 Gilad Antler
Gilad Antler Jennifer V. Mills
Jennifer V. Mills Alec M. Hutchings
Alec M. Hutchings Kelly R. Redeker
Kelly R. Redeker Alexandra V. Turchyn
Alexandra V. Turchyn
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Earth Sci., 25 June 2019
Sec. Microbiological Chemistry and Geomicrobiology
Volume 7 - 2019 | https://doi.org/10.3389/feart.2019.00140
This article is part of the Research TopicMicrobial Sulfur Metabolism - From Symbiosis to Global Nutrient Cycling View all 37 articles
We explore the dynamics of the subsurface sulfur, iron and carbon cycles in salt marsh sediments from East Anglia, United Kingdom. We report measurements of pore fluid and sediment geochemistry, coupled with results from laboratory sediment incubation experiments, and develop a conceptual model to describe the influence of bioturbation on subsurface redox cycling. In the studied sediments the subsurface environment falls into two broadly defined geochemical patterns – iron-rich sediments or sulfide-rich sediments. Within each sediment type nearly identical pore fluid and solid phase geochemistry (in terms of concentrations of iron, sulfate, sulfide, dissolved inorganic carbon (DIC), and the sulfur and oxygen isotope compositions of sulfate) are observed in sediments that are hundreds of kilometers apart. Strictly iron-rich and strictly sulfide-rich sediments, despite their substantive geochemical differences, are observed within spatial distances of less than five meters. We suggest that this bistable system results from a series of feedback reactions that determine ultimately whether sediments will be sulfide-rich or iron-rich. We suggest that an oxidative cycle in the iron-rich sediment, driven by bioirrigation, allows rapid oxidation of organic matter, and that this irrigation impacts the sediment below the immediate physical depth of bioturbation. This oxidative cycle yields iron-rich sediments with low total organic carbon, dominated by microbial iron reduction and no methane production. In the absence of bioirrigation, sediments in the salt marsh become sulfide-rich with high methane concentrations. Our results suggest that the impact of bioirrigation not only drives recycling of sedimentary material but plays a key role in sedimentary interactions among iron, sulfur and carbon.
Salt marshes are highly productive coastal wetlands that are flushed daily or monthly with seawater (Howarth, 1993; Alongi, 1998; Chmura et al., 2003). Due to high nutrient loading from terrestrial run off, primary productivity in salt marsh environments exceeds that of many other ecosystems (Giblin and Weider, 1992). Recently, there has been increased focus on the role of coastal wetlands in the overall storage of transported and locally fixed carbon, called “blue carbon” (McLeod et al., 2011; Pendleton et al., 2012). However, while our understanding of the production of “blue carbon” in coastal wetlands is growing, the processes which prevent coastal ecosystems from storing dead carbon in sediment and soils remain less well understood (Pendleton et al., 2012).
After organic carbon is deposited in sediments, including those found in coastal wetlands, it may either undergo oxidation back to dissolved inorganic carbon (DIC) or fermentation into methane, both of which prevent the ultimate deep burial of this organic carbon (Froelich et al., 1979). During oxidation of organic carbon, bacteria can respire a variety of molecules; the order in which these electron acceptors are used broadly reflects the decrease in free energy yield associated with their reduction coupled to organic carbon oxidation (Froelich et al., 1979). The largest energy yield is associated with aerobic respiration (oxidation of organic carbon with molecular oxygen – O2). In today’s environment, oxic respiration dominates both in the oceanic water column and the very top of the sediment column. Oxygen diffusion into sediments can persist up to a few tens of meters in the deep ocean, but only a few millimeters in coastal wetland sediments before it is consumed (Røy et al., 2012; D’Hondt et al., 2015). Below the depth in sediments where oxygen is depleted, other electron acceptors are utilized by microbial populations to respire and continue to oxidize organic carbon. These alternative electron acceptors are used in order of decreasing energy yield: denitrification, using nitrate (NO3-) as the electron acceptor; manganese and iron reduction, with Mn4+ and Fe3+, in mineral form, being the electron acceptors, respectively; sulfate reduction, using aqueous SO42-, and finally organic matter fermentation into methane (methanogenesis) (Froelich et al., 1979).
These anaerobic processes of organic carbon oxidation intimately link the burial of organic carbon to other redox-sensitive biogeochemical cycles, in particular iron and sulfur. In modern global ocean sediments, microbial iron reduction accounts for about 10% of total sedimentary organic carbon oxidation and, due to the high concentration of sulfate in the ocean, microbial sulfate reduction is responsible for over 50% of sedimentary organic carbon oxidation (Kasten and Jørgensen, 2000). Organic carbon that escapes oxidation may be fermented into methane which, as a relatively insoluble gas, migrates up through the sediment and, if not consumed along the way, is released to the atmosphere. While most of the methane produced in ocean sediments is consumed before reaching the surface (Boetius et al., 2000), in marginal marine sediments and tidal flats, much of the methane produced may escape in situ oxidation and reach the atmosphere via methane ebullition (Middelburg, 1989).
While it is tempting to think of anaerobic organic carbon mineralization as occurring in spatially distinct, largely independent zones dominated by various electron acceptors, increasing evidence has emerged that the biogeochemical cycles of iron and sulfur are tightly linked in the anoxic subsurface, and both of these cycles play key roles in the storage or release of carbon in coastal wetlands (Straub et al., 2004; Flynn et al., 2014; Sivan et al., 2014; Hansel et al., 2015). We can illustrate the complexities and interconnectedness of couplings between the sedimentary sulfur and iron cycles by following the fate of the products of microbial sulfate reduction. The end product of microbial sulfate reduction is hydrogen sulfide (H2S):
Ultimately, this reduced form of sulfur has two possible fates: burial as pyrite, or oxidation back to sulfate (Figure 1). In the presence of ferrous iron, aqueous sulfide will react quickly to form iron monosulfide (FeS) (Pyzik and Sommer, 1981). This metastable product will react with any additional available sulfide to form pyrite, releasing hydrogen (Drobner et al., 1990):
Alternatively, in the presence of an oxidant such as oxygen, nitrate, or iron and manganese oxides, the sulfide or iron monosulfide can undergo chemical or microbial oxidation, producing sulfate and/or a variety of intermediate valence state sulfur species such as thiosulfate– S2O32-, zero-valent sulfur– S0, polysulfides, or sulfite– (Jørgensen, 1990; Zopfi et al., 2004; Kamyshny and Ferdelman, 2010). For example, in anoxic sediments, hydrogen sulfide can reduce iron oxides to form ferrous iron and elemental sulfur (Canfield, 1989; Thamdrup et al., 1994; Canfield and Thamdrup, 1996):
These intermediate-valence state sulfur species produced during sulfide oxidation may undergo further oxidation, reduction, or disproportionation.
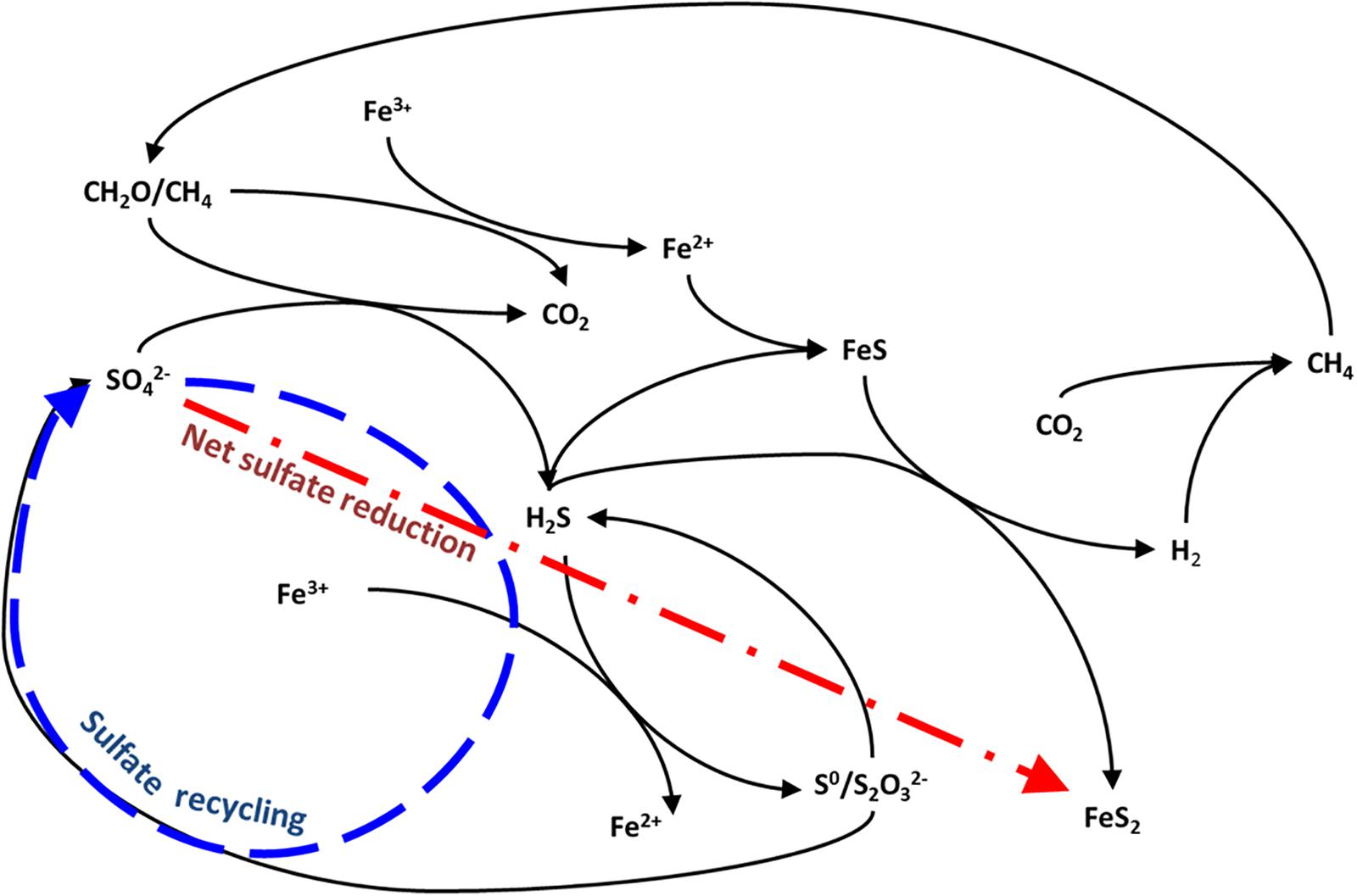
Figure 1. Possible pathways for carbon-sulfur-iron interaction in anoxic sediments after Aller et al. (2010) and Holmkvist et al. (2011). The arrows represent two possible fates for sulfide: burial as pyrite, and reoxidation to sulfate.
