 Nour Ammar
Nour Ammar Magda M. El-Tekeya
Magda M. El-Tekeya- Department of Pediatric Dentistry and Dental Public Health, Faculty of Dentistry, Alexandria University, Alexandria, Egypt
Hallermann–Streiff syndrome (HSS) is a disorder of rare occurrence affecting the craniofacial complex, with approximately 200 cases reported in the literature. Nonetheless, its distinctive facial features render it highly recognizable. We present the case of a 5-year-old girl with this syndrome and review the dental manifestations and management in this patient. In addition to the diagnostic facial features of brachycephaly with frontal bossing, beak-shaped nose, microphthalmia, and mandibular retrusion, multiple dental manifestations were noted, including the absence of the mandibular condyle, ghost teeth, and unusual pulpal calcifications in both the primary and the permanent teeth, which have not been previously reported in a case of HSS. There is no consensus on the suitable treatment plan to be given for HSS patients at a young age due to an underreporting of these cases in the literature. In this report, we discuss pediatric dental management options for a patient with HSS and share her perspective on the treatment.
Introduction
Hallermann–Streiff syndrome (HSS) is an ectodermal dysplasia disorder that affects the development of the craniofacial complex. Its prevalence is very rare, with approximately 200 cases reported in the literature (1). Initially described by Aubury in 1893, it was further studied by Hallermann in 1948 and Francois in 1950 (2, 3). It is also known as Hallermann–Streiff Francois syndrome, Ullrich–Fremery–Dohna syndrome, and oculomandibulofacial syndrome (OMIM %234,100). Although rare the facial features of individuals with this syndrome are highly recognizable. Affected patients present with proportionate nanism, brachycephaly with frontal bossing, microphthalmia, congenital cataracts, a distinctive beak-shaped nose, hypotrichosis, cutaneous atrophy, and a host of dental abnormalities. Along with these classic positive signs, François reported other negative signs to differentiate this syndrome from other syndromes commonly misdiagnosed with HSS, including progeria and mandibulofacial dysostosis (3). There is no reported sex predilection, nor is its genetic basis elucidated; it is widely believed to be caused by dominant sporadic mutations (4). Almost 85% of patients show dental anomalies such as enamel hypoplasia, natal teeth, high and narrow palatal vault, microstomia, and micrognathia (5). The etiology of this syndrome remains unknown and is widely believed to be caused by a de novo dominant mutation. Some studies suggest that the occurrence of HSS is caused by a homozygous mutation in the GJA1 gene (6), but recent molecular sequencing findings contest this theory (4).
The limited number of reported cases of HSS, of which even a fewer number of publications discuss dental management options, and the lack of consensus on how to manage the multifaceted dental malformations caused by this syndrome, add to the clinical challenge of treating such patients. Formulating an effective treatment plan based on evidence from previous literature remains a challenge due to an underreporting of these cases. Specifically, with regard to pediatric dental management, the literature is sparse. In this report, we present the case of a 5-year-old female patient with HSS and discuss dental management options for her.
Case report
This case report has been written according to the case reporting guidelines (CARE) (7). Informed consent for publication has been obtained from the patient's legal guardians. A 5-year-old female patient presented to the Department of Pediatric Dentistry, Faculty of Dentistry, Alexandria University, for dental treatment. She had previously sought dental attention and care, but she had been denied treatment by general dental practitioners due to a lack of knowledge about her syndrome. She had been complaining of dental hypersensitivity and inability to efficiently chew food for a few months.
The patient is the child of healthy non-consanguineous parents and has a normal older sister. The parents reported a negative medical history of any syndrome in their families. The mother had an uneventful pregnancy, for she did not take any medications. The patient was born at full term with a vaginal delivery, but she was born underweight. Immediately after her birth, she was admitted to the neonatal intensive care unit, where she stayed for 45 days due to respiratory difficulties. Upon discharge, she faced problems during breastfeeding by her mother and was often subject to upper respiratory tract infections. She was diagnosed with HSS two months after birth. Since then, the patient has been scheduled for biannual ophthalmic checkup in efforts to mitigate the known risk of bilateral congenital cataracts associated with HSS. Fortunately, she has not developed cataracts thus far.
