
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Earth Sci., 05 August 2021
Sec. Biogeoscience
Volume 9 - 2021 | https://doi.org/10.3389/feart.2021.601194
This article is part of the Research TopicThe Marine Carbon Cycle: From Ancient Storage to Future ChallengesView all 5 articles
 Daniel H. James1*
Daniel H. James1* Harold J. Bradbury1
Harold J. Bradbury1 Gilad Antler2,3
Gilad Antler2,3 Zvi Steiner1,4
Zvi Steiner1,4 Alec M. Hutchings1
Alec M. Hutchings1 Xiaole Sun5
Xiaole Sun5 Raoul Saar3
Raoul Saar3 Mervyn Greaves1
Mervyn Greaves1 Alexandra V. Turchyn1
Alexandra V. Turchyn1We present pore fluid geochemistry, including major ion and trace metal concentrations and the isotopic composition of pore fluid calcium and sulfate, from the uppermost meter of sediments from the Gulf of Aqaba (Northeast Red Sea) and the Iberian Margin (North Atlantic Ocean). In both the locations, we observe strong correlations among calcium, magnesium, strontium, and sulfate concentrations as well as the sulfur isotopic composition of sulfate and alkalinity, suggestive of active changes in the redox state and pH that should lead to carbonate mineral precipitation and dissolution. The calcium isotope composition of pore fluid calcium (δ44Ca) is, however, relatively invariant in our measured profiles, suggesting that carbonate mineral precipitation is not occurring within the boundary layer at these sites. We explore several reasons why the pore fluid δ44Ca might not be changing in the studied profiles, despite changes in other major ions and their isotopic composition, including mixing between the surface and deep precipitation of carbonate minerals below the boundary layer, the possibility that active iron and manganese cycling inhibits carbonate mineral precipitation, and that mineral precipitation may be slow enough to preclude calcium isotope fractionation during carbonate mineral precipitation. Our results suggest that active carbonate dissolution and precipitation, particularly in the diffusive boundary layer, may elicit a more complex response in the pore fluid δ44Ca than previously thought.
In modern marine settings, the layer of the uppermost sediment near the sediment–water interface marks the physical and geochemical transition between the overlying oxidizing water column and the reducing conditions of the deeper sediment column; hereafter, we will call this the boundary layer (Sayles, 1979; Sayles, 1981; Klinkhammer et al., 1982; Rudnicki et al., 2001). This boundary layer is thought to be the most active part of the sediment in terms of changes in geochemical conditions, and related mineral dissolution and precipitation (Sayles, 1979; Sayles, 1981).
One geochemical process that might be particularly important in the boundary layer is the precipitation and dissolution of calcium carbonate minerals (CaCO3). Calcium carbonate minerals are the dominant sink for carbon from the Earth’s surface environment, playing a major role in the global carbon cycle (Berner et al., 1983; Milliman, 1993; Berner, 2003; Ridgwell and Zeebe, 2005). Calcium carbonate mineral dissolution is one of the primary buffers for mitigating changes in ocean pH during ocean acidification, therefore understanding what governs its precipitation and dissolution is a key challenge in the oceanographic community. Seawater is undersaturated with respect to calcium carbonate minerals where the sediment–water interface lies below the lysocline; this is much of the modern deep ocean floor (Feely et al., 2009; Higgins et al., 2009; Carter et al., 2014). The saturation state of carbonate minerals (Ωcc) is given by the following equation:
where [X]SW is the activity of each aqueous species in seawater, and K*sp is the stoichiometric solubility product at equilibrium, which is itself a function of temperature, pressure, salinity, and carbonate allotrope.
Carbonate ions are a part of the total dissolved inorganic carbon (DIC) pool, along with dissolved CO2 and the bicarbonate ion (HCO3−). The relative proportion of the carbonate ion to the total DIC is a function of pH, with protonated species (HCO3− and carbonic acid—H2CO3) dominating below pH ∼7.2 (Millero, 1995). Thus, a higher pH or increased total DIC will increase the concentration of carbonate ions (equivalent to [CO32–]SW in Eq. 1), and thus, by Eq. 1, the saturation state of carbonate minerals will increase in turn—provided [Ca2+]SW and K*SP remain constant—in theory, promoting mineral precipitation. The pH of the modern deep ocean is lowered by the aerobic oxidation of organic carbon in oxygenated bottom waters, resulting in widespread carbonate undersaturation (Palmer et al., 1998; Yu et al., 2014), whereas in marginal oceans, the water column is frequently supersaturated with calcium carbonate minerals from the surface to the sediment–water interface (Zeebe and Wolf-Gladrow, 2001).
The aerobic oxidation of organic matter that influences the pH of the deep ocean persists into the sedimentary boundary layer and continues to reduce the pH of sedimentary pore fluids (Ben-Yaakov, 1973; Froelich et al., 1979; Soetaert et al., 2007). Where this process is active, carbonate mineral dissolution in shallow sediments is likely as pore fluid [CO32–], and consequently, Ωcc (Eq. 1) decreases (Sayles, 1979; Sayles, 1981). In most sediment columns, however, there is a maximum depth of penetration for oxygenated water. This depth of penetration varies extensively, and oxygen can persist to a significant depth, particularly in parts of the abyssal plain (D’Hondt et al., 2015). However, below the depth of oxygen penetration, anaerobic metabolic reactions occur, oxidizing organic matter using other electron acceptors (Froelich et al., 1979; Berner, 1980; Kasten et al., 2003). The range of anaerobic metabolisms influence pH in different ways, either consuming or releasing protons, thus causing different responses in the carbonate mineral saturation state (Soetaert et al., 2007). As such, we expect a complex profile of calcium carbonate mineral dissolution, precipitation, or recrystallization with depth in a sediment column where some or all of these processes operate sequentially (Kasten et al., 2003; Arndt et al., 2006; Arndt et al., 2009). The depth at which these various processes occur below the sediment–water interface is thought to be controlled largely by the organic carbon flux to the seafloor (Berner, 1980; Boudreau, 1997). The interplay of these processes will influence how shallow sediment carbonate dissolution may act to buffer changes in ocean acidification and thus place important constraints on our understanding of the response of the ocean under increased CO2 forcing.
Measurements of reactions involving anaerobic metabolisms and associated changes in the carbonate mineral saturation state are most frequently taken at large depths below the sediment–water interface. For example, the Ocean Drilling Program/Integrated Ocean Drilling Program (ODP/IODP) can sample sediments up to 2,000 mbsf, but the first sediment and pore fluid samples are at least 1 m below the sediment–water interface. At this depth in many marginal (e.g., continental shelf) or high organic-flux locations, boundary layer redox changes have already occurred, and there is potential that any changes in the carbonate saturation state driven by redox geochemistry have already been missed. There have been several attempts to quantify changes in the boundary layer carbonate mineral saturation state, but how these vary geographically, including the changes with the water depth or distance from shore, is less well understood (Sayles, 1979; Sayles, 1981; Steiner et al., 2019).
Pore fluid calcium isotope ratios have been shown to be a useful tracer of carbonate mineral dissolution, precipitation, and particularly recrystallization in sediment columns at longer depth scales (e.g., Fantle and DePaolo, 2007; Fantle, 2015; Huber et al., 2017; Bradbury and Turchyn, 2018), a utility which may be applicable to boundary layer processes. Calcium has five stable isotopes: 40Ca (96.94%), 42Ca (0.65%), 43Ca (0.14%), 44Ca (2.09%), and 46Ca (0.01%) and one quasi-stable isotope, that is, 48Ca (0.19%). The significant mass difference among these isotopes leads to a measurable amount of calcium isotope fractionation in many common biological and chemical processes (Fantle and Tipper, 2014; Gussone et al., 2016). The isotope composition is here expressed in terms of the ratio between 40Ca and 44Ca, the two most abundant isotopes, in delta notation (Eq. 2), with units of parts-per-thousand, or permil (‰), which is as follows:
using the bulk silicate Earth (BSE) reference, with a standard seawater value of 0.92‰ (Skulan et al., 1997).
During the precipitation of carbonate minerals, the 40Ca isotope is preferentially incorporated into the solid phase via a rate-dependent kinetic isotope effect, leading to an enrichment in 44Ca in the remaining fluid (Gussone et al., 2003; Gussone et al., 2005; DePaolo, 2011; Nielsen et al., 2012). Conversely, it is taken that there is no calcium isotope fractionation during carbonate mineral dissolution, which hence releases 44Ca-depleted calcium from the carbonate mineral to the aqueous phase and decreases fluid δ44Ca (Fantle and DePaolo, 2007; Jacobson and Holmden, 2008). During carbonate mineral recrystallization, the calcium isotopic compositions of both phases approach isotopic equilibrium with one another, and mineral and fluid calcium converge to a given equilibrium isotopic difference, or offset, the value of which is widely thought to be 0; that is, the 44/40Ca ratio in the solid phase over that of the pore fluid is 1 (Fantle and DePaolo, 2007; Jacobson and Holmden, 2008). The majority of calcium in the sediment is hosted in solid phases, so this weighted-average isotopic convergence during isotopic equilibration manifests as a significant decrease in the fluid δ44Ca toward the lower δ44Ca of the sediment minerals during sediment-buffered early diagenesis (Fantle and DePaolo, 2007; Fantle, 2015; Huber et al., 2017; Ahm et al., 2018; Higgins et al., 2018).
Here, we present centimeter-scale δ44Ca measurements from the pore fluids and solid phase of the upper boundary layer of sediment in the two locations, the Gulf of Aqaba and the Iberian Margin (Figure 1), along with pore fluid major ion and trace metal concentrations, to elucidate how complex and varied redox conditions affect carbonate mineral precipitation and dissolution in the shallowest boundary layer of the sediment. The objective is to extend the observations that have been made in deeper sediments using changes in δ44Ca to resolve rates of mineral precipitation and dissolution into the shallowest sediments. In doing so, we hope to test whether δ44Ca in the boundary layer pore fluids in conjunction with other geochemical analyses can help resolve the depth distribution of carbonate mineral dissolution, and thus how the buffering capacity for ocean acidification extends from the ocean water column into the uppermost sediments.
