 Brennan Ferguson1
Brennan Ferguson1 Vikas Agrawal
Vikas Agrawal Shikha Sharma
Shikha Sharma J. Alexandra Hakala
J. Alexandra Hakala Wei Xiong
Wei Xiong- 1Department of Geology and Geography, West Virginia University, Morgantown, WV, United States
- 2National Energy Technology Laboratory, U.S. Department of Energy, Pittsburgh, PA, United States
- 3Leidos Research Support Team, Pittsburgh, PA, United States
Natural gas extracted from tight shale formations, such as the Marcellus Shale, represents a significant and developing front in energy exploration. By fracturing these formations using pressurized fracturing fluid, previously unobtainable hydrocarbon reserves may be tapped. While pursuing this resource, hydraulic fracturing operations leave chemically complex fluids in the shale formation for at least two weeks. This provides a substantial opportunity for the hydraulic fracturing fluid (HFF) to react with the shale formation at reservoir temperature and pressure. In this study, we investigated the effects of the carbonates on shale-HFF reactions with a focus on the Marcellus Shale. We performed autoclave experiments at high temperature and pressure reservoir conditions using a carbonate-rich and a decarbonated or carbonate-free version of the same shale sample. We observed that carbonate minerals buffer the pH of the solution, which in turn prevents clay dissolution. Carbonate and bicarbonate ions also scavenge reactive oxidizing species (ROS), which prevents oxidation of shale organic matter and volatile organic compounds (VOCs). Carbonate-free samples also show higher pyrite dissolution compared to the carbonate-rich sample due to chelation reactions. This study demonstrates how carbonate minerals (keeping all other variables constant) affect shale-HFF reactions that can potentially impact porosity, microfracture integrity, and the release of heavy metals and volatile organic contaminants in the produced water.
1 Introduction
The use of hydraulic fracturing to extract natural gas from shale reservoirs has become a widespread practice of considerable importance to America’s energy portfolio (U.S. EIA, 2017). The Marcellus Shale is one of the major shale reservoirs that has been utilized in this boom, making hydraulic fracturing an enormously important industry to West Virginia and the Appalachian region. Hydraulic fracturing, often referred to as fracturing or fracking, is generically the fracturing of rocks around a wellbore in order to increase the permeability of the rock and therefore enable gas flow from the reservoir. Many wells are drilled in close proximity on a well pad and, after reaching a specific depth, the drill pivots to go horizontally into the target formation (e.g., the Marcellus Shale). This multitude of wells creates a wheel-spoke pattern in the gas-bearing shale formation and dramatically increases the yield of hydrocarbons from shale compared to what is available through conventional means (Arthur et al., 2008). Approximately 4.25 million gallons of water per well are used to hydraulically fracture the Marcellus Shale (Kondash and Vengosh, 2015), and is mixed with a variety of chemicals to create fractures and maintain the well integrity. To prop open these fractures, silica proppant is injected along with gelling agents to help push the proppant into place (PA DEP, 2010). This is followed by oxidative breakers, such as persulfates, which break down the gel in order to recover it following a shut-in period of several days to weeks (Marcon et al., 2017). After this shut-in period, a portion of this water and the natural brine in the formation, totaling approximately 1.37 million gallons per well in the Marcellus Shale (Kondash and Vengosh, 2015), are produced in order to recover the natural gas.
The Marcellus Shale is the most productive shale formation for dry gas in the United States (EIA, 2016) and is economically vital to the Appalachian region. However, within the Marcellus shale Formation, there can be significant variations in total organic carbon, organic matter type, thermal maturity, and mineralogy (U.S. EIA, 2017; Agrawal and Sharma, 2018a, 2018b, 2018c, 2020; Pilewski et al., 2019; Sharma et al., 2020a; Sharma et al., 2020b). Due to these variations in physicochemical properties, the chemical reactions that occur within hydraulically fractured unconventional formations can vary significantly, and influence the selection of hydraulic fracturing fluid (HFF) components used at any given well site (Abualfaraj et al., 2014).
During hydraulic fracturing operations, the mixture of water and chemical additives that comprise HFF is in contact with the target shale formation for weeks and reacts with the brine and shale in the formation at high temperature and pressure. During this time, numerous interactions can take place that significantly alter HFF fluid chemistry, the mineral composition and petrophysical properties of shale, and the release of organic and inorganic contaminants (Hoelzer et al., 2016; Harrison et al., 2017; Paukert Vankeuren et al., 2017; Sumner and Plata, 2018a, 2018b, 2019; Pilewski et al., 2019; Hakala et al., 2021). Studies conducted using benchtop reactors showed that multiple shale-HFF reactions could occur, such as mineral precipitation and dissolution, organo-metallic complex formation, ion adsorption onto shale organic matter and clay minerals, and organic matter degradation (Jew et al., 2017; Sumner and Plata, 2018a; Pilewski et al., 2019; Hakala et al., 2021). Among these, mineral dissolution and precipitation reactions impact the porosity and permeability of shale the most. Changes in flow dynamics from precipitation reactions have been implicated as a potential cause for discrepancies between modeled and actual late-stage hydrocarbon production in hydraulically fractured wells (Jew et al., 2017). Dissolution of shale minerals and organic matter degradation can increase the porosity and permeability of shale but increases the toxicity of produced waters (Harrison et al., 2017; Armstrong et al., 2019; Sharma et al., 2020; Donmoyer et al., 2021) and the risks posed by produced water spills.
Carbonate mineral reactions are one of the critical variables controlling reservoir geochemistry (Jew et al., 2017; Pilewski et al., 2019). Carbonate minerals play a dominant role by buffering the pH of shale-HFF interactions, which significantly impacts most reactions taking place in the reservoir and affects the stability of all other reactive components. The Marcellus Shale has a variable composition of carbonate minerals with calcite (CaCO3) ranging from 3 to 48% and dolomite (CaMg(CO3)2) ranging from 0 to 10%, as well as trace amounts of other carbonates such as siderite (Morsy et al., 2013; U.S. EIA, 2017). Carbonates react with acids in HFF, such as HCl, and release Ca2+, Mg2+ (in the case of dolomite), HCO3−, and trace metals such as Sr2+ via dissolution or ion exchange. The reaction neutralizes acid through the formation of HCO3−, which further buffers the chemical system and controls the pH (Wang et al., 2015; Pilewski et al., 2019). The extent of the reaction between HFF and carbonate minerals depends on the carbonate content of the shale formation and acid added in the HFF (Wang et al., 2015, 2016). Previous work on shale-HFF interactions demonstrated a strong relationship between increases in pH, Ca, Mg, and dissolved inorganic carbon (DIC) proportionate to the amount of carbonate in the system (Wang et al., 2015; Pilewski et al., 2019). X-ray diffraction analysis demonstrated commensurate losses of the primary carbonate minerals in reservoir shales, calcite and dolomite, to the fluid chemistry changes (Pilewski et al., 2019). Carbonate mineral dissolution can have either a desirable effect for gas extraction by increasing pore size (Paukert Vankeuren et al., 2017) or cause instability and collapse in microfractures depending on the particular physical dynamics of the well (Harrison et al., 2017; Jew et al., 2017; Pilewski et al., 2019).
Although numerous studies have investigated shale-HFF reactions to understand the influence of these reactions on shale permeability and fluid flow (Wang et al., 2015; Harrison et al., 2017; Jew et al., 2017; Pilewski et al., 2019), the influence of other variables such as mineralogy (e.g., varied clay and pyrite content), organic matter type and amount, and different fracturing fluid composition affected the ability to isolate the specific impact of carbonate minerals on shale-HFF interactions. Evaluating the impact of carbonate content on shale-HFF reactions requires keeping other experimental variables constant. Further, most prior studies were conducted at ambient pressure conditions and may not best represent in situ reservoir conditions. The aim of this study is to isolate the effects of carbonate mineral content on the shale-HFF reactions, keeping all the other variables constant, and to represent reservoir conditions using high pressure temperature reactors. Developing a refined understanding of how carbonates affect the geochemistry of hydraulic fracturing operations will allow operators to create targeted strategies for unconventional reservoir design and management.
