 Renny T. Franceschi
Renny T. Franceschi Shawn A. Hallett
Shawn A. Hallett Chunxi Ge
Chunxi Ge
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Dent. Med., 11 May 2023
Sec. Systems Integration
Volume 4 - 2023 | https://doi.org/10.3389/fdmed.2023.1181817
This article is part of the Research TopicBone and Metabolic ActivitiesView all 10 articles
The extracellular matrix (ECM) niche plays a critical role in determining cellular behavior during bone development including the differentiation and lineage allocation of skeletal progenitor cells to chondrocytes, osteoblasts, or marrow adipocytes. As the major ECM component in mineralized tissues, collagen has instructive as well as structural roles during bone development and is required for bone cell differentiation. Cells sense their extracellular environment using specific cell surface receptors. For many years, specific β1 integrins were considered the main collagen receptors in bone, but, more recently, the important role of a second, more primordial collagen receptor family, the discoidin domain receptors, has become apparent. This review will specifically focus on the roles of discoidin domain receptors in mineralized tissue development as well as related functions in abnormal bone formation, regeneration and metabolism.
As the most abundant class of ECM proteins, collagens provide structural support for connective tissues, skin and, most importantly, bones and teeth, and can convey information about the extracellular mechanical environment via their interaction with cells using specific collagen receptors. The importance of collagen to bone development is well established; collagen synthesis is necessary for differentiation of skeletal progenitors to osteoblasts (1–4) and conditions that interfere with collagen synthesis or structure in vivo such as vitamin C deficiency or osteogenesis imperfecta severely disrupt bone development (5–8).
Until recently, it was generally assumed that bone cells interacted with the collagenous ECM exclusively through integrins, the best-known ECM receptors. Through their linkage with the cytoskeleton, integrins are major force transducers linking the ECM microenvironment with cellular functions including nuclear transcription (9). The collagen-binding integrins all have a common β1 subunit and four different alpha subunits to produce α1β1, α2β1, α10β1 and α11β1 integrins, which are all detected in bone (10–13). Disruption of integrin-collagen binding in cell culture using blocking antibodies to specific integrin subunits inhibits osteoblast differentiation of skeletal progenitor cells including preosteoblast cell lines and primary bone marrow cell cultures (12, 14–16). Because of their shared β1 subunit, the overall requirement for collagen-binding integrins in bone was assessed in vivo using conditional inactivation of the β1 integrin gene (Itgb1). Using this approach, bone phenotypes of varying severity were observed with the strongest effects of Itgb1 inactivation being associated with expression of Cre recombinase early in the bone lineage and milder phenotypes seen at later stages. For example, Itgb1 inactivation in embryonic mesenchymal progenitors using Twist2-Cre was associated with severe bone phenotypes and perinatal lethality (17). Disruption at later stages using Osx-Cre (preosteoblast stage) reduced skeletal growth, mineralization and mechanical properties, effects that became progressively milder with age while disruption of Itgb1 with Bglap-Cre had only minor effects on skeletal development (17, 18). Similarly, Itgb1 inactivation in cartilage using Col2a1-Cre resulted in perinatal lethality in most pups, stunted cartilage growth and disruption of chondrocyte proliferation and polarity (19). Although in some cases loss of Itgb1 function severely retarded bone development, in no case was bone formation and mineralization completely disrupted. This shows that some degree of bone formation can occur in the absence of collagen-binding integrins and suggests the involvement of other collagen receptors.
Interestingly, the collagen-binding integrins appeared relatively late in evolutionary history, being first seen with the emergence of chordates (20). In contrast, collagen-like proteins are present in all metazoan species (21). The discoidin domain receptors (DDRs) are a more ancient class of cell-surface collagen binding proteins than integrins. Like collagens, they are present in most invertebrate metazoans including Caenorhabditis elegans, Drosophila melanogaster, and Hydra vulgaris and so could function as collagen receptors before the collagen-binding integrins appeared on the scene. Although functions of DDRs in invertebrates have not been extensively examined, in C. elegans, specific DDR functions have been described related to axonal guidance which also requires collagen. Since DDRs have likely functioned as collagen receptors over a much longer period of time than integrins, they may have more primordial functions related to collagen signaling [for review, see reference (22)].
As will be discussed, DDRs are very different from integrins in terms of their interaction with collagens, structure, mechanism of action, tissue distribution and activity in specific cell populations. This review will specifically focus on roles of DDRs in mineralized tissues. However, it should be noted that DDRs also have non-skeletal functions in epithelial and connective tissues and have been linked to several diseases including cancer, fibrosis, and kidney disease that will not be discussed here. The reader is referred to several excellent reviews for a comprehensive treatment of these diverse DDR activities (23–26).
Unlike integrins, which lack intrinsic kinase activity, the DDRs are collagen-activated receptor tyrosine kinases (RTKs) that share homology in their kinase domain with growth factor receptors such as the neurotrophin receptor, TrkA (25, 27, 28). DDRs are named for their homology to the Dictyostelium discoideum lectin, discoidin. In mammals, there are two DDR proteins, DDR1 and DDR2, which show different preferences for binding to fibrillar and non-fibrillar collagens. Both DDR1 and 2 bind type I, II, III and V fibrillar collagens. In contrast, DDR1 selectively binds basement membrane type IV collagen while DDR2 binds type X collagen (27–29). The overall structural features of DDR1 and 2 are summarized in Figure 1. Starting from the N-terminus, both proteins have an extracellular DS domain, the region of homology with discoidin, a DS-like domain, a juxtamembrane domain, a single pass transmembrane domain, an intracellular juxtamembrane domain and an intracellular kinase domain. DS and DS-like domains and the kinase domain are highly conserved between DDR1 and DDR2. The DS domain distinguishes the DDRs from other RTKs and contains the binding site for triple-helical collagens (31, 32). DDR1 exists in 5 different spliced forms while only a single DDR2 protein has been described. In DDR1, the extracellular and transmembrane domains are shared between all 5 isoforms while there are several differences in the cytoplasmic domains. Two of the 5 DDR1 splice variants lack a functional kinase domain and could potentially act as decoy receptors for the kinase-containing isoforms (25).
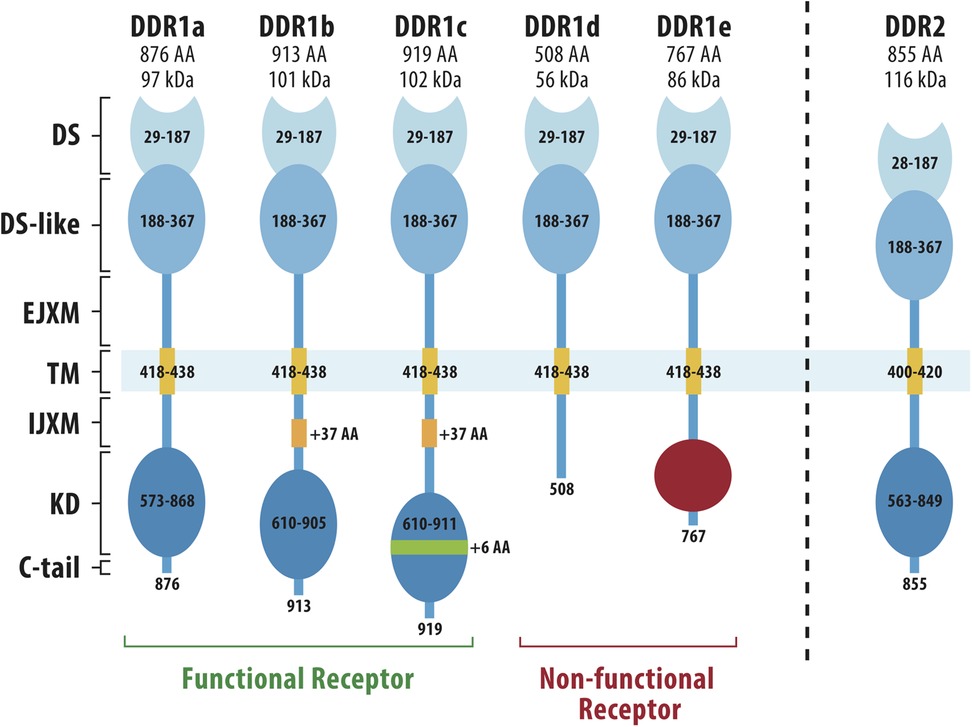
Figure 1. Structures of DDR1 and DDR2. DDR1 has 5 different spliced variants (DDR1a-e) while DDR2 exists only as a single protein. N-terminal DS (discoidin) and DS-like (discoidin-like) globular domains are shared by all DDR1 spliced variants and share high homology with the same domains in DDR2. Other regions are an extracellular juxtamembrane domain (EJXM), a transmembrane domain (TM), an intracellular juxtamembrane domain (IJXM), a kinase domain (KD) and a short C-terminal tail. The collagen-binding pocket is contained within the DS domain. Adapted from Rammal et al. (30).
Like the collagen-binding integrins, the DDRs only bind to native triple-helical collagens [i.e., thermally denatured collagen cannot serve as a binding substrate (21, 28, 31)]. DDR1 and 2 both bind a 6 amino acid sequence present in fibrillar collagens I-III, GVMGFO, where O is hydroxyproline (33, 34). This same sequence is also recognized by two other collagen-binding proteins, Secreted Protein Acidic and Rich in Cysteine (SPARC) and von Willebrand Factor that have functions in collagen mineralization and the blood coagulation cascade, respectively (35, 36). The GVMGFO sequence is distinct from the motif recognized by collagen-binding integrins which has the consensus sequence, GxOGEx (e.g., GFOGER or GAOGER in fibrillar collagens) (37, 38). Interestingly, in the COL1A1, COL2A1 and COL3A1 chains of types I–III collagen, the O of GVMGFO and the G of GFOGER/GAOGER are separated by 96 amino acid residues, a finding with possible implications concerning coupling between DDRs and integrins (see Section 6). The interaction between the DDR2 DS domain and a triple-helical peptide containing the GVMGFO sequence has been examined at atomic resolution using x-ray crystallography (39). These studies identified an amphiphilic binding pocket for the GVMGFO sequence that is conserved between DDR2 and DDR1. One side of this pocket contains apolar amino acid residues (Trp52, Thr56, Asn175, Cys73-Cys177) while the other side contains polar residues forming a salt bridge (Arg105-Glu113, Asp69) (39).
Like other RTKs, the DDRs are ligand-activated tyrosine kinases. However, instead of responding to soluble molecules such as growth factors, the DDRs have high molecular weight triple-helical collagen as a ligand. They differ from classic RTKs in other ways as well. Instead of existing as monomers that dimerize with ligand binding, DDRs are homodimers in the unactivated state (40, 41). Also, instead of being activated by their ligands and undergoing autophosphorylation within seconds to minutes like other RTKs, DDR phosphorylation takes hours and can often persist for days after binding collagen (27, 28). No truly satisfactory explanation for this phenomenon has been advanced although the involvement of secondary cellular processes such as oligomerization or internalization may be important (40, 42). Since DDRs are activated with similar kinetics by small triple-helical peptides containing the GVMGFO core binding sequence, higher order fibrillar structure of native collagen is not required for this unusual behavior (33, 34).