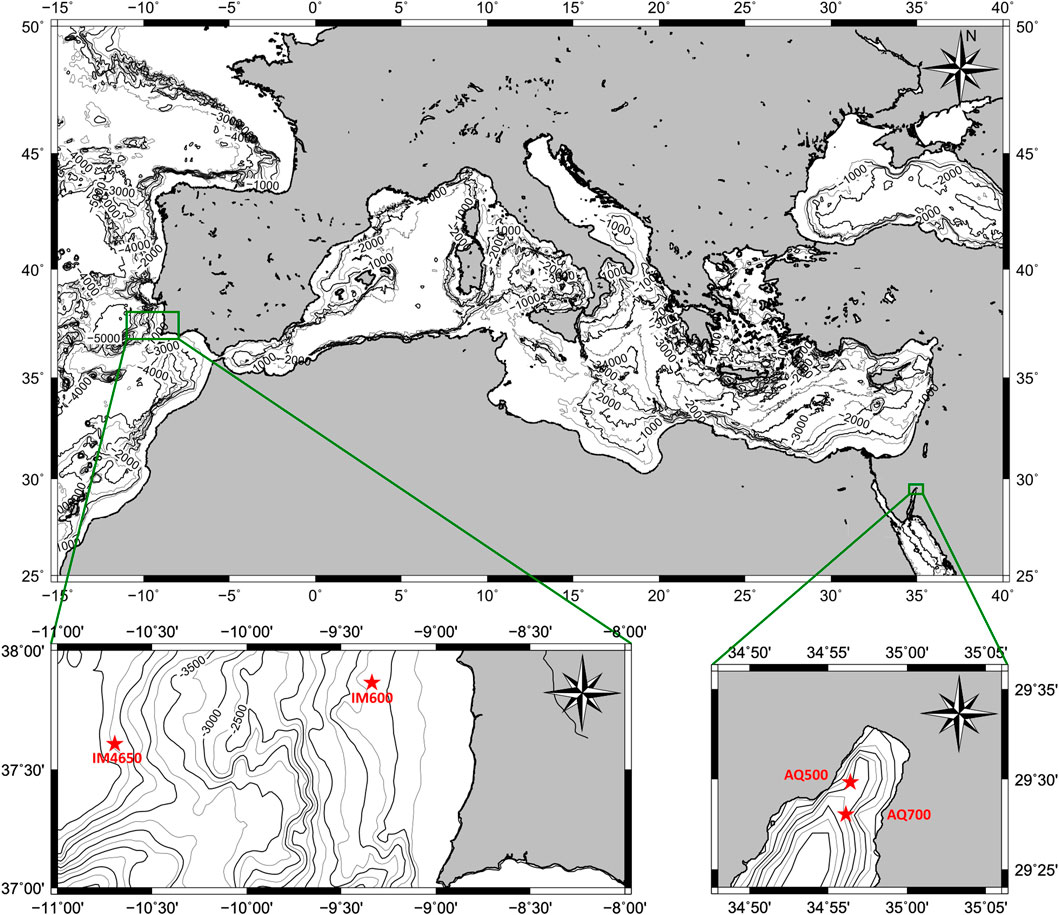
FIGURE 1. Map of southern Europe, North Africa, and the Mediterranean. The Iberian Margin (Top) and Gulf of Aqaba (Bottom) are displayed in zoomed subfigures, with associated sample sites as red stars.
The Gulf of Aqaba is an isolated northeast extension of the Red Sea, connected only by the 13-km-wide Strait of Tiran (Figure 1). The Gulf of Aqaba reaches 1830 m depth and is never more than 25 km wide (Ben-Avraham et al., 1979). There is no major riverine input into the Gulf of Aqaba; instead, seawater is sourced across the Strait, retaining a stratified water column excepting annual mixing events in late winter (Biton and Gildor, 2011). Mixed layer depths regularly extend below 700 m, the maximum sampling depth for this study (Wurgaft et al., 2016), and as such, the water column is considered well mixed with respect to the atmosphere (Krumgalz et al., 1990), maintaining a constant oxygen saturation at the sediment–water interface (Lazar et al., 2008). Sediment in the Gulf of Aqaba is supplied mainly from episodic fluvial input events (Katz et al., 2015; Torfstein et al., 2020), with additional contribution from biogenic precipitation in the photic zone (Steiner et al., 2014), and dust deposition (Chen et al., 2008; Blonder et al., 2017), resulting in surface sediment that is roughly 35% carbonate minerals by weight (Steiner et al., 2019). Steiner et al. (2016) and Steiner et al. (2019) have undertaken an extensive analysis of the solid phase sediment composition and bottom water chemistry in the Gulf of Aqaba, including the quantification of the extent of bioturbation, carbonate mineral saturation state, and pore fluid pH. The Gulf of Aqaba’s average salinity is 40.7 g/L, and bottom water temperatures average 21°C (Biton and Gildor, 2011).
The Iberian Margin, the continental shelf and descending slope west of the Atlantic coast of Portugal (Figure 1), is an open marine location overlain by the North Atlantic Deep Water and Mediterranean Outflow Water in varying proportions. Specifically, cores used here were taken from the “Shackleton Sites” (Hodell et al., 2013a), which has previously been the focus of many paleoclimatic studies, including the seminal work of Nick Shackleton identifying the phase relationships in oxygen isotopes between the planktonic Foraminifera and the Greenland ice cores (Shackleton et al., 2000; Shackleton et al., 2004). Later, studies into the sediment at these sites have focused on developing further high-resolution paleoclimatic records (Hodell et al., 2013b; Hodell et al., 2015; Rodríguez-Tovar and Dorador, 2014; Rodríguez-Tovar et al., 2015).
Short cores in the Red Sea were sampled from the “Sam Rothberg RV,” a 16 m research vessel owned and operated by the Inter-University Institute for Marine Sciences in Eilat (IUI) at two sites in the Gulf of Aqaba in September 2018. Gravity cores (GC1 and GC2) and piston cores (MC1 and MC3) were taken at “Station A” (water depth 696 m, 29o28′03″N, 34o55′64″E), which is regularly sampled and hereafter referred to as AQ700, and piston cores (MC7 and MC8) were taken from a shallower, less frequently sampled site (water depth 518 m, 29o29′50″N, 34o56′24″E), hereafter AQ500. Pore fluids were extracted within 24 h of collection, either by centrifuge or Rhizon (Rhizosphere Research Products—Seeberg-Elverfeldt et al., 2005). Cores processed by centrifuge were sampled at 10 cm (gravity core) or 1 cm (piston core) intervals; all data are reported at the midpoint depth within these intervals. All pore fluid samples (Rhizon and Centrifuge) were passed through a Millipore 0.22 µm syringe filter, acidified with nitric acid to 40 mM (except 5 ml reserved for the measurement of pore fluid alkalinity), and stored under refrigeration prior to the analysis. The carbonate fraction of the solid sediment samples was dissolved in ultrapure 10% acetic acid for 1 h and converted to a nitrate form.
A second set of cores was taken from the southwest Iberian Margin as a part of the JC089 IODP site survey of the Shackleton site, in July and August 2013, as detailed in the cruise report (Hodell et al., 2014). Here we report analyses from three cores from two of the sampled locations: The piston core JC089 SHAK-05-3P and the Megacore JC089 SHAK-05-4M-B from the site “JC089-05 SHAK05” (water depths 4,670 and 4672 m, 37°36′16″N, 10°41′30″W), hereafter IM4650, and the Megacore JC089 SHAK-11-10M-D from the site “JC089-11 SHAK11” (water depth 628 m, 37°51′31″N, 9°20′10″W), hereafter IM600. Both cores had their pore fluids sampled using Rhizons.
The calcium isotope analysis was conducted using a Triton Plus MC-thermal ionization mass spectrometer (TIMS) as per the method reported in the study by Bradbury and Turchyn (2018). A 1:1 42Ca-48Ca double-spike was added to a pore fluid sample containing 6 µg of calcium such that the sample-to-spike ratio was 10:1. These were dried down and then redissolved in 0.5% HNO3−, before calcium was separated from other cations on a Dionex ICS-5000+ HPIC system fitted with a high-capacity carboxylate-functionalized Dionex CS-16 column using a methyl sulfonic acid eluent. Calcium was isolated using a fraction collector by eluent concentration within a predetermined peak-detection window. The procedural blank when collecting 4.4 µg of calcium in 7 ml of solution was 96 ng (2%—determined for this machine by Bradbury and Turchyn, 2018).
Calcium (4 µg) was precipitated as calcium nitrate and then redissolved in 1 µL of 2 M HNO3− for loading onto 0.7 mm outgassed zone-refined rhenium filaments, with 0.5 µL of 10% H3PO4. These were analyzed using a double filament method on the TIMS. Ionization filaments were heated to 1400°C, and evaporation filaments were heated manually until the 40Ca beam stabilized at 5–10 V (on a 1011 Ω resistor). Measurements were taken with an integration time of 8.389 s, consisting of 200 cycles of data per run in blocks of 20. Errors are quoted as the external precision, 2σ, of results where duplicates were measured. Standards measured over the course of the study are as follows: NIST915B mean = –0.281, 2σ = 0.085, and n = 20, and IAPSO seawater mean = 0.929, 2σ = 0.12, and n = 15.
Major element concentrations of the pore fluids were measured using an Agilent Technologies 5,100 inductively coupled plasma–optical emission spectrometer. Samples were diluted at 1:100 with 0.1 M HNO3, and the Iberian Margin samples were then analyzed in duplicate with regularly spaced standards. The Gulf of Aqaba samples were analyzed in duplicate with sample-standard bracketing following the methods of Steiner et al. (2018). Initial calibrations were obtained by running different concentrations of the IAPSO seawater batch P157. The same IAPSO standard, diluted to the same salinity as the samples, was used as the bracketing standard. Results are presented as ratios relative to sodium (Na), which we assume is conservative. Errors are quoted as 2σ from the duplicates run.
Pore fluid δ34SSO4 was analyzed for select cores. Pore fluid sulfate was precipitated as barium sulfate (barite) using a saturated barium chloride solution, acidified with HCl, then sequentially rinsed thrice with deionized water, and dried in an oven. Barite was combusted at 1030°C in a flash element analyzer, and the resulting SO2 was measured for isotopic composition on a GS-IRMS (Thermo Finnigan Delta V+). All results were corrected against NBS 127 (standard value 21.1‰), from which errors were generated (2σ = 0.260‰, n = 16).
Alkalinity was measured at the IUI in Eilat, Israel, using 1.5 ml of sample per titration and in triplicate using a SI Analytics Titrator TitroLine® 7,000 and shipboard on the JC089 cruise. Alkalinity was calculated by the Gran plot. The average precision is below 5 µM for the triplicate samples.
Pore fluid major element concentrations, δ44Ca, δ34SSO4, and alkalinity from the Iberian Margin (Figure 2) and the Gulf of Aqaba (Figure 3) vary over the sampled depth range. Pore fluid major ion concentrations decrease quasi-linearly with depth in both sites, including that of sulfate (commonly consumed through microbial sulfate reduction), calcium (commonly consumed through subsurface carbonate precipitation), strontium, and magnesium (see Supplementary Figures S1, S2). We also note a linear or near-linear increase in sedimentary pore fluid alkalinity. We observe no minimum inflection points in any of the above species’ profiles; if these pore fluid data decrease due to some consumption reaction in the sediment, then the lowest concentrations likely lie outside the sampled depth range in every analyzed core.
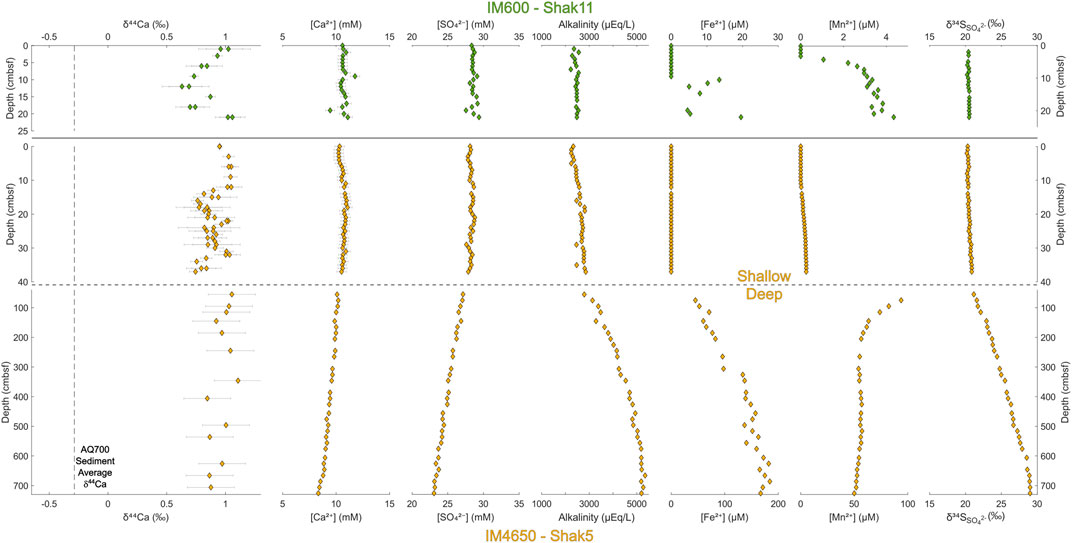
FIGURE 2. Depth profiles for the Iberian Margin sites IM600 and IM4650. For IM4650, shallower cores taken by megacorer are displayed above the deeper profile generated by a piston core. Pore fluid major ion concentrations and isotopic compositions are recorded relative to sample depth in cm below the sea floor. N.B. Iron and manganese results have different axis scales at each site for ease of comparison.