2 Materials and Methods
2.1 Sample Preparation
A series of experiments were conducted to isolate the effects of calcium carbonate on shale-HFF interactions. We utilized a leftover sample of Marcellus Shale that was fully characterized in our laboratory (Pilewski et al., 2019). Sample LM-2 is a relatively immature Marcellus shale sample (VRo = 0.8) with high total organic carbon (15.4 wt%), high carbonate content (21 wt% calcite, 5 wt% dolomite), moderate mixed clay content (42 wt%), and moderate pyrite content (5 wt%) (Pilewski et al., 2019) (Table 1). The minerals wt% were calculated to the nearest unit, and the total sum of the minerals was within 100 ± 1 wt%. This Marcellus Shale sample was chosen because it had the highest carbonate content in our sample collection.

TABLE 1. LM-2 shale depth, thermal maturity, organic content and mineral composition. Error is ±1% (Pilewski et al., 2019).
Sample exteriors were removed using a cleaned Dremel tool before grinding to prevent any contaminating drill mud. Shale samples were ground to 100 mesh to maximize surface area for reaction, with a small amount of shale cores broken into ∼0.25 cm2 chips for SEM-EDS analysis. The ground shale and shale chips were divided into two samples: SH-HFF, which was not altered, and SH-HFF (-CO3), which had carbonate minerals removed. Carbonate removal in SH-HFF (-CO3) was accomplished by selective digestion using trace metal grade hydrochloric acid, following the procedure outlined in (Riley et al., 2012). Hydraulic fracturing fluid, in the absence of shale, was also reacted for 14 days which acted as a control to determine chemical changes caused by elevated temperature and pressure reactor conditions and was named HFF.
All pH measurements were taken with a calibrated YSI Pro Series Instrument equipped with a YSI Pro Series 1,001 pH sensor. A separate portion of the rock chips was also acid digested to collect pre-reaction SEM-EDS for comparison with the post-reaction carbonate-free experiment. This allows for comparison of pre-and post-experimental samples without changing the remaining mass of the primary acid digested sample for the reactor experiment.
The mixture of synthetic brine and HFF was prepared using the methods reported in (Paukert Vankeuren et al., 2017) (Table 2). Some proprietary chemicals that were used as part of the mixture in previous studies were no longer available. These included a gelling agent WGA-15L, a clay stabilizer WCS-631LC, a friction reducer WFR-61LA, and a corrosion inhibitor WAI-251LC. Substitutes for these were based upon MSDS sheet data and common chemicals used in the same region for the same purposes per FracFocus. Petroleum distillates were used in place of the proprietary gelling agent and friction reducer, choline chloride replaced the proprietary clay stabilizer, and cinnamaldehyde replaced the proprietary corrosion inhibitor.

TABLE 2. Fracturing fluid additives and brine salts used to make the synthetic fracturing fluid mixture used in reactions.
2.2 Experimental Set-Up and Analysis
The shale-HFF reactions were conducted in 600 ml Parr 4768 static reactors to simulate high pressure temperature conditions in the reservoir. The reactions were conducted at 100°C and ∼2,500 psi for 14 days to mimic shut-in phase duration and conditions (Marcon et al., 2017; Paukert Vankeuren et al., 2017; Pilewski et al., 2019). Inert N2 (100% pure) was used to pressurize the reactors. A borosilicate glass sleeve containing 420 ml of HFF, 20 g of 100 mesh shale powder, and 1 gram of shale chips at a fixed mass ratio of 20:1 solid: liquid were placed inside the reactors, with the shale and HFF mixed immediately before the pressurization and heating of the reactors, following the methods of Macron et al., 2017. The control experiment was conducted with 420 ml of synthetic HFF mixture with no shale.
Upon completion of the reactions at 14 days, samples were collected and analyzed by ion chromatography (IC), Total Organic Carbon/Total Inorganic Carbon (TOC/TIC) analyzer, gas chromatography-mass spectrometry (GC-MS), and inductively coupled plasma mass spectrometry (ICP-MS). The IC samples were filtered to 0.22 microns using a syringe filter and collected in 10 ml plastic vials with zero headspace. DIC samples were also collected in 10 ml plastic vials with zero headspace. GC-MS samples were collected with zero headspace in 60 ml amber volatile organics analysis (VOA) vials acidified with HCl. Samples for ICP-MS were collected with zero headspace 10 ml plastic vials after being filtered to 0.45 μm and acidified with nitric acid. The reacted shale was vacuum filtered and oven-dried at 50°C. All collected samples were refrigerated immediately after collection.
Ion chromatography (IC), inductively coupled plasma mass spectrometry (ICP-MS), and total dissolved organic and inorganic carbon (TOC/TIC) analyses were performed at the NETL Pittsburgh campus. IC was measured on a ThermoFisher ICS-5000+ with AS11-HC column for anion and CS16 column for cation quantification. This method provided sulfate data with an error of less than 4% in each of the four standards run with the samples. The lower and upper limits of detection for sulfate were 0.2 and 25 mg/L, respectively. At the 1:10 dilution factor, all of the samples were within the calibrated range. Citrate data gathered with this method had 4%, and 3% errors in the two standards. The lower and upper calibration limits for citrate were 0.1–0.5 mg/L. Trace metals present in fluids were measured using a Perkin Elmer Nexion 300D ICPMS instrument. Samples were run at 1:100 dilution. The duplicate analysis of samples showed a relative percent difference of less than 4%. TOC/TIC analysis was conducted using a Shimadzu Total Organic Carbon/Total Inorganic Carbon (TOC/TIC) analyzer. The highest error in TIC/TOC analysis was 14.0%, and the average error was 11.76%.
GC-MS analysis was conducted by Pace Analytical Services following EPA method SW8260B. Samples were put on ice immediately after collection and were brought to Pace Analytical Services’ Morgantown Branch immediately after sampling and were analyzed within 24 h of sampling. In accordance with method SW8260B, samples were purged from the aqueous sample with helium gas flowing at 40 ml/min for 11 min onto a Supelco Trap A, Tenax 24 cm sorbent trap. Volatile organics were liberated from the trap by heating it to 180°C and flushed into the GC-MS.
Qualitative analysis of spatial elemental distribution performed on the shale samples was performed using the Oxford INCA EDS capabilities of the JEOL JSM-7600F SEM at the West Virginia University’s Shared Research Facility. The chip portion of the dried reacted shales was separated and mounted on aluminum pin mounts with carbon tape. Each mounted chip was sputter-coated with a gold-palladium source to prevent charging during SEM analysis.
Mineral saturation indices were calculated using Geochemists’ Workbench with Pitzer and Minteq databases based on measured species concentrations and pH. Geochemical modeling of clay mineral solubility at different pH and reservoir temperatures was performed using the Visual MINTEQ 3.1 modeling program. The SIT method of correction was used for ionic strength, which was set at 0.5 to avoid overloading the program with a higher value. The model was run for kaolinite to determine the mole percent dissolved under the pH conditions of each shale reaction.
3 Results
3.1 pH of the Solution
A contrasting difference was observed in pH of the carbonate-rich and carbonate-free experiments. The pH of the SH-HFF experiment rose from 2.2 to 6.2 after the reaction, whereas in the SH-HFF (-CO3), pH was similar (1.79 before and 1.85 after) as shown in Figure 1. The pH of the control sample (HFF) also remained similar before and after the reaction (2.25 before and 2.32 after).
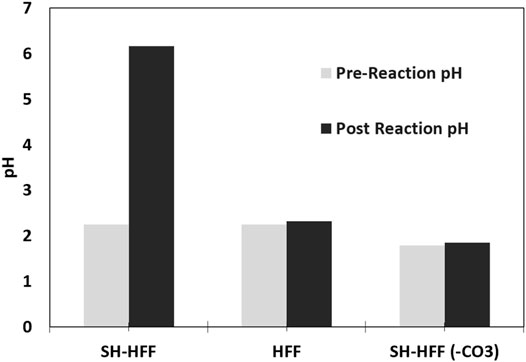
FIGURE 1. pH measured immediately before reaction vs post-reaction pH for each sample.
