venkatesh B. Clinical review: ketones and brain injury. Crit Care (2011) 15(2):219. doi:10.1186/cc10020
91. Marty N, Dallaporta M, Thorens B. Brain glucose sensing, counterregulation, and energy homeostasis. Physiology (Bethesda) (2007) 22:241–51. doi:10.1152/physiol.00010.2007
92. Alvarez E, Roncero I, Chowen JA, Thorens B, Blazquez E. Expression of the glucagon-like peptide-1 receptor gene in rat brain. J Neurochem (1996) 66(3):920–7. doi:10.1046/j.1471-4159.1996.66030920.x
93. Navarro M, Rodriquez de Fonseca F, Alvarez E, Chowen JA, Zueco JA, Gomez R, et al. Colocalization of glucagon-like peptide-1 (GLP-1) receptors, glucose transporter GLUT-2, and glucokinase mRNAs in rat hypothalamic cells: evidence for a role of GLP-1 receptor agonists as an inhibitory signal for food and water intake. J Neurochem (1996) 67(5):1982–91. doi:10.1046/j.1471-4159.1996.67051982.x
94. Roncero I, Alvarez E, Vazquez P, Blazquez E. Functional glucokinase isoforms are expressed in rat brain. J Neurochem (2000) 74(5):1848–57. doi:10.1046/j.1471-4159.2000.0741848.x
95. Thorens B, Mueckler M. Glucose transporters in the 21st century. Am J Physiol Endocrinol Metab (2010) 298(2):E141–5. doi:10.1152/ajpendo.00712.2009
96. Simpson IA, Appel NM, Hokari M, Oki J, Holman GD, Maher F, et al. Blood-brain barrier glucose transporter: effects of hypo- and hyper-glycemia revisited. J Neurochem (1999) 72(1):238–47. doi:10.1046/j.1471-4159.1999.0720238.x
97. Kang L, Routh VH, Kuzhikandathil EV, Gaspers LD, Levin BE. Physiological and molecular characteristics of rat hypothalamic ventromedial nucleus glucosensing neurons. Diabetes (2004) 53(3):549–59. doi:10.2337/diabetes.53.3.549
98. Levin BE, Routh VH, Kang L, Sanders NM, Dunn-Meynell AA. Neuronal glucosensing: what do we know after 50 years? Diabetes (2004) 53(10):2521–8. doi:10.2337/diabetes.53.10.2521
99. Li B, Xi X, Roane DS, Ryan DH, Martin RJ. Distribution of glucokinase, glucose transporter GLUT2, sulfonylurea receptor-1, glucagon-like peptide-1 receptor and neuropeptide Y messenger RNAs in rat brain by quantitative real time RT-PCR. Brain Res Mol Brain Res (2003) 113(1–2):139–42. doi:10.1016/S0169-328X(03)00125-6
100. Nagamatsu S, Kornhauser JM, Burant CF, Seino S, Mayo KE, Bell GI. Glucose transporter expression in brain. cDNA sequence of mouse GLUT3, the brain facilitative glucose transporter isoform, and identification of sites of expression by in situ hybridization. J Biol Chem (1992) 267(1):467–72.
101. Gould GW, Brant AM, Kahn BB, Shepherd PR, McCoid SC, Gibbs EM. Expression of the brain-type glucose transporter is restricted to brain and neuronal cells in mice. Diabetologia (1992) 35(4):304–9. doi:10.1007/BF00401196
102. El Messari S, Leloup C, Quignon M, Brisorgueil MJ, Penicaud L, Arluison M. Immunocytochemical localization of the insulin-responsive glucose transporter 4 (Glut4) in the rat central nervous system. J Comp Neurol (1998) 399(4):492–512. doi:10.1002/(SICI)1096-9861(19981005)399:4<492::AID-CNE4>3.0.CO;2-X
103. Vannucci SJ, Koehler-Stec EM, Li K, Reynolds TH, Clark R, Simpson IA. GLUT4 glucose transporter expression in rodent brain: effect of diabetes. Brain Res (1998) 797(1):1–11. doi:10.1016/S0006-8993(98)00103-6
104. Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest (2012) 122(4):1316–38. doi:10.1172/JCI59903
105. Livingstone C, Lyall H, Gould GW. Hypothalamic GLUT 4 expression: a glucose- and insulin-sensing mechanism? Mol Cell Endocrinol (1995) 107(1):67–70. doi:10.1016/0303-7207(94)03423-Q
106. Wang R, Liu X, Hentges ST, Dunn-Meynell AA, Levin BE, Wang W, et al. The regulation of glucose-excited neurons in the hypothalamic arcuate nucleus by glucose and feeding-relevant peptides. Diabetes (2004) 53(8):1959–65. doi:10.2337/diabetes.53.8.1959
107. Reagan LP, Gorovits N, Hoskin EK, Alves SE, Katz EB, Grillo CA, et al. Localization and regulation of GLUTx1 glucose transporter in the hippocampus of streptozotocin diabetic rats. Proc Natl Acad Sci U S A (2001) 98(5):2820–5. doi:10.1073/pnas.051629798
108. Ibberson M, Riederer BM, Uldry M, Guhl B, Roth J, Thorens B. Immunolocalization of GLUTX1 in the testis and to specific brain areas and vasopressin-containing neurons. Endocrinology (2002) 143(1):276–84. doi:10.1210/endo.143.1.8587
109. Reagan LP, Rosell DR, Alves SE, Hoskin EK, McCall AL, Charron MJ, et al. GLUT8 glucose transporter is localized to excitatory and inhibitory neurons in the rat hippocampus. Brain Res (2002) 932(1–2):129–34. doi:10.1016/S0006-8993(02)02308-9
110. Sankar R, Thamotharan S, Shin D, Moley KH, Devaskar SU. Insulin-responsive glucose transporters-GLUT8 and GLUT4 are expressed in the developing mammalian brain. Brain Res Mol Brain Res (2002) 107(2):157–65. doi:10.1016/S0169-328X(02)00487-4
111. McEwen BS, Reagan LP. Glucose transporter expression in the central nervous system: relationship to synaptic function. Eur J Pharmacol (2004) 490(1–3):13–24. doi:10.1016/j.ejphar.2004.02.041
112. Piroli GG, Grillo CA, Hoskin EK, Znamensky V, Katz EB, Milner TA, et al. Peripheral glucose administration stimulates the translocation of GLUT8 glucose transporter to the endoplasmic reticulum in the rat hippocampus. J Comp Neurol (2002) 452(2):103–14. doi:10.1002/cne.10368
113. Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat Neurosci (2002) 5(6):566–72. doi:10.1038/nn0602-861
114. Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med (2002) 8(12):1376–82. doi:10.1038/nm1202-798
115. Demuro G, Obici S. Central nervous system and control of endogenous glucose production. Curr Diab Rep (2006) 6(3):188–93. doi:10.1007/s11892-006-0033-8
116. Girard J. The inhibitory effects of insulin on hepatic glucose production are both direct and indirect. Diabetes (2006) 55(Suppl 2):S65–9. doi:10.2337/db06-S009
117. Pocai A, Lam TK, Gutierrez-Juarez R, Obici S, Schwartz GJ, Bryan J, et al. Hypothalamic K(ATP) channels control hepatic glucose production. Nature (2005) 434(7036):1026–31. doi:10.1038/nature03439
118. Cotero VE, Routh VH. Insulin blunts the response of glucose-excited neurons in the ventrolateral-ventromedial hypothalamic nucleus to decreased glucose. Am J Physiol Endocrinol Metab (2009) 296(5):E1101–9. doi:10.1152/ajpendo.90932.2008