Upon physical examination, the patient showed proportionate nanism, mild brachycephaly with frontal bossing, bilateral microphthalmos, strabismus (esotropia of the left eye), and a “bird-like” beaked nose with constricted alae nasi. Additionally, the patient had hypotrichosis manifested by alopecia, eyebrow and eyelash loss, and atrophy of the skin. She had a markedly convex facial profile due to mandibular hypoplasia. There was complete bilateral syndactyly between her fourth and fifth fingers (syndactyly type III) (Figure 1). Furthermore, she suffered from mild intellectual disability, which is an uncommon manifestation of the syndrome (5). Despite suffering from recurrent respiratory infections and breathing problems as an infant, the patient did not show or report any signs of breathing difficulties at the time of examination. The patient's blood tests were all within normal ranges, including complete blood counts serum calcium, serum phosphate, and serum alkaline phosphatase.
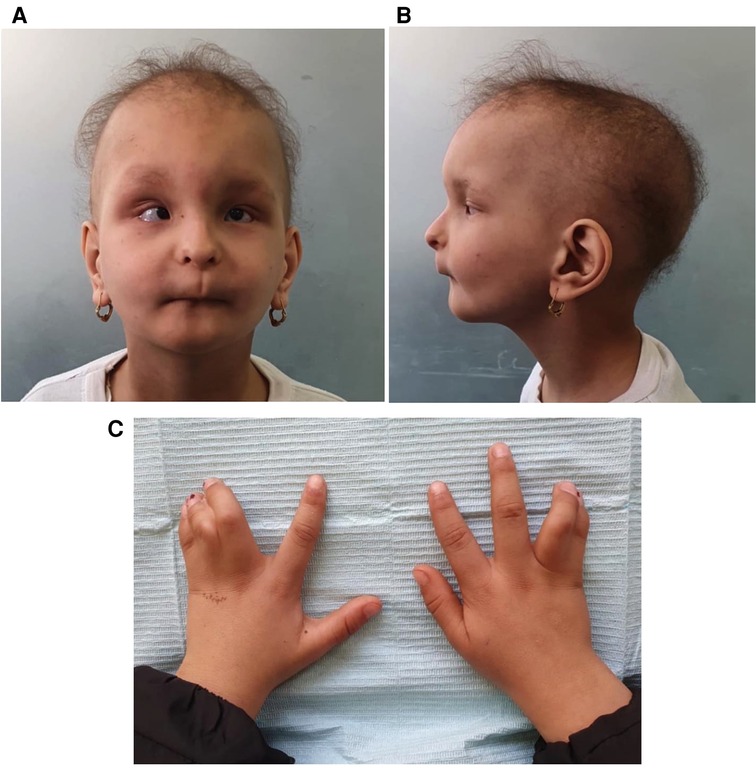
Figure 1. (A) An extraoral frontal photograph showing the distinctive facial features of the syndrome, (B) a lateral view, (C) syndactyly.
A clinical examination of the temporomandibular joint showed that the condyles on both sides were undetectable by palpation. Mouth opening showed slight restriction; it was almost 34-mm wide. The intraoral examination revealed a plethora of abnormalities, including microstomia and a high and narrow palatal vault (Figure 2). All teeth showed microdontia and enamel hypoplasia. The patient had mixed dentition (#16, #55, #54, #53, #11, #21, #62, #64, #26, #36, #73, #72, #71, #41, #82, #46) with severe caries and multiple premature tooth loss (#52, #63, #74, #75, #84, #85). The parents reported that these teeth were never extracted, rather they were so weak and fragile that their crowns gradually chipped away until there was no more left. The presence of the remaining roots (#65, #75, #85) was noted where the parents had indicated that teeth had “chipped away.” The permanent first molars were erupted, hypoplastic, and extensively decayed with open apices and thin roots. They also showed mild taurodontism. Tooth #62 was discolored and necrotic. Considering the chronological age of the patient, it is notable that there were several prematurely erupted permanent teeth (#11, #21, #16, #26, #36, #46). Although congenitally missing teeth are a common manifestation in HSS patients, this patient had no congenitally missing teeth. The permanent tooth buds expected to be seen in a 5-year-old patient were present in the radiographs. The mandibular alveolar ridge was thin and sharp with a very short sulcus depth.