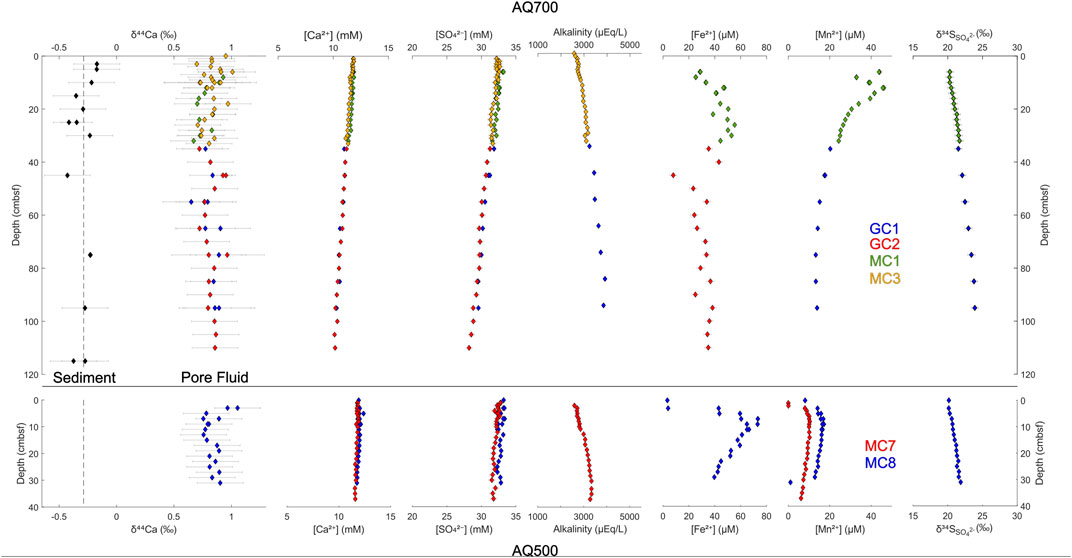
FIGURE 3. Depth profiles for the Gulf of Aqaba sites AQ700 and AQ500. Individual core IDs are color-coded. Pore fluid major ion concentrations and isotopic compositions are recorded relative to sample depth in cm below the sea floor.
The rate of decrease, with depth, of pore fluid calcium and sulfate, and increase in alkalinity and δ34SSO4, is larger in all cases in the Gulf of Aqaba than that at the Iberian Margin. We note that at IM600 (where sampling only extends to 25 cmbsf), there is no observed change in the geochemical data outside of error, while at IM4650, significant changes only begin to occur deeper in the core (up to 700 cmbsf).
In the Gulf of Aqaba cores, local maxima are observed in pore fluid concentrations of iron ([Fe2+]) and manganese ([Mn2+]). Dissolved [Fe2+] reaches its maximum 26 cmbsf in AQ700, and 9 cmbsf in AQ500, before decreasing. Dissolved [Mn2+] reaches a maximum at 12 cmbsf at AQ700 and at 9 cmbsf at AQ500. No [Fe2+] maxima are observed in either core from the Iberian Margin; indeed, [Fe2+] only begins increasing at 75 cmbsf in the piston core at IM4650, reaching its recorded maximum near the base of the core (686 cmbsf). At IM4650, there is an apparent [Mn2+] maximum in the region between the base of the Megacore at 37 cmbsf and the next shallowest sample in the piston core at 55 cmbsf. The sulfur isotope composition of pore fluid sulfate (δ34SSO4) increases with depth in every core, to measured maxima of 24.9‰ in AQ700 and 29.0‰ in IM4650.
Pore fluid δ44Ca does not vary significantly outside of error over the meter-depth scale within the upper sediment in any of the four coring locations. Sedimentary δ44Ca is also invariant (where tested at AQ700) over this interval—retaining a stable offset of ∼1.1‰ from pore fluid δ44Ca (Figure 3, top left). Some smaller scale variations in the pore fluid δ44Ca are recorded in both cores at shallower water depth, AQ500 and IM600; δ44Ca decreases by ∼0.3‰ (to 0.25‰) in the upper 10 cmbsf before increasing again; δ44Ca remains stably at seawater values to 14 cmbsf in IM4650; and δ44Ca has a local maximum (0.95‰) at 45 cmbsf in AQ700.
To better visualize the relevant changes in various concentrations in the measured pore fluids, we plot the data using a cross-correlation matrix, which is here normalized to pore-fluid sodium to account for differences between the two studied sites (Figure 4). Across cores from both the Gulf of Aqaba and the Iberian Margin, we observe strong correlations between certain species, with least-square-regression coefficients of over 0.9 (Figure 4); these correlations imply causal or cogenetic relationships between these species and can be used to geochemically characterize the early diagenetic system in the boundary layer at these sites. First, we can test the extent to which certain species decrease linearly with depth—Ca2+/Na+, SO42–/Na+, and Sr2+/Na+ all strictly display this behavior, with R values of 0.62, 0.85, and 0.67 vs. depth, respectively (Figure 4), with a larger decrease in the Gulf of Aqaba than in the Iberian Margin in all cases. Pore fluid Mg behaves this way in the Gulf of Aqaba (R = 0.58 vs. depth), but no decrease in the Iberian Margin is apparent.
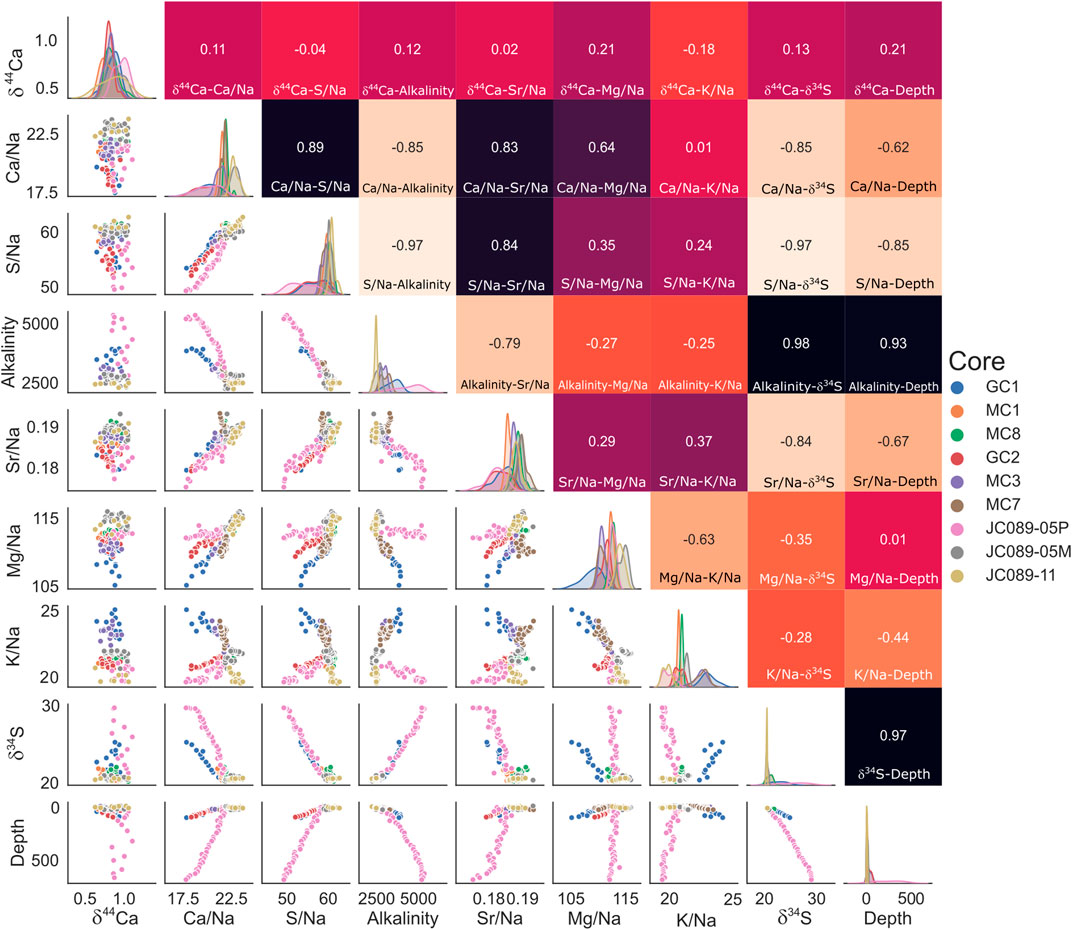
FIGURE 4. Correlations between pore fluid species: all concentrations are here normalized to local salinity to allow comparison between sites. Top right quadrant: least squares regression coefficient (R) between corresponding pore fluid species, color-coded from white (–1) through red (0) to black (1). Bottom left quadrant: Cross plots of species, color-coded by core as per the legend (right). Central diagonal: frequency density of data for the corresponding species in the plotted range, by core.
There is a strong negative correlation between sulfate concentration and alkalinity (R = –0.97), hinting that in our sites, microbial sulfate reduction coupled to either the anaerobic oxidation of methane or organic matter oxidation generates the majority of alkalinity. This is confirmed by the fact that the increase in δ34SSO4 is anticorrelated with the pore fluid sulfate concentration (R = –0.97); the observed decrease in sulfate concentration is due to microbial sulfate reduction, preferentially converting the light 32S isotope to sulfide and leaving the 34S isotope in the remaining sulfate. Alkalinity and calcium concentrations display a strong anticorrelation (R = –0.85), suggesting a link between the alkalinity generated through microbial sulfate reduction and sedimentary carbonate precipitation (Zhang, 2020).
Our data suggest that this carbonate mineral precipitate is associated with the uptake of both Mg and Sr, as there is a strong correlation between pore fluid Mg and Ca (R = 0.64) and Sr and Ca (R = 0.83). The correlation matrix (Figure 4) suggests that magnesium incorporation varies with location, with a higher Mg incorporation in the Gulf of Aqaba sites, which may be due to higher bottom water temperatures; temperature is known to strongly influence the partition coefficient of Mg into the carbonate lattice (Elderfield and Ganssen, 2000).