Once activated, DDRs stimulate several downstream signals including ERK1/2 and p38 mitogen-activated protein kinase, phosphatidylinositol-3-kinase/AKT and NF-Kβ pathways. DDRs may also have functions separate from their kinase activities, possibly related to the control of collagen fibrillogenesis and/or orientation (43, 44). It is not the purpose of this review to provide a comprehensive discussion of DDR2 signaling mechanisms as these have been thoroughly reviewed by others [see ref (23, 25)].
Initial evaluation of Ddr1 and Ddr2 mRNA distribution suggested that Ddr1 is predominantly expressed in epithelial tissues, smooth muscle and immune cells while Ddr2 is in connective tissues (45). More recently, tissue distribution was assessed by immunohistochemistry and in situ hybridization as well as by using a LacZ knock-in Ddr2 mutant where a bacterial β-galactosidase gene was inserted into the Ddr2 locus. The following discussion will emphasize DDR distribution in mineralized tissues.
Although an early study that measured DDR1 binding sites in mice using DDR1 extracellular domain fused with alkaline phosphatase showed binding to all skeletal structures, skin and the urogenital tract because of their high collagen content (46), studies that actually measured the tissue distribution of DDR1 protein or mRNA are quite limited. In neonatal and adult mice, DDR1 was localized by immunohistochemistry to proliferating and hypertrophic chondrocytes of long bone growth plates, cortical and trabecular bone osteocytes, periosteum, and articular chondrocytes (47–49). In situ hybridization analysis was conducted in oral tissues using a Ddr1 probe (50). Consistent with an epithelial pattern of expression, highest Ddr1 mRNA levels were detected in oral epithelium including enamel organs of developing molars and basal cell layers of the oral epithelium, but low expression in ectomesenchymal tissues.
Early in situ hybridization studies localized Ddr2 expression to tibial growth plates (51). Subsequent more detailed analysis using Ddr2+/LacZ mice stained for β-galactosidase activity, first detected Ddr2 expression in bone rudiments at E11.5 (52, 53). Analysis from E13.5 through adulthood showed strong staining in all developing skeletal elements in the appendicular, axial and cranial skeletons including growth plate cartilage, metaphyses, periosteum, cranial sutures and cranial base synchondroses. In general, expression was higher in cells representing earlier stages of each skeletal lineage. For example, in growth plates and synchondroses, expression was higher in resting and proliferating zone cells and lower in hypertrophic layers. Also, while Ddr2 was detected in marrow and periosteal/preosteoblast layers near forming trabecular and cortical bone surfaces, no expression was detected in osteocytes. Similar periosteal localization was reported using immunohistochemistry where DDR2 colocalized with alkaline phosphatase, a preosteoblast marker (54). Notably, this distribution is very different from most of the collagen-binding integrins (α1β1, α2β1, α11β1) that are broadly expressed in connective tissues [reviewed in ref (55)]. However, there may be some overlap with integrin α10β1 which shows preferential expression in chondrocytes (11, 56). Ddr2+/LacZ mice were also used to examine Ddr2 expression during tooth development (57) and in the temporomandibular joint (TMJ) (58). Ddr2 was widely expressed in non-epithelial tooth structures including dental follicle and dental papilla during development and odontoblasts, alveolar bone osteoblast and periodontal ligament fibroblasts of adults. In contrast to the Ddr1 mRNA distribution described above, it was conspicuously absent from epithelial structures including ameloblasts and Hertwig's epithelial root sheath. Strong Ddr2 expression was also detected in the TMJ articular surface of adult mice. Interestingly, at this age, Ddr2 expression in the articular surface of the knee joint was quite low suggesting differences between the fibrocartilage of the TMJ and hyaline cartilage of the knee (58).
To gain further insight into the lineage of Ddr2-expressing cells, Ddr2mer−icre−mer; ROSA26LSLtdTomato mice were developed (52, 53). After tamoxifen-induced recombination, Ddr2-expressing cells are labelled with tdTomato fluorescent protein, thereby allowing these cells to be followed over time. Mice were injected with tamoxifen from P1-P4 and tdTomato+ cells were lineage-traced for up to 2 months. Initially, tdTomato+cells had a similar distribution to that seen in Ddr2+/LacZ mice with labelling in growth plate and synchondrosis resting zone, cranial sutures, perichondrium, trabeculae, and periosteum, but absent in more differentiated cells. Over time, tdTomato+ cells appeared in proliferating and hypertrophic chondrocytes, osteoblasts and, eventually, osteocytes. Osteoclasts were not labelled. This result is what would be expected if Ddr2 was expressed in skeletal progenitor cells (SPCs) whose progeny became the mature cells of each skeletal lineage (hypertrophic chondrocytes for the cartilage lineage, osteocytes for the osteoblast lineage). Consistent with this concept, a high degree of colocalization between DDR2 and the skeletal progentitor/stem cell marker, GLI1 (59, 60), was observed by immunofluorescence in cranial sutures, synchondroses and tibial growth plates (52, 53). Also, CD140α+/CD51+ SPCs purified from bone marrow by FACS were enriched in Ddr2 mRNA (52).
Further evidence for DDR2 being a marker for skeletal stem cells comes from a recent study published in preprint form where DDR2 was detected in a unique cranial suture cell population (61) that could be distinguished from previously described CTSK+ suture stem cells (SSCs) (62). These DDR2+ cells have several stem cell properties including long cycling time, capacity for self-renewal after in vivo implantation, potential to differentiate to osteoblasts, adipocytes and chondrocytes, expression of several SC markers including GLI1 and capacity to generate all DDR2+ cells present in the native suture. Interestingly, conditional ablation of Ctsk-labeled SSCs using diphtheria toxin administration to iDTR; Ctsk-Cre mice led to increased expansion of DDR2+ suture cells and suture fusion via an endochondral mechanism. The authors postulate that DDR2+ suture stem cells contribute to a novel form of endochondral ossification without hematopoietic recruitment, a third potential mechanism of bone formation.
The transcriptional control mechanisms regulating DDR2 levels in bone cells are not well understood. To date they have only been examined in cell culture where Ddr2 is upregulated during osteoblast differentiation (63–65). One possible factor controlling this upregulation is ATF4 which, together with C/EBPβ, interacts with a C/EBP binding site at −1,150 bp in the Ddr2 promoter to stimulate Ddr2 expression and subsequent increases in osteoblast marker mRNAS (65). However, it is not known if these control mechanisms function in vivo or if other factors participate in this regulation.
Experiments of nature (i.e., human genetic diseases) as well as gene inactivation mouse models have been described that, taken together, provide considerable insight into how DDRs function in bone, cartilage and the dentition.
To date, no human mutations in DDR1 have been identified. In contrast, genetic disorders have been described associated with both loss and gain-of-function mutations in DDR2. Spondylo-meta-epiphyseal dysplasia with short limbs and abnormal calcifications (SMED, SL-AC) is a rare autosomal recessive genetic disorder first described in 1993 that is associated with dwarfism, short limbs, reduced bone mass, abnormal skull shape including mid-face hypoplasia and hypertelorism, open fontanelles, micrognathia and tooth abnormalities (66). This disorder was subsequently mapped to chromosome 1q23, the locus of DDR2, and shown to be caused by loss-of-function mutations in the DDR2 tyrosine kinase domain as well as mutations affecting intracellular trafficking (67–70). Unfortunately, individuals with this disorder rarely survive beyond childhood; atlantoaxial instability and resulting spinal cord damage is the most common cause of death (71, 72). The short lifespan of SMED, SL-AC patients compounded with the rarety of this disorder have limited studies in humans.
A second disorder, designated as Warburg-Cinotti Syndrome, was described in 2018 and associated with putative activating mutations in the DDR2 kinase domain (73). Fibroblasts from patients exhibited high levels of DDR2 phosphorylation in the absence of collagen stimulation, suggesting that receptor activation was ligand-independent. This disorder, which is inherited in an autosomal dominant manner, is associated with progressive fibrosis, corneal vascularization, skull abnormalities and osteolysis. In view of the deleterious effects of DDR2 loss-of-function mutations on bone formation in SMED, SL-AC pateints, it is not clear why activating mutations would lead to an osteolytic phenotype. However, since only 6 patients with Warburg-Cinotti Syndrome have been described, the phenotypic variation within this disorder cannot be currently assessed.
DDR2 may also be a determinant of bone mineral density (BMD) and fracture risk in human populations. Analysis of a Chinese Han population and an American Caucasian population identified 28 SNPs in DDR2. Of these, 3 were significantly associated with hip BMD in the Chinese, but not in the American population (74). Although this preliminary finding suggests that certain polymorphisms in DDR2 may be risk factors for osteoporosis, more studies are needed, particularly in diverse populations to assess the significance of these findings.
As will be described below, the phenotypic similarities between SMED, SL-AC patients and Ddr2-deficient mice indicate that mouse models are an appropriate model for studying this disease.
As shown in early studies, global knockout of either Ddr1 or Ddr2 resulted in dwarf phenotypes, particularly for Ddr2-deficient mice (46, 51). However, different bases for the observed growth deficits were proposed. In Ddr1 deficient mice, all organs were proportionally smaller suggesting an overall growth defect (46). However, no differences in growth plate size, chondrocyte proliferation or apoptosis were noted.
In contrast, initial analysis of globally Ddr2 deficient mice showed prominent growth retardation that was attributed to decreased proliferation of growth plate chondrocytes in the absence of changes in apoptosis resulting in shortened growth plates (51). A similar phenotype was subsequently observed in Ddr2slie/slie mice, which have a spontaneous 150 kb deletion in Ddr2 that encompasses exons 1–17 to produce an effective null allele (75). A more detailed analysis of the bone phenotype of Ddr2slie/slie mice revealed that skeletal growth defects were accompanied by large reductions in trabecular bone volume, trabecular thickness and number, changes that were attributed to reduced bone formation rate rather than stimulation of osteoclastic bone resorption (65). Similar changes in vertebral trabecular bone were also seen. However, cortical bone was only slightly affected. Interestingly, the reduction in bone mass with Ddr2 deficiency was accompanied by an increase in marrow fat. Consistent with these changes, bone marrow stromal cells (BMSCs) or calvarial preosteoblasts cultured from Ddr2slie/slie mice exhibited defective osteoblast differentiation while differentiation of BMSCs to adipocytes was enhanced.
