 Roberto Grilli1*
Roberto Grilli1* Dominique Birot2
Dominique Birot2 Mia Schumacher3
Mia Schumacher3 Jean-Daniel Paris4Camille Blouzon1Jean Pierre Donval2
Jean-Daniel Paris4Camille Blouzon1Jean Pierre Donval2 Vivien Guyader2Helene Leau2Thomas Giunta2
Vivien Guyader2Helene Leau2Thomas Giunta2 Marc Delmotte4
Marc Delmotte4 Vlad Radulescu5
Vlad Radulescu5 Sorin Balan5,6
Sorin Balan5,6 Jens Greinert3
Jens Greinert3 Livio Ruffine2
Livio Ruffine2- 1CNRS, Univ Grenoble Alpes, IRD, Grenoble INP, Grenoble, France
- 2Département Ressources Physiques et Ecosystèmes de Fond de Mer (REM), IFREMER, Plouzané, France
- 3GEOMAR Helmholtz Centre for Ocean Research, Kiel, Germany
- 4Laboratoire des Sciences du Climat et de l’Environnement, LSCE/IPSL, CEA-CNRS-UVSQ, Gif-sur-Yvette, France
- 5National Institute of Marine Geology and Geoecology–GeoEcoMar, Bucharest, Romania
- 6Faculty of Geology and Geophysics, Doctoral School of Geology, University of Bucharest, Bucharest, Romania
Understanding the dynamics and fate of methane (CH4) release from oceanic seepages on margins and shelves into the water column, and quantifying the budget of its total discharge at different spatial and temporal scales, currently represents a major scientific undertaking. Previous works on the fate of methane escaping from the seafloor underlined the challenge in both, estimating its concentration distribution and identifying gradients. In April 2019, the Envri Methane Cruise has been conducted onboard the R/V Mare Nigrum in the Western Black Sea to investigate two shallow methane seep sites at ∼120 m and ∼55 m water depth. Dissolved CH4 measurements were conducted with two continuous in-situ sensors: a membrane inlet laser spectrometer (MILS) and a commercial methane sensor (METS) from Franatech GmbH. Additionally, discrete water samples were collected from CTD-Rosette deployment and standard laboratory methane analysis was performed by gas chromatography coupled with either purge-and-trap or headspace techniques. The resulting vertical profiles (from both in situ and discrete water sample measurements) of dissolved methane concentration follow an expected exponential dissolution function at both sites. At the deeper site, high dissolved methane concentrations are detected up to ∼45 m from the seabed, while at the sea surface dissolved methane was in equilibrium with the atmospheric concentration. At the shallower site, sea surface CH4 concentrations were four times higher than the expected equilibrium value. Our results seem to support that methane may be transferred from the sea to the atmosphere, depending on local water depths. In accordance with previous studies, the shallower the water, the more likely is a sea-to-atmosphere transport of methane. High spatial resolution surface data also support this hypothesis. Well localized methane enriched waters were found near the surface at both sites, but their locations appear to be decoupled with the ones of the seafloor seepages. This highlights the need of better understanding the processes responsible for the transport and transformation of the dissolved methane in the water column, especially in stratified water masses like in the Black Sea.
Introduction
Methane is a key climate-relevant trace gas widely encountered in seawater (Reeburgh, 2007; Etiope, 2012; Myhre et al., 2016; Saunois et al., 2017; Weber et al., 2019). Its distribution in the water column is highly heterogeneous, both horizontally and vertically. In the open ocean, dissolved methane concentrations are at level of nmol per litre (10–9 mol L−1), and usually at higher concentrations within the near-surface most-ventilated and most-oxygenated water (Karl et al., 2008; Repeta et al., 2016). However, very high concentrations of methane can also be found in bottom waters at coastal, shelf and margin settings characterized by widespread gas seeps discharging fluids at the seafloor (e.g., Reeburgh et al., 1991; Borges et al., 2016; Mau et al., 2017; Ruffine et al., 2018). In such settings, the water column is considered as a sink for methane in which it is transported at short (meters) and medium (kilometers) distances, degraded or transferred into other earth compartments like the atmosphere. The dynamic of methane in the water column is complex and depends on the properties of the water mass: physical conditions such as currents, layer thickness, temperature, ventilation/exchange with the atmosphere, chemical conditions that control its redox state (e.g. hypoxic/anoxic conditions) and biological activity that might oxidize or even produce methane in the water column (Solomon et al., 2009; Shakhova et al., 2014; Weinstein et al., 2016; Garcia-Tigreros and Kessler, 2018). The multiple factors involved in methane transport and transformation explain why the fate of this compound in the dissolved state is still difficult to capture in the field. However, in a progressively warming climate, in-depth knowledge of the fate of methane is essential, as emissions are expected to increase, particularly from continental shelves and margins due to eutrophication, permafrost thaw and gas hydrate destabilization (Westbrook et al., 2009; Naqvi et al., 2010; James et al., 2016; Riboulot et al., 2018). Accordingly, campaigns of time series and worldwide measurements are indispensable to capture the time evolution of methane.
The methane concentration in seawater can be measured either in the laboratory from previously collected water samples or in situ using specific sensors and analyzers. Laboratory measurements consist of determining the methane concentration either by headspace (HS) or purge-and-trap (PT) methods coupled with gas chromatography (GC) (Lammers and Suess, 1994; Tsurushima et al., 1999; Donval and Guyader, 2017; Wilson et al., 2018). HS-GC is based on the analysis of the gas phase in equilibrium with the seawater sample; it is easy to perform and could also be implemented in the field. The PT-GC method requires a more sophisticated installation in which the originally dissolved methane is extracted by flowing a carrier gas throughout the seawater sample, followed by its entrapment in a cooled material, frequently active carbon or silica. The choice of method depends mainly on the methane concentration and the sample volume available. Although the PT-GC requires a larger volume of water than the HS-GC (>100 ml vs 5–20 ml), it provides a sub-nmol L−1 detection limit, whereas that of the HS technique is usually around few nmol L−1.
Despite the reliability of laboratory measurements, in situ measurements are increasingly needed for both long-term monitoring through remote sensing and observatory, and high-resolution coverage of large areas of methane emissions. The most commonly encountered instruments for in situ methane concentration measurement are optical and chemical sensors (Marinaro et al., 2004; Faure et al., 2006; Krabbenhoeft et al., 2010; Schmidt et al., 2013), as well as optical spectrometers (e.g. Chua et al., 2016; Boulart et al., 2017; Grilli et al., 2018; Hartmann et al., 2018; Yuan et al., 2020). Their measurement range spans from few tens of nmol L−1 to hundreds of μmol L−1, and they can be used up to hundreds of meters water depth.
Furthermore, anoxic environments receiving huge inputs of organic matter provide favourable conditions for the production and preservation of high concentrations of methane, and the Black Sea is well known for being a typical example (e.g. Kessler et al., 2006a; Pape et al., 2008; Reeburgh et al., 1991, 2006). Indeed, it represents the largest thick and permanently anoxic and sulfidic basin on earth, in which methane is discharged abundantly through seeps widely distributed on the shelf and slope, typically at the rim of the gas hydrate stability zone and from mud volcanoes in the deep basins (Artemov et al., 2019). In the anoxic waters below ∼100 m water depth, methane concentrations can reach values of more than 10 µmol L−1 in contrast to the nmol L−1 level observed in the surface layer (e.g. Schmale et al., 2010).
The anoxic layer is successively superimposed by an intermediate suboxic- and an oxic-layer, leading to a distinct vertical stratification of the water column with limited chemical exchanges of redox-sensitive species (Capet et al., 2016; Özsoy and Ünlüata, 1997; Stanev et al., 2018, 2019). The total amount of dissolved methane stored in the Black Sea is estimated at ∼96 Tg (Reeburgh et al., 1991) with the anoxic water layer being more charged (∼72 Tg) than the others (Artemov et al., 2019).
Previous Black Sea methane studies have highlighted a variable vertical concentration distribution (McGinnis et al., 2006; Schmale et al., 2010; Sovga et al., 2008), with increasing values while going from the oxic into the anoxic water layer. So, methane concentrations up to 12 nmol L−1 were measured in the oxic layer, and in some areas, the sea surface was oversaturated in methane with respect to the atmosphere (Malakhova et al., 2010; Reeburgh et al., 1991). Concentrations reaching a few μmol L−1 were measured within the suboxic layer, and up to >10 μmol L−1 in the anoxic water mass (Kessler et al., 2006a; Reeburgh et al., 2006), where the concentration variations are much less pronounced.
Although today we are relatively confident that the methane originating from the Black Sea sediments is not a significant source accounting for the atmospheric CH4 budget of the region, its transfer mechanisms from the seafloor through the three aforementioned water layers and seldom to the atmosphere still remain unknown and controversial in the scientific community (Schmale et al., 2005; McGinnis et al., 2006). Depending on the investigated area, different sources of discharged methane can be identified: deep hydrocarbon-reservoirs, gas-hydrate destabilization, shallow methanogenesis within the sediments, and even methanogenesis in the water column (Reeburgh et al., 1991; Kessler et al., 2006b). The contribution of these sources can be very asymmetric, and Kessler et al. (2006b) showed that the major methane inputs to the Black Sea water column are discharges from seeps and diagenesis within the sediments.
Despite several European and national Black Sea Projects (e.g., CRIMEA, METROL, MSM34 cruise by Geomar in 2013–2014 (EU project MIDAS), Ghass cruise by Ifremer in 2015 (ANR project Blame) and many other German research cruises with R/V Meteor and R/V Maria S. Merian), the distribution and fate of methane emissions in Romanian waters are still not fully constrained (Ghass, 2015; MSM34, 2014; Riboulot et al., 2018). The objective of this study is to present insights from the dissolved methane distribution in the water column from two shallow water emission sites (∼55 m water depth, hereafter referred to as “shallower site” and ∼120 m water depth, hereafter referred to as “deeper site”) in the Romanian sector of the Black Sea using a multi-technique approach. High-resolution, horizontal and vertical profiles of dissolved methane concentration obtained from in situ measurements are reported and correlated to hydro-acoustic studies of gas bubbles. A detailed analysis of the results of the measurement systems is presented, and it emphasises the need to develop reliable and standardized protocols for in situ measurement of dissolved CH4. The two sites are then compared and the dissolved methane distribution in the water column discussed.