Microbial sulfate reduction, coupled with either oxidation of organic matter or methane, is not the only anaerobic metabolism generating alkalinity at these sites. The Gulf of Aqaba has previously been shown to have active iron and manganese cycles (Blonder et al., 2017), with the zones of iron and manganese reduction being visible in the dissolved metal pore fluid profile (Figure 5); our data support these previous results. The similar concentrations of dissolved iron and manganese in the Iberian Margin pore fluid suggest iron and manganese cycling is also occurring at this location. We do not, however, observe distinct horizons of increased alkalinity coincident with the zones of iron and manganese reduction at any site.
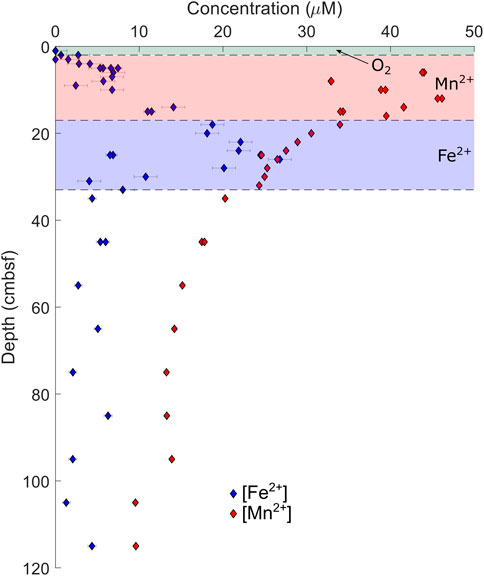
FIGURE 5. Pore fluid iron and manganese concentrations from AQ700, with overlying “zones” as interpreted by the relative depths of the recorded maximum of each ion.
Sedimentary carbonate mineral precipitation or dissolution is driven by chemical reactions that consume and produce protons and thus alter pore fluid pH; these chemical reactions can be driven by both aerobic and anaerobic microbial metabolisms. The anaerobic metabolic pathways that raise the pore fluid pH may counteract the pH decrease caused by calcium carbonate precipitation itself and allow precipitation to continue. These anaerobic pathways include manganese reduction, iron reduction, and the anaerobic oxidation of methane through sulfate reduction, all of which raise pH above the observed “calcification-threshold” of 7.2–7.4 (Soetaert et al., 2007). Conversely, aerobic respiration, organoclastic sulfate reduction, and fermentation via methanogenesis have all been suggested to lower pH below this threshold. The aerobic–anaerobic boundary is often considered a point where conditions switch from carbonate mineral dissolution to carbonate mineral precipitation (Arndt et al., 2013).
In the Gulf of Aqaba and on the Iberian Margin, there is an aerobic–anaerobic transition within the boundary layer, given the changes in the iron and manganese pore fluid concentrations and other indications of anaerobic metabolism such as a decrease in the pore fluid sulfate concentration and increase in δ34SSO4. All our sites are overlain by seawater that is saturated or supersaturated with respect to calcium carbonate (Eq. 1). The Gulf of Aqaba sites are considered to share the same overlying water conditions, with Ωcalcite = 4.4 and Ωaragonite = 2.8. At IM600, Ωcalcite = 3.0 and Ωaragonite = 1.9, and at IM4650—the only site near the saturation horizon—Ωcalcite = 1.3 and Ωaragonite = 0.9.
The primary control on the anaerobic–aerobic transition is the depth of sedimentary oxygen penetration. In turn, one of the primary controls on the oxygen penetration depth is primary productivity, and organic matter production and delivery to the sediments. Sites closer to the shore would experience higher overall levels of primary productivity in the overlying water due to higher nutrient levels supplied from terrestrial environments. Therefore, more organic carbon is delivered to sediments, and there will be a shallower oxygen penetration depth into the sediment, with all other factors being equal. Another major influence on primary productivity in the upper water column is proximity to a zone of upwelling from the deep ocean. The Iberian Margin is impacted by both of these effects, being proximal to shore and in an Eastern boundary upwelling zone, supporting high levels of primary productivity in the surface waters. High organic-matter delivery and a shallow aerobic–anaerobic transition should lead to a shoaling of those anaerobic processes outlined above, which raise pore fluid pH and encourage sedimentary carbonate mineral precipitation, which would lead to a decrease in pore fluid calcium concentrations.
The delivery of carbon decreases further away from the shore as most of the organic carbon is oxidized in the (larger) water column before it reaches the sediment. This lower delivery of organic carbon increases the depths of sedimentary oxygen penetration and the associated underlying aerobic–anaerobic transition (D’Hondt et al., 2015). Aerobic respiration lowers pore fluid pH sufficiently to drive surface sedimentary carbonate mineral dissolution and supply aqueous calcium to the pore fluid, which should manifest as an increase in either pore fluid calcium concentration or diffusive calcium flux to the overlying bottom waters.
The most comprehensive survey of boundary layer calcium concentrations was conducted on a series of cores from the West Atlantic Ocean by Sayles (1979), Sayles (1981). Data from this study demonstrated the concept of this proposed onshore-to-offshore switch from upper sedimentary carbonate mineral precipitation to upper sedimentary carbonate mineral dissolution (Supplementary Figure S3). Closer to the shore in shallower water, pore fluid calcium concentrations decreased with depth below the sediment–water interface, but further from the shore in deeper water, pore fluid calcium concentrations increased. Our chosen sites sampled in this study are close to the shore in shallow water, and we measure a decrease in pore fluid calcium concentrations with depth below the sediment–water interface, similar to the near-shore part of the transect studied by Sayles (1979), Sayles (1981), Supplementary Figure S3.
All of our sampling locations underlie waters at saturation or oversaturated with respect to calcium carbonate; pore fluid calcium concentrations decrease with depth at all sites (albeit at a greater rate in the Gulf of Aqaba), and we know that the isotopic composition of calcium is a sensitive tracer of carbonate mineral precipitation. However, we do not observe the expected changes in pore fluid δ44Ca. This poses the following question: What features of the boundary layer preclude these observations in these locations? Here we are going to explore three nonexclusive possibilities. First, we will explore the idea that calcium carbonate precipitation is occurring deeper in the sediment column and our results capture the uppermost phase where, due to mixing driven by diffusion between the (deeper) precipitation horizon and the overlying water column, there is no change in the pore fluid δ44Ca. Second, we will explore whether an active iron and manganese cycle could be precluding carbonate mineral precipitation. Finally, we will explore whether rates of carbonate mineral precipitation are too slow for the fractionation of calcium isotopes. We hope that these results will help future studies interpret geochemical data acquired from the sedimentary boundary layer.
The fact that we see gradients in the pore fluid concentrations of many species through the boundary layer (but not δ44Ca) suggests that while there is some open system mixing with the overlying water column, advection and rapid diffusive exchange alone cannot explain the broad invariance in δ44Ca. We believe that the upper 12 cmbsf of the pore fluid at IM4650 is in advective exchange with the water column, and here all measured geochemistry is constant at seawater values, which is in clear contrast with the rest of the measured sites.
First, we will explore the idea that most of the carbonate mineral precipitation is happening meters below the depth of our sampling within the uppermost boundary layer, resulting in limited change in boundary layer pore fluid δ44Ca. It has been suggested that microbial sulfate reduction alone can drive supersaturation of carbonate minerals, and hence carbonate mineral precipitation, but only when the majority (approximately half) of the sulfate has been consumed (Meister, 2013). Given that only ∼10% of total pore fluid sulfate is consumed over the sampled range in the Gulf of Aqaba, this saturation threshold is likely not reached in the upper meter of the sediment, and we expect carbonate mineral precipitation would be occurring deeper in the sediment. It is worth noting that the degree of supersaturation induced by sulfate reduction is heavily dependent on pH, which is in turn influenced by the electron donor utilized in the metabolic process (Gallagher et al., 2012; Gallagher et al., 2014; Meister, 2013; Meister, 2014; Fantle and Ridgwell, 2020).
In all of the measured pore fluid profiles, the rate of change of the calcium concentration with depth is similar, hinting that calcium concentrations in the boundary layer are controlled by the balance between carbonate mineral precipitation and the diffusion of calcium from the overlying seawater. Whether carbonate mineral precipitation is controlled by sulfate reduction alone or another process such as the anaerobic oxidation of methane, which has been demonstrated to lead to carbonate precipitation in marine sediments, is not distinguishable from the pore fluid evidence presented here. However, methane is found deeper in the sediment of the Iberian Margin (Turchyn et al., 2016) and would be migrating vertically within the sediment. As there is no evidence of methane at the surface, any methane produced in any of these locations must be consumed through anaerobic metabolism at depth.
It has been shown that in marine sediments with a high organic carbon content, or the presence of methane, the precipitation of carbonate minerals leads to an observable increase in the δ44Ca values measured in the pore fluid (Teichert et al., 2005; Teichert et al., 2009; Bradbury and Turchyn, 2018; Blättler et al., 2021). As discussed above, the recrystallization of carbonate minerals leads to a decrease in the δ44Ca values of the pore fluid toward those in equilibrium with carbonate minerals (Fantle and DePaolo, 2007; Fantle, 2015; Huber et al., 2017; Fantle and Ridgwell, 2020).
To explore how the balance of carbonate mineral precipitation and recrystallization influences changes in calcium concentrations and pore fluid δ44Ca, we use the reactive transport modeling (RTM) software, CrunchTope for AQ700 (Druhan et al., 2013; Druhan et al., 2014; Steefel et al., 2015; Fantle and Ridgwell, 2020). The RTM is set up similar to previous models of sulfate reduction, carbonate mineral recrystallization, and precipitation in marine sediments (Steefel et al., 2014; Huber et al., 2017; Fantle and Ridgwell, 2020; Bradbury et al., 2021). The complete modeling methods for the RTM, including the governing equations, are derived from the work of Bradbury et al. (2021). We use CrunchTope to simulate advection, diffusion, and reaction over a 10 m pore fluid column with a coexisting solid phase, a seawater Dirichlet upper boundary condition, and a Neumann or dC/dx = 0 lower boundary condition. The porosity is fixed at 0.7, with a constant sediment burial modeled using equal burial and fluid flow rate terms of 0.037 cm/year, which is approximately equal to the previously reported sedimentation rate at 700 mbsl in the Gulf of Aqaba (Steiner et al., 2016). The diffusivity was calculated from a molecular diffusion coefficient of 9.19 × 10−6 cm2/s and the porosity of the sediment column (Huber et al., 2017) and was the same for all species. The full list of initial conditions is detailed in Supplementary Table S1. The solid phase has two major constituents: quartz (66%) and primary calcite (33%). The percentage of carbonate minerals is similar to the average of the previously reported carbonate content in the Gulf of Aqaba sediments (Steiner et al., 2019). Primary calcite has a δ44Ca of −0.35‰, similar to the average δ44Ca of the measured sediment, with a recrystallization rate constant of 10−10.0 mol m−2 s−1, which is comparable to previously published calcite recrystallization constants in CrunchTope (10−10.6; Huber et al., 2017).
The sediment also contains trace amounts of secondary calcite written explicitly as two calcite minerals containing either 40Ca or 44Ca, which both follow TST rate laws with the same rate constant as the primary calcite. Primary and secondary calcite are separated to allow the primary calcite to recrystallize with an equilibrium fractionation factor of 1.0, and secondary calcite to precipitate with a variable kinetic fractionation factor ranging from α = 0.9992 to 1, similar to previously reported calcium carbonate fractionation factors in deep marine sediment systems (Bradbury and Turchyn, 2018; Blättler et al., 2021). The system starts from the initial conditions listed in Supplementary Table S1 and is allowed to run until the fluid concentrations and isotopic composition are no longer changing with time, which is assumed to be the steady state. Microbial sulfate reduction is modeled with formaldehyde (CH2O) representing the bulk composition of organic matter (Meister, 2013; Meister, 2014; Meister et al., 2019) using a dual Monod rate law as detailed in Bradbury et al. (2021). The sulfate δ34S profile is fit with a sulfur isotope fractionation factor of α = 0.9955, which is common in deep sea sediments.