Changes in craniofacial morphology in Ddr1 and Ddr2-deficient mice have been compared using a machine learning approach that was able to clearly discriminate between skulls from wildtype, Ddr1 and Ddr2-deficient mice (76). Although Ddr1-deficient skulls are somewhat smaller than wild type controls, they have no substantial alterations in relative skull dimensions. In contrast, skulls from Ddr2-deficient mice are dramatically shorter in the anterior-posterior direction with a more spherical skull shape associated with increased anterior skull width as well as reduced nasal bone length. Subsequent analysis of this phenotype identified a defect in proliferation of synchondrosis chondrocytes, particularly in the intersphenoid synchondrosis, in the absence of changes in apoptosis (53). These changes were associated with a characteristic expansion of the synchondrosis resting zone, possibly related to the defective conversion of these cells into proliferating chondrocytes. Ddr2-deficient skulls also have open fontanelles at birth, thinning of frontal bones and defects in frontal suture fusion that persist into adulthood (53, 65).
Effects of global Ddr1 and Ddr2 inactivation on the dentition were also examined. Ddr1-deficient mice had normal teeth, but age-dependent periodontal degeneration including alveolar bone loss was noted (50). In contrast, teeth from Ddr2slie/slie mice had smaller roots and reduced crown/root ratio resulting in disproportionate tooth size (57). These mice also exhibited gradual alveolar bone loss over a 10-month period due to increased osteoclast activity as well as atypical periodontal ligament collagen fibrils.
In addition to affecting the skeleton, global Ddr1 deficiency inhibits uterine development and embryo implantation as well as mammary epithelium development leading to defective milk production (46). Likewise, Ddr2 deficiency reduces fertility by inhibiting female and male gonadal function and steroid hormone production leading to partial sterility and interferes with certain metabolic activities (75) (see Section 8). Because effects of global inactivation of Ddr1 or Ddr2 are not restricted to the skeleton, specific cell-autonomous functions of these collagen receptors in bone cannot be inferred from global knockout studies. Although several early studies with osteoblast and chondrocyte cell lines and primary cultures suggested direct functions for DDR1 and 2 in bone cells (48, 63, 64), this issue was not resolved until recently when results of tissue-specific Ddr1 and Ddr2 knockouts were reported.
Chondrocyte or osteoblast-selective inactivation of Ddr1 was achieved by crossing Ddr1fl/fl mice with Col2a1CreERT or Col1a1CreERT mice (47–49). Chondrocyte-selective knockout of Ddr1 in tamoxifen-treated Col2a1CreERT;Ddr1fl/fl mice led to a 10–20 percent decrease in body weight and length and delayed formation of a secondary ossification center (47). In contrast to early reports with global Ddr1 knockouts (46), decreases in chondrocyte proliferation, apoptosis and hypertrophy were reported (47). These changes were accompanied by an approximately 20 percent change in trabecular bone volume while cortical thickness was unchanged. In addition, the chondrocyte hypertrophy markers (ColX, MMP13, RUNX2) and hedgehog pathway intermediate, IHH, all decreased. These results suggest that inactivation of Ddr1 in chondrocytes preferentially affects endochondral ossification. Results with Col1a1CreERT;Ddr1fl/fl mice, where Ddr1 was preferentially inactivated in osteoblasts/osteocytes were markedly different from chondrocyte-selective knockouts (48). In this case, minimal changes in endochondral ossification or trabecular bone parameters were noted while cortical thickness was reduced by approximately 50 percent. These changes were accompanied by a loss of mechanical properties and inhibition of osteoblast markers such as RUNX2, ALPL, BGLAP and COLIA1. In a second study with Col1a1CreERT;Ddr1fl/fl mice, the same group examined the consequences of Ddr1 inactivation in adults over extended periods (49). In this case, modest changes in trabecular parameters were noted together with reductions in cortical thickness, osteoblast differentiation markers and cortical bone formation rate. These changes were accompanied by increased apoptosis and autophagy markers. No craniofacial changes were described in any of these studies.
Conditional knockout studies with Ddr2 were informed by results of localization and lineage tracing experiments showing preferential expression of this gene in GLI1+ skeletal progenitor cells, chondrocytes, and osteoblasts (see Sections 3.2, 3.3). To determine functions of Ddr2 in these cells, Ddr2fl/fl mice were crossed with Gli1CreERT, Col2a1Creor BglapCre mice and resulting long bone and craniofacial phenotypes were examined (52, 53). Inactivation of Ddr2 in Gli1-expressing cells, induced by injecting neonatal Gli1CreERT;Ddr2fl/fl mice with tamoxifen, resulted in essentially the same phenotype seen in Ddr2slie/slie mice. Dwarfism was observed in both males and females, and this was associated with an approximately 12 percent reduction of growth plate length at P14. In addition, severe defects in endochondral bone formation were observed, particularly in males where trabecular BV/TV was reduced by approximately 50 percent. Associated reductions in trabecular number and thickness and increased trabecular spacing were also seen at 3 months. However, cortical BV/TV was not affected. The craniofacial phenotype of Gli1CreERT;Ddr2fl/fl mice was also essentially identical to Ddr2slie/slie mice; anterior-posterior skull length was reduced with an associated increase in anterior skull width. Mutants also exhibited frontal bone thinning and shortened nasal bones (53). Also like global knockouts, the anterior portion of frontal sutures failed to mineralize in most mice.
The phenotype of Col2a1Cre;Ddr2fl/fl mice was similar to Gli1CreERT;Ddr2fl/fl and Ddr2slie/slie mice with the important exception that no defects in suture fusion were observed. Although it has been proposed that changes in growth of the cranial base can affect suture fusion (77), this is clearly not an adequate explanation for effects of Ddr2 inactivation on frontal sutures since Col2a1Cre;Ddr2fl/fl mice had the same cranial base growth defects seen in Gli1CreERT;Ddr2fl/fl mice. Based on this result, it was concluded that functions of Ddr2 in synchondrosis endochondral bone formation are independent from its functions in cranial sutures. Consistent with the observed reduction in tibial bone formation, mRNA levels of osteoblast and hypertrophic chondrocyte markers were all reduced in Col2a1Cre;Ddr2fl/fl mice. These changes were accompanied by decreased mRNA levels of the hedgehog pathway intermediates, Ihh and Gli1. Since defects in Hh signaling were also noted with conditional Ddr1 knockout (47) (Section 4.3.1), this pathway may be a common target for DDRs.
Although Ddr2 was expressed in osteoblasts on trabecular and periosteal surfaces, it probably does not have a major function in mature osteoblasts since BglapCre;Ddr2fl/fl mice were essentially identical to wild type control mice. Because this Cre is mainly active in mature osteoblasts and, possibly, osteocytes, it is still possible that Ddr2 may have functions in earlier stages of the osteoblast lineage.
Overall, Ddr2 conditional knockout studies support the concept that this gene functions in earlier stages of bone formation (i.e., in Gli1CreERT-positive skeletal progenitor cells and Col2a1Cre-positive resting zone and proliferative chondrocytes) rather than in terminally differentiated osteoblasts or hypertrophic chondrocytes. Two cell culture studies reinforce this conclusion (52). In the first, E12.5 limb buds from Ddr2fl/fl mice were used to prepare micromass cultures enriched in chondro-osteo progenitors that were treated with control or Cre adenovirus before growth in chondrogenic medium. Ddr2 inactivation strongly inhibited chondrogenesis as measured by Alcian blue staining or expression of chondrocyte markers. In the second study, CD140α+/CD51+ SPCs were prepared from Ddr2fl/fl mice and grown in osteogenic medium after treatment with Cre adenovirus. In this case, Ddr2 inactivation strongly inhibited osteoblast differentiation (mineralization and expression of osteoblast markers).
The studies described above all focused on functions of Ddr2 in chondro-osteo lineage cells which form chondrocytes, osteoblasts and osteocytes. However, there is still some controversy regarding possible Ddr2 functions in osteoclasts. On one hand, lineage tracing studies with Ddr2mer−icre−mer; ROSA26LSLtdTomato mice did not show colocalization of the tdTomato label with TRAP-positive osteoclasts (Section 3.3) and globally Ddr2 deficient mice (Ddr2slie/slie mice) did not have any detectable changes in bone resorption markers or osteoclast differentiation capacity (Section 4.1). On the other hand, evidence was presented that DDR2 has a suppressive effect on osteoclast formation in cell culture models (78). DDR2 protein and mRNA were detected at low levels in the RAW264.7 macrophage cell line and primary cultures of bone marrow macrophage and these levels were further reduced with in vitro induction of osteoclast formation. Also, overexpression of Ddr2 in RAW264.7 was shown to inhibit osteoclast induction while shRNA knockdown of Ddr2 further stimulated this process. Furthermore, adenovirus-mediated overexpression of Ddr2 in the femur marrow cavity partially reversed osteoporosis in ovariectomized mice, a phenotype that is largely due to osteoclast activation. These studies suggest that Ddr2 can function in the monocytic lineage to suppress osteoclastogenesis. Lastly, in a recent study Ddr2fl/fl mice were crossed with LysMCre mice to conditionally inactivate Ddr2 in myeloid lineage cells (79). The resulting animals had a hyperinflammatory phenotype after exposure to either collagen antibody-induced arthritis or a high-fat diet. After arthritis induction, mice had increased ankle inflammation, elevation of inflammatory markers, increased bone resorption and increased osteoclast surface per bone surface as well as an approximately 15 percent decrease in bone mineral density. Also, evidence was presented that loss of DDR2 increased macrophage repolarization from an M2 to M1 phenotype resulting in enhanced inflammation. However, this study did not look for changes in bone density in the absence of an inflammatory stimulus. Nevertheless, this work supports a role for DDR2 in the suppression of osteoclastogenesis through its inhibitory actions on monocytic osteoclast precursors. However, it is still not clear why, in previous studies, changes in bone resorption markers were not detected in Ddr2slie/slie mice or why osteoclasts were not detected as part of the DDR2 lineage (52, 65). It is possible that effects on bone resorption in the absence of induced inflammation may not be large enough to affect bone mass or, alternatively, that in globally Ddr2 deficient mice, interference with other DDR2 dependent processes may compensate for effects on osteoclastogenesis. Another possibility would be that DDR2 is not expressed in the osteoclast lineage and does not have a direct function in these cells, but rather modulates effects of macrophage on osteoclastogenesis. Studies where Ddr2 is more selectively inactivated only in osteoclasts (for example, using Ctsk-Cre or TRAP-Cre) may be necessary to resolve this issue (80).
