 Jayanthi Maniam
Jayanthi Maniam Christopher Antoniadis
Christopher Antoniadis Margaret J. Morris
Margaret J. Morris
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 13 May 2014
Sec. Neuroendocrine Science
Volume 5 - 2014 | https://doi.org/10.3389/fendo.2014.00073
This article is part of the Research Topic Programming HPA-axis by early life experience: Mechanisms of stress susceptibility and adaptation View all 14 articles
Stress activates the hypothalamic–pituitary–adrenal (HPA) axis, which then modulates the degree of adaptation and response to a later stressor. It is known that early-life stress can impact on later health but less is known about how early-life stress impairs HPA axis activity, contributing to maladaptation of the stress–response system. Early-life stress exposure (either prenatally or in the early postnatal period) can impact developmental pathways resulting in lasting structural and regulatory changes that predispose to adulthood disease. Epidemiological, clinical, and experimental studies have demonstrated that early-life stress produces long term hyper-responsiveness to stress with exaggerated circulating glucocorticoids, and enhanced anxiety and depression-like behaviors. Recently, evidence has emerged on early-life stress-induced metabolic derangements, for example hyperinsulinemia and altered insulin sensitivity on exposure to a high energy diet later in life. This draws our attention to the contribution of later environment to disease vulnerability. Early-life stress can alter the expression of genes in peripheral tissues, such as the glucocorticoid receptor and 11-beta hydroxysteroid dehydrogenase (11β-HSD1). We propose that interactions between altered HPA axis activity and liver 11β-HSD1 modulates both tissue and circulating glucocorticoid availability, with adverse metabolic consequences. This review discusses the potential mechanisms underlying early-life stress-induced maladaptation of the HPA axis, and its subsequent effects on energy utilization and expenditure. The effects of positive later environments as a means of ameliorating early-life stress-induced health deficits, and proposed mechanisms underpinning the interaction between early-life stress and subsequent detrimental environmental exposures on metabolic risk will be outlined. Limitations in current methodology linking early-life stress and later health outcomes will also be addressed.
Stress can be defined as any condition including an adverse environment, experience, or perceived threat to alter an organism’s homeostasis, which elicits a physiological response involving both peripheral and central systems via the release of glucocorticoids from the adrenal cortex through activation of the hypothalamic–pituitary–adrenal (HPA) axis (1). Glucocorticoids serve as the critical end product of the HPA axis and negative feedback through glucocorticoid–glucocorticoid receptor binding in the hippocampus promotes adaptation and recovery from stress (2). Activity of the HPA axis plays a critical role in restoring homeostasis following imminent or acute stressor exposure (1). In contrast, recurrent or persistent activation of the HPA axis and the autonomic nervous system are associated with adverse health outcomes (3). Individuals respond differently to stressors, which can reflect a wide range of adversities in life from major events to daily conflicts and pressures. Increased levels of glucocorticoids interfere with energy utilization and modify metabolic hormones such as insulin and glucose, which are key regulators of energy metabolism (4–6). The elicited response affects multiple physiological systems including neuroendocrine, autonomic, and the immune system and is an established risk factor for the development of disease (7–10).
There is longstanding recognition of the impact of stress during critical early developmental periods such as childhood and the consequential association with adverse mental health outcomes and changes in brain development (11–13), however, less is known regarding how this dysfunction may confer increased metabolic disease risk. Emerging epidemiological evidence demonstrates that adverse early-life stress-induced dysregulation of the HPA axis and increases vulnerability for metabolic disorders. In particular, exposure to an adverse environment during prenatal and postnatal periods, such as lack of nutrition or starvation during war, traumatic experiences including childhood physical or sexual abuse, neglect, adverse parenting or medical trauma has been demonstrated to be one of the major risk factors contributing to the development metabolic disorders including insulin resistance, type 2 diabetes mellitus (T2DM), and hyperlipidemia (14–16).
While studies aimed at exploring underlying mechanisms are difficult to achieve in humans, animal studies have shed some light into the effects of early-life stress induced during the prenatal and postnatal periods on metabolic hormones and on peripheral tissues involved in glucose/insulin and lipid metabolism (17–24). In addition, the morphology of the pancreas is also affected by prenatal stress, with reduced beta-cell numbers (25). Thus, stress during early development may incur permanent alterations in morphology and function of key peripheral organs involved in metabolism of insulin, glucose and lipids, and these changes suggest programing effects of early-life stress (26).
This review will unravel how early-life stress induces metabolic derangements; an area that is less well explored. Despite the emergence of human data in the field, mechanisms elucidating the association between stress and adverse metabolic outcomes are lacking. Animal studies permit closer investigation of the stress system and through various manipulations, allow the mechanisms underlying effects of stress on metabolism to be explored. Of particular focus is exploring outcomes of stress during the perinatal and early-life period for later life metabolic outcomes.
As direct examination of the prospective effects of early-life stress is not feasible in humans this had led to the development of numerous animal models to explore the question of whether early-life stress can affect risk for later life metabolic deficit. Animal models of early-life stress allow for controlled environmental manipulation throughout developmental periods and later life. With necessary caution these models can assist our understanding of the link between developmental and environment experiences and the conferred later life metabolic disease vulnerabilities. Inherent differences in human and rodent biological maturation and neuroendocrine development must be considered in study design and translation to human health. Humans give birth to mature young; with the final trimester of pregnancy being a period of rapid brain development (27, 28). Rodent offspring are born relatively immature with maximal growth phase initiated early in the postnatal period. Despite greater maturity of human offspring at the time of birth, development is far from complete with changes in neurological processes, synaptogenesis, synaptic pruning, and plastic changes in key functional areas including the hippocampus occurring until late adolescence (29, 30). The environment has been shown to impact this ongoing development beyond gestation. Children physically healthy at the time of birth who were abused in early life were shown to have reduced brain volume, correlated to age of abuse onset, and duration of the stress (31). Imposed stressors during rodent gestational or early postnatal life are suggested to model the period of gestation, early postnatal, or infancy in humans.
Humans and rodents have a vital dependence on adequate nourishment and care to ensure normal development. This altricial nature and vulnerable perinatal period across both species means that models of maternal care can provide insight into how early support and sensitivity to offspring needs can impact development. Changes in maternal care have been shown to impact rodent development with mother–offspring interactions such as licking and feeding providing critical input for normal neurobiological development and HPA axis function (32, 33). Adequate maternal contact during this period assists in maintaining rodents in their early-life hypo-responsive stress state and adverse experience through physical or psychological means or synthetic glucocorticoid administration can permanently alter HPA axis function (34, 35).
Three popular paradigms of postnatal early-life stress are maternal separation for varied periods of time from 15 min to 8 h, maternal deprivation (absence of the dam for a more extended period) and provision of only limiting nesting material. The maternal separation model has been studied for more than five decades, and is demonstrated in both mice and rats to affect the HPA axis, and behavioral responses in mothers and offspring in a sex dependant manner (36–39). Maternal deprivation is another common form of early-life stress which has been studied over decades (40–44). Twenty-four hours of maternal deprivation in neonatal rodents induced marked elevations in plasma corticosterone and decreases in plasma glucose and leptin, amongst other hormonal and neurotrophic factor changes (40, 43). This model, however appears to reflect a severe nutritional insult rather than psychological disturbances. Therefore, whether maternal deprivation represents adverse early experience in the human context is debatable. The limiting nesting paradigm, a more recently developed model of early-life stress, has been described elsewhere (35) but, briefly, involves limiting the dam’s available material for nest building, resulting in rudimentary and inadequate housing for offspring, a chronic stressor for the dam and pups. Limited nesting (LN) material, attempts to enhance commonalities to the human condition of childhood neglect and maternal stress in which the mother is present, yet care is abnormal and fragmented. Notably, the LN model has been demonstrated to impair HPA axis activity and induce behavioral deficits both in the dam and pups (35, 45, 46).
Recently, early-life stress models in non-human primates have demonstrated a lasting health impact following adverse early experiences. Altering secure attachment relationships during early life in Rhesus macaques significantly elevated prevalence and frequency of illness and increased bodyweight trajectory (47). Stress induced via variable foraging demand in bonnet macaques during lactation affected the metabolic profile of their offspring; a decreased glucose disposal rate was observed during hyperinsulinemic-euglycemic clamps in those exposed to early-life stress (48).
Retrospective and observational studies in humans demonstrate that early-life experiences can influence later life metabolic outcome. Environmental changes during gestation and the early postnatal period may impact development and predict metabolic health outcomes. Manipulations of the early environment can affect the developing nervous system, shaping individual differences in physiological and behavioral responses to environmental insults. For example, disruption of the mother–infant relationship during early life contributes to neuroendocrine, neurochemical, and behavioral changes in the adult organism (49). Experience of adversity during early life and adolescence in the form of parental conflict or parental separation increased the risk of later life obesity (50). Similarly, experience of a range of early-life stressors was positively correlated with increased adult BMI in men, independently of mental health condition (51). These adverse early-life experiences are associated with persisting changes in HPA axis function in adult life with changes in the normal dynamics of the stress system and its end-point hormone cortisol (52). Commonly, a flattened cortisol circadian rhythm and hypo- or hyper-responsiveness to future novel stressors is observed (53, 54).
Barker’s theory postulates that low birth weight predicts increased disease risk later in life, including metabolic vulnerability (55). Given this association, identifying factors that influence gestational growth, and ultimately determine birth weight is important. Reduced birth weight and preterm birth have both been associated with psychosocial stress exposure during pregnancy (56–58). Having a low birth weight baby (<2500 g) was associated with stress-related psychiatric illness in pregnant mothers, such as melancholic depression (59). Prenatal psychological stress such as experiencing bereavement during pregnancy led to an increased risk of developing T2DM in their children during adulthood (60). Degree of social support during pregnancy has also been associated with birth weight (61). Other psychological stressors such as financial, relationship problems, and illness during pregnancy led to elevated glucose, insulin, and C-peptide levels during glucose tolerance test in their children aged 25 (62). Nutritional stress during the neonatal period has also been shown to adversely impact offspring health outcome, as explored below.
Famine exposure during pregnancy is a chronic early-life stress that is also known to affect birth weight and increase risk for metabolic disorders later in life. A well-documented period providing epidemiological evidence linking early-life adversity and health outcomes is the 1944–1945 Dutch Famine (see Table 1). A cohort of 741 subjects exposed to the Dutch Famine prenatally had a reduced birth weight, yet at adulthood these subjects had increased body weight, BMI, fasting proinsulin levels, and glucose intolerance (63). A report of 7557 women exposed to the Dutch famine showed increased risk for T2DM development in their offspring (64). Exposure to the Chinese famine during the 1960s showed similar adverse outcomes for offspring, with women having higher prevalence of metabolic disorders such as diabetes, hypertriglyceridemia, and hypertension (65). A cross-sectional study of subjects exposed to the Biafran famine during the Nigerian Civil War showed derangements in their metabolic profile during adulthood with increased risk for diabetes in both middle-aged men and non-pregnant women (66) (see Table 1). These data highlight that adverse early experiences, whether psychological or nutritional in nature during vulnerable developmental periods can impact offspring insulin and glucose metabolism during adulthood (Table 1).
