
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol., 31 March 2014
Sec. Thyroid Endocrinology
Volume 5 - 2014 | https://doi.org/10.3389/fendo.2014.00040
This article is part of the Research TopicThyroid Hormone in Brain and Brain CellsView all 11 articles
 Amy C. Schroeder
Amy C. Schroeder Martin L. Privalsky*
Martin L. Privalsky*Thyroid hormones (THs) are essential for fetal and post-natal nervous system development and also play an important role in the maintenance of adult brain function. Of the two major THs, T4 (3,5,3′,5′-tetraiodo-l-thyronine) is classically viewed as an pro-hormone that must be converted to T3 (3,5,3′-tri-iodo-l-thyronine) via tissue-level deiodinases for biological activity. THs primarily mediate their effects by binding to thyroid hormone receptor (TR) isoforms, predominantly TRα1 and TRβ1, which are expressed in different tissues and exhibit distinctive roles in endocrinology. Notably, the ability to respond to T4 and to T3 differs for the two TR isoforms, with TRα1 generally more responsive to T4 than TRβ1. TRα1 is also the most abundantly expressed TR isoform in the brain, encompassing 70–80% of all TR expression in this tissue. Conversion of T4 into T3 via deiodinase 2 in astrocytes has been classically viewed as critical for generating local T3 for neurons. However, deiodinase-deficient mice do not exhibit obvious defectives in brain development or function. Considering that TRα1 is well-established as the predominant isoform in brain, and that TRα1 responds to both T3 and T4, we suggest T4 may play a more active role in brain physiology than has been previously accepted.
Thyroid hormones (THs) are synthesized by the thyroid gland and are critical regulatory molecules with important roles in vertebrate physiology and development, including fetal and post-natal nervous system development and the maintenance of adult brain function (1, 2). The TH requirement for development is most apparent in the central nervous system (CNS) where severe TH deficiency in fetal and neonatal periods results in cretinism, a disease characterized by mental retardation, deafness, and ataxia; these consequences are irreversible if not treated soon after birth (3–5). Additionally, untreated hypothyroidism in the adult is associated with severe intellectual defects, abnormal balance and defects in fine motor skills, spasticity, and deafness (6). Correcting TH deficiencies is critical for normal brain development and function.
Thyroid hormones mediate CNS effects primarily through thyroid hormone receptors (TRs), members of the nuclear hormone receptor family (4, 7, 8). TRs bind to the DNA regulatory regions of target genes to activate or repress transcription through interactions with accessory proteins known as coregulators. There are two major THs, which bind to and activate TRs: T3 (3,5,3′-triiodo-l-thyronine) and T4 (3,5,3′,5′-tetraiodo-l-thyronine, also known as thyroxine). T4 differs from T3 by an additional iodine located at the 5′-position of the first thyroxine ring. T3 has been assumed to be the active form of TH, as T3 binds to TRs with a greater affinity than T4. In this model, T4 is thought to simply act as a pro-hormone, existing only to be circulated in the serum and converted at the tissue-level to T3 through an enzymatic reaction involving the removal of the 5′-iodine atom from T4 by local deiodinases (9, 10). Nonetheless, it is notable that most of the TH produced under normal conditions in the thyroid is secreted in the form of T4 and steady-state serum concentrations of T4 are many fold greater than those of T3 (11–14). Notably, iodine intake is important for the maintenance of both of these TH levels in circulation. In fact, during gestation and lactation in females, double the normal iodine intake is required to maintain adequate T3 and T4 in circulation to ensure normal fetal development (15, 16). Under conditions of low iodine intake, the serum T3/T4 ratio is somewhat increased reflecting the reduced abundance of iodine atoms (16). Although the ready availability of dietary iodized salt has largely eliminated these iodine deficiencies for school children in most developed countries today, these advances are often not adequate for pregnant and lactating women (17).
Indeed the primary TH crossing the adult blood–brain barrier (BBB) is believed to be T4; therefore, the adult brain may have access to sufficiently high levels of T4 to allow for direct binding to and transcriptional activation of TRs (18, 19). In fact, we know that both T4 and T3 binding by TRs lead to very similar structural changes in the receptor (12). Several reports have also shown that T4 exhibits non-genomic effects by interacting with integrin cell membrane receptors (20). These studies suggest that T4 may exhibit a greater role in physiology than merely acting as a pro-hormone. Therefore, the precise role of T4 as a pro-hormone and whether T4 might function directly as an active hormone in the CNS, remain incompletely answered questions.
Determining the effective cellular concentrations of T4 and T3 in the brain, or in any tissue, is difficult due to the complexities of TH synthesis, transport, and regulation. Vertebrates have developed multiple mechanisms to ensure delivery of appropriate levels of TH to peripheral tissues such as the brain. These include regulation of secretion of THs from the thyroid into serum (21, 22), control of free versus bound levels of THs determined by reversible binding to serum-binding proteins (22), cell-specific expression of TH cell membrane transporters (23, 24), and finally intracellular deiodination of T4 to form T3 [(22, 25); Figure 1].