A consistent finding from Ddr2 knockout studies is that osteoblast differentiation and associated expression of osteoblast marker genes are suppressed. A limited number of studies have investigated the basis for this suppression. Because of its central role as a master transcriptional regulator of bone formation, studies to date have focused on RUNX2. This transcription factor is expressed at early times during bone development coincident with the formation of cartilage condensations and has roles in both hypertrophic cartilage formation as well as osteoblast differentiation [for review (81)]. RUNX2 activity is subject to several controls including phosphorylation by ERK1/2 and p38 mitogen-activated protein kinases (MAPKs) (82). Both MAPKs are important for bone formation as demonstrated by in vivo gain and loss-of-function studies (83, 84). Once activated, MAPKs translocate to the nucleus where they bind and phosphorylate RUNX2 on the chromatin of target genes (85). MAPKs phosphorylate RUNX2 on several serine residues, the most important being Ser301 and Ser319 (86). Phosphorylated RUNX2 recruits specific histone acetyltransferases and methylases to chromatin resulting in increased H3K9 and H4K5 acetylation and H3K4 di-methylation, histone modifications associated with transcriptional activation, as well as decreased H3K9 mono-, di and tri-methylation, histone marks associated with repression (85). These changes open chromatin structure to allow RNA polymerase II to bind and initiate transcription of osteoblast-related genes. RUNX2 phosphorylation and MAPK activity are obligatory for these changes since transfection of cells with a phosphorylation-resistant S301,319A mutant RUNX2 or treatment with MAPK inhibitors blocks transcription.
Since both ERK1/2 and p38 MAPKs are known downstream responses to DDR2 activation (25), it was hypothesized that this pathway could explain the observed stimulatory effects of DDR2 on osteoblast gene expression. This concept has been tested in cell culture studies with osteoblast cell lines as well as in osteoblasts from Ddr2-deficient mice (64, 65). In early studies with osteoblast cell lines and primary BMSC cultures, DDR2 was shown to stimulate osteoblast differentiation through a pathway involving ERK/MAPK activation and RUNX2 phosphorylation (64). Ddr2 shRNA inhibited differentiation while overexpression was stimulatory. These changes were paralleled by increased or decreased ERK/MAPK activity, RUNX2 phosphorylation and transcriptional activity. Significantly, effects of Ddr2 shRNA knockdown could be overcome by transfecting cells with a phosphomimetic Runx2 S301,319E mutant where replacement of alanine with glutamate mimics a phosphate group. In separate studies referenced in Section 4.1 (65), calvarial preosteoblasts or BMSCs isolated from Ddr2slie/slie mice were found to be deficient in ability to undergo osteoblast differentiation while BMSCs from these mice exhibited enhanced adipogenic differentiation. The reduced osteoblast differentiation in Ddr2-deficient cells was directly related to reduced ERK/MAPK activity and RUNX2-S319 phosphorylation and was rescued by transfection with the RUNX2 S301/319E mutant described above. The ability of DDR2 to stimulate ERK/MAPK activity may also explain the increase in marrow fat observed in Ddr2slie/slie mice. In addition to phosphorylating RUNX2, ERK1/2 can phosphorylate the adipogenic transcription factor, PPARγ, on Ser112. In this case, however, phosphorylation inhibits transcriptional activity. By preventing this inhibitory phosphorylation, Ddr2 knockout would be expected to restore PPARγ activity to permit formation of marrow fat. Consistent with this interpretation, transgenic mice containing a phosphorylation-resistant S112A PPARγ mutant have increased marrow fat and reduced bone mass (87).
Consistent with the marked effects of Ddr2 deficiency on bone development, inactivation of this gene was also shown to inhibit bone regeneration. Two regeneration models were examined, a calvarial bone defect and a tibial fracture (88, 89). For the calvarial model, a 0.5 mm burr hole defect was generated in wild type or Ddr2slie/slie mice and regeneration was examined for increasing times up to 12 weeks. In wild type mice, this type of defect was completely healed after 4 weeks while no bone bridging was seen in mutant mice even after 12 weeks. Ddr2, which was expressed in sutures and periosteal cells before injury, was detected in the injury site within 3 days and expanded during the healing process. Also, inactivation of Ddr2 in calvarial cells in culture reduced osteoblast differentiation. For the fracture model, a mid-shaft tibial fracture was created in wild type or Ddr2slie/slie mice and fracture healing was monitored for 3 weeks. In this case, Ddr2-deficient mice were unable to form complete unions at the fracture site as measured by Radiographic Union Score Tibia (mRUST) (90).
In the studies described above, the reduced linear growth of long bones and skulls in Ddr2-deficient mice was attributed to proliferation defects in growth plate and synchondrosis chondrocytes in the absence of changes in apoptosis (52, 53). Interestingly, an examination of chondrocyte morphology revealed that the normal organization of these cells into columns was disrupted with Ddr2 inactivation. This effect was seen in long bone growth plates but was particularly striking in cranial base synchondroses where the central resting zone was greatly expanded with widely separated disorganized cells (52, 53). In some cases, chondrocytes actually shifted their orientation by 90 degrees to form an ectopic hypertrophic zone at right angles to the normal plane of synchondrosis organization. These changes were accompanied by loss of chondrocyte polarity as measured by disruption of the normally consistent orientation of GM130, a Golgi apparatus marker, relative to the nucleus and anterior-posterior axis of the skull. This may explain the proliferation defect seen in chondrocytes of Ddr2-deficient mice since disruption of GM130 orientation is known to impair spindle assembly and cell division (65). The relevance of these findings to human physiology is emphasized by the observation that collagen matrix distribution is also disrupted in growth plate cartilage from SMED, SL-AC patients (66).
How might DDR2 affect chondrocyte polarity? One possibility is that it is necessary for collagen matrix organization and fibril orientation which would subsequently affect chondrocyte orientation. Examination of the type II collagen distribution in both growth plates and synchondroses by immunofluorescence microscopy revealed a shift from a uniform distribution in the territorial matrix next to chondrocytes and the extraterritorial matrix between cell clusters in wild type mice to an uneven distribution restricted to the pericellular space adjacent to chondrocytes in mutants (53). These changes were accompanied by loss of type II collagen fibril orientation as measured by second harmonic generation (SHG) microscopy. This analysis detected a dramatic shift from a highly oriented matrix (high anisotrophy) in synchondroses of wild type mice to a disorganized matrix (low anisotropy) in mutants where fibrils had a randomized orientation (53). Although primary cilia have been related to cell polarity and collagen orientation in other systems (91), regulation of this important organelle by DDR2 has not been reported.
Another consequence of DDR2 maintaining collagen fibril orientation is an increase in overall ECM stiffness. Although this has not been examined during bone development, there are several examples in other experimental systems. For example, DDR2 in breast cancer-associated fibroblasts (CAFs) increases tumor stiffness by organizing type I collagen fibrils (92). Also, at sites of trauma-induced heterotopic ossification, DDR2 increases collagen fibril orientation as measured by SHG (93) (also see Section 7.3). In both cases, evidence was presented that DDR2 functioned in concert with collagen-binding β1 integrins to stimulate, on one hand, tumor metastasis to the lungs or, on the other, ectopic bone formation. As noted in Section 2, fibrillar collagens I–III containing binding sites for both DDRs and integrins are always separated by 96 amino acid residues. This characteristic spacing may allow collagen to simultaneously regulate both these receptors. For example, in breast tumor metastasis, DDR2 was found to stimulate CAF-mediated mechanotransduction by increasing integrin activation in response to collagen. This was accomplished by stimulating RAP1-mediated Talin1 and Kindlin2 recruitment to integrins in focal adhesions (92). Also, in trauma-induced heterotopic ossification, DDR2 was necessary for full activation of integrin-dependent signals such as focal adhesion kinase (FAK) activation as well as nuclear levels of the Hippo pathway intermediate, TAZ, and its downstream targets (93).
Given the involvement of DDRs in normal bone formation, it is not totally surprising that they are also involved when this process goes awry. It this section, DDR involvement in vascular calcification, osteoarthritis and heterotopic ossification will be discussed.
Initiated by insults such as high levels of circulating LDL cholesterol, diabetes or chronic kidney disease, vascular calcification is a key event in advanced atherosclerosis. Calcium phosphate crystals can be deposited either in the subepithelial intima of blood vessels (intimal calcification) or in the smooth muscle-rich media (medial calcification) (94). This latter process shares many similarities with normal bone formation. It is initiated by differentiation of vascular smooth muscle cells or SMC progenitors into osteochondroprogenitor cells which form bone-like structures in arteries through a process that mimics endochondral bone formation as indicated by formation of cartilage that subsequently is converted into a bone-like structure (95). Like normal bone formation, this process requires interactions of progenitor cells with type I collagen and is mediated by the master transcriptional regulator of bone formation, RUNX2 (96, 97). Vascular calcification can be induced in mice by feeding LDL receptor-deficient animals (Ldlr−/− mice) a high fat, high cholesterol diet. Breeding a Ddr1-null allele into Ldlr−/− mice resulted in animals that were resistant to developing vascular calcification (97). Subsequent analysis showed that calcification was inhibited via a mechanism involving suppression of phosphatidyl inositol-3-kinase/AKT and p38/ERK MAP kinase signaling and inhibition of RUNX2 phosphorylation and activation (98). More recent studies extended this work by showing that DDR1 up-regulates its own synthesis in response to the stiffness of the matrix environment around VSMCs. This is accomplished by stimulating the nuclear translocation of the Hippo pathway intermediates, YAP and TAZ, to increase Ddr1 transcription and subsequent mineralization (99). This may explain the known relationship between arterial stiffening and acceleration of vascular calcification (100).
Osteoarthritis (OA), a primary indicator for joint degeneration, is characterized by cartilage degradation, osteophyte formation and joint mineralization (101). OA can occur in fibrocartilage of the temporomandibular joint (TMJ) or in hyaline cartilage of major joints such as the knee. OA in hyaline cartilage generally increases with age. In contrast, TMJ OA has an earlier onset (102, 103). Interactions between chondrocytes and the ECM of hyaline cartilage and fibrocartilage may be key factors for understanding OA pathogenesis in these two tissues. TMJ fibrocartilage extracellular matrix mainly contains type I collagen while type II collagen predominates in hyaline joints (104). Both DDR1 and DDR2 are involved in OA etiology although they may function through different mechanisms. Unlike DDR1, which is broadly but weakly activated by collagens I to IV, DDR2 is strongly activated by types I and III collagen of TMJ fibrocartilage but is less responsive to type II collagen (28). Ddr2 is expressed at low levels in healthy adult hyaline cartilage joints but is abundant in TMJ fibrocartilage (58). Thus, Ddr2 is normally expressed at highest levels in an ECM environment that is conducive to its activation. Consistent with its distribution, Ddr2 is required for normal TMJ formation; global Ddr2 inactivation disrupts TMJ development beginning in neonates which show an initial delay in condyle mineralization that persists in adults leading to eventual joint degeneration and subchondral bone loss (58). In contrast, knee joints, which are composed of hyaline cartilage, are not affected by Ddr2 deficiency. Ddr1 global knockout mice, in contrast, exhibit a spontaneous rapid-onset TMJ OA that is seen by 9 weeks without involvement of other joints (105). The authors of this study proposed that induction of TMJ OA is related to the observation that loss of DDR1 was accompanied by a compensatory up-regulation of DDR2. This is then activated by the type I collagen in TMJ fibrocartilage to induce OA. It is not known if these changes are seen in Ddr1-deficient neonates although a separate study reported TMJ abnormalities in mice as young as 4 weeks (50).