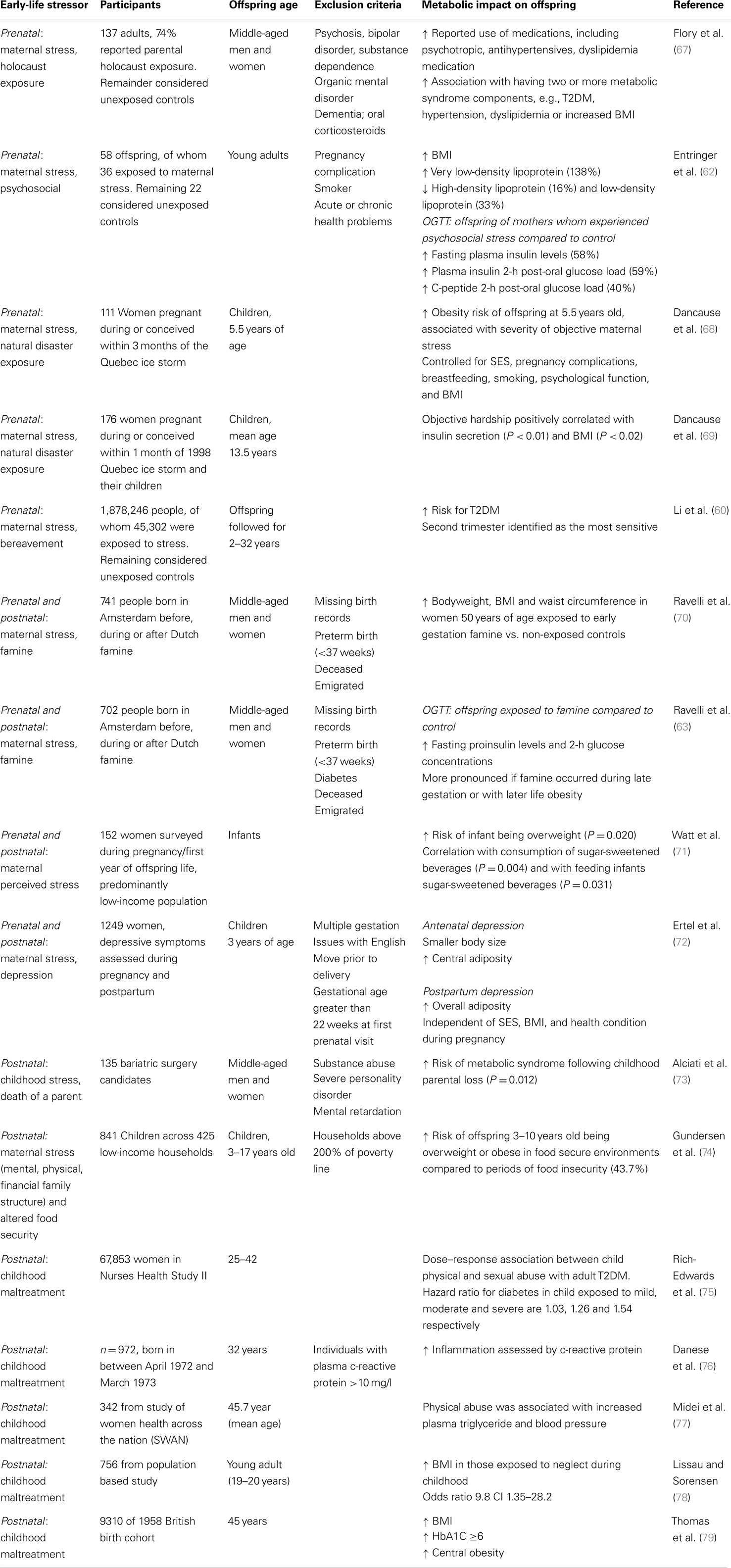
Table 1. Human early-life stress studies exploring metabolic outcomes.
Despite increasingly available epidemiological evidence, the mechanisms driving stress effects to lower birth weight remain largely unknown. Changes in HPA axis function in individuals of low birth weight have been identified. A group of adult men born with low birth weight had increased HPA axis responsiveness, which was shown to be associated with metabolic risk factors including blood pressure and increased triglycerides (80). Thus epidemiological evidence generally suggests early-life stress induces perturbed HPA axis function and alters neuroendocrine axis responsiveness. The increasing evidence demonstrating the risk of early-life stress and later metabolic disorders emphasizes the need to explore the mechanisms underlying this association. Targets for intervention, whether through pharmacological means or through lifestyle modification, need to be identified to reduce this identified risk. It is important to note that not all individuals exposed to adverse early environments develop metabolic deficits later in life; these differences may relate to genetic makeup and the environment to which the individual is exposed throughout life.
Early-life stress in combination with a sub-optimal later environment, such as a sedentary lifestyle, increased consumption of high energy food or persistent adulthood stress may alter the risk for developing metabolic disorders throughout life. Humans and rodents are able to habituate and adapt to the environmental conditions to which they are exposed (81–83). This adaptation occurs in prediction of exposure to similar situations in the future, and under normal conditions is of significant value, improving future resilience and coping in these situations (see Figure 2). However, it is suggested that if adaptation is inadequate, maladaptive, or future environment differs from the programed phenotype, there is increased disease susceptibility (84–87). Determining the long term consequences of early-life stress-induced changes in neuronal structure, and hormonal and nutritional status across different environments is an important public health concern. Although prevention or mitigation of early-life stress is the ideal, if through modulating the later environment (e.g., providing a positive environment) disease risk can be attenuated, important targets for intervention can be identified.
Stress throughout adulthood negatively influences lifestyle choices that are risk factors for metabolic disease, such as alterations in eating behavior, intake of high-fat food (88), drug addiction (89, 90), and reduced physical activity levels (91). Given this, it is critical to determine whether early-life stress can have a lasting influence on adult behavioral choices. Indeed, parental care factors play a key role in developing health behaviors and outcomes in children (92, 93). Parental behaviors are often imitated by children, thus a push to improve attitudes in parents, whether through reducing stress, improving eating attitudes or increasing physical activity may foster improved health status in successive generations. Recent research showed a direct association between activity levels of parents and their preschool-aged children (94). Additionally, the behavioral profile that results from early-life stress heightens the risk for impaired psychosocial function and psychiatric disorders, and this may independently influence metabolic disease risk. Future human studies must focus on these lifestyle factors during critical exposure periods and throughout adult life.
Prenatal exposure to adverse environments such as famine have been associated with poorer lifestyle choices including smoking incidence (66), HFD consumption, and reduced physical activity (95, 96). As explored, a possible consequence of gestational stress is preterm birth or low birth weight. Total physical activity levels (97) and non-conditioning leisure time physical activity levels (98) were not influenced by low birth weight. Despite this, adults born preterm with very low birth weight (i.e., less than 1500 g) had reduced smoking rates, yet were less likely to engage in leisure time physical activity with reduced energy expenditure than normal gestational birth controls (98). Further, healthy children aged 5–8 years old who were born prematurely were shown to have reduced physical ability to normal gestational age controls (99). Physical activity has been shown to improve health outcomes following premature birth, with 4 weeks of passive range of motion and compression exercises in premature infants shown to increase bone mineral density. The authors suggested that early exercise programs may improve physical fitness in later life (100). The unequivocal benefits of physical activity should be considered as a therapeutic tool, with activity levels inversely associated with metabolic syndrome (101). In low birth weight individuals, physical activity has been shown to modestly attenuate the association between low birth weight and insulin resistance, as assessed by HOMA-IR (102). Although this finding has not been replicated by other studies (97, 103), physical activity was identified as a better predictor of HOMA-IR comparative to birth weight a possible target for intervention (103).
There are limited data exploring the metabolic outcomes of postnatal stress and environment interaction. Early-life stress has been associated with later life addiction problems, including drug and food addiction (104). Adult women who were abused as children were significantly heavier and had a marked increase in food addiction risk (105). Early-life maternal stress has been positively correlated with the risk of the infant being overweight in an environment of high food security (see Table 1). Infants exposed to maternal stress during periods of food insecurity showed no significant overweight risk (74) (see Figure 2). One limitation of this study is the low socioeconomic status of the participants, as this is an individual risk factor for the development of childhood and adult obesity, amongst other health issues. Similar to these findings, the number of parental stressors and degree of perceived stress were positively associated with child obesity and increased fast-food consumption (106). Children and adolescents are susceptible to stress, and given the association between perceived stress and increased food consumption frequency and palatability (107–109), environmental factors such as palatable food access and physical activity may play a mediating factor between human early-life stress and poor metabolic outcomes.
Physical activity may facilitate a protective role against childhood stressors, with more active children showing reduced salivary cortisol response to stressful situations, reflective of lower HPA axis reactivity (110). In addition to alterations in stress mediators, childhood maltreatment has been independently associated with elevated levels of inflammatory markers in adulthood (76, 111) (see Table 1). Increased inflammation following early-life stress is clinically relevant and may provide an important causal link between adverse early-life experience and adulthood metabolic risk. The benefits of physical activity on immune function and inflammation are well-established, with reductions in inflammatory biomarkers associated with disease (112).
Given the global epidemic of metabolic disease, examining the growing body of animal evidence linking prenatal stress with impaired metabolic profile when exposed to energy dense, palatable diets is imperative. The effects of stress during pregnancy on metabolic consequences in later life are well characterized relative to stress during the early postnatal period (22). Identifying how programed changes during early life adversely impact susceptibility to later life nutrition is critical and could provide targets for intervention (see Figure 2). Two common models of prenatal stress exposure involve subjecting dams to a single stressor during gestation, for instance restraint stress. Alternatively, dams can be exposed to a combination of variable stressors such as restraint, air puff startle, forced swimming, starvation, and bright light exposure (113).
Evidence to support a programed resilience to later life metabolic deficits following prenatal early-life stress has been observed in some animal studies. For example, independent of body weight, adult female offspring of stressed dams consuming standard laboratory chow, had a lower insulin area under the curve (AUC) during an oral glucose tolerance test (OGTT) (114), reduced plasma insulin, and improvements in HOMA-IR (24) compared to offspring of non-stressed dams. This improvement in insulin sensitivity is suggestive of a prenatal early-life stress programing of glucose handling in insulin sensitive tissues. However, it is unknown if these improvements in insulin sensitivity following prenatal early-life stress will be sustained as animals age. Exposure of offspring to a later negative environment following prenatal stress, such as an HFD, induces maladaptation, rather than resilience (see Figure 2). Offspring of stressed dams weaned onto an HFD showed alterations in metabolic parameters with female rats shown to have an elevated glucose AUC during an OGTT, relative to control and stress-chow groups, whilst HFD fed males of stressed dams required a greater amount of insulin to clear a given glucose load relative to control (114). In a similar study, prenatal stress did not alter glucose clearance as assessed through intraperitoneal glucose tolerance test (IPGTT), yet, female offspring exposed to stress had increased visceral and retroperitoneal fat depot mass after 10 weeks on a high-fat sucrose diet relative to unstressed controls consuming the same diet (24).
The mechanisms by which prenatal stress alters later life metabolic outcome remain unknown. Stress during pregnancy alters maternal hormones, including circulating glucocorticoid levels. Normal physiological levels of glucocorticoids during development are essential for tissue growth and maturation, however, excess levels of glucocorticoids, e.g., through pharmacological interventions, have been shown to affect maturation (115, 116). Elevated maternal glucocorticoid can cross the placenta (117), and this has been shown to affect growth, morphology, and function of brain and peripheral tissues during fetal development (118). Seckl (119) has demonstrated a stress-mediated mechanism that underpins the low birth weight and increased risk for adulthood health deficits; exposure to increased level of glucocorticoids either synthetically (dexamethasone) during late gestation or via stress (malnutrition, adverse environment exposures) reduces birth weight and impacts maturation of major organs (119, 120). Dams exposed to prenatal variable stress were shown to have heavier adrenal mass and lower fecal corticosterone secretion vs. non-stressed controls (121). This is suggestive of a reduced corticosterone clearance in these stressed dams during gestation. Excess glucocorticoid exposure during pregnancy has also been shown to affect glucose and insulin metabolism (17, 122, 123).
To model periods of nutritional stress in humans, such as famine, reduced nutritional availability during gestation can be used, enabling investigation of the developmental and programing outcomes on offspring (124–126). Restricting dams to 30% of normal food intake, led to low birth weight offspring; pups who were undernourished then weaned onto HFD were found to have significantly elevated leptin, C-peptide, insulin, and body fat compared to control-HFD pups. Notably, injection with leptin for 10 days during lactation (PND3–13) completely normalized these markers (127), suggesting normal maternal levels of leptin are important during development. Food intake and activity levels are also influenced by nutritional insult during gestation. Offspring of female Wistar rats undernourished during gestation (30% of ad libitum) showed significantly reduced locomotor activity with marked hyperphagia and hyperleptinemia when consuming either standard chow or a high caloric diet (128). Leptin is a critical adipocyte derived hormone known to play an essential role in the regulation of feeding and in the maintenance of energy homeostasis. Adult rats exposed to a single 24-h deprivation period during the lactation period demonstrated marked reductions in leptin, although this is difficult to interpret due to the nutritional insult that would have accompanied 24 h of starvation (40, 129, 130).
Various models of postnatal early-life stress have explored the influence of later life environmental insults on metabolic function, each of which seem to elicit different outcomes and sex-specific effects (see Table 2). Work in our lab demonstrated HFD fed male rats previously exposed to maternal separation, have marked elevations in plasma insulin, and decreased total white adipose tissue mass, independent of body weight vs. HFD controls (131, 132). In agreement, maternal deprivation induced early adulthood hyperinsulinemia and impairments in insulin sensitivity, measured through HOMA-IR, in male offspring fed with an HFD relative to HFD controls (130). Similar metabolic changes are observed in maternal deprivation exposed female offspring, with HFD shown to cause early adulthood hyperinsulinemia, at PND35 compared to PND102 in control rats consuming an HFD (130). Further, maternally deprived female rats consuming a high-fat sucrose diet had a trend for decreased fat depot mass vs. control (121). Less work has examined the metabolic profile arising subsequent to early-life stress induced by LN material. A recent study explored for the first time the metabolic profile in Wistar female rats; showing reduced body weight at weaning, and reduced food intake, suggesting altered energy utilization and storage. Interestingly, these pups had exaggerated HPA axis activity with delayed clearance of corticosterone from the circulation, and taken together these data further suggest an early-life stress-induced interaction between the HPA axis and metabolic profile (133) (see Table 2).