The aim of the model is to test whether there would be an observable change in δ44Ca values in the pore fluid of the upper boundary layer if there was carbonate mineral precipitation with a non-zero calcium isotope fractionation factor at some depth below the measured section. In the RTM, precipitation is governed by the saturation state of calcite, which in itself is controlled by microbial sulfate reduction, with supersaturation occurring when 50% of sulfate has been consumed, which for these sites is below the sampled depth interval (Meister, 2013). The model was run with a varying seawater calcium isotope composition (δ44CaSW = 0.7–0.9‰) and a constant calcium isotope fractionation factor of α = 0.9992 (upper row), and with a constant δ44CaSW and a varying calcium isotope fractionation factor in the lower row (α = 0.9992 to 1, Δ44Cas-f = −0.8 to 0‰). Our results match the measured pore fluid concentration profiles well and reproduce the measured δ44Ca to within analytical error (Figure 6).
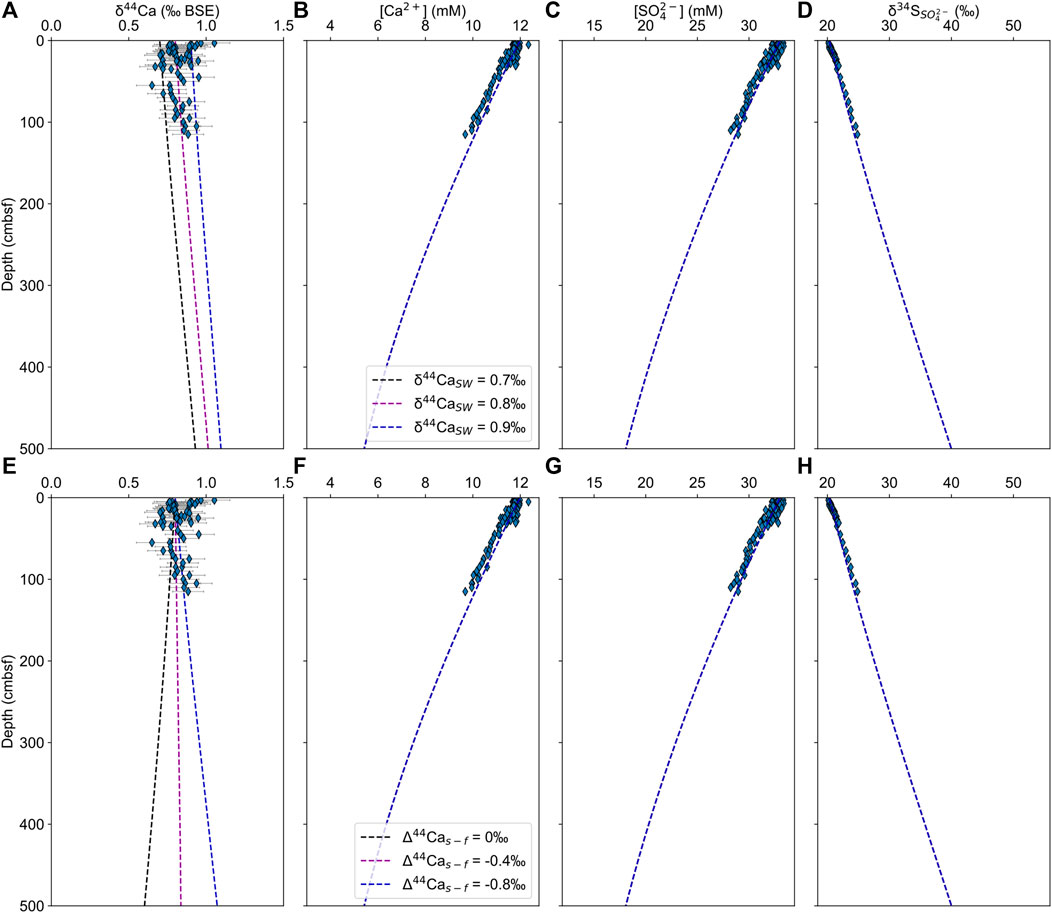
FIGURE 6. CrunchTope reactive transport modeling results for pore fluid δ44Ca (A,E), [Ca2+] (B,F), [SO42–] (C,G), and δ34S (D,H). The top row (A–D) includes the results of runs with varying seawater calcium isotope composition (δ44CaSW = 0.7–0.9‰) and a constant calcium isotope fractionation factor of α = 0.9992. The bottom row (E–H) includes the results of runs with a constant δ44CaSW and a varying calcium isotope fractionation factor (α = 0.9992–1, Δ44Cas-f = –0.8–0‰).
Our results demonstrate that the balance between diffusion of calcium from overlying seawater and the precipitation of carbonate minerals (with a non-zero calcium isotope fractionation factor) at a deeper horizon would produce pore fluids where there are changes in the calcium and sulfate concentrations, and the sulfur isotopic composition of sulfate, but it would produce no change in pore fluid δ44Ca. Specifically, the model shows that the reciprocal nature of the diffusive profile of δ44Ca leads to large isotopic changes close to the horizon of mineral precipitation at depth but less changes near the surface of the sediment. This modeled variation is so small within the upper meter of the sediment that the results are indistinguishable from a straight line within error of δ44Ca.
Previous studies have shown that mixing seawater from the Gulf of Aqaba with the dried local sediment does induce inorganic carbonate mineral precipitation (Wurgaft et al., 2016) and that pore fluids in the Gulf of Aqaba remain stably supersaturated with respect to calcium carbonate in the boundary layer (Steiner et al., 2019). It is possible, therefore, that if no carbonate mineral precipitation occurs in the boundary layer, some process is actively suppressing it.
We now explore whether active iron and manganese cycles, in both localities, could suppress carbonate mineral precipitation. This is initially counterintuitive; it is largely held that iron and manganese reduction, when linked to the oxidation of organic carbon, leads to an increase in pH, which drives precipitation of carbonate minerals (Soetaert et al., 2007; Arndt et al., 2013). The change in pH is, however, dependent on the overall reaction stoichiometry, and the fate of the reduced iron and manganese. If reduced iron and manganese later react with fluid mobile oxidants, they will reoxidize. This is the reverse of the metabolic reduction reaction and produces protons, lowering pH. This is most likely to occur when up-diffusing reduced iron and manganese move into the proximity of dissolved oxygen at the oxic–anoxic transition horizon, with the resulting oxide minerals forming nodular precipitates (Froelich et al., 1979; Kasten et al., 2003). The pH decrease associated with this process creates conditions unfavorable for carbonate mineral precipitation and can counteract the pH increase caused by metabolic iron and manganese reduction if the oxidation and reduction horizons are in close enough proximity.
The Gulf of Aqaba sediment contains relatively high concentrations of manganese and iron minerals due to the flux of aeolian dust from the surrounding deserts; indeed, a previous work has suggested that iron and manganese drive a cryptic sulfur cycle in these sediments (Blonder et al., 2017). The Iberian Margin sediments, given their proximity to terrestrial environments and the presence of iron–manganese nodules and mineralized burrows, are also likely to have increased delivery of terrestrial metals (J. Einsle pers. comm.). At both the Gulf of Aqaba and the Iberian Margin, there is a pore fluid increase in reduced manganese and iron, suggesting that both are being reduced in the sediment (Figures 2, 3, 5).
Although iron and manganese reduction is widely suspected to induce sedimentary carbonate mineral precipitation, we suggest that this does not happen at either studied site because oxidants at the vertically adjacent proximal oxic-anoxic transition reoxidize the up-diffusing reduced iron and manganese, which lowers local pH sufficiently to preclude precipitation throughout (Soetaert et al., 2007).
Furthermore, Steiner et al. (2019) suggest that the incorporation of Mn2+ into the calcium carbonate minerals that do precipitate in the Gulf of Aqaba leads to their subsequent preferential dissolution, as pore fluid [Mn2+] decreases with depth, effectively precluding net carbonate mineral precipitation in the boundary layer. Imaging with SEM of coccolith dissolution in the upper 30 cm of the sediment from the same study indicates that either of the above processes may be sufficient to dissolve biogenic calcium carbonate.
We note that some variation in the pore fluid δ44Ca profile is observed at IM600, which, we propose, may be related to this process. That the decrease in pore fluid δ44Ca over the upper 15 cmbsf is not coupled to a decrease in calcium concentrations seems to suggest recrystallization of carbonate minerals, similar to what is seen in other deep marine locations (Fantle and DePaolo, 2007). At this site, 15 cmbsf is the depth of maximum oxygen penetration and the onset of metabolic iron reduction (as indicated by a non-zero pore fluid ferrous iron content from 10 cmbsf: Figure 2, upper row), and where the pore fluid δ44Ca stops decreasing. The correspondence in these depths implies that upwardly diffusing reduced iron is being oxidized and precipitating out of the pore fluid at this depth, likely in contact with downwardly diffusing dissolved oxygen. The pH decrease associated with the oxidation of ferrous iron would promote back reaction, being the release of low-δ44Ca carbonate into the pore fluid, establishing a recrystallizing system, and creating the observed minimum. Below this depth, both δ44Ca and pore fluid [Fe2+] increase, active iron reduction should increase pH above the calcification threshold, and precipitation driven by the associated release of DIC is therefore no longer inhibited to the same degree. A δ44Ca minimum also immediately overlies an [Fe2+] maximum in AQ500 (5 and 7 cmbsf, respectively)—it may be that the same process is active here. However, in the absence of a more detailed solid sediment analysis, these conclusions cannot be reliably verified.
There are several lines of evidence suggesting both an active iron and manganese cycle at our studied sites as well as suppression of carbonate mineral precipitation in the upper boundary layer; whether these two are linked directly is less clear. Below the measured maxima in pore fluid iron and manganese (>50 cmbsf in AQ500, AQ700, and IM600), pore fluid concentrations and oxidant supply are low enough that the oxidative inhibition of carbonate precipitation is not likely a significant control on δ44Ca. This suppression therefore requires that carbonate mineral precipitation beneath the zones of iron and manganese reduction does occur to explain the quasi-linear diffusive decrease in calcium concentrations with depth.
One final possibility is that there is carbonate mineral precipitation in the boundary layer but that the rate is slow enough that there is little, or no, calcium isotope fractionation during this precipitation (Fantle and DePaolo, 2007; Jacobson and Holmden, 2008). Above, we have described a confluence of factors that we suspect may interfere with and inhibit carbonate mineral precipitation in the boundary layer, in particular the existence of iron and manganese redox cycling that precludes a monotonic increase in pH. It is possible that boundary layer carbonate mineral precipitation is slowed down by the existence of these various processes such that the rate of mineral formation leads to no calcium isotope fractionation during precipitation. This would result in little change in the δ44Ca of pore fluids in affected regions of the boundary layer.