Materials and Methods
Recent previous studies on the distribution of dissolved CH4 at a seepage site have highlighted the need for high-resolution methane concentration measurements to assess the extent of the influence area of a bubble plume and map the spatial concentration variability (Jansson et al., 2019b). In this respect, the fast response membrane inlet laser spectrometer (MILS) prototype (t90 < 30 s, Grilli et al., 2018) is well-suited. To better appreciate its performances, comparison was made with the Franatech METS sensor and against a standard method consisting of sampling with Niskin bottles followed by PT and HS analysis in the laboratory. This multi-technique approach identifies the advantages and drawbacks of the different methods, revealing weaknesses and possible artifacts in the measurements, while making the dataset more robust for the comparison between the two locations reported in the discussion section. It should be noticed here that, since the research vessel was not equipped with dynamic positioning, inter-comparisons on dissolved methane measurements remain challenging.
The Study Areas
The survey was performed on board of the R/V Mare Nigrum, operated by GeoEcoMar Romania, in April 2019 at two shallow sites in the Black Sea Romania territorial water. Over a period of 5 days (3–7 April), we surveyed an area of ∼ 12.5 km2 at the deeper site (44.233°N, 30.737°E, 100-km long survey), with water depths between 110 and 128 m, and ∼ 3.5 km2 at the shallower site (44.057°N, 29.491°E, 19-km long survey), with water depths between 53 and 58 m (Figure 1).
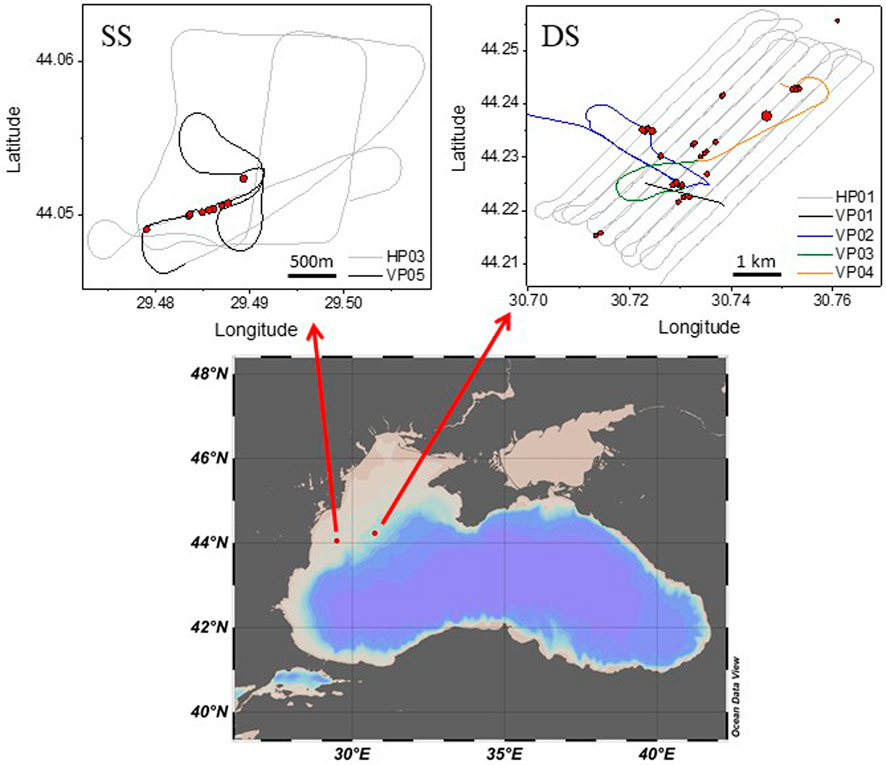
FIGURE 1. Maps of the survey areas. At the bottom a large map of the Black Sea, and at the top two maps of the shallower site (SS, left) and deeper site (DS, right) with the trajectories of the vessel for the profiles of interest for this work. VP are vertical and HP are horizontal near-surface profiles in the water. During the vertical profiles the position of the vessel was drifting due to water current. Red dots are the locations of flares identified during the survey by the echosounder, with the size proportional to the strength estimated from the acoustic signature.
At the deeper site, water temperature and electrical conductivity were on average 9.5°C and 18.5 mS cm−1 at the sea surface, and 9.0°C and 20.5 mS cm−1 near the seafloor. At the shallower site, temperature and electrical conductivity were 9.5°C and 18 mS cm−1 at the sea surface and 8.0°C and 18.5 mS cm−1 at the seafloor. The deeper site showed an oxycline between 60 and 80 m water depth, while the concentration of dissolved oxygen near the seafloor at the shallower site was 17% lower than at the surface (360 mmol L−1) (CTD, Conductivity, Temperature and Depth, data are reported in the Supplementary Datasheet S1).
Description of the Surveys
Two near-surface horizontal profile surveys (HP01, mean depth 5.2 m, min 0.4 m, max 18 m, and HP03, mean depth 4.2 m, min 1.5 m, max 9 m) on dissolved CH4 were conducted. HP01 was performed for 11 h at the deeper site on April 4th and is composed of ten parallel ∼5.5-km long lines spaced by ∼260 m, for a total surface area of ∼ 12.5 km2. HP03 was a 2-h long survey at the shallower site performed on the April 6th, covering an area of ∼3.5 km2 (see Figure 1). During these two surveys, the MILS and METS sensors were deployed simultaneously. The MILS probe was configured to improve the sensitivity of the measurements, by minimizing the flow of carrier gas (see the method description below for further details). This decreased the dynamic range of the measurement, while increasing the precision at low concentrations, to the detriment of a slightly longer response time (t90 of 30 s, corresponding to 75 m resolution at the highest speed of 2.5 m s−1 reached during the surveys).
Vertical profiles (VPs) with the MILS and METS sensors were performed at different locations at the two sites. Because of the lacking in dynamical positioning, the vessel was located in the vicinity of a flare (hydroacoustic anomalies in echosounder records attributed to the presence of gas bubbles) or a cluster of flares, and down- and up-casts were performed. It should be noticed that because of the strong dependency of the METS sensor to dissolved oxygen content and change in hydrostatic pressure, the recorded vertical profiles are not reported in this work.
Hydro-casts (HYs) for discrete water sampling were conducted at different time with respect to in situ measurements. The locations should have been the same as the in situ measurements, but this has proven to be challenging due to the lack of dynamical positioning of the vessel.
The Acoustic Method
During the cruise, a 70 kHz split beam echosounder (Simrad EK80 with ES70 transducer) was used to hydro-acoustically detect and locate bubble releasing methane seeps. With an opening angle of 18°, it has a footprint at the seabed of ∼22 m and ∼48 m diameter at 55 m and 120 m water depths, respectively. The pulse length was set to 0.256 ms over the entire cruise. This turned out to be suitable for the shallower site, while for the deeper site the noise level remained visibly higher. However, since this noise level was acceptable, for a better inter-comparability between the two study sites, the pulse length was unchanged for both surveys. At an average vessel speed of ∼2 m s−1, the distance between two pings was around 32 cm (14 cm) for 120 m (55 m) water depth at a ping rate of 0.16 s (0.07 s). To obtain precise backscatter values for the bubble quantification method, the echosounder was calibrated prior to the cruise with a 38.5 mm Tungsten sphere for the applied pulse length (MacLennan and Svellingen, 1989).
The idea to detect gas seepage locations using echo-sounding techniques was adopted from a series of former studies and has been accepted as an efficient approach to identify submarine gas flares. The method has been described in detail in e.g. Greinert et al. (2010) and Veloso et al. (2015).
The Membrane Inlet Laser Spectrometer in situ Sensor
A membrane inlet laser spectrometer (MILS) prototype allowing the combination of fast response and in situ dissolved methane measurements was used. The instrument relies on a patent-based membrane extraction system (Triest et al., 2017). It is described in detail in a previously published paper (Grilli et al., 2018), where a laboratory comparison with measurements of discrete water samples at different water temperatures and salinities was conducted. It was deployed successfully during two campaigns, over a methane seepage area in western Svalbard in 2015 (Jansson et al., 2019b), and at Lake Kivu in Rwanda in March 2018 (Grilli et al., 2020). The instrument allows a dynamic range from a few nmol L−1 up to a few μmol L−1. The MILS was powered by a battery pack (SeaCell, STR Subsea Technology and Rentals) and deployed together with a CTD SBE 911plus (Seabird) for measuring temperature, conductivity and water depth.
Because of the dynamic and fast profiling capability of the instrument, the spatial and temporal synchronization of measurements needs particular attention. For this, a first dynamic correction of the time-lag due to the flushing time (the time it takes the gas sample to go from the membrane extraction system into the measurement cell) was applied. By knowing the total gas flow (sum of the carrier gas plus the dry and wet gas permeating the membrane) and the volume of the gas line between the extraction system and the measurement cell, this time lag was calculated and varied between 15 and 60 s during the campaign (depending on the total gas flow). The instrument response time is related to the time necessary to replace the gas inside the measurement cell; it depends on the measurement cell volume (∼20 cm3 at standard temperature and pressure), the working pressure (20 mbar) and the total gas flow. This parameter can as well be calculated and it varied between 8 and 30 s during the campaign. Both time-lag and response time are affected by the total gas flow (Grilli et al., 2018). With the instrument towed behind the vessel, the distance between the instrument and the ship also varies as function of the ship speed and water depth of the sensor. Instrument location was therefore dynamically corrected simulating the mooring of our sensor using the “Mooring Design and Dynamics” matlab routine (Dewey, 1999). This dynamic correction allows to apply a time (and therefore a position) correction of the sensor which ranged from a few seconds and a few meters at the sea surface and at minimum speed, up to 100 s when the system was towed at 100 m water depth. This corresponds to maximum horizontal correction of ∼80 m, since typical ship speed during vertical profiles was ∼0.8 m/s. Water currents were neglected for this position correction. The vessel position was provided by a Garmin GPS 18x, with an accuracy of 15 m (1σ).
Measured concentrations are reported in mixing ratio with respect to the total dissolved gas pressure, which is assumed to be 1 atm for this setting. Therefore, a value of partial pressure, pCH4, in the gas mixture can be retrieved, which is then converted into dissolved methane concentrations, CCH4, expressed in mol per liter of water. This conversion is performed by considering the solubility of the gas in the water under given physical conditions as well as its fugacity. CCH4 is related to the pCH4 through the following equation:
where φCH4 is the fugacity coefficient (assumed to be one in this case) and K is the solubility coefficient, i.e. the ratio between the dissolved methane concentration and its fugacity. The solubility coefficient K (mol L−1 atm−1) of CH4 as a function of temperature T (K) and salinity S (g/kg) is calculated using the following equation:
Where Ai and Bi are empirical parameters from Wiesenburg and Guinasso. (1979).