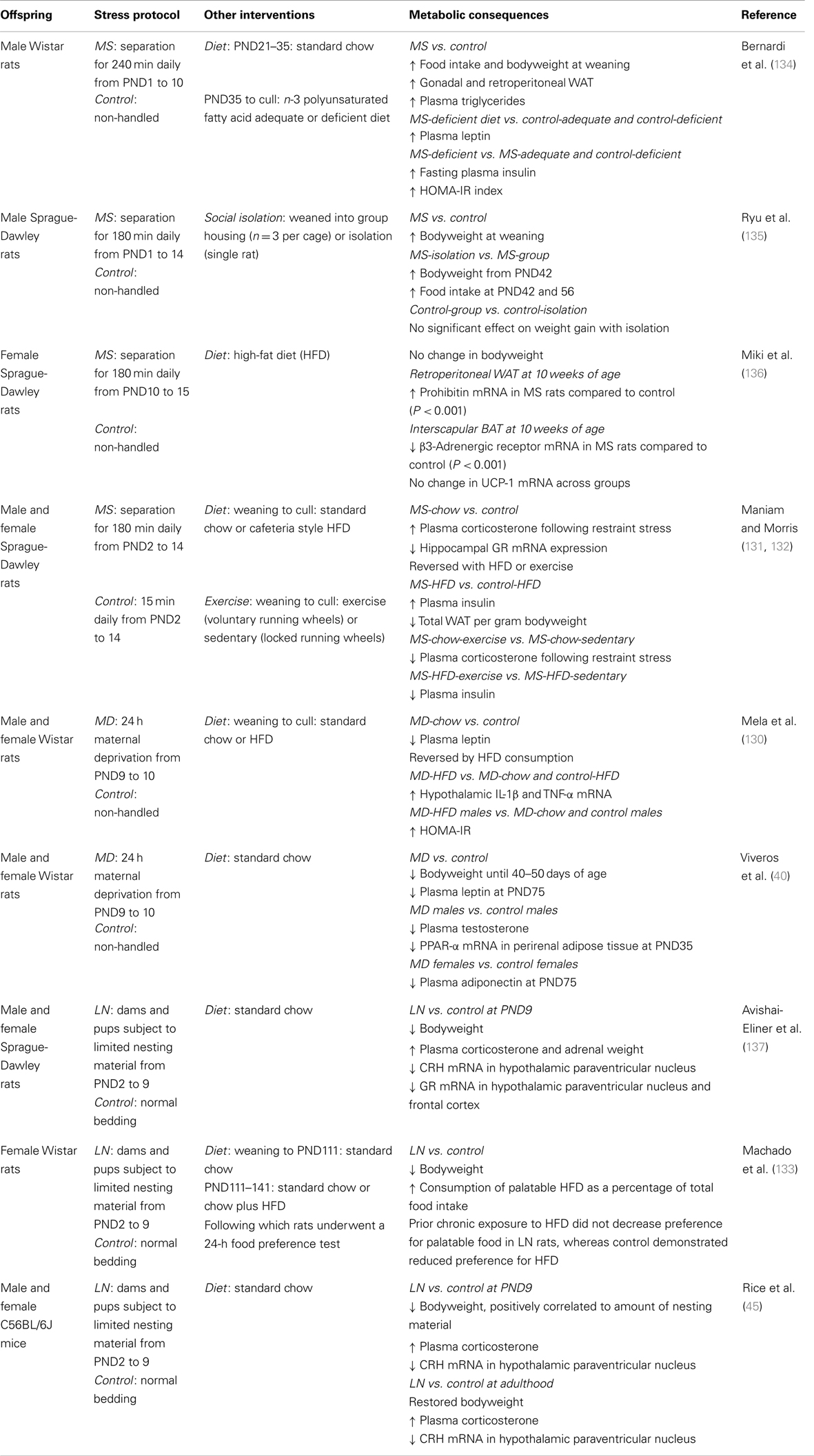
Table 2. Postnatal early-life stress and metabolic consequences in rodents.
Few studies have explored biological characteristics of peripheral tissues following early-life stress. Recently, maternally deprived female rats were shown to have significantly reduced brown adipose tissue β3-adrenergic receptor mRNA expression and increased white adipose tissue prohibitin mRNA relative to control, with no change in UCP-1 (136). The authors concluded these results may facilitate adipose tissue proliferation later in life. An altered response to nutritional challenge following early-life stress is not only observed in high caloric fed states. Maternally separated rats consuming an omega-3 deficient diet demonstrated marked elevations in plasma insulin and impaired insulin sensitivity, as assessed by HOMA-IR, relative to control animals consuming the same diet (134). Functional studies performed by our lab have demonstrated attenuated adulthood outcomes of early-life stress when siblings are provided with a positive environment of voluntary running wheel exercise (see Figure 2). Male rats exposed to maternal separation and weaned onto a standard chow diet demonstrated a hyper-responsive corticosterone response to novel restraint stress, which was dampened with exercise and HFD. Impaired changes in metabolic parameters of insulin and diet-induced obesity were also attenuated with exercise, reversing maternal separation-induced hyperinsulinemia and increased body weight relative to HFD controls and stressed sedentary counterparts (132).
A common aspect across early-life stress models centers around glucocorticoid exposure and HPA axis activation during the stress-hypo-responsive period, a stage of neonatal resilience to mild stressors suggested to likely trigger corticosterone secretion in adult life (84). Synthetic glucocorticoid administration attempts to replicate stress models, as this elicits an HPA axis response during the early postnatal period in rodents (41, 138). This also allows for controlled dose administration and with necessary caution can improve understanding of the impact of glucocorticoid exposure during these critical periods. Early-life corticosterone administration reduced adult female rat body weight and decreased fat depot mass relative to control (139). Studies of glucocorticoid administration highlight the marked influence of combining stress hormones and additional metabolic insults. In young male Sprague-Dawley rats, glucocorticoid administration alone did not alter insulin sensitivity, assessed by HOMA-IR, or glucose disposal following an OGTT (140). Conversely, consumption of a palatable HFD in combination with glucocorticoid exposure is known to induce a marked increase in fasting plasma glucose and insulin, with impaired glucose clearance following OGTT (140, 141). Further investigation is required to determine whether the unaltered or improved insulin sensitivity with chow consumption would prove deleterious in the long term (as shown in Figure 2).
To uncover the mechanisms underlying early-life stress-induced metabolic derangements, it is first essential to understand the action of glucocorticoids at different concentrations on glucose and insulin homeostasis and lipid metabolism. At pharmacological doses, glucocorticoids act as potent anti-inflammatory agents but high levels of circulating glucocorticoids result in metabolic derangements including increased visceral adiposity, dyslipidemia (increased levels of triglycerides), increased non-esterified fatty acids (NEFA) (142–144), and impaired glucose and insulin tolerance (145–147). In contrast to the anabolic actions of insulin, glucocorticoids are predominantly catabolic, decreasing glucose utilization and insulin sensitivity with both human and animal data revealing insulin intolerance with excess exposure (145, 148).
Glucocorticoids exert tissue-specific metabolic effects, directly targeting tissues for insulin metabolism, and they regulate skeletal muscle, liver, and adipocyte insulin signaling (149). Glucocorticoids alter glucose and protein metabolism; increased levels of glucocorticoids induced during stress increase protein degradation, which results in the generation of amino acids that serve as precursor for glucose synthesis in the liver. In addition, excess glucocorticoids inhibit glucose uptake into muscle by inhibiting translocation of glucose transporter-4 (150–152). Chronically increased circulating or tissue glucocorticoid levels may also lead to insulin resistance, hypertriglyceridemia, and hepatic steatosis (see Figure 1). Circulating glucocorticoid concentrations are tightly controlled by activation of the HPA axis, however, tissue-specific availability is regulated by multiple means including glucocorticoid receptor expression, receptor affinity, and alterations in glucocorticoid metabolism and clearance.
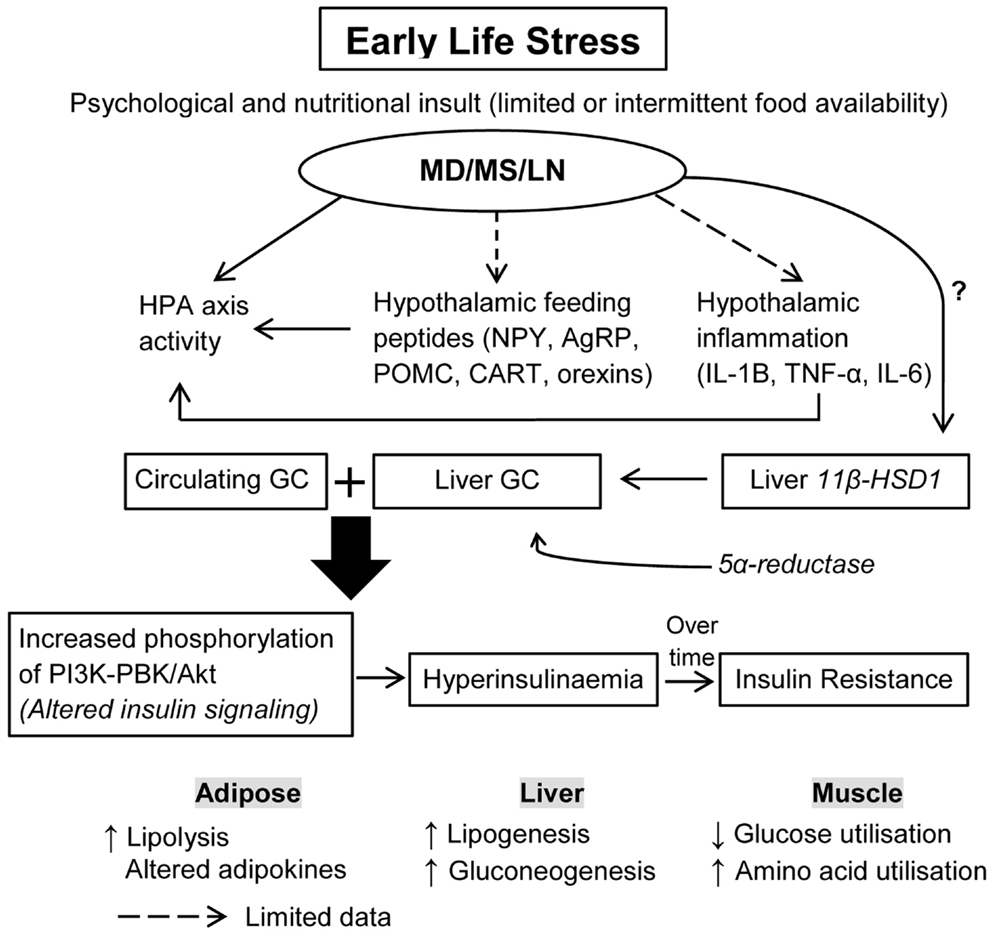
Figure 1. How does ELS increase the risk for insulin resistance and hyperglycemia? Early-life stress (ELS) induced by three different paradigms including maternal deprivation, maternal separation, and limited nesting material are known to dysregulate HPA axis activity, with limited data on the effects of ELS on hypothalamic feeding neuropeptides and inflammation. It is proposed that ELS disturbs circulating glucocorticoids (GC) through a combined action of HPA axis activity, hypothalamic feeding neuropeptides, and inflammatory changes. The effects of ELS on liver 11β-HSD1, an enzyme that converts inactive GC to active GC, and 5α-reductase, an enzyme involved in GC metabolism, is less known. It is proposed that during the maladaptation period, ELS affects tissue levels of these enzymes, thus increasing exposure of peripheral tissues to GC. Excess GC availability can alter insulin signaling, leading to hyperinsulinemia and insulin resistance over time. Thus, increases in circulating and tissue GC induced by ELS act synergistically to exacerbate insulin resistance in peripheral tissues and alter energy expenditure and utilization. ELS, early-life stress; MD, maternal deprivation; MS, maternal separation; LN, limited nesting material; GC, glucocorticoids; TNF-α, tumor necrosis factor alpha; IL-6, interleukin 6; IL-1β, interleukin-1 beta; 11β-HSD1, 11-beta hydroxysteroid dehydrogenase.
