 Sandra Merscher
Sandra Merscher Alessia Fornoni
Alessia Fornoni- Peggy and Harold Katz Family Drug Discovery Center and Division of Nephrology, Department of Medicine, University of Miami, Miami, FL, USA
Sphingolipids are components of the lipid rafts in plasma membranes, which are important for proper function of podocytes, a key element of the glomerular filtration barrier. Research revealed an essential role of sphingolipids and sphingolipid metabolites in glomerular disorders of genetic and non-genetic origin. The discovery that glucocerebrosides accumulate in Gaucher disease in glomerular cells and are associated with clinical proteinuria initiated intensive research into the function of other sphingolipids in glomerular disorders. The accumulation of sphingolipids in other genetic diseases including Tay–Sachs, Sandhoff, Fabry, hereditary inclusion body myopathy 2, Niemann–Pick, and nephrotic syndrome of the Finnish type and its implications with respect to glomerular pathology will be discussed. Similarly, sphingolipid accumulation occurs in glomerular diseases of non-genetic origin including diabetic kidney disease (DKD), HIV-associated nephropathy, focal segmental glomerulosclerosis (FSGS), and lupus nephritis. Sphingomyelin metabolites, such as ceramide, sphingosine, and sphingosine-1-phosphate have also gained tremendous interest. We recently described that sphingomyelin phosphodiesterase acid-like 3b (SMPDL3b) is expressed in podocytes where it modulates acid sphingomyelinase activity and acts as a master modulator of danger signaling. Decreased SMPDL3b expression in post-reperfusion kidney biopsies from transplant recipients with idiopathic FSGS correlates with the recurrence of proteinuria in patients and in experimental models of xenotransplantation. Increased SMPDL3b expression is associated with DKD. The consequences of differential SMPDL3b expression in podocytes in these diseases with respect to their pathogenesis will be discussed. Finally, the role of sphingolipids in the formation of lipid rafts in podocytes and their contribution to the maintenance of a functional slit diaphragm in the glomerulus will be discussed.
Sphingolipids, more precisely sphingomyelin, cerebroside, and cerebrosulfatide were first described in 1884 by Johann L. W. Thudichum who named them for their enigmatic (“Sphinx-like”) properties (1). They are important components of the lipid rafts in plasma membranes of mammalian cells and thus contribute to the proper function of cells. Within the kidney, the function and survival of major cell constituents of the glomerular filtration barrier, i.e., podocytes, heavily depends on the integrity of lipid rafts. Podocytes are differentiated cells of the kidney glomerulus consisting of a cell body, major processes, and foot processes (FP). The FP of podocytes are linked to the glomerular basement membrane with their actin cytoskeleton. Processes from neighboring podocytes form a characteristic interdigitating pattern that leaves filtration slits in between them. The latter are bridged by the slit diaphragm (SD) that together with the glomerular basement membrane and the fenestrated endothelium plays an important role in the selective permeability of the filtration barrier of the glomerulus (2–4). Integrity of this filtration barrier is important in order to prevent the loss of protein into the urine (proteinuria) and mutations in genes coding for SD proteins cause proteinuria-associated nephropathies (5–9). Research of the past two decades has revealed an essential role of sphingolipids in glomerular disorders with podocyte involvement.
This review will focus on different types of sphingolipids and sphingolipid metabolites that have been implicated in the pathogenesis of sphingolipidoses of genetic and non-genetic origin with podocyte involvement. We will also discuss sphingolipid signaling in podocytes and its influence on the actin cytoskeleton.
Biology of Sphingolipids
Sphingolipids are a diverse class of lipids with a varying degree of hydrophobic and hydrophilic properties. The hydrophobic region of sphingolipids consists of a longchain sphingoid base with generally 18 carbons, such as sphingosine, which is linked to a fatty acid via an amide bond. The hydrophilic region consists in the simplest case of a hydroxyl group in the case of ceramide. Fatty acids may vary in their composition but palmitic (C16:0) and stearic (C18:0) are most commonly present. More complex sphingolipids have sugar residues (glycosphingolipids) and phosphates as side chains (phosphosphingolipids) (Figure 1).
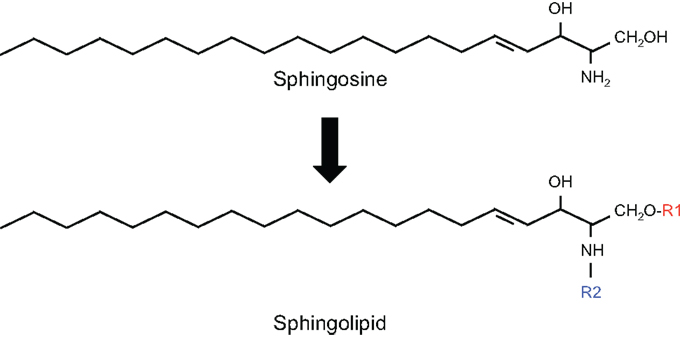
Figure 1. Structure of sphingolipids. In sphingolipids, the hydrophobic region consists of a longchain sphingoid base with generally 18 carbons, such as sphingosine, which is linked to the acyl group of a fatty acid via an amide bond (R2). The hydrophilic region (R1) consists in the simplest case of a hydroxyl group in the case of ceramide.
Glycosphingolipids such as GM1 and phosphosphingolipids such as sphingomyelins are commonly found in eukaryotes, in some prokaryotes, and in viruses as components of plasma membranes and membranes of organelles such as lysosomes, endosomes, endoplasmatic reticulum (ER), and others. The fluidity of the plasma membrane is tightly regulated by ordered packing of cholesterol between the phospholipid molecules, mainly sphingomyelin (SM) and thus sphingolipids have an important structural function. Sphingolipids are localized in the outer leaflet of the plasma membrane where they are asymmetrically distributed. Lipid rafts or raft-related caveolae are sphingomyelin-rich microdomains of the membrane, which are also enriched with cholesterol and membrane-associated proteins. The formation of lipid rafts is critical for proper cell function, protein–protein interactions, and signal transduction. For example, conversion of SM to ceramide, locally at the plasma membrane, by sphingomyelinases (SMases) will have a direct effect on the biophysical properties of the membrane and cell function as ceramide accumulation will lead to the displacement of cholesterol from the plasma membrane thus altering lipid rafts and signaling properties (10–12). Likewise, an interruption of raft-dependent cell signaling or even cell death can occur as a consequence of a cellular depletion of cholesterol that can be achieved by the use of cholesterol-depleting agents such as beta-cyclodextrin, methyl-beta-cyclodextrin, or 2-hydroxy-propyl-beta-cyclodextrin.
In recent years, it has become clear that, besides being integral part of membranes and having a structural function, sphingolipid metabolites such as ceramides, sphingosine, sphingosine-1-phosphate (S1P), and others play also important roles as second messengers in many biological processes including cell growth (13), differentiation, migration, and apoptosis (14). Complex sphingolipids were shown to interact with growth factor receptors, extracellular matrix, and neighboring cells (15). In addition, studies in yeast mutants revealed that sphingolipids have an important role in cellular stress responses as sphingolipid mutants yeast grew normally under usual culture conditions but were unable to survive if challenged or stressed (16).
Ceramide represents the centerpiece of the sphingolipid metabolic pathway reviewed in (17). Ceramide can be synthesized de novo starting with the condensation of l-serine and palmitoyl-CoA by serine palmitoyl transferase (SPT) to generate 3-ketodihydrosphinganine. The latter is then reduced by 3-ketosphinganine reductase to sphinganine, which in turn is N-acylated by ceramide synthetase (CS) to produce dihydroceramide. Finally, dihydroceramide is converted to ceramide by the enzyme dihydroceramide desaturase. Ceramide can also be generated by hydrolysis from sphingomyelin (SM) by SMases producing ceramide and phosphocholine. For sphingolipid biosynthesis, ceramide can be converted to sphingomyelin. This reaction is catalyzed by sphingomyelin synthetase (SMS), an enzyme that transfers the phosphocholine head group from phosphatidylcholine (PC) onto ceramide simultaneously generating diacylglycerol (DAG). Finally, ceramide can be generated by breakdown of glycosphingolipids and galactosylceramide to dihydroceramide and subsequent hydrolyzation (Figure 2).