3.2 Ions in Solution
Post reaction aluminum, iron, arsenic, cadmium, and uranium concentrations were all higher in SH-HFF (-CO3) than in either SH-HFF or HFF (Table 3). Iron in solution was over 800 times higher in SH-HFF (-CO3) than in SH-HFF. Cadmium and arsenic were 8.7 and 10.5 times more concentrated in SH-HFF (-CO3) than in SH-HFF, respectively, as shown in Figure 2. Iron was also detected in the HFF sample at 36.42 mg/L though no shale was added, nor was any iron included in the HFF. This likely indicates some corrosion of the stainless-steel temperature probe in the reactor by the acidic conditions. We also observed aluminum concentrations in SH-HFF (-CO3) with two orders of magnitude higher than the other samples. Based on XRD results, the primary sources of aluminum in our shale samples are aluminosilicate clays which comprised 42% of the mineral content (Pilewski et al., 2019).
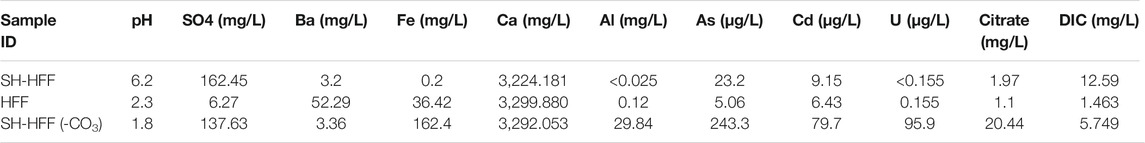
TABLE 3. pH, IC and ICP-MS fluid chemistry results for each reaction and control fracturing fluid (with no shale).
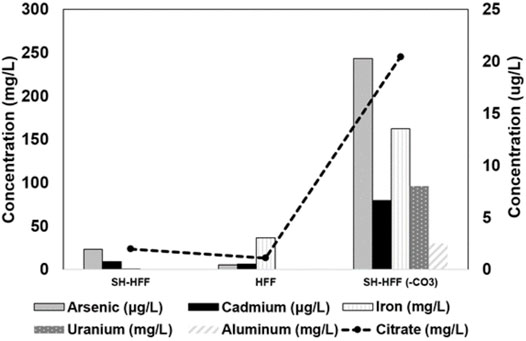
FIGURE 2. Metals in solution released from reactions with shale. HFF serves as a control for contaminants in additives, reactor corrosion (Fe), and instrument error.
Both SH-HFF and SH-HFF (-CO3) had similarly low barium concentrations at 3.20 mg/L and 3.36 mg/L, respectively. HFF had comparatively more barium at 52.29 mg/L but was still well below the 306 mg/L initial concentration (Tables 2 and 3). Sulfate displayed an opposite trend with SH-HFF and SH-HFF (-CO3) at 162.45 mg/L and 137.63 mg/L, respectively, and much lower in HFF at 6.27 mg/L.
Citrate ion concentration was 10 times higher in SH-HFF (-CO3) than in SH-HFF or HFF though still lower than the amount originally added as citric acid (Tables 2 and 3). The IC used to detect organic anions reports total citrate ion concentration as it does not distinguish between citric acid and citrate ion. In the unreacted fluid 33.6 mg/L of citrate ion were initially included, while 20.44 mg/L were detected in SH-HFF (-CO3), and only 1.97 and 1.10 mg/L were detected in SH-HFF and HFF, respectively.
Dissolved inorganic carbon was highest in SH-HFF, followed by SH-HFF (-CO3) and then HFF with 12.59, 5.749, and 1.463 mg/L, respectively (Tables 2 and 3). This indicates that DIC in SH-HFF was 2.19 times higher than SH-HFF (-CO3) and 8.60 times higher than HFF, while SH-HFF (-CO3) was 3.92 times higher than HFF. Based on the pH dependent speciation of carbonate species, SH-HFF (-CO3) and HFF will have predominantly CO2 (aq) and SH-HFF will contain a mixture of CO2 (aq) and HCO3− (Stefansson et al., 2014).
3.3 SEM-EDS of Shale Chips Post-reaction
Characteristic morphologies of pyrite crystals for SH-HFF and SH-HFF (-CO3) are shown in Figure 3. EDS spectra in both samples revealed areas predominantly composed of iron and sulfur, indicating pyrite. In SH-HFF, pyrite areas were mostly found as clusters of tiny crystals, as shown in Figure 3. SH-HFF (-CO3) exclusively displayed pyrite areas in the form of larger single crystals with much lower volume-to-surface area ratios. The crystalline precipitate collected from the HFF reaction primarily gave EDS spectra that indicated barium and sulfur composition, indicating barite precipitation; however, barium was diffused in both shale reactions and insufficiently concentrated to give useful EDS results from the shale.
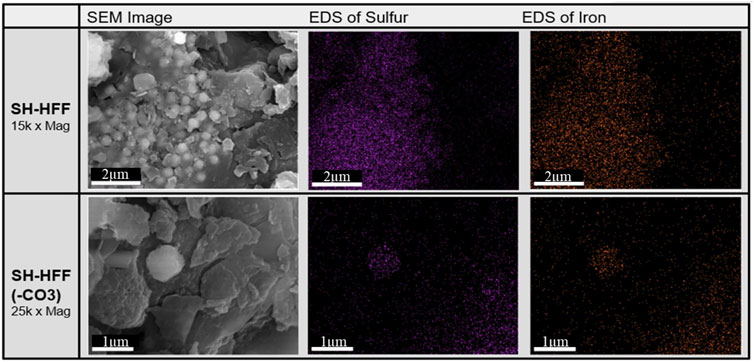
FIGURE 3. SEM and SEM-EDS images of reacted SH-HFF and SH-HFF (-CO3) shale chips. The sulfur and iron dense areas detected by EDS are presumed to be pyrite.
3.4 Volatile Organic Compounds (VOCs)
SH-HFF had significantly higher concentrations of VOCs: benzene, toluene, ethylbenzene, and xylene, relative to the other experiments (Figure 4). It was the only reaction in which ethylbenzene or toluene was detected and had 288% higher benzene, 960% higher m,p-xylene, and 792% higher o-xylene than SH-HFF (-CO3). Only m,p-xylene was above detection in HFF, and there was 1,300% more in SH-HFF. All BTEX compounds detected in SH-HFF (-CO3), and HFF are qualitative concentrations as they were above the method detection limit but below the practical quantification limit for the GC-MS method. Amounts detected in each reaction and limits of detection for each analyte are listed in Table 4.

FIGURE 4. BTEX in solution for each reaction by GC-MS.

TABLE 4. BTEX in solution from GC-MS analysis with detection and quantification limits.
3.5 Saturation Index (SI) Calculation
The geochemical modeling performed to determine SI of minerals is shown in Table 5 and Figure 5. In this study, we discuss the relative SI compared to other groups instead of the absolute value because the geochemical modeling databases do not account for organic-complexation/chelation reactions, sorption reactions, cationic exchange reactions, and organic ions (in Pitzer database). SH-HFF fluid has the highest SI for barite, followed by SH-HFF (-CO3) and HFF. Ca-oxalate hydrates were most saturated in SH-HFF fluid, followed by HFF and SH-HFF (-CO3). Celestite and anhydrite SI values were highest in SH-HFF fluid, followed by SH-HFF (-CO3) fluid and HFF fluid. Fe-related minerals (oxides and hydroxides) and calcite also have the highest SI in SH-HFF fluid, but SI in the SH-HFF (-CO3) fluid and HFF fluid was similar.