The solubility coefficients need to be corrected for local pressure P (Pa) at the sampling water depth (sum of hydrostatic and atmospheric pressure), using the following equation (Weiss, 1974):
where R = 8.31446 J mol−1 K−1 is the ideal gas constant, and νCH4 is the partial molar volume (cm3 mol−1) of CH4 calculated from Rettich et al. (1981).
Calibration of the instrument was performed in the laboratory using the calibration system described in Grilli et al. (2018). Experiments were performed at atmospheric pressure for different water conditions (temperature from 5 to 25°C, and salinity from 0 to 30.5 g/kg) and at different concentrations of CH4 (0–1,000 ppm). The gas mixtures were obtained using two mass flow controllers (Bronkhorst, EL-FLOW) and mixing zero air (ALPHAGAZ 2, Air Liquide) with synthetic air containing CH4 (8920 Labline, 1,000 ppm of CH4 in air, Messer).
The Commercial Sensor Franatech
A Franatech METS sensor was used to measure anomalies in a concentration of dissolved methane in water. The measurement principle is based on a SnO2 semiconductor detector (Seiyama et al., 1962) working at ∼500°C (Ippommatsu et al., 1990). Its principles can be summarized as follows: first, oxygen (O2) is absorbed on the SnO2, then, the dissolved CH4 diffuses through a membrane to the measurement cell and interacts with O2 molecules, causing their desorption and increasing the conductivity of the SnO2 material. This technology is however known for its lack in gas selectivity and its dependency on the amount of O2 present in the measured environment (Boulart et al., 2010; Chua et al., 2016). The METS sensor can be operated at water depths down to 4,000 m and temperature ranging between 2°C and 20°C. Prior to its deployment, the sensor was calibrated by the manufacturer (in February 2019) at atmospheric pressure and methane concentrations ranging between 100 nmol L−1 and 40 µmol L−1. Although the sensor can be operated over a larger methane concentration range, the manufacturer calibrated the sensor in a narrower range in order to preserve its linear response (Franatech Pers. Comm.).
The Discrete Water Measurements
Discrete water sampling was conducted using a CTD-rosette with 16 Niskin sampling bottles (8 L), a SBE 911plus CTD (Seabird), an altimeter (Teledyne Benthos PSA 916), and an oxygen optode (Aanderaa Optode 4831F). The sensors were connected through telemetry for real-time monitoring of the water depth of the assembly. This allowed to adjust the sampling strategy during the profile according to the anomalies recorded by the echosounder. The Niskin bottles were sampled during the up-cast, and water subsampling was performed onboard for laboratory gas analysis (both PT and HS). For all the samples, a few mg of sodium azide (NaN3) were added to the vials and glass bulbs before adding the water sample. Filled vials for HS analysis were then stored upside down.
The samples were analyzed by two different techniques: the purge and trap (PT) and the headspace (HS) techniques. These methods have already been compared and validated in the laboratory (Donval and Guyader, 2017). The PT method used here is based on Swinnerton et al. (1968) and modified by Luc Charlou et al. (1988). Briefly, 125 ml glass bulbs devoted to the analysis of methane by PT method were used. The bulbs were overflowed with twice the volume of seawater. Particular care was taken to exclude air bubbles during sampling and avoid contamination. Once in the laboratory, CH4 was stripped from seawater for 8 min using helium as carrier gas (quality 99.9995%), trapped on activated charcoal at −80°C, and detected and quantified with a flame ionization detector after separation on a packed column (GC Agilent 7890A/column Porapak Q 2 m). The calibration was performed by injection of gas standards containing 108 ppm of CH4 in air ±5% (Restek). The limit of detection was 0.03 nmol L−1, the precision based on five replicates from the same rosette bottle was within ±2% (±1σ), while the accuracy corresponds to that of the gas standards (±5%). The HS method (Donval et al., 2008; Donval and Guyader, 2017) was performed on 10 ml vials by analyzing the composition of the headspace in equilibrium with the water. At the beginning of the cruise, the vials were flushed with zero air (Alphagaz 2, Air Liquide) to avoid introducing methane into the initial gas phase. With a gas tight syringe, 5 ml of seawater were transferred into the vial while a second needle was introduced to keep the pressure close to atmospheric pressure inside the vial. The analysis was performed by means of a headspace sampler connected to the same GC unit used for the PT method. The limit of detection was 5 nmol L−1 and the precision was ≤10% for concentrations below 100 nmol L−1, and ≤5% for higher concentration (±1σ). Further details in the comparison between the results of two methods can be found in the Supplementary Datasheet S1.
Results
Inter-Comparison Between the Techniques
In this section we compare results from different techniques with the aim of testing the reliability of the measurements, identifying possible artefacts or weaknesses of each technique used, and validating the finds on the dissolved methane distribution in the water column at the studied sites. This data validation is important for comparing the distribution of dissolved methane at the two locations as reported in the discussion section.
Qualitative Comparison of Echosounder Data and Membrane Inlet Laser Spectrometer
On the echogram, large areas of high backscatter (>−30 dB) of a flare-like shape have been identified as methane gas seeping areas (Figure 2, color-coded in orange to red). Broader high backscatter areas are related to vessel movement. In our case, the ship remained over the methane seep location for some time and as the echogram displays backscatter over time, the seepage area widens. During the cruise, 36 and 13 gas emissions in the deeper and shallower site, respectively, were identified from the 70 kHz echosounder data. The signal produced by the scattering of the acoustic wave by the gas bubbles provides only qualitative information about the distribution of dissolved CH4 in the water column. This is due to the fact that gas bubbles shrink and change their chemical composition during their rise through the water column, and that free bubble-forming gas is mobile whereas dissolved methane is more stationary. Therefore, a quantitative analysis would require different assumptions on the initial bubble size distribution, the bubble rising speed and the gas exchange ratio between the two phases. Results from the quantitative analysis of the acoustic signal is beyond the scope of this paper, and will be the subject of another study. Here, an example of the qualitative comparison between the acoustic signal and the dissolved CH4 concentrations from MILS is reported in Figure 2 for the VP03 profile performed on April 5th at the deeper location (44.224° N, 30.730° E). The concentrations of dissolved methane determined by MILS were dynamically corrected for the position of the instrument with respect to the vessel. This correction allows to synchronize/match the two time-series, accounting for the fact that the echosounder passed over a target area prior to the towed MILS. A more detailed figure reporting original and synchronised data can be found in the Supplementary Datasheet S1.
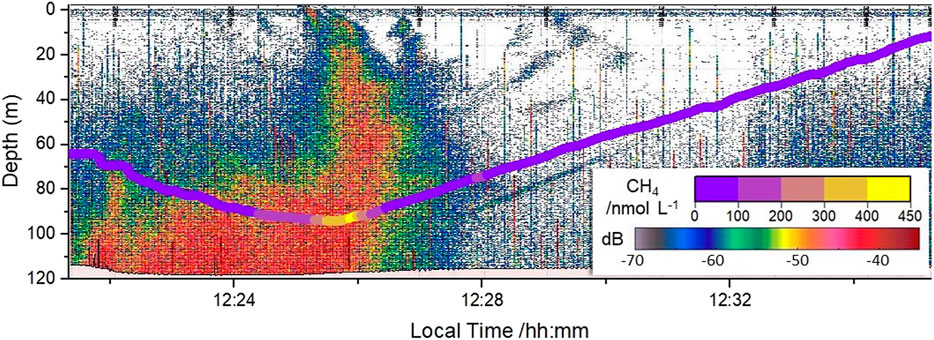
FIGURE 2. Qualitative comparison between the time series from the continuous concentration of dissolved methane measured by MILS (colored line) and the acoustic signal from the echosounder (colored background). The data are plotted against local time. The data are from the VP03 profile performed on April 5th at the deeper site (44.224°N, 30.730°E).
In this inter-comparison, one should consider that the MILS probe is measuring a specific location behind the vessel, while acoustic data has a larger footprint on the seafloor as well as in the water column. This may induce discrepancy between the two signals, for instance in the case of a bubble plume located a few tens of meters on a side of the MILS instrument that would be spotted acoustically but not observed by the MILS (this could be the case for the signature at 12:22 local time in Figure 2, more visible in Supplementary Figure S1-3). The results may inverse if water enriched in CH4 (by a bubble plume that is outside the acoustic lobe) is laterally transported by currents. This may be the case for the increase in dissolved CH4 at 12:28 local time (visible in Supplementary Figure S1-3) that was observed by the MILS sensor without the corresponding acoustic signal.
Despite its evident limitations, this comparison allowed us to verify the qualitative agreements between these two datasets and validate the dynamic correction of the position of the MILS probe with respect to the vessel during the profiles.
Comparison Between the Two in situ Instruments
For comparison, the MILS and the METS sensor were deployed simultaneously during the vertical and horizontal profiles. However, due to dependency of the METS sensor to the hydrostatic pressure, salinity and oxygen (Newman et al., 2008), this comparison only focus on the 12-h long horizontal profile (HP01). The profile was conducted at ∼5 m depth, and the water inlets of the two sensors were ∼10 cm distance from each other. The resulting profiles used for comparison are reported in Figure 3. One can clearly see that most of the peaks in methane concentration for both instruments agree with each other. However, the methane concentrations measured by the METS are more than one order magnitude higher than the concentrations obtained with the MILS. Previous field studies have shown significant differences between measurements of discrete water samples of seawater methane concentration from well-proven laboratory methods and the METS outputs (Heeschen et al., 2005; Newman et al., 2008). This has led Heeschen et al. (2005) to interpret their METS in situ measurements in a qualitative way. Here, the differences observed between the MILS and the METS measurements may be explained by: 1) the fact that the METS sensor was used below the calibration range (100 nmol L−1–40 μmol L−1 at atmospheric pressure) certified by the manufacturer and therefore it cannot provide reliable quantitative measurements at the sea surface; 2) the METS suffers from dependency to hydrostatic pressure, salinity and oxygen content (Newman et al., 2008), which makes near-surface horizontal profile also challenging. Moreover, the wide drifts observed during this 12 h continuous near-surface profile (Figure 3) can be explained by the fact that small height changes in near-surface depth lead to large relative changes in hydrostatic pressure that considerably affect the sensor response. Despite the large discrepancy on the methane concentration from the METS sensor, both in situ instruments agreed on the presence of highly localized peaks of CH4 at the sea surface. The METS sensor, even when it is used outside the calibration range and under severe conditions (changing oxygen concentration that affect its detection system) can provide valuable qualitative information on the location of dissolved methane concentration spots. Further laboratory tests, together with deployment at deeper water depth within the anoxic layer, would be required to provide a thorough assessment of the sensor for quantitative and reliable dissolved methane measurements.