The sulfur and oxygen isotope ratios of sulfate are a powerful tool for tracing the redox cycling of sulfur involved in this sulfur-iron interplay (Böttcher et al., 1998, 1999; Aller et al., 2010; Antler et al., 2017). The lighter 32S isotope is preferentially reduced during sulfate reduction, thus as sulfide is produced and sequestered as iron sulfide minerals, the sulfur isotope composition of the remaining sulfate pool increases (Red line in Figure 1). Oxidation of sulfide back to sulfate returns this preferentially 32S-rich sulfur back to the sulfate pool. If this sulfur is quantitatively recycled back to sulfate there is no change in the sulfur isotopic composition of sulfate as sulfur isotopes are conserved (Blue line in Figure 1). However, the oxygen isotope composition of sulfate changes as a result of this recycling or reoxidation, because it reflects the incorporation of the oxygen isotopes either exchanged with water in intermediate valence state sulfur species or acquired from water during reoxidation. Therefore, the sulfur and oxygen isotope composition of sulfate becomes more or less decoupled depending on whether local biogeochemical conditions favor net sulfate reduction or sulfide oxidation and sulfur recycling (Figure 1; Böttcher et al., 2001; Aller et al., 2010; Mills et al., 2016; Blonder et al., 2017).
Another important control on subsurface geochemistry and carbon storage in marginal marine environments is physical reworking of the sediment, which disrupts the redox processes and linkages introduced above. Sediment reworking and pore water ventilation by flora and fauna, typically plant roots or worms, is often called bioirrigation (or bioturbation) and impacts not only physical characteristics of the sediment-water interface, but also subsurface geochemistry through sediment mixing. Thus, bioturbation has been called “ecosystem engineering” (Meysman et al., 2006; Kristensen et al., 2012). Bioturbation has been suggested to have shaped Earth’s surface redox state through the recycling of iron sulfide minerals, leading to an increase in marine sulfate concentrations (McIlroy and Logan, 1999; Canfield and Farquhar, 2009). The conventional view on bioturbation is that ventilation retards and thereby stretches the zonation of anaerobic microbial respiration, pushing classic redox zones deeper into the sediment (McIlroy and Logan, 1999), thereby increasing the oxidation of reduced compounds such as ferrous iron, aqueous sulfide and iron sulfide minerals (Canfield and Farquhar, 2009). Ultimately it is the interconnected web linking the sedimentary carbon, iron and sulfur redox cycles that is perturbed when sediment reworking changes, even subtly, the redox state of sedimentary fluids. Understanding the impact and feedbacks that bioturbation has on redox sensitive elements, and long term burial or storage of organic carbon, remains challenging.
Salt marshes present a unique opportunity to study the sedimentary cycles of sulfur and iron, the influence of bioturbation on these cycles, and the impact of different redox pathways on carbon stability. Salt marsh sediments have high concentrations of sulfate from daily or monthly inundation with seawater, and most coastal wetlands have exceptionally high concentrations of iron oxides, due to metal trapping in marginal marine environments (Luther et al., 1982, 1992; Luther and Church, 1988; Thamdrup et al., 1994; Canfield and Thamdrup, 1996; Tobias and Neubauer, 2009; Burton et al., 2011; Mortimer et al., 2011; Johnston et al., 2014; Mills et al., 2016; Hutchings et al., 2019). Salt marsh sediments have high levels of organic carbon consumption (from the large quantity of vegetation present) and high levels of bioturbation and bio-irrigation – indicated by the presence of worm burrows at, or near, the sediment interface. As such, these salt marsh systems are ideal places to study the effect of bioturbation on geochemical processes.
In this paper we explore the in situ effect of bioirrigation on the interplay between sulfur and iron and how it is related to the fate of organic carbon in salt marsh sediments. Recently, results from a multi-year coring campaign in the North Norfolk (East Anglia) salt marsh pond sediments were published, along with drone images mapping out the distribution of various pond types and looking for evidence of pond sediments dominated by the sulfur cycle versus those that were dominated by the iron cycle (Hutchings et al., 2019). In this work, it was suggested that one control on the type of sub-surface sediment geochemistry was the position of the pond on the salt marsh platform and therefore the frequency of flushing with oxygenated seawater (Hutchings et al., 2019). Here we present additional sediment cores collected across East Anglia at different times of the year, and one core taken 5 years later and explore an additional control on the subsurface geochemistry; the role of bioirrigation. We report sediment and pore fluid geochemistry, including the concentration of key elements and the sulfur and oxygen isotopes of the sulfate and sulfide. We couple these field measurements with slurry incubations, where we incubate sediments from two depths within the marsh to understand the geochemical evolution of the sediment in the laboratory. We then present a conceptual model for how redox processes are operating and interlinked in these salt marsh sediments.
Six sediments cores were collected from the Stiffkey and Blakeney salt marshes on the north Norfolk coast and the Abbotts Hall salt marsh in Essex between the spring and the fall of 2014 (Map – Supplementary Figure S5), using 50 cm polycarbonate push cores. A further core was taken from Abbotts Hall in 2018. Pore water was extracted using Rhizon® samplers, a syringe-based sampler with an inert polymer membrane that allows high-resolution sampling of sedimentary pore fluids. Rhizon samplers have been shown to be reliable for aqueous phase samples but not as reliable for species such as dissolved carbon dioxide and aqueous sulfide which may degas during sampling. The sampled pore water was filtered (0.2 μm filter) upon extraction; subsamples were taken for dissolved sulfide, sulfate, chloride, and iron concentrations. In addition, subsamples for δ13CDIC and DIC (0.5 mL) were transferred into a capped syringe. The remaining pore fluids were fixed with zinc acetate (ZnAc, 20%) for sulfate concentration and δ18OSO4 and δ34SSO4 analysis. All pore fluid samples were stored at 4°C until measured. For methane concentrations, ∼2 ml of the sediment was sampled using an edge cut syringe and transferred into an N2 flushed bottle containing 5 mL sodium hydroxide (1.5 N) and the bottle was sealed with a crimper.
An additional core was taken for solid phase analysis. The core was brought back to the lab and split vertically. One half of the core was immediately taken to an X-Ray Fluorescence (XRF) Core Scanner to determine the elemental composition of a sample based on the re-emission of secondary X-rays after excitation by a primary X-ray source. Within the other half of the core, sediment samples were taken to determine the concentration of reactive iron, total organic carbon (TOC, through combustion in an element analyzer) and δ34S of Acid Volatile Sulfide (AVS) and Chromium Reducible Sulfide (CRS). For highly reactive Fe(III) oxide measurements, 50 mL of HCl (0.5 M) was added to an 0.5 g sediment sample. Tubes were shaken for 1 h and centrifuged. The supernatant was filtered and iron concentrations were measured on an ICP-OES (Agilent 5100 ICP-OES, University of Cambridge). For acid-volatile sulfur (AVS) and chromium reducible sulfur (CRS), 5 ml of sediment was placed in a 15 mL Falcon tube containing 10 mL zinc acetate in the field (50 g L-1). The sediments were then subjected to a two-step distillation. AVS was extracted over 2 h by boiling in 6 M HCl, followed by an hour boiling with 1 mol L-1 acidic CrCl2 solution to extract CRS. Resulting H2S was trapped with silver nitrate solution. For porosity measurements, 2 mL of sediment was weighed before and after drying.
Worm density was measured by sieving a liter of sediment with a mesh strainer and counting the number of worms. This was repeated in several different ponds (six iron rich and four sulfide rich ponds).
A series of sediment incubation experiments were carried out to further explore iron-sulfur dynamics in an iron-rich core taken from Warham salt marsh in March, 2014. A total of five slurry batch reactors were created by incubating sediments taken from either the discernibly “black” layer (upper 5 cm of core) or from gray-tinted sediments (at 25 cm depth). 10 mL sediment samples were incubated in 100 mL of filtered salt marsh pond water; four of the five vials were incubated in 18O-enriched pond water (δ18OH2O 400‰). High δ18OH2O amplifies the δ18OSO4 signal that may be seen if sulfate recycling occurs in the incubation. One of the samples with sediment from 5 cm depth was autoclaved to kill any microbes present in the sediment and act as an experimental control. All samples were incubated in anoxic conditions, under a N2 atmosphere. Additional experimental setup information and measurement is described by Mills et al. (2016).
Sulfate and chloride concentrations were measured by ion chromatography (IC, Dionex DX-500, University of Cambridge) with an error of 2% between duplicates. Concentration of DIC was measured according to the peak height and calibration curve on a Gas Source Isotopic Ratio Mass Spectrometer (GS-IRMS, Thermo at Ben Gurion University of the Negev, Israel) with an error of 0.2 mM. For this analysis, a 1 mL headspace sample was taken from the crimped vial with a gas-tight pressure lock syringe after the bottle was shaken vigorously. The δ13CDIC was on the same GS-IRMS equipped with a Gas Bench II (GBII) interface with an error of 0.1% (reported versus the Vienna Pee Dee Belemnite (VPDB) standard). Methane was measured from the headspace on a Focus Gas Chromatograph (Thermo) with a ShinCarbon column with precision of 2 μM (After Sela-Adler et al., 2015).
Pore fluid sulfate was precipitated as barium sulfate (barite) using a saturated barium chloride solution. The barite was then washed once with 6 N HCl and twice with deionized water. For the analysis of δ18OSO4, barite was pyrolysed at 1450°C in a temperature conversion element analyzer (TC/EA), producing carbon monoxide. Carbon monoxide was measured by continuous helium flow in a GS-IRMS (Thermo Finnegan Delta V Plus, at the Godwin Laboratory, University of Cambridge). To analyze δ34S, barite or silver sulfide were combusted at 1030°C in a flash element analyzer (EA), and the resulting sulfur dioxide (SO2) was measured by continuous helium flow on a GS-IRMS (Thermo Finnegan Delta V Plus Godwin Laboratory, University of Cambridge). Analyses of δ18OSO4 were conducted in replicates (n = 3–5) and the standard deviation of these replicate analyses is reported (∼0.4% 1σ). The error for δ34S was determined using the standard deviation of multiple standards (n = 6) at the beginning and the end of each run (∼0.3% 1σ). Measurements of δ18OSO4 were corrected to NBS 127, IAEA-SO-6, and IAEA-SO-5 (8.6%, -11.35%, and 12.1%, respectively). Measurements of δ34S were corrected to NBS 127, IAEA-SO-6, IAEA-SO-5, and IAEA-S-3 (20.3%, -34.1%, 0.5%, and -32.4%, respectively). The δ18OSO4 is reported relative to the Vienna Standard Mean Ocean Water (VSMOW) and δ34S is reported with respect to Vienna Canyon Diablo Troilite (VCDT).