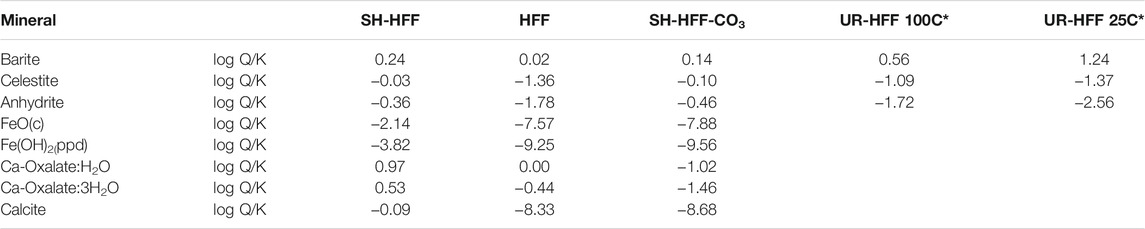
TABLE 5. Saturation indices (SI) of selected minerals. Ca-oxalates and calcite SI were calculated using MINTEQ database and other minerals were calculated using Pitzer database. The temperature of experimental groups is 100°C. *-UR-HFF values are determined using data from (Pilewski et al., 2019).
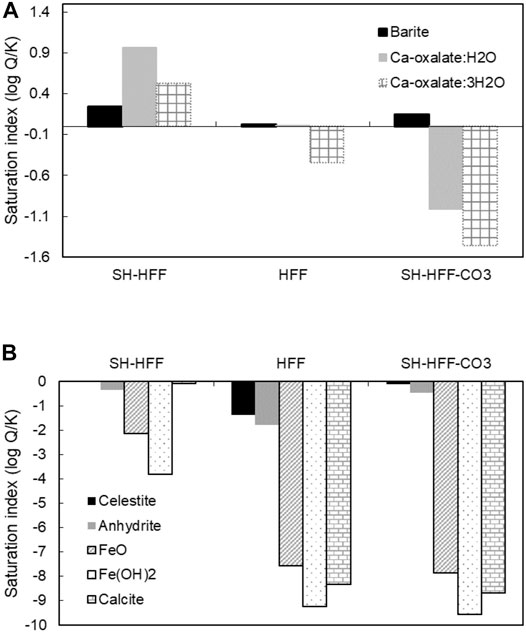
FIGURE 5. Saturation indices (SI) of selected minerals. Ca-oxalates and calcite SI were calculated using MINTEQ database and other minerals were calculated using Pitzer database. The temperature of experimental groups is 100°C.
4 Discussion
4.1 Carbonate Dissolution
SH-HFF experiment demonstrates a significant increase in pH post-reaction (Figure 1). On the other hand, the SH-HFF (-CO3) sample maintained a stable acidic pH before and after the reaction (Figure 1). Since the only difference between the SH-HFF and SH-HFF (-CO3) sample was the lack of carbonate in the latter, and we can conclude that the carbonate minerals are the primary factor controlling the pH of shale-HFF solutions. These results are in agreement with previous studies that reacted shale samples of varying mineral composition with HFF and reported that pH in the post-reaction solution is strongly correlated with the carbonate content of the shale (Harrison et al., 2017; Jew et al., 2017; Pilewski et al., 2019). For example, prior experimental studies show that carbonate-rich Eagle Ford shale samples had a consistently higher pH than the lower carbonate Marcellus Shale following reaction with acidic solutions (Jew et al., 2017). Our results also strongly support that the pH of the SH-HFF reaction increased due to the dissolution of carbonate minerals, while the SH-HFF (-CO3) maintained the lower pH due to lack of buffering by carbonates.
Carbonate dissolution should also increase the Ca ion concentration in the solution (as observed by Harrison et al., 2017). However, in our experiment, the calcium in the solution does not show any trend. We hypothesize that the high amount of calcium added to the brine component of our fracturing fluid mixture as calcium chloride probably swamped out any increasing calcium trend due to CaCO3 dissolution (Table 2 and 3).
4.2 Dissolution of Clay Minerals
In the SH-HFF (-CO3) reaction, the aluminium concentration was 29,835 μg/L, while it was below the detection limit (25.1 μg/L) in SH-HFF (Figure 6). The sources of aluminum in these reactions are the aluminosilicate mixed clay minerals and possibly minor amounts of feldspar present in the shale, indicating that clay dissolution occurred in the SH-HFF (-CO3) reaction. Several clays are most soluble at very low pH values when exposed to longer durations (days to weeks) and least soluble near pH 6 (Oelkers et al., 1994; Takahashi et al., 1995). The Visual MINTEQ 3.1 program shows that kaolinite was slightly soluble at 100°C and pH 1.89 as in SH-HFF (-CO3) and was not soluble when the pH was raised to 6.2 similar to SH-HFF reaction. In this simplified model, kaolinite was added as a finite solid and it dissolved over 1800 times more at pH 1.89 compared to 6.2 pH when the model reached equilibrium. Based on this simple modeling, it seems likely that the absence of carbonates buffers the pH and results in some clay mineral dissolution. Since clay generally constitutes the major fraction of shale, in real field situations dissolution of clays (especially in low carbonate shales) can substantially increase the porosity and permeability of shale. However, large scale clay dissolution could also result in instability and collapse of microfractures decreasing permeability.
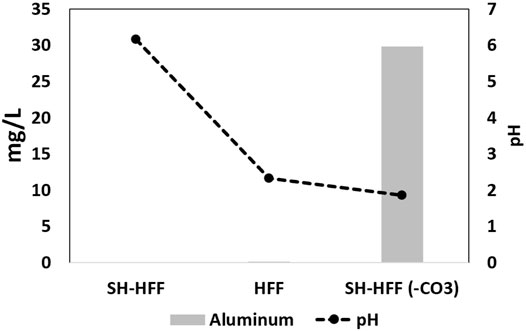
FIGURE 6. Aluminum in solution plotted against pH to highlight the role of pH in aluminum dissolution from clay minerals.
Using stoichiometric calculations, we determined the amount of clay minerals that dissolved in SH-HFF (-CO3) reaction (Supplementary Material). We determined clay dissolution by considering two end-member models: one with 100% kaolinite and other with 100% illite mineral. Based on our calculations, if we assume 100% of clay minerals are comprised of kaolinite, ∼57.02 mg of kaolinite can be dissolved. On the other hand, if 100% of clay is illite, ∼28.66 mg of illite can be dissolved. Since clay minerals in Marcellus shale are primarily composed of illite and kaolinite clay minerals (Hupp and Donovan, 2018), the total amount of clay mineral dissolution should range between 28.66 and 57.02 mg. Such low percent of clay mineral dissolution is likely to increase the porosity/permeability and flow of HFF in shale instead of collapsing microfractures.
4.3 Pyrite Dissolution
Previous studies on shale-HFF reactions have shown that pyrite dissolution is mainly controlled by Eh and pH conditions. Several bench-scale experiments demonstrated that pyrite becomes unstable under oxidizing conditions, leading to oxidative dissolution that releases iron and sulfur into solution (Harrison et al., 2017; Jew et al., 2017; Marcon et al., 2017; Paukert Vankeuren et al., 2017). In this system, numerous reactions are proposed to occur due to diverse oxidizing species in solution. For example, oxidizing breakers, including ammonium persulfate, have been observed to produce a range of oxidizing species such as the sulfate and hydroxide anions in the injected HFF that can oxidize pyrite, in addition to dissolved molecular oxygen (Sumner and Plata, 2019). Several previous studies have also observed that pyrite dissolution is higher under conditions of higher pH and carbonate contents (Harrison et al., 2017; Jew et al., 2017; Paukert Vankeuren et al., 2017). However, the actual impact of pH and carbonate content could not be determined as other factors affecting pyrite dissolution such as Eh and ionic strength were not kept constant.