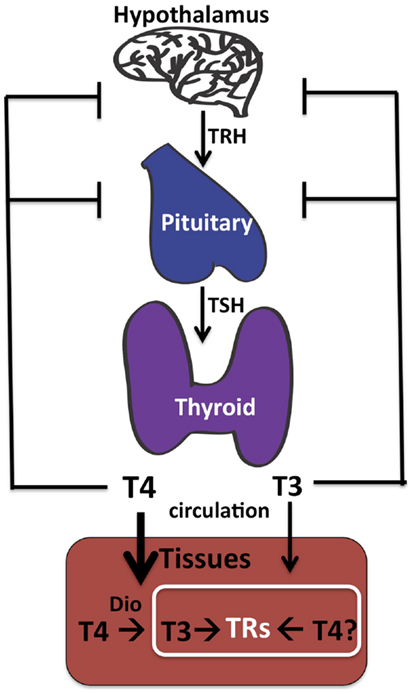
Figure 1. Thyroid hormone synthesis. The thyroid gland makes both T4 and T3, although T4 predominates. The hypothalamus senses low TH in the circulation and responds by stimulating synthesis and secretion of TRH (thyroid releasing hormone), which in turn circulates and stimulates synthesis and secretion of TSH (thyroid stimulating hormone) by the pituitary. Circulating TSH then increases T4 and T3 production by the thyroid and ultimately in the circulation. Tissue-specific deiodinases (“DIO”) are expressed in peripheral tissues such as brain astrocytes to increase local concentrations of T3 from circulating T4. However, we propose that T4 may also act directly on TRs to regulate gene transcription in neurons in the absence of deiodinase 2 conversion to T3.
Transplacental TH transfer from maternal to fetal circulation is particularly important in vertebrate CNS development [reviewed by Ref. (26)] to ensure appropriate levels of TH are available to the fetus throughout development (16). Throughout the first trimester when TH levels are solely obtained through maternal transfer, free T4 levels are high in the fetus, similar to levels of biologically active T4 in adults, whereas fetal concentrations of T3 are at least 10× lower than T4 (16). Notably, T3 levels in the fetal cerebral cortex increase somewhat between 12 and 20 weeks PMA (post-menstrual age) when placental deiodinase 2 levels increase (see below), although maternal serum levels of T3 are still low. Both T4 and T3 in the fetus continue to be transferred from maternal origins through the placenta until half-way through pregnancy when endogenous THs are produced by the fetal thyroid. However, because fetal T4 synthesis is elevated over that of T3 for several weeks at this time, it is possible that an additional window in development exists where fetal circulating T4 is quite high and may act as an active hormone with TRs (16).
Outer ring 5′-monodeiodination via cell-specific deiodinases converts a small fraction of the normal serum T4 pool to T3 (10, 22). Deiodinase 2 is the primary enzyme responsible for intracellular conversion of T4 into T3 in most local tissues including brain, whereas deiodinase 1 is found primarily in the liver (25, 27). Deiodinase 2 is only expressed in selected cell types within the CNS: astrocytes and tanycytes. These are both glial cell-derived and are located in the hypothalamus (28–30). The other deiodinase enzyme expressed in the CNS is deiodinase 3, selectively expressed in neurons. Deiodinase 3 inactivates both T4 and T3 by inner ring deiodination to rT3 and T2 so as to down-regulate local TH concentrations and protect the neuron from supraphysiological levels of TH. Currently it is believed that astrocytes generate active T3 from circulating pro-hormone, T4, whereas neurons degrade both T4 and T3 to inactive rT3 and T2, respectively, and thereby regulate local TH availability within the brain. When levels of TH are low, deiodinase 2 levels in brain increase and contrastingly when there are high levels of TH, deiodinase 3 levels increase (19, 30, 31). This balancing act protects the brain from the detrimental effects of hyper- or hypothyroidism.
T3 concentrations equilibrate rapidly in peripheral tissues such as the liver and kidney but appear to take longer to equilibrate in the brain. In general, TH concentrations in the CNS are approximately 20% that of serum concentrations (32); this is likely due to the added complexity of TH transport across the BBB, which is comprised of the endothelial cells of brain capillaries surrounded by astrocyte end feet. To enter the brain, the THs cross the BBB of the choroid plexus via the MCT8 or OATP1C1 TH transporters. T4 is thought to predominately enter the CNS in preference to T3 as the majority of BBB TH transporters exhibit greater affinities for T4 transport [(19, 33); Figure 2]. As mentioned above, after T4 is taken up into astrocytes likely by OATP1C1, deiodinase 2 can in turn convert it locally to T3. Finally, the astrocyte-generated T3 can enter neuronal cells via the MCT8 transporter to bind and activate TRs. Therefore, it is intriguing that the T4-activating deiodinase is not expressed in the neurons themselves, where the relevant TRs are located, but in the astrocytes. T4 and/or T3 also enter the CNS directly via gaps in the end feet of the astrocytes, which do not completely cover the capillaries in contact with the interstitial spinal fluid (34).