119. Bernard C. Leçons de Physiologie Expérimentale Appliquée à la Médecine. Paris: Baillére (1855).
120. Niswender KD, Morrison CD, Clegg DJ, Olson R, Baskin DG, Myers MG Jr, et al. Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes (2003) 52(2):227–31. doi:10.2337/diabetes.52.2.227
121. Kim YB, Uotani S, Pierroz DD, Flier JS, Kahn BB. In vivo administration of leptin activates signal transduction directly in insulin-sensitive tissues: overlapping but distinct pathways from insulin. Endocrinology (2000) 141(7):2328–39. doi:10.1210/endo.141.7.7536
122. Castellano JM, Roa J, Luque RM, Dieguez C, Aguilar E, Pinilla L, et al. KiSS-1/kisspeptins and the metabolic control of reproduction: physiologic roles and putative physiopathological implications. Peptides (2009) 30(1):139–45. doi:10.1016/j.peptides.2008.06.007
123. Fernandez-Fernandez R, Martini AC, Navarro VM, Castellano JM, Dieguez C, Aguilar E, et al. Novel signals for the integration of energy balance and reproduction. Mol Cell Endocrinol (2006) 25(4–255):127–32. doi:10.1016/j.mce.2006.04.026
124. Arias P, Rodriguez M, Szwarcfarb B, Sinay IR, Moguilevsky JA. Effect of insulin on LHRH release by perifused hypothalamic fragments. Neuroendocrinology (1992) 56(3):415–8. doi:10.1159/000126257
125. Miller DW, Blache D, Martin GB. The role of intracerebral insulin in the effect of nutrition on gonadotrophin secretion in mature male sheep. J Endocrinol (1995) 147(2):321–9. doi:10.1677/joe.0.1470321
126. Dong Q, Lazarus RM, Wong LS, Vellios M, Handelsman DJ. Pulsatile LH secretion in streptozotocin-induced diabetes in the rat. J Endocrinol (1991) 131(1):49–55. doi:10.1677/joe.0.1310049
127. Tanaka T, Nagatani S, Bucholtz DC, Ohkura S, Tsukamura H, Maeda K, et al. Central action of insulin regulates pulsatile luteinizing hormone secretion in the diabetic sheep model. Biol Reprod (2000) 62(5):1256–61. doi:10.1095/biolreprod62.5.1256
128. Bucholtz DC, Chiesa A, Pappano WN, Nagatani S, Tsukamura H, Maeda KI, et al. Regulation of pulsatile luteinizing hormone secretion by insulin in the diabetic male lamb. Biol Reprod (2000) 62(5):1248–55. doi:10.1095/biolreprod62.5.1248
129. Roger LJ, Fellows RE. Stimulation of ornithine decarboxylase activity by insulin in developing rat brain. Endocrinology (1980) 106(2):619–25. doi:10.1210/endo-106-2-619
130. Wozniak M, Rydzewski B, Baker SP, Raizada MK. The cellular and physiological actions of insulin in the central nervous system. Neurochem Int (1993) 22(1):1–10. doi:10.1016/0197-0186(93)90062-A
131. Schubert M, Brazil DP, Burks DJ, Kushner JA, Ye J, Flint CL, et al. Insulin receptor substrate-2 deficiency impairs brain growth and promotes tau phosphorylation. J Neurosci (2003) 23(18):7084–92.
132. Xu QG, Li XQ, Kotecha SA, Cheng C, Sun HS, Zochodne DW. Insulin as an in vivo growth factor. Exp Neurol (2004) 188(1):43–51. doi:10.1016/j.expneurol.2004.03.008
133. Raizada MK, Yang JW, Fellows RE. Binding of [125I]insulin to specific receptors and stimulation of nucleotide incorporation in cells cultured from rat brain. Brain Res (1980) 200(2):389–400. doi:10.1016/0006-8993(80)90929-4
134. Heidenreich KA, de Vellis G, Gilmore PR. Functional properties of the subtype of insulin receptor found on neurons. J Neurochem (1988) 51(3):878–87. doi:10.1111/j.1471-4159.1988.tb01824.x
135. Clarke DW, Boyd FT Jr, Kappy MS, Raizada MK. Insulin stimulates macromolecular synthesis in cultured glial cells from rat brain. Am J Physiol (1985) 249(5 Pt 1):C484–9.