Although many previous studies have shown increased pyrite dissolution with higher carbonate content, higher pH, and higher Eh conditions, no such trends are observed in this study. Interestingly, an opposite trend was observed in carbonate-rich and carbonate-free reactions. Our SEM-EDS results indicate more significant pyrite dissolution in SH-HFF (-CO3) than in SH-HFF by depicting different morphologies of pyrite crystals on the surface of shale chips after reactions (Figure 3). Under the SEM in the SH-HFF chips, most pyrite crystals were found as framboids of many small crystals with large surface area to volume ratios, while in the SH-HFF (-CO3) chips, the pyrite was found exclusively as larger single crystals. This suggests that the greater extent of pyrite dissolution in the SH-HFF (-CO3) dissolved the smaller framboidal crystals (seen in SH-HFF) due to their greater surface area to volume ratio, leaving only the most robust pyrite crystals intact. We postulate the pyrite dissolution in this study was mainly controlled by chelation reactions by citrate (and possibly by bitumen) instead of pH and Eh conditions.
The pH buffering by carbonate minerals can influence the ability of dissolved organic compounds to chelate and oxidize dissolved iron. Organic iron chelation can accelerate pyrite dissolution by 2 to 3 orders of magnitude depending on conditions (Jones et al., 2015). This has been demonstrated both for citric acid, which is added to HFF to control iron, and bitumen mobilized from the shale formation (Jones et al., 2015; Jew et al., 2017). Citric acid’s level of protonation varies with pH, which affects its reactivity. When carbonates raise the solution pH, the citric acid molecule loses hydrogen and becomes a more reactive anion. Bitumen has an indeterminate chemical composition as it is a blend of soluble organic components found in shales. However, the organic acids present in bitumen are affected by pH similar to citric acid. The citrate ion concentration was an order of magnitude greater in SH-HFF (-CO3) than in SH-HFF (Figure 2). Therefore, the higher citrate in SH-HFF (-CO3) promoted higher pyrite dissolution through iron chelation and oxidation. During this process, citric acid chelates iron, increasing the iron II rate to iron III oxidation. Additionally, iron III in solution could promote further oxidation of sulfide in pyrite to sulfate, resulting in a cyclical breakdown of pyrite catalyzed by citric acid (Bonnissel-Gissinger et al., 1998). Another factor that can influence iron II oxidation in SH-HFF (-CO3) could be the presence of bitumen mobilized from the shale by acid treatment (Jew et al., 2017). However, this possibility needs to be further investigated.
Heavy metals such as arsenic and cadmium are commonly associated with pyrite in organic-rich shales (Wang et al., 2016; Armstrong et al., 2019) and are released when pyrite is dissolved. This indicates that pyrite dissolution can pose a challenge for produced water treatment. The LM-2 shale sample was comprised of 5% pyrite by weight (Pilewski et al., 2019), and pyrite is the presumptive primary source of heavy metals in the shale (Salomons, 1995; Rimstidt and Vaughan, 2003; Jew et al., 2017; Mehta and D. Kocar, 2019). Our data also demonstrate this trend and show that under acidic conditions (sample SH-HFF(-CO32-)), a higher concentration of metals such as As, Cd, and Fe are released due to pyrite dissolution (as shown in Figure 2 and Table 3).
Another possibility is that pyrite dissolution occurred in both SH-HFF (-CO3) and SH-HFF, but the precipitation reactions (e.g. iron oxyhydroxides precipitation) did not occur in SH-HFF (-CO3) experiment (e.g., Jew et al., 2017; Hakala et al., 2021). The secondary precipitation reactions are also expected to scavenge trace metals from the solution. This observation is consistent with the calculated low saturation indices of iron (oxy) hydroxides in SH-HFF (-CO3) as compared to the SH-HFF sample (Figure 5B). However, since no evidence of such precipitation of iron (oxy) hydroxides was observed in SEM-EDS analysis of SH-HFF sample, iron mineral precipitation most likely played a minor role in controlling fluid chemistry in these experiments.
Our results reveal the importance of understanding organic complexation/chelation reactions in pyrite dissolution and heavy metals release. Further, our geochemical modeling calculations failed to predict such chelation-based reactions and demonstrate the limitations of geochemical software in modeling complex shale-HFF interactions. Therefore, results from geochemical modeling should be used with caution as several of the thermodynamics-based software packages do not incorporate extensive organic complexation/chelation reactions.
4.4 Carbonate Effect on Fracturing Fluid Chemistry
IC analysis of fluids post-reaction indicates that the citric acid added to the HFF was retained at 61% of the initial concentration in SH-HFF (-CO3) and 10 times less in SH-HFF and HFF (Figure 7). The disparity in citric acid left in the solution could have been controlled by 1) different reactivity of citrate ions at different pH and 2) calcium citrate precipitation. Citric acid has a pKa1 of 2.79, indicating it remains protonated at pH below 2.79 and deprotonated above it (Al-Khaldi et al., 2007). In the presence of calcite, the deprotonated citrate anions attach to positive sites on the calcite surface, resulting in a decrease in citrate concentrations in solution. Additionally, calcium citrate precipitation has been previously reported to increase further when the pH of the solution is greater than 6 (Al-Khaldi et al., 2007). Since the SH-HFF had a final pH of 6.2, calcium citrate precipitation seems to be the most likely scenario that controlled total citrate concentration. On the other hand, the SH-HFF (-CO3) sample had a pH of 1.8, which would not allow for calcium citrate precipitation (Figure 7). Additionally, SH-HFF (-CO3) had a pH below citric acid’s pKa1 of 2.79 (Al-Khaldi et al., 2007), and citric acid was less likely to react with other chemical species while protonated.
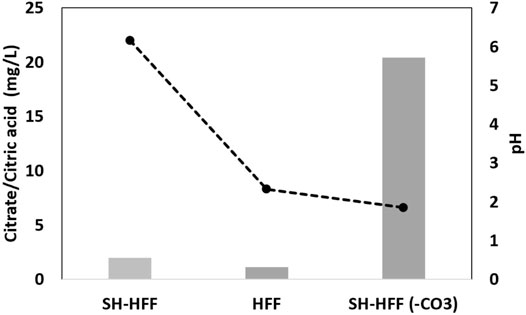
FIGURE 7. Citrate plotted against pH demonstrating the pH dependency of calcium citrate.
Although calcium citrate precipitation is not predicted by geochemical modeling, this is probably due to the unaccountability of chelation reactions by citrate ions in the Pitzer and Minteq databases. However, Ca-oxalate hydrates were predicted to be supersaturated in the SH-HFF (Figure 5A; Table 5), supporting the hypothesis of Ca -organic ions precipitation. The oxalate ions are potentially derived from shale organic matter or from petroleum distillate added in the fracturing fluid (Pilewski et al., 2019; Donmoyer et al., 2021).
In the control reaction, although HFF had a similar pH and lacked any carbonate minerals, the detected citrate was similar to SH-HFF rather than SH-HFF (-CO3). This is likely a result of the ammonium persulfate breaker reacting with citric acid added in the fracturing fluid in the absence of minerals and organic matter. Al-Khaldi et al., 2007 found that oxidizing breakers in fracturing fluid predominantly reacted with pyrite when oxidizer concentrations were low and more aggressively oxidized shale organic matter when the amount of oxidizer needed to dissolve pyrite was exceeded. It is likely that ammonium persulfate preferentially reacted with shale mineral and organic phases in the reactions containing shale; however, in the HFF-only experiment, ammonium persulfate reacted with citric acid and decreased their net concentration.
Carbonate minerals in the shale-HFF experiment also seem to affect oxidation strength of the fluid by impacting the oxidizing ability of the ammonium persulfate in the fracturing fluid. Ammonium persulfate is a highly soluble salt ((NH4)2S2O8) and dissociates into separate ions in solution. The persulfate ion (S2O8−2) is strongly oxidizing and reduces to two sulfate ions which are also strongly oxidizing. This step requires activation energy and can be limiting in ambient conditions (Liang et al., 2008). However, at 100°C for 14 days, the persulfate is exposed to conditions that enhance its oxidation kinetics; for example, persulfate was found to fully decompose in 2 h at 90°C and decompose more quickly at higher temperatures (Babu et al., 2002). The sulfate ions react with water to produce HSO4− and OH, the latter of which is a reactive oxidizing species (ROS) (Deng and Ezyske, 2011). Carbonate dissolution produces bicarbonate ions (HCO3−), which are ROS scavengers and react with oxidizing hydroxide ions to produce water and carbonate ions, which are not oxidizing. Because of this, alkalinity from carbonate minerals probably reduced ammonium persulfate’s overall ability to oxidize (Deng and Ezyske, 2011).