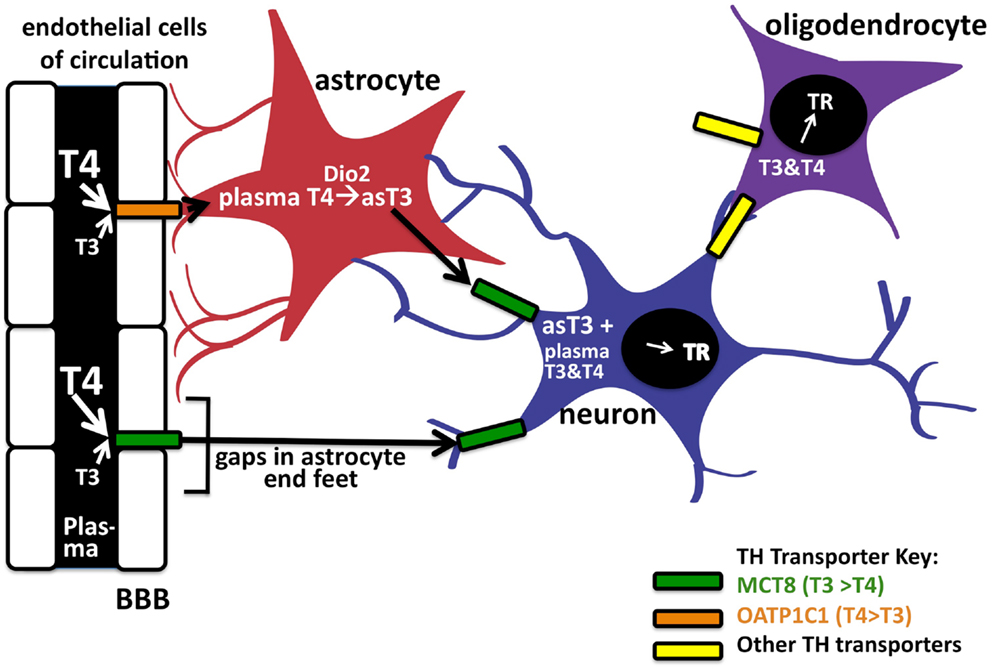
Figure 2. Entry of TH into brain via the blood–brain barrier. TH can enter neurons by two pathways. The first is by crossing the endothelial cells of the blood–brain barrier (BBB) by the OAT1P1C transporter to enter astrocyte end feet (in red). After entering astrocytes, T4 can be converted into T3 via deiodinase 2, to enter the neuron (in blue) by the MCT8 transporter. Circulating T4 and T3 may also enter neurons (and astrocytes) directly via these transporters through gaps in the astrocyte end feet. Oligodendrocytes (purple), which express TRs, are also known TH cell targets in the CNS. There is also evidence of as-yet unknown TH transporters in the brain; the TH transporters, and their known preferences for T4 or T3, are indicated in color codes on the right.
Thyroid hormones bind TRs, ligand-regulated transcription factors, which bind to specific target DNA sequences and repress or activate target genes through the recruitment and release of accessory proteins. TRs contact their DNA-binding elements as protein dimers, heterodimerizing with another member of the nuclear receptor family, RXRs (primarily Retinoid X Receptors), or homodimerizing with themselves (35–39). TRs exhibit bimodal regulation, typically binding corepressors to repress transcription of target genes in the absence of TH, but releasing corepressors and recruiting coactivators to activate transcription of these “positive response” target genes in the presence of TH (40, 41). These corepressor and coactivator proteins alter the chromatin template or interact with the general transcription machinery to produce the appropriate transcriptional outputs. However, many TR target genes display the opposite properties in that they are expressed in the absence of TH and are repressed in the presence of TH; the molecular mechanisms involved in this “negative response” is not well-understood.
Thyroid hormone receptors are encoded by two distinct genetic loci, denoted THRA and THRB, which are each expressed as alternatively spliced mRNAs to create additional receptor diversity [reviewed in Ref. (42)]. Two of the major TR isoforms are referred to as TRα1 and TRβ1; both bind TH and yet exhibit distinct biological roles [reviewed in Ref. (43)]. TRα1 is expressed early in embryonic development and then widely in adults whereas TRβ1 is expressed later in embryonic development and exhibits a more restricted tissue-expression pattern in adults (31, 44–49). Genetic disruption in mice of TRα1 or TRβ1 indicates that these isoforms have somewhat overlapping, yet distinct roles in normal physiology (45–47, 49, 50).