Our sampling presented here and in recent work demonstrates that sediments in salt marshes across East Anglia in the United Kingdom can be divided into two types: iron-rich and sulfide-rich sediment (Figures 2, 3; Hutchings et al., 2019). In Figure 2 we present one iron-rich pond and one sulfide-rich pond sampled in Norfolk in October 2014, for which we have complete datasets including the concentration of pore fluid DIC and its carbon isotope composition (δ13CDIC). Figure 3 shows partial datasets from five further cores taken at different sampling times (four throughout 2014 and a further core taken in 2018) and locations (both Norfolk and Essex) – and includes the concentration of dissolved iron, aqueous sulfide, sulfate, and the sulfur and oxygen isotope composition of sulfate. A further 16 pore fluid profiles are presented in Hutchings et al. (2019), but are not duplicated here. Porewater data in Figures 2, 3, and in the Hutchings et al. (2019) larger dataset are remarkably similar despite the vast temporal and spatial differences in sampling times and locations (>100 km and ∼4 years). Iron-rich sediments (circles — Figure 2, bottom panel Figure 3) are characterized by high pore fluid ferrous iron concentrations (up to 1.5 mM). In these sediments, sulfate concentrations and δ34SSO4 remain constant with depth and both dissolved sulfide and methane concentrations are below detection limits. However, δ18OSO4 shows significant increases of up to 5% below 10 cm depth. In addition, the concentration of DIC increases from 4.2 mM at the sediment surface to 6.9 mM at 27 cm depth while the δ13CDIC decreases from -5.8% to -9.7% over the same depth interval. In contrast, in sulfide-rich sediments (Squares—Figure 2, top panel Figure 3) the pore fluid geochemistry is characterized by high sulfide concentrations (up to 5 mM), depletion of sulfate, the presence of methane (up to 0.4 mM), and a complete lack of ferrous iron. Unlike in the iron-rich sediments, both sulfur and oxygen isotopes in sulfate increase with depth. From the sediment surface to ∼30 cm depth, the concentration of DIC increases from 15.7 mM to 35 mM while the δ13CDIC decreases from -10.3 to -12.6%.
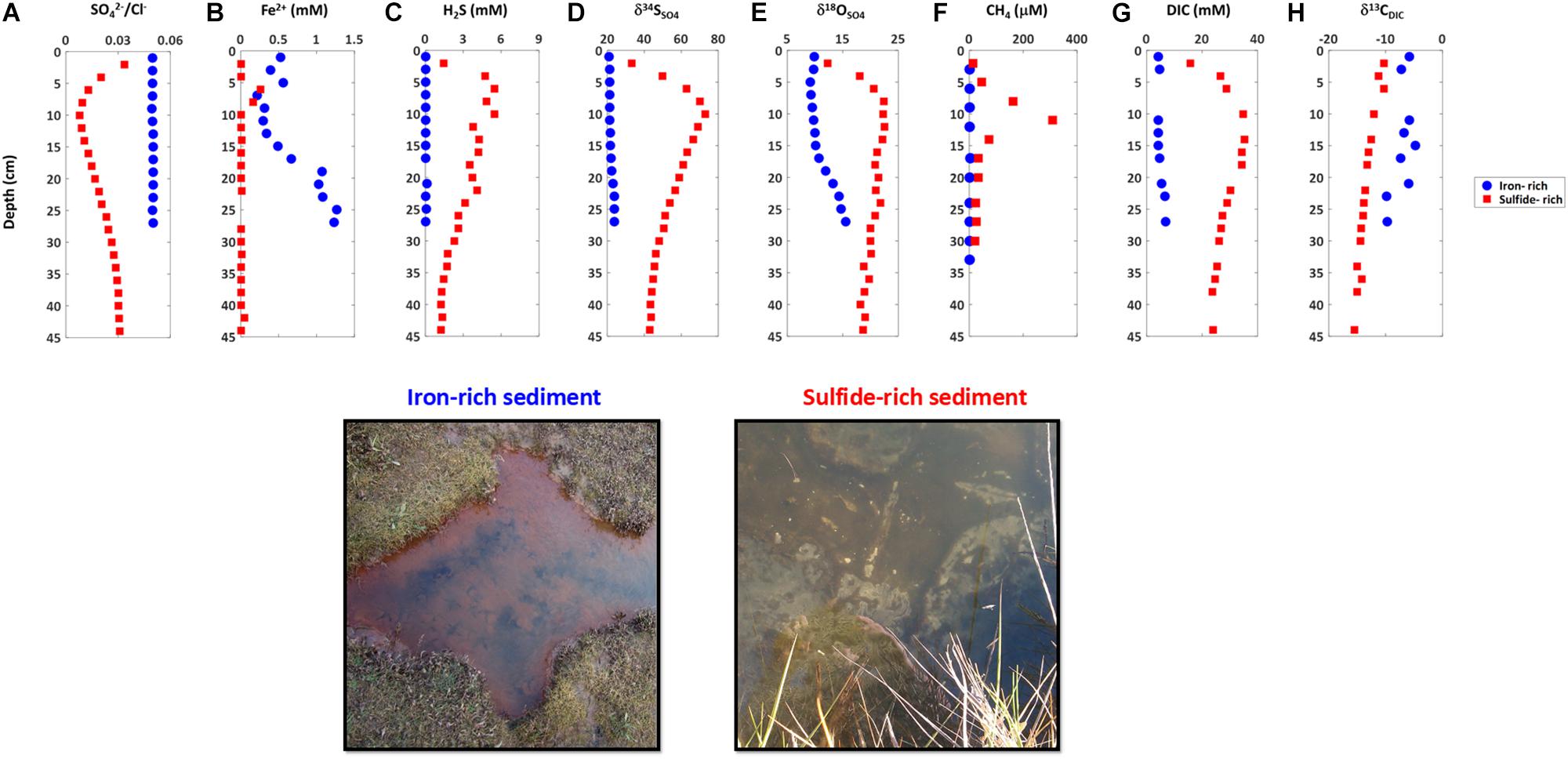
Figure 2. Pore water chemical profiles and picture of the surface sediment of iron-rich (blue circles) and sulfide-rich sediment cores (red squares) taken from Norfolk in October 2014. (A) Sulfate to chloride ratio, (B) ferrous iron, (C) sulfide, (D) sulfur isotope ratio in sulfate, (E) oxygen isotope ratio in sulfate, (F) methane, (G) dissolved inorganic carbon, (H) carbon isotope ratio in dissolved inorganic carbon.
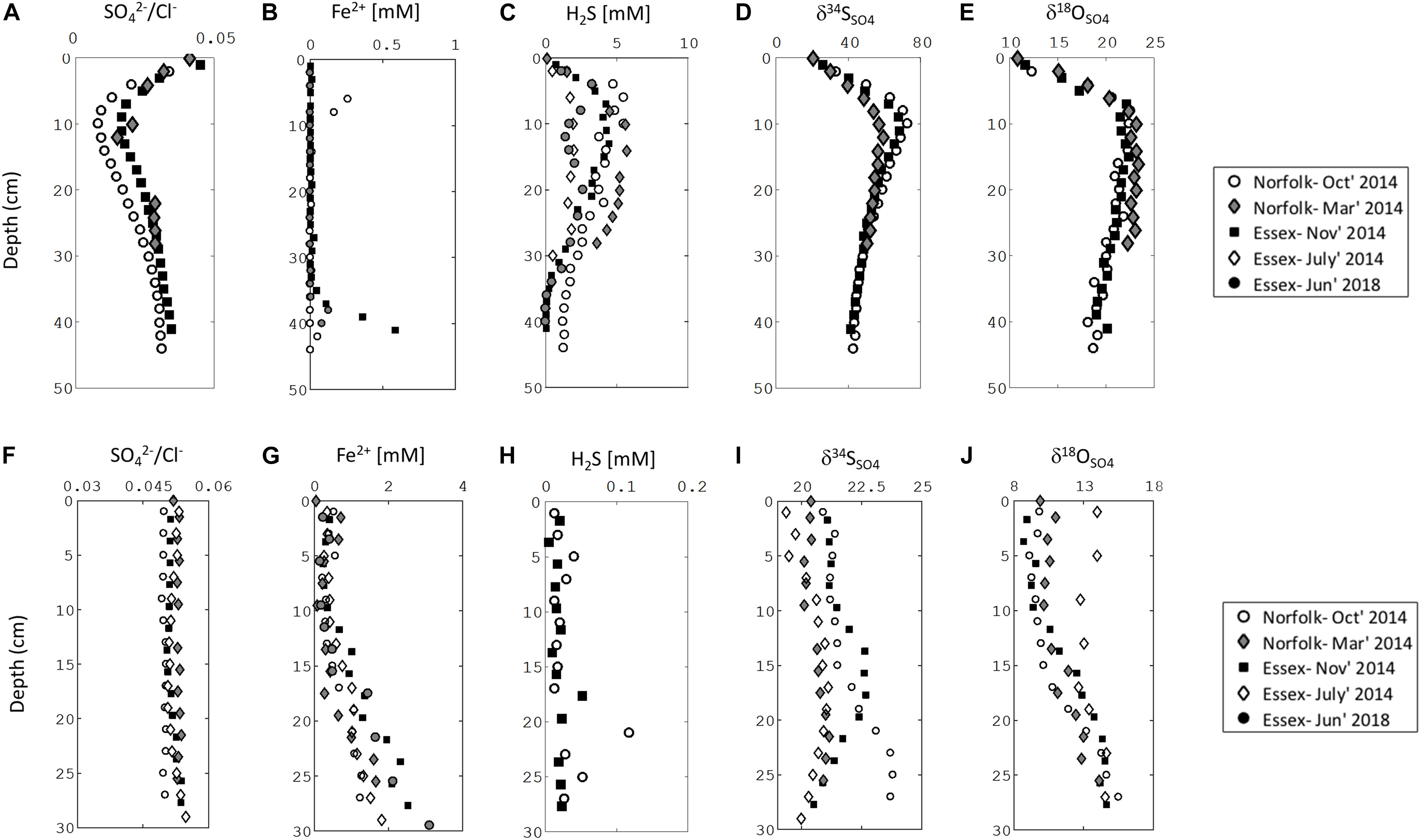
Figure 3. Pore water chemical profiles of 3 iron-rich and 3 sulfide-rich sediment cores taken from Norfolk and Essex. Sulfide-rich sediment: (A) sulfate to chloride ratio, (B) ferrous iron, (C) sulfide, (D) sulfur isotope ratio in sulfate, (E) oxygen isotope ratio in sulfate. Iron-rich sediment: (F) sulfate to chloride ratio, (G) ferrous iron, (H) sulfide, (I) sulfur isotope ratio in sulfate, (J) oxygen isotope ratio in sulfate.
Observationally, we note that the surface of iron-rich ponds often differed from that of sulfide-rich ponds (Figure 2, photo), with the iron-rich ponds characterized by ferric iron coloration and frequent worm burrows and the sulfidic ponds frequently associated with biofilms of sulfur-oxidizing bacteria. These observations have been used to classify ponds over large spatial areas (Hutchings et al., 2019).
Marked differences between the iron-rich and sulfide-rich sediments are also observed in the solid phase (Figure 4). We observe an increase in highly reactive iron (extracted with 0.5 M HCl) in the upper 10 cm (up to 14 ppm- Figure 4A) of iron-rich sediments. Below this depth highly reactive iron decreases to between 4 and 5 ppm (Figure 4A). In sulfide-rich sediments, highly reactive iron is constant at 6 ± 1 ppm throughout the sediment column (Figure 4A). Similar trends appear from XRF measurements of iron (normalized to titanium) (Figure 4B). XRF-measured sulfur concentrations (again normalized to titanium—Figure 4C) are constant in the upper 15 cm for both sulfide-rich and iron-rich sediments. Below 15 cm sulfur concentrations decrease in the iron-rich sediments and remain constant in the sulfide-rich sediments. Total organic carbon (TOC—Figure 4D) gradually decreases in sulfide-rich sediments, from about 10% at 3 cm to 5%, at 30 cm. In iron-rich sediments there is less TOC, and it remains constant (6 ± 0.5%) over the upper 20 cm, gradually decreasing to 2% at 35 cm. The sulfur isotopic composition of the AVS and the CRS in the iron-rich core are similar, around -27 ± 5%. In contrast, in the sulfide-rich core the δ34SAV S gradually decreases from -5% near the sediment surface to -15% at 25 cm depth while the δ34SCRS decreases from -20% within the upper 20 cm to -30% at the deepest sediments (>30 cm depth) (Figure 4E).