Mechanisms regulating intracellular glucocorticoid concentrations are critical to understand the impact of stress on energy metabolism including energy expenditure, storage, and utilization. Intracellular levels of glucocorticoids are influenced by 11β-HSD1 with the type-1 isoform predominantly expressed in the liver (153) and to a lesser degree in adipose and skeletal muscle (154). Evidence shows that tissue glucocorticoid levels are regulated by 11-beta hydroxysteroid dehydrogenase (11β-HSD1) in target tissues (155) as 11β-HSD1 converts inactive cortisone to biologically active cortisol (156, 157). The liver is a major site of glucocorticoid metabolism where 11β-HSD1 regulates the access of glucocorticoid to the glucocorticoid receptor, leading to glucocorticoid metabolism which, is regulated by 5-alpha/beta reductase levels (155, 158). Fat, liver, and muscle-specific increases in 11β-HSD1 are known to increase the risk for metabolic disorders such as insulin resistance, hyperglycemia, and hyperlipidemia (159, 160). Tissue glucocorticoid amplifies the action of insulin to promote lipogenesis within hepatocytes (161). 11β-HSD1 in the liver increases glucocorticoid action in liver to stimulate gluconeogenesis and inhibit beta-oxidation of fat, thus promoting lipid synthesis (162–164). A very recent study demonstrated reduced 5-alpha reductase was associated with fatty liver (165). Thus liver-specific glucocorticoid synthesis and clearance regulated by 11β-HSD1 and 5-alpha reductase appear to affect hepatic lipid accumulation. Interestingly, animal studies demonstrate a link between hepatic glucocorticoid metabolism with regulation of HPA axis activity and lipid synthesis in the liver. For example, transgenic overexpression of 11β-HSD1 in liver of null mice normalized the exaggerated HPA axis activity in response to stress, and led to fatty liver (166, 167). On the other hand, hepatic deletion of 11β-HSD1 led to hyperactivity of the HPA axis (168). These studies suggest liver 11β-HSD1 greatly contributes to amplify circulating glucocorticoid levels, and thus likely mediates the negative feedback activity to dampen HPA axis activity, a concept previously proposed by Chapman et al. (169). Several studies have demonstrated a blunted activity of the HPA axis in response to novel stress following high energy diets such as high sugar or HFD either after adulthood chronic stress or chronic stress exposure during early life (131, 132, 170). As these studies did not report measures of liver 11β-HSD1, it is not clear whether under-activity of the HPA axis following early-life stress and postnatal high energy diet is modulated by liver 11β-HSD1 levels.
Thus, liver 11β-HSD1-HPA axis is a potential pathway in early-life stress-mediated metabolic disturbances, particularly insulin sensitivity, glucose metabolism and lipid synthesis, and mobilization as outlined in Box 1. This hypothesis, however, needs systematic examination in the future. Early-life stress, modeled through prenatal dexamethasone treatment, has been shown to upregulate 11β-HSD1 in peripheral tissues such as liver, pancreas, and subcutaneous fat in rat offspring at 4 months with persistent increases at 1 year of age (171). Interestingly, it was previously shown that early-life stress alters the expression of liver 5-alpha reductase mRNA (172, 173). Thus, this suggests a programing effect of early-life stress on tissue 11β-HSD1 expression, glucocorticoid metabolism, and glucocorticoid signaling. There has been limited evidence regarding early postnatal stress effects on 11β-HSD1 expression and glucocorticoid metabolism of peripheral tissues, which needs to be explored in future studies (Figure 1).
Box 1. Outstanding research questions/proposals.
1. Does chronic exposure to negative postnatal environments such as HFD and adulthood stressors lead to maladaptation following early-life stress?
2. Is the stress-induced perturbation of metabolic profile mediated by the liver 11β-HSD1-HPA axis interaction?
3. How can positive postnatal environments reverse the early-life stress-induced metabolic damage?
4. What are the mechanism(s) underlying early-life stress-induced improvement of insulin sensitivity and improved glucose metabolism if rodents are maintained on chow diet?
We propose that early-life stress may alter availability of tissue glucocorticoids and glucocorticoid signaling. Specifically, we propose that during the maladaptation period (see Figure 2), early-life stress enhances availability of tissue glucocorticoids via increases in liver 11β-HSD1. The tissue glucocorticoid-induced insulin resistance may involve glucocorticoids altering insulin signaling via increasing phosphorylation of PKB/akt, which stimulates insulin secretion, that is, glucocorticoids work synergistically with insulin which increases adipocyte lipolysis and liver lipogenesis (see Figure 1). Another possibility is that early-life stress impairs glucocorticoid signaling involving glucocorticoid–glucocorticoid receptor binding and phosphorylation of the glucocorticoid receptor. Impairment in glucocorticoid signaling leads to alteration in glucocorticoid targeted genes that regulate hepatic glucose production, and hepatic lipogenesis including peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC1-alpha), phosphoenolpyruvate carboxykinase (PEPCK), glucose 6-phosphate (G6P), and diacylglycerol acyltransferase (DGAT). Genes known to mediate hepatic lipid accumulation such as PGC1-alpha and adipose DGAT1 were altered in rat pups from dams that had been subjected to prenatal stress (174). PGC1-alpha plays an essential role in fatty acid oxidation while increased DGAT1 in adipose tissue increases lipogenesis. DGAT1 transgenic mice fed with an HFD demonstrated a 300% increase in liver triglycerides suggesting a redistribution of the fat from adipose tissue to liver via re-esterification of fatty acid with glycerol (175). Another study showed that prenatal stress induced through manipulation of the availability of food, that is either by limiting intake, or exposing to a high energy food during pregnancy led to fatty liver in pups relative to controls (176).
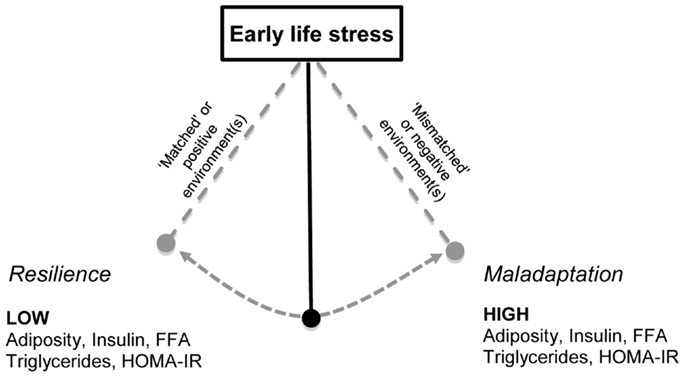
Figure 2. The combination of early-life stress exposure with altered later environment may determine metabolic outcomes. Early-life stress (ELS) exposure during gestation or the postnatal period is hypothesized to influence an offspring’s response to later environments (85, 87). This programing occurs in an attempt to facilitate habituation and resilience to future similar situations. Offspring exposed to environments that do not differ to that to which they were exposed during early life, i.e., “matched” or positive environments, such as exposure to exercise have been shown to adapt and demonstrate resilience (74, 114, 131, 132). Conversely, exposure to a negative environment, i.e., “mismatched,” such as a sub-optimal diet (131, 132, 134) or chronic stress following ELS may lead to maladaptation, and metabolic deficits, with increased levels of triglycerides, free fatty acids, adiposity, and insulin resistance as measured by HOMA-IR. Thus, there is a pendulum of vulnerability and the trajectory following ELS is influenced by the later life environment.
In conclusion, the role of liver 11β-HSD1 in regulating HPA axis activity, and whether it is modulated by stress early in life, warrants investigation. We propose that early-life stress induces changes in glucocorticoid metabolism and signaling, likely mediating the metabolic consequences reported. The question of how these affect glucocorticoid-induced insulin dependent processes, including hyperinsulinemia and lipid metabolism following an early-life stress exposure will be addressed in the following section.
It has been known for decades that stress early in life in both humans and animals can affect HPA axis activity in later life (131, 132, 177–179), however, the effects of early-life stress on hypothalamic neuropeptides involved in feeding are less well known, with only a few studies exploring hypothalamic neuropeptides following maternal separation or deprivation (131, 180). Feeding regulation, which is tightly regulated by orexigenic and anorexigenic hypothalamic neuropeptides, is also influenced by HPA axis activity and circulating glucocorticoid concentrations (181, 182). Glucocorticoids stimulate feeding responses by increasing the release of neuropeptide Y (NPY) and inhibiting that of corticotrophin releasing hormone in the hypothalamus however the orexigenic effect of glucocorticoids may be counteracted by leptin (183).
Mela and colleagues explored the effect of 24 h of maternal deprivation on postnatal day 9 in rats on hypothalamic feeding neuropeptides measured at 14 weeks of age in rats fed with chow or HFD (130). No significant differences in orexigenic (NPY, agouti-related peptide (AgRP) and anorexigenic (pro-opiomelanocortin and cocaine- and amphetamine-regulated transcript) neuropeptides were observed between the chow and HFD fed rats. An interesting finding in this study was that while HFD did not alter these neuropeptides in control females, rats that experienced maternal deprivation had significantly decreased hypothalamic NPY and AgRP mRNA expression and a significant increase in hypothalamic pro-opiomelanocortin and cocaine- and amphetamine-regulated transcript relative to their chow counterparts. The increased caloric intake by HFD fed rats relative to their chow counterparts was similar between the maternally deprived and control rats. But an increased plasma leptin concentration in the maternally deprived HFD fed female rats suggests a programing effect of maternal deprivation which dysregulated feeding homeostasis. Whether this may be linked to either the HPA axis activity or tissue-specific availability of glucocorticoids needs to be explored in the future. The significantly reduced fasting triglycerides and lack of change in fasting insulin by HFD feeding is puzzling, and these may be related to either the strain or the diet used. Thus, use of appropriate controls and dietary conditions are critical to enable exploration of the underlying mechanisms linking combined early-life stress and high-fat feeding with later metabolic risk. Overall, limited studies have directly investigated whether early-life stress affects hypothalamic neuropeptides and inflammation.
The increased availability of glucocorticoids discussed above may play a role in altering tissue insulin signaling by increasing the phosphorylation of PKB/akt. Animals exposed to early-life stress may be resilient to the increased availability of tissue glucocorticoids and thus subsequently show a dampened secretion of insulin. While there are no systematic studies on early-life stress and metabolic consequences, there is evidence in rodents showing early-life stress does not affect basal metabolic hormones if animals consume chow, including non-fasted plasma insulin and leptin (131, 132). Another study demonstrated improved insulin sensitivity following early-life stress when measured during adulthood (170). Taken together, we propose that early-life stress may lead to resilience, and thus the organism potentially may adapt to any changes in the postnatal environment, such as exposure to high energy diet, or later stressors through enhanced negative feedback sensitivity of the HPA axis activity and reprograming of the peripheral tissue sensitivity to glucocorticoid exposure (see Figure 2). However, if an organism exposed to early-life stress is chronically exposed to these negative postnatal environments (high energy diet and later stressors), the enhanced negative feedback sensitivity maybe dysregulated, resulting in perturbed HPA axis activity, leading to a phase known as maladaptation (see Figure 2). Indeed there is evidence that such maladaptations can influence glucose/insulin homeostasis, resulting in the manifestation of metabolic disorders including insulin resistance, hyperglycemia, hyperlipidemia, as suggested in Figure 2.
During periods of maladaptation, increased tissue glucocorticoids and circulating corticosterone will impair the insulin signaling pathway, leading to hypersecretion of insulin; a condition leading to insulin resistance (see Figure 2). This will mediate tissue specific effects, that is, increased adipose lipolysis through beta-oxidation which releases free fatty acids into the circulation and re-esterification in the liver to promote lipogenesis. In addition, increased tissue glucocorticoid levels via insulin stimulation increases hepatic glucose production. Prenatal stress has been shown to increase hepatic PEPCK mRNA and this was enhanced with high energy feeding (174). There are no data thus far on the impact of early postnatal stress on hepatic glucose production, which is an important measure to be considered for future studies to improve understanding of the link between postnatal stress and risk of pre- or diabetes. In addition, increased glucocorticoid-induced hyperinsulinemia also alters the muscle glucose utilization via affecting the glucose transporter gene and expression, including the GLUT4-transporter.
Despite significant progress in the field of early life programing and metabolic disease risk many challenges exist. Human studies are vital in providing evidence of the association between early-life adversity and disease incidence but data must be interpreted with necessary caution. A majority of human evidence is based on parental or offspring self-report raising the possibility of confounding due to issues with information recall, lack of accuracy, and the potential for bias.
Future studies in humans should seek to better quantify stress exposure during the early-life period and at the time of assessment to improve knowledge of how different stress types may alter disease risk. Given both environment and genetic predisposition determine health outcome, studies should not only consider stressor experience during early life but also control for socioeconomic status, food and education availability, ethnicity, and lifestyle factors such as nutritional status, smoking, and physical activity throughout their lifetime.
According to the hypothesis explored throughout this review, programed adaptation during early life occurs in an attempt to adapt to the predicted later life environment and hence even seemingly trivial variations in stressors during these periods may vary the observed outcomes. Inconsistency in findings across experiments could be due to the marked differences in study design. Procedural variations in maternal separation have been reviewed elsewhere (177). In the literature, there is no consistent procedure, rather multiple experimental conditions fall under the broad term of maternal separation. Thus, duration, and age at separation, temperature in which pups are separated, whether pups are isolated from litter mates during separation and whether pups are removed or remain in their home cage have all been shown to influence behavioral outcomes and brain function. Animal models must consider the influence of maternal care on long term outcomes, which is a major aspect the novel model of limiting nesting material attempts to explore. Work exploring prenatal or gestational stress exposure could benefit from cross-fostering to better delineate effects of maternal stress and the influence of received postnatal care. Offspring of low care dams that were cross-fostered by high-licking high arched back nursing dams were resilient in terms of the decline in hippocampal synaptogenesis and spatial learning seen in offspring reared by low care dams (184).
