 Beatriz Bezerra
Beatriz Bezerra Sepehr Monajemzadeh
Sepehr Monajemzadeh Davi Silva
Davi Silva Flavia Q. Pirih
Flavia Q. Pirih- University of California Los Angeles School of Dentistry, Los Angeles, CA, United States
Periodontitis is a chronic inflammatory condition initiated by the accumulation of bacterial biofilm. It is highly prevalent and when left untreated can lead to tooth loss. The presence of bacterial biofilm is essential for the initiation of the inflammatory response but is not the sole initiator. Currently it is unknown which mechanisms drive the dysbiosis of the bacterial biofilm leading to the dysregulation of the inflammatory response. Other players in this equation include environmental, systemic, and genetic factors which can play a role in exacerbating the inflammatory response. Treatment of periodontal disease consists of removal of the bacterial biofilm with the goal of resolving the inflammatory response; however, this does not occur in every case. Understanding the way the inflammatory response does not return to a state of homeostasis has led investigators to consider both systemic and local pharmacological interventions. Nonetheless, a better understanding of the impact that genetics and environmental factors may have on the inflammatory response could be key to helping identify how inflammation can be modulated therefore stopping the destruction of the periodontium. In this article, we will explore the current evidence associating the microbial dysbiosis and the dysregulation of the immune response, potential mechanisms or pathways that may be targeted for the modulation of the inflammatory response, and discuss the advantages and drawbacks associated with local and systemic inflammatory modulation in the management of periodontal disease. This information will be valuable for those interested in understanding potential adjunct methods for managing periodontal diseases, but not limited to, dental professionals, clinical researchers and the public at large.
Introduction
Periodontal disease is a chronic inflammatory disease of the oral cavity with its primary etiological factor being bacterial plaque. In the US, almost half of the population presents with some form of periodontal disease (1). Management of periodontal disease consists of control of the bacterial biofilm, the primary etiological factor, however, the progression of the disease is highly dependent on the inflammatory response triggered by the bacterial plaque.
The host response to the supra- and subgingival biofilm accumulation can be a double-edge sword. It can be potentially protective, in which symbiosis between host-response and the biofilm is maintained, thus maintaining the tissue hemostasis where no periodontal tissue destruction is noted. Or it can be destructive, where a dysregulated immune response can assist in a shift in biofilm composition, giving way to growth of more pathogenic bacteria, which will continue to activate the host immune response leading to a positive feedback loop of continued tissue destruction and maintenance of a dysbiotic microbial community (2). Studies have demonstrated a relationship between dysbiosis and inflammation, where inflammation fuels growth of dysbiotic communities and this dysbiosis exacerbates inflammation leading to destruction of periodontal tissues (3, 4).
Even though periodontitis is highly prevalent, its severity varies widely, and only a small proportion of the population presents with severe forms of the disease (1). This variation has been demonstrated by several studies (5, 6), however, it is still poorly understood why some individuals are more susceptible than others in developing periodontitis. Susceptibility has been proposed to be determined by several factors such as genetics, epigenetic, environmental, aging and systemic conditions which can modify the host immune response toward a protective or destructive pathway.
As a complex chronic inflammatory disease (7), periodontitis presents with a non-linear progression, where it has been demonstrated that the disease presents periods of remission, or stability, and progression (8, 9). These cycles between stability and progression are still poorly understood, as several factors could be simultaneously playing a role in dysregulating the host immune response. One thing is certain, the immune response is the main culprit for the tissue damage observed in periodontitis, and it is reasonable to propose host-modulating approaches for the management of periodontal disease as adjunct therapies for mechanical debridement.
In this review we will summarize the current concept of dysbiosis as it applies to the pathogenesis of periodontal disease, discuss potential mechanisms targeting modulation of the inflammatory response, and finally discuss advantages and drawbacks associated with host modulation therapy (HMT) for the management of periodontal disease.
Periodontal Disease Pathogenesis and the Role of the Host Immune Response on Dysbiosis
The role of bacteria as the primary etiological factor for periodontal disease was initially established by the classic study of Loe et al. (5) which confirmed that presence of plaque was necessary to elicit inflammatory changes to the gingival tissues in humans. Several studies followed which attempted to expose the role of specific bacteria in the progression of periodontal disease (10). As evidence accumulated, the role of the host immune response on the pathogenesis of periodontal disease has now extended beyond the model proposed by Page and Schroeder (11). Longitudinal studies have demonstrated that the presence of bacteria is necessary but not sufficient for development of periodontal disease, and support that a dysregulated immune response along with uncontrolled inflammation are responsible for the tissue destruction observed in periodontitis (12–14).
The polymicrobial synergy and dysbiosis theory (PSD) proposes that a synergistic polymicrobial community is responsible for initiating disease as several factors within this community lead to a dysregulation of the host inflammatory response (15). As disease progresses, the interactions between the polymicrobial community and the host immune response become a continuous cycle that perpetuates the dysbiosis leading to the tissue damage observed in periodontitis.
As bacterial biofilm accumulates in the supra and subgingival environment, the host responds with an inflammatory response aimed at maintaining homeostasis. If this inflammatory response is not properly resolved its persistence leads to the dysbiosis of the polymicrobial community. As a consequence of the dysbiotic community, a positive feedback loop is established where inflammatory tissue breakdown products serve as nutrients for the outgrowth of pathobionts (3, 16) and the dysbiotic microbial community continues to activate the immune response (4) leading to tissue damage. This self-sustained positive feedback loop is consistent with the chronic characteristic of periodontitis (Figure 1).