Multiple studies have linked calcium isotope fractionation during carbonate mineral precipitation to the rate of precipitation, both through experimental studies (e.g., Gussone et al., 2003; Gussone et al., 2005; Tang et al., 2008, 2012; AlKhatib and Eisenhauer, 2017), studies in the natural environment (Fantle and DePaolo, 2007; Fantle, 2015; Huber et al., 2017), and modeling (DePaolo, 2011; Nielsen et al., 2012; Lammers et al., 2020). With the exception of one of the earliest studies in this field (Lemarchand et al., 2004), all of these studies have found that at high rates of carbonate mineral precipitation, the calcium isotope fractionation is highest, at what has been termed the “kinetic limit.” This kinetic limit for calcite mineral precipitation is around Δ44Cas-f = –1.4‰ (α = 0.9986) (the mineral is 1.4‰ lower than the fluid from which it precipitates) and for aragonite mineral precipitation is ∼ –1.9‰ (α = 0.9981) (Gussone et al., 2005; DePaolo, 2011).
Conversely, as the rate of carbonate mineral formation slows down, carbonate minerals can, in theory, grow nearly in (calcium) isotopic equilibrium with the fluid (Fantle and DePaolo, 2007; Jacobson and Holmden, 2008; DePaolo, 2011). This slow growth rate is difficult to be achieved in the laboratory; therefore, the calcium isotope equilibrium fractionation factor has been determined largely through modeling studies of deep sea pore fluids (which have some of the slowest rates of mineral growth and recrystallization on the planet). Through these studies, it is understood that the equilibrium calcium isotope fractionation factor is 0‰ (α = 1; Fantle and DePaolo, 2007). This value means that at isotope equilibrium, the fluid and solid tend toward the same δ44Ca, and there is effectively no calcium isotope partitioning between them. Given that in the boundary layer in the Gulf of Aqaba the solid phase and pore fluid δ44Ca do not begin to converge over the sampled depth range, we believe we are not observing strict isotopic equilibrium behavior between pore fluid and solid. This is reasonable to assume particularly in the Gulf of Aqaba, where high bottom water temperatures should strongly enhance reaction rates relative to deep ocean sites. Yet, we would expect to observe an increase in pore fluid δ44Ca with depth if mineral precipitation rates were sufficiently high that kinetic isotope fractionation was occurring, establishing a “kinetic limit” offset.
The reactive transport model discussed above requires a maximum calcite precipitation rate of 0.65 μmol m−2 h−1 (1.8 × 10−10 mol m−2 s−1) in the lower part of the sulfate reduction zone at AQ700 to match the observed calcium concentration decrease. This rate lies within error of those modeled by Blättler et al. (2021), 1.4 and 3.5 μmol m−2 h−1 (both ±10 μmol m−2 h−1), for two sediment cores shown to be actively precipitating calcium carbonate. The observed calcite fractionation factors from Blättler et al. (2021) of Δ44Cas-f = –0.15‰ (α = 0.99985) and Δ44Cas-f = –0.4‰ (α = 0.9996) lie intermediately between the endmember calcium isotope fractionation factors of equilibrium isotope fractionation close to Δ44Cas-f = 0‰, and the kinetic limit for calcite at Δ44Cas-f = –1.4‰. Given the similar rates, we could anticipate similar calcium isotope fractionation factors at AQ700.
This intermediate rate of carbonate mineral precipitation—slowed by the inhibiting factors discussed in the previous sections—could explain the observed invariance in pore fluid δ44Ca. A precipitation rate close to, but not at, the equilibrium endmember fractionation factor could produce a cumulative pore fluid isotopic effect that is not sufficiently developed over the sampled depth range to be indistinguishable from a straight line within error of δ44Ca.
Pore fluid δ44Ca does not change outside of analytical error over the 1 m depth scale of the boundary layer in the sediments studied from neither the Iberian Margin nor the Gulf of Aqaba. This is despite evidence for changes in redox conditions within the boundary layer that should lead to carbonate mineral precipitation in the sediment. If calcium carbonate mineral precipitation occurs, it is either happening at a depth below the sampled range, with the resultant increased δ44Ca not visible due to offset by back-reaction and/or the reciprocal nature of the upper part of the mixing trend (Figure 6), or precipitation is so slow that a rate-dependent calcium isotopic response is precluded. This confluence of processes lessens the magnitude of the resultant signal such that it is masked by the analytical measurement error of δ44Ca. Any given pore fluid horizon may have its δ44Ca impacted by bioirrigative supply of oxygenated water (as discussed above) or by any other localized geochemical microenvironment, introducing further analytical noise.
Over submeter length scales, however, pore fluid δ44Ca may reflect the complex relationship between redox cycling of iron and manganese and the suitability of resultant fluids for precipitation of carbonate minerals. In near-shore sites where terrigenous iron and manganese concentrations are high, such as those studied here, the δ44Ca appears to change to reflect horizons where iron and manganese are reduced (raising pH, favoring precipitation) or subsequently oxidized (lowering pH, favoring dissolution).
Any future work on boundary layer pore fluid δ44Ca used to assess carbonate mineral dissolution and precipitation should take precautions to help increase the utility of the measurement beyond this study. Sampling that merges both high-resolution boundary layer processes with sediments down to 10–15 m would mean that the continuum of processes can be better resolved; with the sampling limitations of this study, we are unable to conclude what is happening meters below the boundary layer.
The pore fluid major ion concentrations and sulfur isotopic composition data from the boundary layer in both the Gulf of Aqaba and the Iberian Margin show strong correlations between decreasing calcium and sulfate concentrations, increasing alkalinity and changes in the sulfur isotopic composition of sulfate, and decreasing magnesium and strontium concentrations. These indicate that anaerobic metabolisms, especially microbial sulfate reduction, are altering the redox state of the pore fluid and inducing calcium carbonate mineral precipitation. Despite this, we do not observe the expected meter-scale change in pore fluid calcium isotope composition, which remains within error of seawater values throughout the sampled range.
The lack of meter-scale change in δ44Ca is a consequence of interaction between much shorter-depth-scale processes near the oxic–anoxic boundary, and much longer-depth-scale processes below the sampled horizons where sulfate is consumed.
In the uppermost boundary layer, immediately below the maximum depth of oxygen penetration, iron and manganese reduction are expected to induce carbonate precipitation. However, the reoxidation of reduced iron and manganese in contact with dissolved oxygen lowers pH sufficiently to preclude further carbonate mineral precipitation and even promote dissolution, leading to the δ44Ca minimum at the oxic–anoxic boundary and subsequent maximum at the horizon of peak iron reduction observed in two of our sampled sites.
Our results hint that carbonate mineral precipitation occurs deeper in the sediment than we sampled; we use a reactive transport model to show that this deeper precipitation would not necessarily cause a change in boundary layer pore fluid δ44Ca, although changes could still be observed in other pore fluid properties, such as calcium and sulfate concentrations, and δ34SSO4. We suggest that when modeling or measuring geochemistry in the sedimentary boundary layer, one should consider processes that occur at greater depth and that diffusion through the boundary can dominate the geochemistry of boundary layer pore fluids, as has been previously concluded.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
DJ wrote the manuscript and analyzed calcium isotopes for the Gulf of Aqaba. HB, GA, ZS, AH, XS, and AT contributed feedback on the said manuscript. DJ, HB, GA, ZS, and AH collected and processed the Gulf of Aqaba cores, and DJ, HB, ZS, and AH measured major ion concentrations. HB and XS analyzed calcium isotopes and measured major ion concentrations for the Iberian Margin. Pore fluid alkalinity was measured by RS (Gulf of Aqaba) and MG (Iberian Margin). Field sampling in Israel was coordinated by GA. HB supervised all laboratory work in 2018, produced the reactive transport model (Figures 1, 4, 6; Supplementary Figure S3), and assisted DJ throughout. HB, GA, ZS, and AT had the original idea for this study. AT supervised and guided the direction of this work, and first suggested research in this field.
This work was supported by NERC NE/R013519/1 to HB and ERC 307582 StG (CARBONSINK) to AT. The JC089 Cruise was funded by NERC grant NE/J00653X/1 to David Hodell. DJ's fieldwork in Israel in 2018 was funded by Pembroke College, the University of Cambridge.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Adam Bateson for conducting the sulfur isotope analysis for IM600 and IM4650 on a NERC-funded REP, and Josephine Clegg for her assistance with the sulfur isotope analysis for AQ700. We also thank all the participants of cruise JC089 on board the R/V James Cook for recovering the Iberian Margin core material and pore waters used in this study with support from NERC grant NE/J00653X/1 to David Hodell. DJ thanks Pembroke College, the University of Cambridge, for partially funding the fieldwork in Eilat in September 2018.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/feart.2021.601194/full#supplementary-material
Ahm, A.-S. C., Bjerrum, C. J., Blättler, C. L., Swart, P. K., and Higgins, J. A. (2018). Quantifying Early marine Diagenesis in Shallow-Water Carbonate Sediments. Geochimica et Cosmochimica Acta. 236, 140–159. doi:10.1016/j.gca.2018.02.042
AlKhatib, M., and Eisenhauer, A. (2017). Calcium and Strontium Isotope Fractionation in Aqueous Solutions as a Function of Temperature and Reaction Rate; I. Calcite. Geochimica et Cosmochimica Acta. 209, 296–319. doi:10.1016/j.gca.2016.09.035
Arndt, S., Brumsack, H.-J., and Wirtz, K. W. (2006). Cretaceous Black Shales as Active Bioreactors: A Biogeochemical Model for the Deep Biosphere Encountered during ODP Leg 207 (Demerara Rise). Geochimica et Cosmochimica Acta. 70, 408–425. doi:10.1016/j.gca.2005.09.010
Arndt, S., Hetzel, A., and Brumsack, H.-J. (2009). Evolution of Organic Matter Degradation in Cretaceous Black Shales Inferred from Authigenic Barite: A Reaction-Transport Model. Geochimica et Cosmochimica Acta. 73, 2000–2022. doi:10.1016/j.gca.2009.01.018
Arndt, S., Jørgensen, B. B., LaRowe, D. E., Middelburg, J. J., Pancost, R. D., and Regnier, P. (2013). Quantifying the Degradation of Organic Matter in marine Sediments: A Review and Synthesis. Earth-Science Rev. 123, 53–86. doi:10.1016/j.earscirev.2013.02.008
Ben-Avraham, Z., Almagor, G., and Garfunkel, Z. (1979). Sediments and Structure of the Gulf of Elat (Aqaba)-Northern Red Sea. Sediment. Geology 23, 239–267. doi:10.1016/0037-0738(79)90016-2
Ben-Yaakov, S. (1973). pH BUFFERING OF PORE WATER OF RECENT ANOXIC MARINE SEDIMENTS1. Limnol. Oceanogr. 18, 86–94. doi:10.4319/lo.1973.18.1.0086
Berner, R. A. (1980). Early Diagenesis: A Theoretical Approach. Princeton, NJ: Princeton University Press. doi:10.1515/9780691209401
Berner, R. A., Lasaga, A. C., and Garrels, R. M. (1983). The Carbonate-Silicate Geochemical Cycle and its Effect on Atmospheric Carbon Dioxide over the Past 100 Million Years. Am. J. Sci. 283, 641–683. doi:10.2475/ajs.283.7.641
Berner, R. A. (2003). The Long-Term Carbon Cycle, Fossil Fuels and Atmospheric Composition. Nature 426, 323–326. doi:10.1038/nature02131
Biton, E., and Gildor, H. (2011). Stepwise Seasonal Restratification and the Evolution of Salinity Minimum in the Gulf of Aqaba (Gulf of Eilat). J. Geophys. Res. 116, C08022. doi:10.1029/2011JC007106
Blättler, C. L., Hong, W.-L., Kirsimäe, K., Higgins, J. A., and Lepland, A. (2021). Small Calcium Isotope Fractionation at Slow Precipitation Rates in Methane Seep Authigenic Carbonates. Geochimica et Cosmochimica Acta 298, 227–239. doi:10.1016/j.gca.2021.01.001
Blonder, B., Boyko, V., Turchyn, A. V., Antler, G., Sinichkin, U., Knossow, N., et al. (2017). Impact of Aeolian Dry Deposition of Reactive Iron Minerals on Sulfur Cycling in Sediments of the Gulf of Aqaba. Front. Microbiol. 8, 1131. doi:10.3389/fmicb.2017.01131
Boudreau, B. P. (1997). Diagenetic Models and Their Implementation: Modelling Transport and Reactions in Aquatic Sediments. Berlin: Springer.