136. Heidenreich KA, Toledo SP. Insulin receptors mediate growth effects in cultured fetal neurons. II. Activation of a protein kinase that phosphorylates ribosomal protein S6. Endocrinology (1989) 125(3):1458–63. doi:10.1210/endo-125-3-1451
137. Recio-Pinto E, Lang FF, Ishii DN. Insulin and insulin-like growth factor II permit nerve growth factor binding and the neurite formation response in cultured human neuroblastoma cells. Proc Natl Acad Sci U S A (1984) 81(8):2562–6. doi:10.1073/pnas.81.8.2562
138. Ang LC, Bhaumick B, Juurlink BH. Neurite promoting activity of insulin, insulin-like growth factor I and nerve growth factor on spinal motoneurons is astrocyte dependent. Brain Res Dev Brain Res (1993) 74(1):83–8. doi:10.1016/0165-3806(93)90086-P
139. Velazquez E, Blazquez E, Ruiz-Albusac JM. Synergistic effect of glucagon-like peptide 2 (GLP-2) and of key growth factors on the proliferation of cultured rat astrocytes. Evidence for reciprocal upregulation of the mRNAs for GLP-2 and IGF-I receptors. Mol Neurobiol (2009) 40(2):183–93. doi:10.1007/s12035-009-8080-1
140. Heni M, Hennige AM, Peter A, Siegel-Axel D, Ordelheide AM, Krebs N, et al. Insulin promotes glycogen storage and cell proliferation in primary human astrocytes. PLoS One (2011) 6(6):e21594. doi:10.1371/journal.pone.0021594
141. Heidenreich KA, Toledo SP, Brunton LL, Watson MJ, Daniel-Issakani S, Strulovici B. Insulin stimulates the activity of a novel protein kinase C, PKC-epsilon, in cultured fetal chick neurons. J Biol Chem (1990) 265(25):15076–82.
142. Vanhems E, Delbos M, Girardie J. Insulin and neuroparsin promote neurite outgrowth in cultured locust CNS. Eur J Neurosci (1990) 2(9):776–82. doi:10.1111/j.1460-9568.1990.tb00468.x
143. Heidenreich KA, Toledo SP, Kenner KA. Regulation of protein phosphorylation by insulin and insulin-like growth factors in cultured fetal neurons. Adv Exp Med Biol (1991) 293:379–84. doi:10.1007/978-1-4684-5949-4_33
144. Patel RA, Kurian P, Raizada MK, Crews FT. Insulin stimulates phosphatidylinositol 3-kinase activity in rat neuronal primary cultures. J Neurochem (1993) 61(1):360–3. doi:10.1111/j.1471-4159.1993.tb03578.x
145. Lee CC, Huang CC, Wu MY, Hsu KS. Insulin stimulates postsynaptic density-95 protein translation via the phosphoinositide 3-kinase-Akt-mammalian target of rapamycin signaling pathway. J Biol Chem (2005) 280(18):18543–50. doi:10.1074/jbc.M414112200
146. Lee CC, Huang CC, Hsu KS. Insulin promotes dendritic spine and synapse formation by the PI3K/Akt/mTOR and Rac1 signaling pathways. Neuropharmacology (2011) 61(4):867–79. doi:10.1016/j.neuropharm.2011.06.003
147. Nemoto T, Yanagita T, Satoh S, Maruta T, Kanai T, Murakami M, et al. Insulin-induced neurite-like process outgrowth: acceleration of tau protein synthesis via a phosphoinositide 3-kinase mammalian target of rapamycin pathway. Neurochem Int (2011) 59(6):880–8. doi:10.1016/j.neuint.2011.08.002
148. Schechter R, Yanovitch T, Abboud M, Johnson G III, Gaskins J. Effects of brain endogenous insulin on neurofilament and MAPK in fetal rat neuron cell cultures. Brain Res (1998) 808(2):270–8. doi:10.1016/S0006-8993(98)00842-7
149. Rhee YH, Choi M, Lee HS, Park CH, Kim SM, Yi SH, et al. Insulin concentration is critical in culturing human neural stem cells and neurons. Cell Death Dis (2013) 4:e766. doi:10.1038/cddis.2013.295
150. Leopold P. Neuronal differentiation: TOR and insulin receptor pathways set the tempo. Cell (2004) 119(1):4–5. doi:10.1016/j.cell.2004.09.024
151. Ryu BR, Ko HW, Jou I, Noh JS, Gwag BJ. Phosphatidylinositol 3-kinase-mediated regulation of neuronal apoptosis and necrosis by insulin and IGF-I. J Neurobiol (1999) 39(4):536–46. doi:10.1002/(SICI)1097-4695(19990615)39:4<536::AID-NEU7>3.3.CO;2-A
152. Rensink AA, Otte-Holler I, de Boer R, Bosch RR, ten Donkelaar HJ, de Waal RM, et al. Insulin inhibits amyloid beta-induced cell death in cultured human brain pericytes. Neurobiol Aging (2004) 25(1):93–103. doi:10.1016/S0197-4580(03)00039-3
153. Garg R, Chaudhuri A, Munschauer F, Dandona P. Hyperglycemia, insulin, and acute ischemic stroke: a mechanistic justification for a trial of insulin infusion therapy. Stroke (2006) 37(1):267–73. doi:10.1161/01.STR.0000195175.29487.30
154. Duarte AI, Proenca T, Oliveira CR, Santos MS, Rego AC. Insulin restores metabolic function in cultured cortical neurons subjected to oxidative stress. Diabetes (2006) 55(10):2863–70. doi:10.2337/db06-0030
155. Duarte AI, Santos MS, Seica R, de Oliveira CR. Insulin affects synaptosomal GABA and glutamate transport under oxidative stress conditions. Brain Res (2003) 977(1):23–30. doi:10.1016/S0006-8993(03)02679-9
156. Sevanian A, Davies KJ, Hochstein P. Serum urate as an antioxidant for ascorbic acid. Am J Clin Nutr (1991) 54(6 Suppl):1129S–34S.