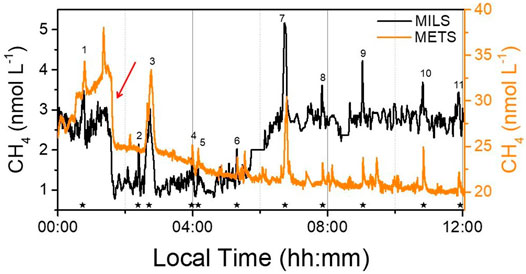
FIGURE 3. Comparison between the dissolved methane measurements from the MILS and the METS sensor which were deployed simultaneously during the 12 h HP01 survey. The red arrow indicates a sharp concentration decrease observed by both instruments. Well localized peaks of dissolved CH4 were observed by both instruments (main identified methane peaks are numbered and identified by the stars at the bottom).
Eleven well localized dissolved CH4 peaks (numerated and marked with stars in Figure 3) were identified during the horizontal profile HP01 and are discussed in the next section. Furthermore, the sharp concentration decrease recorded at 01:35 local time (highlighted in Figure 3 by the red arrow and also visible in the 2D colored map graph of the dissolved methane concentrations measured by the MILS instrument and reported in Figure 4) that could have been associated with a possible instrumental (MILS) drift, was confirmed as a real signal since observed by both in situ sensors.
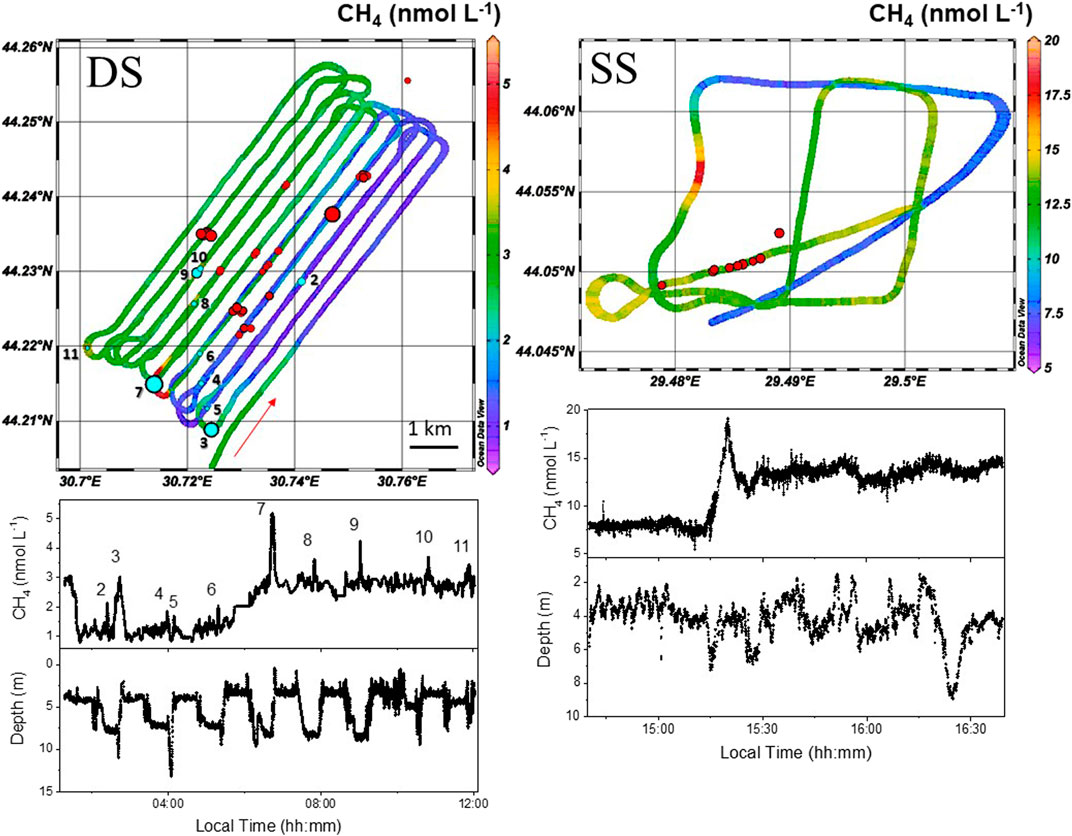
FIGURE 4. 2D surface dissolved methane distribution (color map) at the deeper (DS, left) and shallower (SS, right) site. Both surveys were conducted near surface. The thickness of the color map (∼60 m) was chosen accordingly to the 2σ accuracy of the GPS positioning. Red arrows indicate the time direction. Red and light-blue dots show the locations of the flares (echosounder) and surface dissolved CH4 (MILS) peaks, respectively, both with the size proportional to the estimated intensity. Peak 1 is only visible in Figure 6, because it is located at a southern position with respect to the performed grid. At the bottom, the time series of the data together with the water depth information. Localized peaks of dissolved CH4 were observed at certain locations, which seems uncorrelated with the flare locations.
Membrane Inlet Laser Spectrometer Vs Measurements of Discrete Water Samples at the Deeper and Shallower Sites
Comparison on Vertical Profiles
The size of the MILS prototype did not allow to be mounted on the CTD for continuous in situ measurements simultaneous to discrete water sampling. For this reason, measurements with the MILS probe and discrete water samplings could not be conducted simultaneously. Moreover, because the research vessel was not equipped with dynamic positioning, inter-comparisons on dissolved methane measurements were challenging. As mentioned above, due to the high spatial variability of dissolved methane relative to the location and intensity of bubble streams, a few hundreds or even a few tens of meters could be significant for correctly reproducing the same spatial distribution of dissolved CH4. However, we identified a few vertical profiles at the shallower and deeper sites, where the position of the MILS was relatively close to the hydro-casts (HYs) for discrete water sampling. These profiles are reported in Figure 5. The trajectories of the vessel during the measurements are shown in the inserts. The arrows indicate the time direction of the deployment, while the location of the bottom of the profile is indicated by green dots. The VP03 profile performed by the MILS sensor started with the probe at 60 m water depth, and it went first down to 93 m, and then back up to the surface. Red dots are the positions of the flares identified during the campaign. For discrete water sample measurements average values between the PT and HS analysis were used. Dissimilarities between the two techniques at the two sites ranged from 8 nmol L−1 (0–70 m water depth) to 86 nmol L−1 (>70 m water depth) at the deeper site, and from 8 nmol L−1 (0–25 m water depth) to 62 nmol L−1 (>25 m water depth) at the shallower site. At the shallower site, the MILS curves systematically show lower concentrations of dissolved CH4 with respect to the measurement from discrete water sample analysis. This may be due to the strong spatial variability caused by the ascent of the multiple methane bubbles of variable size and trajectory within the water column, or by an unidentified bias in either the MILS or discrete sampling methods. Our results highlight the limitations of current in situ instrumentation and laboratory measurement techniques. Nevertheless, a larger number of measurements together with improved position maintaining or simultaneous deployment of the in situ instruments and Niskin bottle sampler would have been required for a finer comparison of the two methods. Further information on this comparison can be found in the Supplementary Datasheet S1.
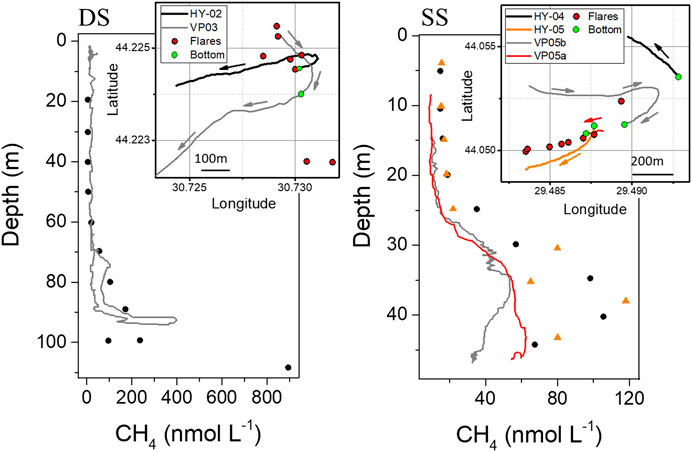
FIGURE 5. Comparison between the dissolved methane measurements from MILS (VP) and discrete water sample (HY) techniques at the two sites (average concentrations between PT and HS methods are reported). The inserts show the trajectory of the vessel during the measurements, which were performed at different time and without dynamic positioning of the vessel. Red circles in the maps report the location of the flares identified from the acoustic survey. Arrows in the inserts represent the time direction of the measurement, and green dots the location of the bottom of the profile.
Comparison on Near-Surface Measurements
MILS continuous sea-surface measurements were compared with the results from discrete water sampling performed during the HYs. The results from the shallowest measurements of the HY profiles were used (average concentrations between the PT and HS method; depths and accuracies of the measurements are reported in Table 1), and compared against the closest data from the MILS sensor. In the two maps of Figure 6, the trajectories during the MILS survey and the location of the near-surface discrete water sampling (HYs) are reported for the deeper and shallower site. The concentration of dissolved methane measured by MILS closest to the locations of the HYs were selected and highlighted with thick black lines. The selected MILS data were averaged at each location, and reported in the bar graph of Figure 6 together with the results from the concentrations in dissolved methane by the analysis of discrete water samples.
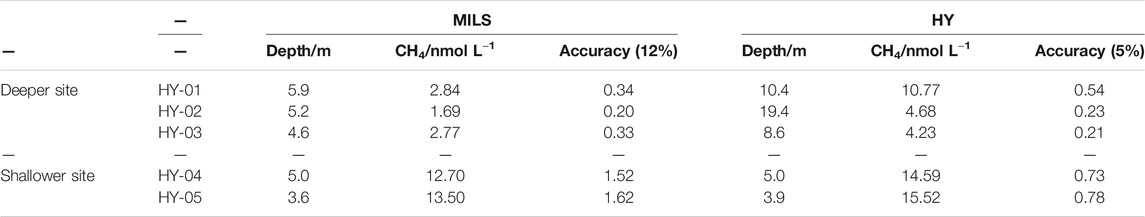
TABLE 1. Data used in Figure 6 for the deeper and shallower site. MILS (Membrane inlet laser spectrometer) data are an average from measurements which were closest to the discrete water sampling (HY) locations. HY data are averages between the PT and HS analysis. Water depths and estimated accuracies of the measurements are also reported.