4.5 BTEX Oxidation
All BTEX compounds were present at higher concentrations in SH-HFF compared to SH-HFF (-CO3) or HFF (Figure 4). A major reason for the discrepancy between the shale reactions is the oxidizing strengths of the solutions. In environmental remediation, ammonium persulfate is used as a remediation strategy (Deng and Ezyske, 2011; Hilles et al., 2016) to oxidize BTEX compounds; we expect it is performing the same function in our experiments. The greater efficacy of ammonium persulfate as an oxidizer in SH-HFF (-CO3), due to the absence of ROS scavenging bicarbonate, resulted in higher oxidation of BTEX compounds in solution.
Concurrent oxidation of BTEX and shale organic matter in the SH-HFF (-CO3) experiment is supported by evidence of increased shale organic matter oxidation. Uranium in shale is associated with organic matter and is released into solution under oxidizing conditions when organic matter is broken down (Armstrong et al., 2019). Elevated concentration of uranium in the SH-HFF (-CO3) fluids relative to the other samples suggests that it is released from the shale (Figure 2). Enhanced organic matter oxidation in the SH-HFF (-CO3) experiment is also supported by the increased level of TIC in the SH-HFF (-CO3) experiment compared to the HFF experiment (5.749 mg/L and 1.463 mg/L, respectively: Figure 8). Since the SH-HFF (-CO3) experiment contained shale with carbonate phases removed, the only source of DIC would have been CO2 released by the oxidation of organic matter. The oxidative decomposition of BTEX has also been shown to produce carbon dioxide, which could have also increased DIC (Lovley, 1997). HFF had a similar pH of 2.3 to SH-HFF (-CO3) at 1.8 and experienced the same pressure and temperature effects on CO2 solubility; however, since it did not have the shale organic matter to oxidize, it had a lower DIC in solution. The uranium and dissolved inorganic carbon in solution both indicate that SH-HFF (-CO3) experienced organic matter oxidation, which would also explain the low BTEX in the solution.

FIGURE 8. The change in pH due to carbonate dissolution plotted with DIC.
4.6 Barium and Sulfate
Evidence for barite precipitation is found in all three experiments, as barium concentration is much lower in all three post-reaction solutions than the initial concentration added to the fracturing fluid (Figure 9). The geochemical modeling results also show the SI values > 0 for all three reactions indicating barite precipitation (Figure 5; Table 5). This matches with observations from previous workers as barite is typically found oversaturated under well conditions (Paukert Vankeuren et al., 2017; Pilewski et al., 2019).
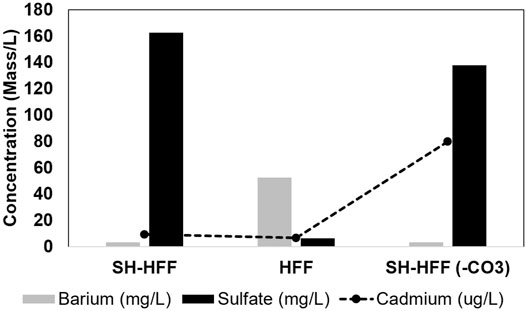
FIGURE 9. Barium and sulfate ions in solution indicating limiting reagent or barite precipitation. Cadmium is plotted as a proxy for pyrite dissolution vs sulfate in solution.
Using geochemical data from Pilewski et al., 2019, we determined that barite SI values were much higher in unreacted HFF (Table 5). The higher SI value of unreacted HFF indicates the HFF was not thermodynamically stable, and it led to the precipitation of barite even before the high T-P reaction started. Similar evidence of barite precipitation was observed in field-collected HFF (Xiong et al., 2020b) and similar synthetic HFF (Xiong et al., 2020a). Interestingly, in the three high T-P reactions, the barite SI and sulfate concentrations were generally higher in both shale-containing batches as compared to reacted HFF (Figure 5; Table 5). Moreover, the sulfate and SI trends in these three reactions were opposite to the Ba trend. This is probably due to the addition of sulfate released from pyrite dissolution compared to the HFF batch. In HFF, sulfate was the limiting factor as its only source was the breakdown of persulfates, and there was more barium added to the fracturing fluid stoichiometrically. In the shale reactions, sulfate was released from pyrite dissolution, but there was relatively less barium released, which was likely the limiting reagent (Xiong et al., 2020a). Similar to barite, celestite and anhydrite SI were higher in the two batches with shale than the HFF batch due to sulfate released from the shale (Figure 5A; Table 5).
It is noteworthy that despite showing other evidence of higher pyrite oxidation, SH-HFF (-CO3) had lower sulfate than SH-HFF, though it is a product of pyrite oxidation. It is likely that the oxidation reactions with organics led to the binding of the sulfur species with shale organic matter. Evidence of sulfur uptake by organic matter in shale-HFF reactions has been reported previously, particularly during kerogen oxidation (Yan et al., 2013; Hull et al., 2019). It is also possible that some of the sulfur reacted with BTEX compounds or the oxidized products of BTEX compounds. However, these proposed reaction mechanisms need to be further investigated.
5 Conclusion
The results of our study demonstrate that carbonate mineral content controls several critical shale-HFF reactions in the reservoir and need to be accounted for in designing the chemical make-up of HFF. The key observations of our study are:
1) The presence of carbonate minerals in shale increased the pH of the solution dramatically. The pH increase is accompanied by a lack of aluminum released to the solution, indicating that carbonates prevented clay mineral dissolution. Carbonate free experiments had lower pH which led to the dissolution of clay minerals.
2) Lower citrate concentration in carbonate rich shale (SH-HFF experiment) indicated that citric acid included in the HFF was removed via precipitation of calcium citrate.
3) Pyrite dissolution is mainly controlled via chelation by citrate ions. The lower concentration of citrate in the carbonate rich shale limited the pyrite dissolution and vice versa in the carbonate free sample. Pyrite dissolution in carbonate free sample led to release of inorganic contaminants such as As and Cd.
4) The oxidizing breaker ammonium persulfate was less effective at dissolving pyrite in carbonate rich shale due to higher ROS scavenging by carbonate and bicarbonate anions.
5) ROS scavenging by carbonate and bicarbonate anions led to lower uranium and higher BTEX in solution in the carbonate rich shale experiment, indicating that less organic matter is impacted by ROS.
6) A higher DIC in the carbonate free shale experiments indicate that shale organic matter was possibly oxidized in the presence of a greater abundance of ROS.
The key limitations of this study were that 1) due to limited sample size, duplicate experiments could not be performed and, 2) there could be minor artifacts related to the carbonate dissolution, such as increased specific surface area and release of carbonate bound metals/organics.
6 Implications
The combination of the amount and type of carbonate minerals in the shale formation and the acid added to the HFF controls the pH, Eh, and ionic strength of downhole conditions. In high carbonate shale formations, carbonate dissolution by HCl is the dominant dissolution process that can significantly increase the porosity/permeability while there is little or no effect on the clay minerals. However, in low carbonate shale, HCl can dissolve substantial amount of clay. Clays are generally hard to fracture due to their ductile nature, so dissolving clay minerals could improve permeability and stimulate fracture propagation. Generally during drilling in lower carbonate shales, HCl is either not added or added in lower amounts to the HFF. Our study shows that HCl could be added to HFF even for low carbonate high clay shales to improve permeability of the formation.