157. Auer RN. Insulin, blood glucose levels, and ischemic brain damage. Neurology (1998) 51(3 Suppl 3):S39–43. doi:10.1212/WNL.51.3_Suppl_3.S39
158. Shuaib A, Ijaz MS, Waqar T, Voll C, Kanthan R, Miyashita H, et al. Insulin elevates hippocampal GABA levels during ischemia. This is independent of its hypoglycemic effect. Neuroscience (1995) 67(4):809–14. doi:10.1016/0306-4522(95)00093-X
159. Grunstein HS, James DE, Storlien LH, Smythe GA, Kraegen EW. Hyperinsulinemia suppresses glucose utilization in specific brain regions: in vivo studies using the euglycemic clamp in the rat. Endocrinology (1985) 116(2):604–10. doi:10.1210/endo-116-2-604
160. Voll CL, Auer RN. Insulin attenuates ischemic brain damage independent of its hypoglycemic effect. J Cereb Blood Flow Metab (1991) 11(6):1006–14. doi:10.1038/jcbfm.1991.168
161. Kovacs P, Hajnal A. In vivo electrophysiological effects of insulin in the rat brain. Neuropeptides (2009) 43(4):283–93. doi:10.1016/j.npep.2009.05.006
162. Wang Q, Liu L, Pei L, Ju W, Ahmadian G, Lu J, et al. Control of synaptic strength, a novel function of Akt. Neuron (2003) 38(6):915–28. doi:10.1016/S0896-6273(03)00356-8
163. Plum L, Schubert M, Bruning JC. The role of insulin receptor signaling in the brain. Trends Endocrinol Metab (2005) 16(2):59–65. doi:10.1016/j.tem.2005.01.008
164. Jonas EA, Knox RJ, Smith TC, Wayne NL, Connor JA, Kaczmarek LK. Regulation by insulin of a unique neuronal Ca2+ pool and of neuropeptide secretion. Nature (1997) 385(6614):343–6. doi:10.1038/385343a0
165. Boyd FT Jr, Clarke DW, Muther TF, Raizada MK. Insulin receptors and insulin modulation of norepinephrine uptake in neuronal cultures from rat brain. J Biol Chem (1985) 260(29):15880–4.
166. Masters BA, Shemer J, Judkins JH, Clarke DW, Le Roith D, Raizada MK. Insulin receptors and insulin action in dissociated brain cells. Brain Res (1987) 417(2):247–56. doi:10.1016/0006-8993(87)90449-5
167. Lozovsky D, Saller CF, Kopin IJ. Dopamine receptor binding is increased in diabetic rats. Science (1981) 214(4524):1031–3. doi:10.1126/science.6458088
168. Lozovsky DB, Kopin IJ, Saller CF. Modulation of dopamine receptor supersensitivity by chronic insulin: implication in schizophrenia. Brain Res (1985) 343(1):190–3. doi:10.1016/0006-8993(85)91178-3
169. Levin BE, Israel P, Lattemann DP. Insulin selectively downregulates alpha2-adrenoceptors in the arcuate and dorsomedial nucleus. Brain Res Bull (1998) 45(2):179–81. doi:10.1016/S0361-9230(97)00336-5
170. Rhoads DE, DiRocco RJ, Osburn LD, Peterson NA, Raghupathy E. Stimulation of synaptosomal uptake of neurotransmitter amino acids by insulin: possible role of insulin as a neuromodulator. Biochem Biophys Res Commun (1984) 119(3):1198–204. doi:10.1016/0006-291X(84)90903-3
171. Park CR, Seeley RJ, Craft S, Woods SC. Intracerebroventricular insulin enhances memory in a passive-avoidance task. Physiol Behav (2000) 68(4):509–14. doi:10.1016/S0031-9384(99)00220-6
172. Zhao W, Chen H, Xu H, Moore E, Meiri N, Quon MJ, et al. Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats. J Biol Chem (1999) 274(49):34893–902. doi:10.1074/jbc.274.49.34893
173. Voll CL, Whishaw IQ, Auer RN. Postischemic insulin reduces spatial learning deficit following transient forebrain ischemia in rats. Stroke (1989) 20(5):646–51. doi:10.1161/01.STR.20.5.646
174. Lannert H, Hoyer S. Intracerebroventricular administration of streptozotocin causes long-term diminutions in learning and memory abilities and in cerebral energy metabolism in adult rats. Behav Neurosci (1998) 112(5):1199–208. doi:10.1037/0735-7044.112.5.1199
175. Kern W, Peters A, Fruehwald-Schultes B, Deininger E, Born J, Fehm HL. Improving influence of insulin on cognitive functions in humans. Neuroendocrinology (2001) 74(4):270–80. doi:10.1159/000054694
176. Feldman DE. Synaptic mechanisms for plasticity in neocortex. Annu Rev Neurosci (2009) 32:33–55. doi:10.1146/annurev.neuro.051508.135516
177. Song I, Huganir RL. Regulation of AMPA receptors during synaptic plasticity. Trends Neurosci (2002) 25(11):578–88. doi:10.1016/S0166-2236(02)02270-1
178. Huang CC, Lee CC, Hsu KS. An investigation into signal transduction mechanisms involved in insulin-induced long-term depression in the CA1 region of the hippocampus. J Neurochem (2004) 89(1):217–31. doi:10.1111/j.1471-4159.2003.02307.x
179. Ahmadian G, Ju W, Liu L, Wyszynski M, Lee SH, Dunah AW, et al. Tyrosine phosphorylation of GluR2 is required for insulin-stimulated AMPA receptor endocytosis and LTD. EMBO J (2004) 23(5):1040–50. doi:10.1038/sj.emboj.7600126
180. Choopani S, Moosavi M, Naghdi N. Involvement of nitric oxide in insulin induced memory improvement. Peptides (2008) 29(6):898–903. doi:10.1016/j.peptides.2008.01.005
181. Ramsey MM, Adams MM, Ariwodola OJ, Sonntag WE, Weiner JL. Functional characterization of des-IGF-1 action at excitatory synapses in the CA1 region of rat hippocampus. J Neurophysiol (2005) 94(1):247–54. doi:10.1152/jn.00768.2004