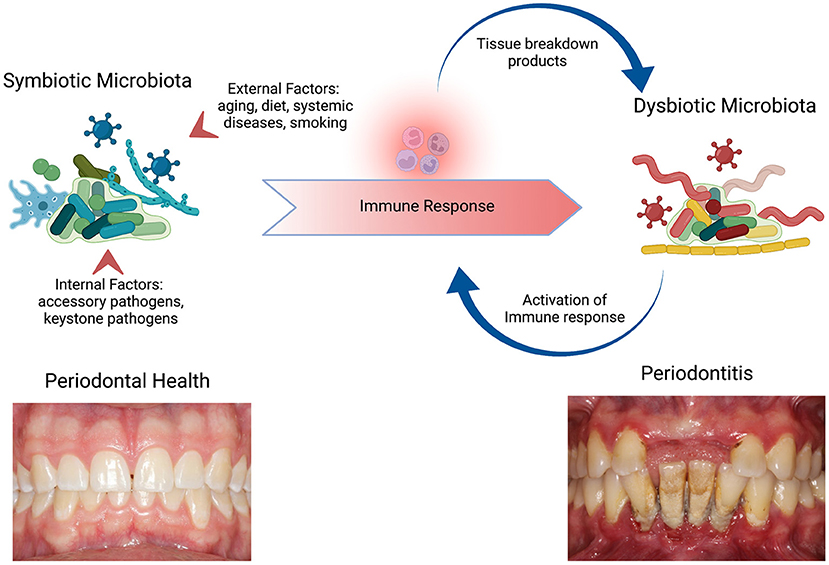
Figure 1. Periodontal disease pathogenesis and the role of the immune response on dysbiosis. Periodontal health is maintained by a homeostasis between the symbiotic microbiota and the host immune response. The internal factors to the polymicrobial community, such as the colonization by keystone pathogens, and external factors, such as smoking, leads to alterations within the microbiota and the immune response to the new polymicrobial community, that is dysbiotic. The availability of nutrients (tissue breakdown products) promotes further growth of pathobionts leading to continuous activation of the immune response and consequently further tissue breakdown perpetuating the cycle. As a result, a self-sustaining loop is established and underlies the chronicity of periodontitis. Adapted from “The Oral Microbiome,” by http://BioRender.com (2022). Retrieved from https://app.Biorender.com/biorender-templates.
In vitro studies have shown that the availability of products derived from inflammatory tissue breakdown, such as amino acids, iron, etc., drives the outgrowth of pathogens which perpetuate the dysbiotic environment in the periodontal pocket (4, 15, 17). The inflammatory changes in the subgingival environment also have been known to elevate the expression of certain genes in subgingival bacteria that assist these pathobionts in exploiting these changes for their benefit (15, 18). Animal studies demonstrated that the inflammatory lesion observed in periodontitis contributes to the shift in the microbial community to one that is more gram-negative, anaerobic and proteolytic (19, 20).
As previously noted, the host response to the bacterial stimulus varies amongst individuals and this variation is due to a wide variety of determinants that play a role in the regulation of the immune response. A combination of genetic, environmental and lifestyle factors, and more recently, epigenetic factors, can affect an individual's susceptibility to and severity of periodontal disease (21, 22).
The site-specific presentation of periodontitis suggests that local factors could be playing a role in regulating genes associated with inflammation. The local factors present at the biofilm-gingival interface of the tooth could be driving the epigenetic modifications locally and resulting in an hyperinflammatory response (23). These modifications can not only be a direct result of bacteria and their products on epithelial cells, but also on inflammatory cells recruited to the area (24, 25). Currently, alterations in several genes involved in the inflammatory response in gingival tissue (23), peripheral blood and buccal mucosa (26–28) of periodontitis patients have been identified.
It is clear that a dysregulated inflammatory response is responsible for the tissue destruction observed in periodontitis. Current therapeutic methods for the management of periodontal disease focus mainly on the removal of bacterial plaque, however, it has been shown that this approach is not enough to halter the destruction of the periodontal tissues in some cases. With the current knowledge of the role of the inflammatory response on the destruction of these tissues, alternative methods targeting the immune response could provide clinicians with additional tools to manage periodontal disease.
Host-Modulation Strategies in the Management of Periodontitis
The goal of HMT is to manipulate the immune response in order to prevent or ameliorate tissue damage, where hemostasis is achieved between the polymicrobial community and the host immune response. As we increase our understanding of the different inflammatory mechanisms involved in periodontal inflammation and tissue destruction, we can potentially identify therapeutic alternatives to be employed as adjuncts to mechanical debridement. On this review we will focus on therapies that present in vivo evidence, both animal and/or clinical studies, of improvement and/or prevention of periodontal tissue destruction. HTM strategies will be presented in a chronological order as we discuss the evolution of these strategies and their applications in the management of Periodontitis. A summary of these strategies can is available on Table 1.

Table 1. Host Modulation Therapy (HTM) agents in the treatment of periodontitis.
Modulation of Arachidonic Acid Metabolites
Tissue damage caused by bacteria and/or the host immune response exposes phospholipids from plasma membranes which interact with phospholipase A2 promoting the release of arachidonic acid (AA). This cascade of events leads to the production of prostaglandins (PG) via the action of the cyclooxygenase enzyme (COX). Elevated levels of PGE2 have been identified in gingival crevicular fluid of patients with gingivitis, periodontitis and periimplantitis. PGE2 has even been reported to correlate with disease progression (75). The blockade of COX enzymes via use of NSAIDs has demonstrated the ability to inhibit bone resorption in both animal (33–36) and clinical studies (9, 37). The effects of these drugs, however, did not remain after withdrawal of the drug (29).
The use of both non-selective and selective COX inhibitors come with a range of side effects, as will any therapy that is provided systemically. With the current knowledge that this agent's benefits rapidly decline after stopping its use, its long-term use is not recommended due to its adverse effects. Adverse effects include damage to the gastrointestinal mucosa, renal toxicity and pro-thrombotic side effects (30–32).
Matrix Metalloproteinase Inhibitors
Matrix metalloproteinases (MMPs) are proteolytic enzymes responsible for the breakdown of collagen. MMPs have been implicated in a variety of pathological conditions including periodontitis (76) and several resident and infiltrating cells within the extracellular matrix of the periodontium are able to secrete them. A balance between collagen breakdown and synthesis is achieved by endogenous tissue inhibitors of MMP (TIMPs) under healthy conditions (77). MMP-8 and MMP-9 are the predominant MMPs responsible for collagen breakdown in periodontitis, which are normally secreted by neutrophils (45, 46), however, gingival fibroblasts, epithelial cells and monocyte/macrophages are also able to secrete them (78, 79).
