 Alexandra V. Turchyn
Alexandra V. Turchyn Harold J. Bradbury
Harold J. Bradbury Kathryn Walker1
Kathryn Walker1 Xiaole Sun
Xiaole Sun
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Earth Sci., 26 February 2021
Sec. Biogeoscience
Volume 9 - 2021 | https://doi.org/10.3389/feart.2021.618311
This article is part of the Research TopicThe Marine Carbon Cycle: From Ancient Storage to Future ChallengesView all 5 articles
The vast majority of carbonate minerals in modern marine sediments are biogenic, derived from the skeletal remains of organisms living in the ocean. However, carbonate minerals can also precipitate abiotically within marine sediments, and this carbonate mineral precipitation within sediments has been suggested as a third major, and isotopically distinct, sink in the global carbon cycle, particularly important earlier in Earth history. Here we present a global compilation of pore fluid data and compare the sulfate, calcium, phosphate and magnesium concentrations with pore fluid alkalinity to explore the emerging relationships and explore what drives carbonate mineral precipitation in sediments. Our data compilation shows that the gradient of pore fluid sulfate concentrations correlates strongly with the gradient of alkalinity as well as with the gradient of calcium, and that these correlations improve dramatically in sediments where methane is present. We also note that sedimentary pore fluids that are high in phosphate concentration are also high in alkalinity, which may indicate suppression of carbonate mineral precipitation in the presence of sedimentary phosphate. Our data can be used to highlight sediments where both dolomite formation and dolomitization of previously deposited calcium carbonate minerals is occurring. We explore how carbonate mineral saturation state changes as a function of calcium concentrations, alkalinity, and pH, and suggest a reason why calcium concentrations are never fully depleted in sedimentary pore fluids. We conclude that carbonate minerals precipitate in sediments with methane, where the anaerobic oxidation of this methane helps promote particularly high saturation states for carbonate minerals.
Carbon burial in marine sediments as organic carbon or calcium carbonate minerals represents the largest removal path for carbon from Earth’s surface environment (Berner and Caldeira, 1997; Berner, 2003; Martin, 2017). Calcium carbonate minerals, largely biogenic but also detrital, are delivered to marine sediments from the water column, however the majority of deep marine sediments are bathed in water that is undersaturated with respect to calcium carbonate minerals (Burdige, 2006; Burdige et al., 2008). Within marine sediments, changing environmental conditions such as changes in pH and the concentration of dissolved inorganic carbon may allow carbonate minerals to precipitate within the sediment column (Middelburg et al., 1997; Soetaert et al., 2007; Arndt et al., 2009). This carbonate mineral precipitation in sediments has been called ‘authigenic carbonate,’ or ‘sedimentary carbonate,’ and is found in the form of cements and disseminated particles within the marine sedimentary pile (Ussler and Paull, 2008; Higgins et al., 2009). Precipitation of calcium carbonate minerals in sediments is driven by the generation of subseafloor alkalinity, which is typically the result of microbial respiration of organic carbon in the absence of oxygen; the oxygen penetration depth for the majority of the seafloor is less than 1 m (D’Hondt et al., 2015).
In theory, the precipitation of carbonate minerals in sediments can be traced by studying the chemistry of pore fluids within the sediment column. Over the last 50 years of Ocean Research Drilling, the chemistry of sedimentary pore fluids from over 700 locations has been acquired and measured (Figure 1). The premise of this work is that this dataset holds much information about the global sedimentary carbon cycle. Throughout this paper we will use the term ‘sedimentary carbon cycle’ to refer to the transformation of carbon among various mineralized, dissolved and gaseous forms within marine sediments. Specifically, carbon deposited in marine sediments in the form of biogenic calcium carbonate minerals or organic material, may end up oxidized, buried, dissolved or converted to methane. Our principal aim is to investigate processes controlling carbonate mineral precipitation in sediments in the modern ocean, focusing on the relationships between the concentrations and fluxes of the main species (sulfate and alkalinity) involved in the generation of subsurface alkalinity, which drives precipitation of carbonate minerals in global sediments.
FIGURE 1. Map of the global dataset from which data presented in this paper are presented.
Microbial respiration in anoxic sediments is the key factor in carbonate mineral precipitation in sediments because oxidation of organic carbon yields alkalinity—in the form of bicarbonate ions—which is required to precipitate calcium carbonate. Unlike aerobic respiration, some anaerobic metabolisms also consume protons, raising pH, which helps supersaturate calcium carbonate minerals in anoxic sediments (Soetaert et al., 2007; Arndt et al., 2009). ‘Organic carbon’ comprises proteins, lipids, nucleic acids and decomposition products of marine and terrestrial life. Organic carbon is consumed and thus oxidized back to dissolved inorganic carbon (DIC) by bacteria, archaea and eukaryotes within the ocean, and mostly by bacteria and archaea in sediments. The type of organic carbon that a microbial community consumes influences both the change in pH as well as the amount of DIC generated through their metabolism (Gallagher et al., 2012).
Oxic respiration of organic carbon yields the greatest free energy change per mole of organic carbon oxidized (Froelich et al., 1979):
Aerobic respiration dominates in ocean and surface sediments, although the penetration depth of oxygen is typically less than 1 m (Kasten and Jørgensen, 2000; D’Hondt et al., 2015). Nitrate is released during aerobic respiration. Nitrate is the second most efficient oxidant, and is used when dissolved oxygen concentration falls below 5% of background seawater concentration (Prokopenko et al., 2011):
When nitrate has been depleted, iron and manganese oxides are typically respired. The lower abundance of iron and manganese oxides in marine sediments (most are trapped in marginal marine environments) means their contribution to organic carbon oxidation is small in the modern ocean (Froelich et al., 1979; Aller and Rude, 1988; Shaw et al., 1990). Manganese oxides are more energetically favorable for organic carbon oxidation than iron oxides, although iron oxides span a wide range in their energetic favorability (Froelich et al., 1979).
Microbial sulfate reduction dominates anaerobic organic carbon oxidation in the modern ocean because ocean sulfate concentrations are typically orders of magnitude greater than that of other commonly used oxidants (Kasten and Jørgensen, 2000; Egger et al., 2018):
When sulfate concentrations become rate-limiting, any remaining organic carbon may be converted to methane (Sivan et al., 2007):
Methane is a gas and is less dense than fluids, so migrates upwards toward the surface. Methane may be anaerobically oxidized by sulfate (through a process called sulfate-driven anaerobic methane oxidation or AOM), producing DIC and hydrogen sulfide (Whiticar and Faber, 1986; Niewöhner et al., 1998; Whiticar, 1999). The anaerobic oxidation of methane is mediated by a consortium of microorganisms involving anaerobic methanotrophs and sulfate-reducing bacteria (Knittel and Boetius, 2009; Schubert et al., 2011) and is governed by the following reaction stoichiometry:
Reactions 2–4 generate dissolved inorganic carbon (Froelich et al., 1979; Wang and Van Cappellen, 1996). This in turn increases total alkalinity, which is defined as:
The generation of alkalinity through the subsurface oxidation of organic carbon (Eqs. 1–4) can lead to the subsurface precipitation of calcium carbonate:
In order for carbonate mineral precipitation to occur, calcium ions are supplied from overlying seawater, while the concentration of bicarbonate ions is determined by the concentration of total dissolved inorganic carbon and the in situ pH (Zeebe and Wolf-Gladrow, 2001). The saturation state (Ω) of the pore fluid informs the likelihood of carbonate mineral precipitation. When Ω > 1, the fluid is supersaturated with respect to calcium carbonate, and precipitation should occur. Fluid is undersaturated when Ω < 1, and should lead to mineral dissolution (Zeebe and Westbroek, 2003).
The reaction stoichiometry detailed above predicts that 1 mol of calcium ion reacts with 2 mol of bicarbonate ion to precipitate calcium carbonate (Eq. 7), while for every mole of sulfate reduced during organic-matter consuming (organoclastic) microbial sulfate reduction, 2 mol of bicarbonate ion are produced (Eq. 3), while 1 mol of bicarbonate ion is produced when sulfate reduction is coupled to anaerobic methane oxidation (Eq. 5). This defines stoichiometric relationships among alkalinity, sulfate, and calcium that together inform carbonate mineral precipitation in sediments (Figure 1, column 1). For example, in theory, calcium should be consumed in a one-to-one molar ratio with sulfate. In the modern ocean, sulfate concentrations are three times higher than those of calcium. We would predict, therefore, that excess alkalinity should be produced relative to calcium, which should allow all calcium in pore fluid to form carbonate minerals (Eq. 7).