182. Le Grevès M, Zhou Q, Berg M, Le Grevès P, Fhölenhag K, Meyerson B, et al. Growth hormone replacement in hypophysectomized rats affects spatial performance and hippocampal levels of NMDA receptor subunit and PSD-95 gene transcript levels. Exp Brain Res (2006) 173(2):267–73. doi:10.1007/s00221-006-0438-2
183. Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging (2000) 21(3):383–421. doi:10.1016/S0197-4580(00)00124-X
184. Montine TJ, Kaye JA, Montine KS, McFarland L, Morrow JD, Quinn JF. Cerebrospinal fluid abeta42, tau, and f2-isoprostane concentrations in patients with Alzheimer disease, other dementias, and in age-matched controls. Arch Pathol Lab Med (2001) 125(4):510–2. doi:10.1043/0003-9985(2001)125<0510:CFATAF>2.0.CO;2
185. Sheng JG, Bora SH, Xu G, Borchelt DR, Price DL, Koliatsos VE. Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol Dis (2003) 14(1):133–45. doi:10.1016/S0969-9961(03)00069-X
186. Dandona P. Endothelium, inflammation, and diabetes. Curr Diab Rep (2002) 2(4):311–5. doi:10.1007/s11892-002-0019-0
187. Soop M, Duxbury H, Agwunobi AO, Gibson JM, Hopkins SJ, Childs C, et al. Euglycemic hyperinsulinemia augments the cytokine and endocrine responses to endotoxin in humans. Am J Physiol Endocrinol Metab (2002) 282(6):E1276–85. doi:10.1152/ajpendo.00535.2001
188. Fishel MA, Watson GS, Montine TJ, Wang Q, Green PS, Kulstad JJ, et al. Hyperinsulinemia provokes synchronous increases in central inflammation and beta-amyloid in normal adults. Arch Neurol (2005) 62(10):1539–44. doi:10.1001/archneur.62.10.noc50112
189. Johnston H, Boutin H, Allan SM. Assessing the contribution of inflammation in models of Alzheimer’s disease. Biochem Soc Trans (2011) 39(4):886–90. doi:10.1042/BST0390886
190. Rogers J, Mastroeni D, Leonard B, Joyce J, Grover A. Neuroinflammation in Alzheimer’s disease and Parkinson’s disease: are microglia pathogenic in either disorder? Int Rev Neurobiol (2007) 82:235–46. doi:10.1016/S0074-7742(07)82012-5
191. Pistell PJ, Morrison CD, Gupta S, Knight AG, Keller JN, Ingram DK, et al. Cognitive impairment following high fat diet consumption is associated with brain inflammation. J Neuroimmunol (2010) 219(1–2):25–32. doi:10.1016/j.jneuroim.2009.11.010
192. Neumann KF, Rojo L, Navarrete LP, Farias G, Reyes P, Maccioni RB. Insulin resistance and Alzheimer’s disease: molecular links & clinical implications. Curr Alzheimer Res (2008) 5(5):438–47. doi:10.2174/156720508785908919
193. Zhao M, Cribbs DH, Anderson AJ, Cummings BJ, Su JH, Wasserman AJ, et al. The induction of the TNFalpha death domain signaling pathway in Alzheimer’s disease brain. Neurochem Res (2003) 28(2):307–18. doi:10.1023/A:1022337519035
194. Dzienis-Straczkowska S, Straczkowski M, Szelachowska M, Stepien A, Kowalska I, Kinalska I. Soluble tumor necrosis factor-alpha receptors in young obese subjects with normal and impaired glucose tolerance. Diabetes Care (2003) 26(3):875–80. doi:10.2337/diacare.26.3.875
195. Bastard JP, Jardel C, Bruckert E, Vidal H, Hainque B. Variations in plasma soluble tumour necrosis factor receptors after diet-induced weight loss in obesity. Diabetes Obes Metab (2000) 2(5):323–5. doi:10.1046/j.1463-1326.2000.00090.x
196. Wrighten SA, Piroli GG, Grillo CA, Reagan LP. A look inside the diabetic brain: contributors to diabetes-induced brain aging. Biochim Biophys Acta (2009) 1792(5):444–53. doi:10.1016/j.bbadis.2008.10.013
197. Ghasemi R, Dargahi L, Haeri A, Moosavi M, Mohamed Z, Ahmadiani A. Brain insulin dysregulation: implication for neurological and neuropsychiatric disorders. Mol Neurobiol (2013) 47(3):1045–65. doi:10.1007/s12035-013-8404-z
198. Avila J, Wandosell F, Hernandez F. Role of glycogen synthase kinase-3 in Alzheimer’s disease pathogenesis and glycogen synthase kinase-3 inhibitors. Expert Rev Neurother (2010) 10(5):703–10. doi:10.1586/ern.10.40
199. Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature (1995) 378(6559):785–9. doi:10.1038/378785a0
200. Lee J, Kim MS. The role of GSK3 in glucose homeostasis and the development of insulin resistance. Diabetes Res Clin Pract (2007) 77(Suppl 1):S49–57. doi:10.1016/j.diabres.2007.01.033
201. Beurel E, Jope RS. Differential regulation of STAT family members by glycogen synthase kinase-3. J Biol Chem (2008) 283(32):21934–44. doi:10.1074/jbc.M802481200
202. Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol (2005) 6(8):777–84. doi:10.1038/ni1221
203. Agostinho P, Cunha RA, Oliveira C. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Curr Pharm Des (2010) 16(25):2766–78. doi:10.2174/138161210793176572
204. Najem D, Bamji-Mirza M, Chang N, Liu QY, Zhang W. Insulin resistance, neuroinflammation, and Alzheimer’s disease. Rev Neurosci (2014) 25(4):509–25. doi:10.1515/revneuro-2013-0050
205. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol (2010) 72:219–46. doi:10.1146/annurev-physiol-021909-135846
206. Lee YS, Li P, Huh JY, Hwang IJ, Lu M, Kim JI, et al. Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes (2011) 60(10):2474–83. doi:10.2337/db11-0194
207. Hotamisligil GS, Budavari A, Murray D, Spiegelman BM. Reduced tyrosine kinase activity of the insulin receptor in obesity-diabetes. Central role of tumor necrosis factor-alpha. J Clin Invest (1994) 94(4):1543–9.