These two different TR isoforms differ in their ability to respond to T4, with TRα1 generally exhibiting a much stronger response to T4 than TRβ1. We suggest that different cell types may modulate their relative ability to respond to T4 versus T3 by altering the relative abundance of different coactivators and corepressors that have distinct responses to T4 and T3, raising the possibility that T4 may be able to function as a direct-acting hormone agonist with TRα1 (Amy C. Schroeder and Martin L. Privalsky, unpublished observations).
Notably, TRα1 encompasses 70–80% of all TR expression in the adult vertebrate brain (2) and TRα1 is present in nearly all neurons (51). Intriguingly, TRα1 is also the predominating TR isoform early in fetal brain development (detected by 8.1 weeks and increasing until 13.9 weeks post-menstrual age). Critical roles in CNS development are known to be mediated by TRα1 including TH-dependent oligodendrocyte differentiation (52). If TRα1 is inactivated, the number of mature oligodendrocytes after T3 treatment is decreased (52). The commitment of these cells as oligodendrocytes is therefore believed to be linked to cell-specific TRα1 expression while the availability of TH regulates the timing of differentiation (52). In fact, maturation of several cell types in the brain in development may depend on specific windows of TRα1 expression and involve a complicated interplay between TRs, THs, and coregulators (2). Additionally, TRα1 is known to exhibit important roles in later stages of neurodevelopment and its expression persists in adult neurons. Therefore, it is interesting that expression of the TRα1 isoform predominates in both fetal and in adult brain at the same times when free T4 levels appear to be at biologically active levels (16), suggesting windows in brain development may exist where T4 may act on TRα1.
As noted above, deiodinase 2 expression does not overlap TR receptor expression in the brain. Deiodinase 2 is expressed instead in astrocytes whereas the TRs are expressed in neurons along with deiodinase 3 [(28, 29); Figure 2]. The current theory therefore suggests astrocytes are involved with T4 uptake from capillaries to subsequently generate a source of locally generated T3. Conversion of T4 into T3 via deiodinase 2 in astrocytes has been estimated to produce as much as 80% of the T3 bound to the TRs in the brain (18), suggesting astrocyte deiodinase 2 is important for generating local T3 concentrations. Therefore, many argue that deiodinase 2 likely plays a critical role in developing brain by providing the necessary amount of T3. If this were in fact the case, one would predict the absence of deiodinase 2 would result in detrimental defects in CNS development similar to that seen in hypothyroidism.
However, the Galton lab produced a deiodinase 2-deficient and a deiodinase 2/deiodinase 1 dual-deficient mouse (KOs) without any evident defects in brain development or function (27, 53). The deiodinase KO mice demonstrated slightly elevated circulating T4 and TSH levels, and normal thyroid-secretion of T3 but no tissue-level production of T3 from T4 (27). Notably, these mice did not display any signs of hypothyroidism and have no gross physiological or behavioral abnormalities (27). The deiodinase KO was also combined with an MCT8 TH transporter knockout (54, 55); this combination resulted in minor neuronal defects mostly noted by decreased expression of genes in the neural cortex, which are usually positively regulated by T3, however, most neural development and function was normal. KO mice studies suggest that T3 transport into the brain and local conversion of T4 to T3 in the brain are not essential for normal brain function in mice, and suggest that CNS T3-defects do not produce syndromes as severe as that seen in the hypothyroid mice (27).
Many suggest that there might be compensation in the deiodinase KO mice through the absorption of more T3 directly from circulation via the MCT8 transporter in endothelial cells of the BBB, but it should be again noted that the parallel transporters such as OATP1C1 and OATP2 favor T4 transport (56, 57) and it is unlikely that T3 can be transported into the brain at rate equivalent to T4 transport. We suggest that in the absence of available T3, T4 can act as an active TH in the brain working on, most likely, TRα1. Interestingly, in the absence of deiodinase 1 and 2, positively regulated TH genes in the cerebral cortex remain unaffected but negatively regulated TH genes appear to be impaired in a way that parallel the hypothyroid mice (27, 58). Perhaps in the absence of deiodinase 2, T4 can act as an active hormone in brain cells to activate positively regulated TH genes, but not to repress negatively regulated TH genes.
It should be noted that humans with MCT8 mutations display severe neurodevelopmental defects with psychomotor retardation and abnormal serum TH levels (57, 59). Contrastingly, MCT8 KO mice mimic the human MCT8 mutations in their thyroid phenotype but display no obvious brain developmental defects (57, 59). It is therefore possible that the need for locally produced T3, and/or the presence of alternative T3-specific transporters, differ in mice and in humans (55).