In this paper, the relative relationship among sulfate, calcium, and alkalinity is explored using pore fluid data from the global ODP/IODP database (Figure 1). Data was sourced from the Janus/LIMS Web database (https://web.iodp.tamu.edu/OVERVIEW/), collected on and measured during various ODP and IODP cruises; the methods of collection and measurement of various aqueous geochemical data discussed here can be found in the original cruise reports. Pore fluid concentrations of sulfate, calcium and alkalinity were cross-plot to reveal the relationship among these key species (Figure 2) and to compare them to those predicted from the first-order relationships from the stoichiometry of the various chemical reactions (Eqs. 1–7).
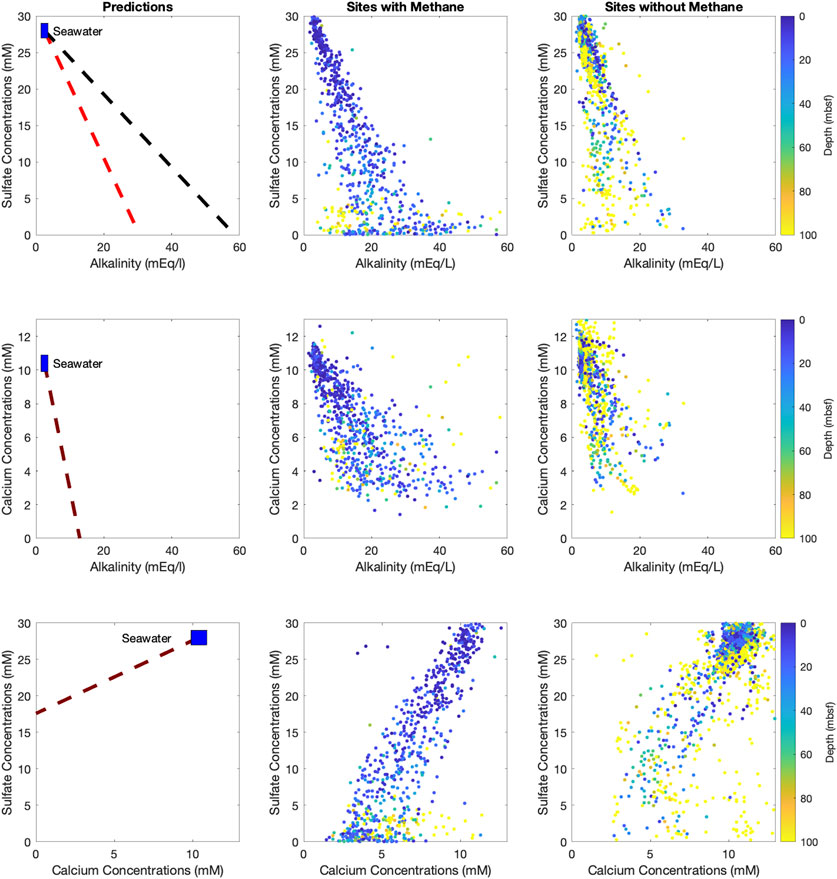
FIGURE 2. Left column—predicted relationships between sulfate and alkalinity, with red denoting anaerobic methane oxidation and black denoting organoclastic sulfate reduction (top), calcium and alkalinity (middle) and sulfate and calcium (bottom) based on the stoichiometry given in Eqs 1–7. Middle and Right columns—the measured pore fluid sulfate, alkalinity and calcium concentrations for sites where methane is present (middle) and where methane is absent (right). The data in the middle and right columns are color-coded according to the depth of the sample, pore fluid data deeper than 100 mbsf all plot in yellow.
The relationships between sulfate and alkalinity, calcium and alkalinity, and sulfate and calcium in the global pore fluid database are in the same direction and sense as those predicted from the stoichiometric relationships (Figure 2). We note that when methane is present, measured alkalinity spans close to 40 mEq/L at the depth where sulfate concentrations are zero (between ∼8 and 45 mEq/L), while when methane is not present sulfate concentrations rarely reach zero and alkalinity is much lower (between 2 and 28 mEq/L). Similar to previous work, our compilation shows that when methane is present, sulfate is depleted far higher in the sediment column than if methane is not present (Egger et al., 2018; Bradbury and Turchyn, 2019). Our second observation is that in sediment where methane is not present, we note that the relationship between pore fluid calcium concentrations and alkalinity is close to that predicted from stoichiometry (Figure 2, middle row). We also note that at none of the study sites calcium concentrations reach zero. Finally, our results show that sulfate and calcium are tightly correlated when methane is present, but less so when methane is absent (Figure 2, bottom row). However, the tight correlation has a gradient that is steeper than that predicted from the stoichiometry of microbial sulfate reduction coupled to carbonate mineral precipitation.
To better understand the cause of the correlations that emerge in Figure 2, we also explore the data color-coded by the reported pore fluid phosphate concentration (Figure 3) and by the reported pore fluid magnesium concentration (Figure 4). These results show that sites without methane have far lower pore fluid phosphate concentrations (D’Hondt et al., 2015), while at sites with methane, pore fluid alkalinity and pore fluid phosphate are positively correlated (Figure 3). Pore fluid phosphate concentrations reach their maximum at the depth with the lowest sulfate and calcium concentrations when methane is present (Figure 3). Pore fluid magnesium concentrations are lowest when alkalinity is lowest regardless of whether methane is present (Figure 4). We also note in Figure 4 an anomalous cluster of low pore fluid magnesium concentrations with high pore fluid calcium concentrations in sites without methane.
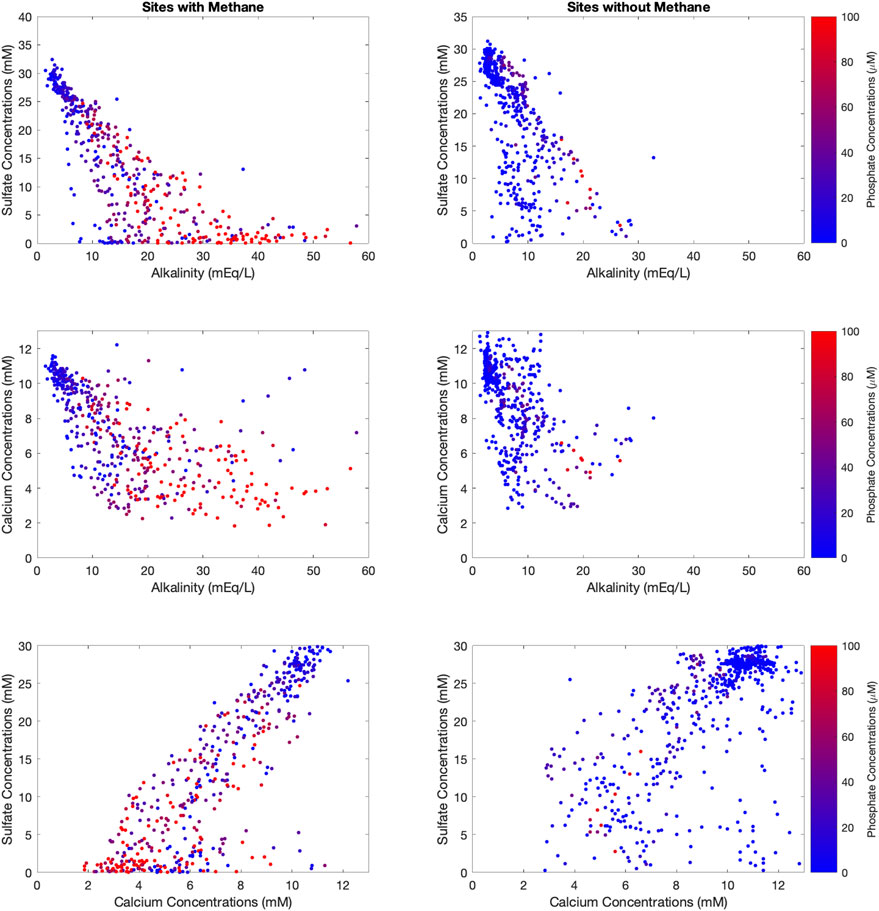
FIGURE 3. Cross plots of sulfate vs. alkalinity (top row), calcium vs. alkalinity (middle row) and sulfate vs. calcium (bottom row) with the data color-coded by the concentration of pore fluid phosphate. The data are split into sites that have methane (left column) vs. sites that do not (right column).