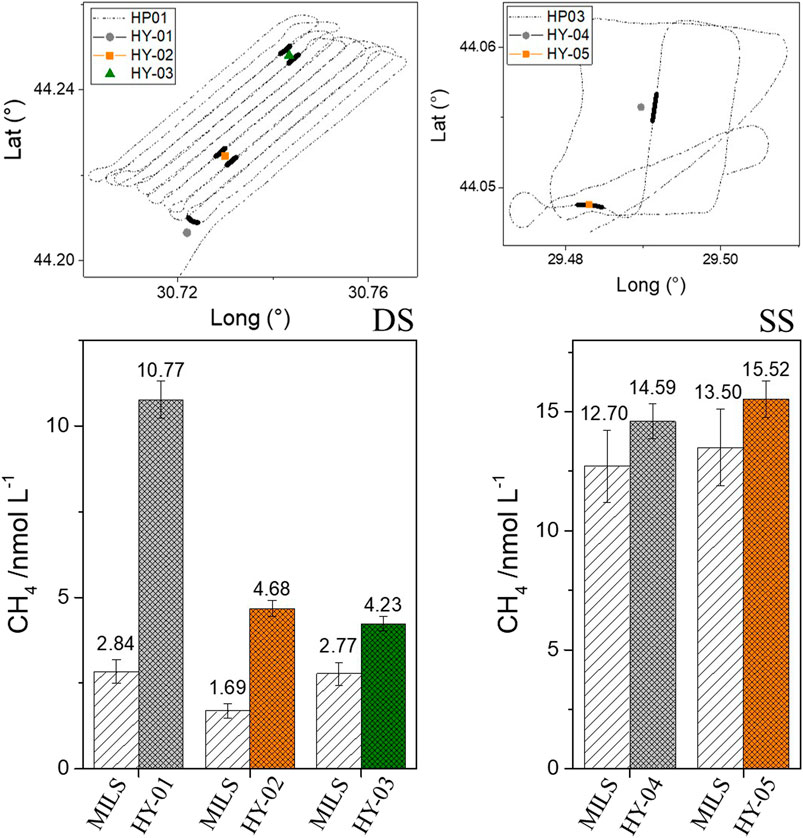
FIGURE 6. Bottom: Comparison between near-surface water depth measurements made by the MILS sensor and the analysis of discrete samples (HY) at the deeper (DS, left) and shallower (SS, right) site. Data from discrete samples are reported as the average between the two analytical techniques used (PT and HS). Maps with locations are reported at the top, with the trajectories of the MILS surveys (dotted lines) and locations of the near sea surface sampling of the HYs. In thick black lines are the selected dissolved CH4 MILS data closest to the discrete sampling locations which were averaged and used for the comparison.
The location of the HY-02 and HY-03 measurements lies between two horizontal profile lines, therefore data from both lines were selected and averaged. HY-05 was very close to the horizontal profile trajectories, while for HY-01 and HY-04 the closest dissolved CH4 MILS data were 300 and 140 m away, respectively.
The comparison between MILS and measurements of discrete water samples at the shallower site (HY-04 and HY-05) shows a discrepancy of ∼13% (([CH4]MILS - [CH4]HY)/[CH4]HY), which lies within the accuracy of the measurements, as reported in Table 1. At the deeper site, the differences are larger, with 34% discrepancy for the HY-03 and even larger for HY-01 and HY-02. Different hypotheses can explain these discrepancies. As mentioned above, the measurements were not conducted at the same time. At the shallower location, HP03, HY-04, HY-05 were conducted on the same day (April 6th), while the measurements at the deeper site were spread over two and half days (April 3rd to 5th). Thus 1) The spatio-temporal variability of dissolved CH4 at the sea surface can be affected by water currents producing different distribution patterns as well as meteorological conditions (change in wind direction or speed, humidity, water temperature, etc.) (Shakhova et al., 2014) that modify gas exchange/equilibration with the atmosphere and degassing activities at the seafloor. Although, this last hypothesis would require significant changes in meteorological conditions in order to explain the reported discrepancies; 2) the discrepancy could be due to the analytical techniques itself. The MILS measurements seem to be systematically lower than the measurements of discrete water samples. Apart from the HY-01 location where a 7.9 nmol L−1 difference was found, for the other locations the offset between the two techniques is ∼2 nmol L−1, which remains within the order of magnitude of the best precision one could expect today on dissolved CH4 measurements in the ocean.
Discussion: Comparison Between the Two Sites of Study
Discussion on Vertical Profiles
The MILS vertical profiles recorded at the two sites during the campaign are reported by grey and orange dots in Figures 7A,B. For the deeper site, a total of four down- and up-casts (VP01, 02, 03, and 04) were used, whereas for the shallower location, the vertical profile VP05, composed of a series of seven down- and up-casts over the seepage area was used. The 2-m average curves are in black and blue for the deeper and shallower sites, respectively. Because the gas bubbles originate from the seafloor, the vertical profiles were compiled in distance from seabed (rather than in water depth), allowing a better comparison of the vertical distribution of the dissolved CH4 within the two sites. For each datapoint, the distance from the seafloor was calculated using the depth of the seafloor measured by the echosounder and the water depth of the instrument provided by MILS-implemented CTD and the MILS instrument itself. The systematic decrease in dissolved CH4 concentration near the seafloor may be due to the fact that the position of the vessel was not dynamically maintained. In most of the vertical profiles recorded, the instrument passed over the bubble plume either during its descent or ascent (this is visible in the time series reported in Figure 2). The MILS instrument was therefore within the uppermost part of the gas bubble plume most of the time a few tens of meters above the seafloor. For a finer visualization of this effect, the stronger profile recorded by the MILS at the deeper location (corresponding to the VP04) is reported in orange dots (Figure 6A). The two black arrows indicate the time direction during the descent. The maximum dissolved CH4 concentration of 924.5 nmol L−1 was reached at 15 m from the seafloor (or 93 m water depth). Then, despite the probe continuing its descent, the concentration decreased, probably due to the fact that the probe was moving out of the bubble plume. Another possible reason for this trend may be directly related to bubble dissolution. The seafloor topography can influence bottom current (Weber et al., 2000; Stow et al., 2009), which in turn affect bubble trajectory in the water column and shape the plume morphology. Hence, the rim of the bubble plume is widened few meters above the seafloor, enhancing the spreading of dissolved CH4 at this height, and placing the maximum level of dissolved methane concentration further above the seafloor. Lastly, note that the distance between the instrument and the ship was calculated by mooring simulation (as mentioned in the materials and methods section) which may add uncertainty on the position of the MILS sensor with respect to the one of the bubble plumes. From the maximum concentrations of the averaged curves, a difference in emission intensity of ∼80 fold between the shallower and the deeper site was estimated.
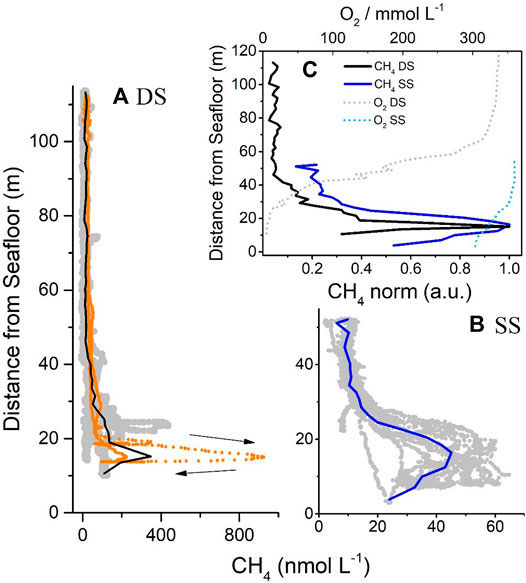
FIGURE 7. (A) A compilation of vertical profiles acquired with the MILS probe at the deeper location (VP01, 02, 03 and 04 for a total of four down- and up-casts) and (B) at the shallower location (VP05 which is a series of seven down- and up-casts) (grey dots). The profile VP04 is represented with orange dots, highlighting the temporal evolution of the measurement during the descent and ascent (black arrows). Black and blue lines are 2-m average values. (C) the average curves of dissolved CH4 (solid lines), normalized by the maximum concentration at each site, are plotted against the distance from the seafloor, showing a similar and expected exponential dissolution trend. While at the DS dissolved CH4 concentrations reached background value at 50 m from the seafloor, at the SS, methane concentration at the sea surface was ∼20% of the maximum of the average concentration measured at the sea bottom. The average variations in dissolved O2 at both locations are also reported (dotted lines).
In Figure 6C, all the vertical profiles were averaged producing one data point every 2 m water depth, and the concentration of dissolved CH4 was normalized with respect to the maximum averaged dissolved CH4 concentration. At both sites, an exponential trend was observed which follows the expected dissolution function of the bubbles into the water column (Jansson et al., 2019a). By fitting the exponential curves on the whole vertical profile, exponential time constants (corresponding to the distance from the seafloor required for decreasing the intensity by 1/e – e-folding – of the value at the sea bottom) of 6.8 and 6.3 m were obtained for the deeper and shallower sites, respectively. They are close, but the difference remains visible in Figure 6C, with a faster decreasing in concentration of the profile at the deeper site while moving away from the seafloor. This emphasizes the larger storage capability of dissolved CH4 at the bottom water level of the deeper site (i.e. a better tendency of CH4 to escape towards to the sea surface at the shallower site). The reason for this difference is likely a combination of factors, including: 1) the difference in hydrostatic pressure and bubble-size distribution, leading to a different bubble/water exchange (more precisely related to the difference between the buoyant rise time of the bubble and its diffusive equilibrium time (Leifer and Patro, 2002)); 2) decoupling between bottom and surface water at the deeper location marked by the presence of the oxycline, which prevents the rise of CH4 towards the sea surface; 3) a possible local production of CH4 in the anoxic water of the deeper site (Artemov et al., 2019), which would increase the concentration of dissolved CH4 below the thermocline. Discriminating between the different scenarios would involve a more intensive investigation of methane distribution in all three water layers, combining a larger number of horizontal and vertical profiles, and both molecular and isotopic concentration measurements of CH4; this was not performed during this campaign. From this analysis, we can conclude that at the deeper site, going from the seafloor towards the surface, the dissolved CH4 rapidly decreases within the first 45 m (∼7 times the exponential time constant), and remains uniform in the upper part. On the other hand, at the shallower site, dissolved CH4 concentrations corresponding to ∼20% of the maximum average dissolved CH4 concentration on the water column are still present at the sea surface (at ∼52 m from the seafloor).