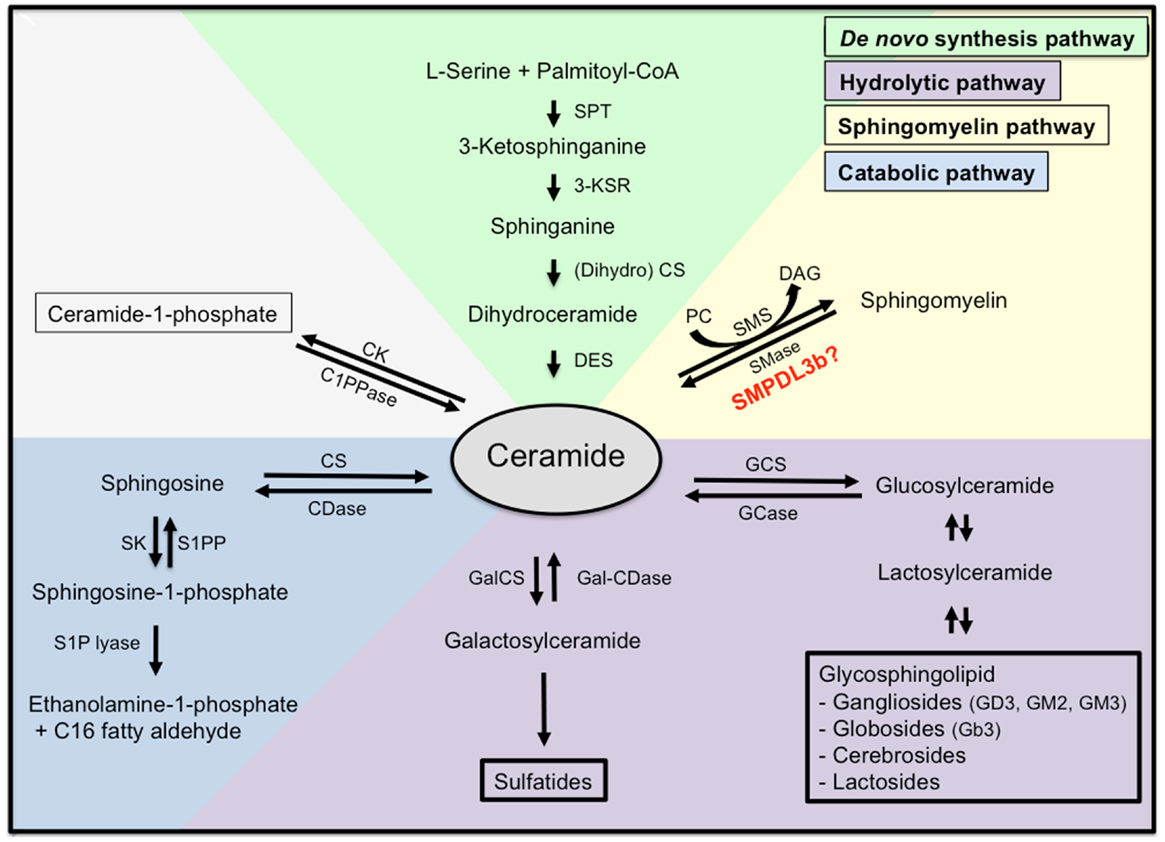
Figure 2. Sphingolipid metabolism. Ceramide is the centerpiece of the sphingolipid metabolic pathway and can be synthesized de novo from l-serine and palmitoyl-CoA (green), through hydrolysis of sphingomyelin (yellow), or through hydrolysis of glycosphingolipids and sulfatites (purple). Ceramide can also be synthesized from sphingomyelin through the action of sphingomyelinases, or from ceramide-1-phosphate through the action of ceramide-1-phosphate phosphatase. Finally, ceramide can be further catabolized (blue) to sphingosine and sphingosine-1-phosphate, which are biologically active metabolites and finally to ethanolamine-1-phosphate and C16 fatty aldehydes. SPT, serine palmitoyl transferase; 3-KSR, 3-ketosphinganine reductase; CS, ceramide synthetase; DES, dihydroceramide desaturase; SMase, sphingomyelinase; SMS, sphingomyelin synthetase; PC, phosphatidylcholine; DAG, diacylglycerol; C1PPase, ceramide-1-phosphate phosphatase; CK, ceramide kinase; CDase, ceramidase; CS, ceramide synthase; SK, spingosine kinase; S1PP, spingosine-1-phosphate phosphatase; GCS, glycosylceramide synthase; GCase, glycosylceramidase; GalCS, galactosylceramide synthase; Gal-CDase, galactosylceramidase.
Once ceramide is generated, it can accumulate in the cell or it may be further metabolized. Phosphorylation by ceramide kinase will generate ceramide-1-phosphate whereas deacylation by neutral or acid ceramidases will generate sphingosine, which can be phosphorylated by sphingosine kinase to yield S1P. Endproducts of the ceramide catabolic pathway are ethanolamine-1-phosphate and C16 fatty aldehydes, which are generated from S1P lyase from S1P.
Sphingolipids in Glomerular Diseases
In the past decade, it has become clear that there is an association between the accumulation of sphingolipids in the kidney and glomerular disease. The accumulation of sphingolipids in form of glycosphingolipids, ceramide, and ceramide metabolites has been described in several models of experimental and clinical nephropathy and is characteristic of some rare genetic glycosphingolipid (GSL) storage disorders. The observation that intracellular accumulation of sphingolipids in glomerular cells such as podocytes is also observed in the absence of genetic mutations and is associated with the development and progression of kidney disease suggests the existence of “acquired” sphingolipid storage disorders.
Most mammalian GSLs are synthesized from glucosylceramide (Figure 2) and are primarily present in the outer leaflet of the plasma membrane where they have important functions in mediating cell–cell interactions and modulating activities of proteins in their proximity. They are usually not uniformly distributed within the plasma membrane but cluster in lipid rafts (18, 19). Gangliosides are sialic acid-containing glycosphingolipids in which one or more N-acetlyneuraminic acids (NANA) is linked to the sugar chain and are essential components of plasma membranes (20). Gangliosides with one NANA include GM1, GM2, GM3, gangliosides with two NANAs are GD1a, GD1b, GD2, GD3, and the gangliosides GT1b and GQ1 are characterized by three and four NANAs, respectively. Gangliosides were first identified in nervous tissue but are also abundantly present in the kidney (21). GM1, GM2, GM3, GD1a, GD1b, GD2, GD3, GT1a, and GT1b are gangliosides present in normal rat glomeruli (22–24). GM3, GD3, and disialosyllactosylceramide (O-acetyl GD3) are the most abundant gangliosides present in kidney and 9-O-acetyl GD3 is a podocyte specific ganglioside (25, 26).
Sphingolipid Accumulation and Glomerular Disease of Genetic Origin
Sphingolipidoses are inherited disorders leading to defects in the sphingolipid metabolism resulting in the accumulation of excess glycosphingolipids and phosphosphingolipids. It is interesting to note that different metabolites will tend to accumulate in different cell types, therefore leading to highly variable clinical–pathological findings.
Gaucher disease type 1 (OMIM #230800) is the most prevalent GSL storage disease and is characterized by an accumulation of glucocerebroside (GlcCer) in the affected tissues and cells, mainly in red blood cells, liver, and spleen. Gaucher disease is of autosomal recessive inheritance and it is caused by mutations in the acid beta-glucosidase 1 (GBA1) gene on chromosome 1q22 in the vast majority of patients (Table 1). This gene encodes for the enzyme that cleaves the beta-glucosidic linkage of glycosylceramide and mutations in this gene lead to accumulation of GlcCer. However, in some patients GlcCer accumulation is due to a lack of saposin C. Enzyme replacement therapy with macrophage-targeted recombinant human glucocerebrosidase is successfully used to treat patients with Gaucher disease (27–29) and drugs that block GlcCer synthesis are currently being tested in clinical trials (30). Although the presence of renal pathology in Gaucher disease is rather rare, it has been described in some patients (31) and it is associated with the accumulation of GlcCer in form of Gaucher bodies in glomerular mesangial and endothelial cells and in interstitial cells of the kidney (31). Sphingolipid activator proteins (saposins A, B, C, and D) are glycoproteins that are encoded in tandem and are derived from a common precursor protein (prosaposin, PSAP). Saposins stimulate the degradation of GSLs by lysosomal enzymes. Defects in saposins are associated with the accumulation of lipids in affected tissues in lysosomal storage disorders. Humans with saposin C deficiency exhibit the clinical presentation of Gaucher-like disease (32). Combined deficiency of Saposin C and D in mice led to accumulation of GSLs and ceramide in brain and kidney due to decreased β-glucosidase activity (33).
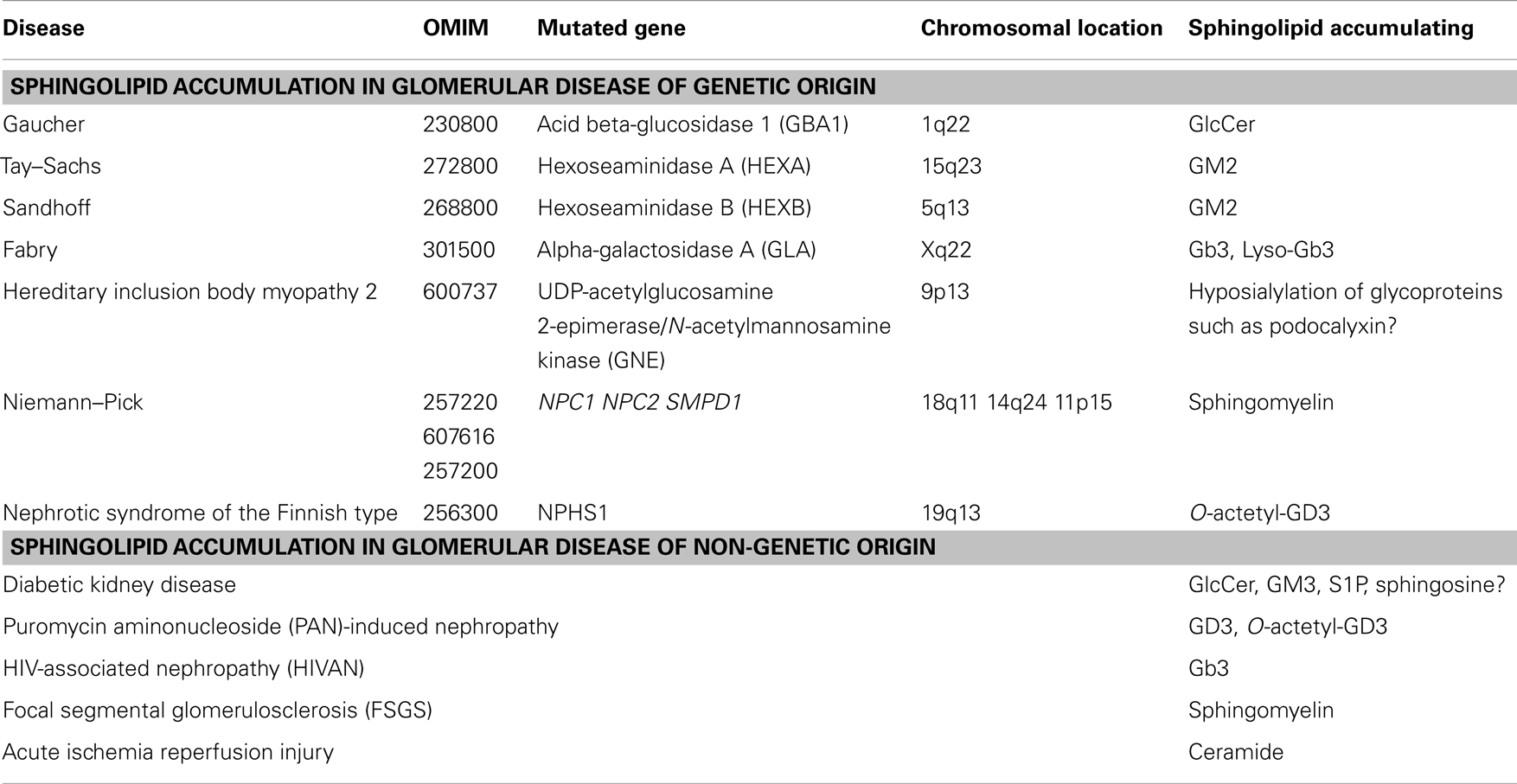
Table 1. Sphingolipid accumulation in glomerular diseases of genetic and non-genetic origin.