208. Kalupahana NS, Moustaid-Moussa N. The renin-angiotensin system: a link between obesity, inflammation and insulin resistance. Obes Rev (2012) 13(2):136–49. doi:10.1111/j.1467-789X.2011.00942.x
209. Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest (2005) 115(5):1111–9. doi:10.1172/JCI25102
210. Tu YF, Tsai YS, Wang LW, Wu HC, Huang CC, Ho CJ. Overweight worsens apoptosis, neuroinflammation and blood-brain barrier damage after hypoxic ischemia in neonatal brain through JNK hyperactivation. J Neuroinflammation (2011) 8:40. doi:10.1186/1742-2094-8-40
211. Sartorius T, Lutz SZ, Hoene M, Waak J, Peter A, Weigert C, et al. Toll-like receptors 2 and 4 impair insulin-mediated brain activity by interleukin-6 and osteopontin and alter sleep architecture. FASEB J (2012) 26(5):1799–809. doi:10.1096/fj.11-191023
212. Arruda AP, Milanski M, Coope A, Torsoni AS, Ropelle E, Carvalho DP, et al. Low-grade hypothalamic inflammation leads to defective thermogenesis, insulin resistance, and impaired insulin secretion. Endocrinology (2011) 152(4):1314–26. doi:10.1210/en.2010-0659
213. Konner AC, Bruning JC. Toll-like receptors: linking inflammation to metabolism. Trends Endocrinol Metab (2011) 22(1):16–23. doi:10.1016/j.tem.2010.08.007
214. Frolich L, Blum-Degen D, Riederer P, Hoyer S. A disturbance in the neuronal insulin receptor signal transduction in sporadic Alzheimer’s disease. Ann N Y Acad Sci (1999) 893:290–3. doi:10.1111/j.1749-6632.1999.tb07839.x
215. Zhao WQ, Lacor PN, Chen H, Lambert MP, Quon MJ, Krafft GA, et al. Insulin receptor dysfunction impairs cellular clearance of neurotoxic oligomeric a{beta}. J Biol Chem (2009) 284(28):18742–53. doi:10.1074/jbc.M109.011015
216. Xie L, Helmerhorst E, Taddei K, Plewright B, Van Bronswijk W, Martins R. Alzheimer’s beta-amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci (2002) 22(10):RC221.
217. Lee HK, Kumar P, Fu Q, Rosen KM, Querfurth HW. The insulin/Akt signaling pathway is targeted by intracellular beta-amyloid. Mol Biol Cell (2009) 20(5):1533–44. doi:10.1091/mbc.E08-07-0777
218. Holscher C. Diabetes as a risk factor for Alzheimer’s disease: insulin signalling impairment in the brain as an alternative model of Alzheimer’s disease. Biochem Soc Trans (2011) 39(4):891–7. doi:10.1042/BST0390891
219. Bosco D, Fava A, Plastino M, Montalcini T, Pujia A. Possible implications of insulin resistance and glucose metabolism in Alzheimer’s disease pathogenesis. J Cell Mol Med (2011) 15(9):1807–21. doi:10.1111/j.1582-4934.2011.01318.x
220. Citron M. Alzheimer’s disease: treatments in discovery and development. Nat Neurosci (2002) 5(Suppl):1055–7. doi:10.1038/nn940
221. Correia SC, Santos RX, Carvalho C, Cardoso S, Candeias E, Santos MS, et al. Insulin signaling, glucose metabolism and mitochondria: major players in Alzheimer’s disease and diabetes interrelation. Brain Res (2012) 1441:64–78. doi:10.1016/j.brainres.2011.12.063
222. Efendic S, Luft R, Wajngot A. Aspects of the pathogenesis of type 2 diabetes. Endocr Rev (1984) 5(3):395–410. doi:10.1210/edrv-5-3-395
223. Chen Z, Zhong C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies. Prog Neurobiol (2013) 108:21–43. doi:10.1016/j.pneurobio.2013.06.004
224. Janson J, Ashley RH, Harrison D, McIntyre S, Butler PC. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes (1999) 48(3):491–8. doi:10.2337/diabetes.48.3.491
225. Luchsinger JA, Tang MX, Shea S, Mayeux R. Hyperinsulinemia and risk of Alzheimer disease. Neurology (2004) 63(7):1187–92. doi:10.1212/01.WNL.0000140292.04932.87
226. Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes (2004) 53(2):474–81. doi:10.2337/diabetes.53.2.474
227. Hokama M, Oka S, Leon J, Ninomiya T, Honda H, Sasaki K, et al. Altered expression of diabetes-related genes in Alzheimer’s disease brains: the Hisayama study. Cereb Cortex (2013) 24(9):2476–88. doi:10.1093/cercor/bht101
228. de la Monte SM, Wands JR. Alzheimer’s disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol (2008) 2(6):1101–13. doi:10.1177/193229680800200619
229. Correia SC, Santos RX, Perry G, Zhu X, Moreira PI, Smith MA. Insulin-resistant brain state: the culprit in sporadic Alzheimer’s disease? Ageing Res Rev (2011) 10(2):264–73. doi:10.1016/j.arr.2011.01.001
230. Baker LD, Cross DJ, Minoshima S, Belongia D, Watson GS, Craft S. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch Neurol (2011) 68(1):51–7. doi:10.1001/archneurol.2010.225
231. Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol (2006) 5(1):64–74. doi:10.1016/S1474-4422(05)70284-2
232. Kuusisto J, Koivisto K, Mykkanen L, Helkala EL, Vanhanen M, Hanninen T, et al. Association between features of the insulin resistance syndrome and Alzheimer’s disease independently of apolipoprotein E4 phenotype: cross sectional population based study. BMJ (1997) 315(7115):1045–9. doi:10.1136/bmj.315.7115.1045
233. Matsuzaki T, Sasaki K, Tanizaki Y, Hata J, Fujimi K, Matsui Y, et al. Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology (2010) 75(9):764–70. doi:10.1212/WNL.0b013e3181eee25f
234. Tolppanen AM, Lavikainen P, Solomon A, Kivipelto M, Uusitupa M, Soininen H, et al. History of medically treated diabetes and risk of Alzheimer disease in a nationwide case-control study. Diabetes Care (2013) 36(7):2015–9. doi:10.2337/dc12-1287
235. Kim B, Backus C, Oh S, Hayes JM, Feldman EL. Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology (2009) 150(12):5294–301. doi:10.1210/en.2009-0695
236. Ho L, Qin W, Pompl PN, Xiang Z, Wang J, Zhao Z, et al. Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J (2004) 18(7):902–4. doi:10.1096/fj.03-0978fje
237. Papon MA, El Khoury NB, Marcouiller F, Julien C, Morin F, Bretteville A, et al. Deregulation of protein phosphatase 2A and hyperphosphorylation of tau protein following onset of diabetes in NOD mice. Diabetes (2013) 62(2):609–17. doi:10.2337/db12-0187
238. Planel E, Tatebayashi Y, Miyasaka T, Liu L, Wang L, Herman M, et al. Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms. J Neurosci (2007) 27(50):13635–48. doi:10.1523/JNEUROSCI.3949-07.2007
239. Takeda S. Pathological interaction between diabetes mellitus and Alzheimer’s disease. Nihon Shinkei Seishin Yakurigaku Zasshi (2012) 32(5–6):239–44.