DDR2 has also been related to OA in hyaline cartilage joints. In this case, the normally low levels of DDR2 in adults are increased with injuries such as trauma or surgical destabilization of the medial meniscus, which subsequently induce OA (106). In this case, globally Ddr2-deficient mice or mice where Ddr2 is selectively inactivated in articular cartilage are resistant to surgically-induced OA indicating that DDR2 is required for OA induction in this tissue (107, 108). However, overexpressing Ddr2 in hyaline cartilage does not lead to spontaneous OA formation unless hyaline cartilage ECM is altered by trauma (106, 108). It has been proposed that trauma-induced damage to the ECM may disrupt the pericellular matrix around chondrocytes and allow them to interact with type II collagen fibrils resulting in DDR2 activation and OA (107).
Heterotopic ossification (HO) is a debilitating condition that occurs after many traumatic injuries. In HO, PDGFRα+ connective tissue cells present in soft tissue adjacent to the injury site change their differentiation trajectory to form ectopic cartilage and bone (109). Ddr2 has been recently shown to play a role in the pathogenesis of HO (93). Using single cell RNA sequencing, Ddr2 was discovered to be highly expressed by PDGFRα+ cells, that form the major cell lineages involved in HO formation. In HO, both DDR2 and phospho-DDR2, a marker of active DDR2, were shown to be significantly upregulated in PDGFRα+ cells within the tendon, peritendon, and soft tissue areas surrounding the HO site. Interestingly, DDR2 mediates HO formation after injury, as both Ddr2slie/slie mice (global knockout) and tamoxifen-treated Pdgfa-CreER;Ddr2fl/fl mice (conditional knockout in progenitor cells) display significant reductions in Sox9 expressing chondrocytes, safranin O-labeled cells and reductions in ectopic bone formation due to extracellular matrix disorganization and FAK/YAP/TAZ dysregulation (described in Section 5). This study highlights how extracellular matrix alignment can have profound effects on HO progression and how DDR2 is an important regulator of this process.
In addition to inhibiting skeletal growth, global Ddr2 deficiency also affects metabolism. For example, Ddr2slie/slie mice have elevated blood glucose levels, reduced body fat and increased lean body mass (75), elevated levels of circulating adiponectin and decreased serum leptin (65). It is not known if there is a relationship between these metabolic changes and the bone phenotype of these mice. However, as discussed in Sections 4.1, 4.3, the decrease in bone mass in Ddr2slie/slie mice is paralleled by an increase in marrow fat, a change that may be related to the reduced ERK/MAPK activity in mutant mice. The consequences of this reduced MAPK activity would include suppression of RUNX2 and PPARγ phosphorylation, decreased osteoblast and increased marrow adipocyte gene expression and differentiation. Since marrow adipocytes are a major source of serum adiponectin (110), the increase in marrow adipocytes in Ddr2slie/slie mice may explain the observed increase in serum adiponectin. However, specific knockout of the Adipoq gene in marrow adipocytes using a recently described double recombination strategy (111) would be necessary to definitively test this hypothesis.
Interestingly, Ddr2 is expressed in adipocytes. Early studies suggested possible direct effects of DDR2 on these cells such as suppression of insulin stimulated tyrosine phosphorylation of the insulin receptor in the 3T3-L1 adipocyte cell line (112). More recently, direct effects of DDR2 on adipocytes in vivo were examined using AdipoCre;Ddr2fl/fl mice, where Ddr2 is inactivated in peripheral as well as marrow fat (113). In this study, mutant mice were protected from high fat diet-induced weight gain, a response that was attributed to decreased adipocyte size. Significantly, these animals also had a high bone mass phenotype accompanied by increases in both bone formation rate and resorption. These changes were explained by a DDR2-specific repression of adenylate cyclase 5 (Adcy5) in adipocytes removed in mutant mice leading to increased cAMP production and lipolysis in marrow adipocytes. The released fatty acids in the marrow cavity then promote increased oxidative metabolism in osteoblast leading to increased osteoblast and osteoclast activity. Therefore, by modulating lipolysis in adipocytes, DDR2 can indirectly control bone formation. This mechanism may complement the more direct effects of DDR2 on skeletal progenitor cells described in Section 4.3.2.
The study of DDR functions in bone is a relatively new research area and many questions remain about what these collagen receptors do and how they do it. As shown in this review, both DDR1 and DDR2 have functions in mineralized tissues with DDR2 perhaps having a greater role under physiological conditions. However, clear functions for DDR1 are also seen, particularly in pathological conditions such as vascular calcification.
Although tissue distribution studies, particularly for DDR1, are incomplete, the original conclusion that DDR1 functions in epithelia while DDR2 is in connective tissues may need revision, particularly for DDR1, which has clear functions in connective tissues like cartilage and bone. More detailed DDR1 localization and lineage tracing studies will be required to more fully understand where this collagen receptor functions. The observation that DDR2 is present in GLI1-positive skeletal progenitor cells of cranial sutures and, possibly, cartilage where it controls cell proliferation and differentiation to chondrocytes and osteoblasts is of particular interest. These studies suggest that DDR2, together with collagen binding integrins, allows certain classes of skeletal progenitor/stem cells to sense their ECM environment and modulate their differentiation state according to ECM stiffness and mechanical loads. As the more ancient of the two collagen receptors, the DDRs were likely complemented by the newly emerging collagen-binding integrins when the vertebrate skeleton first evolved so that these two receptors now work in concert. Another intriguing area is the possible function of DDR2 in osteoimmunology where it may modulate activities of various myleloid lineages to control inflammation and bone resorption.
Although conditional knockout studies showed that DDR2 functions in skeletal progenitor cells and chondrocytes, little is known about its actual mechanism of action in these tissues. Current, albeit incomplete, knowledge in this area is summarized in Figure 2. Some of its activities may be explained by modulation of MAP kinases which subsequently control osteogenic and adipogenic transcription factors through phosphorylation. However, this is likely only part of the story. The dramatic effects of DDR2 on collagen fibril orientation, matrix stiffness and cell polarity may also be an important part of an overall mechanism that still needs to be discerned. By modulating matrix stiffness-associated pathways including the Hippo pathway, DDR2 and integrins may work together to control stiffness-associated nuclear changes and transcription. These matrix signals may also modify the response of cells to soluble signals coming from growth factors or morphogens. All these topics are clearly fruitful areas for future investigations.
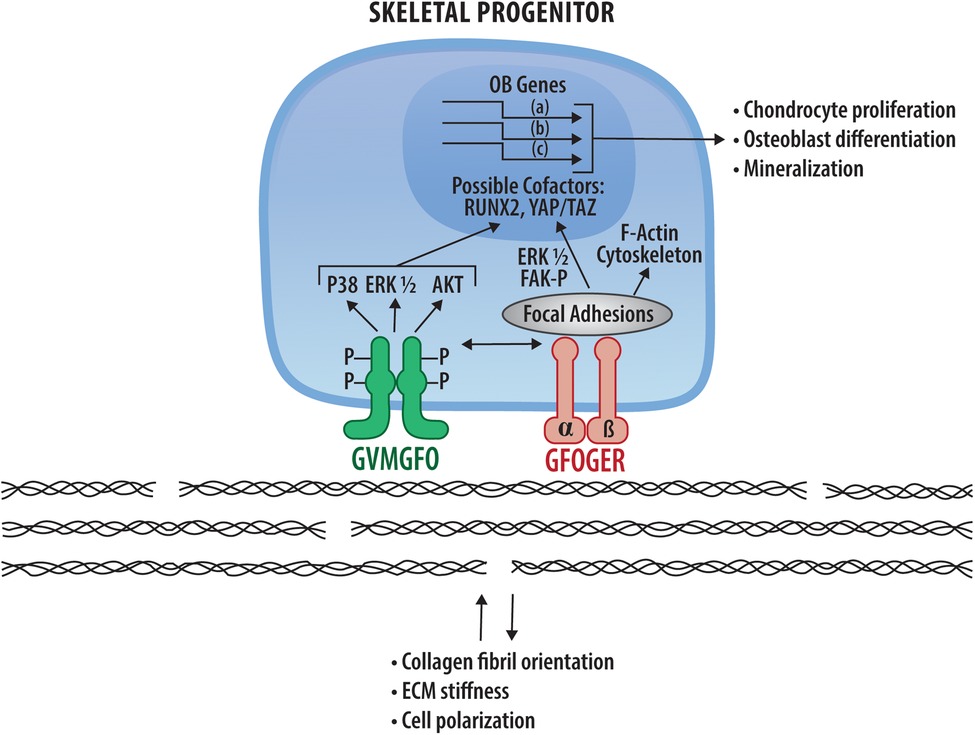
Figure 2. DDR2 signaling mechanisms in bone and potential interactions with collagen-binding integrins. DDR2 (green) and integrins (pink) bind and are activated by the specific sequences in fibrillar collagens indicated. Downstream signals arising from DDR2 and integrin activation are indicated as are nuclear changes in RUNX2 and HIPPO pathway intermediates, YAP and TAZ, resulting in transcriptional changes necessary for chondrocyte proliferation, osteoblast differentiation and mineralization (see Sections 4.4). Also shown are DDR-dependent changes in collagen matrix organization and cell polarity described in Section 6.
Recent discoveries on DDR function may also have important implications for the treatment of disease. For example, the demonstrated role of DDR1 in vascular calcification and of DDR2 in osteoarthritis and heterotopic ossification suggest that specific DDR2 inhibitors already under development could be used to treat these disorders (114). Also, the recent discovery that DDR2 is required for skeletal regeneration may open new directions for therapy through the development of either DDR-activating tissue engineering scaffolds or other treatments that modify DDR activity.
Clearly, the study of DDRs in bone will continue to be a growing area of musculoskeletal research that holds much promise for exciting future discoveries.
RF wrote and edited the article. SH wrote portions of the article and edited the entire article CG wrote portions of the article and edited the entire article. All authors contributed to the article and approved the submitted version.