Tay–Sachs disease (OMIM #272800) is a genetic disorder with autosomal recessive inheritance that is caused by mutations in the alpha subunit of the hexoseaminidase A (HEXA) gene on chromosome 15q23. Sandhoff disease (OMIM #268800) is caused by mutations in the beta subunit of the hexosaminidase gene B (HEXB) on chromosome 5q13 (Table 1). Both disorders are characterized by an accumulation of the ganglioside GM2 in the affected tissues and are clinically indistinguishable from each other. GM2 accumulation occurs mainly in the brain and liver but has also been found in the kidney (34, 35). Unlike in humans, targeted inactivation of the Hexa and Hexb gene in mice revealed phenotypical differences between the two models. Whereas Hexa knockout mice showed GM2 accumulation in the brain and membranous cytoplasmatic bodies in neurons in the absence of neurological manifestations, Hexb knockout mice showed profound neurological disturbances. These differences, which are not found in patients with the two diseases, are due to differences in the ganglioside degradation pathway between humans and mice (36).
Fabry disease (OMIM #301500) is caused by mutations in the gene encoding alpha-galactosidase A (GLA) on chromosome Xq22, which leads to the systemic accumulation of globotriaoslyceramide (Gb3) (Table 1) and related glycosphingolipids in body fluids and affected tissues (37), mainly brain, heart, and kidney. Elevated levels of Gb3 are detected in plasma or urine of patients with Fabry disease (38, 39) and more recently, highly increased levels of globotriaosylsphingosine (lyso-Gb3) were also described (40, 41). In the kidney, Gb3 accumulation occurred mainly within lysosomal, ER, and nuclear markers of renal cells (42). The renal pathology observed in Fabry disease includes hypertrophic podocytes with foamy appearing vacuoles, characteristic inclusion bodies of glycolipids in podocytes (zebrabodies), and mesangial widening (43). Progressive podocyte injury due to accumulation of Gb3 and related glycosphingolipids was shown to be associated with albuminuria and foot process effacement. Thereby, podocyte Gb3 inclusion volume density and foot process effacement increased with age when compared with controls and correlated directly with proteinuria (44, 45). Enzyme replacement therapy using recombinant human α-GalA is the primary treatment for patients with Fabry disease and was shown to attenuate renal complications, halt the progression of renal pathology, and prevent renal failure in patients with Fabry disease (44, 46). Studies in the alpha GalA knockout mouse, a mouse model of Fabry disease, revealed reduced levels of glycosylceramide and ceramide in plasma, liver, spleen, kidney, and heart possibly a consequence of Gb3 accumulation. The observation that enzyme replacement therapy in this model normalized glycosylceramide levels possible via increased Gb3 degradation further supported the hypothesis that Gb3 accumulation contributes to the phenotype observed in these mice (46). Thereby, targeting of recombinant α-GalA requires the expression of endocytic receptors, megalin, sortilin, and mannose-6-phosphate receptor (M6PR), which are expressed in human glomerular podocytes (47). Interestingly, lentiviral knockdown of α-GalA in human podocytes led to intracellular Gb3 accumulation, which was associated with a loss of mTOR kinase activity and dysregulated autophagy suggesting a link between autophagy and glomerular injury in Fabry disease (48).
Hereditary inclusion body myopathy 2 (HIBM2) (OMIM #600737) is a genetic disorder with autosomal recessive inheritance that is caused by mutations in the gene encoding UDP-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) on chromosome 9p13 (Table 1). GNE is a key enzyme in the sialic acid biosynthetic pathway, which catalyzes the first two steps in NANA biosynthesis, which are main components of gangliosides (49). The disease is a progressive neuromuscular disorder but no renal disorders have been reported in patients with HIBM2. Interestingly, knockin mice carrying a homozygous M712T Gne/Mnk mutation died perinatally in the absence of myopathic features but were characterized by a renal phenotype. The renal pathology observed included glomerular hematuria, segmental splitting of the glomerular basement membrane, proteinuria, and podocytopathy, including effacement of podocyte FP, and reduced sialylation of the major podocyte sialoprotein, podocalyxin. ManNAc administration to homozygous knockin mice was associated with improved renal histology, increased sialylation of podocalyxin, and increased Gne/Mnk protein expression and Gne-epimerase activities (50). Likewise, mice with a V572L point mutation in the GNE kinase domain show no apparent myopathies or motor dysfunctions but exhibited renal impairment accompanied by massive albuminuria. Histologically, kidneys of the mutant mice showed enlarged glomeruli with mesangial matrix deposition, leading to glomerulosclerosis, and abnormal podocyte foot process morphologies. This phenotype was partially prevented by the administration of N-acetylneuraminic acid (Neu5Ac) to the mutant mice (51). These studies indicate that hyposylation of podocyte glycoproteins including podocalyxin may contribute to albuminuria, foot process effacement, and splitting of the glomerular basement membrane and that treatment with sialic acid may represent a new strategy to prevent or treat glomerular phenotypes associated with HIBM2.
Farber disease or Farber lipogranulomatosis (OMIM #228000) is a genetic disorder with autosomal recessive inheritance that is caused by mutations in the gene encoding acid ceramidase (ASAH1) on chromosome 8p22, the enzyme, which is responsible for the degradation of ceramide into sphingosine and free fatty acids (Table 1). Lipid accumulation is mainly seen in the joints, tissues, and the central nervous system, but also in liver, heart, and kidneys. Ceramide accumulation in the kidney leading to a particular phenotype of lipogranlulomatosis was described (52).
Niemann–Pick is a genetic disorder with autosomal recessive inheritance that is caused by mutations in the NPC1 gene on chromosome 18q11 (OMIM #257220), by mutations in the NPC2 gene on chromosome 14q24 (OMIM #607616), and by mutations in the sphingomyelin phosphodiesterase-1 (SMPD1) gene on chromosome 11p15 (OMIM #257200). Mutations in these genes lead to the accumulation of lipids in form of cholesterol (NPC1 and NPC2 mutations) and sphingomyelin (SMPD1 mutation) (Table 1). Acid sphingomyelinase (ASMase) deficiency in Niemann–Pick disease due to mutations in the SMPD1 gene leads accumulation of sphingomyelin in the affected tissues including the kidney. The occurrence of lipid-laden macrophages resembling foam cells was described in the bone marrow, liver, and kidney in patients with Niemann–Pick disease and in SMPD1 knockout mice (53, 54). Enzyme replacement therapy using recombinant human ASM in SMPD1 knockout mice led to significant improvements in the organs of the reticuloendothelial system but neurological deficits remained (55).
Nephrotic syndrome of the Finnish type (OMIM #256300) is a genetic disorder caused by homozygous or compound heterozygous mutations in the gene NPHS1 encoding for the SD protein nephrin (Table 1). Nephrotic syndrome of the Finnish type occurs in association with deposits of disialoganglioside O-acetyl GD3 (56). Accumulation of galactosylceramides, mainly sulfatides, was also described in nephrotic syndrome of non-genetic, idiopathic orgin (57). However, what causes the accumulation of O-acetyl GD3 in nephrotic syndrome remains unclear. Saposins do not seem to play an important role as mRNA expression in diseased kidneys was found normal and no mutations in the PSAP gene were found in cDNA clones (56). It has been suggested that tumor necrosis factor alpha (TNFα) and CD95 may play an important role in the pathogenesis of nephrotic syndrome. TNFα and CD95 were found significantly increased in patients with nephrotic syndrome (58). It was shown in lymphoid and myeloid tumor cells that accumulation of GD3 induced Fas (APO-1/CD95)-mediated apoptosis in a caspase-independent manner that was the consequence of the disruption of the mitochondrial transmembrane potential. This phenotype was prevented by pharmacological inhibition of GD3 synthesis (59). In other studies, an important role of membrane-associated ASMase was suggested in Fas-mediated apoptosis as activation of ASMase leads to the generation of free ceramide, which then can be converted to GD3 (60, 61). A similar pathway has been recently described in TNFα-mediated apoptosis (62, 63). Significant 9-O-acetyl GD3 accumulation, together with increases in GM2 and GM4 gangliosides in glomerular cells was also observed after low level and long term lead exposure and was associated with decreased apoptosis in glomerular cells suggesting that GD3-O acetylation could represent a new strategy to attenuate apoptosis in renal glomerular cells and contribute to cell survival as observed during lead exposure (64).
Sphingolipid Accumulation in Glomerular Disease of Non-Genetic Origin
Diabetic kidney disease (DKD) is the most common cause of end-stage renal disease and renal failure in the US and podocyte injury and the consequent loss of podocytes (podocytopenia) is an important feature of DKD in patients with type 1 and type 2 diabetes (65–69). Increased levels of sphingolipids such as glycosphingolipids (70), ceramide (71, 72), sphingosine (73), and sphinganine (72, 73) have been described in the plasma of patients with diabetes. More recently, it has become clear that the intracellular sphingolipid composition in podocytes and other cells of the kidney glomerulus may contribute to the pathogenesis and progression of the disease (Table 1).