The modulation of the host's collagenolytic response as an HMT targets the action of MMPs against the periodontium. As collagen is the most abundant component of the extracellular matrix in the periodontal tissues, targeting the mechanism responsible for collagen breakdown would be beneficial for managing periodontal tissue destruction.
Tetracyclines have demonstrated inhibitory activity against MMPs at low concentrations, called subantimicrobial doses (80). A series of in vitro, in vivo, and clinical studies have demonstrated effects of doxycycline in the inhibition of MMPs in gingival tissues and gingival crevicular fluid and concomitant improvements in periodontal status (45–47). Of the available tetracyclines, doxycycline has demonstrated to be more effective at inhibiting MMP-8, which is highly predominant in periodontitis (46, 81). Subantimicrobial dose doxycycline (SDD) has demonstrated no adverse side effects, including no impact on antibiotic resistance on either subgingival (48, 80) nor intestinal microbiota (82). The effect of SDD lies on the modulation of pathologic levels of MMPs which are secreted in excess as a result of the inflammatory process.
Clinical studies investigating the use of SDD as an adjunct to nonsurgical periodontal treatment have demonstrated that for benefits to be noted, at least a 3-month regimen is necessary (49). Among the clinically relevant benefits demonstrated by these studies is an increased percentage of sites presenting probing depth reductions of 2 mm or more (50, 51). Long-term use of this HMT, treatments ranging from 3 months to 2 years, has shown to provide prolonged benefits, where studies have demonstrated the clinical benefits were maintained for at least 3 months after stopping treatment (38–43). Benefits were also noted when using SDD as an adjunct to periodontal surgery, where significant pocket depth reduction was achieved and maintained even after 6 months of discontinuing the medication (44).
As the only FDA-approved medication for the management of periodontitis, clinical studies have shown promising long-term effects on probing depth reduction and stability of clinical attachment loss. However, it is still not clear when to utilize it as an adjunct to periodontal treatment. Another aspect is patient compliance. As studies demonstrate that at least 3 months is required to see clinical benefits, patients must be committed to this regimen, including maintaining proper oral hygiene along with participating in maintenance recalls.
Most recently, chemically modified tetracyclines, which have been modified to eliminate the antibacterial activity of tetracycline while retaining the MMP inhibitory effects, have been developed. Chemically modified tetracycline-3 (CMT-3) showed major inhibitory activity against MMPs. Only one pilot clinical trial is available demonstrating the effects of low dose CMT-3 on periodontitis (52). This pilot study utilized 10mg of CMT-3 per day, instead of 50-100 mg per day as in previous cancer trials (53, 54). The authors observed a reduction in IL-1β and MMP-8 levels in the GCF of patients receiving scaling and root planing, when compared to placebo, with no adverse side effects.
Based on the limitations associated with tetracyclines, such as allergic reactions and adverse effects associated with pregnancy, the scope of use is limited. Recently, a novel compound derived from curcumin, has been developed at Stony Brook University, which has exhibited MMP inhibiting effects, including effects on MMP-9,−13, as well as suppressing inflammatory mediators IL-1β, IL-6 and PGE2 (55, 83). Chemically-modified curcumin-2.24 (CMC-2.24) has demonstrated anti-inflammatory effects as well as decreased bone loss in animal models of periodontal disease, with substantial impacts on IL-1β, MMP-9 and MMP-8 levels both locally (gingival tissues) and systemically (55–57). Currently no clinical trials are available testing the effects of CMC-2.24 as an adjunct for the treatment of periodontitis. Based on pre-clinical trials, the application of such molecules as HMT shows promises in the treatment and, potentially, prevention of periodontitis-induced bone loss.
Modulation of Cytokines
The activation of the immune response by the subgingival biofilm leads to the release of several pro-inflammatory cytokines which have been associated with the progression of periodontal bone loss. A several cytokines have been shown to play a role in the pathogenesis of periodontitis with its hallmark signs of bone loss and soft tissue destruction. These cytokines are secreted by different cells such as gingival epithelial cells, fibroblasts, endothelial cells, neutrophils among others present within the periodontal pocket environment.
The investigation on the use of anti-cytokine drugs for the management of periodontitis was by chance, as the goal of studies targeting the effects of these drugs was to investigate their efficacy on rheumatoid arthritis (RA) outcomes. Periodontitis and RA share similar inflammatory pathways, where overproduction of pro-inflammatory cytokines such as TNF-α and IL-6 have been implicated in the pathogenesis of both conditions (84). Some studies even suggesting a bidirectional link between these two chronic inflammatory conditions (85, 86). With such information is only natural to suggest that drugs currently used for the management of RA can potentially be used as an adjunctive therapy in the management of periodontal disease.
The investigation on the effects of anti-cytokine therapies on periodontal disease currently presents minimal evidence. The use of monoclonal antibodies, or receptor antagonists, to block the action of specific pro-inflammatory cytokines has been employed in the treatment of RA and findings on periodontal inflammation have been consequential as reported in the systematic review by Han and Reynolds (87).
More recently, an update on the topic provided by a systematic review and meta-analysis (88) investigating the influence of anti-rheumatic agents on the periodontal condition of RA patients with periodontitis demonstrated improvements in clinical parameters (bleeding on probing-BOP, probing depth-PD and clinical attachment levels-CAL) in patients who did not received periodontal treatment, and were treated with anti-IL-6R therapy (58, 59) and anti-TNF-α agents (60–62). This systematic review (88) points out that the studies included measured several periodontal parameters (PD, BOP, CAL) in patient with periodontitis, whereas the previous SR (87) included studies reporting only on BOP and GI, and RA patients with and without periodontitis.