Research from the authors laboratory described in this article was supported by NIH/NIDCR grants DE11723, DE029012, DE029465, DE030675, Department of Defense grant PR190899, research funds from the Department of Periodontics and Oral Medicine, University of Michigan School of Dentistry and the Michigan Musculoskeletal Health Core Center (NIH/NIAMS P30 AR069620).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Franceschi RT, Iyer BS. Relationship between collagen synthesis and expression of the osteoblast phenotype in MC3T3-E1 cells. J Bone Miner Res. (1992) 7(2):235–46. doi: 10.1002/jbmr.5650070216
2. Franceschi RT. The developmental control of osteoblast-specific gene expression: role of specific transcription factors and the extracellular matrix environment. Crit Rev Oral Biol Med. (1999) 10(1):40–57. doi: 10.1177/10454411990100010201
3. Lynch MP, Stein JL, Stein GS, Lian JB. The influence of type I collagen on the development and maintenance of the osteoblast phenotype in primary and passaged rat calvarial osteoblasts: modification of expression of genes supporting cell growth, adhesion, and extracellular matrix mineralization. Exp Cell Res. (1995) 216(1):35–45. doi: 10.1006/excr.1995.1005
4. D'Alonzo RC, Kowalski AJ, Denhardt DT, Nickols GA, Partridge NC. Regulation of collagenase-3 and osteocalcin gene expression by collagen and osteopontin in differentiating MC3T3-E1 cells. J Biol Chem. (2002) 277(27):24788–98. doi: 10.1074/jbc.M202604200
5. Togari A, Arai M, Nakagawa S, Banno A, Aoki M, Matsumoto S. Alteration of bone status with ascorbic acid deficiency in ODS (osteogenic disorder shionogi) rats. Jpn J Pharmacol. (1995) 68(3):255–61. doi: 10.1254/jjp.68.255
6. Mahmoodian F, Gosiewska A, Peterkofsky B. Regulation and properties of bone alkaline phosphatase during vitamin C deficiency in Guinea pigs. Arch Biochem Biophys. (1996) 336(1):86–96. doi: 10.1006/abbi.1996.0535
7. Fedarko NS, Moerike M, Brenner R, Robey PB. Extracellular matrix formation by osteoblasts from patients with osteogenesis imperfecta. J Bone Miner Res. (1992) 7(8):921–30. doi: 10.1002/jbmr.5650070809
8. Fedarko NS, Sponseller PD, Shapiro JR. Long-term extracellular matrix metabolism by cultured human osteogenesis imperfecta osteoblasts. J Bone Miner Res. (1996) 11(6):800–5. doi: 10.1002/jbmr.5650110611
9. Kechagia JZ, Ivaska J, Roca-Cusachs P. Integrins as biomechanical sensors of the microenvironment. Nat Rev Mol Cell Biol. (2019) 20(8):457–73. doi: 10.1038/s41580-019-0134-2
10. Saito T, Albelda SM, Brighton CT. Identification of integrin receptors on cultured human bone cells. J Orthop Res. (1994) 12(3):384–94. doi: 10.1002/jor.1100120311
11. Lundgren-Akerlund E, Aszodi A. Integrin alpha10beta1: a collagen receptor critical in skeletal development. Adv Exp Med Biol. (2014) 819:61–71. doi: 10.1007/978-94-017-9153-3_4
12. Xiao G, Wang D, Benson MD, Karsenty G, Franceschi RT. Role of the alpha2-integrin in osteoblast-specific gene expression and activation of the Osf2 transcription factor. J Biol Chem. (1998) 273(49):32988–94. doi: 10.1074/jbc.273.49.32988
13. Brunner M, Jurdic P, Tuckerman JP, Block MR, Bouvard D. New insights into adhesion signaling in bone formation. Int Rev Cell Mol Biol. (2013) 305:1–68. doi: 10.1016/B978-0-12-407695-2.00001-9
14. Ekholm E, Hankenson KD, Uusitalo H, Hiltunen A, Gardner H, Heino J, et al. Diminished callus size and cartilage synthesis in alpha 1 beta 1 integrin-deficient mice during bone fracture healing. Am J Pathol. (2002) 160(5):1779–85. doi: 10.1016/S0002-9440(10)61124-8
15. Takeuchi Y, Nakayama K, Matsumoto T. Differentiation and cell surface expression of transforming growth factor-beta receptors are regulated by interaction with matrix collagen in murine osteoblastic cells. J Biol Chem. (1996) 271(7):3938–44. doi: 10.1074/jbc.271.7.3938
16. Mizuno M, Fujisawa R, Kuboki Y. Type I collagen-induced osteoblastic differentiation of bone-marrow cells mediated by collagen-alpha2beta1 integrin interaction. J Cell Physiol. (2000) 184(2):207–13. doi: 10.1002/1097-4652(200008)184:2%3C207::AID-JCP8%3E3.0.CO;2-U
17. Shekaran A, Shoemaker JT, Kavanaugh TE, Lin AS, LaPlaca MC, Fan Y, et al. The effect of conditional inactivation of beta 1 integrins using twist 2 cre, osterix cre and osteocalcin cre lines on skeletal phenotype. Bone. (2014) 68:131–41. doi: 10.1016/j.bone.2014.08.008
18. Brunner M, Mandier N, Gautier T, Chevalier G, Ribba AS, Guardiola P, et al. Beta1 integrins mediate the BMP2 dependent transcriptional control of osteoblast differentiation and osteogenesis. PLoS One. (2018) 13(4):e0196021. doi: 10.1371/journal.pone.0196021
19. Aszodi A, Hunziker EB, Brakebusch C, Fassler R. Beta1 integrins regulate chondrocyte rotation, G1 progression, and cytokinesis. Genes Dev (2003) 17(19):2465–79. doi: 10.1101/gad.277003
20. Huhtala M, Heino J, Casciari D, de Luise A, Johnson MS. Integrin evolution: insights from ascidian and teleost fish genomes. Matrix Biol. (2005) 24(2):83–95. doi: 10.1016/j.matbio.2005.01.003
21. Heino J. The collagen family members as cell adhesion proteins. Bioessays. (2007) 29(10):1001–10. doi: 10.1002/bies.20636
22. Unsoeld T, Taylor J, Hutter H. Discoidin domain receptors in invertebrates. In: Fridman R, Huang P, editors. Discoidin domain receptors in health and disease. New York, N.Y: Springer (2016). p. 87–105.
23. Chen L, Kong X, Fang Y, Paunikar S, Wang X, Brown JAL, et al. Recent advances in the role of discoidin domain receptor tyrosine kinase 1 and discoidin domain receptor tyrosine kinase 2 in breast and ovarian cancer. Front Cell Dev Biol. (2021) 9:747314. doi: 10.3389/fcell.2021.747314
24. Majo S, Auguste P. The yin and yang of discoidin domain receptors (DDRs): implications in tumor growth and metastasis development. Cancers. (2021) 13(7):1725–51. doi: 10.3390/cancers13071725
25. Leitinger B. Discoidin domain receptor functions in physiological and pathological conditions. Int Rev Cell Mol Biol. (2014) 310:39–87. doi: 10.1016/B978-0-12-800180-6.00002-5
26. Marquez J, Olaso E. Role of discoidin domain receptor 2 in wound healing. Histol Histopathol. (2014) 29(11):1355–64. doi: 10.14670/HH-29.1355
27. Shrivastava A, Radziejewski C, Campbell E, Kovac L, McGlynn M, Ryan TE, et al. An orphan receptor tyrosine kinase family whose members serve as nonintegrin collagen receptors. Mol Cell. (1997) 1(1):25–34. doi: 10.1016/S1097-2765(00)80004-0
28. Vogel W, Gish GD, Alves F, Pawson T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell. (1997) 1(1):13–23. doi: 10.1016/S1097-2765(00)80003-9
29. Leitinger B, Kwan AP. The discoidin domain receptor DDR2 is a receptor for type X collagen. Matrix Biol. (2006) 25(6):355–64. doi: 10.1016/j.matbio.2006.05.006
30. Rammal H, Saby C, Magnien K, Van-Gulick L, Garnotel R, Buache E, et al. Discodin domain receptors: potential actors and targets in cancer. Front Pharmacol. (2016) 7:55. doi: 10.3389/fphar.2016.00055
31. Leitinger B. Molecular analysis of collagen binding by the human discoidin domain receptors, DDR1 and DDR2. Identification of collagen binding sites in DDR2. J Biol Chem. (2003) 278(19):16761–9. doi: 10.1074/jbc.M301370200
32. Leitinger B, Steplewski A, Fertala A. The D2 period of collagen II contains a specific binding site for the human discoidin domain receptor, DDR2. J Mol Biol. (2004) 344(4):993–1003. doi: 10.1016/j.jmb.2004.09.089
33. Konitsiotis AD, Raynal N, Bihan D, Hohenester E, Farndale RW, Leitinger B. Characterization of high affinity binding motifs for the discoidin domain receptor DDR2 in collagen. J Biol Chem. (2008) 283(11):6861–8. doi: 10.1074/jbc.M709290200
34. Xu H, Raynal N, Stathopoulos S, Myllyharju J, Farndale RW, Leitinger B. Collagen binding specificity of the discoidin domain receptors: binding sites on collagens II and III and molecular determinants for collagen IV recognition by DDR1. Matrix Biol. (2011) 30(1):16–26. doi: 10.1016/j.matbio.2010.10.004
35. Lisman T, Raynal N, Groeneveld D, Maddox B, Peachey AR, Huizinga EG, et al. A single high-affinity binding site for von willebrand factor in collagen III, identified using synthetic triple-helical peptides. Blood. (2006) 108(12):3753–6. doi: 10.1182/blood-2006-03-011965
36. Hohenester E, Sasaki T, Giudici C, Farndale RW, Bachinger HP. Structural basis of sequence-specific collagen recognition by SPARC. Proc Natl Acad Sci U S A. (2008) 105(47):18273–7. doi: 10.1073/pnas.0808452105
37. Knight CG, Morton LF, Peachey AR, Tuckwell DS, Farndale RW, Barnes MJ. The collagen-binding A-domains of integrins alpha(1)beta(1) and alpha(2)beta(1) recognize the same specific amino acid sequence, GFOGER, in native (triple-helical) collagens. J Biol Chem. (2000) 275(1):35–40. doi: 10.1074/jbc.275.1.35
38. Barczyk M, Carracedo S, Gullberg D. Integrins. Cell Tissue Res. (2010) 339(1):269–80. doi: 10.1007/s00441-009-0834-6
39. Carafoli F, Bihan D, Stathopoulos S, Konitsiotis AD, Kvansakul M, Farndale RW, et al. Crystallographic insight into collagen recognition by discoidin domain receptor 2. Structure. (2009) 17(12):1573–81. doi: 10.1016/j.str.2009.10.012
40. Mihai C, Chotani M, Elton TS, Agarwal G. Mapping of DDR1 distribution and oligomerization on the cell surface by FRET microscopy. J Mol Biol. (2009) 385(2):432–45. doi: 10.1016/j.jmb.2008.10.067
41. Noordeen NA, Carafoli F, Hohenester E, Horton MA, Leitinger B. A transmembrane leucine zipper is required for activation of the dimeric receptor tyrosine kinase DDR1. J Biol Chem. (2006) 281(32):22744–51. doi: 10.1074/jbc.M603233200
42. Agarwal G, Smith AW, Jones B. Discoidin domain receptors: micro insights into macro assemblies. Biochim Biophys Acta Mol Cell Res. (2019) 1866(11):118496. doi: 10.1016/j.bbamcr.2019.06.010
43. Blissett AR, Garbellini D, Calomeni EP, Mihai C, Elton TS, Agarwal G. Regulation of collagen fibrillogenesis by cell-surface expression of kinase dead DDR2. J Mol Biol. (2009) 385(3):902–11. doi: 10.1016/j.jmb.2008.10.060
44. Flynn LA, Blissett AR, Calomeni EP, Agarwal G. Inhibition of collagen fibrillogenesis by cells expressing soluble extracellular domains of DDR1 and DDR2. J Mol Biol. (2010) 395(3):533–43. doi: 10.1016/j.jmb.2009.10.073