Several studies investigated the effects of streptozotocin (STZ)-induced diabetes in rats on intracellular sphingolipid accumulation and its association with glomerular cell proliferation and glomerular hypertrophy. Accumulation of S1P was observed in rat glomeruli after 4 days of diabetes induction and was associated with an increase in neutral ceramidase and sphingosine kinase activity, the two enzymes involved in the conversion of ceramide to S1P (74). In another study, accumulation of GlcCer and GM3 occurred in the kidneys of rats 16 days after STZ-induced diabetes (75) whereas reduced GM3 and sialic acid content was detected in the glomeruli of rats 15 days after STZ-induced diabetes (76). Increased ceramide production due to increased expression of SPT, a key enzyme in the ceramide de novo synthesis pathway (Figure 2), was described in tubular epithelial cells and microvascular endothelial cells and was associated with increased apoptosis. Rapamycin treatment significantly reduced apoptosis and proteinuria indicating an important function of the Akt/mTOR pathway in STZ-induced DKD (77). We recently showed that the expression of sphingomyelin phosphodiesterase acid-like 3b (SMPDL3b) is increased in glomeruli from patients with DKD, in DKD sera treated human podocytes and in glomeruli of diabetic mice (db/db). Because SMPDL3b is a protein with homology to ASMase, we hypothesized that SMPDL3b may activate SM metabolic pathways leading to the accumulation of sphingolipids other than sphingomyelin. Increased SMPDL3b expression was associated with increased RhoA activity and apoptosis but prevented αVβ3 integrin activation via its interaction with soluble urokinase plasminogen activating receptor (suPAR) in human podocytes cultured in the presence of sera from patients with DKD and in db/db mice (78). Because ceramide, sphingosine, and S1P are known sphingolipid metabolites to accumulate in apoptotic cells, we determined the ceramide content in kidney cortexes of db/db mice and found ceramide levels to be decreased in kidney cortexes of these mice. We therefore concluded that increased SMPDL3b levels may lead to increased cellular sphingosine or S1P content in the kidneys of db/db mice as it is the case in glomerular mesangial and tubular cells in db/db mice (79, 80), and in adipocytes of ob/ob mice (81). Taken together, these studies indicate a possible link between sphingolipid accumulation in form of S1P, GlcCer, and GM3 and glomerular proliferation and hypertrophy in DKD whereas the accumulation of ceramide and other ceramide metabolites such as sphingosine may contribute to podocytopenia observed in DKD. Thus, targeting sphingolipids and sphingolipid metabolites may represent a new strategy to treat patients with DKD.
Puromycin aminonucleoside (PAN) induced nephropathy is a model for human minimal change disease. Following PAN injection in rats, significant decreases in kidney GD3 and O-acetyl GD3 occurred in a dose- and time-dependent manner and preceded the development of proteinuria indicating a possible causative effect (82). Because sialoglycoproteins contribute significantly to the negative charge of the glomerular filtration barrier, it seems possible that decreases of GD3 and O-acetyl GD3 contribute to decreases in the negative charge of the filtration barrier and to the changes in glomerular permeability observed in PAN-induced nephropathy (83). Likewise, it was shown that PAN treatment of human podocytes led to a loss of sialic acid which was accompanied by increased generation of superoxide anions, a phenotype that was prevented by sialic acid supplementation (84).
HIV-associated nephropathy (HIVAN) is the classic renal disease associated with HIV infection. HIV-1 infection of renal tubular and glomerular podocytes leads to dedifferentiation and increased proliferation of podocytes (85, 86). Because podoyctes do not express HIV-1 receptors, it has been suggested lipid raft mediated endocytosis may facilitate the viral entry (87) underlining an important role for sphingolipids in mediating viral entry into the host cell. Most of our understanding of the pathogenesis of HIVAN has come from the Tg26 transgenic mouse model in which the gag/pol-deleted HIV-1 provirus is expressed. Transgenic mice show glomerular epithelial cell dedifferentiation and proliferation that is associated with proteinuria and renal failure. Renal histology revealed focal segmental glomerulosclerosis (FSGS) and microcystic tubular dilatation, resembling human HIVAN (85, 88). Studies in human podocytes in culture and transgenic mice showed that stable expression of Nef was sufficient to induce increased proliferation and loss of contact inhibition (89–92). In addition, recent studies have shown a strong association between HIVAN and the APOL1 gene on human chromosome 22 (93) and, although not found in glomeruli, significant accumulation of Gb3 was found in renal tubular epithelial cells of HIV transgenic mice (94) (Table 1) indicating a possible role for (sphingo-) lipid metabolism in HIVAN.
Focal segmental glomerulosclerosis is a glomerular disease that is characterized by proteinuria and progression to end-stage renal disease. FSGS is the leading cause of nephrotic syndrome and the most common cause of primary glomerular disease in adults (95). Several mutations in genes coding for proteins that are expressed in podocytes have been shown to cause FSGS. We will focus in this paragraph on non-genetic forms of FSGS, mainly FSGS recurrence after transplantation, which occurs in about one third of patients (96–98) and on primary (idiopathic) forms of FSGS. We recently reported an important role of sphingomyelin-like phosphodiesterase 3b (SMPDL3b) gene in FSGS. Studying 41 patients at high risk for recurrent FSGS, we showed that the number of SMPDL3b-positive podocytes in post-reperfusion biopsies was decreased in patients who developed recurrent FSGS. As mentioned above, SMPDL3b is a protein with homology to ASMase and we hypothesized that decreased expression of SMPDL3b may lead to decreased ASMase activity and accumulation of sphingomyelin contributing to the pathogenesis of FSGS (Table 1). Indeed, we were able to show that human podocytes treated with the sera from patients with FSGS had decreased SMPDL3b expression and decreased ASMase activity. In addition, this was associated with actin cytoskeleton remodeling and apoptosis, a phenotype that was prevented by overexpression of SMPDL3b in podocytes or by treatment with rituximab, a monoclonal antibody directed against CD20 that we have found to also bind SMPDL3b in podocytes. The percentage of cells characterized by actin cytoskeleton remodeling in form of a loss of stress fibers correlated with proteinuria suggesting an important role of sphingomyelin in the pathogenesis of FSGS (99). Because decreased SMPDL3b expression in podocytes per se does not cause actin cytoskeleton remodeling and apoptosis, it seems possible that accumulation of sphingomyelin renders podocytes more susceptible to apoptosis and may act as a master modulator of danger signaling in podocytes. Supporting an important role of SMPDL3b in actin cytoskeleton remodeling and apoptosis, it was recently demonstrated that administration of rituximab to baboons after xeno-kidney transplantation from pigs delayed, but did not prevent, the post-transplant occurrence of proteinuria. As in the case of our study investigating the role of SMPDL3b in FSGS, this study demonstrated that rituximab was also able to prevent pig podocyte injury and prevented decreases in SMPDL3b expression in podoytes after exposure to naive baboon sera in association with preservation of cell viability (100). Finally, sequestration of plasma membrane lipids by cyclodextrin was shown to prevent suPAR-mediated αVβ3 integrin activation in podocytes (101), a pathway that may be causative of proteinuria in FSGS. Because circulating suPAR levels are elevated in FSGS patients, associate with decreased SMPDL3b expression, and suPAR-dependent αVβ3 integrin activation in podocytes (78, 99, 101, 102), whereas cyclodextrin protects podoctyes from injury in DKD where SMPDL3b expression is increased in podocytes (78, 103), we investigated if SMPDL3b expression modulates the podocyte injury phenotype in these two kidney diseases. We demonstrated that contrary to what is observed in FSGS, increased SMPDL3b expression in DKD prevented αVβ3 integrin activation via its interaction with suPAR and led to increased RhoA activity rendering podocytes more susceptible to apoptosis (78). These observations suggest that SMPDL3b and thus sphingomyelin or sphingomyelin catabolites are important modulators of podocyte function shifting suPAR-mediated podocyte injury from a migratory to an apoptotic phenotype. Therefore, modulating sphingolipids in podocytes may represent a new strategy to prevent or treat podocyte injury in FSGS and DKD.
Special Considerations Focusing on S1P and S1P Receptors in Renal Disease
Sphingosine-1-phosphate is generated by phosphorylation of sphingosine by sphingosine kinases (SPHK1, SPHK2) in response to various stimuli including growth factors, cytokines, G-protein-coupled receptor agonists, antigens, and others (Figure 2). Examples of factors that can transiently increase levels of S1P are TNFα and factors such as angiogenic growth factor, platelet derived growth factor (PDGF), and vascular endothelial growth factor (VEGF) all of which have been implicated in the pathogenesis of glomerular diseases. S1P signaling governs important cellular processes that determine cell fate. Thereby, extracellular S1P signaling is mediated via binding of S1P to G-protein-coupled-receptors (GPCRs). A family of five GPCRs termed S1P1–S1P5 [reviewed in Ref. (104, 105)] has been identified to date. Depending on the receptor subtype being expressed in the target cell, exogenous S1P can bind, and regulate a variety of important cellular functions including cell survival, cytoskeletal rearrangment, mitogenesis, cell differentiation, migration, and apoptosis. In the kidney, the receptors S1P1 (EDG1), S1P2 (EDG5), S1P3 (EDG3), and S1P5 (EDG8) are expressed in glomerular mesangial cells (106, 107) and whereas expression of S1P1, S1P2, S1P3, and S1P4, but not S1P5, was shown to be expressed in an immortalized mouse podocyte cell line (108). Increases in S1P synthesis mediated by sphingosine kinase, the use of S1P1 receptor agonists, such as FTY720 (an unselective S1P receptor agonist) and SEW2871 (a selective S1P1 receptor agonists) or by SPHK1 gene delivery were shown to protect from renal ischemia reperfusion injury (109–112), which is associated with increased ceramide expression (113–115), from DKD (108), and from various forms of glomerulonephritis (116–118) (Table 1). In addition, FTY720 and KRP-203, another S1P1 receptor agonist, have proven highly effective in preventing graft rejection in preclinical models of renal transplantation (119, 120). Whereas the activation of the S1P/S1P1 receptor pathway seems to be beneficial in the context of kidney disease, it was suggested that excessive S1P/S1P2 receptor pathway in renal tubular cells in DKD may play an important role in Rho kinase activation and renal fibrosis (80). Such mechanism could also explain activation of RhoA and increased apoptosis in podocytes in DKD as we previously described (78). In patients with lupus nephritis (LN), an inflammation of the kidney caused by systemic lupus erythematosus (SLE), a disease of the immune system, circulating S1P levels are increased (121). Likewise, S1P and dihydro-S1P levels in serum and kidney tissues from a mouse model of LN were elevated and treatment of these mice with a specific SPHK2 inhibitor, ABC294640 improved renal injury (122). It was suggested that in the case of renal inflammatory disease, extracellular S1P induces COX-2 expression via activation of S1P2, subsequently leading to Gi and p42/p44 MAPK-dependent signaling in renal mesangial cells. Although research of the past two decades has greatly advanced our understanding of the role of S1P and S1P/S1P receptor signaling in the pathogenesis and/or treatment of kidney diseases, more studies are needed to obtain a better and more detailed understanding of their physiological and pathophysiological significance in vivo. Certainly, targeting S1P/S1P receptor signaling pathways may represent a novel strategy to treat renal diseases.