Overall, the availability of clinical trials investigating the use of this drug class for the management of periodontal disease is low and this can be explained by the potential adverse effects that these drugs may pose to the patient's immune response, especially when being administered systematically. Among the adverse effects reported increased risk for serious bacterial, fungal and viral infections, drug-induced Lupus, lymphoma, just to list a few (89). Tissue destruction in periodontitis is the result of network of cytokines working in concert to activate different aspects of the immune response and blocking a single cytokine may not provide the expected effects ones is looking for.
Lipid Mediators of Resolution of Inflammation
Recent studies have demonstrated the presence of a pathway associated with the termination of acute inflammation (90, 91). Resolution of inflammation involves a sequence of events where pro-inflammatory mediators induce the production of specialized proresolving mediators, which in turn stimulate their receptor targets to inhibit different cells involved in the acute inflammatory response. This process seems to be initiated at the peak of the acute inflammatory response, and if the resolution pathway fails to resolve inflammation, chronicity ensues.
Specialized proresolving mediators (SPM) are lipid mediators derived from dietary omega-3 polyunsaturated fatty-acids (resolvins, protectins) or endogenous fatty acids (lipoxins) (92). These mediators function as receptor agonists that initiate proresolving pathways and consequently promote the resolution of inflammation and return to homeostasis.
Resolvins can be divided into two major groups based on their chemical structures: E-series, which is derived from eicosapentaenoic acid (EPA), and D-series, derived from docosahexaenoic acid (DHA). E-series have demonstrated effects on neutrophils (93, 94), whereas D-series affects platelet-leukocyte adhesion and production of molecules with anti-inflammatory and proresolution functions (95). Overall, Resolvins activate the hallmark functions of resolution of inflammation by stopping neutrophil infiltration, activating neutrophil apoptosis, recruiting macrophages to phagocytose apoptotic neutrophils to clear the lesion thus enhancing clearance of inflammation in the lesion to promote tissue regeneration (19, 96, 97).
Investigation of the effects of SPM on periodontitis have shown a protective effect of these mediators on experimentally-induced periodontal bone loss as well as the ability to regenerate periodontal tissues (19, 20, 64–66). These studies investigated the effects of topically delivered SPM on experimental periodontitis with successful outcomes. These studies also demonstrated a shift in biofilm composition after treatment with Resolvin E-1 (RvE1), which the authors hypothesize to be a result of the proresolution effects of RvE1 via the release of antimicrobial peptides, reduced inflammatory mediators and increase bacterial clearance by macrophages, which promotes a change in the biofilm environment.
Clinical studies utilizing these molecules are scarce as they have not yet received approval for clinical use. There is, however, clinical data on the use of dietary supplementation with omega-3 PUFAs in combination with aspirin and its impact on treatment outcomes on periodontal disease. The addition of aspirin promotes the acetylation of and changes the enzymatic function of COX2 triggering the production of E- and D-resolvins, instead of prostaglandins (98). These studies have demonstrated beneficial effects when used as adjuncts to non-surgical periodontal treatment, showing significant improvements in probing depth reduction, gain in clinical attachment levels in both systemically healthy (99, 100) and Type 2 diabetic patients (101, 102).
The first clinical trial to investigate the effects of SPM on gingival inflammation (63) utilized a topical delivery method (oral rinse) to investigate the efficacy of Lipoxin A4 mimetic (BLXA4). This Phase 1 clinical trial demonstrated that once a day rinse with BLXA4 was safe and well tolerated by the study participants, with no adverse effects. It also significantly reduced modified gingival index as compared to both placebo and no rinse group. Data also showed a steady and more pronounced reduction in bleeding on probing, probing depth and gain in clinical attachment, however these results were not significant.
The application of SPM as an HMT is an evolving field with promising effects on controlling periodontal tissue destruction and potentially leading to regeneration of lost tissues. As studies continue to dissect the mechanisms involved in resolution of inflammation via use of SPMs, this appears to be the most promising therapy as it has demonstrated thus far no adverse side effects with its application.
Complement Inhibitors
The complement system is responsible for regulating immunity and inflammation and it comprises of a network of over 50 proteins (103, 104). The activation of the complement system can follow three distinct pathways, however all three converge to the central complement component C3. C3 has been linked to periodontitis, where elevated levels have been noted in an experimental gingivitis study in humans (105) and successful periodontal treatment resulted in decreased C3 activation in gingival crevicular fluid (GCF) (106) and saliva (107). Animal studies also demonstrated that mice deficient in C3 did not exhibit periodontal bone loss in an experimental periodontitis model (108, 109).
Data from preclinical studies indicate that targeting the complement cascade could be a novel method for controlling the immune response in periodontitis and thus preventing tissue destruction. Different antagonists have been studied and demonstrated inhibition of alveolar bone loss as well as suppression of pro-inflammatory cytokines (67, 110, 111). A novel complement inhibitor targeting C3 (Cp40) has demonstrated inhibition of inflammation and bone loss in a non-human primate model of periodontitis when applied intragingivally (109). Other effects included inhibition of naturally occurring periodontitis in aging non-human primates even in the absence of periodontal treatment (68, 69), with effects of drug still present 6 weeks after session of treatment. The observed effects of inhibiting C3 component occurs via suppression of T-helper 17 cells. By blocking the action of the complement in activating Th17 cells, secretion of IL-17 is blocked and MMPs and TNF ligand superfamily cannot promote degradation of connective tissues and alveolar bone (112).
Safety concerns have been raised with this therapeutic application, as the complement system plays a role in antimicrobial defenses, which may increase the risk of infections. The studies evaluating systemic treatment with Cp40 demonstrated no significant differences in biochemical, immunological, and hematological parameters in blood or tissues when compared to vehicle alone (113). Intragingival application of the Cp40 result in no local irritation (68). Recently, the safety of this drug was tested in healthy volunteers and was demonstrated to be safe and well tolerated (114).