Discussion on Horizontal Near-Surface Profiles
Two near-surface horizontal profiles were conducted during the cruise: HP01 at the deeper site and HP03 at the shallower site. The 2D distributions of dissolved CH4 are reported in Figure 4. The average concentration at the deeper site was 2.23 ± 0.78 nmol L−1 (1σ, min 0.78, max 5.16 nmol L−1). For the measured temperature and electrical conductivity of the surface water (9.5°C and 18 mS cm−1), the atmospheric CH4 concentration of 2 ppm (part per million) would correspond to an equilibrium dissolved CH4 concentration of 3.5 nmol L−1 (calculated from Equations 1, 2 and 3). This is slightly higher than the average concentration measured by the MILS, but it still lays within the range of the measurements made by the MILS sensor near the surface. On the other hand, from the comparison with measurements of discrete water samples (Figure 6), concentrations measured by the MILS seems to be systematically ∼2 nmol L−1 lower. We can therefore conclude that at the deeper site, dissolved CH4 in the water is close to equilibrium with the atmosphere. At the shallower site, the average dissolved CH4 concentration was 5.6 times higher than observed at the deeper site (average concentration 12.5 ± 2.76 nmol L−1 1σ; min 5.16, max 19.7 nmol L−1) and almost four times higher than the expected concentration in equilibrium with the atmosphere. These concentrations are close to previously reported measurements by Amouroux et al. (2002) and Reeburgh et al. (1991) in north-western and central part of the Black sea, respectively. This, together with what we observed in the vertical profiles (i.e. the fact that, at the shallower site, 20% of the amount of dissolved CH4 present at the bottom was found near the surface), confirms the occurrence of methane fluxes from the oxic layer to the atmosphere at shallow water depths, which is also in agreement with previous findings (Reeburgh et al., 1991; Schmale et al., 2005; McGinnis et al., 2006). The difference in dissolved CH4 concentration at the sea surface of the two sites may support the hypothesis of an efficient vertical transport of dissolved methane from the seafloor up to the surface at shallow depths. Alternatively, other unidentified sources may be the cause of this oversaturation of the surface water, either through lateral diffusion or CH4 generation in the upper water layer.
Average concentrations from measurements of discrete water samples (reported in Table 1) were obtained from only three data points at the deeper site and two at the shallower, with values of 6.6 and 15.1 nmol L−1, respectively. This confirms the results of the in situ technique showing a generally higher concentration of methane at the shallower location.
From the near-surface dissolved CH4 data of the deeper site, eleven isolated peaks were identified (light-blue dots in Figure 4 corresponding to the peaks marked with stars in Figure 3, also numerated on both figures) with peak intensities ranging from 0.6 to 2.3 nmol L−1 and average width of ∼500 m (full width at half maximum, FWHM). All the selected peaks were observed by both in situ instruments except for the second peak in the time series, which was recorded only by the MILS instrument but with a peak intensity of 1.26 nmol L−1 and therefore a relatively good signal to noise ratio. Peaks observed by the METS but not by the MILS sensor were disregarded, since they may be due to a measurement artefact because of the low selectivity of the sensor. The intensities were calculated after subtraction of the background concentration, by means of a Gaussian fit. The two stronger and larger peaks (FWHM of ∼1,500 and ∼640 m) are the N° 3 and 7, both located at the southern edge of the grid survey. These events at the sea surface are however difficult to correlate with the locations of the identified flares (red dots in Figure 4), and the mismatch suggests that: 1) the vertical transport of dissolved CH4 from the seafloor up to the surface may strongly be affected by lateral transport of methane through currents; 2) other factors or other unidentified sources (i.e. microbial activities) may play a role in the occurrence spots of high methane concentration near the sea surface. Despite our achievements, we are still far from computing all the processes for conclusively assessing the fate of methane in the Black Sea water column. The fact that the shallower site has a higher dissolved CH4 concentration at the sea surface with respect to the deeper site agrees with previous findings (Reeburgh et al., 1991; Amouroux et al., 2002). This reinforces the hypothesis of methane transport from the seafloor to the atmosphere at shallow sites, although contributions of other sources cannot be firmly discarded. This is further supported by the fact that the methane oxidation rate is lower in the oxic layer (Reeburgh et al., 1991), promoting its persistence in the water during the ascent. Our results agree with Schmale et al. who, in 2005, concluded that only shallow seeps (depths <100 m) seem to affect the methane concentration of surface water and direct local emissions into the atmosphere, while high-intensity seep sites below this boundary do not show regional influence on the surface layer. Greinert et al. (2010) highlighted how methane fluxes rapidly increase with increasing bubble size and decreasing water depth, underlining that shallow sites may represent a significant source of methane to the atmosphere. Other factors have been proposed to explain marine methane transfer to the atmosphere. Indeed, Shakhova et al., 2017 and Pohlman et al., 2017 also discussed the large release of CH4 from the sediments at shallow sites in the Arctic regions and its potential impact on the climate. They claimed that upwelling and erosion are, for instance, possible mechanisms promoting gas transfer to the atmosphere.
Conclusion
We have presented a multi-technique approach (using acoustic, in situ and laboratory methods) capturing dissolved methane distribution in the water column at the Romanian sector of the Western Black Sea. The results from the cruise allowed the comparison of data from different sensors and techniques, highlighting the challenge for reliable dissolved gas measurements. Thanks to this multi-technique approach we have obtained new insights into the vertical and horizontal distribution of dissolved methane at two different sites.
Relatively good agreement between continuous, in situ, high resolution MILS measurements and discrete sampling followed by laboratory analysis (purge-and-trap and headspace technique followed by gas chromatography analysis) was found on vertical as well as horizontal profiles, despite the difficulties of the comparison due to the lack of dynamic positioning of the vessel. The METS sensor is compact, low-power and easy to use, and allowed to confirm the presence of localized sea surface ‘hot spots’ of dissolved methane observed by the MILS sensor. Nevertheless, it showed limitations for providing quantitative measurement on both vertical and horizontal surface profiles due to its low selectivity and strong dependency to changes in the physical conditions: hydrostatic pressure, water temperature, salinity and dissolved oxygen content. Concentrations of dissolved methane measured by MILS show good agreement with the acoustic data (qualitatively) and measurements of discrete water samples (quantitatively), supporting the reliability of this in situ sensor.
The vertical profiles highlighted a similar distribution of the dissolved CH4 that follows an expected dissolution function. Concentrations at the seafloor were on average ∼80 times larger at the deeper site with respect to the shallower site. At the sea surface of the deeper location, dissolved CH4 was present at a concentration close to that expected from equilibrium with the atmosphere, while it was four times higher at the shallower site. Localized peaks of dissolved CH4 were observed at the sea surface, but a direct correlation with the position of flares at the seafloor was difficult to make. Due to the continuous decreasing trend (bottom to top) obtained from the dissolved methane concentration vertical profiles at the two investigated sites, we hypothesized that higher concentrations of dissolved methane near the surface at the shallower site can be explained by a methane transfer from the seafloor. However, we do not yet have undisputable evidence that would prove this transfer while methane may also be supplied from other sources. This underlines the need of further investigations for better understanding the methane dynamics in the Black Sea. For such a study, dynamic positioning of the vessel or a deployment using a Remotely Operated underwater Vehicle or a submersible will be crucial for accurately capturing the vertical distribution of CH4 in the bubble plume. This would allow for easier comparison between different sensors and techniques, to better evaluate their accuracy, and eventually identify possible instrumental bias for future improvements. Furthermore, following the isotopic signature of methane together with its concentration variability in the water column would provide key information on the fate of methane released from the seafloor and eventually identify the processes mitigating or increasing its concentration at the different water layers of the Black Sea.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, upon request.
Author Contributions
All authors except JG participated in the cruise. LR was chief of the expedition, JP was the leader of the WP4 of the ENVRI + project. RG developed and calibrated the MILS sensor, and conceived and performed the experiments of deployments during the cruise. CB deployed the MILS. DB worked on the calibration and deployment of the METS sensor. JD and VG were in charge of the CTD-Rosette and sampled the seawater. JD performed the measurement of water samples in the laboratory. MS and HL installed the echosounder and performed the acoustic survey. MS performed the analysis of the acoustic data. RG and LR wrote the first draft, and all the authors contributed to the manuscript.