Special Considerations Focusing on the Actin Cytoskeleton in Renal Disease
The kidney glomerulus is a highly specialized structure ensuring the selective ultrafiltration of plasma so that essential proteins are retained in the blood (3). Podocytes are glomerular epithelial cells consisting of a cell body, major processes, and FP. FP from neighboring cells are bridged by a 40-μm wide extracellular structure known as the SD (123, 124). Podocyte injury is an important feature of several renal diseases, including FSGS and DKD, in which independent of the underlying disease, a reorganization of the FP structure with fusion of filtration slits and apical displacement of the SD occurs (3, 125, 126). The SD is also required to control actin dynamics, response to injury, endocytosis, and cell viability. These observations make actin the common denominator in podocyte function and dysfunction (127, 128). Regulation of the podocyte actin cytoskeleton is therefore of critical importance for sustained function of the glomerular filter (129, 130). The connection of the actin cytoskeleton to the SD is mediated by several podocyte proteins such as CD2AP, Nephrin, ZO-1, and Podocin (131–134). Lipid rafts in podocytes are critical for the dynamic functional organization of the SD. Nephrin is partially associated with podocyte lipid rafts and co-immunoprecipitates with a podocyte specific 9-O-acetylated ganglioside. Injection of an antibody against the 9-O-acetylated ganglioside causes morphological changes of the filtration slits, resembling FP effacement (135) further underlining the important of intact lipid rafts and sphingolipids in the organization of the SD. Other sphingolipids such as S1P have also been implicated in cytoskeletal remodeling. S1P was shown to induce rapid reorganization of the actin cytoskeleton resulting in stress-fiber formation in 3T3 fibroblast, which was accompanied by transient tyrosine phosphorylation of focal adhesion kinase (FAK) and of the cytoskeleton-associated protein paxillin in association with RhoA activation in 3T3 fibroblasts (136). In renal mesangial cells, the serine/threonine protein kinase LIM kinase-1 (LIMK-1) was identified, which is involved in the regulation of cytoskeletal organization, as a ceramide-induced protein (137). Shiga toxin is a bacterial toxin that induces intracellular signals in a manner that is dependent on glycolipid-enriched membrane domains, or lipid rafts. Shiga-toxin-mediated intracellular signals were shown to induce cytoskeleton remodeling in renal tubular epithelial carcinoma cells (138). VEGF and its receptors, FLK1/KDR and FLT1, are key regulators of angiogenesis. However, recently a new role for FLT1, i.e., the soluble form of FLT, sFLT has been described in podocytes where it binds to the glycosphingolipid GM3 in lipid rafts, promoting adhesion, and rapid actin reorganization (139). Taken together, these studies underline the important function of sphingolipids in the formation of lipid rafts in podocytes thus contributing to the maintenance of a functional SD under physiological conditions.
Concluding Remarks
Sphingolipids play an important role in modulating podocyte function in glomerular disorders of genetic and non-genetic origin. Several genetic diseases are characterized by genetic mutations in genes that code for enzymes involved in the sphingolipid metabolism and are characterized by the accumulation of sphingolipids and sphingolipid metabolites in glomerular cells resulting in glomerular pathology. Thus, targeting sphingolipid metabolism in glomerular disease may prove beneficial in the treatment of proteinuric kidney diseases with glomerular involvement. Enzyme replacement therapy has proven to ameliorate disease progression in sphingolipid associated disorders of genetic origin such as Gaucher and Fabry disease. However, less is known about sphingolipid associated disorders of non-genetic origin. While ManNAc and rituximab are promising available therapeutic strategies for sphingolipid associated disorders of non-genetic origin, additional therapeutic strategies specifically targeting proteins such as SMPDL3b remain to be developed. Because sphingolipidoses of non-genetic origin seem to be more complex, additional research needs to be completed in order to elucidate the exact mechanisms by which sphingolipids cause injury to renal cells and thus contribute to the pathology of glomerular diseases.
Conflict of Interest Statement
Sandra Merscher and Alessia Fornoni are inventors on pending or issued patents aimed to diagnose or treat proteinuric renal diseases. They stand to gain royalties from their future commercialization. Alessia Fornoni is consultant for Hoffman-La Roche, Alexion, and Mesoblast on subject matters that are unrelated to this publication.
Acknowledgments
Sandra Merscher and Alessia Fornoni are supported by US National Institutes of Health (NIH, http://www.nih.gov/) (DK090316), by the Diabetes Research Institute Foundation, by the Nephcure Foundation, by the Peggy and Harold Katz Family Foundation, and by the Diabetic Complications Consortium (DiaComp). Sandra Merscher is supported by Stanley J. Glaser Foundation Research Award. Alessia Fornoni is also supported by the NIH/NIDDK (5U24DX076169) and by the National Center for Advancing Translational Sciences (1UL1TR000460).
References
1. McIlwain H. The second thudichum lecture. Cerebral isolates and neurochemical discovery. Biochem Soc Trans (1975) 3:579–90.
2. Pavenstadt H, Kriz W, Kretzler M. Cell biology of the glomerular podocyte. Physiol Rev (2003) 83:253–307. doi: 10.1152/physrev.00020.2002
3. Somlo S, Mundel P. Getting a foothold in nephrotic syndrome. Nat Genet (2000) 24:333–5. doi:10.1038/71724
4. Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol (2007) 17:428–37. doi:10.1016/j.tcb.2007.06.006
5. Kestilä M, Lenkkeri U, Männikkö M, Lamerdin J, McCready P, Putaala H, et al. Positionally cloned gene for a novel glomerular protein – nephrin – is mutated in congenital nephrotic syndrome. Mol Cell (1998) 1:575–82. doi:10.1016/S1097-2765(00)80057-X
6. Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, et al. NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet (2000) 24:349–54. doi:10.1038/74166
7. Li C, Ruotsalainen V, Tryggvason K, Shaw AS, Miner JH. CD2AP is expressed with nephrin in developing podocytes and is found widely in mature kidney and elsewhere. Am J Physiol Renal Physiol (2000) 279:F785–92.
8. Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science (2005) 308:1801–4. doi:10.1126/science.1106215
9. Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, et al. Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet (2000) 24:251–6. doi:10.1038/73456
10. Goni FM, Alonso A. Effects of ceramide and other simple sphingolipids on membrane lateral structure. Biochim Biophys Acta (2009) 1788:169–77. doi:10.1016/j.bbamem.2008.09.002
11. van Blitterswijk WJ, van der Luit AH, Veldman RJ, Verheij M, Borst J. Ceramide: second messenger or modulator of membrane structure and dynamics? Biochem J (2003) 369:199–211. doi:10.1042/BJ20021528
12. Zhang Y, Li X, Becker KA, Gulbins E. Ceramide-enriched membrane domains – structure and function. Biochim Biophys Acta (2009) 1788:178–83. doi:10.1016/j.bbamem.2008.07.030
13. Olivera A, Spiegel S. Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature (1993) 365:557–60. doi:10.1038/365557a0
14. Kaipia A, Chun SY, Eisenhauer K, Hsueh AJ. Tumor necrosis factor-alpha and its second messenger, ceramide, stimulate apoptosis in cultured ovarian follicles. Endocrinology (1996) 137:4864–70. doi:10.1210/endo.137.11.8895358
15. Merrill AH. Ceramide: a new lipid “second messenger”? Nutr Rev (1992) 50:78–80. doi:10.1111/j.1753-4887.1992.tb01273.x
16. Nagiec MM, Wells GB, Lester RL, Dickson RC. A suppressor gene that enables Saccharomyces cerevisiae to grow without making sphingolipids encodes a protein that resembles an Escherichia coli fatty acyltransferase. J Biol Chem (1993) 268:22156–63.
17. Merrill AH Jr. De novo sphingolipid biosynthesis: a necessary, but dangerous, pathway. J Biol Chem (2002) 277:25843–6. doi:10.1074/jbc.R200009200
18. Mondal S, Mukhopadhyay C. Molecular level investigation of organization in ternary lipid bilayer: a computational approach. Langmuir (2008) 24:10298–305. doi:10.1021/la8015589
19. Hall A, Rog T, Karttunen M, Vattulainen I. Role of glycolipids in lipid rafts: a view through atomistic molecular dynamics simulations with galactosylceramide. J Phys Chem B (2010) 114:7797–807. doi:10.1021/jp912175d
20. Hakomori S. Bifunctional role of glycosphingolipids. Modulators for transmembrane signaling and mediators for cellular interactions. J Biol Chem (1990) 265:18713–6.
21. Shayman JA, Radin NS. Structure and function of renal glycosphingolipids. Am J Physiol (1991) 260:F291–302.
22. Iwamori M, Shimomura J, Tsuyuhara S, Nagai Y. Gangliosides of various rat tissues: distribution of ganglio-N-tetraose-containing gangliosides and tissue-characteristic composition of gangliosides. J Biochem (1984) 95:761–70.
23. Saito M, Sugiyama K. Gangliosides in rat kidney: composition, distribution, and developmental changes. Arch Biochem Biophys (2001) 386:11–6. doi:10.1006/abbi.2000.2206
24. Hoon DS, Okun E, Neuwirth H, Morton DL, Irie RF. Aberrant expression of gangliosides in human renal cell carcinomas. J Urol (1993) 150:2013–8.
25. Reivinen J, Holthofer H, Miettinen A. A cell-type specific ganglioside of glomerular podocytes in rat kidney: an O-acetylated GD3. Kidney Int (1992) 42:624–31. doi:10.1038/ki.1992.327
26. Holthofer H, Reivinen J, Miettinen A. Nephron segment and cell-type specific expression of gangliosides in the developing and adult kidney. Kidney Int (1994) 45:123–30. doi:10.1038/ki.1994.14
27. Barton NW, Brady RO, Dambrosia JM, Di Bisceglie AM, Doppelt SH, Hill SC, et al. Replacement therapy for inherited enzyme deficiency – macrophage-targeted glucocerebrosidase for Gaucher’s disease. N Engl J Med (1991) 324:1464–70. doi:10.1056/NEJM199105233242104
28. Barton NW, Furbish FS, Murray GJ, Garfield M, Brady RO. Therapeutic response to intravenous infusions of glucocerebrosidase in a patient with Gaucher disease. Proc Natl Acad Sci U S A (1990) 87:1913–6. doi:10.1073/pnas.87.5.1913
29. Weinreb NJ, Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, et al. Effectiveness of enzyme replacement therapy in 1028 patients with type 1 Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher registry. Am J Med (2002) 113:112–9. doi:10.1016/S0002-9343(02)01150-6
30. Lukina E, Watman N, Arreguin EA, Banikazemi M, Dragosky M, Iastrebner M, et al. A phase 2 study of eliglustat tartrate (Genz-112638), an oral substrate reduction therapy for Gaucher disease type 1. Blood (2010) 116:893–9. doi:10.1182/blood-2010-03-273151
31. Chander PN, Nurse HM, Pirani CL. Renal involvement in adult Gaucher’s disease after splenectomy. Arch Pathol Lab Med (1979) 103:440–5.