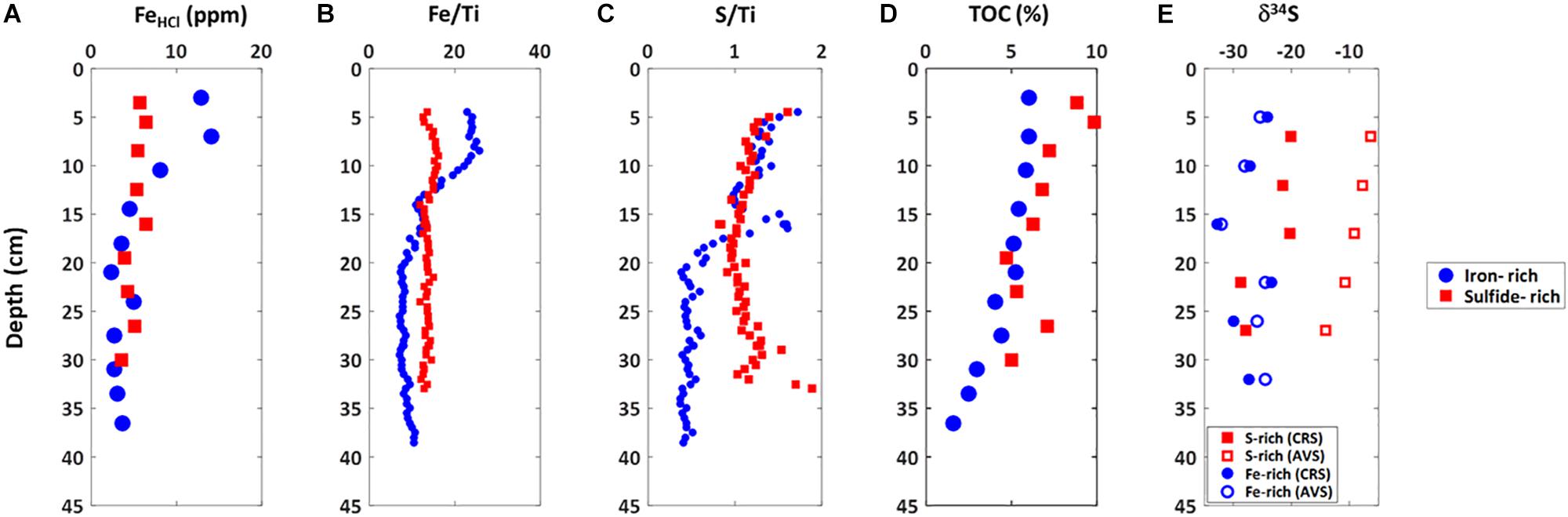
Figure 4. Solid phase profiles of iron-rich (blue circles) and sulfide-rich sediment (red squares) cores taken from Norfolk in October 2014. (A) Reactive iron, (B) Iron to titanium ratio, (C) Sulfur to titanium ratio, (D) total organic carbon, (E) Sulfur isotope ratio in AVS and CRS.
Ferrous iron concentration increased in the slurry experiments in the sediment incubated from 5 cm and 25 cm depth over the first 30 days (Figure 5). However, after 30 days, the ferrous iron concentrations decreased in the slurry taken from 5 cm depth while they increased steadily in the slurry with sediments taken from 25 cm, up to 0.4 mM. The ferrous iron concentration in the control (autoclaved) experiment remained constant throughout the experiment at 0.05 mM. Sulfate concentrations remained constant within error for both the control and the 25 cm depth sediment incubation, while they decreased, from 25 mM to 20 mM, in the incubation with sediment from 5 cm suggesting the onset of microbial sulfate reduction in the sediment incubated from 5 cm depth. The sulfur isotopes in sulfate (δ34SSO4) mirrored sulfate concentrations, increasing about 7% in the slurry with sediment from 5 cm. The oxygen isotopes of sulfate (δ18OSO4) increased in the slurry experiments from both depths, increasing from 9‰ to 2‰ (5 cm) and 9‰ to 100‰ (25 cm) when incubated with 18O-enriched water. No change was observed in the δ18OSO4 in the control experiment.
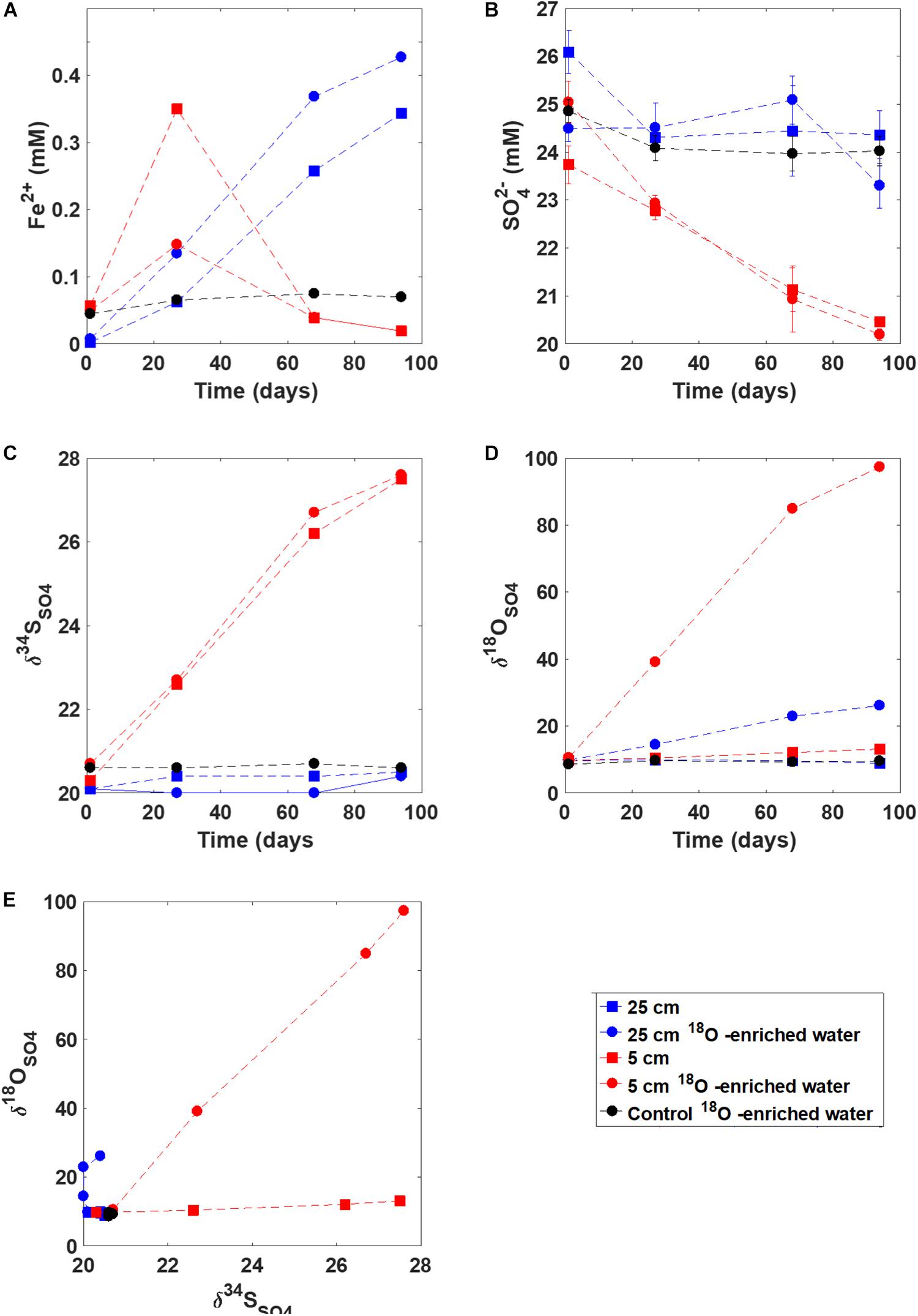
Figure 5. Results from the slurry experiment. Panels (A–D) show the evolution of (A) ferrous iron concentration, (B) sulfate concentration, (C) d34SSO4, and (D) d18OSO4; Panel (E) shows the d18OSO4 vs. d34SSO4 cross-plot for these experiments.
Our cores were taken from ponds that are sometimes only a few meters apart (See Google Earth image in Supplementary Figure S1; see further examples in Hutchings et al. (2019)). However, the bi-modal distribution of geochemical behavior described above shows geographic and temporal consistency (Hutchings et al., 2019). Cores taken from Essex salt marshes (160 km from the Norfolk salt marshes) are geochemically identical to those taken from Norfolk, as are cores taken from the same Norfolk location during different times of the year (Figure 3). When we revisited the same ponds in Essex 4.5 years later we found that the subsurface geochemistry did not change in any significant manner (Figure 3).
In our study area, we found that in iron-rich sediments worm density (Polychaete worm (Nereis spp.)) was 3 ± 3 (n = 6) worms per liter of sediment in the upper 5 centimeters of sediment (or 150 worms-per m2) and 9 ± 2 (n = 6) worms per liter of sediment between 5 cm to 15 cm (or 900 worms per m2). No worms were observed in sulfide-rich sediments at any depth. Further evidence for ongoing burrowing was observed on the sediment surface of iron-rich ponds which typically contained mounds resulting from bioturbation (see photos in Supplementary Figure S2). Worm burrows surrounded by rust-colored rims were also observed in iron-rich sediment cores (Supplementary Figure S2). Polychaete worms (Nereis spp.) can grow to a length of several centimeters and are the most dominant burrowing macrofauna in these and other shallow salt marsh pond sediments. Along with vegetation nearby the ponds and associated rhizosphere, worms perturb local subsurface redox geochemistry by introducing oxygenated waters through their burrowing behavior.
This burrowing process was reflected in the pore fluid geochemistry, which in the uppermost 15 cm in the iron-rich ponds was geochemically invariant, as expected in a mixed, well-aerated sediment (Figures 2, 3 and Supplementary Figure S3). Geochemical and isotopic analyses in the iron-rich sediment showed a kink in the pore fluid profile at around 15 cm (Figure 3), including in species that act purely as mixing tracers such as chloride (Supplementary Figure S3). We therefore divide the iron-rich sediments into an upper part of the sediment that appeared to be well mixed and aerated, either by bioturbation or advection, and a deeper zone in which a limited subset of geochemical characteristics deviated from surface conditions. Conversely, in sulfide-rich sediments, no worms were found and there were no regions of the sediment that displayed uniform geochemical characteristics and behavior.