Funding
The research leading to these results has received funding from the ENVRIPlusH2020 project (call 597 Environment, project number 654182) the European Community’s Seventh Framework Programme 598 ERC-2015-PoC under grant agreement no. 713619 (ERC OCEAN-IDs) and from the Agence 599 Nationale de la Recherche (ANR) under grant agreement ANR-18-CE04-0003-01.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer TP declared a past co-authorship with one of the authors LR to the handling editor.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to thank all members of the team who took part in the cruise, colleagues from INGV-Palermo for their fruitful discussions and exchanges during the cruise, the captain and staff of the R/V Mare Nigrum and all the logistic support from the Romanian GeoEcoMar.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/feart.2021.626372/full#supplementary-material
References
Amouroux, D., Roberts, G., Rapsomanikis, S., and Andreae, M. O. (2002). Biogenic Gas (CH4, N2O, DMS) Emission to the Atmosphere from Near-Shore and Shelf Waters of the North-western Black Sea. Estuarine Coastal Shelf Sci. 54 (3), 575–587. doi:10.1006/ecss.2000.0666
Artemov, Y. G., Egorov, V. N., and Gulin, S. B. (2019). Influx of Streaming Methane into Anoxic Waters of the Black Sea Basin. Oceanology 59 (6), 860–870. doi:10.1134/S0001437019060018
Borges, A. V., Champenois, W., Gypens, N., Delille, B., and Harlay, J. (2016). Massive marine Methane Emissions from Near-Shore Shallow Coastal Areas. Sci. Rep. 6, 2–9. doi:10.1038/srep27908
Boulart, C., Briais, A., Chavagnac, V., Révillon, S., Ceuleneer, G., Donval, J.-P., et al. (2017). Contrasted Hydrothermal Activity along the South-East Indian Ridge (130°E-140°E): From Crustal to Ultramafic Circulation. Geochem. Geophys. Geosyst. 18 (7), 2446–2458. doi:10.1002/2016GC006683
Boulart, C., Connelly, D. P., and Mowlem, M. C. (2010). Sensors and Technologies for In Situ Dissolved Methane Measurements and Their Evaluation Using Technology Readiness Levels. Trac - Trends Anal. Chem. 29 (2), 186–195. doi:10.1016/j.trac.2009.12.001
Capet, A., Stanev, E. V., Beckers, J.-M., Murray, J. W., and Grégoire, M. (2016). Decline of the Black Sea Oxygen Inventory. Biogeosciences 13 (4), 1287–1297. doi:10.5194/bg-13-1287-2016
Chua, E. J., Savidge, W., Short, R. T., Cardenas-valencia, A. M., and Fulweiler, R. W. (2016). A Review of the Emerging Field of Underwater Mass Spectrometry. Front. Mar. Sci. 3 (209). doi:10.3389/fmars.2016.00209
Dewey, R. K. (1999). Mooring Design & Dynamics-A Matlab Package for Designing and Analyzing Oceanographic Moorings. Mar. Models 1 (1–4), 103–157. doi:10.1016/S1369-9350(00)00002-X
Donval, J. P., Charlou, J. L., and Lucas, L. (2008). Analysis of Light Hydrocarbons in marine Sediments by Headspace Technique: Optimization Using Design of Experiments. Chemometrics Intell. Lab. Syst. 94 (2), 89–94. doi:10.1016/j.chemolab.2008.06.010
Donval, J. P., and Guyader, V. (2017). Analysis of Hydrogen and Methane in Seawater by “Headspace” Method: Determination at Trace Level with an Automatic Headspace Sampler. Talanta 162, 408–414. doi:10.1016/j.talanta.2016.10.034
Faure, K., Greinert, J., Pecher, I. A., Graham, I. J., Massoth, G. J., de Ronde, C. E. J., et al. (2006). Methane Seepage and its Relation to Slumping and Gas Hydrate at the Hikurangi Margin, New Zealand. New Zealand J. Geology. Geophys. 49 (4), 503–516. doi:10.1080/00288306.2006.9515184
Garcia-Tigreros, F., and Kessler, J. D. (2018). Limited Acute Influence of Aerobic Methane Oxidation on Ocean Carbon Dioxide and pH in Hudson Canyon, Northern U.S. Atlantic Margin. J. Geophys. Res. Biogeosciences 123 (7), 2135–2144. doi:10.1029/2018JG004384
Ghass (2015). Ghass. Available at: https://campagnes.flotteoceanographique.fr/campagnes/15000500/.
Greinert, J., McGinnis, D. F., Naudts, L., Linke, P., and De Batist, M. (2010). Atmospheric Methane Flux from Bubbling Seeps: Spatially Extrapolated Quantification from a Black Sea Shelf Area. J. Geophys. Res. 115 (1), 1–18. doi:10.1029/2009JC005381
Grilli, R., Darchambeau, F., Chappellaz, J., Mugisha, A., Triest, J., and Umutoni, A. (2020). Continuous In Situ Measurement of Dissolved Methane in Lake Kivu Using a Membrane Inlet Laser Spectrometer. Geosci. Instrum. Method. Data Syst. 9 (1), 141–151. doi:10.5194/gi-9-141-2020
Grilli, R., Triest, J., Chappellaz, J., Calzas, M., Desbois, T., Jansson, P., et al. (2018). Sub-Ocean: Subsea Dissolved Methane Measurements Using an Embedded Laser Spectrometer Technology. Environ. Sci. Technol. 52 (18), 10543–10551. doi:10.1021/acs.est.7b06171
Hartmann, J. F., Gentz, T., Schiller, A., Greule, M., Grossart, H.-P., Ionescu, D., et al. (2018). A Fast and Sensitive Method for the Continuous In Situ Determination of Dissolved Methane and its δ13C-isotope Ratio in Surface Waters. Limnol. Oceanogr. Methods 16 (5), 273–285. doi:10.1002/lom3.10244
Heeschen, K. U., Collier, R. W., de Angelis, M. A., Suess, E., Rehder, G., Linke, P., et al. (2005). Methane Sources, Distributions, and Fluxes from Cold Vent Sites at Hydrate Ridge, Cascadia Margin. Glob. Biogeochem. Cycles 19 (2), 1–19. doi:10.1029/2004GB002266
Ippommatsu, M., Sasaki, H., and Yanagida, H. (1990). Sensing Mechanism of SnO2 Gas Sensors. J. Mater. Sci. 25 (1), 259–262. doi:10.1007/BF00544217
James, R. H., Bousquet, P., Bussmann, I., Haeckel, M., Kipfer, R., Leifer, I., et al. (2016). Effects of Climate Change on Methane Emissions from Seafloor Sediments in the Arctic Ocean: A Review. Limnol. Oceanogr. 61, S283–S299. doi:10.1002/lno.10307
Jansson, P., Ferré, B., Silyakova, A., Dølven, K. O., and Omstedt, A. (2019a). A New Numerical Model for Understanding Free and Dissolved Gas Progression toward the Atmosphere in Aquatic Methane Seepage Systems. Limnol. Oceanogr. Methods 17 (3), 223–239. doi:10.1002/lom3.10307
Jansson, P., Triest, J., Grilli, R., Ferré, B., Silyakova, A., Mienert, J., et al. (2019b). High-resolution Underwater Laser Spectrometer Sensing Provides New Insights into Methane Distribution at an Arctic Seepage Site. Ocean Sci. 15 (4), 1055–1069. doi:10.5194/os-15-1055-2019
Karl, D. M., Beversdorf, L., Björkman, K. M., Church, M. J., Martinez, A., and Delong, E. F. (2008). Aerobic Production of Methane in the Sea. Nat. Geosci. 1 (7), 473–478. doi:10.1038/ngeo234
Kessler, J. D., Reeburgh, W. S., Southon, J., Seifert, R., Michaelis, W., and Tyler, S. C. (2006a). Basin-wide Estimates of the Input of Methane from Seeps and Clathrates to the Black Sea. Earth Planet. Sci. Lett. 243 (3–4), 366–375. doi:10.1016/j.epsl.2006.01.006
Kessler, J. D., Reeburgh, W. S., and Tyler, S. C. (2006b). Controls on Methane Concentration and Stable Isotope (δ2H-CH4andδ13C-CH4) Distributions in the Water Columns of the Black Sea and Cariaco Basin. Glob. Biogeochem. Cycles 20 (4), 1–13. doi:10.1029/2005GB002571
Krabbenhoeft, A., Netzeband, G. L., Bialas, J., and Papenberg, C. (2010). Episodic Methane Concentrations at Seep Sites on the Upper Slope Opouawe Bank, Southern Hikurangi Margin, New Zealand. Mar. Geology 272 (1–4), 71–78. doi:10.1016/j.margeo.2009.08.001
Lammers, S., and Suess, E. (1994). An Improved Head-Space Analysis Method for Methane in Seawater. Mar. Chem. 47 (2), 115–125. doi:10.1016/0304-4203(94)90103-1
Leifer, I., and Patro, R. K. (2002). The Bubble Mechanism for Methane Transport from the Shallow Sea Bed to the Surface: A Review and Sensitivity Study. Continental Shelf Res. 22 (16), 2409–2428. doi:10.1016/S0278-4343(02)00065-1
Luc Charlou, J., Dmitriev, L., Bougault, H., and Needham, H. D. (1988). Hydrothermal CH4 between 12°N and 15°N over the Mid-Atlantic Ridge. Deep Sea Res. A. Oceanographic Res. Pap. 35 (1), 121–131. doi:10.1016/0198-0149(88)90061-1
MacLennan, D. N., and Svellingen, I. (1989). Simple Calibration Technique for the Split-Beam echo-sounder. Fiskdir. Skr. Ser. Havunders 18 (9), 365–379.
Malakhova, L. V., Egorov, V. N., Malakhova, T. V., Gulin, S. B., and Artemov, Y. G. (2010). Methane in the Sevastopol Coastal Area, Black Sea. Geo-Mar Lett. 30 (3–4), 391–398. doi:10.1007/s00367-010-0198-7
Marinaro, G., Etiope, G., Gasparoni, F., Calore, D., Cenedese, S., Furlan, F., et al. (2004). GMM?a Gas Monitoring Module for Long-Term Detection of Methane Leakage from the Seafloor. Env Geol. 46 (8), 1053–1058. doi:10.1007/s00254-004-1092-2
Mau, S., Römer, M., Torres, M. E., Bussmann, I., Pape, T., Damm, E., et al. (2017). Widespread Methane Seepage along the continental Margin off Svalbard - from Bjørnøya to Kongsfjorden. Sci. Rep. 7, 1–13. doi:10.1038/srep42997
McGinnis, D. F., Greinert, J., Artemov, Y., Beaubien, S. E., and Wüest, a. (2006). Fate of Rising Methane Bubbles in Stratified Waters: How Much Methane Reaches the Atmosphere?. J. Geophys. Res. 111 (9), 1–15. doi:10.1029/2005JC003183
MSM34 (2014). MSM34. Available at: http://eprints.uni-kiel.de/29989/.
Myhre, C. L., Ferré, B., Platt, S. M., Silyakova, A., Hermansen, O., Allen, G., et al. (2016). Extensive Release of Methane from Arctic Seabed West of Svalbard during Summer 2014 Does Not Influence the Atmosphere. Geophys. Res. Lett. 43 (9), 4624–4631. doi:10.1002/2016GL068999
Naqvi, S. W. A., Bange, H. W., Farías, L., Monteiro, P. M. S., Scranton, M. I., and Zhang, J. (2010). Marine Hypoxia/anoxia as a Source of CH4 and N2O. Biogeosciences 7 (7), 2159–2190. doi:10.5194/bg-7-2159-2010
Newman, K. R., Cormier, M.-H., Weissel, J. K., Driscoll, N. W., Kastner, M., Solomon, E. A., et al. (2008). Active Methane Venting Observed at Giant Pockmarks along the U.S. Mid-Atlantic Shelf Break. Earth Planet. Sci. Lett. 267, 341–352. doi:10.1016/j.epsl.2007.11.053
Özsoy, E., and Ünlüata, Ü. (1997). Oceanography of the Black Sea: A Review of Some Recent Results. Earth-Science Rev. 42 (4), 231–272. doi:10.1016/S0012-8252(97)81859-4
Pape, T., Blumenberg, M., Seifert, R., Bohrmann, G., and Michaelis, W. (2008). “Marine Methane Biogeochemistry of the Black Sea: A Review,” in Links Between Geological Processes, Microbial Activities&Evolution of Life (Dordrecht Netherlands: Springer), 281–311.