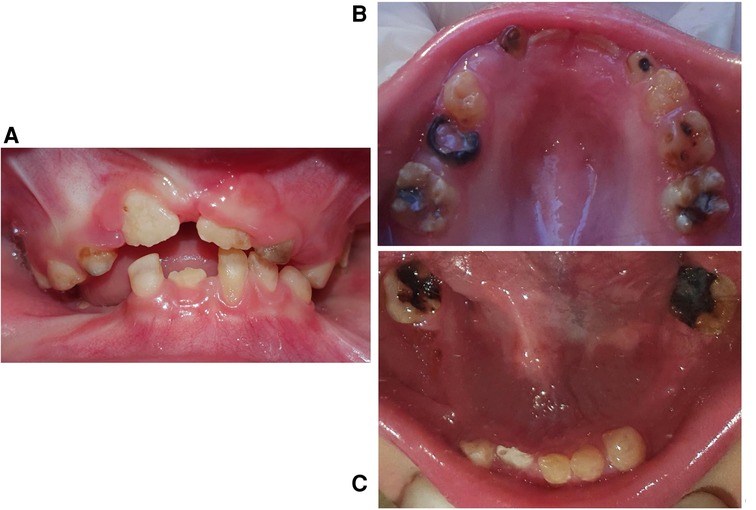
Figure 2. Intraoral photographs. (A) A frontal view in occlusion, (B) maxillary arch, (C) mandibular arch.
Radiographs were taken, which revealed that all of the dentitions had a ghost teeth-like appearance. An inspection of the panoramic radiograph revealed the absence of bilateral condylar and an obtuse mandibular angle (Figure 3). Periapical x-rays revealed that the roots were extremely thin and poorly calcified. Furthermore, the pulpal chambers contained multiple separate pulp calcifications (Figure 4).
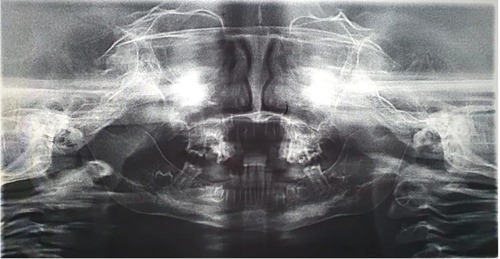
Figure 3. A panoramic radiograph showing the absence of mandibular condyles and ghost teeth appearance.