32. Vaccaro AM, Motta M, Tatti M, Scarpa S, Masuelli L, Bhat M, et al. Saposin C mutations in Gaucher disease patients resulting in lysosomal lipid accumulation, saposin C deficiency, but normal prosaposin processing and sorting. Hum Mol Genet (2010) 19:2987–97. doi:10.1093/hmg/ddq204
33. Sun Y, Witte DP, Zamzow M, Ran H, Quinn B, Matsuda J, et al. Combined saposin C and D deficiencies in mice lead to a neuronopathic phenotype, glucosylceramide and alpha-hydroxy ceramide accumulation, and altered prosaposin trafficking. Hum Mol Genet (2007) 16:957–71. doi:10.1093/hmg/ddm040
34. Sandhoff K, Andreae U, Jatzkewitz H. Deficient hexozaminidase activity in an exceptional case of Tay-Sachs disease with additional storage of kidney globoside in visceral organs. Life Sci (1968) 7:283–8. doi:10.1016/0024-3205(68)90024-6
35. Tatematsu M, Imaida K, Ito N, Togari H, Suzuki Y, Ogiu T. Sandhoff disease. Acta Pathol Jpn (1981) 31:503–12.
36. Sango K, Yamanaka S, Hoffmann A, Okuda Y, Grinberg A, Westphal H, et al. Mouse models of Tay-Sachs and Sandhoff diseases differ in neurologic phenotype and ganglioside metabolism. Nat Genet (1995) 11:170–6. doi:10.1038/ng1095-170
37. Nance CS, Klein CJ, Banikazemi M, Dikman SH, Phelps RG, McArthur JC, et al. Later-onset Fabry disease: an adult variant presenting with the cramp-fasciculation syndrome. Arch Neurol (2006) 63:453–7. doi:10.1001/archneur.63.3.453
38. Krüger R, Bruns K, Grünhage S, Rossmann H, Reinke J, Beck M, et al. Determination of globotriaosylceramide in plasma and urine by mass spectrometry. Clin Chem Lab Med (2010) 48:189–98. doi:10.1515/CCLM.2010.048
39. Young E, Mills K, Morris P, Vellodi A, Lee P, Waldek S, et al. Is globotriaosylceramide a useful biomarker in Fabry disease? Acta Paediatr Suppl (2005) 94:51–4; discussion 37–58. doi:10.1111/j.1651-2227.2005.tb02112.x
40. Gold H, Mirzaian M, Dekker N, Joao Ferraz M, Lugtenburg J, Codée JD, et al. Quantification of globotriaosylsphingosine in plasma and urine of fabry patients by stable isotope ultraperformance liquid chromatography-tandem mass spectrometry. Clin Chem (2012) 59:547–56. doi:10.1373/clinchem.2012.192138
41. Auray-Blais C, Ntwari A, Clarke JT, Warnock DG, Oliveira JP, Young SP, et al. How well does urinary lyso-Gb3 function as a biomarker in Fabry disease? Clin Chim Acta (2010) 411:1906–14. doi:10.1016/j.cca.2010.07.038
42. Askari H, Kaneski CR, Semino-Mora C, Desai P, Ang A, Kleiner DE, et al. Cellular and tissue localization of globotriaosylceramide in Fabry disease. Virchows Arch (2007) 451:823–34. doi:10.1007/s00428-007-0468-6
43. Alroy J, Sabnis S, Kopp JB. Renal pathology in Fabry disease. J Am Soc Nephrol (2002) 13(Suppl 2):S134–8. doi:10.1097/01.ASN.0000016684.07368.75
44. Thurberg BL, Rennke H, Colvin RB, Dikman S, Gordon RE, Collins AB, et al. Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int (2002) 62:1933–46. doi:10.1046/j.1523-1755.2002.00675.x
45. Najafian B, Svarstad E, Bostad L, Gubler MC, Tøndel C, Whitley C, et al. Progressive podocyte injury and globotriaosylceramide (GL-3) accumulation in young patients with Fabry disease. Kidney Int (2011) 79:663–70. doi:10.1038/ki.2010.484
46. Quinta R, Rodrigues D, Assunção M, Macedo MF, Azevedo O, Cunha D, et al. Reduced glucosylceramide in the mouse model of Fabry disease: correction by successful enzyme replacement therapy. Gene (2014) 536:97–104. doi:10.1016/j.gene.2013.11.073
47. Prabakaran T, Nielsen R, Larsen JV, Sørensen SS, Feldt-Rasmussen U, Saleem MA, et al. Receptor-mediated endocytosis of alpha-galactosidase A in human podocytes in Fabry disease. PLoS One (2011) 6:e25065. doi:10.1371/journal.pone.0025065
48. Liebau MC, Braun F, Höpker K, Weitbrecht C, Bartels V, Müller RU, et al. Dysregulated autophagy contributes to podocyte damage in Fabry’s disease. PLoS One (2013) 8:e63506. doi:10.1371/journal.pone.0063506
49. Keppler OT, Hinderlich S, Langner J, Schwartz-Albiez R, Reutter W, Pawlita M. UDP-GlcNAc 2-epimerase: a regulator of cell surface sialylation. Science (1999) 284:1372–6. doi:10.1126/science.284.5418.1372
50. Galeano B, Klootwijk R, Manoli I, Sun M, Ciccone C, Darvish D, et al. Mutation in the key enzyme of sialic acid biosynthesis causes severe glomerular proteinuria and is rescued by N-acetylmannosamine. J Clin Invest (2007) 117:1585–94. doi:10.1172/JCI30954
51. Ito M, Sugihara K, Asaka T, Toyama T, Yoshihara T, Furuichi K, et al. Glycoprotein hyposialylation gives rise to a nephrotic-like syndrome that is prevented by sialic acid administration in GNE V572L point-mutant mice. PLoS One (2012) 7:e29873. doi:10.1371/journal.pone.0029873
52. Samuelsson K, Zetterstrom R. Ceramides in a patient with lipogranulomatosis (Farber’s disease) with chronic course. Scand J Clin Lab Invest (1971) 27:393–405. doi:10.3109/00365517109080235
53. Brière J, Calman F, Lageron A, Hinglais N, Emerit J, Bernard J. Adult Niemann-Pick disease: a 26 years follow-up. Report of a case with isolated visceral involvement, excess of tissue sphingomyelin, and deficient sphingomyelinase activity (author’s transl). Nouv Rev Fr Hematol Blood Cells (1976) 16:185–202.
54. Kuemmel TA, Thiele J, Schroeder R, Stoffel W. Pathology of visceral organs and bone marrow in an acid sphingomyelinase deficient knock-out mouse line, mimicking human Niemann-Pick disease type A. A light and electron microscopic study. Pathol Res Pract (1997) 193:663–71. doi:10.1016/S0344-0338(97)80025-8
55. Miranda SR, He X, Simonaro CM, Gatt S, Dagan A, Desnick RJ, et al. Infusion of recombinant human acid sphingomyelinase into Niemann-pick disease mice leads to visceral, but not neurological, correction of the pathophysiology. FASEB J (2000) 14:1988–95. doi:10.1096/fj.00-0014com
56. Haltia A, Solin ML, Jalanko H, Holmberg C, Miettinen A, Holthöfer H. Sphingolipid activator proteins in a human hereditary renal disease with deposition of disialogangliosides. Histochem J (1996) 28:681–7. doi:10.1007/BF02409005
57. Tamaoki A, Kikkawa Y. The role of sulfatides in autoimmunity in children with various glomerular disease. Nihon Jinzo Gakkai Shi (1991) 33:1045–54.
58. Twfeek DM, Zaki SM. Role of tumour necrosis factor alpha and CD95 as markers of apoptosis in pathogenesis of pediatrics renal diseases. Egypt J Immunol (2005) 12:155–65.
59. De Maria R, Lenti L, Malisan F, d’Agostino F, Tomassini B, Zeuner A, et al. Requirement for GD3 ganglioside in CD95- and ceramide-induced apoptosis. Science (1997) 277:1652–5. doi:10.1126/science.277.5332.1652
60. De Maria R, Rippo MR, Schuchman EH, Testi R. Acidic sphingomyelinase (ASM) is necessary for fas-induced GD3 ganglioside accumulation and efficient apoptosis of lymphoid cells. J Exp Med (1998) 187:897–902. doi:10.1084/jem.187.6.897
61. Cifone MG, De Maria R, Roncaioli P, Rippo MR, Azuma M, Lanier LL, et al. Apoptotic signaling through CD95 (Fas/Apo-1) activates an acidic sphingomyelinase. J Exp Med (1994) 180:1547–52. doi:10.1084/jem.180.4.1547
62. Omran OM, Saqr HE, Yates AJ. Molecular mechanisms of GD3-induced apoptosis in U-1242 MG glioma cells. Neurochem Res (2006) 31:1171–80. doi:10.1007/s11064-006-9147-2
63. Wiegmann K, Schwandner R, Krut O, Yeh WC, Mak TW, Krönke M. Requirement of FADD for tumor necrosis factor-induced activation of acid sphingomyelinase. J Biol Chem (1999) 274:5267–70. doi:10.1074/jbc.274.9.5267
64. Aguilar RP, Genta S, Sanchez S. Renal gangliosides are involved in lead intoxication. J Appl Toxicol (2008) 28:122–31. doi:10.1002/jat.1256
65. Meyer TW, Bennett PH, Nelson RG. Podocyte number predicts long-term urinary albumin excretion in Pima Indians with type II diabetes and microalbuminuria. Diabetologia (1999) 42:1341–4. doi:10.1007/s001250051447
66. Steffes MW, Schmidt D, McCrery R, Basgen JM. Glomerular cell number in normal subjects and in type 1 diabetic patients. Kidney Int (2001) 59:2104–13. doi:10.1046/j.1523-1755.2001.0590062104.x
67. Verzola D, Gandolfo MT, Ferrario F, Rastaldi MP, Villaggio B, Gianiorio F, et al. Apoptosis in the kidneys of patients with type II diabetic nephropathy. Kidney Int (2007) 72(10):1262–72. doi:10.1038/sj.ki.5002531
68. White KE, Bilous RW, Marshall SM, El Nahas M, Remuzzi G, Piras G, et al. Podocyte number in normotensive type 1 diabetic patients with albuminuria. Diabetes (2002) 51:3083–9. doi:10.2337/diabetes.51.10.3083
69. Pagtalunan ME, Miller PL, Jumping-Eagle S, Nelson RG, Myers BD, Rennke HG, et al. Podocyte loss and progressive glomerular injury in type II diabetes. J Clin Invest (1997) 99:342–8. doi:10.1172/JCI119163
70. Kremer GJ, Atzpodien W, Schnellbacher E. Plasma glycosphingolipids in diabetics and normals. Klin Wochenschr (1975) 53:637–8. doi:10.1007/BF01469685
71. Haus JM, Kashyap SR, Kasumov T, Zhang R, Kelly KR, Defronzo RA, et al. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes (2009) 58:337–43. doi:10.2337/db08-1228
72. Błachnio-Zabielska AU, Pułka M, Baranowski M, Nikołajuk A, Zabielski P, Górska M, et al. Ceramide metabolism is affected by obesity and diabetes in human adipose tissue. J Cell Physiol (2011) 227:550–7. doi:10.1002/jcp.22745
73. Gorska M, Dobrzyn A, Baranowski M. Concentrations of sphingosine and sphinganine in plasma of patients with type 2 diabetes. Med Sci Monit (2005) 11:CR35–8.