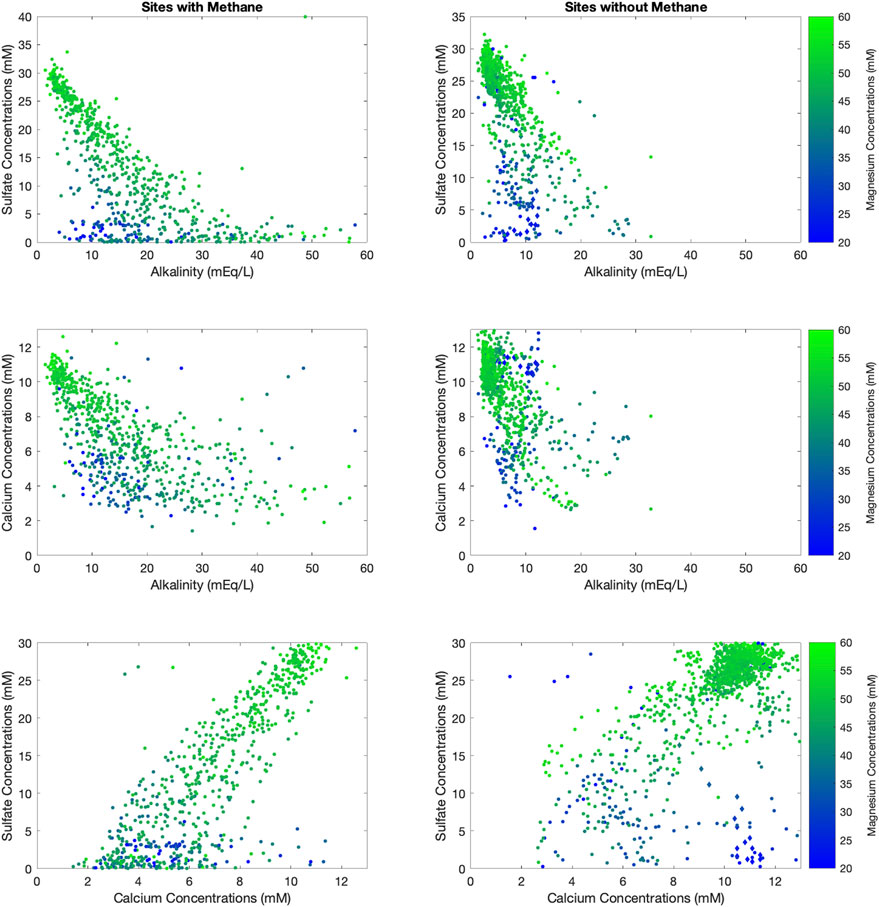
FIGURE 4. Cross plots of sulfate vs. alkalinity (top row), calcium vs. alkalinity (middle row) and sulfate vs. calcium (bottom row) with the data color-coded by the concentration of pore fluid magnesium. The data are split into sites that have methane (left column) vs. sites that do not (right column).
Concentration-depth plots were produced for all sites to determine the concentration gradient (mM/meter or mEq/L/meter for alkalinity). Where profiles displayed strong curvature—indicating the predominance of chemical reactions over diffusion—the steepest slope was used for the determination of the chemical gradient. We present the relative changes in the concentration gradient among the key species in Figure 5. There is a strong correlation between the gradient of alkalinity and the gradient of sulfate (Figure 5A), regardless of whether methane is present or not. However, as seen in Figure 2, sites with methane have a stronger correlation between pore fluid sulfate and calcium and also between pore fluid calcium and alkalinity. Although visually the correlation between pore fluid calcium and alkalinity looks strong in sites without methane, there is such little change in calcium or alkalinity in sites without methane that there is not much correlation between the two.
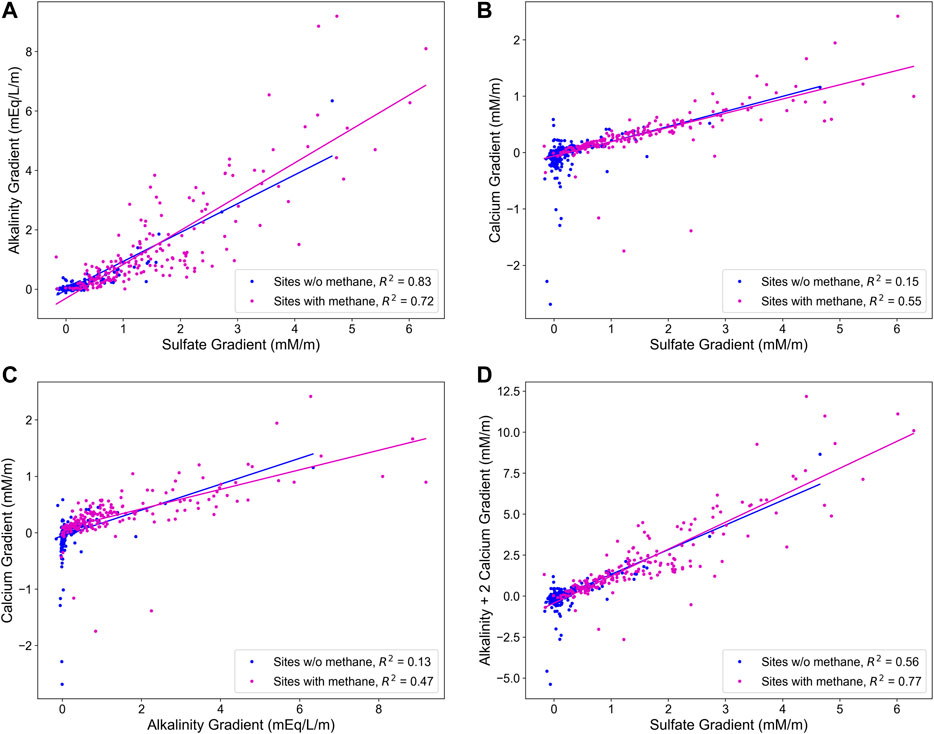
FIGURE 5. From top left, correlations between the concentration gradients of sulfate vs. alkalinity, sulfate vs. calcium, calcium vs. alkalinity, and sulfate vs. alkalinity + 2*Calcium.
The linear arrays between the concentration of sulfate and pore fluid alkalinity (Figure 2), as well as the tight correlation between the gradient of sulfate and the gradient of alkalinity (Figure 5) suggest that microbial sulfate reduction plays a key role in sedimentary alkalinity generation. While the exact relationship between the decrease in sulfate and increase in alkalinity varies among the sites, we note that, on the whole, a wider range of alkalinity is measured for a given change in pore fluid sulfate concentration than predicted for anaerobic methane oxidation (where the change should be 1:1) or organoclastic sulfate reduction (where the change should be 2:1, that is 2 mol of alkalinity produced for each mol of sulfate reduced). This data points to other sources and sinks for alkalinity within marine sediments.
Possible sinks for alkalinity include precipitation of carbonate minerals or apatite in sediments (e.g., calcite—Eq 7 or dolomite or calcium fluorapatite), sedimentary authigenic clay precipitation, or exchange with underlying basalt or basement rocks (Wang and Van Cappellen, 1996; Moore et al., 2004; Chatterjee et al., 2011; Dunlea et al., 2017). We will discuss each of these in term. Sedimentary mineral precipitation, including calcite, dolomite, and fluorapatite, could be inferred from pore fluid profiles of particular elements, including calcium or magnesium (Higgins and Schrag, 2010; Fantle and Higgins, 2014; Higgins et al., 2018). For example, dolomite is a calcium magnesium carbonate mineral: CaMg(CO3)2, that requires calcium and magnesium in an equal molar ratio. The sites with low magnesium that also have low alkalinity could indicate dolomite formation in sediments, consuming both dissolved inorganic carbon (carbonate alkalinity) and magnesium (Figure 3). Dolomite can also form from recrystallization of deposited calcium carbonate minerals to dolomite or protodolomite, where the replacement of calcium ions by magnesium ions releases calcium to the pore fluid (Fantle and Higgins, 2014; Higgins et al., 2018). This may explain the sites where low concentrations of pore fluid magnesium correlate with high concentrations of pore fluid calcium (Figure 4). A clear example of this process is shown at Site 1000 located on the Bahama Banks, which is plotted in diamonds in Figure 4, where active dolomite precipitation has been previously documented (Sigurdsson et al., 1997).