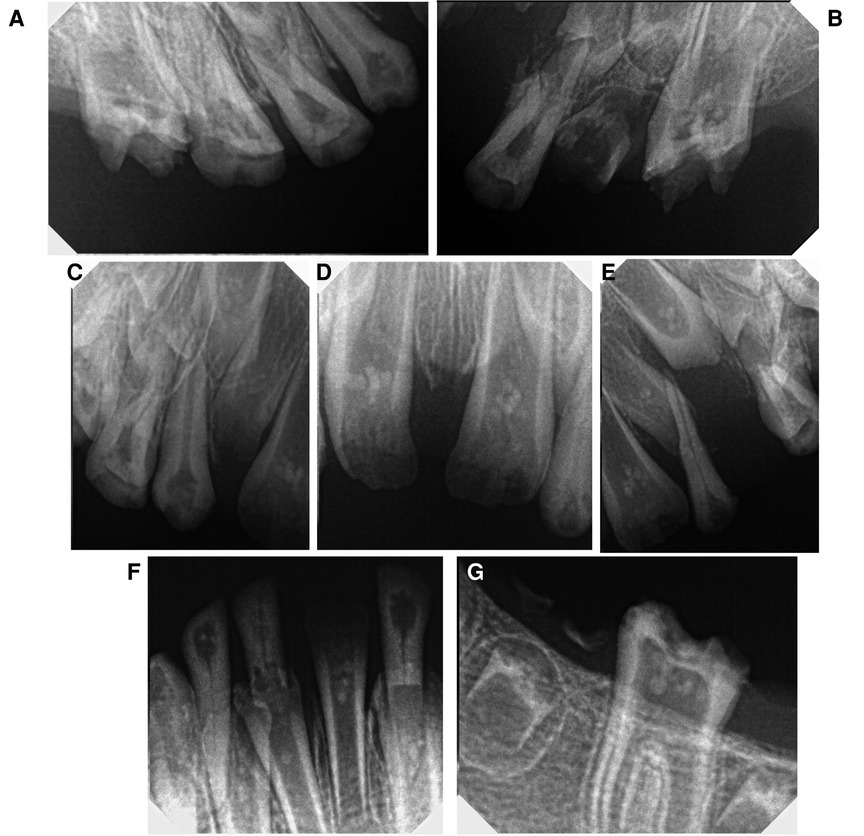
Figure 4. Periapical radiographs showing pulp calcifications. (A) Left maxillary, (B) right maxillary, (C–E) anterior maxillary region, (F) anterior mandibular, (G) right mandibular.
The child showed tense cooperative behavior in the first dental visit, and some difficulties were encountered with communication. However, due to the known obstacles in anesthetizing and intubating HSS patients caused by their upper airway malformations, treatment under general anesthesia was excluded (8).
Management proceeded with the regular sequence of preventive, restorative, surgical, prosthetic, and maintenance phases. Pediatric dental behavior management techniques were used, including tell, show and do, distraction, modeling, positive reinforcement, and voice modulation, to which the patient responded favorably. The preventive phase included oral hygiene instructions, dietary counseling, prophylaxis, and topical fluoride varnish application. Then, the restorative phase included pulp capping of the permanent first molars with Mineral Trioxide Aggregate (MTA) and restoration with stainless-steel crowns, and the same was done for #55. The Silver Modified Atraumatic Restorative Technique (SMART) was used to restore #53 as the patient had become uncooperative after several visits (Figure 5).
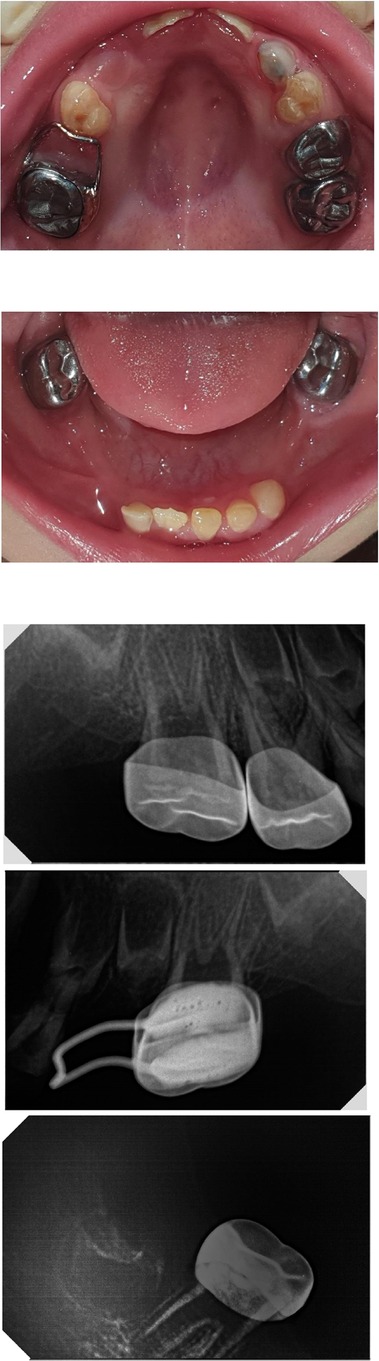
Figure 5. Postoperative radiographs and photographs.