45. Alves F, Vogel W, Mossie K, Millauer B, Hofler H, Ullrich A. Distinct structural characteristics of discoidin I subfamily receptor tyrosine kinases and complementary expression in human cancer. Oncogene. (1995) 10(3):609–18. PMID: 7845687
46. Vogel WF, Aszodi A, Alves F, Pawson T. Discoidin domain receptor 1 tyrosine kinase has an essential role in mammary gland development. Mol Cell Biol. (2001) 21(8):2906–17. doi: 10.1128/MCB.21.8.2906-2917.2001
47. Chou LY, Chen CH, Lin YH, Chuang SC, Chou HC, Lin SY, et al. Discoidin domain receptor 1 regulates endochondral ossification through terminal differentiation of chondrocytes. FASEB J. (2020) 34(4):5767–81. doi: 10.1096/fj.201901852RR
48. Chou LY, Chen CH, Chuang SC, Cheng TL, Lin YH, Chou HC, et al. Discoidin domain receptor 1 regulates Runx2 during osteogenesis of osteoblasts and promotes bone ossification via phosphorylation of p38. Int J Mol Sci. (2020) 21(19):7210–26. PMID: 33003599
49. Chou HC, Lin SY, Chou LY, Ho ML, Chuang SC, Cheng TL, et al. Ablation of discoidin domain receptor 1 provokes an osteopenic phenotype by regulating osteoblast/osteocyte autophagy and apoptosis. Biomedicines. (2022) 10(9):2173–87. doi: 10.3390/biomedicines10092173
50. Chavez MB, Kolli TN, Tan MH, Zachariadou C, Wang C, Embree MC, et al. Loss of discoidin domain receptor 1 predisposes mice to periodontal breakdown. J Dent Res. (2019) 98(13):1521–31. doi: 10.1177/0022034519881136
51. Labrador JP, Azcoitia V, Tuckermann J, Lin C, Olaso E, Manes S, et al. The collagen receptor DDR2 regulates proliferation and its elimination leads to dwarfism. EMBO Rep. (2001) 2(5):446–52. doi: 10.1093/embo-reports/kve094
52. Mohamed FF, Ge C, Cowling RT, Lucas D, Hallett SA, Ono N, et al. The collagen receptor, discoidin domain receptor 2, functions in Gli1-positive skeletal progenitors and chondrocytes to control bone development. Bone Res. (2022) 10(1):11. doi: 10.1038/s41413-021-00182-w
53. Mohamed FF, Ge C, Hallett SA, Bancroft AC, Cowling RT, Ono N, et al. Control of craniofacial development by the collagen receptor, discoidin domain receptor 2. Elife. (2023) 12:e77257. doi: 10.7554/eLife.77257
54. Yang H, Sun L, Cai W, Gu J, Xu D, Deb A, et al. DDR2, A discoidin domain receptor, is a marker of periosteal osteoblast and osteoblast progenitors. J Bone Miner Metab. (2020) 38:670–7. doi: 10.1007/s00774-020-01108-y
55. Zeltz C, Gullberg D. The integrin-collagen connection–a glue for tissue repair? J Cell Sci. (2016) 129(4):653–64. doi: 10.1242/jcs.180992
56. Camper L, Holmvall K, Wangnerud C, Aszodi A, Lundgren-Akerlund E. Distribution of the collagen-binding integrin alpha10beta1 during mouse development. Cell Tissue Res. (2001) 306(1):107–16. doi: 10.1007/s004410100385
57. Mohamed FF, Ge C, Binrayes A, Franceschi RT. The role of discoidin domain receptor 2 in tooth development. J Dent Res. (2020) 99(2):214–22. doi: 10.1177/0022034519892563
58. Ge C, Mohamed F, Binrayes A, Kapila S, Franceschi RT. Selective role of discoidin domain receptor 2 in murine temporomandibular joint development and aging. J Dent Res. (2018) 97(3):321–8. doi: 10.1177/0022034517738190
59. Shi Y, He G, Lee WC, McKenzie JA, Silva MJ, Long F. Gli1 identifies osteogenic progenitors for bone formation and fracture repair. Nat Commun. (2017) 8(1):2043. doi: 10.1038/s41467-017-02171-2
60. Zhao H, Feng J, Ho TV, Grimes W, Urata M, Chai Y. The suture provides a niche for mesenchymal stem cells of craniofacial bones. Nat Cell Biol. (2015) 17(4):386–96. doi: 10.1038/ncb3139
61. Greenblatt M, Bok S, Yallowitz A, McCormick J, Cung M, Sun J, et al. A multi-stem cell basis for craniosynostosis and calvarial mineralization. Res Sq. (2021). doi: 10.21203/rs.3.rs-1061838/v1
62. Debnath S, Yallowitz AR, McCormick J, Lalani S, Zhang T, Xu R, et al. Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature. (2018) 562(7725):133–9. doi: 10.1038/s41586-018-0554-8
63. Lin KL, Chou CH, Hsieh SC, Hwa SY, Lee MT, Wang FF. Transcriptional upregulation of DDR2 by ATF4 facilitates osteoblastic differentiation through p38 MAPK-mediated Runx2 activation. J Bone Miner Res. (2010) 25(11):2489–503. doi: 10.1002/jbmr.159
64. Zhang Y, Su J, Yu J, Bu X, Ren T, Liu X, et al. An essential role of discoidin domain receptor 2 (DDR2) in osteoblast differentiation and chondrocyte maturation via modulation of Runx2 activation. J Bone Miner Res. (2011) 26(3):604–17. doi: 10.1002/jbmr.225
65. Ge C, Wang Z, Zhao G, et al. Discoidin receptor 2 controls bone formation and marrow adipogenesis. J Bone Miner Res. (2016) 31(12):2193–203. doi: 10.1002/jbmr.2893
66. Borochowitz Z, Langer LO Jr, Gruber HE, Lachman R, Katznelson MB, Rimoin DL. Spondylo-meta-epiphyseal dysplasia (SMED), short limb-hand type: a congenital familial skeletal dysplasia with distinctive features and histopathology. Am J Med Genet. (1993) 45(3):320–6. doi: 10.1002/ajmg.1320450308
67. Bargal R, Cormier-Daire V, Ben-Neriah Z, Le Merrer M, Sosna J, Melki J, et al. Mutations in DDR2 gene cause SMED with short limbs and abnormal calcifications. Am J Hum Genet. (2009) 84(1):80–4. doi: 10.1016/j.ajhg.2008.12.004
68. Al-Kindi A, Kizhakkedath P, Xu H, John A, Sayegh AA, Ganesh A, et al. A novel mutation in DDR2 causing spondylo-meta-epiphyseal dysplasia with short limbs and abnormal calcifications (SMED-SL) results in defective intra-cellular trafficking. BMC Med Genet. (2014) 15:42. doi: 10.1186/1471-2350-15-42
69. Urel-Demir G, Simsek-Kiper PO, Akgun-Dogan O, Gocmen R, Wang Z, Matsumoto N, et al. Further expansion of the mutational spectrum of spondylo-meta-epiphyseal dysplasia with abnormal calcification. J Hum Genet. (2018) 63(9):1003–7. doi: 10.1038/s10038-018-0473-4
70. Ali BR, Xu H, Akawi NA, John A, Karuvantevida NS, Langer R, et al. Trafficking defects and loss of ligand binding are the underlying causes of all reported DDR2 missense mutations found in SMED-SL patients. Hum Mol Genet. (2010) 19(11):2239–50. doi: 10.1093/hmg/ddq103
71. Langer LO Jr, Wolfson BJ, Scott CI Jr, Reid CS, Schidlow DV, Millar EA, et al. Further delineation of spondylo-meta-epiphyseal dysplasia, short limb-abnormal calcification type, with emphasis on diagnostic features. Am J Med Genet. (1993) 45(4):488–500. doi: 10.1002/ajmg.1320450419
72. Dias C, Cairns R, Patel MS. Sudden death in spondylo-meta-epiphyseal dysplasia, short limb-abnormal calcification type. Clin Dysmorphol. (2009) 18(1):25–9. doi: 10.1097/MCD.0b013e3283186907
73. Xu L, Jensen H, Johnston JJ, Di Maria E, Kloth K, Cristea I, et al. Recurrent, activating variants in the receptor tyrosine kinase DDR2 cause warburg-cinotti syndrome. Am J Hum Genet. (2018) 103(6):976–83. doi: 10.1016/j.ajhg.2018.10.013
74. Guo Y, Yang TL, Dong SS, Yan H, Hao RH, Chen XF, et al. Genetic analysis identifies DDR2 as a novel gene affecting bone mineral density and osteoporotic fractures in Chinese population. PLoS One. (2015) 10(2):e0117102. doi: 10.1371/journal.pone.0117102
75. Kano K, Marin de Evsikova C, Young J, Wnek C, Maddatu TP, Nishina PM, et al. A novel dwarfism with gonadal dysfunction due to loss-of-function allele of the collagen receptor gene, Ddr2, in the mouse. Mol Endocrinol. (2008) 22(8):1866–80. doi: 10.1210/me.2007-0310
76. Dullin C, Missbach-Guentner J, Vogel WF, Grabbe E, Alves F. Semi-automatic classification of skeletal morphology in genetically altered mice using flat-panel volume computed tomography. PLoS Genet. (2007) 3(7):e118. doi: 10.1371/journal.pgen.0030118
77. Kreiborg S, Marsh JL, Cohen MM Jr, Liversage M, Pedersen H, Skovby F, et al. Comparative three-dimensional analysis of CT-scans of the calvaria and cranial base in apert and crouzon syndromes. J Craniomaxillofac Surg. (1993) 21(5):181–8. doi: 10.1016/S1010-5182(05)80478-0
78. Zhang Y, Su J, Wu S, Teng Y, Yin Z, Guo Y, et al. DDR2 (discoidin domain receptor 2) suppresses osteoclastogenesis and is a potential therapeutic target in osteoporosis. Sci Signal. (2015) 8(369):ra31. doi: 10.1126/scisignal.2005835
79. Liu Q, Wang X, Chen Y, Ma X, Kang X, He F, et al. Ablation of myeloid discoidin domain receptor 2 exacerbates arthritis and high fat diet induced inflammation. Biochem Biophys Res Commun. (2023) 649:47–54. doi: 10.1016/j.bbrc.2023.01.074
80. Dallas SL, Xie Y, Shiflett LA, Ueki Y. Mouse cre models for the study of bone diseases. Curr Osteoporos Rep. (2018) 16(4):466–77. doi: 10.1007/s11914-018-0455-7
81. Komori T. Runx2, an inducer of osteoblast and chondrocyte differentiation. Histochem Cell Biol. (2018) 149(4):313–23. doi: 10.1007/s00418-018-1640-6
82. Franceschi RT, Ge C. Control of the osteoblast lineage by mitogen-activated protein kinase signaling. Curr Mol Biol Rep. (2017) 3:122–32. doi: 10.1007/s40610-017-0059-5
83. Ge C, Xiao G, Jiang D, Franceschi RT. Critical role of the extracellular signal-regulated kinase-MAPK pathway in osteoblast differentiation and skeletal development. J Cell Biol. (2007) 176(5):709–18. doi: 10.1083/jcb.200610046
84. Greenblatt MB, Shim JH, Zou W, Sitara D, Schweitzer M, Hu D, et al. The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. J Clin Invest. (2010) 120(7):2457–73. doi: 10.1172/JCI42285
85. Li Y, Ge C, Franceschi RT. MAP kinase-dependent RUNX2 phosphorylation is necessary for epigenetic modification of chromatin during osteoblast differentiation. J Cell Physiol. (2017) 232(9):2427–35. doi: 10.1002/jcp.25517
86. Ge C, Xiao G, Jiang D, Yang Q, Hatch NE, Roca H, et al. Identification and functional characterization of ERK/MAPK phosphorylation sites in the Runx2 transcription factor. J Biol Chem. (2009) 284(47):32533–43. doi: 10.1074/jbc.M109.040980
87. Ge C, Zhao G, Li B, Li Y, Cawthorn WP, MacDougald OA, et al. Genetic inhibition of PPARgamma S112 phosphorylation reduces bone formation and stimulates marrow adipogenesis. Bone. (2018) 107:1–9. doi: 10.1016/j.bone.2017.10.023
88. Binrayes A, Ge C, Mohamed F, Kozloff KM, Franceschi RT. Role of discoidin domain receptor 2 in bone regeneration. J Bone MinerRes. (2018) 33(Abstract):S0448.