Animal research should ultimately aim to improve public health outcomes. To ensure this, analysis of behavioral, physiological, and molecular parameters is required. The current literature lacks assessment of whole body insulin sensitivity measures or assessment of β-cell structural changes – key factors that influence metabolic outcome. Models need to reflect the human condition upon which they are based, which brings the need for valid controls. Given the complex heterogeneity of both the stress system and metabolic disease the phenotype of experimental animals would ideally be comprehensively assessed, rather than examining single factors. Species and strain differences must be considered and these animals must be valid models for the environmental conditions that the study aims to investigate.
Investigation of the environmental influence provides us with an opportunity to better identify factors determining vulnerability and resilience to early-life experience. A better understanding of factors driving the association between genetic predisposition with both early and adult environment will help with the identification of targets for intervention with the hope of minimizing disease incidence. As known, high energy feeding impairs glucose and insulin tolerance and affects lipid metabolism, thus chronic HFD could serve as a good model to study the altered effects of early-life stress on metabolism. Our lab and others have shown early evidence that HFD or altered nutrition lead to altered insulin levels in early-life stressed animals relative to control (131, 132, 134, 170). Together these studies appear to suggest that both short term diet and long term diet exaggerate plasma insulin. Despite this, a single marker or circulating hormone levels limit ability to draw conclusions of how early-life stress may exert negative impact on insulin sensitivity and overall metabolic risk. Future studies need to adopt a mechanistic approach, examining animals using appropriate metabolic tests that will provide answers to the outstanding questions outlined in Box 1. An alternate model of early-life stress, variable foraging demand, demonstrated impaired insulin resistance in non-human primates as measured by hyperglycemic-insulin clamp (185). Functional studies in rodent early-life stress models, such as glucose or insulin tolerance tests, or preferably glucose clamp, would be useful to explore this further.
In conclusion, the mechanisms whereby adverse early-life events accelerate metabolic deficits have received little attention to date. When combined with a sub-optimal subsequent environment (e.g., poor diet, stress, physical inactivity) early-life stress may exacerbate the risk of metabolic disease. One potential mechanism underlying early-life stress-induced metabolic deficits is the interaction between the HPA axis and liver 11β-HSD1. Positive later environments may modulate the negative impact of early-life stress not only on behavioral outcomes, but also on metabolism. Given the burgeoning issues of metabolic and mental health disorders, the question of how early-life stress impacts subsequent disease risk warrants further investigation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Chrousos GP. Stress and disorders of the stress system. Nat Rev Endocrinol (2009) 5:374–81. doi: 10.1038/nrendo.2009.106
2. De Kloet ER, Derijk R. Signaling pathways in brain involved in predisposition and pathogenesis of stress-related disease: genetic and kinetic factors affecting the MR/GR balance. Ann N Y Acad Sci (2004) 1032:14–34. doi:10.1196/annals.1314.003
3. McEwen BS, Gianaros PJ. Central role of the brain in stress and adaptation: links to socioeconomic status, health, and disease. Ann N Y Acad Sci (2010) 1186:190–222. doi:10.1111/j.1749-6632.2009.05331.x
4. Dallman MF, Akana SF, Laugero KD, Gomez F, Manalo S, Bell ME, et al. A spoonful of sugar: feedback signals of energy stores and corticosterone regulate responses to chronic stress. Physiol Behav (2003) 79:3–12. doi:10.1016/S0031-9384(03)00100-8
5. Dallman MF, La Fleur SE, Pecoraro NC, Gomez F, Houshyar H, Akana SF. Minireview: glucocorticoids – food intake, abdominal obesity, and wealthy nations in 2004. Endocrinology (2004) 145:2633–8. doi:10.1210/en.2004-0037
6. Dallman MF, Akana SF, Pecoraro NC, Warne JP, La Fleur SE, Foster MT. Glucocorticoids, the etiology of obesity and the metabolic syndrome. Curr Alzheimer Res (2007) 4:199–204. doi:10.2174/156720507780362236
7. Oda N, Nakai A, Mokuno T, Sawai Y, Nishida Y, Mano T, et al. Dexamethasone-induced changes in glucose transporter 4 in rat heart muscle, skeletal muscle and adipocytes. Eur J Endocrinol (1995) 133:121–6. doi:10.1530/eje.0.1330121
8. McEwen BS. Steroid hormones: effect on brain development and function. Horm Res (1992) 37(Suppl 3):1–10. doi:10.1159/000182393
9. Munck A, Koritz SB. Studies on the mode of action of glucocorticoids in rats. I. Early effects of cortisol on blood glucose and on glucose entry into muscle, liver and adipose tissue. Biochim Biophys Acta (1962) 57:310–7. doi:10.1016/0006-3002(62)91124-1
10. Issekutz B Jr, Shaw WA. Glucose turnover in the exercising dog with chemically induced diabetes and the effect of methylprednisolone. Diabetes (1975) 24:915–21. doi:10.2337/diab.24.10.915
11. Calabrese F, Molteni R, Racagni G, Riva MA. Neuronal plasticity: a link between stress and mood disorders. Psychoneuroendocrinology (2009) 34(Suppl 1):S208–16. doi:10.1016/j.psyneuen.2009.05.014
12. Pruessner JC, Dedovic K, Pruessner M, Lord C, Buss C, Collins L, et al. Stress regulation in the central nervous system: evidence from structural and functional neuroimaging studies in human populations – 2008 Curt Richter Award Winner. Psychoneuroendocrinology (2010) 35:179–91. doi:10.1016/j.psyneuen.2009.02.016
13. Heim C, Binder EB. Current research trends in early life stress and depression: review of human studies on sensitive periods, gene-environment interactions, and epigenetics. Exp Neurol (2012) 233:102–11. doi:10.1016/j.expneurol.2011.10.032
14. Bjorntorp P, Holm G, Rosmond R. Hypothalamic arousal, insulin resistance and type 2 diabetes mellitus. Diabet Med (1999) 16:373–83. doi:10.1046/j.1464-5491.1999.00067.x
15. Bjorntorp P, Rosmond R. Hypothalamic origin of the metabolic syndrome X. Ann N Y Acad Sci (1999) 892:297–307. doi:10.1111/j.1749-6632.1999.tb07803.x
16. Buchmann AF, Kopf D, Westphal S, Lederbogen F, Banaschewski T, Esser G, et al. Impact of early parental child-rearing behavior on young adults’ cardiometabolic risk profile: a prospective study. Psychosom Med (2010) 72:156–62. doi:10.1097/PSY.0b013e3181c88343
17. Lesage J, Del-Favero F, Leonhardt M, Louvart H, Maccari S, Vieau D, et al. Prenatal stress induces intrauterine growth restriction and programmes glucose intolerance and feeding behaviour disturbances in the aged rat. J Endocrinol (2004) 181:291–6. doi:10.1677/joe.0.1810291
18. Nemeroff CB. Neurobiological consequences of childhood trauma. J Clin Psychiatry (2004) 65(Suppl 1):18–28.
19. Cohen RA, Grieve S, Hoth KF, Paul RH, Sweet L, Tate D, et al. Early life stress and morphometry of the adult anterior cingulate cortex and caudate nuclei. Biol Psychiatry (2006) 59:975–82. doi:10.1016/j.biopsych.2005.12.016
20. Foscolo DR, Foscolo RB, Marubayashi U, Reis AM, Coimbra CC. Neonatal maternal separation affects endocrine and metabolic stress responses to ether exposure but not to restraint exposure in adult rats. Metab Brain Dis (2008) 23:375–85. doi:10.1007/s11011-008-9102-9
21. Carpenter LL, Tyrka AR, Ross NS, Khoury L, Anderson GM, Price LH. Effect of childhood emotional abuse and age on cortisol responsivity in adulthood. Biol Psychiatry (2009) 66:69–75. doi:10.1016/j.biopsych.2009.02.030
22. Cottrell EC, Seckl JR. Prenatal stress, glucocorticoids and the programming of adult disease. Front Behav Neurosci (2009) 3:19. doi:10.3389/neuro.08.019.2009
23. Brunton PJ, Sullivan KM, Kerrigan D, Russell JA, Seckl JR, Drake AJ. Sex-specific effects of prenatal stress on glucose homoeostasis and peripheral metabolism in rats. J Endocrinol (2013) 217:161–73. doi:10.1530/JOE-12-0540
24. Paternain L, de la Garza AL, Batlle MA, Milagro FI, Martinez JA, Campion J. Prenatal stress increases the obesogenic effects of a high-fat-sucrose diet in adult rats in a sex-specific manner. Stress (2013) 16:220–32. doi:10.3109/10253890.2012.707708
25. de Vries A, Holmes MC, Heijnis A, Seier JV, Heerden J, Louw J, et al. Prenatal dexamethasone exposure induces changes in nonhuman primate offspring cardiometabolic and hypothalamic-pituitary-adrenal axis function. J Clin Invest (2007) 117:1058–67. doi:10.1172/JCI30982
26. Seckl JR, Holmes MC. Mechanisms of disease: glucocorticoids, their placental metabolism and fetal “programming” of adult pathophysiology. Nat Clin Pract Endocrinol Metab (2007) 3:479–88. doi:10.1038/ncpendmet0515
28. Giedd JN, Snell JW, Lange N, Rajapakse JC, Casey BJ, Kozuch PL, et al. Quantitative magnetic resonance imaging of human brain development: ages 4-18. Cereb Cortex (1996) 6:551–60. doi:10.1093/cercor/6.4.551
29. Lenroot RK, Giedd JN. Brain development in children and adolescents: insights from anatomical magnetic resonance imaging. Neurosci Biobehav Rev (2006) 30:718–29. doi:10.1016/j.neubiorev.2006.06.001
30. Tau GZ, Peterson BS. Normal development of brain circuits. Neuropsychopharmacology (2010) 35:147–68. doi:10.1038/npp.2009.115
31. De Bellis MD, Keshavan MS, Clark DB, Casey BJ, Giedd JN, Boring AM, et al. A.E. Bennett Research Award. Developmental traumatology. Part II: brain development. Biol Psychiatry (1999) 45:1271–84. doi:10.1016/S0006-3223(99)00045-1
32. Suchecki D, Nelson DY, van Oers H, Levine S. Activation and inhibition of the hypothalamic-pituitary-adrenal axis of the neonatal rat: effects of maternal deprivation. Psychoneuroendocrinology (1995) 20:169–82. doi:10.1016/0306-4530(94)00051-B
33. van Oers HJ, de Kloet ER, Whelan T, Levine S. Maternal deprivation effect on the infant’s neural stress markers is reversed by tactile stimulation and feeding but not by suppressing corticosterone. J Neurosci (1998) 18:10171–9.