Calcium fluorapatite
An additional sink for alkalinity, as suggested by Figure 4, could be through sedimentary authigenic clay mineral formation. Authigenic clay formation, also called reverse weathering, within sediments involves the conversion of detrital particles into new clay minerals, consuming protons and thus reducing alkalinity (Garrels, 1965; Mackenzie and Kump, 1995; Dunlea et al., 2017). Authigenic clay formation includes reactions that have stoichiometries such as:
However, authigenic clay precipitation is difficult to infer from pore fluid profiles alone because these cations may also be involved in other reactions and the stoichiometries of which protons are produced or consumed is complicated. Furthermore, authigenic clay precipitation requires a source of reactive silica; in the modern ocean reactive silica is supplied by the tests of siliceous algae or volcanic ash, which are unevenly distributed globally within sediments (Olgun et al., 2011; Dunlea et al., 2017). Indeed, we do not see any correlation between pore fluid silica concentrations and pore fluid alkalinity or calcium, although it appears that pore fluid silica concentrations are higher in the absence of methane (Figure 6). Authigenic clay formation also consumes cations other than magnesium and therefore may not be apparent in the magnesium pore fluid concentrations (Mackenzie and Kump, 1995). We do not see a region of Figure 4 where the combination of changes in pore fluid magnesium, sulfate, calcium or alkalinity suggest, definitively, that there is authigenic clay formation or that it is dominating the relative pore fluid behavior of the various cations.
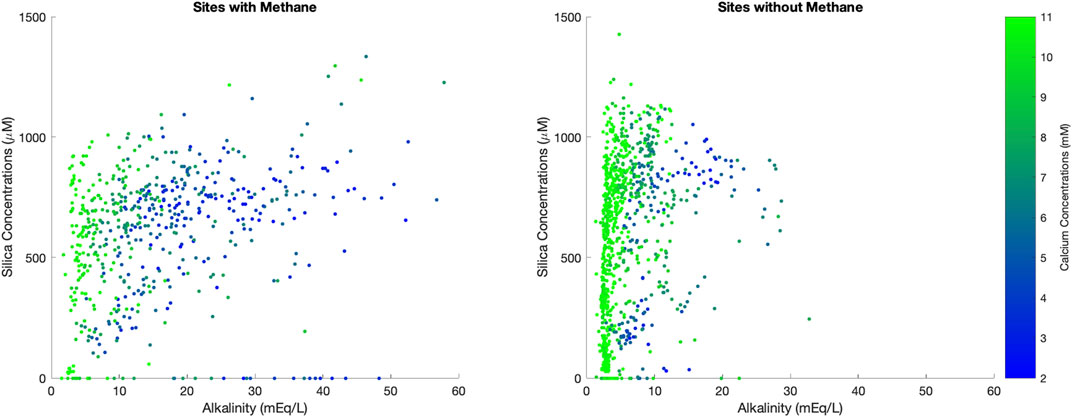
FIGURE 6. Pore fluid silica concentrations vs. pore fluid alkalinity, shaded as a function of calcium concentrations. Data are split into sediment that has methane vs. sediment that does not.
A final sink for alkalinity could be chemical reaction with underlying basalt, which can lead to mineral carbonation of the basalt, consuming alkalinity. However, the diffusion reaction length for alkalinity is very short in marine sediments, and not dominated by the long length-scales required for diffusion through the sediment pile to reaction with the underlying crust. Therefore, we do not think that chemical reaction with underlying basalt and mineral carbonation in this basalt is influencing the alkalinity profiles in global marine sediments.
Another possibility for why microbial sulfate reduction does not lead to a predictable change in sediment alkalinity, in that sulfate reducing microbial communities may consume a range of organic compounds that have a different stoichiometry per mol of sulfate consumed (Gallagher et al., 2012). It is tempting to simplify microbial sulfate reduction as either having a 1:1 stoichiometry if sulfate reduction is coupled to anaerobic methane oxidation or 1:2 if it is coupled to organic matter oxidation, however, the true stoichiometry will vary as a function of the nature of oxidation (complete vs. incomplete) and the precise carbon compound that is oxidized (Visscher et al., 2000; Gallagher et al., 2012, 2014; Meister, 2013; Meister, 2014). Thus, it may not be possible to predict the increase in sedimentary alkalinity from simply knowing the number of mol of sulfate that have been reduced. We can see from the global data compilation (Figure 2) that sulfate concentrations that have been depleted by 20 mM could have generated alkalinity between 10 and 45 mEq/L, suggesting a complicated array of processes are influencing the sedimentary carbon cycle.
We note that the sites that have the largest alkalinity also are sites that have sulfate being depleted in the shallowest depths and the highest concentrations of pore fluid phosphate (Figure 3). This supports the idea that the more labile the organic carbon, the quicker it is consumed, releasing its dissolved inorganic carbon to the pore fluid as well as its phosphate. This shallow release of phosphate may also promote formation of calcium fluorapatite in the shallowest sediments with carbonate minerals precipitating in deeper sediments. The deeper in the sediment column that sulfate is consumed, the less phosphate is released, and the lower the total measured alkalinity. We suggest this may be pointing to the range of organic compounds of varying degrees of oxidation state contributing to feed the sedimentary subsurface sulfate reducing communities (Berner, 1978; Canfield, 1994; Anderson et al., 2001).
Ultimately our question is whether we can see the geochemical fingerprint of the production of subsurface alkalinity leading to carbonate mineral precipitation in sediments, to understand the governing environmental conditions under which this happens. In theory, this should be a relatively more straightforward relationship. Calcium is not consumed in authigenic clay formation and is thought to have an exceptionally short diffusion-reaction length within marine sediments, due to the rapid reaction of pore fluids and carbonate minerals during carbonate mineral recrystallization (Fantle and DePaolo, 2007). Therefore, calcium concentrations in marine sediments are thought to reflect chemical, and isotopic, equilibrium between pore fluid and sediment (Fantle and DePaolo, 2007). In sediments undergoing rapid carbonate mineral precipitation, pore fluid calcium concentrations are often decreasing and carbonate minerals are being added to the sediment (Teichert et al., 2009; Bradbury and Turchyn, 2018).
Global pore fluids suggest that the relationship between the generation of alkalinity and the depletion of calcium is not as simple as would be expected (Figure 2). While in sediments with methane, the gradient in alkalinity is strongly correlated with the gradient in calcium (R2 = 0.65—Figure 5), for a given change in alkalinity, there is no prediction as to how much calcium would have been consumed. What we notice instead is that for a given amount of sulfate reduced, there is a nearly fixed amount of calcium consumed, particularly in sites where there is AOM (Figure 2, bottom row) and that the gradient between sulfate and calcium is more tightly correlated (R2 = 0.71). We note that when methane is not present, the correlation between the gradients in alkalinity and calcium concentrations, as well as the correlation between the gradients in sulfate and calcium concentrations break down (R2 = 0.19 and 0.09, respectively—Figure 5).
Another way to interpret this lack of correlation between calcium and alkalinity is that alkalinity is generated not only through microbial sulfate reduction, but also through in situ silicate mineral weathering (Wallmann et al., 2008; Wallmann et al., 2012). Previous studies have suggested that the production of methane leads to lower pH and accelerates sedimentary alteration of silicate minerals (e.g., detrital clay minerals) that releases alkalinity to the pore fluids. This hypothesis would mean that carbonate mineral precipitation in sediments occurs in part because of alkalinity produced by microbial communities within sediments, and in part because of the alkalinity generated by methane production (Visscher et al., 2000; Soetaert et al., 2007; Arndt et al., 2013; Hong et al., 2020; Torres et al., 2020). This could explain the enhanced correlation between calcium and alkalinity in sites with methane, which is much weaker in sites without methane (Figure 5). To explore this further, we plot the amount of alkalinity gained, or lost, below the sulfate minimum in global sedimentary pore fluids (Figure 7). We note that there are sites that have both high pore fluid silica concentrations and high pore fluid alkalinity in the zone of methane production; these may be locations where there is sedimentary silicate weathering producing a deep excess of alkalinity. However, most data below the sulfate minimum zone shows a decrease in pore fluid alkalinity.