Bradbury, H. J., Turchyn, A. V., Bateson, A., Antler, G., Fotherby, A., Druhan, J. L., et al. (2021). The Carbon-Sulfur Link in the Remineralization of Organic Carbon in Surface Sediments. Front. Earth Sci. 9, 301–315. doi:10.3389/feart.2021.652960
Bradbury, H. J., and Turchyn, A. V. (2018). Calcium Isotope Fractionation in Sedimentary Pore Fluids from ODP Leg 175: Resolving Carbonate Recrystallization. Geochimica et Cosmochimica Acta 236, 121–139. doi:10.1016/j.gca.2018.01.040
Carter, B. R., Toggweiler, J. R., Key, R. M., and Sarmiento, J. L. (2014). Processes Determining the marine Alkalinity and Calcium Carbonate Saturation State Distributions. Biogeosciences. 11, 7349–7362. doi:10.5194/bg-11-7349-2014
Chen, Y., Paytan, A., Chase, Z., Measures, C., Beck, A. J., Sañudo-Wilhelmy, S. A., et al. (2008). Sources and Fluxes of Atmospheric Trace Elements to the Gulf of Aqaba, Red Sea. J. Geophys. Res. 113, a–n. doi:10.1029/2007JD009110
DePaolo, D. J. (2011). Surface Kinetic Model for Isotopic and Trace Element Fractionation during Precipitation of Calcite from Aqueous Solutions. Geochimica et Cosmochimica Acta. 75, 1039–1056. doi:10.1016/j.gca.2010.11.020
D’Hondt, S., Inagaki, F., Zarikian, C. A., Abrams, L. J., Dubois, N., Engelhardt, T., et al. (2015). Presence of Oxygen and Aerobic Communities from Sea Floor to Basement in Deep-Sea Sediments. Nat. Geosci. 8, 299–304. doi:10.1038/ngeo2387
Druhan, J. L., Steefel, C. I., Conrad, M. E., and DePaolo, D. J. (2014). A Large Column Analog experiment of Stable Isotope Variations during Reactive Transport: I. A Comprehensive Model of Sulfur Cycling and δ34S Fractionation. Geochimica et Cosmochimica Acta. 124, 366–393. doi:10.1016/j.gca.2013.08.037
Druhan, J. L., Steefel, C. I., Williams, K. H., and DePaolo, D. J. (2013). Calcium Isotope Fractionation in Groundwater: Molecular Scale Processes Influencing Field Scale Behavior. Geochimica et Cosmochimica Acta. 119, 93–116. doi:10.1016/j.gca.2013.05.022
Elderfield, H., and Ganssen, G. (2000). Past Temperature and δ18O of Surface Ocean Waters Inferred from Foraminiferal Mg/Ca Ratios. Nature 405, 442–445. doi:10.1038/35013033
Fantle, M. S. (2015). Calcium Isotopic Evidence for Rapid Recrystallization of Bulk marine Carbonates and Implications for Geochemical Proxies. Geochimica et Cosmochimica Acta. 148, 378–401. doi:10.1016/j.gca.2014.10.005
Fantle, M. S., and DePaolo, D. J. (2007). Ca Isotopes in Carbonate Sediment and Pore Fluid from ODP Site 807A: The Ca2+(aq)-Calcite Equilibrium Fractionation Factor and Calcite Recrystallization Rates in Pleistocene Sediments. Geochimica et Cosmochimica Acta. 71, 2524–2546. doi:10.1016/j.gca.2007.03.006
Fantle, M. S., and Ridgwell, A. (2020). Towards an Understanding of the Ca Isotopic Signal Related to Ocean Acidification and Alkalinity Overshoots in the Rock Record. Chem. Geology 547, 119672. doi:10.1016/j.chemgeo.2020.119672
Fantle, M. S., and Tipper, E. T. (2014). Calcium Isotopes in the Global Biogeochemical Ca Cycle: Implications for Development of a Ca Isotope Proxy. Earth-Science Rev. 129, 148–177. doi:10.1016/j.earscirev.2013.10.004
Feely, R., Doney, S., and Cooley, S. (2009). Ocean Acidification: Present Conditions and Future Changes in a High-CO2 World. Oceanog. 22, 36–47. doi:10.5670/oceanog.2009.95
Froelich, P. N., Klinkhammer, G. P., Bender, M. L., Luedtke, N. A., Heath, G. R., Cullen, D., et al. (1979). Early Oxidation of Organic Matter in Pelagic Sediments of the Eastern Equatorial Atlantic: Suboxic Diagenesis. Geochimica et Cosmochimica Acta. 43, 1075–1090. doi:10.1016/0016-7037(79)90095-4
Gallagher, K. L., Dupraz, C., and Visscher, P. T. (2014). Two Opposing Effects of Sulfate Reduction on Carbonate Precipitation in normal marine, Hypersaline, and Alkaline Environments: COMMENT. Geology 42, e313–e314. doi:10.1130/G34639C.1
Gallagher, K. L., Kading, T. J., Braissant, O., Dupraz, C., and Visscher, P. T. (2012). Inside the Alkalinity Engine: the Role of Electron Donors in the Organomineralization Potential of Sulfate-Reducing Bacteria. Geobiology. 10, 518–530. doi:10.1111/j.1472-4669.2012.00342.x
Gussone, N., Böhm, F., Eisenhauer, A., Dietzel, M., Heuser, A., Teichert, B. M. A., et al. (2005). Calcium Isotope Fractionation in Calcite and Aragonite. Geochimica et Cosmochimica Acta. 69, 4485–4494. doi:10.1016/j.gca.2005.06.003
Gussone, N., Eisenhauer, A., Heuser, A., Dietzel, M., Bock, B., Böhm, F., et al. (2003). Model for Kinetic Effects on Calcium Isotope Fractionation (δ44Ca) in Inorganic Aragonite and Cultured Planktonic Foraminifera. Geochimica et Cosmochimica Acta. 67, 1375–1382. doi:10.1016/S0016-7037(02)01296-6
Gussone, N., Schmitt, A.-D., Heuser, A., Wombacher, F., Dietzel, M., Tipper, E., et al. (2016). Calcium Stable Isotope Geochemistry. Berlin: Springer. doi:10.1007/978-3-540-68953-9
Higgins, J. A., Blättler, C. L., Lundstrom, E. A., Santiago-Ramos, D. P., Akhtar, A. A., Crüger Ahm, A.-S., et al. (2018). Mineralogy, Early marine Diagenesis, and the Chemistry of Shallow-Water Carbonate Sediments. Geochimica et Cosmochimica Acta. 220, 512–534. doi:10.1016/j.gca.2017.09.046
Higgins, J. A., Fischer, W. W., and Schrag, D. P. (2009). Oxygenation of the Ocean and Sediments: Consequences for the Seafloor Carbonate Factory. Earth Planet. Sci. Lett. 284, 25–33. doi:10.1016/j.epsl.2009.03.039
Hodell, D. A., Lourens, L., Stow, D. A. V., Hernández-Molina, J., and Alvarez Zarikian, C. A. (2013a). The “Shackleton Site” (IODP Site U1385) on the Iberian Margin. Sci. Dril. 16, 13–19. doi:10.5194/sd-16-13-2013
Hodell, D. A., Crowhurst, S., Skinner, L., Tzedakis, P. C., Margari, V., Channell, J. E. T., et al. (2013b). Response of Iberian Margin Sediments to Orbital and Suborbital Forcing over the Past 420 Ka. Paleoceanography. 28, 185–199. doi:10.1002/palo.20017
Hodell, D. A., Elderfield, H., Greaves, M., McCave, I. N., Skinner, L., Thomas, A., et al. (2014). The JC089 Scientific Party, JC089 Cruise Report—IODP Site Survey of the Shackleton Sites, SW Iberian Margin. Liverpool: British ocean data Centre.
Hodell, D. A., Lourens, L., Crowhurst, S., Konijnendijk, T., Tjallingii, R., Jiménez-Espejo, F., et al. (2015). A Reference Time Scale for Site U1385 (Shackleton Site) on the SW Iberian Margin. Glob. Planet. Change. 133, 49–64. doi:10.1016/j.gloplacha.2015.07.002
Huber, C., Druhan, J. L., and Fantle, M. S. (2017). Perspectives on Geochemical Proxies: The Impact of Model and Parameter Selection on the Quantification of Carbonate Recrystallization Rates. Geochimica et Cosmochimica Acta. 217, 171–192. doi:10.1016/j.gca.2017.08.023
Jacobson, A. D., and Holmden, C. (2008). δ44Ca Evolution in a Carbonate Aquifer and its Bearing on the Equilibrium Isotope Fractionation Factor for Calcite. Earth Planet. Sci. Lett. 270, 349–353. doi:10.1016/j.epsl.2008.03.039
Kasten, S., Zabel, M., Heuer, V., and Hensen, C. (2003). “Processes and Signals of Nonsteady-State Diagenesis in Deep-Sea Sediments and Their Pore Waters,” in The South Atlantic in the Late Quaternary: Reconstruction of Material Budgets and Current Systems. Editors G. Wefer, S. Mulitza, and V. Ratmeyer (Berlin, Heidelberg, New York: Springer), 431–459. doi:10.1007/978-3-642-18917-3_20
Katz, T., Ginat, H., Eyal, G., Steiner, Z., Braun, Y., Shalev, S., et al. (2015). Desert Flash Floods Form Hyperpycnal Flows in the Coral-Rich Gulf of Aqaba, Red Sea. Earth Planet. Sci. Lett. 417, 87–98. doi:10.1016/j.epsl.2015.02.025
Klinkhammer, G., Heggie, D. T., and Graham, D. W. (1982). Metal Diagenesis in Oxic marine Sediments. Earth Planet. Sci. Lett. 61, 211–219. doi:10.1016/0012-821X(82)90054-1
Krumgalz, B. S., Erez, J., and Chen, C.-T. (1990). Anthropogenic CO2 Penetration in the Northern Red Sea and in the Gulf of Elat (Aqaba). Oceanol. Acta 13, 283–290.