34. Sapolsky RM, Meaney MJ. Maturation of the adrenocortical stress response: neuroendocrine control mechanisms and the stress hyporesponsive period. Brain Res (1986) 396:64–76. doi:10.1016/0165-0173(86)90010-X
35. Ivy AS, Brunson KL, Sandman C, Baram TZ. Dysfunctional nurturing behavior in rat dams with limited access to nesting material: a clinically relevant model for early-life stress. Neuroscience (2008) 154:1132–42. doi:10.1016/j.neuroscience.2008.04.019
36. Hofer MA. Maternal separation affects infant rats’ behavior. Behav Biol (1973) 9:629–33. doi:10.1016/S0091-6773(73)80057-4
37. Meaney MJ, Aitken DH. The effects of early postnatal handling on hippocampal glucocorticoid receptor concentrations: temporal parameters. Brain Res (1985) 22:301–4. doi:10.1016/0165-3806(85)90183-X
38. Meaney MJ, Diorio J, Francis D, Widdowson J, Laplante P, Caldji C, et al. Early environmental regulation of forebrain glucocorticoid receptor gene expression: implications for adrenocortical responses to stress. Dev Neurosci (1996) 18:49–72. doi:10.1159/000111395
39. Liu D, Caldji C, Sharma S, Plotsky PM, Meaney MJ. Influence of neonatal rearing conditions on stress-induced adrenocorticotropin responses and norepinepherine release in the hypothalamic paraventricular nucleus. J Neuroendocrinol (2000) 12:5–12. doi:10.1046/j.1365-2826.2000.00422.x
40. Viveros MP, Diaz F, Mateos B, Rodriguez N, Chowen JA. Maternal deprivation induces a rapid decline in circulating leptin levels and sexually dimorphic modifications in hypothalamic trophic factors and cell turnover. Horm Behav (2010) 57:405–14. doi:10.1016/j.yhbeh.2010.01.009
41. Schmidt MV, Enthoven L, van der Mark M, Levine S, de Kloet ER, Oitzl MS. The postnatal development of the hypothalamic-pituitary-adrenal axis in the mouse. Int J Dev Neurosci (2003) 21:125–32. doi:10.1016/S0736-5748(03)00030-3
42. Schmidt M, Enthoven L, van Woezik JH, Levine S, de Kloet ER, Oitzl MS. The dynamics of the hypothalamic-pituitary-adrenal axis during maternal deprivation. J Neuroendocrinol (2004) 16:52–7. doi:10.1111/j.1365-2826.2004.01123.x
43. Schmidt MV, Levine S, Alam S, Harbich D, Sterlemann V, Ganea K, et al. Metabolic signals modulate hypothalamic-pituitary-adrenal axis activation during maternal separation of the neonatal mouse. J Neuroendocrinol (2006) 18:865–74. doi:10.1111/j.1365-2826.2006.01482.x
44. Viveros MP, Llorente R, Lopez-Gallardo M, Suarez J, Bermudez-Silva F, De la Fuente M, et al. Sex-dependent alterations in response to maternal deprivation in rats. Psychoneuroendocrinology (2009) 34(Suppl 1):S217–26. doi:10.1016/j.psyneuen.2009.05.015
45. Rice CJ, Sandman CA, Lenjavi MR, Baram TZ. A novel mouse model for acute and long-lasting consequences of early life stress. Endocrinology (2008) 149:4892–900. doi:10.1210/en.2008-0633
46. Baram TZ, Davis EP, Obenaus A, Sandman CA, Small SL, Solodkin A, et al. Fragmentation and unpredictability of early-life experience in mental disorders. Am J Psychiatry (2012) 169:907–15. doi:10.1176/appi.ajp.2012.11091347
47. Conti G, Hansman C, Heckman JJ, Novak MF, Ruggiero A, Suomi SJ. Primate evidence on the late health effects of early-life adversity. Proc Natl Acad Sci U S A (2012) 109:8866–71. doi:10.1073/pnas.1205340109
48. Kaufman D, Banerji MA, Shorman I, Smith ELP, Coplan JD, Rosenblum LA, et al. Early-life stress and the development of obesity and insulin resistance in juvenile bonnet macaques. Diabetes (2007) 56:1382–6. doi:10.2337/db06-1409
49. Cirulli F, Berry A, Alleva E. Early disruption of the mother-infant relationship: effects on brain plasticity and implications for psychopathology. Neurosci Biobehav Rev (2003) 27:73–82. doi:10.1016/S0149-7634(03)00010-1
50. D’Argenio A, Mazzi C, Pecchioli L, Di Lorenzo G, Siracusano A, Troisi A. Early trauma and adult obesity: is psychological dysfunction the mediating mechanism? Physiol Behav (2009) 98:543–6. doi:10.1016/j.physbeh.2009.08.010
51. Gunstad J, Paul RH, Spitznagel MB, Cohen RA, Williams LM, Kohn M, et al. Exposure to early life trauma is associated with adult obesity. Psychiatry Res (2006) 142:31–7. doi:10.1016/j.psychres.2005.11.007
52. Ward AM, Syddall HE, Wood PJ, Chrousos GP, Phillips DI. Fetal programming of the hypothalamic-pituitary-adrenal (HPA) axis: low birth weight and central HPA regulation. J Clin Endocrinol Metab (2004) 89:1227–33. doi:10.1210/jc.2003-030978
53. Wust S, Entringer S, Federenko IS, Schlotz W, Hellhammer DH. Birth weight is associated with salivary cortisol responses to psychosocial stress in adult life. Psychoneuroendocrinology (2005) 30:591–8. doi:10.1016/j.psyneuen.2005.01.008
54. Miller AL, Clifford C, Sturza J, Rosenblum K, Vazquez DM, Kaciroti N, et al. Blunted cortisol response to stress is associated with higher body mass index in low-income preschool-aged children. Psychoneuroendocrinology (2013) 38:2611–7. doi:10.1016/j.psyneuen.2013.06.014
55. Mi J, Law C, Zhang KL, Osmond C, Stein C, Barker D. Effects of infant birthweight and maternal body mass index in pregnancy on components of the insulin resistance syndrome in China. Ann Intern Med (2000) 132:253–60. doi:10.7326/0003-4819-132-4-200002150-00002
56. Class QA, Lichtenstein P, Langstrom N, D’Onofrio BM. Timing of prenatal maternal exposure to severe life events and adverse pregnancy outcomes: a population study of 2.6 million pregnancies. Psychosom Med (2011) 73:234–41. doi:10.1097/PSY.0b013e31820a62ce
57. Rondo PH, Ferreira RF, Nogueira F, Ribeiro MC, Lobert H, Artes R. Maternal psychological stress and distress as predictors of low birth weight, prematurity and intrauterine growth retardation. Eur J Clin Nutr (2003) 57:266–72. doi:10.1038/sj.ejcn.1601526
58. Wainstock T, Anteby E, Glasser S, Shoham-Vardi I, Lerner-Geva L. The association between prenatal maternal objective stress, perceived stress, preterm birth and low birthweight. J Matern Fetal Neonatal Med (2013) 26:973–7. doi:10.3109/14767058.2013.766696
59. Kajantie E. Fetal origins of stress-related adult disease. Ann N Y Acad Sci (2006) 1083:11–27. doi:10.1196/annals.1367.026
60. Li J, Olsen J, Vestergaard M, Obel C, Kristensen JK, Virk J. Prenatal exposure to bereavement and type-2 diabetes: a Danish longitudinal population based study. PLoS One (2012) 7:e43508. doi:10.1371/journal.pone.0043508
61. Feldman PJ, Dunkel-Schetter C, Sandman CA, Wadhwa PD. Maternal social support predicts birth weight and fetal growth in human pregnancy. Psychosom Med (2000) 62:715–25.
62. Entringer S, Wüst S, Kumsta R, Layes IM, Nelson EL, Hellhammer DH, et al. Prenatal psychosocial stress exposure is associated with insulin resistance in young adults. Am J Obstet Gynecol (2008) 199:498.e1–7. doi:10.1016/j.ajog.2008.03.006
63. Ravelli AC, van der Meulen JH, Michels RP, Osmond C, Barker DJ, Hales CN, et al. Glucose tolerance in adults after prenatal exposure to famine. Lancet (1998) 351:173–7. doi:10.1016/S0140-6736(05)79096-6
64. van Abeelen AFM, Elias SG, Bossuyt PMM, Grobbee DE, van der Schouw YT, Roseboom TJ, et al. Famine exposure in the young and the risk of type 2 diabetes in adulthood. Diabetes (2012) 61:2255–60. doi:10.2337/db11-1559
65. Zheng X, Wang Y, Ren W, Luo R, Zhang S, Zhang JH, et al. Risk of metabolic syndrome in adults exposed to the great Chinese famine during the fetal life and early childhood. Eur J Clin Nutr (2012) 66:231–6. doi:10.1038/ejcn.2011.161
66. Hult M, Tornhammar P, Ueda P, Chima C, Bonamy AK, Ozumba B, et al. Hypertension, diabetes and overweight: looming legacies of the Biafran famine. PLoS One (2010) 5:e13582. doi:10.1371/journal.pone.0013582
67. Flory JD, Bierer LM, Yehuda R. Maternal exposure to the holocaust and health complaints in offspring. Dis Markers (2011) 30:133–9. doi:10.3233/DMA-2011-0748
68. Dancause KN, Laplante DP, Fraser S, Brunet A, Ciampi A, Schmitz N, et al. Prenatal exposure to a natural disaster increases risk for obesity in 5(1/2)-year-old children. Pediatr Res (2012) 71:126–31. doi:10.1038/pr.2011.18
69. Dancause KN, Veru F, Andersen RE, Laplante DP, King S. Prenatal stress due to a natural disaster predicts insulin secretion in adolescence. Early Hum Dev (2013) 89:773–6. doi:10.1016/j.earlhumdev.2013.06.006
70. Ravelli AC, van der Meulen JH, Osmond C, Barker DJ, Bleker OP. Obesity at the age of 50 y in men and women exposed to famine prenatally. Am J Clin Nutr (1999) 70:811–6.
71. Watt TT, Appel L, Roberts K, Flores B, Morris S. Sugar, stress, and the supplemental nutrition assistance program: early childhood obesity risks among a clinic-based sample of low-income Hispanics. J Community Health (2013) 38:513–20. doi:10.1007/s10900-012-9641-1
72. Ertel KA, Koenen KC, Rich-Edwards JW, Gillman MW. Antenatal and postpartum depressive symptoms are differentially associated with early childhood weight and adiposity. Paediatr Perinat Epidemiol (2010) 24:179–89. doi:10.1111/j.1365-3016.2010.01098.x
73. Alciati A, Gesuele F, Casazza G, Foschi D. The relationship between childhood parental loss and metabolic syndrome in obese subjects. Stress Health (2013) 29:5–13. doi:10.1002/smi.1435
74. Gundersen C, Lohman BJ, Garasky S, Stewart S, Eisenmann J. Food security, maternal stressors, and overweight among low-income US children: results from the National Health and Nutrition Examination Survey (1999-2002). Pediatrics (2008) 122:e529–40. doi:10.1542/peds.2008-0556
75. Rich-Edwards JW, Spiegelman D, Lividoti Hibert EN, Jun HJ, Todd TJ, Kawachi I, et al. Abuse in childhood and adolescence as a predictor of type 2 diabetes in adult women. Am J Prev Med (2010) 39:529–36. doi:10.1016/j.amepre.2010.09.007
76. Danese A, Pariante CM, Caspi A, Taylor A, Poulton R. Childhood maltreatment predicts adult inflammation in a life-course study. Proc Natl Acad Sci U S A (2007) 104:1319–24. doi:10.1073/pnas.0610362104
77. Midei AJ, Matthews KA, Chang YF, Bromberger JT. Childhood physical abuse is associated with incident metabolic syndrome in mid-life women. Health Psychol (2013) 32:121–7. doi:10.1037/a0027891
78. Lissau I, Sorensen TI. Parental neglect during childhood and increased risk of obesity in young adulthood. Lancet (1994) 343:324–7. doi:10.1016/S0140-6736(94)91163-0
79. Thomas C, Hypponen E, Power C. Obesity and type 2 diabetes risk in midadult life: the role of childhood adversity. Pediatrics (2008) 121:e1240–9. doi:10.1542/peds.2007-2403
80. Reynolds RM, Walker BR, Syddall HE, Andrew R, Wood PJ, Whorwood CB, et al. Altered control of cortisol secretion in adult men with low birth weight and cardiovascular risk factors. J Clin Endocrinol Metab (2001) 86:245–50. doi:10.1210/jc.86.1.245
81. Kant GJ, Eggleston T, Landman-Roberts L, Kenion CC, Driver GC, Meyerhoff JL. Habituation to repeated stress is stressor specific. Pharmacol Biochem Behav (1985) 22:631–4. doi:10.1016/0091-3057(85)90286-2
82. Chrousos GP. Stressors, stress, and neuroendocrine integration of the adaptive response. The 1997 Hans Selye Memorial Lecture. Ann N Y Acad Sci (1998) 851:311–35. doi:10.1111/j.1749-6632.1998.tb09006.x
83. Grissom N, Bhatnagar S. Habituation to repeated stress: get used to it. Neurobiol Learn Mem (2009) 92:215–24. doi:10.1016/j.nlm.2008.07.001
84. Daskalakis NP, Bagot RC, Parker KJ, Vinkers CH, de Kloet ER. The three-hit concept of vulnerability and resilience: toward understanding adaptation to early-life adversity outcome. Psychoneuroendocrinology (2013) 38:1858–73. doi:10.1016/j.psyneuen.2013.06.008
85. Gluckman PD, Hanson MA, Beedle AS. Early life events and their consequences for later disease: a life history and evolutionary perspective. Am J Hum Biol (2007) 19:1–19. doi:10.1002/ajhb.20590
86. McEwen BS. Stress, adaptation, and disease. Allostasis and allostatic load. Ann N Y Acad Sci (1998) 840:33–44. doi:10.1111/j.1749-6632.1998.tb09546.x
87. Schmidt MV. Animal models for depression and the mismatch hypothesis of disease. Psychoneuroendocrinology (2011) 36:330–8. doi:10.1016/j.psyneuen.2010.07.001
88. Ng DM, Jeffery RW. Relationships between perceived stress and health behaviors in a sample of working adults. Health Psychol (2003) 22:638–42. doi:10.1037/0278-6133.22.6.638
89. Keyes KM, Hatzenbuehler ML, Grant BF, Hasin DS. Stress and alcohol: epidemiologic evidence. Alcohol Res (2012) 34:391–400.