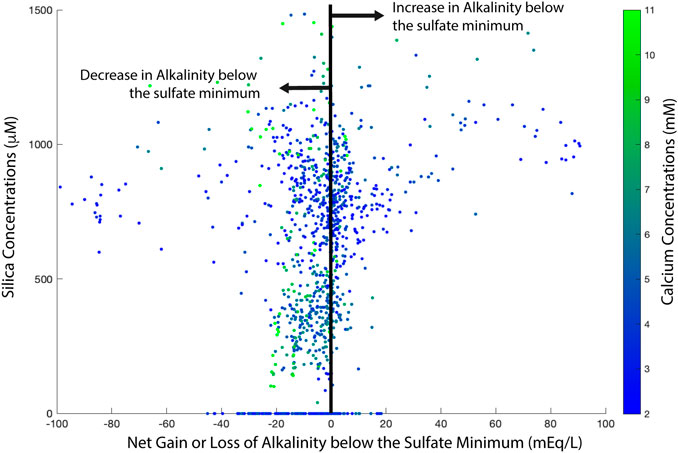
FIGURE 7. Below the sulfate minimum in sediments, the amount of alkalinity gained or loss in sedimentary pore fluids. Data to the right of the black line suggest an increase in alkalinity, possibly through silicate mineral weathering while data to the left of the black line suggest consumption of alkalinity below the sulfate minimum.
On the whole, the large variability in calcium concentration for a given alkalinity (Figure 2), suggests that alkalinity is a poor predictor of the balance of processes influencing sedimentary carbon dynamics. This is ironic given that alkalinity is the only direct measure of the subsurface carbon cycle, given that in most sediments carbonate alkalinity dominates total alkalinity (Soetaert et al., 2007; Gallagher et al., 2012). However, decades of work have shown that few processes are able to properly predict exactly how much alkalinity to expect in different conditions (Chatterjee et al., 2011; Sauvage et al., 2014; Middelburg et al., 2020). We suggest that reading the patterns that arise from the global pore fluid database suggests that there are locations where there is excess alkalinity generation from methane production driving sedimentary silicate weathering, but that these are less common.
The strong correlation between sulfate concentrations and calcium concentrations (Figure 2), and between the gradient in sulfate concentration and the gradient in calcium concentration, particularly when corrected for changes in alkalinity (Figure 5), suggests these two elements are strongly coupled in sediments. Indeed, in sediments with methane, sulfate concentrations are a strong predictor of calcium concentrations; if sulfate concentrations have been reduced by half, then the pore fluid calcium concentrations are in a narrow range between 6 and 8 mM (Figure 2). In sites with methane this correlation is improved by considering the alkalinity gradient as well (Figure 5D). We conclude that this suggests a direct coupling between sulfate and calcium in marine sediments. It has been previously suggested that sulfate reducing bacteria can directly nucleate the precipitation of calcium carbonate minerals, as they release extracellular polymeric substances (EPS) (Visscher et al., 2000; Meister, 2013). We also suggest that in sediments with methane and the anaerobic oxidation of methane (AOM), there are strong diffusion gradients of sulfate, which are often quasi linear (Niewöhner et al., 1998; Sivan et al., 2007), and similarly strong gradients of calcium as both are being driven by diffusion to the depth of AOM, where carbonate mineral precipitation is strongly favored. This may also help explain why there is such a tight correlation between sulfate and calcium concentrations in global pore fluids when methane is present. When methane is not present and sulfate is reduced through organoclastic sulfate reduction, the sulfate concentration profile is often concave down, reflecting progressive depletion through the sediment column (Bradbury and Turchyn, 2019; Zhang, 2020). As noted above, calcium concentrations in this instance often are not changing very rapidly, and not necessarily systematically, if at all as they reflect a combination of diffusion, precipitation, recrystallization and equilibrium between the pore fluid and the sediment. This complicates the ability to interpret correlations between pore fluid profiles in the absence of AOM.
Despite the alkalinity gradients being five times greater than the calcium gradients (Figure 4), calcium concentrations never reach zero (Figure 2), even in the presence of excess alkalinity. Calcium carbonate mineral precipitation in sediments must be inhibited at depth. Carbonate mineral saturation state determines whether carbonate minerals are likely to precipitate or dissolve at a given temperature and pressure, as a function of the concentrations of calcium and carbonate ions at that particular location (Eq. 8). Sediment pH is critical in determining the concentration of carbonate ion, and an accurate pore fluid pH profile is therefore required to model changes in the saturation state with depth below the sediment-water interface (Soetaert et al., 2007). Unfortunately, pH measurements cited in ODP and IODP initial reports are often unreliable. Core exhumation significantly reduces pressure, causing dissolved inorganic carbon species to re-equilibrate and carbon dioxide to exsolve (Sauvage et al., 2014). This may significantly change the measured and thus reported pH. We note from Figure 2 that even in sites where there is methane, and likely AOM, leading to high pH and alkalinity, calcium concentrations in the pore fluids are never fully depleted. Here we ask the question, what is limiting precipitation of calcium carbonate minerals in marine sediments?
Ultimately the pH change in marine sediments will be one critical determinant of whether carbonate minerals are likely to precipitate, along with the total amount of dissolved inorganic carbon and the amount of calcium in the pore fluid. For example, Meister (2013) suggested that during microbial sulfate reduction there would be an initial decrease in pH, as most pathways for microbial sulfate reduction produce protons. However, he then showed through a model that as microbial sulfate reduction continued, the increased production of dissolved inorganic carbon meant that in spite of the lower pH, the saturation state of calcium carbonate minerals increased to oversaturation yielding the possibility of carbonate mineral precipitation. Gallagher et al. (2012), Gallagher et al. (2014) did counter that the source of the organic carbon plays a key role in the overall balance of pH production and consumption, as we touched upon above.
We present a cross plot of the concentration of pore fluid calcium vs. alkalinity, color-coded as a function of the pH required such that the pore fluids are saturated (Ω = 1 in Eq 8; Figure 8). For an example of how to read this graph, if pore fluid calcium concentration is 4 mM, and the alkalinity is 15 mEq/L, then the sediment pH must be 7.6 or higher for calcium carbonate minerals to be supersaturated and potentially precipitate. What is apparent in Figure 7 is that if pore fluid calcium concentrations and alkalinity are high, then the sediment pH can be quite low and carbonate minerals are still supersaturated and may precipitate, similar to the result presented in Meister (2013). On the other hand, when pore fluid calcium concentrations are low, for any range of alkalinity present in the pore fluid, the pH has to be very high for there to be enough dissolved carbonate ion for calcium carbonate minerals to be supersaturated and potentially precipitate.
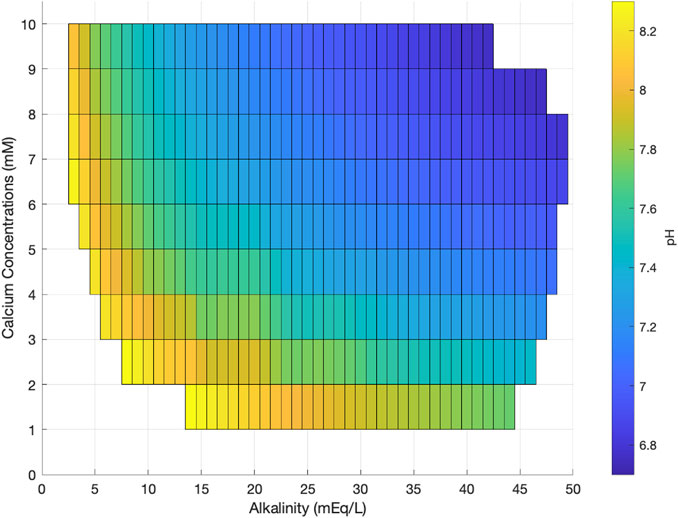
FIGURE 8. The pH required for calcite saturation (shaded color bar) as a function of the pore fluid calcium and alkalinity at any given sediment depth.
Sediment pH has been predicted to vary over a wide range (∼5–9) depending on the biogeochemical processes that dominate (Soetaert et al., 2007) and the organic carbon that is used to fuel microbial activity (Gallagher et al., 2012). It is thought from previous modeling that sediment pore fluid pH can reach as high as 8.2 during sedimentary anaerobic methane oxidation (Soetaert et al., 2007). However, it is unlikely that such high pH is achieved in situ because of the active sedimentary iron cycle that takes place along with methane production and anaerobic methane oxidation, as well as diffusive exchange of the potentially high pH pore fluid with lower pH pore fluid above and below the zone of anaerobic methane oxidation. Therefore, we think it would be difficult to get sediment pore fluid pH as high as 7.8–8 that our model suggests would be required for carbonate minerals to remain at or supersaturated in the zone of anaerobic methane oxidation (Figure 8). This may explain why calcium concentrations decrease in sedimentary pore fluids but reach a lower limit that is in a narrow range among a large variety of sites, and then do not decrease further.