240. Vandal M, White PJ, Tremblay C, St-Amour I, Chevrier G, Emond V, et al. Insulin reverses the high-fat diet-induced increase in brain abeta and improves memory in an animal model of Alzheimer disease. Diabetes (2014). doi:10.2337/db14-0375
241. Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: the Rotterdam study. Neurology (1999) 53(9):1937–42. doi:10.1212/WNL.53.9.1937
242. Cukierman T, Gerstein HC, Williamson JD. Cognitive decline and dementia in diabetes – systematic overview of prospective observational studies. Diabetologia (2005) 48(12):2460–9. doi:10.1007/s00125-005-0023-4
243. Schoenle EJ, Schoenle D, Molinari L, Largo RH. Impaired intellectual development in children with type I diabetes: association with HbA(1c), age at diagnosis and sex. Diabetologia (2002) 45(1):108–14. doi:10.1007/s125-002-8250-6
244. Dahlquist G, Kallen B. School performance in children with type 1 diabetes – a population-based register study. Diabetologia (2007) 50(5):957–64. doi:10.1007/s00125-007-0615-2
245. Musen G, Lyoo IK, Sparks CR, Weinger K, Hwang J, Ryan CM, et al. Effects of type 1 diabetes on gray matter density as measured by voxel-based morphometry. Diabetes (2006) 55(2):326–33. doi:10.2337/diabetes.55.02.06.db05-0520
246. Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease – is this type 3 diabetes? J Alzheimers Dis (2005) 7(1):63–80.
247. Serbedzija P, Ishii DN. Insulin and insulin-like growth factor prevent brain atrophy and cognitive impairment in diabetic rats. Indian J Endocrinol Metab (2012) 16(Suppl 3):S601–10. doi:10.4103/2230-8210.105578
248. MacKnight C, Rockwood K, Awalt E, McDowell I. Diabetes mellitus and the risk of dementia, Alzheimer’s disease and vascular cognitive impairment in the Canadian study of health and aging. Dement Geriatr Cogn Disord (2002) 14(2):77–83. doi:10.1159/000064928
249. Hassing LB, Johansson B, Nilsson SE, Berg S, Pedersen NL, Gatz M, et al. Diabetes mellitus is a risk factor for vascular dementia, but not for Alzheimer’s disease: a population-based study of the oldest old. Int Psychogeriatr (2002) 14(3):239–48. doi:10.1017/S104161020200844X
250. de la Monte SM, Wands JR. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J Alzheimers Dis (2006) 9(2):167–81.
251. Mosconi L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease. FDG-PET studies in MCI and AD. Eur J Nucl Med Mol Imaging (2005) 32(4):486–510. doi:10.1007/s00259-005-1762-7
252. Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Ann Neurol (1997) 42(1):85–94. doi:10.1002/ana.410420114
253. Simpson IA, Chundu KR, Davies-Hill T, Honer WG, Davies P. Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann Neurol (1994) 35(5):546–51. doi:10.1002/ana.410350507
254. Liu F, Shi J, Tanimukai H, Gu J, Grundke-Iqbal I, Iqbal K, et al. Reduced O-GlcNAcylation links lower brain glucose metabolism and tau pathology in Alzheimer’s disease. Brain (2009) 132(Pt 7):1820–32. doi:10.1093/brain/awp099
255. Mastrogiacoma F, Bettendorff L, Grisar T, Kish SJ. Brain thiamine, its phosphate esters, and its metabolizing enzymes in Alzheimer’s disease. Ann Neurol (1996) 39(5):585–91. doi:10.1002/ana.410390507
256. Brunham LR, Kruit JK, Verchere CB, Hayden MR. Cholesterol in islet dysfunction and type 2 diabetes. J Clin Invest (2008) 118(2):403–8. doi:10.1172/JCI33296
257. Osborne AR, Pollock VV, Lagor WR, Ness GC. Identification of insulin-responsive regions in the HMG-CoA reductase promoter. Biochem Biophys Res Commun (2004) 318(4):814–8. doi:10.1016/j.bbrc.2004.04.105
258. Nelson TJ, Alkon DL. Insulin and cholesterol pathways in neuronal function, memory and neurodegeneration. Biochem Soc Trans (2005) 33(Pt 5):1033–6. doi:10.1042/BST20051033
259. Nelson TJ, Alkon DL. Oxidation of cholesterol by amyloid precursor protein and beta-amyloid peptide. J Biol Chem (2005) 280(8):7377–87. doi:10.1074/jbc.M409071200
260. Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer’s disease. Proc Natl Acad Sci U S A (2004) 101(29):10804–9. doi:10.1073/pnas.0400348101
261. Dias WB, Hart GW. O-GlcNAc modification in diabetes and Alzheimer’s disease. Mol Biosyst (2007) 3(11):766–72. doi:10.1039/b704905f
262. Liu Y, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Brain glucose transporters, O-GlcNAcylation and phosphorylation of tau in diabetes and Alzheimer’s disease. J Neurochem (2009) 111(1):242–9. doi:10.1111/j.1471-4159.2009.06320.x
263. Finder VH, Glockshuber R. Amyloid-beta aggregation. Neurodegener Dis (2007) 4(1):13–27. doi:10.1159/000100355
264. Li ZG, Zhang W, Sima AA. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes (2007) 56(7):1817–24. doi:10.2337/db07-0171
265. Zhao L, Teter B, Morihara T, Lim GP, Ambegaokar SS, Ubeda OJ, et al. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer’s disease intervention. J Neurosci (2004) 24(49):11120–6. doi:10.1523/JNEUROSCI.2860-04.2004
266. Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A (2003) 100(7):4162–7. doi:10.1073/pnas.0230450100
267. Johnson GV. Tau phosphorylation and proteolysis: insights and perspectives. J Alzheimers Dis (2006) 9(3 Suppl):243–50.