Another important implication of the study is that dissolution/precipitation of minerals such as pyrite, calcium citrate/oxalate, barite is controlled directly or indirectly by carbonate content. The dissolution/precipitation of these minerals could also significantly impact hydrocarbon recovery. For example, we found that pyrite dissolution increases in absence of carbonates due to “chelation” by citrate ions. Pyrite accounts for 5% by weight in our sample and its dissolution could result in significant increase of porosity/permeability in the formation. This study therefore reflects light on the role of “chelation” in fluid chemistry and dissolution precipitation reactions that has been generally overlooked in previous studies. Understanding these dissolution mechanisms are also critical for reservoir stimulation.
This study also developed an understanding of the role of carbonate minerals and HFF interactions with shale organic matter on the release of heavy metals and volatile organic compounds in produced water. High carbonate sample generated higher organic contaminants, but lower inorganic contaminants as compared to low carbonate content sample. Using this information, fracturing fluids could be engineered to reduce/remediate contaminants in different types of shales.
Our study demonstrates that a better understanding of how carbonate minerals affect shale-HFF interactions will enable shale-gas operators to improve long-term yield and reduce environmental risks posed by the operation.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material further inquiries can be directed to the corresponding author.
Author Contributions
BF, VA and SS designed, conducted the experiments and wrote the initial draft of the manuscript. VA, SS, JAH, and WX reviewed and edited the manuscript. WX and VA performed geochemical modeling.
Funding
The research was supported by two Department of Energy’s National Energy Technology Laboratory grants (DE#FE0024297; DE#FE0004000) awarded to Sharma, and the Onshore Unconventional Resources Portfolio through the NETL Research and Innovation Center (Field Work Proposal 1022415, Task 3).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank the NETL Pittsburgh Analytical Laboratory for performing ICP-MS analyses and Dr. Mengling Stuckman and Mr. Joshua Miller for performing IC and TOC/TIC analyses. We also thank West Virginia University’s Shared Research Facility and Dr. Marcela Redigolo for helping in the SEM-EDS analysis. SWN Energy are acknowledged for providing sample for the study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/feart.2021.695978/full#supplementary-material
References
Abualfaraj, N., Gurian, P. L., and Olson, M. S. (2014). Characterization of Marcellus Shale Flowback Water. Environ. Eng. Sci. 31, 514–524. doi:10.1089/ees.2014.0001
Agrawal, V., and Sharma, S. (2020). Are We Modeling the Properties of Unconventional Shales Correctly? Fuel 267, 117316. doi:10.1016/j.fuel.2020.117316
Agrawal, V., and Sharma, S. (2018a). Improved Kerogen Models for Determining Thermal Maturity and Hydrocarbon Potential of Shale. Sci. Rep. 8, 17465. doi:10.1038/s41598-018-35560-8
Agrawal, V., and Sharma, S. (2018b). Molecular Characterization of Kerogen and its Implications for Determining Hydrocarbon Potential, Organic Matter Sources and Thermal Maturity in Marcellus Shale. Fuel 228, 429–437. doi:10.1016/j.fuel.2018.04.053
Agrawal, V., and Sharma, S. (2018c). Testing Utility of Organogeochemical Proxies to Assess Sources of Organic Matter, Paleoredox Conditions, and Thermal Maturity in Mature Marcellus Shale. Front. Energ. Res. 6, 42. doi:10.3389/fenrg.2018.00042
Al-Khaldi, M. H., Nasr-El-Din, H. A., Mehta, S., and Al-Aamri, A. D. (2007). Reaction of Citric Acid with Calcite. Chem. Eng. Sci. 62, 5880–5896. doi:10.1016/j.ces.2007.06.021
Armstrong, J. G. T., Parnell, J., Bullock, L. A., Boyce, A. J., Perez, M., and Feldmann, J. (2019). Mobilisation of Arsenic, Selenium and Uranium from Carboniferous Black Shales in West Ireland. Appl. Geochem. 109, 104401. doi:10.1016/j.apgeochem.2019.104401
Arthur, J. D., Bohmand, B., and Layne, M. (2008). Hydraulic fracturing considerations for natural gas wells of the Marcellus shale. in Proceedings of ground water protection council 2008 annual forum, Cincinnati; 21-24 September 2008.
Babu, M. N., Sahu, K. K., and Pandey, B. D. (2002). Zinc Recovery from Sphalerite Concentrate by Direct Oxidative Leaching with Ammonium, Sodium and Potassium Persulphates. Hydrometallurgy 64, 119–129. doi:10.1016/S0304-386X(02)00030-0
Bonnissel-Gissinger, P., Alnot, M., Ehrhardt, J.-J., and Behra, P. (1998). Surface Oxidation of Pyrite as a Function of pH. Environ. Sci. Technol. 32, 2839–2845. doi:10.1021/es980213c
Deng, Y., and Ezyske, C. M. (2011). Sulfate Radical-Advanced Oxidation Process (SR-AOP) for Simultaneous Removal of Refractory Organic Contaminants and Ammonia in Landfill Leachate. Water Res. 45, 6189–6194. doi:10.1016/j.watres.2011.09.015
Dep, P. A. (2010). Hydraulic Fracturing Overview. Available at: http://files.dep.state.pa.us/OilGas/BOGM/BOGMPortalFiles/MarcellusShale/DEP%20Fracing%20overview.pdf.
Donmoyer, S., Agrawal, V., Sharma, S., and Hakala, J. A. (2021). Effect of Oxidative Breakers on Organic Matter Degradation and Metals Release during Shale-Fracturing Fluid Interactions in the Marcellus Shale. Manuscript in preparation.
EIA (2016). Hydraulically Fractured wells Provide Two-Thirds of U.S. Natural Gas Production - Today in Energy - U.S. Energy Information Administration (EIA). Available at: https://www.eia.gov/todayinenergy/detail.php?id=26112 (Accessed November 4, 2018).