The surgical phase included the extraction of #62 and the remaining roots. This was followed by the prosthetic phase, where a band and loop space maintainer was fabricated for the lost #65. A removable partial denture (RPD) was the best choice for restoring the masticatory function and aesthetics for the mandibular arch.
The patient was scheduled for a monthly follow-up for the first 3 months and then for a follow-up at 3-month intervals. At each follow-up visit, the integrity of the restorations was checked and the remaining teeth closely examined for the development of new caries lesions. Oral hygiene instructions were reinforced at every visit, and the patient and her parents were also provided with age-appropriate anticipatory guidance. As for the partial denture, it will be continuously adjusted and refitted to conform to the developing arches of the child in a non-restrictive manner and to accommodate for the eruption of the successor teeth. To ensure this, a new partial denture will be fabricated at yearly intervals. After 2 months of follow-up, all restorations remained successful, no new caries lesions developed, and the patient was content and compliant with the use of the RPD. Her improved oral hygiene reflected her compliance with the given oral hygiene instructions and dental health education.
From the patient's perspective, the girl was excited to receive her new set of teeth (the RPD) to help her in eating and so that she could have teeth like her friends. She developed an interest in her oral health and became more involved with her oral hygiene. Now, she feels a lot of relief after a decrease in her dental hypersensitivity decreased (due to restoration with stainless-steel crowns and the use of sodium fluoride varnish). Similarly, from the parents' perspective, they felt relieved after finding specialized dental care for their daughter, as they faced a hard time finding a dentist willing to accept their child's case. They live in a relatively rural area, and the availability of specialized pediatric dental care is scarce, especially for a child with a rare syndrome. They had to travel for 4 h every time to reach the clinics, where their daughter would receive the needed specialized dental care. They are also relieved that their child can now eat more solid foods with the help of her RPD and restorations, as the patient has been relying on semisolid foods and snacks for nutrition, which proved to be a great obstacle for them in providing her with a balanced non-cariogenic diet.
Discussion
HSS is a rare genetic syndrome classified under disorders of ectodermal dysplasia. The diagnosis is made based on a host of recognizable classic craniofacial features, including the following: brachycephaly, frontal bossing, strabismus, microphthalmia, congenital cataracts, beak-shaped nose, malar and mandibular hypoplasia, hypotrichosis, and skin atrophy (9), which were all present in our patient.
Notably, this patient manifested multiple pulpal calcifications of unknown origin in both primary and permanent dentitions. To our knowledge, these pulp calcifications had not been previously reported in a case of HSS (10–14). They are also not a common finding in young-age patients. In our attempt to investigate the etiology of these calcifications, we requested that the results of the calcium, phosphate, and alkaline phosphatase tests be given for our examination, following which we found that serum levels were all within normal range. Similar calcifications are known to arise consequent to chronic irritation of the dentino-pulpal complex caused by chronic caries activity or trauma (15). We postulate that these pulp stones may have arisen due to chronic irritation of the non-differentiated ectomesenchymal cells in the pulp complex. It is possible that the severe hypersensitivity (which was part of the patient's chief complaint) would have triggered these cells present in the coronal pulp to react upon the formation of such calcifications (reactionary dentin). This theory is supported by the fact that these calcifications were found in sound and carious teeth alike and with no history of trauma reported by the parents. However, the exact etiology of the development of pulp stones remains unknown and may be of idiopathic origin (16).