268. Rissman RA, Poon WW, Blurton-Jones M, Oddo S, Torp R, Vitek MP, et al. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest (2004) 114(1):121–30. doi:10.1172/JCI20640
269. Holmes C, Cotterell D. Role of infection in the pathogenesis of Alzheimer’s disease: implications for treatment. CNS Drugs (2009) 23(12):993–1002. doi:10.2165/11310910-000000000-00000
270. Alonso R, Pisa D, Marina AI, Morato E, Rabano A, Carrasco L. Fungal infection in patients with Alzheimer’s disease. J Alzheimers Dis (2014) 41(1):301–11. doi:10.3233/JAD-132681
271. Alonso R, Pisa D, Rabano A, Carrasco L. Alzheimer’s disease and disseminated mycoses. Eur J Clin Microbiol Infect Dis (2014) 33(7):1125–32. doi:10.1007/s10096-013-2045-z
272. Asai M, Hattori C, Iwata N, Saido TC, Sasagawa N, Szabo B, et al. The novel beta-secretase inhibitor KMI-429 reduces amyloid beta peptide production in amyloid precursor protein transgenic and wild-type mice. J Neurochem (2006) 96(2):533–40. doi:10.1111/j.1471-4159.2005.03576.x
273. Noble W, Planel E, Zehr C, Olm V, Meyerson J, Suleman F, et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci U S A (2005) 102(19):6990–5. doi:10.1073/pnas.0500466102
274. Blass JP, Gleason P, Brush D, DiPonte P, Thaler H. Thiamine and Alzheimer’s disease. A pilot study. Arch Neurol (1988) 45(8):833–5. doi:10.1001/archneur.1988.00520320019008
275. Nolan KA, Black RS, Sheu KF, Langberg J, Blass JP. A trial of thiamine in Alzheimer’s disease. Arch Neurol (1991) 48(1):81–3. doi:10.1001/archneur.1991.00530130093025
276. Radhakrishnan ML, Tidor B. Optimal drug cocktail design: methods for targeting molecular ensembles and insights from theoretical model systems. J Chem Inf Model (2008) 48(5):1055–73. doi:10.1021/ci700452r
277. Kickstein E, Krauss S, Thornhill P, Rutschow D, Zeller R, Sharkey J, et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc Natl Acad Sci U S A (2010) 107(50):21830–5. doi:10.1073/pnas.0912793107
278. Hsu CC, Wahlqvist ML, Lee MS, Tsai HN. Incidence of dementia is increased in type 2 diabetes and reduced by the use of sulfonylureas and metformin. J Alzheimers Dis (2011) 24(3):485–93. doi:10.3233/JAD-2011-101524
279. Kintscher U, Law RE. PPARgamma-mediated insulin sensitization: the importance of fat versus muscle. Am J Physiol Endocrinol Metab (2005) 288(2):E287–91. doi:10.1152/ajpendo.00440.2004
280. Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE. Inflammatory mechanisms in Alzheimer’s disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J Neurosci (2000) 20(2):558–67.
281. Delerive P, Fruchart JC, Staels B. Peroxisome proliferator-activated receptors in inflammation control. J Endocrinol (2001) 169(3):453–9. doi:10.1677/joe.0.1690453
282. Watson GS, Cholerton BA, Reger MA, Baker LD, Plymate SR, Asthana S, et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am J Geriatr Psychiatry (2005) 13(11):950–8. doi:10.1176/appi.ajgp.13.11.950
283. McClean PL, Parthsarathy V, Faivre E, Holscher C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J Neurosci (2011) 31(17):6587–94. doi:10.1523/JNEUROSCI.0529-11.2011
284. Holscher C. The role of GLP-1 in neuronal activity and neurodegeneration. Vitam Horm (2010) 84:331–54. doi:10.1016/B978-0-12-381517-0.00013-8
285. Holscher C. Incretin analogues that have been developed to treat type 2 diabetes hold promise as a novel treatment strategy for Alzheimer’s disease. Recent Pat CNS Drug Discov (2010) 5(2):109–17. doi:10.2174/157488910791213130
286. Li Y, Duffy KB, Ottinger MA, Ray B, Bailey JA, Holloway HW, et al. GLP-1 receptor stimulation reduces amyloid-beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer’s disease. J Alzheimers Dis (2010) 19(4):1205–19. doi:10.3233/JAD-2010-1314
287. Hamilton A, Patterson S, Porter D, Gault VA, Holscher C. Novel GLP-1 mimetics developed to treat type 2 diabetes promote progenitor cell proliferation in the brain. J Neurosci Res (2011) 89(4):481–9. doi:10.1002/jnr.22565
288. Craft S. Insulin resistance syndrome and Alzheimer’s disease: age- and obesity-related effects on memory, amyloid, and inflammation. Neurobiol Aging (2005) 26(Suppl 1):65–9. doi:10.1016/j.neurobiolaging.2005.08.021
289. Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A, et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol (2012) 69(1):29–38. doi:10.1001/archneurol.2011.233
290. Ott V, Benedict C, Schultes B, Born J, Hallschmid M. Intranasal administration of insulin to the brain impacts cognitive function and peripheral metabolism. Diabetes Obes Metab (2012) 14(3):214–21. doi:10.1111/j.1463-1326.2011.01490.x
291. Nakazato M. Development of the novel delivery system of GLP-1 administration for the treatment of diabetes mellitus. Nihon Rinsho (2011) 69(5):918–22.
Keywords: brain, insulin, receptors, biological actions, pathophysiological implications, central insulin resistance, type 2 diabetes, Alzheimer’s disease
Citation: Blázquez E, Velázquez E, Hurtado-Carneiro V and Ruiz-Albusac JM (2014) Insulin in the brain: its pathophysiological implications for states related with central insulin resistance, type 2 diabetes and alzheimer’s disease. Front. Endocrinol. 5:161. doi: 10.3389/fendo.2014.00161
Received: 14 July 2014; Accepted: 21 September 2014;
Published online: 09 October 2014.
Edited by:
Pierrette Gaudreau, Centre Hospitalier de l’Université de Montréal Research, CanadaReviewed by:
Cheng-Xin Gong, New York State Institute for Basic Research in Developmental Disabilities, USACopyright: © 2014 Blázquez, Velázquez, Hurtado-Carneiro and Ruiz-Albusac. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Enrique Blázquez, Departamento de Bioquímica y Biología Molecular III, Facultad de Medicina, Universidad Complutense de Madrid, Plaza Ramón y Cajal s/n, Madrid 28040, Spain e-mail:ZWJsYXpxdWV6QG1lZC51Y20uZXM=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.