Current results of a Phase IIa clinical trial utilizing Cp40 for the treatment of periodontal inflammation (70) demonstrated that local delivery of Cp40, via intragingival injections, significantly reduced gingival inflammation with effects lasting at least 3 months after initial treatment. The authors noted that this improvement was mainly due to the host-modulating effect of the drug rather than a reduction in the amount of accumulated plaque. Treatment also led to a significant reduction in MMP-8 and−9 levels in the gingival crevicular fluid, which are key proteases associated with periodontal tissue destruction. Unfortunately, this trial was not able to assess the effects of the drug on periodontitis, as most of the patients included presented gingivitis.
Thus far, the safety and efficacy of the local application of Cp40 suggest that this could be a promising HMT for the treatment of periodontitis. Further studies are warranted to investigate its therapeutic effects on periodontitis and any potential systemic effects with its long-term application.
Epigenetic Drugs
As previously discussed, environmental and lifestyle factors, such as aging, systemic diseases and smoking, can induce epigenetic changes, modifying the DNA structure, which can contribute to the expression of genes associated with a hyperinflammatory state. DNA methylation and posttranslational modifications (PTMs) of histones have been implicated in alterations of the immune response (115) and demonstrated in gingival samples obtained from patients with periodontitis (26, 116–118).
Epigenetic drugs, or epidrugs, are drugs that target epigenetic markers responsible for the epigenetic alterations leading to inhibition or activation of disease-associated epigenetic proteins leading to improvement, cure, or prevention of the disease (119). These drugs have been widely studied in the treatment of cancer and information has emerged of their anti-inflammatory properties (71). Data from in vitro and animal studies have thus far demonstrated effects on suppression of alveolar bone loss (72), inhibition of pro-inflammatory cytokine production (73), and enhanced osteogenic differentiation (74).
These drugs target DNA methylation, histone acetylation and methylation, among other proteins associated with transcription-related processes. Some of them include histone deacetylase inhibitors (HDACi), histone methyltransferase inhibitors (HMTi), DNA methyltransferase inhibitors (DNMTi), bromodomain and extra-terminal domain protein inhibitors (BETi). Currently several epidrugs have received FDA approval for cancer therapy, including HDACis Vorinostat and Romidepsin, as well as DNMTis Vidaza and Decitabine, while several others are at pre-clinical and clinical trial phases (120).
The field of epigenetics in Periodontitis is expanding but very limited information on the role of epigenetic modifications on the pathogenesis of periodontal disease is available. As more studies become available and help elucidate the role of specific epigenetic modifications in the pathogenesis of periodontitis, we will be better equipped to apply the knowledge obtained from the use of epidrugs as an adjunct therapy in periodontitis.
Conclusions
As our knowledge of the pathogenesis of periodontitis increases, with a better understanding that the host immune response is the main culprit for the tissue destruction observed with periodontal disease, as well as its role in the dysbiosis of the microbial community, we are drawn to consider alternative methods to aid in the treatment of periodontitis. Understanding the different inflammatory mechanisms responsible for both soft and hard tissue destruction associated with periodontal disease allows us to target these processes implicated in tissue destruction. However, when modulating the host immune response, one must keep in mind that a single target may not be sufficient to halt the inflammatory events associated with periodontal attachment loss.
In this review we have discussed the available evidence regarding different methods of host modulation. Due to the chronicity of periodontitis, use of these medications many times need to occur for long periods of time for them to provide any clinical benefits, and this may lead to unwanted and, sometimes, serious adverse effects. HMT may be of particular interest for those patients who are at greater risk for experiencing attachment loss, such as patients with diabetes, smokers, or who do not respond well to non-surgical therapy.
Some of the available HMTs have demonstrated not only an impact on the inflammatory response, but also on the microbial community associated with periodontitis. By resolving inflammation, the dysbiotic community that was previously established returned to homeostatic conditions. These findings suggest that HMTs can also potentially be used to prevent periodontal disease, especially in high-risk populations. Future clinical studies investigating the effects of these drugs should not only address the clinical benefits as it reflects on reduced BOP, PD, and CAL, but also the long-term effects these drugs may possess, as the chronic nature of periodontal disease must be kept in mind. Delivery methods should also be investigated, as systemically administered drugs can potentially result in more adverse effects compared to local delivery.
Author Contributions
BB and FP contributed to the conceptualization of the manuscript and revision of the manuscript. BB, SM, and DS contributed to literature review and writing of the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fdmed.2022.879131/full#supplementary-material
References
1. Eke PI, Thornton-Evans GO, Wei L, Borgnakke WS, Dye BA, Genco RJ. Periodontitis in Us adults: national health and nutrition examination survey 2009-2014. J Am Dent Assoc. (2018) 149:576-88 e6. doi: 10.1016/j.adaj.2018.04.023
2. Hajishengallis G, Chavakis T, Lambris JD. Current understanding of periodontal disease pathogenesis and targets for host-modulation therapy. Periodontol 2000. (2020) 84:14-34. doi: 10.1111/prd.12331
3. Hajishengallis G. The inflammophilic character of the periodontitis-associated microbiota. Mol Oral Microbiol. (2014) 29:248-57. doi: 10.1111/omi.12065
4. Herrero ER, Fernandes S, Verspecht T, Ugarte-Berzal E, Boon N, Proost P, et al. Dysbiotic biofilms deregulate the periodontal inflammatory response. J Dent Res. (2018) 97:547-55. doi: 10.1177/0022034517752675
5. Loe H, Theilade E, Jensen SB. Experimental gingivitis in man. J Periodontol. (1965) 36:177-87. doi: 10.1902/jop.1965.36.3.177
6. Ramseier CA, Anerud A, Dulac M, Lulic M, Cullinan MP, Seymour GJ, et al. Natural history of periodontitis: disease progression and tooth loss over 40 years. J Clin Periodontol. (2017) 44:1182-91. doi: 10.1111/jcpe.12782
7. Bartold PM, Van Dyke TE. Host modulation: controlling the inflammation to control the infection. Periodontol 2000. (2017) 75:317-29. doi: 10.1111/prd.12169
8. Goodson JM, Haffajee AD, Socransky SS. The relationship between attachment level loss and alveolar bone loss. J Clin Periodontol. (1984) 11:348-59. doi: 10.1111/j.1600-051X.1984.tb01331.x
9. Jeffcoat MK, Williams RC, Reddy MS, English R, Goldhaber P. Flurbiprofen treatment of human periodontitis: effect on alveolar bone height and metabolism. J Periodontal Res. (1988) 23:381-5. doi: 10.1111/j.1600-0765.1988.tb01617.x
11. Page RC, Schroeder HE. Pathogenesis of inflammatory periodontal disease. a summary of current work. Lab Invest. (1976) 34:235-49.