90. Sinha R. Chronic stress, drug use, and vulnerability to addiction. Ann N Y Acad Sci (2008) 1141:105–30. doi:10.1196/annals.1441.030
91. Stults-Kolehmainen MA, Sinha R. The effects of stress on physical activity and exercise. Sports Med (2014) 44:81–121. doi:10.1007/s40279-013-0090-5
92. Brown R, Ogden J. Children’s eating attitudes and behaviour: a study of the modelling and control theories of parental influence. Health Educ Res (2004) 19:261–71. doi:10.1093/her/cyg040
93. Ornelas IJ, Perreira KM, Ayala GX. Parental influences on adolescent physical activity: a longitudinal study. Int J Behav Nutr Phys Act (2007) 4:3. doi:10.1186/1479-5868-4-3
94. Hesketh KR, Goodfellow L, Ekelund U, McMinn AM, Godfrey KM, Inskip HM, et al. Activity levels in mothers and their preschool children. Pediatrics (2014) 133:e973–80. doi:10.1542/peds.2013-3153
95. Lussana F, Painter RC, Ocke MC, Buller HR, Bossuyt PM, Roseboom TJ. Prenatal exposure to the Dutch famine is associated with a preference for fatty foods and a more atherogenic lipid profile. Am J Clin Nutr (2008) 88:1648–52. doi:10.3945/ajcn.2008.26140
96. te Velde SJ, Twisk JW, van Mechelen W, Kemper HC. A birth-weight questionnaire indicated that life style modifies the birth weight and metabolic syndrome relationship at age 36. J Clin Epidemiol (2005) 58:1172–9. doi:10.1016/j.jclinepi.2005.03.013
97. Ridgway CL, Brage S, Anderssen SA, Sardinha LB, Andersen LB, Ekelund U. Do physical activity and aerobic fitness moderate the association between birth weight and metabolic risk in youth? The European Youth Heart Study. Diabetes Care (2011) 34:187–92. doi:10.2337/dc10-1178
98. Kaseva N, Wehkalampi K, Strang-Karlsson S, Salonen M, Pesonen AK, Raikkonen K, et al. Lower conditioning leisure-time physical activity in young adults born preterm at very low birth weight. PLoS One (2012) 7:e32430. doi:10.1371/journal.pone.0032430
99. Falk B, Eliakim A, Dotan R, Liebermann DG, Regev R, Bar-Or O. Birth weight and physical ability in 5- to 8-yr-old healthy children born prematurely. Med Sci Sports Exerc (1997) 29:1124–30. doi:10.1097/00005768-199709000-00002
100. Nemet D, Dolfin T, Litmanowitz I, Shainkin-Kestenbaum R, Lis M, Eliakim A. Evidence for exercise-induced bone formation in premature infants. Int J Sports Med (2002) 23:82–5. doi:10.1055/s-2002-20134
101. Franks PW, Ekelund U, Brage S, Wong MY, Wareham NJ. Does the association of habitual physical activity with the metabolic syndrome differ by level of cardiorespiratory fitness? Diabetes Care (2004) 27:1187–93. doi:10.2337/diacare.27.5.1187
102. Ortega FB, Ruiz JR, Hurtig-Wennlof A, Meirhaeghe A, Gonzalez-Gross M, Moreno LA, et al. Physical activity attenuates the effect of low birth weight on insulin resistance in adolescents: findings from two observational studies. Diabetes (2011) 60:2295–9. doi:10.2337/db10-1670
103. Aoyama T, Tsushita K, Miyatake N, Numata T, Miyachi M, Tabata I, et al. Does cardiorespiratory fitness modify the association between birth weight and insulin resistance in adult life? PLoS One (2013) 8:e73967. doi:10.1371/journal.pone.0073967
104. Volkow ND, Wise RA. How can drug addiction help us understand obesity? Nat Neurosci (2005) 8:555–60. doi:10.1038/nn1452
105. Mason SM, Flint AJ, Field AE, Austin SB, Rich-Edwards JW. Abuse victimization in childhood or adolescence and risk of food addiction in adult women. Obesity (Silver Spring) (2013) 21:E775–81. doi:10.1002/oby.20500
106. Parks EP, Kumanyika S, Moore RH, Stettler N, Wrotniak BH, Kazak A. Influence of stress in parents on child obesity and related behaviors. Pediatrics (2012) 130:e1096–104. doi:10.1542/peds.2012-0895
107. Dallman MF, Pecoraro NC, La Fleur SE. Chronic stress and comfort foods: self-medication and abdominal obesity. Brain Behav Immun (2005) 19:275–80. doi:10.1016/j.bbi.2004.11.004
108. Gibson EL. Emotional influences on food choice: sensory, physiological and psychological pathways. Physiol Behav (2006) 89:53–61. doi:10.1016/j.physbeh.2006.01.024
109. Adam TC, Epel ES. Stress, eating and the reward system. Physiol Behav (2007) 91:449–58. doi:10.1016/j.physbeh.2007.04.011
110. Martikainen S, Pesonen AK, Lahti J, Heinonen K, Feldt K, Pyhala R, et al. Higher levels of physical activity are associated with lower hypothalamic-pituitary-adrenocortical axis reactivity to psychosocial stress in children. J Clin Endocrinol Metab (2013) 98:E619–27. doi:10.1210/jc.2012-3745
111. Matthews KA, Chang YF, Thurston RC, Bromberger JT. Child abuse is related to inflammation in mid-life women: role of obesity. Brain Behav Immun (2014) 36:29–34. doi:10.1016/j.bbi.2013.09.013
112. Gleeson M, Bishop NC, Stensel DJ, Lindley MR, Mastana SS, Nimmo MA. The anti-inflammatory effects of exercise: mechanisms and implications for the prevention and treatment of disease. Nat Rev Immunol (2011) 11:607–15. doi:10.1038/nri3041
113. Koenig JI, Elmer GI, Shepard PD, Lee PR, Mayo C, Joy B, et al. Prenatal exposure to a repeated variable stress paradigm elicits behavioral and neuroendocrinological changes in the adult offspring: potential relevance to schizophrenia. Behav Brain Res (2005) 156:251–61. doi:10.1016/j.bbr.2004.05.030
114. Tamashiro KL, Terrillion CE, Hyun J, Koenig JI, Moran TH. Prenatal stress or high-fat diet increases susceptibility to diet-induced obesity in rat offspring. Diabetes (2009) 58:1116–25. doi:10.2337/db08-1129
115. Bian XP, Seidler FJ, Slotkin TA. Promotional role for glucocorticoids in the development of intracellular signalling: enhanced cardiac and renal adenylate cyclase reactivity to beta-adrenergic and non-adrenergic stimuli after low-dose fetal dexamethasone exposure. J Dev Physiol (1992) 17:289–97.
116. Fowden AL. Endocrine regulation of fetal growth. Reprod Fertil Dev (1995) 7:351–63. doi:10.1071/RD9950351
117. Cottrell EC, Holmes MC, Livingstone DE, Kenyon CJ, Seckl JR. Reconciling the nutritional and glucocorticoid hypotheses of fetal programming. FASEB J (2012) 26:1866–74. doi:10.1096/fj.12-203489
118. Duthie L, Reynolds RM. Changes in the maternal hypothalamic-pituitary-adrenal axis in pregnancy and postpartum: influences on maternal and fetal outcomes. Neuroendocrinology (2013) 98:106–15. doi:10.1159/000354702
119. Seckl JR. Prenatal glucocorticoids and long-term programming. Eur J Endocrinol (2004) 151(Suppl 3):U49–62. doi:10.1530/eje.0.151U049
120. Lesage J, Blondeau B, Grino M, Breant B, Dupouy JP. Maternal undernutrition during late gestation induces fetal overexposure to glucocorticoids and intrauterine growth retardation, and disturbs the hypothalamo-pituitary adrenal axis in the newborn rat. Endocrinology (2001) 142:1692–702. doi:10.1210/endo.142.5.8139
121. Paternain L, Batlle MA, De la Garza AL, Milagro FI, Martinez JA, Campion J. Transcriptomic and epigenetic changes in the hypothalamus are involved in an increased susceptibility to a high-fat-sucrose diet in prenatally stressed female rats. Neuroendocrinology (2012) 96:249–60. doi:10.1159/000341684
122. Nyirenda MJ, Lindsay RS, Kenyon CJ, Burchell A, Seckl JR. Glucocorticoid exposure in late gestation permanently programs rat hepatic phosphoenolpyruvate carboxykinase and glucocorticoid receptor expression and causes glucose intolerance in adult offspring. J Clin Invest (1998) 101:2174–81. doi:10.1172/JCI1567
123. Cleasby ME, Livingstone DE, Nyirenda MJ, Seckl JR, Walker BR. Is programming of glucocorticoid receptor expression by prenatal dexamethasone in the rat secondary to metabolic derangement in adulthood? Eur J Endocrinol (2003) 148:129–38. doi:10.1530/eje.0.1480129
124. Jones AP, Simson EL, Friedman MI. Gestational undernutrition and the development of obesity in rats. J Nutr (1984) 114:1484–92.
125. Bertram CE, Hanson MA. Animal models and programming of the metabolic syndrome. Br Med Bull (2001) 60:103–21. doi:10.1093/bmb/60.1.103
126. Remacle C, Bieswal F, Bol V, Reusens B. Developmental programming of adult obesity and cardiovascular disease in rodents by maternal nutrition imbalance. Am J Clin Nutr (2011) 94:1846S–52S. doi:10.3945/ajcn.110.001651
127. Vickers MH, Gluckman PD, Coveny AH, Hofman PL, Cutfield WS, Gertler A, et al. Neonatal leptin treatment reverses developmental programming. Endocrinology (2005) 146:4211–6. doi:10.1210/en.2005-0581
128. Vickers MH, Breier BH, McCarthy D, Gluckman PD. Sedentary behavior during postnatal life is determined by the prenatal environment and exacerbated by postnatal hypercaloric nutrition. Am J Physiol Regul Integr Comp Physiol (2003) 285:R271–3.
129. Llorente-Berzal A, Fuentes S, Gagliano H, Lopez-Gallardo M, Armario A, Viveros MP, et al. Sex-dependent effects of maternal deprivation and adolescent cannabinoid treatment on adult rat behaviour. Addict Biol (2011) 16:624–37. doi:10.1111/j.1369-1600.2011.00318.x
130. Mela V, Llorente-Berzal A, Diaz F, Argente J, Viveros MP, Chowen JA. Maternal deprivation exacerbates the response to a high fat diet in a sexually dimorphic manner. PLoS One (2012) 7(11):e48915. doi:10.1371/journal.pone.0048915
131. Maniam J, Morris MJ. Palatable cafeteria diet ameliorates anxiety and depression-like symptoms following an adverse early environment. Psychoneuroendocrinology (2010) 35:717–28. doi:10.1016/j.psyneuen.2009.10.013
132. Maniam J, Morris MJ. Voluntary exercise and palatable high-fat diet both improve behavioural profile and stress responses in male rats exposed to early life stress: role of hippocampus. Psychoneuroendocrinology (2010) 35:1553–64. doi:10.1016/j.psyneuen.2010.05.012
133. Machado TD, Molle RD, Laureano DP, Portella AK, Werlang ICR, Benetti CD, et al. Early life stress is associated with anxiety, increased stress responsivity and preference for “comfort foods” in adult female rats. Stress (2013) 16:549–56. doi:10.3109/10253890.2013.816841
134. Bernardi JR, Ferreira CF, Senter G, Krolow R, de Aguiar BW, Portella AK, et al. Early life stress interacts with the diet deficiency of omega-3 fatty acids during the life course increasing the metabolic vulnerability in adult rats. PLoS One (2013) 8(4):e62031. doi:10.1371/journal.pone.0062031
135. Ryu V, Yoo SB, Kang DW, Lee JH, Jahng JW. Post-weaning isolation promotes food intake and body weight gain in rats that experienced neonatal maternal separation. Brain Res (2009) 1295:127–34. doi:10.1016/j.brainres.2009.08.006
136. Miki T, Liu JQ, Ohta K, Suzuki S, Kusaka T, Warita K, et al. Early postnatal maternal separation causes alterations in the expression of beta3-adrenergic receptor in rat adipose tissue suggesting long-term influence on obesity. Biochem Biophys Res Commun (2013) 442:68–71. doi:10.1016/j.bbrc.2013.11.005
137. Avishai-Eliner S, Gilles EE, Eghbal-Ahmadi M, Bar-El Y, Baram TZ. Altered regulation of gene and protein expression of hypothalamic-pituitary-adrenal axis components in an immature rat model of chronic stress. J Neuroendocrinol (2001) 13:799–807. doi:10.1046/j.1365-2826.2001.00698.x
138. Pihoker C, Owens MJ, Kuhn CM, Schanberg SM, Nemeroff CB. Maternal separation in neonatal rats elicits activation of the hypothalamic-pituitary-adrenocortical axis: a putative role for corticotropin-releasing factor. Psychoneuroendocrinology (1993) 18:485–93. doi:10.1016/0306-4530(93)90042-J
139. Nilsson C, Jennische E, Ho HP, Eriksson E, Bjorntorp P, Holmang A. Increased insulin sensitivity and decreased body weight in female rats after postnatal corticosterone exposure. Eur J Endocrinol (2002) 146:847–54. doi:10.1530/eje.0.1460847
140. Shpilberg Y, Beaudry JL, D’Souza A, Campbell JE, Peckett A, Riddell MC. A rodent model of rapid-onset diabetes induced by glucocorticoids and high-fat feeding. Dis Model Mech (2012) 5:671–80. doi:10.1242/dmm.008912
141. D’Souza AM, Beaudry JL, Szigiato AA, Trumble SJ, Snook LA, Bonen A, et al. Consumption of a high-fat diet rapidly exacerbates the development of fatty liver disease that occurs with chronically elevated glucocorticoids. Am J Physiol Gastrointest Liver Physiol (2012) 302:G850–63. doi:10.1152/ajpgi.00378.2011
142. Taskinen MR, Nikkila EA, Pelkonen R, Sane T. Plasma lipoproteins, lipolytic enzymes, and very low density lipoprotein triglyceride turnover in Cushing’s syndrome. J Clin Endocrinol Metab (1983) 57:619–26. doi:10.1210/jcem-57-3-619
143. Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr Rev (2000) 21:697–738. doi:10.1210/edrv.21.6.0415
144. Rockall AG, Sohaib SA, Evans D, Kaltsas G, Isidori AM, Monson JP, et al. Computed tomography assessment of fat distribution in male and female patients with Cushing’s syndrome. Eur J Endocrinol (2003) 149:561–7. doi:10.1530/eje.0.1490561
145. Buren J, Liu HX, Jensen J, Eriksson JW. Dexamethasone impairs insulin signalling and glucose transport by depletion of insulin receptor substrate-1, phosphatidylinositol 3-kinase and protein kinase B in primary cultured rat adipocytes. Eur J Endocrinol (2002) 146:419–29. doi:10.1530/eje.0.1460419
146. Schacke H, Docke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther (2002) 96:23–43. doi:10.1016/S0163-7258(02)00297-8
147. Bernal-Mizrachi C, Weng S, Feng C, Finck BN, Knutsen RH, Leone TC, et al. Dexamethasone induction of hypertension and diabetes is PPAR-alpha dependent in LDL receptor-null mice. Nat Med (2003) 9:1069–75. doi:10.1038/nm898
148. Reynolds RM, Walker BR. Human insulin resistance: the role of glucocorticoids. Diabetes Obes Metab (2003) 5:5–12. doi:10.1046/j.1463-1326.2003.00221.x
149. Kuo T, Lew MJ, Mayba O, Harris CA, Speed TP, Wang JC. Genome-wide analysis of glucocorticoid receptor-binding sites in myotubes identifies gene networks modulating insulin signaling. Proc Natl Acad Sci U S A (2012) 109:11160–5. doi:10.1073/pnas.1111334109
150. Hajduch E, Hainault I, Meunier C, Jardel C, Hainque B, Guerre-Millo M, et al. Regulation of glucose transporters in cultured rat adipocytes: synergistic effect of insulin and dexamethasone on GLUT4 gene expression through promoter activation. Endocrinology (1995) 136:4782–9. doi:10.1210/endo.136.11.7588207
151. Dimitriadis G, Leighton B, Parry-Billings M, Sasson S, Young M, Krause U, et al. Effects of glucocorticoid excess on the sensitivity of glucose transport and metabolism to insulin in rat skeletal muscle. Biochem J (1997) 321(Pt 3):707–12.