89. Binrayes A, Ge C, Mohamed FF, Franceschi RT. Role of discoidin domain receptor 2 in craniofacial bone regeneration. J Dent Res. (2021) 100(12):1359–66. doi: 10.1177/00220345211007447
90. Cooke ME, Hussein AI, Lybrand KE, Wulff A, Simmons E, Choi JH, et al. Correlation between RUST assessments of fracture healing to structural and biomechanical properties. J Orthop Res. (2018) 36(3):945–53. doi: 10.1002/jor.23710
91. Donnelly E, Ascenzi MG, Farnum C. Primary cilia are highly oriented with respect to collagen direction and long axis of extensor tendon. J Orthop Res. (2010) 28(1):77–82. doi: 10.1002/jor.20946
92. Bayer SV, Grither WR, Brenot A, Hwang PY, Barcus CE, Ernst M, et al. DDR2 controls breast tumor stiffness and metastasis by regulating integrin mediated mechanotransduction in CAFs. Elife. (2019) 8:e45508–32. doi: 10.7554/eLife.45508
93. Pagani CA, Bancroft AC, Tower RJ, Livingston N, Sun Y, Hong JY, et al. Discoidin domain receptor 2 regulates aberrant mesenchymal lineage cell fate and matrix organization. Sci Adv. (2022) 8(51):eabq6152. doi: 10.1126/sciadv.abq6152
94. Block GA. Control of serum phosphorus: implications for coronary artery calcification and calcific uremic arteriolopathy (calciphylaxis). Curr Opin Nephrol Hypertens. (2001) 10(6):741–7. doi: 10.1097/00041552-200111000-00003
95. Speer MY, Yang HY, Brabb T, Leaf E, Look A, Lin WL, et al. Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circ Res. (2009) 104(6):733–41. doi: 10.1161/CIRCRESAHA.108.183053
96. Sun Y, Byon CH, Yuan K, Chen J, Mao X, Heath JM, et al. Smooth muscle cell-specific Runx2 deficiency inhibits vascular calcification. Circ Res. (2012) 111(5):543–52. doi: 10.1161/CIRCRESAHA.112.267237
97. Ahmad PJ, Trcka D, Xue S, Franco C, Speer MY, Giachelli CM, et al. Discoidin domain receptor-1 deficiency attenuates atherosclerotic calcification and smooth muscle cell-mediated mineralization. Am J Pathol. (2009) 175(6):2686–96. doi: 10.2353/ajpath.2009.080734
98. Lino M, Wan MH, Rocca AS, Ngai D, Shobeiri N, Hou G, et al. Diabetic vascular calcification mediated by the collagen receptor discoidin domain receptor 1 via the phosphoinositide 3-kinase/akt/runt-related transcription factor 2 signaling axis. Arterioscler Thromb Vasc Biol. (2018) 38(8):1878–89. doi: 10.1161/ATVBAHA.118.311238
99. Ngai D, Mohabeer AL, Mao A, Lino M, Bendeck MP. Stiffness-responsive feedback autoregulation of DDR1 expression is mediated by a DDR1-YAP/TAZ axis. Matrix Biol. (2022) 110:129–40. doi: 10.1016/j.matbio.2022.05.004
100. Chen Y, Zhao X, Wu H. Arterial stiffness: a focus on vascular calcification and its link to bone mineralization. Arterioscler Thromb Vasc Biol. (2020) 40(5):1078–93. doi: 10.1161/ATVBAHA.120.313131
101. Arden N, Nevitt MC. Osteoarthritis: epidemiology. Best Pract Res Clin Rheumatol. (2006) 20(1):3–25. doi: 10.1016/j.berh.2005.09.007
102. Liu F, Steinkeler A. Epidemiology, diagnosis, and treatment of temporomandibular disorders. Dent Clin North Am. (2013) 57(3):465–79. doi: 10.1016/j.cden.2013.04.006
103. Wiberg B, Wanman A. Signs of osteoarthrosis of the temporomandibular joints in young patients: a clinical and radiographic study. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. (1998) 86(2):158–64. doi: 10.1016/S1079-2104(98)90118-4
104. Benjamin M, Ralphs JR. Biology of fibrocartilage cells. Int Rev Cytol. (2004) 233:1–45. doi: 10.1016/S0074-7696(04)33001-9
105. Schminke B, Muhammad H, Bode C, Sadowski B, Gerter R, Gersdorff N, et al. A discoidin domain receptor 1 knock-out mouse as a novel model for osteoarthritis of the temporomandibular joint. Cell Mol Life Sci. (2014) 71(6):1081–96. doi: 10.1007/s00018-013-1436-8
106. Xu L, Polur I, Servais JM, Hsieh S, Lee PL, Goldring MB, et al. Intact pericellular matrix of articular cartilage is required for unactivated discoidin domain receptor 2 in the mouse model. Am J Pathol. (2011) 179(3):1338–46. doi: 10.1016/j.ajpath.2011.05.023
107. Xu L, Servais J, Polur I, Kim D, Lee PL, Chung K, et al. Attenuation of osteoarthritis progression by reduction of discoidin domain receptor 2 in mice. Arthritis Rheum. (2010) 62(9):2736–44. doi: 10.1002/art.27582
108. Manning LB, Li Y, Chickmagalur NS, Li X, Xu L. Discoidin domain receptor 2 as a potential therapeutic target for development of disease-modifying osteoarthritis drugs. Am J Pathol. (2016) 186(11):3000–10. doi: 10.1016/j.ajpath.2016.06.023
109. Dey D, Wheatley BM, Cholok D, Agarwal S, Yu PB, Levi B, et al. The traumatic bone: trauma-induced heterotopic ossification. Transl Res. (2017) 186:95–111. doi: 10.1016/j.trsl.2017.06.004
110. Cawthorn WP, Scheller EL, Learman BS, Parlee SD, Simon BR, Mori H, et al. Bone marrow adipose tissue is an endocrine organ that contributes to increased circulating adiponectin during caloric restriction. Cell Metab. (2014) 20(2):368–75. doi: 10.1016/j.cmet.2014.06.003
111. Li Z, Bowers E, Zhu J, Yu H, Hardij J, Bagchi DP, et al. Lipolysis of bone marrow adipocytes is required to fuel bone and the marrow niche during energy deficits. Elife. (2022) 11:e78496. doi: 10.7554/eLife.78496
112. Zurakowski H, Gagnon A, Landry A, Layne MD, Sorisky A. Discoidin domain receptor 2 impairs insulin-stimulated insulin receptor substrate-1 tyrosine phosphorylation and glucose uptake in 3T3-L1 adipocytes. Horm Metab Res. (2007) 39(8):575–81. doi: 10.1055/s-2007-985132
113. Yang X, Li J, Zhao L, Chen Y, Cui Z, Xu T, et al. Targeting adipocytic discoidin domain receptor 2 impedes fat gain while increasing bone mass. Cell Death Differ. (2022) 29(4):737–49. doi: 10.1038/s41418-021-00887-9
114. Wang Z, Zhang Y, Pinkas DM, Fox AE, Luo J, Huang H, et al. Design, synthesis, and biological evaluation of 3-(imidazo[1,2- a]pyrazin-3-ylethynyl)-4-isopropyl- N-(3-((4-methylpiperazin-1-yl)methyl)-5-(trifluoromethyl)phenyl)benzamide as a dual inhibitor of discoidin domain receptors 1 and 2. J Med Chem. (2018) 61(17):7977–90. doi: 10.1021/acs.jmedchem.8b01045
Keywords: extracellular matrix, collagen receptor, differentiation, stem cell, bone, cartilage
Citation: Franceschi RT, Hallett SA and Ge C (2023) Discoidin domain receptors; an ancient family of collagen receptors has major roles in bone development, regeneration and metabolism. Front. Dent. Med 4:1181817. doi: 10.3389/fdmed.2023.1181817
Received: 7 March 2023; Accepted: 20 April 2023;
Published: 11 May 2023.
Edited by:
Martha J. Somerman, Frontiers in Dental Medicine, United StatesReviewed by:
Pamela Gehron Robey, National Institutes of Health (NIH), United States© 2023 Franceschi, Hallett and Ge. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renny T. Franceschi, cmVubnlmQHVtaWNoLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.