Carbonate minerals are notorious for disobeying the laws of thermodynamics, and just because they are supersaturated for a given pH-DIC-temperature combination, it is not predetermined that they will precipitate (Schott et al., 2009). Often carbonate minerals require a nucleation site that allows for the initial mineral to begin to grow and on which future minerals can grow (Bischoff, 1968). In addition to nucleation, carbonate minerals have a variety of kinetic inhibitors to nucleation that interfere with the classic nucleation pathways for carbonate mineral formation. One of the classic inhibitors for calcium carbonate precipitation is the presence of phosphate (House, 1987; Burton and Walter, 1990). In our compilation, the sites with the highest phosphate concentrations also have the highest alkalinity (Figure 3), which we previously attributed to labile organic carbon releasing phosphate into the pore fluids. Another way to interpret the phosphate data is that the release of phosphate from organic carbon oxidation inhibits carbonate mineral precipitation in sediments and thus prevents this sedimentary sink for alkalinity generated through microbial sulfate reduction.
The presence of pore fluid phosphate as a global inhibitor of carbonate mineral precipitation in sediments may help explain why calcium concentrations in global pore fluid seem higher than they should be for a given change in either alkalinity or sulfate. The release of phosphate and organic acids during organic carbon oxidation may act as an inhibitor, or retardant, to carbonate mineral formation (House, 1987; Reddy and Hoch, 2002). If there are iron oxides in the sediment the phosphate released can be reabsorbed, however, iron oxides are not abundant in global marine sediments today because they are mostly trapped in marginal marine environments. We observe (Figure 2, middle and bottom row) that for a given change in sulfate or alkalinity, there is more pore fluid calcium than we would otherwise expect. This may be because there is not a straight exchange of alkalinity generated for carbonate precipitated and may be because of complicating factors during burial diagenesis in the deep biosphere.
The global pore fluid database allows us to explore the correlations among the various elements involved in the sedimentary carbon cycle. Our results suggest that the presence of methane within sediments plays a key role in the sedimentary carbon cycle. The presence of methane leads to tight correlation between pore fluid sulfate and calcium, demonstrating the importance of the zone of anaerobic methane oxidation in the consumption of sulfate and carbonate mineral precipitation in sediments. Our results also suggest that limited locations have deep alkalinity generation possibly coupled to methanogenesis-driven silicate weathering (Torres et al., 2020), however this is not as pervasive as may have been previously thought. Overall, the modern sedimentary database points to the importance of sedimentary methane in the global sedimentary carbon cycle.
Methane is produced in marine sediments either via acetoclastic methanogenesis (the breakdown or fermentation of small chained organic molecules) or by hydrogenotrophic methanogenesis (the direct reduction of CO2) (Meister et al., 2019). The consumption of sulfate in marine pore fluids leads to the formation of sedimentary sulfide minerals such as pyrite, which is the largest sink in the global sulfur cycle, and produces hydrogen that can fuel hydrogenoclastic methanogenesis (Garrels and Lerman, 1981; Berner, 1989). What controls whether there is a large sedimentary methane reservoir or not? One argument has been that short chained organic compounds delivered to sediment or generated within sediment through incomplete oxidation is the key lever controlling the amount of methane production. Another argument points to the importance of the marine sulfate reservoir in restricting methane production. Sulfate reducing bacteria consume many of the short-chained organic molecules that otherwise would go to fuel methanogenesis, and sulfate-reducing bacteria should outcompete methanogenic archaea for organic carbon (Froelich et al., 1979; Berner, 1980). A large marine sulfate reservoir pushes the zone of methane production deeper in the sediment column and creates a deep sulfate-methane transition zone. We speculate that the role of changing sulfate concentrations in the sedimentary methane cycle and, by extension, the sedimentary carbon cycle may be more nuanced, involving the distribution of organic carbon that can fuel methane production.
If methane is produced but at shallower depths in the sediment column, and in larger volumes due to the higher lability and availability of organic carbon (either because of more delivery or less sulfate), this would drive a stronger flux of calcium into the sediment and lead to less of the shoaling of the calcium concentration profile seen today. A shallower zone of AOM coupled to either sulfate or other electron acceptors such as iron or nitrate would lead to higher precipitation rates of calcium carbonate given the faster rates of diffusion that would occur due to steeper diffusion gradients. It has been previously suggested that anoxic conditions allow enhanced carbonate mineral precipitation in sediments (Higgins et al., 2009), but our dataset suggests that carbonate mineral precipitation in sediments is occurring mainly where anaerobic methane oxidation coupled to sulfate reduction occurs.
A further consideration is the role of sedimentary phosphate in determining if and when carbonate mineral precipitation in sediments may occur. Phosphate concentrations remain low in oxic sedimentary pore fluids, because phosphate is largely absent due to adsorption onto iron oxides and other iron minerals. When sediments become anoxic, the iron oxides can be reduced and the phosphate released into the anoxic pore fluids (D’Hondt et al., 2015). However, phosphate is an inhibitor to carbonate mineral precipitation, and may interfere with the ability of marine sediments to turn dissolved inorganic carbon into carbonate minerals.
We propose a simple, qualitative, model for the carbonate mineral precipitation in sediments that ties these observations together. The formation of methane in sedimentary environments is the key driver of carbonate mineral precipitation in sediments and the presence of methane accelerates the sedimentary carbon cycle. When sedimentary methane production is enhanced, a larger amount of (and larger range in) alkalinity is generated in sediments, through a combination of processes which either create or consume alkalinity as discussed throughout the paper. There is also larger release of phosphate in sedimentary pore fluids in the presence of methane. This will lead to a combination of carbonate and phosphate mineralization at shallow depths in the sediment column, and conversion of a large amount of calcium into mineralized forms. In contrast to previous work, we suggest that calcium concentrations in the overlying ocean are not a key driver of sedimentary mineral formation, because of the short diffusion reaction length for calcium in marine sediments. Instead of a driver from within the water column, we suggest instead that it is the formation and release of methane that plays a key role in the depth and intensity of mineral formation in the sediment column.
The global pore fluid data base allows us to look for correlations among various elements to explore sedimentary biogeochemistry. Our primary findings are:
• There is a correlation between sulfate and calcium concentrations and alkalinity in pore fluids over the zone of sulfate reduction. However, the relationships deviate from the expected stoichiometries.
• Calcium concentration is near-constant for a given sulfate concentration, whereas alkalinity is highly variable. This suggests that other subsurface processes are consuming alkalinity in addition to carbonate mineral precipitation in sediments.
• Microbial sulfate reduction is a key factor in mediating carbonate mineral precipitation in sediments because it provides the majority of alkalinity required. We find evidence, if limited, for sedimentary silicate weathering driving pervasive alkalinity generation.
• Methane is a key variable in the sedimentary carbon cycle. The presence of methane is tightly linked to enhanced precipitation of carbonate minerals in marine sediments through AOM and methanogenesis-driven silicate weathering.
• Exploring the relative controls on the formation of methane and how this may vary over geological time will offer better constraints on the role of carbonate mineral precipitation in sediments as a major player in the global carbon cycle.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
AVT designed the study and with HJB wrote most of the manuscript. KW did the initial research in a Masters project under the supervision of AVT and XS. HJB did the data analysis and prepared all the figures. All four authors, AVT, HJB, KW, and XS contributed to refining the manuscript.