T4 efficiently recruits many coactivators to TRα1, with certain well-established TR coactivators (SRC1 and TRAP220) exhibiting a T4 response equal or near equal to that induced by T3 (Amy C. Schroeder and Martin L. Privalsky, unpublished data). SRC1 mRNA is expressed in many tissues during development including the CNS (60). TRAP220 is also expressed in the developing brain and is thought to play a regulatory role in the process of cell proliferation and differentiation, in learning, and in memory formation (61). The widespread expression of TRAP220 in the developing brain appears to parallel TRα1 expression. Therefore, CNS development correlates with a high level of expression of TRα1 together with TRAP220 and/or SRC1 and may provide an opportunity for T4 to directly regulate gene transcription. CNS cell-specific differences in TR isoform and cofactor levels or function are likely to contribute to differences in T4 hormone response and may suggest a means by which the T4 sensitivity of a given CNS cell type can be regulated in response to internal or external signals.
Several recent studies have led to the view that T4 exhibits non-genomic roles that do not require conversion to T3 (20) but which have not challenged the general view that T3, not T4, is the only direct, biologically relevant agonist for nuclear TR function. Our own experiments indicate that TRα1 has the potential to act as a dual sensor of both T4 and T3 (Amy C. Schroeder and Martin L. Privalsky, unpublished observations).
Although the effective concentration of T4 in the brain is difficult to determine, it is plausible that T4 levels are sufficient to induce activation of TRα1-regulated genes in the brain even in the absence of T3. We suggest that the normal mix of T4 and T3 in the brain may actually confer a mixed T4/T3 transcription response mediated primarily by TRα1, together with a more pure T3 response mediated primarily by TRβ1. Notably, mice in which both deiodinase 1 and 2 have been genetically ablated, and thus lack astrocyte deiodinase conversion of T4 to T3, display only very mild defects in their physiological with little to no neurological defects (27). If, as indicated by these knockouts, T4 is not absolutely required in its traditional role as a pro-hormone, the dominance of T4 to T3 in the circulation and transport into the CNS may instead reflect a novel role of T4 as a direct-acting hormone and this direct role may be helping to ameliorate the effects of the deiodinase knockouts in the CNS.
In conclusion, TH endocrinology in the CNS is tightly regulated at multiple tiers. Negative feedback loops in the hypothalamus and the pituitary control T3 and T4 output by the thyroid gland itself. Further, multiple phenomenon functions together to modulate the transport of circulating TH through the BBB, and multiple transporters act together to directly alter TH availability in the CNS itself. Additionally, conversion of intracellular T4 into T3 by deiodinase 2, inactivation of both T3 and T4 by deiodinase 3, and, the ability of different TR isoforms and different coregulators to respond directly to T4 versus T3 further regulate the CNS response to TH. Operating together, we propose these mechanisms serve to maintain proper endocrine homeostasis while permitting the CNS to respond to developmental and physiological needs.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Morreale de Escobar G, Obregon MJ, Escobar Del Rey F. Role of thyroid hormone during early brain development. Eur J Endocrinol (2004) 151(Suppl 3):U25–37. doi:10.1530/eje.0.151U025
2. Wallis K, Dudazy S, Van Hogerlinden M, Nordstrom K, Mittag J, Vennstrom B. The thyroid hormone receptor alpha1 protein is expressed in embryonic postmitotic neurons and persists in most adult neurons. Mol Endocrinol (2010) 24:1904–16. doi:10.1210/me.2010-0175
3. Galton VA. The roles of the iodothyronine deiodinases in mammalian development. Thyroid (2005) 15:823–34. doi:10.1089/thy.2005.15.823
4. Laurberg P. Thyroid function: thyroid hormones, iodine and the brain-an important concern. Nat Rev Endocrinol (2009) 5:475–6. doi:10.1038/nrendo.2009.155
5. Arrojo EDR, Fonseca TL, Werneck-De-Castro JP, Bianco AC. Role of the type 2 iodothyronine deiodinase (D2) in the control of thyroid hormone signaling. Biochim Biophys Acta (2013) 1830:3956–64. doi:10.1016/j.bbagen.2012.08.019
6. DeLong GR, Stanbury JB, Fierro-Benitez R. Neurological signs in congenital iodine-deficiency disorder (endemic cretinism). Dev Med Child Neurol (1985) 27:317–24. doi:10.1111/j.1469-8749.1985.tb04542.x
7. Thompson CC, Potter GB. Thyroid hormone action in neural development. Cereb Cortex (2000) 10:939–45. doi:10.1093/cercor/10.10.939
8. Quignodon L, Legrand C, Allioli N, Guadano-Ferraz A, Bernal J, Samarut J, et al. Thyroid hormone signaling is highly heterogeneous during pre- and postnatal brain development. J Mol Endocrinol (2004) 33:467–76. doi:10.1677/jme.1.01570
9. Gereben B, Zeold A, Dentice M, Salvatore D, Bianco AC. Activation and inactivation of thyroid hormone by deiodinases: local action with general consequences. Cell Mol Life Sci (2008) 65:570–90. doi:10.1007/s00018-007-7396-0
10. St Germain DL, Galton VA, Hernandez A. Minireview: defining the roles of the iodothyronine deiodinases: current concepts and challenges. Endocrinology (2009) 150:1097–107. doi:10.1210/en.2008-1588
11. Yen PM. Thorough physiological review – physiological and molecular basis of thyroid hormone action. Physiol Rev (2001) 81:1097–142.