Lammers, L. N., Kulasinski, K., Zarzycki, P., and DePaolo, D. J. (2020). Molecular Simulations of Kinetic Stable Calcium Isotope Fractionation at the Calcite-Aqueous Interface. Chem. Geology 532, 119315. doi:10.1016/j.chemgeo.2019.119315
Lazar, B., Erez, J., Silverman, J., Rivlin, T., Rivlin, A., Dray, M., et al. (2008). “Recent Environmental Changes in the Chemical-Biological Oceanography of the Gulf of Aqaba (Eilat),” in Aqaba-Eilat, the Improbable Gulf. Environment, Biodiversity and Preservation (Jerusalem: Magnes Press), 49–61.
Lemarchand, D., Wasserburg, G. J., and Papanastassiou, D. A. (2004). Rate-controlled Calcium Isotope Fractionation in Synthetic Calcite. Geochimica et Cosmochimica Acta. 68, 4665–4678. doi:10.1016/j.gca.2004.05.029
Meister, P., Liu, B., Khalili, A., Böttcher, M. E., and Jørgensen, B. B. (2019). Factors Controlling the Carbon Isotope Composition of Dissolved Inorganic Carbon and Methane in marine Porewater: An Evaluation by Reaction-Transport Modelling. J. Mar. Syst. 200, 103227. doi:10.1016/j.jmarsys.2019.103227
Meister, P. (2013). Two Opposing Effects of Sulfate Reduction on Carbonate Precipitation in normal marine, Hypersaline, and Alkaline Environments. Geology 41, 499–502. doi:10.1130/G34185.1
Meister, P. (2014). Two Opposing Effects of Sulfate Reduction on Carbonate Precipitation in normal marine, Hypersaline, and Alkaline Environments: REPLY. Geology 42, e315. doi:10.1130/G35240Y.1
Millero, F. J. (1995). Thermodynamics of the Carbon Dioxide System in the Oceans. Geochimica et Cosmochimica Acta. 59, 661–677. doi:10.1016/0016-7037(94)00354-O
Milliman, J. D. (1993). Production and Accumulation of Calcium Carbonate in the Ocean: Budget of a Nonsteady State. Glob. Biogeochem. Cycles. 7, 927–957. doi:10.1029/93GB02524
Nielsen, L. C., DePaolo, D. J., and De Yoreo, J. J. (2012). Self-consistent Ion-By-Ion Growth Model for Kinetic Isotopic Fractionation during Calcite Precipitation. Geochimica et Cosmochimica Acta. 86, 166–181. doi:10.1016/j.gca.2012.02.009
Palmer, M. R., Pearson, P. N., and Cobb, S. J. (1998). Reconstructing Past Ocean pH-Depth Profiles. Science. 282, 1468–1471. doi:10.1126/science.282.5393.1468
Ridgwell, A., and Zeebe, R. (2005). The Role of the Global Carbonate Cycle in the Regulation and Evolution of the Earth System. Earth Planet. Sci. Lett. 234, 299–315. doi:10.1016/j.epsl.2005.03.006
Rodríguez-Tovar, F. J., Dorador, J., Grunert, P., and Hodell, D. (2015). Deep-sea Trace Fossil and Benthic Foraminiferal Assemblages across Glacial Terminations 1, 2 and 4 at the "Shackleton Site" (IODP Expedition 339, Site U1385). Glob. Planet. Change. 133, 359–370. doi:10.1016/j.gloplacha.2015.05.003
Rodríguez-Tovar, F. J., and Dorador, J. (2014). Ichnological Analysis of Pleistocene Sediments from the IODP Site U1385 “Shackleton Site” on the Iberian Margin: Approaching Paleoenvironmental Conditions. Palaeogeogr. Palaeoclimatol. Palaeoecol. 409, 24–32. doi:10.1016/j.palaeo.2014.04.027
Rudnicki, M. D., Wilson, P. A., and Anderson, W. T. (2001). Numerical Models of Diagenesis, Sediment Properties, and Pore Fluid Chemistry on a Paleoceanographic Transect: Blake Nose, Ocean Drilling Program Leg 171B. Paleoceanography. 16, 563–575. doi:10.1029/2000PA000551
Sayles, F. L. (1979). The Composition and Diagenesis of Interstitial Solutions-I. Fluxes across the Seawater-Sediment Interface in the Atlantic Ocean. Geochimica et Cosmochimica Acta. 43, 527–545. doi:10.1016/0016-7037(79)90163-7
Sayles, F. L. (1981). The Composition and Diagenesis of Interstitial Solutions-II. Fluxes and Diagenesis at the Water-Sediment Interface in the High Latitude North and South Atlantic. Geochimica et Cosmochimica Acta. 45, 1061–1086. doi:10.1016/0016-7037(81)90132-0
Seeberg-Elverfeldt, J., Schlüter, M., Feseker, T., and Kölling, M. (2005). Rhizon Sampling of Porewaters Near the Sediment-Water Interface of Aquatic Systems. Limnology and Oceanography: Methods. 3, 361–371. doi:10.4319/lom.2005.3.361
Shackleton, N. J., Fairbanks, R. G., Chiu, T.-c., and Parrenin, F. (2004). Absolute Calibration of the Greenland Time Scale: Implications for Antarctic Time Scales and for Δ14C. Quat. Sci. Rev. 23, 1513–1522. doi:10.1016/j.quascirev.2004.03.006
Shackleton, N. J., Hall, M. A., and Vincent, E. (2000). Phase Relationships between Millennial-Scale Events 64,000-24,000 Years Ago. Paleoceanography. 15, 565–569. doi:10.1029/2000PA000513
Skulan, J., DePaolo, D. J., and Owens, T. L. (1997). Biological Control of Calcium Isotopic Abundances in the Global Calcium Cycle. Geochim. Cosmochim. Acta. 61 (12), 2505–2510.
Soetaert, K., Hofmann, A. F., Middelburg, J. J., Meysman, F. J. R., and Greenwood, J. (2007). The Effect of Biogeochemical Processes on pH. Mar. Chem. 105, 30–51. doi:10.1016/j.marchem.2006.12.012
Steefel, C. I., Appelo, C. A. J., Arora, B., Jacques, D., Kalbacher, T., Kolditz, O., et al. (2015). Reactive Transport Codes for Subsurface Environmental Simulation. Comput. Geosci. 19, 445–478. doi:10.1007/s10596-014-9443-x
Steefel, C. I., Druhan, J. L., and Maher, K. (2014). Modeling Coupled Chemical and Isotopic Equilibration Rates. Proced. Earth Planet. Sci. 10, 208–217. doi:10.1016/j.proeps.2014.08.022
Steiner, Z., Erez, J., Shemesh, A., Yam, R., Katz, A., and Lazar, B. (2014). Basin-scale Estimates of Pelagic and Coral Reef Calcification in the Red Sea and Western Indian Ocean. Proc. Natl. Acad. Sci. USA. 111, 16303–16308. doi:10.1073/pnas.1414323111
Steiner, Z., Lazar, B., Levi, S., Tsroya, S., Pelled, O., Bookman, R., et al. (2016). The Effect of Bioturbation in Pelagic Sediments: Lessons from Radioactive Tracers and Planktonic Foraminifera in the Gulf of Aqaba, Red Sea. Geochimica et Cosmochimica Acta. 194, 139–152. doi:10.1016/j.gca.2016.08.037
Steiner, Z., Lazar, B., Reimers, C. E., and Erez, J. (2019). Carbonates Dissolution and Precipitation in Hemipelagic Sediments Overlaid by Supersaturated Bottom-Waters - Gulf of Aqaba, Red Sea. Geochimica et Cosmochimica Acta. 246, 565–580. doi:10.1016/j.gca.2018.12.007
Steiner, Z., Turchyn, A. V., Harpaz, E., and Silverman, J. (2018). Water Chemistry Reveals a Significant Decline in Coral Calcification Rates in the Southern Red Sea. Nat. Commun. 9, 3615. doi:10.1038/s41467-018-06030-6
Tang, J., Dietzel, M., Böhm, F., Köhler, S. J., and Eisenhauer, A. (2008). Sr2+/Ca2+ and 44Ca/40Ca Fractionation during Inorganic Calcite Formation: II. Ca Isotopes. Geochimica et Cosmochimica Acta. 72, 3733–3745. doi:10.1016/j.gca.2008.05.033
Tang, J., Niedermayr, A., Köhler, S. J., Böhm, F., Kısakürek, B., Eisenhauer, A., et al. (2012). Sr2+/Ca2+ and 44Ca/40Ca Fractionation during Inorganic Calcite Formation: III. Impact of Salinity/ionic Strength. Geochimica et Cosmochimica Acta. 77, 432–443. doi:10.1016/j.gca.2011.10.039
Teichert, B. M. A., Gussone, N., Eisenhauer, A., and Bohrmann, G. (2005). Clathrites: Archives of Near-Seafloor Pore-Fluid Evolution (δ44/40Ca, δ13C, δ18O) in Gas Hydrate Environments. Geol. 33, 213–216. doi:10.1130/G21317.1
Teichert, B. M. A., Gussone, N., and Torres, M. E. (2009). Controls on Calcium Isotope Fractionation in Sedimentary Porewaters. Earth Planet. Sci. Lett. 279, 373–382. doi:10.1016/j.epsl.2009.01.011
Torfstein, A., Kienast, S. S., Yarden, B., Rivlin, A., Isaacs, S., and Shaked, Y. (2020). Bulk and export Production Fluxes in the Gulf of Aqaba, Northern Red Sea. ACS Earth Space Chem. 4, 1461–1479. doi:10.1021/acsearthspacechem.0c00079
Turchyn, A. V., Antler, G., Byrne, D., Miller, M., and Hodell, D. A. (2016). Microbial Sulfur Metabolism Evidenced from Pore Fluid Isotope Geochemistry at Site U1385. Glob. Planet. Change. 141, 82–90. doi:10.1016/j.gloplacha.2016.03.004
Wurgaft, E., Steiner, Z., Luz, B., and Lazar, B. (2016). Evidence for Inorganic Precipitation of CaCO3 on Suspended Solids in the Open Water of the Red Sea. Mar. Chem. 186, 145–155. doi:10.1016/j.marchem.2016.09.006
Yu, J., Anderson, R., and Rohling, E. (2014). Deep Ocean Carbonate Chemistry and Glacial-Interglacial Atmospheric CO2 Change. oceanog. 27, 16–25. doi:10.5670/oceanog.2014.04
Zeebe, R., and Wolf-Gladrow, D. (2001). CO2 in Seawater: Equilibrium, Kinetics, Isotopes. Amsterdam: Elsevier Oceanography Book Series, Vol. 65, 346.
Keywords: carbonate precipitation, calcium isotopes, early diagenesis, microbial sulfate reduction, microbial iron reduction, sedimentary boundary layer, carbonate dissolution
Citation: James DH, Bradbury HJ, Antler G, Steiner Z, Hutchings AM, Sun X, Saar R, Greaves M and Turchyn AV (2021) Assessing Sedimentary Boundary Layer Calcium Carbonate Precipitation and Dissolution Using the Calcium Isotopic Composition of Pore Fluids. Front. Earth Sci. 9:601194. doi: 10.3389/feart.2021.601194
Received: 31 August 2020; Accepted: 25 June 2021;
Published: 05 August 2021.
Edited by:
Christian Marz, University of Leeds, United KingdomReviewed by:
Catherine Elisabeth Pierre, UMR7159 Laboratoire d'océanographie et du climat expérimentations et approches numériques, FranceCopyright © 2021 James, Bradbury, Antler, Steiner, Hutchings, Sun, Saar, Greaves and Turchyn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel H. James, ZGozMzFAY2FtLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.