12. Hajishengallis G, Darveau RP, Curtis MA. The keystone-pathogen hypothesis. Nat Rev Microbiol. (2012) 10:717-25. doi: 10.1038/nrmicro2873
13. Lamont RJ, Koo H, Hajishengallis G. The oral microbiota: dynamic communities and host interactions. Nat Rev Microbiol. (2018) 16:745-59. doi: 10.1038/s41579-018-0089-x
14. Page RC, Kornman KS. The pathogenesis of human periodontitis: an introduction. Periodontol 2000. (1997) 14:9-11. doi: 10.1111/j.1600-0757.1997.tb00189.x
15. Hajishengallis G, Lamont RJ. Breaking bad: manipulation of the host response by porphyromonas gingivalis. Eur J Immunol. (2014) 44:328-38. doi: 10.1002/eji.201344202
16. Cugini C, Klepac-Ceraj V, Rackaityte E, Riggs JE, Davey ME. Porphyromonas gingivalis: keeping the pathos out of the biont. J Oral Microbiol. (2013) 5. doi: 10.3402/jom.v5i0.19804
17. Diaz PI, Hoare A, Hong BY. Subgingival microbiome shifts and community dynamics in periodontal diseases. J Calif Dent Assoc. (2016) 44:421-35.
18. Duran-Pinedo AE, Chen T, Teles R, Starr JR, Wang X, Krishnan K, et al. Community-wide transcriptome of the oral microbiome in subjects with and without periodontitis. ISME J. (2014) 8:1659-72. doi: 10.1038/ismej.2014.23
19. Hasturk H, Kantarci A, Goguet-Surmenian E, Blackwood A, Andry C, Serhan CN, et al. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J Immunol. (2007) 179:7021-9. doi: 10.4049/jimmunol.179.10.7021
20. Lee CT, Teles R, Kantarci A, Chen T, McCafferty J, Starr JR, et al. Resolvin E1 reverses experimental periodontitis and dysbiosis. J Immunol. (2016) 197:2796-806. doi: 10.4049/jimmunol.1600859
21. Meyle J, Chapple I. Molecular aspects of the pathogenesis of periodontitis. Periodontol 2000. (2015) 69:7-17. doi: 10.1111/prd.12104
22. Offenbacher S, Barros SP, Beck JD. Rethinking periodontal inflammation. J Periodontol. (2008) 79(8 Suppl):1577-84. doi: 10.1902/jop.2008.080220
23. Barros SP, Offenbacher S. Modifiable risk factors in periodontal disease: epigenetic regulation of gene expression in the inflammatory response. Periodontol 2000. (2014) 64:95-110. doi: 10.1111/prd.12000
24. Barros SP, Hefni E, Nepomuceno R, Offenbacher S, North K. Targeting epigenetic mechanisms in periodontal diseases. Periodontol 2000. (2018) 78:174-84. doi: 10.1111/prd.12231
25. Diomede F, Thangavelu SR, Merciaro I, D'Orazio M, Bramanti P, Mazzon E, et al. Porphyromonas gingivalis lipopolysaccharide stimulation in human periodontal ligament stem cells: role of epigenetic modifications to the inflammation. Eur J Histochem. (2017) 61:2826. doi: 10.4081/ejh.2017.2826
26. Andia DC, de Oliveira NF, Casarin RC, Casati MZ, Line SR, de Souza AP. DNA methylation status of the Il8 gene promoter in aggressive periodontitis. J Periodontol. (2010) 81:1336-41. doi: 10.1902/jop.2010.100082
27. Kobayashi T, Ishida K, Yoshie H. Increased expression of interleukin-6 (Il-6) gene transcript in relation to Il-6 promoter hypomethylation in gingival tissue from patients with chronic periodontitis. Arch Oral Biol. (2016) 69:89-94. doi: 10.1016/j.archoralbio.2016.05.018
28. Shaddox LM, Mullersman AF, Huang H, Wallet SM, Langaee T, Aukhil I. Epigenetic regulation of inflammation in localized aggressive periodontitis. Clin Epigenetics. (2017) 9:94. doi: 10.1186/s13148-017-0385-8
29. Heasman PA, Offenbacher S, Collins JG, Edwards G, Seymour RA. Flurbiprofen in the prevention and treatment of experimental gingivitis. J Clin Periodontol. (1993) 20:732-8. doi: 10.1111/j.1600-051X.1993.tb00699.x
30. Dogne JM, Hanson J, Supuran C, Pratico D. Coxibs and cardiovascular side-effects: from light to shadow. Curr Pharm Des. (2006) 12:971-5. doi: 10.2174/138161206776055949
31. Hawkey CJ. Gastroduodenal problems associated with non-steroidal, anti-inflammatory drugs (Nsaids). Scand J Gastroenterol Suppl. (1993) 200:94-5. doi: 10.3109/00365529309101583
32. Lindsley CB, Warady BA. Nonsteroidal antiinflammatory drugs. renal toxicity. review of pediatric issues. Clin Pediatr. (1990) 29:10-3. doi: 10.1177/000992289002900101
33. Azoubel MC, Menezes AM, Bezerra D, Oria RB, Ribeiro RA, Brito GA. Comparison of etoricoxib and indomethacin for the treatment of experimental periodontitis in rats. Braz J Med Biol Res. (2007) 40:117-25. doi: 10.1590/S0100-879X2007000100015
34. Howell TH, Fiorellini J, Weber HP, Williams RC. Effect of the nsaid piroxicam, topically administered, on the development of gingivitis in beagle dogs. J Periodontal Res. (1991) 26(3 Pt 1):180-3. doi: 10.1111/j.1600-0765.1991.tb01643.x