Hakala, J. A., Paukert Vankeuren, A. N., Scheuermann, P. P., Lopano, C., and Guthrie, G. D. (2021). Predicting the Potential for mineral Scale Precipitation in Unconventional Reservoirs Due to Fluid-Rock and Fluid Mixing Geochemical Reactions. Fuel 284, 118883. doi:10.1016/j.fuel.2020.118883
Harrison, A. L., Jew, A. D., Dustin, M. K., Thomas, D. L., Joe-Wong, C. M., Bargar, J. R., et al. (2017). Element Release and Reaction-Induced Porosity Alteration during Shale-Hydraulic Fracturing Fluid Interactions. Appl. Geochem. 82, 47–62. doi:10.1016/j.apgeochem.2017.05.001
Hilles, A. H., Abu Amr, S. S., Hussein, R. A., El-Sebaie, O. D., and Arafa, A. I. (2016). Performance of Combined Sodium persulfate/H2O2 Based Advanced Oxidation Process in Stabilized Landfill Leachate Treatment. J. Environ. Manage. 166, 493–498. doi:10.1016/j.jenvman.2015.10.051
Hoelzer, K., Sumner, A. J., Karatum, O., Nelson, R. K., Drollette, B. D., O’Connor, M. P., et al. (2016). Indications of Transformation Products from Hydraulic Fracturing Additives in Shale-Gas Wastewater. Environ. Sci. Technol. 50, 8036–8048. doi:10.1021/acs.est.6b00430
Hull, K. L., Jacobi, D., and Abousleiman, Y. N. (2019). Oxidative Kerogen Degradation: A Potential Approach to Hydraulic Fracturing in Unconventionals. Energy Fuels 33, 4758–4766. doi:10.1021/acs.energyfuels.9b00104
Hupp, B. N., and Donovan, J. J. (2018). Quantitative Mineralogy for Facies Definition in the Marcellus Shale (Appalachian Basin, USA) Using XRD-XRF Integration. Sediment. Geology. 371, 16–31. doi:10.1016/j.sedgeo.2018.04.007
Jew, A. D., Dustin, M. K., Harrison, A. L., Joe-Wong, C. M., Thomas, D. L., Maher, K., et al. (2017). Impact of Organics and Carbonates on the Oxidation and Precipitation of Iron during Hydraulic Fracturing of Shale. Energy Fuels 31, 3643–3658. doi:10.1021/acs.energyfuels.6b03220
Jones, A. M., Griffin, P. J., and Waite, T. D. (2015). Ferrous Iron Oxidation by Molecular Oxygen under Acidic Conditions: The Effect of Citrate, EDTA and Fulvic Acid. Geochimica et Cosmochimica Acta 160, 117–131. doi:10.1016/j.gca.2015.03.026
Kondash, A., and Vengosh, A. (2015). Water Footprint of Hydraulic Fracturing. Environ. Sci. Technol. Lett. 2, 276–280. doi:10.1021/acs.estlett.5b00211
Liang, C., Huang, C.-F., and Chen, Y.-J. (2008). Potential for Activated Persulfate Degradation of BTEX Contamination. Water Res. 42, 4091–4100. doi:10.1016/j.watres.2008.06.022
Lovley, D. R. (1997). Potential for Anaerobic Bioremediation of BTEX in Petroleum-Contaminated Aquifers. J. Ind. Microbiol. Biotechnol. 18, 75–81. doi:10.1038/sj.jim.2900246
Marcon, V., Joseph, C., Carter, K. E., Hedges, S. W., Lopano, C. L., Guthrie, G. D., et al. (2017). Experimental Insights into Geochemical Changes in Hydraulically Fractured Marcellus Shale. Appl. Geochem. 76, 36–50. doi:10.1016/j.apgeochem.2016.11.005
Mehta, N., and Kocar, B. D. (2019). Geochemical Conditions Conducive for Retention of Trace Elements and Radionuclides during Shale-Fluid Interactions. Environ. Sci. Process. Impacts 21, 1764–1776. doi:10.1039/C9EM00244H
Morsy, S., Sheng, J. J., Hetherington, C. J., Soliman, M. Y., and Ezewu, R. O. (2013). “Impact of Matrix Acidizing on Shale Formations,” in SPE Nigeria Annual International Conference And Exhibition (Lagos, Nigeria: Society of Petroleum Engineers). doi:10.2118/167568-MS
Oelkers, E. H., Schott, J., and Devidal, J.-L. (1994). The Effect of Aluminum, pH, and Chemical Affinity on the Rates of Aluminosilicate Dissolution Reactions. Geochimica et Cosmochimica Acta 58, 2011–2024. doi:10.1016/0016-7037(94)90281-X
Paukert Vankeuren, A. N., Hakala, J. A., Jarvis, K., and Moore, J. E. (2017). Mineral Reactions in Shale Gas Reservoirs: Barite Scale Formation from Reusing Produced Water as Hydraulic Fracturing Fluid. Environ. Sci. Technol. 51, 9391–9402. doi:10.1021/acs.est.7b01979
Pilewski, J., Sharma, S., Agrawal, V., Hakala, J. A., and Stuckman, M. Y. (2019). Effect of Maturity and Mineralogy on Fluid-Rock Reactions in the Marcellus Shale. Environ. Sci. Process. Impacts 21, 845–855. doi:10.1039/C8EM00452H
Riley, K. W., French, D. H., Farrell, O. P., Wood, R. A., and Huggins, F. E. (2012). Modes of Occurrence of Trace and Minor Elements in Some Australian Coals. Int. J. Coal Geology. 94, 214–224. doi:10.1016/j.coal.2011.06.011
Rimstidt, J. D., and Vaughan, D. J. (2003). Pyrite Oxidation: a State-Of-The-Art Assessment of the Reaction Mechanism. Geochimica et Cosmochimica Acta 67, 873–880. doi:10.1016/S0016-7037(02)01165-1
Salomons, W. (1995). Environmental Impact of Metals Derived from Mining Activities: Processes, Predictions, Prevention. J. Geochemical Exploration 52, 5–23. doi:10.1016/0375-6742(94)00039-E
Sharma, S., Agrawal, V., and Akondi, R. N. (2020a). Role of biogeochemistry in efficient shale oil and gas production. Fuel 259, 116207–47. doi:10.1016/j.fuel.2019.116207
Sharma, S., Agrawal, V., Akondi, R. N., Wang, Y., and Hakala, A. (2020b). Understanding Controls on the Geochemistry of Hydrocarbon Produced Waters from Different Basins across the US. Environ. Sci. Process. Impacts 23, 28–47. doi:10.1039/D0EM00388C
Stefansson, A., Benezeth, P., and Schott, J. (2014). Potentiometric and spectrophotometric study of the stability of magnesium carbonate and bicarbonate ion pairs to 150°C and aqueous inorganic carbon speciation and magnesite solubility. Geochimica et Cosmochimica Acta 138, 21–31. doi:10.1016/j.gca.2014.04.008
Sumner, A. J., and Plata, D. L. (2018a). Exploring the Hydraulic Fracturing Parameter Space: a Novel High-Pressure, High-Throughput Reactor System for Investigating Subsurface Chemical Transformations. Environ. Sci. Process. Impacts 20, 318–331. doi:10.1039/C7EM00470B
Sumner, A. J., and Plata, D. L. (2018b). Halogenation Chemistry of Hydraulic Fracturing Additives under Highly Saline Simulated Subsurface Conditions. Environ. Sci. Technol. 52, 9097–9107. doi:10.1021/acs.est.8b01591
Sumner, A. J., and Plata, D. L. (2019). Oxidative Breakers Can Stimulate Halogenation and Competitive Oxidation in Guar-Gelled Hydraulic Fracturing Fluids. Environ. Sci. Technol. 53, 8216–8226. doi:10.1021/acs.est.9b01896
U.S. EIA (2017). Updates to the Marcellus Shale Play Maps. Washington, DC: U.S. Department of Energy, 14.
Wang, L., Burns, S., Giammar, D. E., and Fortner, J. D. (2016). Element Mobilization from Bakken Shales as a Function of Water Chemistry. Chemosphere 149, 286–293. doi:10.1016/j.chemosphere.2016.01.107
Wang, L., Fortner, J. D., and Giammar, D. E. (2015). Impact of Water Chemistry on Element Mobilization from Eagle Ford Shale. Environ. Eng. Sci. 32, 310–320. doi:10.1089/ees.2014.0342
Xiong, W., Gill, M., Moore, J., Crandall, D., Hakala, J. A., and Lopano, C. (2020a). Influence of Reactive Flow Conditions on Barite Scaling in Marcellus Shale during Stimulation and Shut-In Periods of Hydraulic Fracturing. Energy Fuels 34, 13625–13635. doi:10.1021/acs.energyfuels.0c02156
Xiong, W., Lopano, C., Hakala, A., and Carney, B. (2020b). “Investigation of Barite Scaling during Reaction between Pre-treated Hydraulic Fracturing Fluid from the Field and Marcellus Shale,” in Proceedings of the Unconventional Resources Technology Conference July 20–22, 2020(URTeC), 2275–2284. doi:10.15530/urtec-2020-2734
Keywords: marcellus shale, hydraulic fracturing, carbonate minerals, chemical additives, hydrocarbons, dissolution, precipitation, oxidation
Citation: Ferguson B, Agrawal V, Sharma S, Hakala JA and Xiong W (2021) Effects of Carbonate Minerals on Shale-Hydraulic Fracturing Fluid Interactions in the Marcellus Shale. Front. Earth Sci. 9:695978. doi: 10.3389/feart.2021.695978
Received: 15 April 2021; Accepted: 26 October 2021;
Published: 22 November 2021.
Edited by:
Dongdong Wang, Shandong University of Science and Technology, ChinaReviewed by:
Cheng Shen, Petrochina South Oil and Gas Field Company, ChinaJihua Cai, China University of Geosciences Wuhan, China
Copyright © 2021 Ferguson, Agrawal, Sharma, Hakala and Xiong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vikas Agrawal, dmlrYXMuYWdyYXdhbEBtYWlsLnd2dS5lZHU=, dmEwMDEyQG1peC53dnUuZWR1