12. Sandler B, Webb P, Apriletti JW, Huber BR, Togashi M, Cunha Lima ST, et al. Thyroxine-thyroid hormone receptor interactions. J Biol Chem (2004) 279:55801–8. doi:10.1074/jbc.M410124200
13. Larsen PR, Davies TF, Schlumberger M-J, Hay ID. Thyroid physiology and diagnostic evaluation of patients with thyroid disorders. In: Larsen PR, Kronenberg HM, Melmed S, Polonsky KS, editors. Williams Textbook of Endocrinology. Philadelphia: Saunders (2003). p. 331–73.
14. Galton VA. The role of 3,5,3′-triiodothyronine in the physiological action of thyroxine in the premetamorphic tadpole. Endocrinology (1989) 124:2427–33. doi:10.1210/endo-124-5-2427
15. Zimmermann MB. The impact of iodised salt or iodine supplements on iodine status during pregnancy, lactation and infancy. Public Health Nutr (2007) 10:1584–95. doi:10.1017/S1368980007360965
16. de Escobar GM, Ares S, Berbel P, Obregon MJ, Del Rey FE. The changing role of maternal thyroid hormone in fetal brain development. Semin Perinatol (2008) 32:380–6. doi:10.1053/j.semperi.2008.09.002
17. Stagnaro-Green A, Abalovich M, Alexander E, Azizi F, Mestman J, Negro R, et al. Guidelines of the American Thyroid Association for the diagnosis and management of thyroid disease during pregnancy and postpartum. Thyroid (2011) 21:1081–125. doi:10.1089/thy.2011.0087
18. Crantz FR, Silva JE, Larsen PR. An analysis of the sources and quantity of 3,5,3′-triiodothyronine specifically bound to nuclear receptors in rat cerebral cortex and cerebellum. Endocrinology (1982) 110:367–75. doi:10.1210/endo-110-2-367
19. Heuer H. The importance of thyroid hormone transporters for brain development and function. Best Pract Res Clin Endocrinol Metab (2007) 21:265–76. doi:10.1016/j.beem.2007.03.003
20. Davis PJ, Davis FB, Lin HY, Mousa SA, Zhou M, Luidens MK. Translational implications of nongenomic actions of thyroid hormone initiated at its integrin receptor. Am J Physiol Endocrinol Metab (2009) 297:E1238–46. doi:10.1152/ajpendo.00480.2009
21. O’Shea PJ, Williams GR. Insight into the physiological actions of thyroid hormone receptors from genetically modified mice. J Endocrinol (2002) 175:553–70. doi:10.1677/joe.0.1750553
23. Friesema EC, Ganguly S, Abdalla A, Manning Fox JE, Halestrap AP, Visser TJ. Identification of monocarboxylate transporter 8 as a specific thyroid hormone transporter. J Biol Chem (2003) 278:40128–35. doi:10.1074/jbc.M300909200
24. Friesema EC, Jansen J, Jachtenberg JW, Visser WE, Kester MH, Visser TJ. Effective cellular uptake and efflux of thyroid hormone by human monocarboxylate transporter 10. Mol Endocrinol (2008) 22:1357–69. doi:10.1210/me.2007-0112
25. Bianco AC, Kim BW. Deiodinases: implications of the local control of thyroid hormone action. J Clin Invest (2006) 116:2571–9. doi:10.1172/JCI29812
26. Chan SY, Vasilopoulou E, Kilby MD. The role of the placenta in thyroid hormone delivery to the fetus. Nat Clin Pract Endocrinol Metab (2009) 5:45–54. doi:10.1038/ncpendmet1026
27. Galton VA, Schneider MJ, Clark AS, St Germain DL. Life without thyroxine to 3,5,3′-triiodothyronine conversion: studies in mice devoid of the 5′-deiodinases. Endocrinology (2009) 150:2957–63. doi:10.1210/en.2008-1572
28. Rodriguez EM, Gonzalez CB, Delannoy L. Cellular organization of the lateral and postinfundibular regions of the median eminence in the rat. Cell Tissue Res (1979) 201:377–408. doi:10.1007/BF00236998
29. Tu HM, Kim SW, Salvatore D, Bartha T, Legradi G, Larsen PR, et al. Regional distribution of type 2 thyroxine deiodinase messenger ribonucleic acid in rat hypothalamus and pituitary and its regulation by thyroid hormone. Endocrinology (1997) 138:3359–68. doi:10.1210/endo.138.8.5318
30. Guadano-Ferraz A, Escamez MJ, Rausell E, Bernal J. Expression of type 2 iodothyronine deiodinase in hypothyroid rat brain indicates an important role of thyroid hormone in the development of specific primary sensory systems. J Neurosci (1999) 19:3430–9.