This study was supported by ERC 307582 to AVT and NERC NE/R013519/1 to HJB.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Aller, R. C., and Rude, P. D. (1988). Complete oxidation of solid phase sulfides by manganese and bacteria in anoxic marine sediments. Geochem. Cosmochim. Acta. 52, 751–765. doi:10.1016/0016-7037(88)90335-3
Anderson, L. D., Delaney, M. L., and Faul, K. L. (2001). Carbon to phosphorus ratios in sediments: implications for nutrient cycling. Global Biogeochem. Cycles. 15, 65–79. doi:10.1029/2000GB001270
Arndt, S., Hetzel, A., and Brumsack, H.-J. (2009). Evolution of organic matter degradation in Cretaceous black shales inferred from authigenic barite: a reaction-transport model. Geochem. Cosmochim. Acta. 73, 2000–2022. doi:10.1016/j.gca.2009.01.018
Arndt, S., Jørgensen, B. B., LaRowe, D. E., Middelburg, J. J., Pancost, R. D., and Regnier, P. (2013). Quantifying the degradation of organic matter in marine sediments: a review and synthesis. Earth Sci. Rev. 123, 53–86. doi:10.1016/j.earscirev.2013.02.008
Berner, R. A. (1978). Sulfate reduction and the rate of deposition of marine sediments. Earth Planet Sci. Lett. 37, 492–498. doi:10.1016/0012-821X(78)90065-1
Berner, R. A. (1989). Biogeochemical cycles of carbon and sulfur and their effect on atmospheric oxygen over phanerozoic time. Global Planet. Change. 1, 97–122. doi:10.1016/0921-8181(89)90018-0
Berner, R. A. (2003). The long-term carbon cycle, fossil fuels and atmospheric composition. Nature 426, 323–326. doi:10.1038/nature02131
Berner, R. A., and Caldeira, K. (1997).The need for mass balance and feedback in the geochemical carbon cycle. Geology 25, 955–956. doi:10.1130/0091-7613(1997)025<0955:TNFMBA>2.3
Bischoff, J. L. (1968). Kinetics of calcite nucleation: magnesium ion inhibition and ionic strength catalysis. J. Geophys. Res. 73, 3315–3322. doi:10.1029/JB073i010p03315
Bradbury, H. J., and Turchyn, A. V. (2018). Calcium isotope fractionation in sedimentary pore fluids from ODP Leg 175: resolving carbonate recrystallization. Geochem. Cosmochim. Acta. 236, 121–139. doi:10.1016/j.gca.2018.01.040
Bradbury, H. J., and Turchyn, A. V. (2019). Reevaluating the carbon sink due to sedimentary carbonate formation in modern marine sediments. Earth Planet Sci. Lett. 519, 40–49. doi:10.1016/j.epsl.2019.04.044
Burdige, D. J., Zimmerman, R. C., and Hu, X. (2008). Rates of carbonate dissolution in permeable sediments estimated from pore-water profiles: the role of sea grasses. Limnol. Oceanogr. 53, 549–565. doi:10.4319/lo.2008.53.2.0549
Burton, E. A., and Walter, L. M. (1990). The role of pH in phosphate inhibition of calcite and aragonite precipitation rates in seawater. Geochem. Cosmochim. Acta. 54, 797–808. doi:10.1016/0016-7037(90)90374-T
Canfield, D. E. (1994). Factors influencing organic carbon preservation in marine sediments. Chem. Geol. 114, 315–329. doi:10.1016/0009-2541(94)90061-2
Chatterjee, S., Dickens, G. R., Bhatnagar, G., Chapman, W. G., Dugan, B., Snyder, G. T., et al. (2011). Pore water sulfate, alkalinity, and carbon isotope profiles in shallow sediment above marine gas hydrate systems: a numerical modeling perspective. J. Geophys. Res.: Solid Earth 116. doi:10.1029/2011JB008290
Dunlea, A. G., Murray, R. W., Santiago Ramos, D. P., and Higgins, J. A. (2017). Cenozoic global cooling and increased seawater Mg/Ca via reduced reverse weathering. Nat. Commun. 8, 844. doi:10.1038/s41467-017-00853-5
D’Hondt, S., Inagaki, F., Zarikian, C. A., Abrams, L. J., Dubois, N., Engelhardt, T., et al. (2015). Presence of oxygen and aerobic communities from sea floor to basement in deep-sea sediments. Nat. Geosci. 8, 299–304. doi:10.1038/ngeo2387
Egger, M., Riedinger, N., Mogollón, J. M., and Jørgensen, B. B. (2018). Global diffusive fluxes of methane in marine sediments. Nat. Geosci. 11, 421–425. doi:10.1038/s41561-018-0122-8
Fantle, M. S., and DePaolo, D. J. (2007). Ca isotopes in carbonate sediment and pore fluid from ODP Site 807A: the Ca2+(aq)–calcite equilibrium fractionation factor and calcite recrystallization rates in Pleistocene sediments. Geochem. Cosmochim. Acta. 71, 2524–2546. doi:10.1016/j.gca.2007.03.006
Fantle, M. S., and Higgins, J. (2014). The effects of diagenesis and dolomitization on Ca and Mg isotopes in marine platform carbonates: implications for the geochemical cycles of Ca and Mg. Geochem. Cosmochim. Acta. 142, 458–481. doi:10.1016/j.gca.2014.07.025
Froelich, P. N., Klinkhammer, G. P., Bender, M. L., Luedtke, N. A., Heath, G. R., Cullen, D., et al. (1979). Early oxidation of organic matter in pelagic sediments of the eastern equatorial Atlantic: suboxic diagenesis. Geochem. Cosmochim. Acta. 43, 1075–1090. doi:10.1016/0016-7037(79)90095-4
Gallagher, K. L., Kading, T. J., Braissant, O., Dupraz, C., and Visscher, P. T. (2012). Inside the alkalinity engine: the role of electron donors in the organomineralization potential of sulfate-reducing bacteria. Geobiology 10, 518–530. doi:10.1111/j.1472-4669.2012.00342.x
Gallagher, K. L., Dupraz, C., and Visscher, P. T. (2014). Two opposing effects of sulfate reduction on carbonate precipitation in normal marine, hypersaline, and alkaline environments: comment. Geology 42, e313–e314. doi:10.1130/G34639C.1
Garrels, R. M., and Lerman, A. (1981). Phanerozoic cycles of sedimentary carbon and sulfur. Proc. Natl. Acad. Sci. U.S.A. 78, 4652–4656. doi:10.1073/pnas.78.8.4652
Garrels, R. M. (1965). Silica: role in the buffering of natural waters. Science 148, 69. doi:10.1126/science.148.3666.69
Goldhammer, T., Brüchert, V., Ferdelman, T. G., and Zabel, M. (2010). Microbial sequestration of phosphorus in anoxic upwelling sediments. Nat. Geosci. 3, 557–561. doi:10.1038/ngeo913
Higgins, J. A., Blättler, C. L., Lundstrom, E. A., Santiago-Ramos, D. P., Akhtar, A. A., Crüger Ahm, A.-S., et al. (2018). Mineralogy, early marine diagenesis, and the chemistry of shallow-water carbonate sediments. Geochem. Cosmochim. Acta. 220, 512–534. doi:10.1016/j.gca.2017.09.046
Higgins, J. A., Fischer, W. W., and Schrag, D. P. (2009). Oxygenation of the ocean and sediments: consequences for the seafloor carbonate factory. Earth Planet Sci. Lett. 284, 25–33. doi:10.1016/j.epsl.2009.03.039
Higgins, J. A., and Schrag, D. P. (2010). Constraining magnesium cycling in marine sediments using magnesium isotopes. Geochem. Cosmochim. Acta. 74, 5039–5053. doi:10.1016/j.gca.2010.05.019
Hong, W.-L., Torres, M. E., and Kutterolf, S. (2020). Towards a global quantification of volcanogenic aluminosilicate alteration rates through the mass balance of strontium in marine sediments. Chem. Geol. 550, 119743. doi:10.1016/j.chemgeo.2020.119743
House, W. A. (1987). Inhibition of calcite crystal growth by inorganic phosphate. J. Colloid Interface Sci. 119, 505–511. doi:10.1016/0021-9797(87)90296-7
Kasten, S., and Jørgensen, B. B. (2000). “Sulfate reduction in marine sediments,” in Marine geochemistry. (Berlin, Heidelberg: Springer), 263–281. doi:10.1007/978-3-662-04242-7_8
Knittel, K., and Boetius, A. (2009). Anaerobic oxidation of methane: progress with an unknown process. Annu. Rev. Microbiol. 63, 311–334. doi:10.1146/annurev.micro.61.080706.093130
Mackenzie, F. T., and Kump, L. R. (1995). Reverse weathering, clay mineral formation, and oceanic element cycles. Science 270, 586. doi:10.1126/science.270.5236.586