In the Hutchings et al. (2019) paper, there was a detailed discussion of the role of subsurface hydrology and surface nutrient supply to driving the geochemical heterogeneity in the salt marsh pond sediment system. Here we summarize our thoughts on the impact of hydrology and local geology, given that it is key to resolve how much variation we observe in the pore water chemistry is due to local biological and chemical processes and how much is due to regionally complex hydrological dynamics driving heterogeneity within sediments across broad regional zones. Salt marshes are hydrologically dynamic environments, exposed to evaporation and precipitation, as well as frequent flooding and subsurface inundation by seawater tides (Gardner, 1973; Martin, 1997). Temperature and average surface wind speeds can change throughout the year, altering evaporation rates (Abdollahi and Nedwell, 1979; Hines et al., 1989), and tidal pumping can lead to complicated advective flow patterns.
Furthermore, it is our contention that the delivery of iron and other nutrients, as well as seawater, will vary across East Anglia. The nearby Crag formation, which is where most of the sediment in East Anglia is sourced, is highly rich in iron. Furthermore the Cretaceous Carstone formation both underlies and outcrops throughout East Anglia. So while there are sources of iron in outcrop throughout East Anglia, it is certain that the delivery of reactive iron to various coastal wetlands around East Anglia will vary. North Norfolk likely gets different amounts of riverine and terrestrial input of iron than Essex, and this should vary throughout the year. Furthermore, the southern part of East Anglia receives different storm surges and different tidal heights than the northern part of East Anglia, suggesting that the terrestrial and seawater input to each coastal wetland is likely different.
In spite of these differences cores taken from sulfide-rich sediment and from the iron-rich sediment are remarkably similar, despite significant spatial and temporal separation (Figure 3). Given the likely differences in regional hydrology and geology, it is surprising that sediments separated by hundreds of kilometers, as between the Norfolk and Essex salt marshes, show near-identical pore water geochemistry, whilst sediment cores taken only a few meters apart can be so markedly different. We propose that the subsurface geochemistry in both locations is shaped by a set of common, fundamental processes independent of regional nutrient (e.g., iron) supply or local salt marsh hydrology.
In the iron-rich sediments the dominant process controlling the subsurface geochemistry is microbial iron reduction. The elevated sedimentary iron and sulfur concentrations (Figure 4 – and often observed as a black layer near the top of the core) and the presence of both chromium-reducible and AVS (with low δ34S) indicate that microbial sulfate reduction also contributes to organic matter mineralization in the near-surface zone of iron-rich sediments, ironically given they are “iron-rich”. The activity of microbial sulfate reduction in surface layers of iron-rich sediments is supported by our slurry experiments. When sediment from the near-surface black layer was incubated (without oxygen), microbial sulfate reduction began quickly (indicated by the drop in ferrous iron and sulfate concentrations coupled to increases in δ34SSO4 and δ18OSO4 – Figure 5). In the natural environment, however, in iron-rich salt marsh sediments any sulfide that would be formed through microbial sulfate reduction is quickly re-oxidized, or is sequestered via titration with the measurable excess of ferrous iron. We observe that the black layer of sediment often occurs at depths where pore water is ventilated, either through worm burrows, or by vegetated root systems accessing salt marsh ponds from the verges. This ventilation continually introduces oxygen to the sediment, recycling iron and sulfide back to their oxidized forms, Fe(III) and sulfate.
In the sulfide-rich sediments, the depletion of sulfate and the increase in hydrogen sulfide, along with the increase in the δ34SSO4, support the dominance of microbial sulfate reduction controlling the subsurface geochemistry. Sediment methane concentrations suggest the relative rate of methanogenesis to methanotrophy peaks at 12 cm (Supplementary Figure S4), while sulfate concentrations are still high, a process which has been observed in other organic-rich systems (e.g., Dale et al., 2008; Sela-Adler et al., 2017). Although methanogenesis is often accompanied by an increase in δ13CDIC, in our case the methane concentration is two orders of magnitude smaller than the concentration of DIC and therefore has little effect on the δ13CDIC.
As mentioned in the introduction, we can use the sulfur and oxygen isotope composition of sulfate to explore sulfur redox cycling in these sediments. Notably, the sulfur and oxygen isotope composition of pore fluid sulfate is remarkably different between the sulfide-rich and iron-rich pond sediments. The relationship between δ18OSO4 and δ34SSO4 has been shown to reflect the pathway by which sulfur is reduced and reoxidized in pore fluids, pure cultures, and other environmental settings (Böttcher et al., 1998, 2005; Aharon and Fu, 2000, 2003; Böttcher and Thamdrup, 2001; Brunner et al., 2005, 2012; Turchyn et al., 2006, 2010, 2016; Pellerin et al., 2018). The consensus is when δ18OSO4 and δ34SSO4 are cross plot, the relative slope between δ18OSO4 and δ34SSO4 reveals the dynamics of microbial sulfate reduction – the higher the slope between δ18OSO4 and δ34SSO4, the lower the sulfate reduction rate, and the higher amount of sulfur recycling (Böttcher et al., 1998, 1999; Brunner et al., 2005, 2012; Wortmann et al., 2007; Antler et al., 2013).
Our data can be placed in a global context by plotting the δ18OSO4 vs. δ34SSO4 from our iron-rich and sulfide-rich cores with data from other global locations, including gas seeps from the Levantine basin and data from deep ocean sediment (Figure 6; Rubin-Blum et al., 2014a). The data from gas seeps are characterized by high methane concentrations (∼2 mM) and relatively fast depletion of sulfate with depth (∼mM/cm). The deep sea sediments (from ODP site 1225 in the Peru Basin, 3760 m water depth) are characterized by low organic carbon content (0–0.9%) and low levels of dissolved ferrous iron (20 μM) (Blake et al., 2006; Böttcher et al., 2006). We also plot compiled data from 22 marine and marginal-marine sites (gray dots, Figure 6).
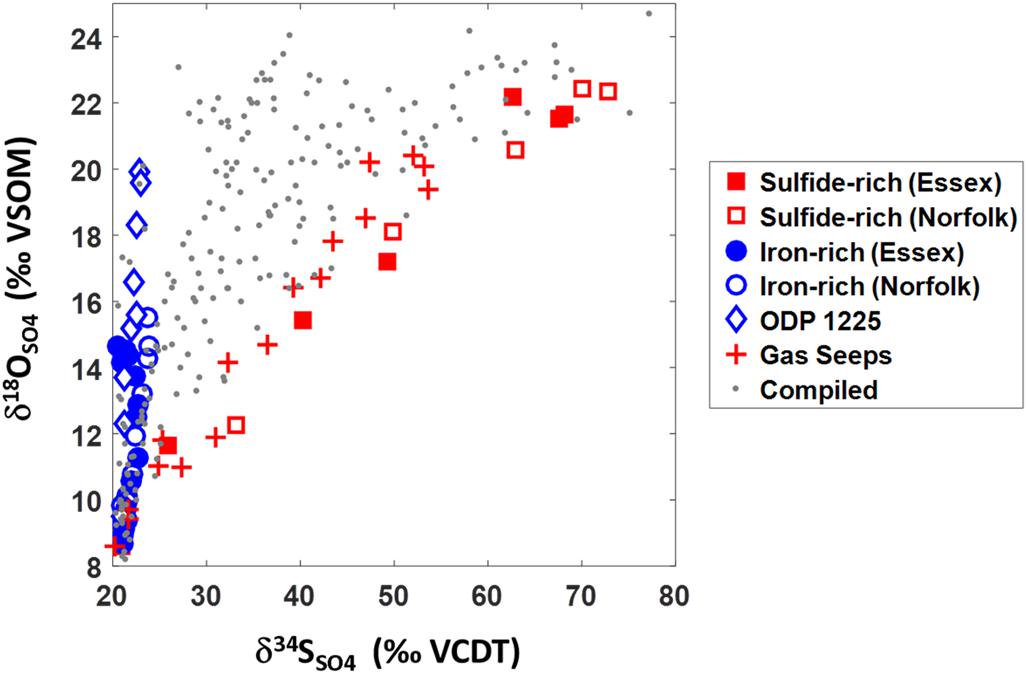
Figure 6. δ18OSO4 vs. δ34SSO4 data from this study together with data from gas seeps (Rubin-Blum et al., 2014a), ODP site 1225 at the Peru Basin (Blake et al., 2006; Böttcher et al., 2006), and compiled data from 22 marine and marginal-marine sites (Böttcher et al., 1998, 1999; Aharon and Fu, 2000, 2003; Turchyn et al., 2006, 2016; Aller et al., 2010; Antler et al., 2013, 2014, 2015).
The δ18OSO4 vs. δ34SSO4 results from our reported salt marsh pore fluids represent end-members that encompass the entire range of δ18OSO4 vs. δ34SSO4 results from all previously reported marine sediments. Over vast differences in environmental settings (from sulfide-rich salt marsh ponds to gas seeps and from iron-rich salt marsh ponds to deep-sea sediments) the sulfur versus oxygen isotope cross-plots are virtually identical (Figure 6). The similarity between our collected iron-rich salt marsh sediments and deep-sea ODP Site 1225 combined with the similarity between our collected salt marsh sulfide-rich sediments and shallow marine gas seeps suggests that similar processes dominate sulfur redox cycling in each environment. From the trajectories in the δ18OSO4 vs. δ34SSO4 space we deduce that in the iron-rich sediment, a high percentage of the sulfate that is reduced is ultimately re-oxidized (“sulfate recycling”— blue line, Figure 1; Böttcher and Thamdrup, 2001; Böttcher et al., 2001, 2005; Aller et al., 2010; Riedinger et al., 2010; Mills et al., 2016). The δ18OSO4 vs. δ34SSO4 trajectory in the sulfide-rich sediment in turn indicates high rates of sulfate reduction with only a negligible portion of sulfate reoxidized (“net sulfate reduction” – red line, Figure 1; Antler et al., 2014, 2015; Antler and Pellerin, 2018).
Our laboratory incubation study provides additional insight into the type of sulfur redox cycling occurring in the iron-rich pond sediments. First, when the upper part of the sediment (5 cm below the sediment-water interface) was incubated in a closed bottle, sulfate concentrations quickly decreased and the δ34SSO4 increased (in a matter of days). This strongly suggests that the capacity for microbial sulfate reduction is present in the upper part of the iron-rich sediment yet there is no discernible evidence of active sulfate reduction in the pore water geochemistry of the natural environment. Second, when the deeper part of the sediment (25 cm) was incubated, δ18OSO4 increased substantially while δ34SSO4 remained constant. Thus, the pattern in δ18OSO4 vs. δ34SSO4 was similar to that observed in the iron-rich sediment pore water, with a very steep slope indicative of slow rates of microbial sulfate reduction and near complete sulfur recycling. This indicates that the full range of δ18OSO4 vs. δ34SSO4 slopes (defining local sulfur metabolism) can be generated in situ, and are more likely to be determined by in situ conditions than by microbial metabolic capacity.