31. Calvo R, Obregon MJ, Ruiz De Ona C, Escobar Del Rey F, Morreale de Escobar G. Congenital hypothyroidism, as studied in rats. Crucial role of maternal thyroxine but not of 3,5,3′-triiodothyronine in the protection of the fetal brain. J Clin Invest (1990) 86:889–99. doi:10.1172/JCI114790
32. Dratman MB, Crutchfield FL, Schoenhoff MB. Transport of iodothyronines from bloodstream to brain: contributions by blood:brain and choroid plexus:cerebrospinal fluid barriers. Brain Res (1991) 554:229–36. doi:10.1016/0006-8993(91)90194-Z
33. Chatonnet F, Picou F, Fauquier T, Flamant F. Thyroid hormone action in cerebellum and cerebral cortex development. J Thyroid Res (2011) 2011:145762. doi:10.4061/2011/145762
34. Mathiisen TM, Lehre KP, Danbolt NC, Ottersen OP. The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia (2010) 58:1094–103. doi:10.1002/glia.20990
35. Lazar MA, Berrodin TJ, Harding HP. Differential DNA binding by monomeric, homodimeric, and potentially heteromeric forms of the thyroid hormone receptor. Mol Cell Biol (1991) 11:5005–15.
36. Naar AM, Boutin JM, Lipkin SM, Yu VC, Holloway JM, Glass CK, et al. The orientation and spacing of core DNA-binding motifs dictate selective transcriptional responses to three nuclear receptors. Cell (1991) 65:1267–79. doi:10.1016/0092-8674(91)90021-P
37. Forman BM, Casanova J, Raaka BM, Ghysdael J, Samuels HH. Half-site spacing and orientation determines whether thyroid hormone and retinoic acid receptors and related factors bind to DNA response elements as monomers, homodimers, or heterodimers. Mol Endocrinol (1992) 6:429–42. doi:10.1210/mend.6.3.1316541
38. Wahlstrom GM, Sjoberg M, Andersson M, Nordstrom K, Vennstrom B. Binding characteristics of the thyroid hormone receptor homo- and heterodimers to consensus AGGTCA repeat motifs. Mol Endocrinol (1992) 6:1013–22. doi:10.1210/me.6.7.1013
39. Kurokawa R, Yu VC, Naar A, Kyakumoto S, Han Z, Silverman S, et al. Differential orientations of the DNA-binding domain and carboxy-terminal dimerization interface regulate binding site selection by nuclear receptor heterodimers. Genes Dev (1993) 7:1423–35. doi:10.1101/gad.7.7b.1423
40. Privalsky ML, Lee S, Hahm JB, Young BM, Fong RN, Chan IH. The p160 coactivator PAS-B motif stabilizes nuclear receptor binding and contributes to isoform-specific regulation by thyroid hormone receptors. J Biol Chem (2009) 284:19554–63. doi:10.1074/jbc.M109.007542
41. Cheng SY, Leonard JL, Davis PJ. Molecular aspects of thyroid hormone actions. Endocr Rev (2010) 31:139–70. doi:10.1210/er.2009-0007
42. Rosen MD, Privalsky ML. Thyroid hormone receptor mutations found in renal clear cell carcinomas alter corepressor release and reveal helix 12 as key determinant of corepressor specificity. Mol Endocrinol (2009) 23:1183–92. doi:10.1210/me.2009-0126
43. Chan IH, Privalsky ML. Isoform-specific transcriptional activity of overlapping target genes that respond to thyroid hormone receptors alpha1 and beta1. Mol Endocrinol (2009) 23:1758–75. doi:10.1210/me.2009-0025
44. Chatonnet F, Guyot R, Benoit G, Flamant F. Genome-wide analysis of thyroid hormone receptors shared and specific functions in neural cells. Proc Natl Acad Sci U S A (2013) 110:E766–75. doi:10.1073/pnas.1210626110
45. Murata Y. Multiple isoforms of thyroid hormone receptor: an analysis of their relative contribution in mediating thyroid hormone action. Nagoya J Med Sci (1998) 61:103–15.