35. Moro MG, Oliveira M, Oliveira LR, Teixeira SA, Muscara MN, Spolidorio LC, et al. Effects of selective versus non-selective cox-2 inhibition on experimental periodontitis. Braz Dent J. (2019) 30:133-8. doi: 10.1590/0103-6440201902241
36. Holzhausen M, Spolidorio DM, Muscara MN, Hebling J, Spolidorio LC. Protective effects of etoricoxib, a selective inhibitor of cyclooxygenase-2, in experimental periodontitis in rats. J Periodontal Res. (2005) 40:208-11. doi: 10.1111/j.1600-0765.2005.00787.x
37. Williams RC, Jeffcoat MK, Howell TH, Rolla A, Stubbs D, Teoh KW, et al. Altering the progression of human alveolar bone loss with the non-steroidal anti-inflammatory drug flurbiprofen. J Periodontol. (1989) 60:485-90. doi: 10.1902/jop.1989.60.9.485
38. Caton JG, Ciancio SG, Blieden TM, Bradshaw M, Crout RJ, Hefti AF, et al. Subantimicrobial dose doxycycline as an adjunct to scaling and root planing: post-treatment effects. J Clin Periodontol. (2001) 28:782-9. doi: 10.1034/j.1600-051X.2001.280810.x
39. Emingil G, Gurkan A, Atilla G, Kantarci A. Subantimicrobial-dose doxycycline and cytokine-chemokine levels in gingival crevicular fluid. J Periodontol. (2011) 82:452-61. doi: 10.1902/jop.2010.100036
40. Golub LM, Lee HM, Stoner JA, Reinhardt RA, Sorsa T, Goren AD, et al. Doxycycline effects on serum bone biomarkers in post-menopausal women. J Dent Res. (2010) 89:644-9. doi: 10.1177/0022034510363367
41. Golub LM, Lee HM, Stoner JA, Sorsa T, Reinhardt RA, Wolff MS, et al. Subantimicrobial-dose doxycycline modulates gingival crevicular fluid biomarkers of periodontitis in postmenopausal osteopenic women. J Periodontol. (2008) 79:1409-18. doi: 10.1902/jop.2008.070623
42. Payne JB, Reinhardt RA. Potential application of low-dose doxycycline to treat periodontitis in post-menopausal women. Adv Dent Res. (1998) 12:166-9. doi: 10.1177/08959374980120011401
43. Payne JB, Stoner JA, Nummikoski PV, Reinhardt RA, Goren AD, Wolff MS, et al. Subantimicrobial dose doxycycline effects on alveolar bone loss in post-menopausal women. J Clin Periodontol. (2007) 34:776-87. doi: 10.1111/j.1600-051X.2007.01115.x
44. Gapski R, Barr JL, Sarment DP, Layher MG, Socransky SS, Giannobile WV. Effect of systemic matrix metalloproteinase inhibition on periodontal wound repair: a proof of concept trial. J Periodontol. (2004) 75:441-52. doi: 10.1902/jop.2004.75.3.441
45. Golub LM, Lee HM, Greenwald RA, Ryan ME, Sorsa T, Salo T, et al. A matrix metalloproteinase inhibitor reduces bone-type collagen degradation fragments and specific collagenases in gingival crevicular fluid during adult periodontitis. Inflamm Res. (1997) 46:310-9. doi: 10.1007/s000110050193
46. Golub LM, Sorsa T, Lee HM, Ciancio S, Sorbi D, Ramamurthy NS, et al. Doxycycline inhibits neutrophil (Pmn)-type matrix metalloproteinases in human adult periodontitis gingiva. J Clin Periodontol. (1995) 22:100-9. doi: 10.1111/j.1600-051X.1995.tb00120.x
47. Caton JG, Ciancio SG, Blieden TM, Bradshaw M, Crout RJ, Hefti AF, et al. Treatment with subantimicrobial dose doxycycline improves the efficacy of scaling and root planing in patients with adult periodontitis. J Periodontol. (2000) 71:521-32. doi: 10.1902/jop.2000.71.4.521
48. Ciancio S, Ashley R. Safety and efficacy of sub-antimicrobial-dose doxycycline therapy in patients with adult periodontitis. Adv Dent Res. (1998) 12:27-31. doi: 10.1177/08959374980120011501
49. Caton J, Ryan ME. Clinical studies on the management of periodontal diseases utilizing subantimicrobial dose doxycycline (Sdd). Pharmacol Res. (2011) 63:114-20. doi: 10.1016/j.phrs.2010.12.003
50. Preshaw PM, Hefti AF, Novak MJ, Michalowicz BS, Pihlstrom BL, Schoor R, et al. Subantimicrobial dose doxycycline enhances the efficacy of scaling and root planing in chronic periodontitis: a multicenter trial. J Periodontol. (2004) 75:1068-76. doi: 10.1902/jop.2004.75.8.1068
51. Preshaw PM, Novak MJ, Mellonig J, Magnusson I, Polson A, Giannobile WV, et al. Modified-release subantimicrobial dose doxycycline enhances scaling and root planing in subjects with periodontal disease. J Periodontol. (2008) 79:440-52. doi: 10.1902/jop.2008.070375
52. Ryan M, Lee H, Tenzler R, Carnu O, Eftekhari S, Dhami A. Effects of short-term Col-3 on local biomarkers of periodontitis. J Dent Res. (2008) 87(special issue A: abstract number 0040).