Pohlman, J. W., Greinert, J., Ruppel, C., Silyakova, A., Vielstädte, L., Casso, M., et al. (2017). Enhanced CO2 Uptake at a Shallow Arctic Ocean Seep Field Overwhelms the Positive Warming Potential of Emitted Methane. Proc. Natl. Acad. Sci. USA 114 (21), 5355–5360. doi:10.1073/pnas.1618926114
Reeburgh, W. S. (2007). Oceanic Methane Biogeochemistry. Chem. Rev. 107, 486–513. doi:10.1021/cr050362v
Reeburgh, W. S., Tyler, S. C., and Carroll, J. (2006). Stable Carbon and Hydrogen Isotope Measurements on Black Sea Water-Column Methane. Deep Sea Res. Part Topical Stud. Oceanography 53 (17–19), 1893–1900. doi:10.1016/j.dsr2.2006.03.018
Reeburgh, W. S., Ward, B. B., Whalen, S. C., Sandbeck, K. A., Kilpatrickt, K. A., and Kerkhof, L. J. (1991). Black Sea Methane Geochemistry. Deep Sea Res. Part A. Oceanographic Res. Pap. 38 (Suppl. 2), S1189–S1210. doi:10.1016/s0198-0149(10)80030-5
Repeta, D. J., Ferrón, S., Sosa, O. A., Johnson, C. G., Repeta, L. D., Acker, M., et al. (2016). Marine Methane Paradox Explained by Bacterial Degradation of Dissolved Organic Matter. Nat. Geosci. 9 (12), 884–887. doi:10.1038/ngeo2837
Rettich, T. R., Handa, Y. P., Battino, R., and Wilhelm, E. (1981). Solubility of Gases in Liquids. 13. High-Precision Determination of Henry's Constants for Methane and Ethane in Liquid Water at 275 to 328 K. J. Phys. Chem. 85 (22), 3230–3237. doi:10.1021/j150622a006
Riboulot, V., Ker, S., Sultan, N., Thomas, Y., Marsset, B., Scalabrin, C., et al. (2018). Freshwater lake to Salt-Water Sea Causing Widespread Hydrate Dissociation in the Black Sea. Nat. Commun. 9 (1), 1–8. doi:10.1038/s41467-017-02271-z
Ruffine, L., Donval, J.-P., Croguennec, C., Burnard, P., Lu, H., Germain, Y., et al. (2018). Multiple Gas Reservoirs Are Responsible for the Gas Emissions along the Marmara Fault Network. Deep Sea Res. Part Topical Stud. Oceanography 153, 48–60. doi:10.1016/j.dsr2.2017.11.011
Saunois, M., Bousquet, P., Poulter, B., Peregon, A., Ciais, P., Canadell, J. G., et al. (2017). Variability and Quasi-Decadal Changes in the Methane Budget over the Period 2000-2012. Atmos. Chem. Phys. 17 (18), 11135–11161. doi:10.5194/acp-17-11135-2017
Schmale, O., Beaubien, S. E., Rehder, G., Greinert, J., and Lombardi, S. (2010). Gas Seepage in the Dnepr Paleo-delta Area (NW-Black Sea) and its Regional Impact on the Water Column Methane Cycle. J. Mar. Syst. 80 (1–2), 90–100. doi:10.1016/j.jmarsys.2009.10.003
Schmale, O., Greinert, J., and Rehder, G. (2005). Methane Emission from High-Intensity marine Gas Seeps in the Black Sea into the Atmosphere. Geophys. Res. Lett. 32 (7), 1–4. doi:10.1029/2004GL021138
Schmidt, M., Linke, P., and Esser, D. (2013). Recent Development in IR Sensor Technology for Monitoring Subsea Methane Discharge. Mar. Technol. Soc. J. 47 (3), 27–36. doi:10.4031/mtsj.47.3.8
Seiyama, T., Kato, A., Fujiishi, K., and Nagatani, M. (1962). A New Detector for Gaseous Components Using Semiconductive Thin Films. Anal. Chem. 34 (11), 1502–1503. doi:10.1021/ac60191a001
Shakhova, N., Semiletov, I., Gustafsson, O., Sergienko, V., Lobkovsky, L., Dudarev, O., et al. (2017). Current Rates and Mechanisms of Subsea Permafrost Degradation in the East Siberian Arctic Shelf. Nat. Commun. 8 (1), 15872. doi:10.1038/ncomms15872
Shakhova, N., Semiletov, I., Leifer, I., Sergienko, V., Salyuk, A., Kosmach, D., et al. (2014). Ebullition and Storm-Induced Methane Release from the East Siberian Arctic Shelf. Nat. Geosci. 7 (1), 64–70. doi:10.1038/ngeo2007
Solomon, E. A., Kastner, M., MacDonald, I. R., and Leifer, I. (2009). Considerable Methane Fluxes to the Atmosphere from Hydrocarbon Seeps in the Gulf of Mexico. Nat. Geosci. 2 (8), 561–565. doi:10.1038/ngeo574
Sovga, E. E., Lyubartseva, S. P., and Lyubitsky, A. A. (2008). Investigation of the Biogeochemistry of Methane and Mechanisms of its Transfer in the Black Sea. Phys. Oceanogr. 18 (5), 272–287. doi:10.1007/s11110-009-9024-z
Stanev, E. V., Peneva, E., and Chtirkova, B. (2019). Climate Change and Regional Ocean Water Mass Disappearance: Case of the Black Sea. J. Geophys. Res. Oceans 124 (7), 4803–4819. doi:10.1029/2019JC015076
Stanev, E. V., Poulain, P. M., Grayek, S., Johnson, K. S., Claustre, H., and Murray, J. W. (2018). Understanding the Dynamics of the Oxic‐Anoxic Interface in the Black Sea. Geophys. Res. Lett. 45 (2), 864–871. doi:10.1002/2017GL076206
Stow, D. A. V., Hernández-Molina, F. J., Llave, E., Sayago-Gil, M., Díaz del Río, V., and Branson, A. (2009). Bedform-velocity Matrix: The Estimation of Bottom Current Velocity from Bedform Observations. Geology 37 (4), 327–330. doi:10.1130/g25259a.1
Swinnerton, J. W., Linnenbom, V. J., and Cheek, C. H. (1968). A Sensitive Gas Chromatographic Method for Determining Carbon Monoxide in Seawater. Limnol. Oceanogr. 13 (1), 193–195. doi:10.4319/lo.1968.13.1.0193
Triest, J., Chappellaz, J., and Grilli, R. (2017). Patent 08276-01: System for Fast and In-Situ Sampling of Dissolved Gases in the Ocean. Grenoble FRANCE: CNRS.
Tsurushima, N., Watanabe, S., and Tsunogai, S. (1999). Determination of Light Hydrocarbons Dissolved in Seawater. Talanta 50 (3), 577–583. doi:10.1016/S0039-9140(99)00144-7
Veloso, M., Greinert, J., Mienert, J., and De Batist, M. (2015). A New Methodology for Quantifying Bubble Flow Rates in Deep Water Using Splitbeam Echosounders: Examples from the A Rctic Offshore NW‐ S Valbard. Limnol. Oceanogr. Methods 13 (6), 267–287. doi:10.1002/lom3.10024
Weber, M. E., von Stackelberg, U., Marchig, V., Wiedicke, M., and Grupe, B. (2000). Variability of Surface Sediments in the Peru basin : Dependence on Water Depth , Productivity , Bottom Water Flow , and Seafloor Topography. Mar. Geol. 163 (1–4), 163169–163184. doi:10.1016/s0025-3227(99)00103-6
Weber, T., Wiseman, N. A., and Kock, A. (2019). Global Ocean Methane Emissions Dominated by Shallow Coastal Waters. Nat. Commun. 10 (1), 1–10. doi:10.1038/s41467-019-12541-7
Weinstein, A., Navarrete, L., Ruppel, C., Weber, T. C., Leonte, M., Kellermann, M. Y., et al. (2016). Determining the Flux of Methane into Hudson Canyon at the Edge of Methane Clathrate Hydrate Stability. Geochem. Geophys. Geosyst. 17 (10), 3882–3892. doi:10.1002/2016GC006421
Weiss, R. F. (1974). Carbon Dioxide in Water and Seawater: the Solubility of a Non-ideal Gas. Mar. Chem. 2 (3), 203–215. doi:10.1016/0304-4203(74)90015-2
Westbrook, G. K., Thatcher, K. E., Rohling, E. J., Piotrowski, A. M., Pälike, H., Osborne, A. H., et al. (2009). Escape of Methane Gas from the Seabed along the West Spitsbergen continental Margin. Geophys. Res. Lett. 36 (15), 1–5. doi:10.1029/2009GL039191
Wiesenburg, D. A., and Guinasso, N. L. (1979). Equilibrium Solubilities of Methane, Carbon Monoxide, and Hydrogen in Water and Sea Water. J. Chem. Eng. Data 24 (4), 356–360. doi:10.1021/je60083a006
Wilson, S. T., Bange, H. W., Arévalo-Martínez, D. L., Barnes, J., Borges, A. V., Brown, I., et al. (2018). An Intercomparison of Oceanic Methane and Nitrous Oxide Measurements. Biogeosciences 15 (19), 5891–5907. doi:10.5194/bg-15-5891-2018
Keywords: dissolved gas, methane, black sea, in situ measurements, gas seepages, instrumental inter-comparison
Citation: Grilli R, Birot D, Schumacher M, Paris J-D, Blouzon C, Donval JP, Guyader V, Leau H, Giunta T, Delmotte M, Radulescu V, Balan S, Greinert J and Ruffine L (2021) Inter-Comparison of the Spatial Distribution of Methane in the Water Column From Seafloor Emissions at Two Sites in the Western Black Sea Using a Multi-Technique Approach. Front. Earth Sci. 9:626372. doi: 10.3389/feart.2021.626372
Received: 05 November 2020; Accepted: 06 July 2021;
Published: 28 July 2021.
Edited by:
Martin Scherwath, University of Victoria, CanadaReviewed by:
Xiaole Sun, Baltic Sea Centre, Stockholm University, SwedenThomas Pape, University of Bremen, Germany
Copyright © 2021 Grilli, Birot, Schumacher, Paris, Blouzon, Donval, Guyader, Leau, Giunta, Delmotte, Radulescu, Balan, Greinert and Ruffine. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roberto Grilli, cm9iZXJ0by5ncmlsbGlAY25ycy5mcg==