46. Forrest D, Vennstrom B. Functions of thyroid hormone receptors in mice. Thyroid (2000) 10:41–52. doi:10.1089/thy.2000.10.41
47. Zhang J, Lazar MA. The mechanism of action of thyroid hormones. Annu Rev Physiol (2000) 62:439–66. doi:10.1146/annurev.physiol.62.1.439
48. Wondisford FE. Thyroid hormone action: insight from transgenic mouse models. J Investig Med (2003) 51:215–20. doi:10.2310/6650.2003.39191
49. Cheng SY. Isoform-dependent actions of thyroid hormone nuclear receptors: lessons from knockin mutant mice. Steroids (2005) 70:450–4. doi:10.1016/j.steroids.2005.02.003
50. Flamant F, Samarut J. Thyroid hormone receptors: lessons from knockout and knock-in mutant mice. Trends Endocrinol Metab (2003) 14:85–90. doi:10.1016/S1043-2760(02)00043-7
51. Schwartz HL, Strait KA, Ling NC, Oppenheimer JH. Quantitation of rat tissue thyroid hormone binding receptor isoforms by immunoprecipitation of nuclear triiodothyronine binding capacity. J Biol Chem (1992) 267:11794–9.
52. Heuer H, Mason CA. Thyroid hormone induces cerebellar Purkinje cell dendritic development via the thyroid hormone receptor alpha1. J Neurosci (2003) 23:10604–12.
53. Schneider MJ, Fiering SN, Pallud SE, Parlow AF, St Germain DL, Galton VA. Targeted disruption of the type 2 selenodeiodinase gene (DIO2) results in a phenotype of pituitary resistance to T4. Mol Endocrinol (2001) 15:2137–48. doi:10.1210/me.15.12.2137
54. Liao XH, Di Cosmo C, Dumitrescu AM, Hernandez A, Van Sande J, St Germain DL, et al. Distinct roles of deiodinases on the phenotype of MCT8 defect: a comparison of eight different mouse genotypes. Endocrinology (2011) 152:1180–91. doi:10.1210/en.2010-0900
55. Morte B, Ceballos A, Diez D, Grijota-Martinez C, Dumitrescu AM, Di Cosmo C, et al. Thyroid hormone-regulated mouse cerebral cortex genes are differentially dependent on the source of the hormone: a study in monocarboxylate transporter-8- and deiodinase-2-deficient mice. Endocrinology (2010) 151:2381–7. doi:10.1210/en.2009-0944
56. Roberts LM, Woodford K, Zhou M, Black DS, Haggerty JE, Tate EH, et al. Expression of the thyroid hormone transporters monocarboxylate transporter-8 (SLC16A2) and organic ion transporter-14 (SLCO1C1) at the blood-brain barrier. Endocrinology (2008) 149:6251–61. doi:10.1210/en.2008-0378
57. Ceballos A, Belinchon MM, Sanchez-Mendoza E, Grijota-Martinez C, Dumitrescu AM, Refetoff S, et al. Importance of monocarboxylate transporter 8 for the blood-brain barrier-dependent availability of 3,5,3′-triiodo-l-thyronine. Endocrinology (2009) 150:2491–6. doi:10.1210/en.2008-1616
58. Hernandez A, Morte B, Belinchon MM, Ceballos A, Bernal J. Critical role of types 2 and 3 deiodinases in the negative regulation of gene expression by T(3)in the mouse cerebral cortex. Endocrinology (2012) 153:2919–28. doi:10.1210/en.2011-1905
59. Ferrara AM, Liao XH, Gil-Ibanez P, Marcinkowski T, Bernal J, Weiss RE, et al. Changes in thyroid status during perinatal development of MCT8-deficient male mice. Endocrinology (2013) 154:2533–41. doi:10.1210/en.2012-2031
60. Iannacone EA, Yan AW, Gauger KJ, Dowling AL, Zoeller RT. Thyroid hormone exerts site-specific effects on SRC-1 and NCoR expression selectively in the neonatal rat brain. Mol Cell Endocrinol (2002) 186:49–59. doi:10.1016/S0303-7207(01)00672-4
Keywords: T4 thyronine, T3 thyronine, thyroid hormone receptor, brain, coregulator, deiodinase 2
Citation: Schroeder AC and Privalsky ML (2014) Thyroid hormones, T3 and T4, in the brain. Front. Endocrinol. 5:40. doi: 10.3389/fendo.2014.00040
Received: 10 January 2014; Accepted: 17 March 2014;
Published online: 31 March 2014.
Edited by:
Frédéric Flamant, Ecole Normale Supérieure de Lyon, FranceReviewed by:
Maria Moreno, University of Sannio, ItalyCopyright: © 2014 Schroeder and Privalsky. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin L. Privalsky, Department of Microbiology and Molecular Genetics, College of Biological Sciences, University of California Davis, One Shields Avenue, Davis, CA 95616, USA e-mail:bWxwcml2YWxza3lAdWNkYXZpcy5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.