As mentioned above, the differences we observe in the sediment geochemistry over small spatial scales (<5 m) are not readily explained through surface environmental conditions such as temperature, sediment export, or nutrient supply. While these factors necessarily vary over the East Anglian region, there is no evidence to suggest that they would be significantly different at small spatial scales, yet similar across radically different and largely geographically separated environments and over multiple seasons and years. Indeed, we note that the total sedimentary iron concentration as measured by core-scanning XRF is the same in the sulfide rich and iron rich cores, suggesting they start with similar inputs and then evolve in different geochemical directions (Figure 4B). In order to explain these behavioral changes over such small distances we propose that the subsurface geochemistry in these ponds represent two different stable (or metastable) states of carbon-sulfur-iron interaction. This bistable system must be driven by feedback reactions that determine ultimately if the sediment will be sulfide-rich, with high organic carbon content and methane production, or iron-rich, and relatively organic carbon poor.
Here we present a conceptual model exploring how we think bioirrigation influences the redox geochemistry in the East Anglian salt marsh sediments. It has been demonstrated in many different environments that bioirrigation impacts the rates and pathways of organic matter oxidation coupled either to iron or sulfate reduction and acts to redistribute iron and sulfur in the solid phase (e.g., Goldhaber et al., 1977; Aller, 1980; Berner and Westrich, 1985; Kostka et al., 2002; Wijsman et al., 2002; Canfield and Farquhar, 2009; Aller et al., 2010; Severmann et al., 2010; Kristensen et al., 2014; Rubin-Blum et al., 2014b; Dale et al., 2015; Quintana et al., 2015; van de Velde and Meysman, 2016; Vasquez-Cardenas et al., 2016). In our study area, we find evidence for bioturbation only in the iron-rich sediment and not in the sulfide-rich sediments. Over larger spatial scales, 50% of iron-rich ponds (n = 140) had surface evidence of this bioturbation (Hutchings et al., 2019). We suggest that this process plays a role in shaping the interplay between carbon, sulfur, and iron in these sediments.
Although in this paper we have presented circumstantial evidence that bioturbation plays a role in dictating subsurface geochemistry (iron-rich or sulfide-rich), it is not clear why bioturbation would “activate” one type of pond and not the other. One possibility is differences in sedimentation rate (Boudreau, 1994), however, it is hard to imagine how this would vary dramatically and systematically over the short (<5 meter) distances between ponds. A more defensible hypothesis is that in iron-rich sediments the supply of ferrous iron, transiently, outcompetes the supply of aqueous sulfide. This ferrous iron can react quickly with any hydrogen sulfide that is produced, sequestering it as iron monosulfide. As hydrogen sulfide is toxic to many eukaryotes, the excess of ferrous iron effectively detoxifies the sediment, enabling bioturbation. This results in a renewed supply of oxygen (and sulfate) from the overlying oxygenated pond water and allows DIC that has been produced in the sediment through organic carbon oxidation to escape across the sediment-water interface. This worm-driven bioirrigation thus helps to oxidize ferrous iron, regenerating ferric iron which, when buried below the oxic zone, allows further bacterial iron reduction to occur, producing more ferrous iron to continue to titrate any sulfide produced. The oxidation of ferrous iron with oxygen will outcompete aerobic respiration, and any remaining oxygen will be used to sustain aerobic respiration in the upper, ventilated part of the iron-rich ponds (Burdige, 1993). The end result of this bioturbation-driven, iron-dominated sediment cycle is the enhanced oxidation of organic carbon in the upper part of the sediment, while in the lower part of the sediment, iron reduction dominates along with the previously proposed “cryptic sulfur cycling” in East Anglian salt marshes (Mills et al., 2016). This yields a sedimentary system that is dominated by iron reduction and therefore contains iron-rich subsurface conditions. The theoretical outcomes of this bioturbation and ferrous iron-driven process fits all observations for these iron-rich sediment types. Overall, the turnover of organic carbon is much faster in iron-rich sediments due to enhanced ventilation, which is supported by the lower levels of sedimentary organic carbon (TOC, Figure 4), (DIC, Figure 2), and relatively 13C enriched DIC (high δ13CDIC, Figure 2) in the iron-rich sediments.
On the other hand, in sulfide-rich sediments, the supply of hydrogen sulfide from microbial sulfate reduction outcompetes the delivery of iron. As a result, hydrogen sulfide concentrations increase and the sediment becomes poisonous to bioturbating organisms. The lack of ventilation allows microbial sulfate reduction to proceed and prevents reoxidation of reduced iron. Any iron that does reach the sediment rapidly precipitates as pyrite with excess sulfide, and this precipitation produces hydrogen gas (Drobner et al., 1990) which reacts with CO2 to fuel hydrogenotrophic methanogenesis (Whiticar et al., 1986). This methane is consumed partly or completely via sulfate-driven anaerobic oxidation of methane.
Bioturbation-driven alternate environmental states with respect to the sulfur-iron-carbon interplay have been suggested and investigated over the past few years (Wijsman et al., 2002; Aller et al., 2010; van de Velde and Meysman, 2016). Figure 7 summarizes the two proposed cycles for sulfur, iron and carbon in the iron-rich and sulfide-rich sediments. Each of the two cycles are stand-alone and together they comprise the diagram presented in Figure 1A. The driving force in cycling within the iron-rich sediment is bioturbation, which replenishes the oxygen and hence replenishes the ferric iron concentrations. In the sulfide-rich sediment, methane is the driving force of the cycle and its formation supports sulfate-driven anaerobic oxidation of methane. We suggest that the organic matter remineralisation rate in the iron-rich sediment (Figure 7A) is more rapid than in the sulfide-rich sediment (Figure 7B) since it is driven by pore water ventilation. This ventilation brings more oxygen into the sediment, which in turn reoxidizes ferrous iron to reactive ferric-iron oxides. This cycle leaves the sediment over more organic-carbon poor but with a replenished iron-oxide pool and generally speaking, more oxidized. The more rapid organic carbon respiration is not seen in the pore water geochemistry due to rapid replacement (or ventilation) of the pore water with the overlying pond water.
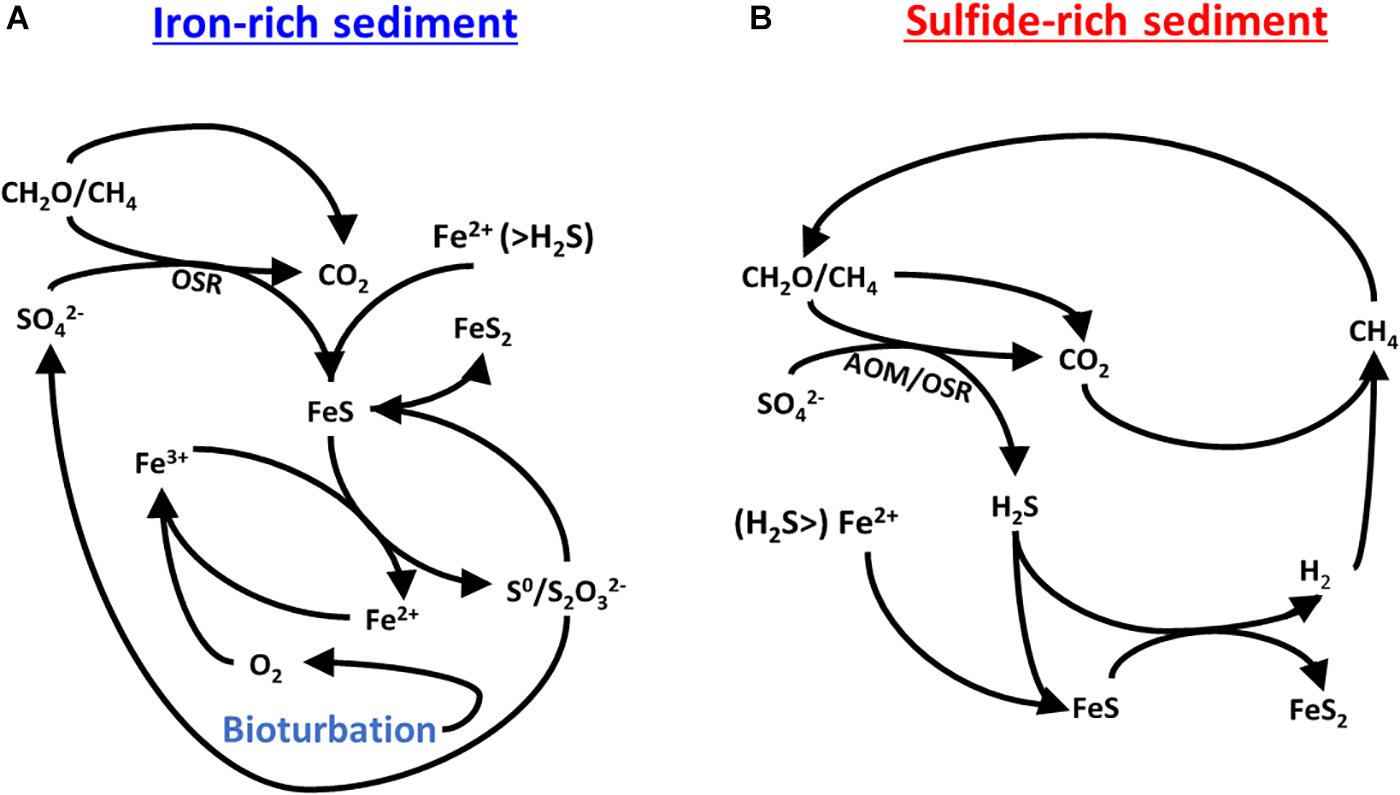
Figure 7. A proposed mechanism for the possible carbon-sulfur-iron interplay in (A) the iron-rich sediment and (B) the sulfide-rich sediment. After Aller et al. (2010).
Currently, it is not clear how one type of sediment could switch to another, under which conditions, and over what timescale. An intermediate state appears to be possible between sulfidic and iron rich sediments and there appears to be long term geochemical changes in some pond sediments, as discussed in Hutchings et al. (2019). Iron-rich sediments have less available energy (in the form of electrons supplied by organic carbon) and the inventory of total organic carbon is lower in the iron-rich sediment than in the sulfide-rich sediment. Mills et al. (2016) showed that by adding available organic carbon to iron-rich sediments in an incubation, they quickly turn sulfidic and methanic. However, a scenario where concentrated, labile organic matter (equivalent to 10 mM of lactate) would be added naturally to salt marsh sediments is unlikely. The transition from sulfide-rich sediment to iron-rich sediment appears to be more probable as it is only requires the introduction of bioturbation. Therefore, it is possible that sulfide-rich sediments could be present in a metastable condition whereas the iron-rich sediment represents a more stable state.