The dental abnormalities manifested in HSS patients are similar to those seen in other hereditary diseases, an example being the clinical presentation of dentin dysplasia. Dentin dysplasia type I (radicular type) can cause the formation of underdeveloped roots with a pointed apex. In most instances, teeth have normally colored enamel. Occasionally, a bluish-brown shine can be seen in the crowns. Radiographically, crescent-shaped calcifications can be seen in the pulp chamber, as well as abnormally radiolucent roots. These calcifications increase in size progressively and can cause the obliteration of the pulp space. However, the morphology and color of the coronal part appears normal, and occasionally teeth appear slightly amber-colored or bluish-brown. In contrast, type II dental dysplasia (coronal type) causes the crowns to have a characteristic opalescence with an abnormal color (brownish-blue, brown, amber). Teeth show normal roots containing enlarged pulp chambers with abnormal extensions toward the root, often described as “thistle tube” shaped on dental radiographs. Moreover, many pulp stones can be often found in the pulp chambers (17, 18).
Dentinogenesis imperfecta is yet another hereditary disorder of dentin formation and tooth development. Three distinct types are recognized. Type I occurs concomitant with osteogenesis imperfecta, type II shows characteristic bulbous crowns with a constricted cervix, and type III manifests with variable clinical presentations similar to types I and II. Generally, dentinogenesis imperfecta is characterized by bluish-brown or amber opalescent teeth. The coronal tooth structure is often weak and liable to chipping and fracture, often leading to a complete wear of the crown till only the roots remain. Pulpal obliteration is a common finding in radiographs, often seen before or soon after eruption (18). Although the phenotype of these hereditary disorders may overlap, especially in the dental manifestations, it is imperative that a complete clinical picture of the patient, medical history, and family history is taken into consideration when formulating a diagnosis (19).
Due to the root condition and the abnormal pulp calcifications in this case, every effort was made to avoid pulp exposure during careful caries removal. It was determined that preservation of the pulp vitality was of utmost importance to allow the completion of root formation and to avoid complete pulpal extirpation if there was a need to endodontically treat the permanent molars. In the case of accessing the pulp chambers, hemorrhage control would be difficult to achieve, and the instrumentation and obturation of such weak roots will likely lead to root fractures. MTA was the best choice for pulp capping as it has shown superior outcomes in vital pulp therapy (20).
Restoration of the posterior teeth was done using stainless-steel crowns. A definitive permanent restoration will be done after the full emergence of the crowns and root formation. Stainless-steel crowns have the highest success rates in the restoration of molars in children and adolescents (21). Their use will provide the additional benefit of partially restoring the patient's lost vertical dimension caused by premature tooth loss. A severely decreased vertical dimension leads to an overclosure of the jaws and chronically strains the temporomandibular joint.
The prosthetic phase focused on space maintenance and rehabilitating the dental arches to restore function and vertical dimension of occlusion. Due to the thin hypoplastic ridge of our patient, denture stability and retention were expected to be compromised. A removable partial denture was chosen for the mandibular arch, as the patient had been complaining of inefficiency in mastication. Flexible dentures and overdentures are a possible treatment modality for patients with similar condition (22).
HSS patients have deformed teeth with unusual root morphology and enamel hypoplasia; they are highly predisposed to severe dental caries, making excellent dental hygiene of paramount importance. Due to the technical difficulties associated with the endodontic treatment of teeth with underdeveloped roots, which would be particularly compounded while operating on pediatric patients with no possibility of general anesthesia, these patients require thorough and periodic dental checkups. Our patient was closely followed up at regular intervals after the treatment was completed. The importance of oral hygiene and regular dental checkups has been clearly explained and repeatedly reinforced to both the parents and the child.
The strength of this case report lies in the reporting of the pediatric dental management of this rare syndrome, as well as reporting atypical pulpal calcification, for which no local or systemic etiology was found. Regarding the limitations of this report, it can be stated that there was no long-term follow-up of this case to build on the success of the provided treatments and to monitor the dental and oral growth and development of children with HSS. There is a gap in the knowledge regarding the pediatric treatment modalities offered for HSS patients. No consensus on the dental management needed for these patients starting from a young age is available. Although this syndrome is usually diagnosed at infancy by its characteristic facial features, there is a deficient number of publications discussing the pediatric dental treatment of these patients (10–14). The limited pool of patients with HSS provides a great obstacle to clinicians and researchers to adequately study the dental features of this syndrome and accordingly agree on the most suitable treatment options. Invariably, prevention is the best strategy, and treatment plans need to be modified on a case-to-case basis.