152. Weinstein SP, Wilson CM, Pritsker A, Cushman SW. Dexamethasone inhibits insulin-stimulated recruitment of GLUT4 to the cell surface in rat skeletal muscle. Metabolism (1998) 47:3–6. doi:10.1016/S0026-0495(98)90184-6
153. Moisan MP, Seckl JR, Edwards CR. 11 Beta-hydroxysteroid dehydrogenase bioactivity and messenger RNA expression in rat forebrain: localization in hypothalamus, hippocampus, and cortex. Endocrinology (1990) 127:1450–5. doi:10.1210/endo-127-3-1450
154. Whorwood CB, Donovan SJ, Flanagan D, Phillips DI, Byrne CD. Increased glucocorticoid receptor expression in human skeletal muscle cells may contribute to the pathogenesis of the metabolic syndrome. Diabetes (2002) 51:1066–75. doi:10.2337/diabetes.51.4.1066
155. Yehuda R, Seckl J. Minireview: stress-related psychiatric disorders with low cortisol levels: a metabolic hypothesis. Endocrinology (2011) 152:4496–503. doi:10.1210/en.2011-1218
156. Jamieson PM, Chapman KE, Edwards CR, Seckl JR. 11 Beta-hydroxysteroid dehydrogenase is an exclusive 11 beta- reductase in primary cultures of rat hepatocytes: effect of physicochemical and hormonal manipulations. Endocrinology (1995) 136:4754–61. doi:10.1210/en.136.11.4754
157. Gathercole LL, Lavery GG, Morgan SA, Cooper MS, Sinclair AJ, Tomlinson JW, et al. 11Beta-hydroxysteroid dehydrogenase 1: translational and therapeutic aspects. Endocr Rev (2013) 34:525–55. doi:10.1210/er.2012-1050
158. Tomlinson JW, Walker EA, Bujalska IJ, Draper N, Lavery GG, Cooper MS, et al. 11β-Hydroxysteroid dehydrogenase type 1: a tissue-specific regulator of glucocorticoid response. Endocr Rev (2004) 25:831–66. doi:10.1210/er.2003-0031
159. Cleasby ME, Kelly PA, Walker BR, Seckl JR. Programming of rat muscle and fat metabolism by in utero overexposure to glucocorticoids. Endocrinology (2003) 144:999–1007. doi:10.1210/en.2002-220559
160. Masuzaki H, Flier JS. Tissue-specific glucocorticoid reactivating enzyme, 11 beta-hydroxysteroid dehydrogenase type 1 (11 beta-HSD1) – a promising drug target for the treatment of metabolic syndrome. Curr Drug Targets Immune Endocr Metabol Disord (2003) 3:255–62. doi:10.2174/1568008033340135
161. Amatruda JM, Danahy SA, Chang CL. The effects of glucocorticoids on insulin-stimulated lipogenesis in primary cultures of rat hepatocytes. Biochem J (1983) 212:135–41.
162. Berdanier CD. Role of glucocorticoids in the regulation of lipogenesis. FASEB J (1989) 3:2179–83.
163. Noguchi T, Iritani N, Tanaka T. Molecular mechanism of induction of key enzymes related to lipogenesis. Proc Soc Exp Biol Med (1992) 200:206–9. doi:10.3181/00379727-200-43419
164. Vasiljevic A, Velickovic N, Bursac B, Djordjevic A, Milutinovic DV, Nestorovic N, et al. Enhanced prereceptor glucocorticoid metabolism and lipogenesis impair insulin signaling in the liver of fructose-fed rats. J Nutr Biochem (2013) 24:1790–7. doi:10.1016/j.jnutbio.2013.04.001
165. Dowman JK, Hopkins LJ, Reynolds GM, Armstrong MJ, Nasiri M, Nikolaou N, et al. Loss of 5alpha-reductase type 1 accelerates the development of hepatic steatosis but protects against hepatocellular carcinoma in male mice. Endocrinology (2013) 154(12):4536–47. doi:10.1210/en.2013-1592
166. Paterson JM, Morton NM, Fievet C, Kenyon CJ, Holmes MC, Staels B, et al. Metabolic syndrome without obesity: hepatic overexpression of 11beta-hydroxysteroid dehydrogenase type 1 in transgenic mice. Proc Natl Acad Sci U S A (2004) 101:7088–93. doi:10.1073/pnas.0305524101
167. Paterson JM, Holmes MC, Kenyon CJ, Carter R, Mullins JJ, Seckl JR. Liver-selective transgene rescue of hypothalamic-pituitary-adrenal axis dysfunction in 11beta-hydroxysteroid dehydrogenase type 1-deficient mice. Endocrinology (2007) 148:961–6. doi:10.1210/en.2006-0603
168. Carter RN, Paterson JM, Tworowska U, Stenvers DJ, Mullins JJ, Seckl JR, et al. Hypothalamic-pituitary-adrenal axis abnormalities in response to deletion of 11beta-HSD1 is strain-dependent. J Neuroendocrinol (2009) 21:879–87. doi:10.1111/j.1365-2826.2009.01899.x
169. Chapman K, Holmes M, Seckl J. 11Beta-hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiol Rev (2013) 93:1139–206. doi:10.1152/physrev.00020.2012
170. Paternain L, Martisova E, Milagro FI, Ramirez MJ, Martinez JA, Campion J. Postnatal maternal separation modifies the response to an obesogenic diet in adulthood in rats. Dis Model Mech (2012) 5:691–7. doi:10.1242/dmm.009043
171. Nyirenda MJ, Carter R, Tang JI, de Vries A, Schlumbohm C, Hillier SG, et al. Prenatal programming of metabolic syndrome in the common marmoset is associated with increased expression of 11beta-hydroxysteroid dehydrogenase type 1. Diabetes (2009) 58:2873–9. doi:10.2337/db09-0873
172. Gustafsson JA, Stenberg A. Irreversible androgenic programming at birth of microsomal and soluble rat liver enzymes active on androstene-3,17-dione and 5alpha-androstane-3alpha,17beta-diol. J Biol Chem (1974) 249:711–8.
173. Stimson RH, Lobley GE, Maraki I, Morton NM, Andrew R, Walker BR. Effects of proportions of dietary macronutrients on glucocorticoid metabolism in diet-induced obesity in rats. PLoS One (2010) 5:e8779. doi:10.1371/journal.pone.0008779
174. Drake AJ, Raubenheimer PJ, Kerrigan D, McInnes KJ, Seckl JR, Walker BR. Prenatal dexamethasone programs expression of genes in liver and adipose tissue and increased hepatic lipid accumulation but not obesity on a high-fat diet. Endocrinology (2010) 151:1581–7. doi:10.1210/en.2009-1088
175. Chen N, Liu L, Zhang Y, Ginsberg HN, Yu Y-H. Whole-body insulin resistance in the absence of obesity in FVB mice with overexpression of Dgat1 in adipose tissue. Diabetes (2005) 54:3379–86. doi:10.2337/diabetes.54.12.3379
176. Bruce KD, Cagampang FR, Argenton M, Zhang J, Ethirajan PL, Burdge GC, et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology (2009) 50:1796–808. doi:10.1002/hep.23205
177. Lehmann J, Feldon J. Long-term biobehavioral effects of maternal separation in the rat: consistent or confusing? Rev Neurosci (2000) 11:383–408. doi:10.1515/REVNEURO.2000.11.4.383
178. Lippmann M, Bress A, Nemeroff CB, Plotsky PM, Monteggia LM. Long-term behavioural and molecular alterations associated with maternal separation in rats. Eur J Neurosci (2007) 25:3091–8. doi:10.1111/j.1460-9568.2007.05522.x
179. Macri S, Chiarotti F, Wurbel H. Maternal separation and maternal care act independently on the development of HPA responses in male rats. Behav Brain Res (2008) 191:227–34. doi:10.1016/j.bbr.2008.03.031
180. Wortwein G, Husum H, Andersson W, Bolwig TG, Mathe AA. Effects of maternal separation on neuropetide Y and calcitonin gene-related peptide in “depressed” Flinders sensitive line rats: a study of gene-environment interactions. Prog Neuropsychopharmacol Biol Psychiatry (2006) 30:684–93. doi:10.1016/j.pnpbp.2006.01.027
181. Krysiak R, Obuchowicz E, Herman Z. Interactions between the neuropeptide Y system and the hypothalamic-pituitary-adrenal axis. Eur J Endocrinol (1999) 140:130–6. doi:10.1530/eje.0.1400130
182. Maniam J, Morris MJ. The link between stress and feeding behaviour. Neuropharmacology (2012) 63:97–110. doi:10.1016/j.neuropharm.2012.04.017
183. Cavagnini F, Croci M, Putignano P, Petroni ML, Invitti C. Glucocorticoids and neuroendocrine function. Int J Obes Relat Metab Disord (2000) 24(Suppl 2):S77–9. doi:10.1038/sj.ijo.0801284
184. Liu D, Diorio J, Day JC, Francis DD, Meaney MJ. Maternal care, hippocampal synaptogenesis and cognitive development in rats. Nat Neurosci (2000) 3:799–806. doi:10.1038/77702
Keywords: early-life stress, metabolic disorders, 11-beta hydroxysteroid dehydrogenase 1, hyperinsulinemia, liver, insulin signaling, glucocorticoids
Citation: Maniam J, Antoniadis C and Morris MJ (2014) Early-life stress, HPA axis adaptation, and mechanisms contributing to later health outcomes. Front. Endocrinol. 5:73. doi: 10.3389/fendo.2014.00073
Received: 31 December 2013; Accepted: 28 April 2014;
Published online: 13 May 2014.
Edited by:
Rachel Yehuda, Icahn School of Medicine at Mount Sinai, USAReviewed by:
Aldo Lucion, Universidade Federal do Rio Grande do Sul, BrazilCopyright: © 2014 Maniam, Antoniadis and Morris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Margaret J. Morris, Department of Pharmacology, School of Medical Sciences, UNSW Australia, Sydney, NSW 2052, Australia e-mail:bS5tb3JyaXNAdW5zdy5lZHUuYXU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.