We suggest that the East Anglia sedimentary environment provides a unique opportunity to study the carbon-sulfur-iron coupling with bioturbation. The ongoing ventilation changes the sediment completely, from sediment that is sulfidic and methanic to sediment with iron reduction and a complex sulfur cycle. Bioturbation has a dramatic effect on the subsurface geochemistry in our studied area allowing for a tipping point between sulfidic and ferruginous conditions. This effect does not only change the supply of oxygenated water to sediment but fundamentally alters the way sulfur, iron, and carbon interact. This might have a dramatic effect on the way organic matter is mineralized and on the export of methane out of the sediment, thus acting as a primary control on the amount of carbon stored in salt marsh sediments and associated greenhouse gas fluxes.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This study was funded in part by European Research Council Starting Grant (CARBONSINK, 307582) to AVT.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We are grateful to O. Sivan and E. Eliani-Russak from Ben Gurion University of the Negev for DIC, δ13CDIC and methane measurements. We are indebted to J. Rolfe and S. Crowhurst from the Godwin Laboratory at the University of Cambridge for the technical support in the laboratory.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/feart.2019.00140/full#supplementary-material
Abdollahi, H., and Nedwell, D. B. (1979). Seasonal temperature as a factor influencing bacterial sulfate reduction in a saltmarsh sediment. Microbial Ecol. 5, 73–79. doi: 10.1007/BF02010581
Aharon, P., and Fu, B. (2000). Microbial sulfate reduction rates and sulfur and oxygen isotope fractionations at oil and gas seeps in deepwater Gulf of Mexico. Geochimica et Cosmochimica Acta 64, 233–246.
Aharon, P., and Fu, B. (2003). Sulfur and oxygen isotopes of coeval sulfate–sulfide in pore fluids of cold seep sediments with sharp redox gradients. Chem. Geol. 195, 201–218.
Aller, R. C. (1980). Quantifying solute distributions in the bioturbated zone of marine sediments by defining an average microenvironment. Geochimica et Cosmochimica Acta 44, 1955–1965.
Aller, R. C., Madrid, V., Chistoserdov, A., Aller, J. Y., and Heilbrun, C. (2010). Unsteady diagenetic processes and sulfur biogeochemistry in tropical deltaic muds: implications for oceanic isotope cycles and the sedimentary record. Geochimica et Cosmochimica Acta 74, 4671–4692.
Antler, G., and Pellerin, A. (2018). A critical look at the combined use of sulfur and oxygen isotopes to study microbial metabolisms in methane-rich environments. Front. Microbiol. 9:519. doi: 10.3389/fmicb.2018.00519
Antler, G., Turchyn, A. V., Herut, B., Davies, A., Rennie, V. C., and Sivan, O. (2014). Sulfur and oxygen isotope tracing of sulfate driven anaerobic methane oxidation in estuarine sediments. Estuar. Coast. Shelf Sci. 142, 4–11.
Antler, G., Turchyn, A. V., Herut, B., and Sivan, O. (2015). A unique isotopic fingerprint of sulfate-driven anaerobic oxidation of methane. Geology 43, 619–622.
Antler, G., Turchyn, A. V., Ono, S., Sivan, O., and Bosak, T. (2017). Combined 34S, 33S and 18O isotope fractionations record different intracellular steps of microbial sulfate reduction. Geochimica et Cosmochimica Acta 203, 364–380.
Antler, G., Turchyn, A. V., Rennie, V., Herut, B., and Sivan, O. (2013). Coupled sulfur and oxygen isotope insight into bacterial sulfate reduction in the natural environment. Geochimica et Cosmochimica Acta 118, 98–117.
Berner, R. A., and Westrich, J. T. (1985). Bioturbation and the early diagenesis of carbon and sulfur. Am. J. Sci. 285, 193–206.
Blake, R. E., Surkov, A. V., Böttcher, M. E., Ferdelman, T. G., and Jørgensen, B. B. (2006). “Oxygen isotope composition of dissolved sulfate in deep-sea sediments: eastern equatorial Pacific Ocean,” in Proceedings of the Ocean Drilling Programm, Scientific Results, Vol. 201, College Station, TX, 1–24.
Blonder, B., Boyko, V., Turchyn, A. V., Antler, G., Sinichkin, U., Knossow, N., et al. (2017). Impact of Aeolian dry deposition of reactive iron minerals on sulfur cycling in sediments of the Gulf of Aqaba. Front. Microbiol. 8:1131. doi: 10.3389/fmicb.2017.01131
Boetius, A., Ravenschlag, K., Schubert, C. J., Rickert, D., Widdel, F., Gieseke, A., et al. (2000). A marine microbial consortium apparently mediating anaerobic oxidation of methane. Nature 407, 623–626.
Böttcher, M. E., Bernasconi, S. M., and Brumsack, H.-J. (1999). “Carbon, sulfur, and oxygen isotope geochemistry of interstitial waters from the western Mediterranien,” in Proceedings of the Ocean Drilling Program, Scientific Results, Vol. 161, College Station, TX, 413–421.
Böttcher, M. E., Brumsack, H.-J., and De Lange, G. J. (1998). Sulfate reduction and related stable isotope (34S,18O) variations in interstitial waters from the Eastern Mediterranean. Proc. Ocean Drill. Progr. Sci. Results 160, 365–376.
Böttcher, M. E., Ferdelman, T. G., Jørgensen, B. B., Blake, R. E., Surkov, A. V., and Claypool, G. E. (2006). Sulfur isotope fractionation by the deep biosphere within sediments of the eastern equatorial Pacific and Peru margin. Proc. Ocean Drill. Progr. Sci. Results 201, 1–21.
Böttcher, M. E., and Thamdrup, B. (2001). Anaerobic sulfide oxidation and stable isotope fractionation associated with bacterial sulfur disproportionation in the presence of MnO2. Geochimica et Cosmochimica Acta 65, 1573–1581.
Böttcher, M. E., Thamdrup, B., Gehre, M., and Theune, A. (2005). 34S/32S and 18O/16O fractionation during sulfur disproportionation by Desulfobulbus propionicus. Geomicrobiol. J. 22, 219–226.
Böttcher, M. E., Thamdrup, B., and Vennemann, T. W. (2001). Oxygen and sulfur isotope fractionation during anaerobic bacterial disproportionation of elemental sulfur. Geochimica et Cosmochimica Acta 65, 1601–1609.
Boudreau, B. P. (1994). Is burial velocity a master parameter for bioturbation? Geochimica et Cosmochimica Acta 58, 1243–1249.
Brunner, B., Bernasconi, S. M., Kleikemper, J., and Schroth, M. H. (2005). A model for oxygen and sulfur isotope fractionation in sulfate during bacterial sulfate reduction processes. Geochimica et Cosmochimica Acta 69, 4773–4785. doi: 10.1186/1467-4866-15-7
Brunner, B., Einsiedl, F., Arnold, G. L., Müller, I., Templer, S., and Bernasconi, S. M. (2012). The reversibility of dissimilatory sulphate reduction and the cell-internal multi-step reduction of sulphite to sulphide: insights from the oxygen isotope composition of sulphate. Isotopes Environ. Health Stud. 48, 33–54. doi: 10.1080/10256016.2011.608128
Burdige, D. J. (1993). The biogeochemistry of manganese and iron reduction in marine sediments. Earth Sci. Rev. 35, 249–284.
Burton, E. D., Bush, R. T., Johnston, S. G., Sullivan, L. A., and Keene, A. F. (2011). Sulfur biogeochemical cycling and novel Fe–S mineralization pathways in a tidally re-flooded wetland. Geochimica et Cosmochimica Acta 75, 3434–3451.
Canfield, D. E. (1989). Reactive iron in marine sediments. Geochimica et Cosmochimica Acta 53, 619–632.
Canfield, D. E., and Farquhar, J. (2009). Animal evolution, bioturbation, and the sulfate concentration of the oceans. Proc. Acad. Sci. U.S.A. 106, 8123–8127. doi: 10.1073/pnas.0902037106
Canfield, D. E., and Thamdrup, B. (1996). Fate of elemental sulfur in an intertidal sediment. FEMS Microbiol. Ecol. 19, 95–103.
Chmura, G. L., Anisfeld, S. C., Cahoon, D. R., and Lynch, J. C. (2003). Global carbon sequestration in tidal, saline wetland soils. Glob. Biogeochem. Cycles 17:1111. doi: 10.13227/j.hjkx.2016.06.049
Dale, A. W., Nickelsen, L., Scholz, F., Hensen, C., Oschlies, A., and Wallmann, K. (2015). A revised global estimate of dissolved iron fluxes from marine sediments. Glob. Biogeochem. Cycles 29, 691–707.
Dale, A. W., Regnier, P., Knab, N., Jørgensen, B. B., and Van Cappellen, P. (2008). Anaerobic oxidation of methane (AOM) in marine sediments from the Skagerrak (Denmark): II. Reaction-transport modeling. Geochim. Cosmochim. Acta 72, 2880–2894.
D’Hondt, S., Inagaki, F., Zarikian, C. A., Abrams, L. J., Dubois, N., Engelhardt, T., et al. (2015). Presence of oxygen and aerobic communities from sea floor to basement in deep-sea sediments. Nat. Geosci. 8, 299–304.
Drobner, E., Huber, H., Wächtershäuser, G., Rose, D., and Stetter, K. O. (1990). Pyrite formation linked with hydrogen evolution under anaerobic conditions. Nature 346, 742–744.
Flynn, T. M., O’Loughlin, E. J., Mishra, B., DiChristina, T. J., and Kemner, K. M. (2014). Sulfur-mediated electron shuttling during bacterial iron reduction. Science 344, 1039–1042. doi: 10.1126/science.1252066
Froelich, P. N., Klinkhammer, G., Bender, M. A., Luedtke, N., Heath, G. R., Cullen, D., et al. (1979). Early oxidation of organic matter in pelagic sediments of the eastern equatorial Atlantic: suboxic diagenesis. Geochimica et Cosmochimica Acta 43, 1075–1090.
Gardner, L. R. (1973). The effect of hydrologic factors on the pore water chemistry of intertidal marsh sediments. Southeast Geol. 15, 17–28.
Giblin, A. E., and Weider, R. K. (1992). “Sulfur Cycling in Marine and Freshwater Wetlands,” in SCOP 48– Sulphur Cycling on the Continents, eds R. W. Howarth, J. W. B. Stewart, and M. V. Ivanot (Hoboken NJ: Wiley).
Goldhaber, M., Aller, R., Cochran, J., Rosenfeld, J., Martens, C., and Berner, R. (1977). Sulfate reduction, diffusion, and bioturbation in Long Island Sound sediments; report of the FOAM Group. Am. J. Sci. 277, 193–237.
Hansel, C. M., Lentini, C. J., Tang, Y., Johnston, D. T., Wankel, S. D., and Jardine, P. M. (2015). Dominance of sulfur-fueled iron oxide reduction in low-sulfate freshwater sediments. ISME J. 9:2400. doi: 10.1038/ismej.2015.50
Hines, M. E., Knollmeyer, S. L., and Tugel, J. B. (1989). Sulfate reduction and other sedimentary biogeochemistry in a northern New England salt marsh. Limnol. Oceanogr. 34, 578–590.
Holmkvist, L., Ferdelman, T. G., and Jørgensen, B. B. (2011). A cryptic sulfur cycle driven by iron in the methane zone of marine sediment (Aarhus Bay, Denmark). Geochimica et Cosmochimica Acta 75, 3581–3599.
Howarth, R. (1993). “Microbial processes in salt-marsh sediments,” in Aquatic Microbiology, ed. T. E. Ford (Boston: Blackwell Scientific), 239–260.