Conclusion
Patients presenting with rare syndromes such as HSS require close dental follow-up from an early age. Factors such as the patient’s age, past dental experience, and the overall state of the dentition and arches, as well as the psychological factors, such as the patient's and parents' needs and expectations, must be taken into consideration during treatment planning. The available reports in the literature are scant and rarely focus on pediatric dental management.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, and further inquiries can be directed to the corresponding author/s.
Ethics statement
Ethical review and approval were not required for the study on human participants in accordance with the local legislation and institutional requirements where this case has been reported. Written informed consent for publication was obtained from the minor's legal guardian for the publication of any potentially identifiable images or data included in this article.
Author contributions
NA conceptualized the case report and performed the clinical treatment. MME supervised the clinical treatment and contributed to treatment planning. NA wrote the initial draft of this manuscript. MME critically reviewed the manuscript. NA and MME approved the final manuscript submission. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Genetic and Rare Diseases Information Center. Hallermann–Streiff syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program (2020). Available at: https://rarediseases.info.nih.gov/diseases/288/hallermann-streiff-syndrome#ref_5346 (cited April 29, 2022).
2. Cohen MM. Hallermann–Streiff syndrome: a review. Am J Med Genet. (1991) 41(4):488–99. doi: 10.1002/ajmg.1320410423
3. François J. A new syndrome: dyscephalia with bird face and dental anomalies, nanism, hypotrichosis, cutaneous atrophy, microphthalmia, and congenital cataract. AMA Arch Ophthalmol. (1958) 60(5):842–62. doi: 10.1001/archopht.1958.00940080862005
4. Schmidt J, Wollnik B. Hallermann–Streiff syndrome: a missing molecular link for a highly recognizable syndrome. Am J Med Genet Part C: Semin Med Genet. (2018) 178(4):398–406. doi: 10.1002/ajmg.c.31668
5. Preudhomme R, Veyssiere A, Ambroise B, Benateau H. Hallermann–Streiff syndrome: cranio-facial manifestations systematic review and report of two cases. J Stomatol Oral Maxillofac Surg. (2021) S2468-7855(21)00242-1. doi: 10.1016/j.jormas.2021.11.002
6. Pizzuti A, Flex E, Mingarelli R, Salpietro C, Zelante L, Dallapiccola B. A homozygous GJA1 gene mutation causes a Hallermann–Streiff/ODDD spectrum phenotype. Hum Mutat. (2004) 23(3):286. doi: 10.1002/humu.9220
7. Gagnier JJ, Kienle G, Altman DG, Moher D, Sox H, Riley D, et al. The CARE guidelines: consensus-based clinical case reporting guideline development. J Med Case Rep. (2013) 7(1):1–6. doi: 10.1186/1752-1947-7-223
8. Srinivasan LP, Viswanathan J. Hallermann–Streiff syndrome: difficulty in airway increases with increasing age. J Clin Anesth. (2018) 50:1. doi: 10.1016/j.jclinane.2018.06.024.