Martin, J. B. (2017). Carbonate minerals in the global carbon cycle. Chem. Geol. 449, 58–72. doi:10.1016/j.chemgeo.2016.11.029
Meister, P. (2013). Two opposing effects of sulfate reduction on carbonate precipitation in normal marine, hypersaline, and alkaline environments. Geology 41, 499–502. doi:10.1130/G34185.1
Meister, P. (2014). Two opposing effects of sulfate reduction on carbonate precipitation in normal marine, hypersaline, and alkaline environments: reply. Geology 42, e315. doi:10.1130/G35240Y.1
Meister, P., Liu, B., Khalili, A., Böttcher, M. E., and Jørgensen, B. B. (2019). Factors controlling the carbon isotope composition of dissolved inorganic carbon and methane in marine polywater: an evaluation by reaction-transport modelling. J. Mar. Syst. 200, 103227. doi:10.1016/j.jmarsys.2019.103227
Middelburg, J. J., Soetaert, K., and Hagens, M. (2020). Ocean alkalinity, buffering and biogeochemical processes. Rev. Geophys. 58, e2019RG000681. doi:10.1029/2019RG000681
Middelburg, J. J., Soetaert, K., and Herman, P. M. J. (1997). Empirical relationships for use in global diagenetic models. Deep Sea Res. Oceanogr. Res. Pap. 44, 327–344. doi:10.1016/S0967-0637(96)00101-X
Moore, T. S., Murray, R. W., Kurtz, A. C., and Schrag, D. P. (2004). Anaerobic methane oxidation and the formation of dolomite. Earth Planet Sci. Lett. 229, 141–154. doi:10.1016/j.epsl.2004.10.015
Niewöhner, C., Hensen, C., Kasten, S., Zabel, M., and Schulz, H. D. (1998). Deep sulfate reduction completely mediated by anaerobic methane oxidation in sediments of the upwelling area off Namibia. Geochem. Cosmochim. Acta. 62, 455–464. doi:10.1016/S0016-7037(98)00055-6
Olgun, N., Duggen, S., Croot, P. L., Delmelle, P., Dietze, H., Schacht, U., et al. (2011). Surface ocean iron fertilization: the role of airborne volcanic ash from subduction zone and hot spot volcanoes and related iron fluxes into the Pacific Ocean. Global Biogeochem. Cycles. 25. doi:10.1029/2009GB003761
Prokopenko, M. G., Sigman, D. M., Berelson, W. M., Hammond, D. E., Barnett, B., Chong, L., et al. (2011). Denitrification in anoxic sediments supported by biological nitrate transport. Geochem. Cosmochim. Acta. 75, 7180–7199. doi:10.1016/j.gca.2011.09.023
Reddy, M. M., and Hoch, A. R. (2002). “Calcite crystal growth rate inhibition by aquatic humic substances,” in Advances in crystal growth inhibition technologies. Editor Z. Amjad (Boston, MA: Springer US), 107–121. doi:10.1007/0-306-46924-3_8
Sauvage, J., Spivack, A. J., Murray, R. W., and D’Hondt, S. (2014). Determination of in situ dissolved inorganic carbon concentration and alkalinity for marine sedimentary polywater. Chem. Geol. 387, 66–73. doi:10.1016/j.chemgeo.2014.06.010
Schott, J., Pokrovsky, O. S., and Oelkers, E. H. (2009). The link between mineral dissolution/precipitation kinetics and solution chemistry. Rev. Mineral. Geochem. 70, 207–258. doi:10.2138/rmg.2009.70.6
Schubert, C. J., Vazquez, F., Lösekann-Behrens, T., Knittel, K., Tonolla, M., and Boetius, A. (2011). Evidence for anaerobic oxidation of methane in sediments of a freshwater system (Lago di Cadagno). FEMS Microbiol. Ecol. 76, 26–38. doi:10.1111/j.1574-6941.2010.01036.x
Shaw, T. J., Gieskes, J. M., and Jahnke, R. A. (1990). Early diagenesis in differing depositional environments: the response of transition metals in pore water. Geochem. Cosmochim. Acta. 54, 1233–1246. doi:10.1016/0016-7037(90)90149-F
Sigurdsson, H., Leckie, R. M., and Acton, G. D. (1997). Proceedings of the ocean drilling program, vol. 165. 865. Initial reports Caribbean Ocean history and the cretaceous/tertiary boundary event. Ocean Drilling Program.
Sivan, O., Schrag, D. P., and Murray, R. W. (2007). Rates of methanogenesis and methanotrophy in deep-sea sediments. Geobiology 5, 141–151. doi:10.1111/j.1472-4669.2007.00098.x
Soetaert, K., Hofmann, A. F., Middelburg, J. J., Meysman, F. J. R., and Greenwood, J. (2007). The effect of biogeochemical processes on pH. Mar. Chem. 105, 30–51. doi:10.1016/j.marchem.2006.12.012
Teichert, B. M. A., Gussone, N., and Torres, M. E. (2009). Controls on calcium isotope fractionation in sedimentary porewaters. Earth Planet Sci. Lett. 279, 373–382. doi:10.1016/j.epsl.2009.01.011
Torres, M. E., Hong, W.-L., Solomon, E. A., Milliken, K., Kim, J.-H., Sample, J. C., et al. (2020). Silicate weathering in anoxic marine sediment as a requirement for authigenic carbonate burial. Earth Sci. Rev. 200, 102960. doi:10.1016/j.earscirev.2019.102960
Ussler, W., and Paull, C. K. (2008). Rates of anaerobic oxidation of methane and authigenic carbonate mineralization in methane-rich deep-sea sediments inferred from models and geochemical profiles. Earth Planet Sci. Lett. 266, 271–287. doi:10.1016/j.epsl.2007.10.056
Veeh, H. H., Burnett, W. C., and Soutar, A. (1973). Contemporary phosphorites on the continental margin of Peru. Science 181, 844–845. doi:10.1126/science.181.4102.844
Visscher, P. T., Reid, R. P., and Bebout, B. M. (2000).Microscale observations of sulfate reduction: correlation of microbial activity with lithified micritic laminae in modern marine stromatolites. Geology 28, 919–922. doi:10.1130/0091-7613(2000)28<919
Wallmann, K., Aloisi, G., Haeckel, M., Tishchenko, P., Pavlova, G., Greinert, J., et al. (2008). Silicate weathering in anoxic marine sediments. Geochem. Cosmochim. Acta. 72, 2895–2918. doi:10.1016/j.gca.2008.03.026
Wallmann, K., Pinero, E., Burwicz, E., Haeckel, M., Hensen, C., Dale, A., et al. (2012). The global inventory of methane hydrate in marine sediments: a theoretical approach. Energies 5, 2449–2498. doi:10.3390/en5072449
Wang, Y., and Van Cappellen, P. (1996). A multicomponent reactive transport model of early diagenesis: application to redox cycling in coastal marine sediments. Geochem. Cosmochim. Acta. 60, 2993–3014. doi:10.1016/0016-7037(96)00140-8
Whiticar, M. J. (1999). Carbon and hydrogen isotope systematics of bacterial formation and oxidation of methane. Chem. Geol. 161, 291–314. doi:10.1016/S0009-2541(99)00092-3
Whiticar, M. J., and Faber, E. (1986). Methane oxidation in sediment and water column environments—isotope evidence. Org. Geochem. 10, 759–768. doi:10.1016/S0146-6380(86)80013-4
Zeebe, R. E., and Westbroek, P. (2003). A simple model for the CaCO3 saturation state of the ocean: the “Strangelove,” the “Neritan,” and the “Cretan” Ocean. Geochem. Geophys. Geosyst. 4, 1104. doi:10.1029/2003GC000538
Zeebe, R. E., and Wolf-Gladrow, D. (2001). CO2 in seawater: equilibrium, kinetics, isotopes: equilibrium, kinetics, isotopes. Elsevier.
Zhang, S. (2020). The relationship between organoclastic sulfate reduction and carbonate precipitation/dissolution in marine sediments. Mar. Geol. 428, 106284. doi:10.1016/j.margeo.2020.106284
Keywords: carbonate minerals, sediments, sulfate, alkalinity, methane
Citation: Turchyn AV, Bradbury HJ, Walker K and Sun X (2021) Controls on the Precipitation of Carbonate Minerals Within Marine Sediments. Front. Earth Sci. 9:618311. doi: 10.3389/feart.2021.618311
Received: 16 October 2020; Accepted: 21 January 2021;
Published: 26 February 2021.
Edited by:
Paul Butler, University of Exeter, United KingdomReviewed by:
Ola Kwiecien, Northumbrian University, United KingdomCopyright © 2021 Turchyn, Bradbury, Walker and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandra V. Turchyn, YXZ0MjVAY2FtLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.