74. Geoffroy K, Troncy L, Wiernsperger N, Lagarde M, El Bawab S. Glomerular proliferation during early stages of diabetic nephropathy is associated with local increase of sphingosine-1-phosphate levels. FEBS Lett (2005) 579:1249–54. doi:10.1016/j.febslet.2004.12.094
75. Zador IZ, Deshmukh GD, Kunkel R, Johnson K, Radin NS, Shayman JA. A role for glycosphingolipid accumulation in the renal hypertrophy of streptozotocin-induced diabetes mellitus. J Clin Invest (1993) 91:797–803. doi:10.1172/JCI116299
76. Kwak DH, Rho YI, Kwon OD, Ahan SH, Song JH, Choo YK, et al. Decreases of ganglioside GM3 in streptozotocin-induced diabetic glomeruli of rats. Life Sci (2003) 72:1997–2006. doi:10.1016/S0024-3205(03)00090-0
77. Liu G, Han F, Yang Y, Xie Y, Jiang H, Mao Y, et al. Evaluation of sphingolipid metabolism in renal cortex of rats with streptozotocin-induced diabetes and the effects of rapamycin. Nephrol Dial Transplant (2011) 26:1493–502. doi:10.1093/ndt/gfq633
78. Yoo TH, Pedigo CE, Guzman J, Correa-Medina M, Wei C, Villarreal R, et al. SMPDL3b expression levels determine podocyte injury phenotypes in glomerular disease. J Am Soc Nephrol (2014) 25(4):737–44. doi:10.1681/ASN.2013111213
79. Brunskill EW, Potter SS. Changes in the gene expression programs of renal mesangial cells during diabetic nephropathy. BMC Nephrol (2012) 13:70. doi:10.1186/1471-2369-13-70
80. Ishizawa S, Takahashi-Fujigasaki J, Kanazawa Y, Matoba K, Kawanami D, Yokota T, et al. Sphingosine-1-phosphate induces differentiation of cultured renal tubular epithelial cells under Rho kinase activation via the S1P2 receptor. Clin Exp Nephrol (2014). doi:10.1007/s10157-014-0933-x
81. Samad F, Hester KD, Yang G, Hannun YA, Bielawski J. Altered adipose and plasma sphingolipid metabolism in obesity: a potential mechanism for cardiovascular and metabolic risk. Diabetes (2006) 55:2579–87. doi:10.2337/db06-0330
82. Holthofer H, Reivinen J, Solin ML, Haltia A, Miettinen A. Decrease of glomerular disialogangliosides in puromycin nephrosis of the rat. Am J Pathol (1996) 149:1009–15.
83. Andrews PM. Glomerular epithelial alterations resulting from sialic acid surface coat removal. Kidney Int (1979) 15:376–85. doi:10.1038/ki.1979.49
84. Pawluczyk IZ, Ghaderi Najafabadi M, Patel S, Desai P, Vashi D, Saleem MA, et al. Sialic acid attenuates puromycin aminonucleoside-induced desialylation and oxidative stress in human podocytes. Exp Cell Res (2013) 320:258–68. doi:10.1016/j.yexcr.2013.10.017
85. Barisoni L, Bruggeman LA, Mundel P, D’Agati VD, Klotman PE. HIV-1 induces renal epithelial dedifferentiation in a transgenic model of HIV-associated nephropathy. Kidney Int (2000) 58:173–81. doi:10.1046/j.1523-1755.2000.00152.x
86. Bruggeman LA, Dikman S, Meng C, Quaggin SE, Coffman TM, Klotman PE. Nephropathy in human immunodeficiency virus-1 transgenic mice is due to renal transgene expression. J Clin Invest (1997) 100:84–92. doi:10.1172/JCI119525
87. Mikulak J, Singhal PC. HIV-1 entry into human podocytes is mediated through lipid rafts. Kidney Int (2010) 77:72–3; author reply 73–4. doi:10.1038/ki.2009.366
88. Kopp JB, Klotman ME, Adler SH, Bruggeman LA, Dickie P, Marinos NJ, et al. Progressive glomerulosclerosis and enhanced renal accumulation of basement membrane components in mice transgenic for human immunodeficiency virus type 1 genes. Proc Natl Acad Sci U S A (1992) 89:1577–81. doi:10.1073/pnas.89.5.1577
89. Husain M, Gusella GL, Klotman ME, Gelman IH, Ross MD, Schwartz EJ, et al. HIV-1 Nef induces proliferation and anchorage-independent growth in podocytes. J Am Soc Nephrol (2002) 13:1806–15. doi:10.1097/01.ASN.0000019642.55998.69
90. Kajiyama W, Kopp JB, Marinos NJ, Klotman PE, Dickie P. Glomerulosclerosis and viral gene expression in HIV-transgenic mice: role of nef. Kidney Int (2000) 58:1148–59. doi:10.1046/j.1523-1755.2000.00271.x
91. Sunamoto M, Husain M, He JC, Schwartz EJ, Klotman PE. Critical role for Nef in HIV-1-induced podocyte dedifferentiation. Kidney Int (2003) 64:1695–701. doi:10.1046/j.1523-1755.2003.00283.x
92. Hanna Z, Priceputu E, Hu C, Vincent P, Jolicoeur P. HIV-1 Nef mutations abrogating downregulation of CD4 affect other Nef functions and show reduced pathogenicity in transgenic mice. Virology (2006) 346:40–52. doi:10.1016/j.virol.2005.10.010
93. Kopp JB, Nelson GW, Sampath K, Johnson RC, Genovese G, An P, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol (2011) 22:2129–37. doi:10.1681/ASN.2011040388
94. Liu XH, Lingwood CA, Ray PE. Recruitment of renal tubular epithelial cells expressing verotoxin-1 (Stx1) receptors in HIV-1 transgenic mice with renal disease. Kidney Int (1999) 55:554–61. doi:10.1046/j.1523-1755.1999.00278.x
95. Kitiyakara C, Eggers P, Kopp JB. Twenty-one-year trend in ESRD due to focal segmental glomerulosclerosis in the United States. Am J Kidney Dis (2004) 44:815–25. doi:10.1053/j.ajkd.2004.07.008
96. Baum MA. Outcomes after renal transplantation for FSGS in children. Pediatr Transplant (2004) 8:329–33. doi:10.1111/j.1399-3046.2004.00181.x
97. Hubsch H, Montané B, Abitbol C, Chandar J, Shariatmadar S, Ciancio G, et al. Recurrent focal glomerulosclerosis in pediatric renal allografts: the Miami experience. Pediatr Nephrol (2005) 20:210–6. doi:10.1007/s00467-004-1706-7
98. Senggutuvan P, Cameron JS, Hartley RB, Rigden S, Chantler C, Haycock G, et al. Recurrence of focal segmental glomerulosclerosis in transplanted kidneys: analysis of incidence and risk factors in 59 allografts. Pediatr Nephrol (1990) 4:21–8. doi:10.1007/BF00858431
99. Fornoni A, Sageshima J, Wei C, Merscher-Gomez S, Aguillon-Prada R, Jauregui AN, et al. Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Sci Transl Med (2011) 3:85ra46. doi:10.1126/scitranslmed.3002231
100. Tasaki M, Shimizu A, Hanekamp I, Torabi R, Villani V, Yamada K. Rituximab treatment prevents the early development of proteinuria following pig-to-baboon xeno-kidney transplantation. J Am Soc Nephrol (2014) 25:737–44. doi:10.1681/ASN.2013040363
101. Wei C, Möller CC, Altintas MM, Li J, Schwarz K, Zacchigna S, et al. Modification of kidney barrier function by the urokinase receptor. Nat Med (2008) 14:55–63. doi:10.1038/nm1696
102. Wei C, Trachtman H, Li J, Dong C, Friedman AL, Gassman JJ, et al. Circulating suPAR in two cohorts of primary FSGS. J Am Soc Nephrol (2012) 23:2051–9. doi:10.1681/ASN.2012030302
103. Merscher-Gomez S, Guzman J, Pedigo CE, Lehto M, Aguillon-Prada R, Mendez A, et al. Cyclodextrin protects podocytes in diabetic kidney disease. Diabetes (2013) 62(11):3817–27. doi:10.2337/db13-0399
104. Pyne NJ, Long JS, Lee SC, Loveridge C, Gillies L, Pyne S. New aspects of sphingosine 1-phosphate signaling in mammalian cells. Adv Enzyme Regul (2009) 49:214–21. doi:10.1016/j.advenzreg.2009.01.011
105. Rosen H, Goetzl EJ. Sphingosine 1-phosphate and its receptors: an autocrine and paracrine network. Nat Rev Immunol (2005) 5:560–70. doi:10.1038/nri1650
106. Imasawa T, Kitamura H, Ohkawa R, Satoh Y, Miyashita A, Yatomi Y. Unbalanced expression of sphingosine 1-phosphate receptors in diabetic nephropathy. Exp Toxicol Pathol (2010) 62:53–60. doi:10.1016/j.etp.2009.02.068
107. Koch A, Völzke A, Puff B, Blankenbach K, Meyer Zu Heringdorf D, Huwiler A, et al. PPARgamma agonists upregulate sphingosine 1-phosphate (S1P) receptor 1 expression, which in turn reduces S1P-induced [Ca(2+)]i increases in renal mesangial cells. Biochim Biophys Acta (2013) 1831:1634–43. doi:10.1016/j.bbalip.2013.07.011
108. Awad AS, Rouse MD, Khutsishvili K, Huang L, Bolton WK, Lynch KR, et al. Chronic sphingosine 1-phosphate 1 receptor activation attenuates early-stage diabetic nephropathy independent of lymphocytes. Kidney Int (2011) 79:1090–8. doi:10.1038/ki.2010.544
109. Park SW, Kim M, Chen SW, Brown KM, D’Agati VD, Lee HT. Sphinganine-1-phosphate protects kidney and liver after hepatic ischemia and reperfusion in mice through S1P1 receptor activation. Lab Invest (2010) 90:1209–24. doi:10.1038/labinvest.2010.102
110. Kim M, Park SW, Pitson SM, Lee HT. Isoflurane protects human kidney proximal tubule cells against necrosis via sphingosine kinase and sphingosine-1-phosphate generation. Am J Nephrol (2010) 31:353–62. doi:10.1159/000298339
111. Awad AS, Ye H, Huang L, Li L, Foss FW Jr, Macdonald TL, et al. Selective sphingosine 1-phosphate 1 receptor activation reduces ischemia-reperfusion injury in mouse kidney. Am J Physiol Renal Physiol (2006) 290:F1516–24. doi:10.1152/ajprenal.00311.2005
112. Park SW, Kim M, D’Agati VD, Lee HT. Sphingosine kinase 1 protects against renal ischemia-reperfusion injury in mice by sphingosine-1-phosphate1 receptor activation. Kidney Int (2011) 80:1315–27. doi:10.1038/ki.2011.281
113. Zager RA, Conrad S, Lochhead K, Sweeney EA, Igarashi Y, Burkhart KM. Altered sphingomyelinase and ceramide expression in the setting of ischemic and nephrotoxic acute renal failure. Kidney Int (1998) 53:573–82. doi:10.1046/j.1523-1755.1998.00772.x
114. Kalhorn T, Zager RA. Renal cortical ceramide patterns during ischemic and toxic injury: assessments by HPLC-mass spectrometry. Am J Physiol (1999) 277:F723–33.