9. Cohen MM. Hallermann–Streiff syndrome: a review. Am J Med Genet. (1991) 41(4):488–99. doi: 10.1002/ajmg.1320410423
10. von Marttens A, Perilla A, Wen S, Acuña P, Beltrán V. Implant-supported maxillary and mandibular rehabilitation in a patient with Hallermann–Streiff syndrome. J Craniofac Surg. (2021) 32(1):e20–3. doi: 10.1097/SCS.0000000000006832
11. Defraia E, Marinelli A, Alarashi M. Case report: orofacial characteristics of Hallermann–Streiff syndrome. Eur J Paediatr Dent. (2003) 4(3):155–8.14529338
12. Damasceno JX, Pimenta Couto JL, Da Silva Alves KS, Chaves CM, Gurgel Costa FW, De Menezes Vieira Pimenta A, et al. Generalized odontodysplasia in a 5-year-old patient with Hallermann–Streiff syndrome: clinical aspects, cone beam computed tomography findings, and conservative clinical approach. Oral Surg Oral Med Oral Pathol Oral Radiol. (2014) 118(2):e58–64. doi: 10.1016/j.oooo.2014.04.013
13. Vadiakas G, Oulis C, Tsianos E, Mavridou S. A typical Hallermann–Streiff syndrome in a 3 year old child. J Clin Pediatr Dent. (1995) 20(1):63–8.8634201
14. Dulong A, Bornert F, Gros CI, Garnier JF, Van Bellinghen X, Fioretti F, et al. Diagnosis and innovative multidisciplinary management of Hallermann–Streiff syndrome: 20-year follow-up of a patient. Cleft Palate-Craniofacial J. (2018) 55(10):1458–66. doi: 10.1177/1055665618765829
15. Borkar N, Jaggi S, Pandit V, Shetty S. Calcific metamorphosis: a review. Int J Health Sci (Qassim). (2016) 10(3):437. doi: 10.17159/2519-0105/2020/v75no6a5
16. Goga R, Chandler NP, Oginni AO. Pulp stones: a review. Int Endod J. (2008) 41(6):457–68. doi: 10.1111/j.1365-2591.2008.01374.x
17. Carroll MKO, Duncan WK, Perkins TM. Dentin dysplasia: review of the literature and a proposed subclassification based on radiographic findings. Oral surgery. Oral Med Oral Pathol. (1991) 72(1):119–25. doi: 10.1016/0030-4220(91)90202-N
18. Barron MJ, McDonnell ST, MacKie I, Dixon MJ. Hereditary dentine disorders: dentinogenesis imperfecta and dentine dysplasia. Orphanet J Rare Dis. (2008) 3(1):1–10. doi: 10.1186/1750-1172-3-31
19. Beattie ML, Kim JW, Gong SG, Murdoch-Kinch CA, Simmer JP, Hu JCC. Phenotypic variation in dentinogenesis imperfecta/dentin dysplasia linked to 4q21. J Dent Res. (2006) 85(4):329–33. doi: 10.1177/154405910608500409
20. Mente J, Hufnagel S, Leo M, Michel A, Gehrig H, Panagidis D, et al. Treatment outcome of mineral trioxide aggregate or calcium hydroxide direct pulp capping: long-term results. J Endod. (2014) 40(11):1746–51. doi: 10.1016/j.joen.2014.07.019
21. Felemban O, Alagl H, Aloufi W, El Meligy O. Success rate of stainless-steel crowns placed on permanent molars among adolescents. Int J Clin Pediatr Dent. (2021) 14(4):488. doi: 10.5005/jp-journals-10005-1982
Keywords: Hallermann–Streiff syndrome, pediatric, dental, case report, management, pediatric dentistry, endodontic, treatment
Citation: Ammar N and El-Tekeya MM (2022) Hallermann–Streiff syndrome: Case report with abnormal pulp calcifications. Front. Dent. Med 3:965560. doi: 10.3389/fdmed.2022.965560
Received: 9 June 2022; Accepted: 25 July 2022;
Published: 12 August 2022.
Edited by:
Yelda Kasimoglu, Istanbul University, TurkeyReviewed by:
Maryam Tofangchiha, Other, IranPriyam Jani, National Institute of Dental and Craniofacial Research (NIH), United States
© 2022 Ammar and El-Tekeya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nour Ammar, bm91ci5hbW1hckBhbGV4dS5lZHUuZWc=
†ORCID:
Nour Ammar
orcid.org/0000-0002-7654-293
Specialty Section: This article was submitted to Pediatric Dentistry, a section of the journal Frontiers in Dental Medicine