Hutchings, A. M., Antler, G., Wilkening, J., Basu, A., Bradbury, H. J., Clegg, J. A., et al. (2019). Creek dynamics determine pond subsurface geochemical heterogeneity in East Anglian (UK) salt marshes. Front. Earth Sci. 7:41. doi: 10.3389/feart.2019.00041
Johnston, S. G., Burton, E. D., Aaso, T., and Tuckerman, G. (2014). Sulfur, iron and carbon cycling following hydrological restoration of acidic freshwater wetlands. Chem. Geol. 371, 9–26.
Jørgensen, B. B. (1990). A thiosulfate shunt in the sulfur cycle of marine sediments. Science 249, 152–154.
Kamyshny, A. Jr., and Ferdelman, T. G. (2010). Dynamics of zero-valent sulfur species including polysulfides at seep sites on intertidal sand flats (Wadden Sea, North Sea). Mar. Chem. 121, 17–26.
Kasten, S., and Jørgensen, B. B. (2000). Sulfate Reduction in Marine Sediments. Marine Geochemistry. Berlin: Springer, 263–281.
Kostka, J. E., Gribsholt, B., Petrie, E., Dalton, D., Skelton, H., and Kristensen, E. (2002). The rates and pathways of carbon oxidation in bioturbated saltmarsh sediments. Limnol. Oceanogr. 47, 230–240.
Kristensen, E., Delefosse, M., Quintana, C. O., Flindt, M. R., and Valdemarsen, T. (2014). Influence of benthic macrofauna community shifts on ecosystem functioning in shallow estuaries. Front. Mar. Sci. 1:41. doi: 10.3389/fmars.2014.00041
Kristensen, E., Penha-Lopes, G., Delefosse, M., Valdemarsen, T., Quintana, C. O., and Banta, G. T. (2012). What is bioturbation? The need for a precise definition for fauna in aquatic sciences. Mar. Ecol. Progress Ser. 446, 285–302.
Luther, G. W., and Church, T. M. (1988). Seasonal cycling of sulfur and iron in porewaters of a Delaware salt marsh. Mar. Chem. 23, 295–309.
Luther, G. W., Giblin, A., Howarth, R. W., and Ryans, R. A. (1982). Pyrite and oxidized iron mineral phases formed from pyrite oxidation in salt marsh and estuarine sediments. Geochimica et Cosmochimica Acta 46, 2665–2669.
Luther, G. W., Kostka, J. E., Church, T. M., Sulzberger, B., and Stumm, W. (1992). Seasonal iron cycling in the salt-marsh sedimentary environment: the importance of ligand complexes with Fe (II) and Fe (III) in the dissolution of Fe (III) minerals and pyrite, respectively. Mar. Chem. 40, 81–103.
Martin, J. E. (1997). Hydrology and Pore-Water Chemistry of a Tidal Marsh, Fraser River Estuary. M.Sc. Thesis, Simon Fraser University, Burnaby.
McIlroy, D., and Logan, G. A. (1999). The impact of bioturbation on infaunal ecology and evolution during the Proterozoic-Cambrian transition. Palaios 14, 58–72.
McLeod, E., Chmura, G. L., Bouillon, S., Salm, R., Bjork, M., Duarte, C. M., et al. (2011). A blueprint for blue carbon: toward an improved understanding of the role of vegetated coastal habitats in sequestering CO2. Front. Ecol. Environ. 9:4. doi: 10.1890/110004
Meysman, F. J., Middelburg, J. J., and Heip, C. H. (2006). Bioturbation: a fresh look at Darwin’s last idea. Trends Ecol. Evol. 21, 688–695.
Middelburg, J. J. (1989). A simple rate model for organic matter decomposition in marine sediments. Geochim. Cosmochim. Acta 53, 1577–1581. doi: 10.1016/0016-7037(89)90239-1
Mills, J. V., Antler, G., and Turchyn, A. V. (2016). Geochemical evidence for cryptic sulfur cycling in salt marsh sediments. Earth Planetary Sci. Lett. 453, 23–32.
Mortimer, R. J., Galsworthy, A. M., Bottrell, S. H., Wilmot, L. E., and Newton, R. J. (2011). Experimental evidence for rapid biotic and abiotic reduction of Fe (III) at low temperatures in salt marsh sediments: a possible mechanism for formation of modern sedimentary siderite concretions. Sedimentology 58, 1514–1529.
Pellerin, A., Antler, G., Røy, H., Findlay, A., Beulig, F., Scholze, C., et al. (2018). The sulfur cycle below the sulfate-methane transition of marine sediments. Geochimica et Cosmochimica Acta. 239, 74–89. doi: 10.1038/s41396-018-0273-z
Pendleton, L., Donato, D. C., Murray, B. C., Crooks, S., Jenkins, W. A., Silfleet, S., et al. (2012). Estimating global “blue carbon” emissions from conversion and degradation of vegetated coastal ecosystems. PLoS One 7:e43542. doi: 10.1371/journal.pone.0043542
Pyzik, A. J., and Sommer, S. E. (1981). Sedimentary iron monosulfides: kinetics and mechanism of formation. Geochimica et Cosmochimica Acta 45, 687–698.
Quintana, C. O., Shimabukuro, M., Pereira, C. O., Alves, B. G., Moraes, P. C., Valdemarsen, T., et al. (2015). Carbon mineralization pathways and bioturbation in coastal Brazilian sediments. Sci. Rep. 5:16122. doi: 10.1038/srep16122
Riedinger, N., Brunner, B., Formolo, M. J., Solomon, E., Kasten, S., Strasser, M., et al. (2010). Oxidative sulfur cycling in the deep biosphere of the Nankai Trough. Japan. Geology. 38, 851–854.
Røy, H., Kallmeyer, J., Adhikari, R. R., Pockalny, R., Jørgensen, B. B., and D’hondt, S. (2012). Aerobic microbial respiration in 86-million-year-old deep-sea red clay. Science 336, 922–925. doi: 10.1126/science.1219424
Rubin-Blum, M., Antler, G., Turchyn, A. V., Tsadok, R., Goodman-Tchernov, B. N., Shemesh, E., et al. (2014a). Hydrocarbon-related microbial processes in the deep sediments of the Eastern Mediterranean Levantine Basin. FEMS Microbiol. Ecol. 87, 780–796. doi: 10.1111/1574-6941.12264
Rubin-Blum, M., Antler, G., Tsadok, R., Shemesh, E., Austin, J. A. Jr., Coleman, D. F., et al. (2014b). First evidence for the presence of iron oxidizing Zetaproteobacteria at the Levantine continental margins. PLoS One 9:e91456. doi: 10.1371/journal.pone.0091456
Sela-Adler, M., Herut, B., Bar-Or, I., Antler, G., Eliani-Russak, E., Levy, E., et al. (2015). Geochemical evidence for biogenic methane production and consumption in the shallow sediments of the SE Mediterranean shelf (Israel). Continental Shelf Research. 101, 117–124.
Sela-Adler, M., Ronen, Z., Herut, B., Antler, G., Vigderovich, H., Eckert, W., et al. (2017). Co-existence of methanogenesis and sulfate reduction with common substrates in sulfate-rich estuarine sediments. Front. Microbiol. 8:766. doi: 10.3389/fmicb.2017.00766
Severmann, S., McManus, J., Berelson, W. M., and Hammond, D. E. (2010). The continental shelf benthic iron flux and its isotope composition. Geochimica et Cosmochimica Acta 74, 3984–4004.
Sivan, O., Antler, G., Turchyn, A. V., Marlow, J. J., and Orphan, V. J. (2014). Iron oxides stimulate sulfate-driven anaerobic methane oxidation in seeps. Proc. Natl. Acad. Sci. U.S.A. 111, E4139–E4147. doi: 10.1073/pnas.1412269111
Straub, K. L., Schönhuber, W. A., Buchholz-Cleven, B. E., and Schink, B. (2004). Diversity of ferrous iron-oxidizing, nitrate-reducing bacteria and their involvement in oxygen-independent iron cycling. Geomicrobiol. J. 21, 371–378.
Thamdrup, B., Fossing, H., and Jørgensen, B. B. (1994). Manganese, iron and sulfur cycling in a coastal marine sediment, Aarhus bay, Denmark. Geochimica et Cosmochimica Acta 58, 5115–5129.
Tobias, C., and Neubauer, S. C. (2009). Salt marsh biogeochemistry–an overview. Coastal Wetlands 1, 445–492. doi: 10.1007/s004420100814
Turchyn, A. V., Antler, G., Byrne, D., Miller, M., and Hodell, D. A. (2016). Microbial sulfur metabolism evidenced from pore fluid isotope geochemistry at Site U1385. Glob. Planetary Chang. 141, 82–90.
Turchyn, A. V., Brüchert, V., Lyons, T. W., Engel, G. S., Balci, N., Schrag, D. P., et al. (2010). Kinetic oxygen isotope effects during dissimilatory sulfate reduction: a combined theoretical and experimental approach. Geochimica et Cosmochimica Acta. 74, 2011–2024.
Turchyn, A. V., Sivan, O., and Schrag, D. P. (2006). Oxygen isotopic composition of sulfate in deep sea pore fluid: evidence for rapid sulfur cycling. Geobiology 4, 191–201.
van de Velde, S., and Meysman, F. J. R. (2016). The influence of bioturbation on iron and sulphur cycling in marine sediments: a model analysis. Aquat. Geochem. 22, 469–504. doi: 10.1007/s10498-016-9301-7
Vasquez-Cardenas, D., Quintana, C. O., Meysman, F. J., Kristensen, E., and Boschker, H. T. (2016). Species-specific effects of two bioturbating polychaetes on sediment chemoautotrophic bacteria. Mar. Ecol. Progress Ser. 549, 55–68.
Whiticar, M. J., Faber, E., and Schoell, M. (1986). Biogenic methane formation in marine and freshwater environments: CO 2 reduction vs. acetate fermentation—isotope evidence. Geochimica et Cosmochimica Acta 50, 693–709.
Wijsman, J. W. M., Herman, P. M. J., Middelburg, J. J., and Soetaert, K. (2002). A model for early diagenetic processes in sediments of the continental shelf of the Black Sea. Estuar. Coast. Shelf Sci. 54, 403–421.
Wortmann, U. G., Chernyavsky, B., Bernasconi, S. M., Brunner, B., Böttcher, M. E., and Swart, P. K. (2007). Oxygen isotope biogeochemistry of pore water sulfate in the deep biosphere: dominance of isotope exchange reactions with ambient water during microbial sulfate reduction (ODP Site 1130). Geochimica et Cosmochimica Acta 71, 4221–4232.
Keywords: bioturbation, sulfur, iron, carbon, salt marsh, isotopes
Citation: Antler G, Mills JV, Hutchings AM, Redeker KR and Turchyn AV (2019) The Sedimentary Carbon-Sulfur-Iron Interplay – A Lesson From East Anglian Salt Marsh Sediments. Front. Earth Sci. 7:140. doi: 10.3389/feart.2019.00140
Received: 10 July 2018; Accepted: 16 May 2019;
Published: 25 June 2019.
Edited by:
Bradley M. Tebo, Oregon Health & Science University, United StatesReviewed by:
Mustafa Yucel, Middle East Technical University, TurkeyCopyright © 2019 Antler, Mills, Hutchings, Redeker and Turchyn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gilad Antler, Z2lsYWRhbnRsZXJAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.