115. Zager RA, Iwata M, Conrad DS, Burkhart KM, Igarashi Y. Altered ceramide and sphingosine expression during the induction phase of ischemic acute renal failure. Kidney Int (1997) 52:60–70. doi:10.1038/ki.1997.304
116. Peters H, Martini S, Wang Y, Shimizu F, Kawachi H, Krämer S, et al. Selective lymphocyte inhibition by FTY720 slows the progressive course of chronic anti-thy 1 glomerulosclerosis. Kidney Int (2004) 66:1434–43. doi:10.1111/j.1523-1755.2004.00906.x
117. Martini S, Krämer S, Loof T, Wang-Rosenke Y, Daig U, Budde K, et al. S1P modulator FTY720 limits matrix expansion in acute anti-thy1 mesangioproliferative glomerulonephritis. Am J Physiol Renal Physiol (2007) 292:F1761–70. doi:10.1152/ajprenal.00253.2006
118. Schwalm S, Pfeilschifter J, Huwiler A. Targeting the sphingosine kinase/sphingosine 1-phosphate pathway to treat chronic inflammatory kidney diseases. Basic Clin Pharmacol Toxicol (2014) 114:44–9. doi:10.1111/bcpt.12103
119. Ferguson R. FTY720 immunomodulation: optimism for improved transplant regimens. Transplant Proc (2004) 36:549S–53S. doi:10.1016/j.transproceed.2004.01.061
120. Fujishiro J, Kudou S, Iwai S, Takahashi M, Hakamata Y, Kinoshita M, et al. Use of sphingosine-1-phosphate 1 receptor agonist, KRP-203, in combination with a subtherapeutic dose of cyclosporine A for rat renal transplantation. Transplantation (2006) 82:804–12. doi:10.1097/01.tp.0000232687.78242.cd
121. Watson L, Tullus K, Marks SD, Holt RC, Pilkington C, Beresford MW. Increased serum concentration of sphingosine-1-phosphate in juvenile-onset systemic lupus erythematosus. J Clin Immunol (2012) 32:1019–25. doi:10.1007/s10875-012-9710-3
122. Snider AJ, Ruiz P, Obeid LM, Oates JC. Inhibition of sphingosine kinase-2 in a murine model of lupus nephritis. PLoS One (2013) 8:e53521. doi:10.1371/journal.pone.0053521
123. Ruotsalainen V, Ljungberg P, Wartiovaara J, Lenkkeri U, Kestilä M, Jalanko H, et al. Nephrin is specifically located at the slit diaphragm of glomerular podocytes. Proc Natl Acad Sci U S A (1999) 96:7962–7. doi:10.1073/pnas.96.14.7962
124. Tryggvason K. Unraveling the mechanisms of glomerular ultrafiltration: nephrin, a key component of the slit diaphragm. J Am Soc Nephrol (1999) 10:2440–5.
125. Saleem MA, O’Hare MJ, Reiser J, Coward RJ, Inward CD, Farren T, et al. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol (2002) 13:630–8.
126. Smoyer WE, Mundel P. Regulation of podocyte structure during the development of nephrotic syndrome. J Mol Med (Berl) (1998) 76:172–83. doi:10.1007/s001090050206
127. Kerjaschki D. Caught flat-footed: podocyte damage and the molecular bases of focal glomerulosclerosis. J Clin Invest (2001) 108:1583–7. doi:10.1172/JCI200114629
128. Asanuma K, Mundel P. The role of podocytes in glomerular pathobiology. Clin Exp Nephrol (2003) 7:255–9. doi:10.1007/s10157-003-0259-6
129. Ichimura K, Kurihara H, Sakai T. Actin filament organization of foot processes in rat podocytes. J Histochem Cytochem (2003) 51:1589–600. doi:10.1177/002215540305101203
130. Ichimura K, Kurihara H, Sakai T. Actin filament organization of foot processes in vertebrate glomerular podocytes. Cell Tissue Res (2007) 329:541–57. doi:10.1007/s00441-007-0440-4
131. Yuan H, Takeuchi E, Salant DJ. Podocyte slit-diaphragm protein nephrin is linked to the actin cytoskeleton. Am J Physiol Renal Physiol (2002) 282:F585–91. doi:10.1152/ajprenal.00290.2001
132. Huber TB, Simons M, Hartleben B, Sernetz L, Schmidts M, Gundlach E, et al. Molecular basis of the functional podocin-nephrin complex: mutations in the NPHS2 gene disrupt nephrin targeting to lipid raft microdomains. Hum Mol Genet (2003) 12:3397–405. doi:10.1093/hmg/ddg360
133. Huber TB, Kottgen M, Schilling B, Walz G, Benzing T. Interaction with podocin facilitates nephrin signaling. J Biol Chem (2001) 276:41543–6. doi:10.1074/jbc.C100452200
134. Fanning AS, Ma TY, Anderson JM. Isolation and functional characterization of the actin binding region in the tight junction protein ZO-1. FASEB J (2002) 16:1835–7. doi:10.1096/fj.02-0121fje
135. Simons M, Schwarz K, Kriz W, Miettinen A, Reiser J, Mundel P, et al. Involvement of lipid rafts in nephrin phosphorylation and organization of the glomerular slit diaphragm. Am J Pathol (2001) 159:1069–77. doi:10.1016/S0002-9440(10)61782-8
136. Wang F, Nobes CD, Hall A, Spiegel S. Sphingosine 1-phosphate stimulates rho-mediated tyrosine phosphorylation of focal adhesion kinase and paxillin in Swiss 3T3 fibroblasts. Biochem J (1997) 324(Pt 2):481–8.
137. Shabahang S, Liu YH, Huwiler A, Pfeilschifter J. Identification of the LIM kinase-1 as a ceramide-regulated gene in renal mesangial cells. Biochem Biophys Res Commun (2002) 298:408–13. doi:10.1016/S0006-291X(02)02483-X
138. Takenouchi H, Kiyokawa N, Taguchi T, Matsui J, Katagiri YU, Okita H, et al. Shiga toxin binding to globotriaosyl ceramide induces intracellular signals that mediate cytoskeleton remodeling in human renal carcinoma-derived cells. J Cell Sci (2004) 117:3911–22. doi:10.1242/jcs.01246
Keywords: sphingolipid, podocyte, kidney disease, glomerular disease, S1P, ASMase, SMPDL3b, ceramide
Citation: Merscher S and Fornoni A (2014) Podocyte pathology and nephropathy – sphingolipids in glomerular diseases. Front. Endocrinol. 5:127. doi: 10.3389/fendo.2014.00127
Received: 08 May 2014; Paper pending published: 09 June 2014;
Accepted: 14 July 2014; Published online: 30 July 2014.
Edited by:
Gavin Iain Welsh, University of Bristol, UKReviewed by:
Tadeusz Pawelczyk, Medical University of Gdansk, PolandKarlhans Endlich, Universitätsmedizin Greifswald, Germany
Copyright: © 2014 Merscher and Fornoni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sandra Merscher, Peggy and Harold Katz Family Drug Discovery Center and Division of Nephrology, Department of Medicine, University of Miami, 1580 NW 10th Avenue, Batchelor Building, Room 628, Miami, FL 33136, USA e-mail:c21lcnNjaGVyQG1lZC5taWFtaS5lZHU=;
Alessia Fornoni, Peggy and Harold Katz Family Drug Discovery Center and Division of Nephrology, Department of Medicine, University of Miami, 1580 NW 10th Avenue, Batchelor Building, Room 633, Miami, FL 33136, USA e-mail:YWZvcm5vbmlAbWVkLm1pYW1pLmVkdQ==