53. Dezube BJ, Krown SE, Lee JY, Bauer KS, Aboulafia DM. Randomized phase Ii trial of matrix metalloproteinase inhibitor col-3 in aids-related kaposi's sarcoma: an aids malignancy consortium study. J Clin Oncol. (2006) 24:1389-94. doi: 10.1200/JCO.2005.04.2614
54. Richards C, Pantanowitz L, Dezube BJ. Antimicrobial and non-antimicrobial tetracyclines in human cancer trials. Pharmacol Res. (2011) 63:151-6. doi: 10.1016/j.phrs.2010.10.008
55. Elburki MS, Moore DD, Terezakis NG, Zhang Y, Lee HM, Johnson F, et al. A novel chemically modified curcumin reduces inflammation-mediated connective tissue breakdown in a rat model of diabetes: periodontal and systemic effects. J Periodontal Res. (2017) 52:186-200. doi: 10.1111/jre.12381
56. Elburki MS, Rossa C Jr., Guimaraes-Stabili MR, Lee HM, Curylofo-Zotti FA, Johnson F, et al. A chemically modified curcumin (Cmc 2.24) inhibits nuclear factor kappab activation and inflammatory bone loss in murine models of lps-induced experimental periodontitis and diabetes-associated natural periodontitis. Inflammation. (2017) 40:1436-49. doi: 10.1007/s10753-017-0587-4
57. de Almeida Brandao D, Spolidorio LC, Johnson F, Golub LM, Guimaraes-Stabili MR, Rossa C Jr. Dose-response assessment of chemically modified curcumin in experimental periodontitis. J Periodontol. (2019) 90:535-45. doi: 10.1002/JPER.18-0392
58. Ancuta C, Chirieac R, Ancuta E, Tanculescu O, Solomon SM, Fatu AM, et al. Exploring the role of interleukin-6 receptor inhibitor tocilizumab in patients with active rheumatoid arthritis and periodontal disease. J Clin Med. (2021) 10. doi: 10.3390/jcm10040878
59. Kobayashi T, Okada M, Ito S, Kobayashi D, Ishida K, Kojima A, et al. Assessment of interleukin-6 receptor inhibition therapy on periodontal condition in patients with rheumatoid arthritis and chronic periodontitis. J Periodontol. (2014) 85:57-67. doi: 10.1902/jop.2013.120696
60. Kobayashi T, Yokoyama T, Ito S, Kobayashi D, Yamagata A, Okada M, et al. Periodontal and serum protein profiles in patients with rheumatoid arthritis treated with tumor necrosis factor inhibitor adalimumab. J Periodontol. (2014) 85:1480-8. doi: 10.1902/jop.2014.140194
61. Mayer Y, Balbir-Gurman A, Machtei EE. Anti-tumor necrosis factor-alpha therapy and periodontal parameters in patients with rheumatoid arthritis. J Periodontol. (2009) 80:1414-20. doi: 10.1902/jop.2009.090015
62. Pers JO, Saraux A, Pierre R, Youinou P. Anti-Tnf-alpha immunotherapy is associated with increased gingival inflammation without clinical attachment loss in subjects with rheumatoid arthritis. J Periodontol. (2008) 79:1645-51. doi: 10.1902/jop.2008.070616
63. Hasturk H, Schulte F, Martins M, Sherzai H, Floros C, Cugini M, et al. Safety and preliminary efficacy of a novel host-modulatory therapy for reducing gingival inflammation. Front Immunol. (2021) 12:704163. doi: 10.3389/fimmu.2021.704163
64. Hasturk H, Kantarci A, Ohira T, Arita M, Ebrahimi N, Chiang N, et al. Rve1 protects from local inflammation and osteoclast- mediated bone destruction in periodontitis. FASEB J. (2006) 20:401-3. doi: 10.1096/fj.05-4724fje
65. Herrera BS, Ohira T, Gao L, Omori K, Yang R, Zhu M, et al. An endogenous regulator of inflammation, resolvin E1, modulates osteoclast differentiation and bone resorption. Br J Pharmacol. (2008) 155:1214-23. doi: 10.1038/bjp.2008.367
66. Van Dyke TE, Hasturk H, Kantarci A, Freire MO, Nguyen D, Dalli J, et al. Proresolving nanomedicines activate bone regeneration in periodontitis. J Dent Res. (2015) 94:148-56. doi: 10.1177/0022034514557331
67. Breivik T, Gundersen Y, Gjermo P, Taylor SM, Woodruff TM, Opstad PK. Oral treatment with complement factor C5a receptor (Cd88) antagonists inhibits experimental periodontitis in rats. J Periodontal Res. (2011) 46:643-7. doi: 10.1111/j.1600-0765.2011.01383.x
68. Kajikawa T, Briones RA, Resuello RRG, Tuplano JV, Reis ES, Hajishengallis E, et al. Safety and efficacy of the complement inhibitor amy-101 in a natural model of periodontitis in non-human primates. Mol Ther Methods Clin Dev. (2017) 6:207-15. doi: 10.1016/j.omtm.2017.08.001
69. Maekawa T, Briones RA, Resuello RR, Tuplano JV, Hajishengallis E, Kajikawa T, et al. Inhibition of pre-existing natural periodontitis in non-human primates by a locally administered peptide inhibitor of complement C3. J Clin Periodontol. (2016) 43:238-49. doi: 10.1111/jcpe.12507
70. Hasturk H, Hajishengallis G, Forsyth Institute Center for C, Translational Research s, Lambris JD, Mastellos DC, et al. Phase Iia clinical trial of complement C3 inhibitor Amy-101 in adults with periodontal inflammation. J Clin Invest. (2021) 131. doi: 10.1172/JCI152973
71. Cantley MD, Bartold PM, Fairlie DP, Rainsford KD, Haynes DR. Histone deacetylase inhibitors as suppressors of bone destruction in inflammatory diseases. J Pharm Pharmacol. (2012) 64:763-74. doi: 10.1111/j.2042-7158.2011.01421.x
72. Cantley MD, Bartold PM, Marino V, Fairlie DP, Le GT, Lucke AJ, et al. Histone deacetylase inhibitors and periodontal bone loss. J Periodontal Res. (2011) 46:697-703. doi: 10.1111/j.1600-0765.2011.01392.x
73. Kim TI, Han JE, Jung HM, Oh JH, Woo KM. Analysis of histone deacetylase inhibitor-induced responses in human periodontal ligament fibroblasts. Biotechnol Lett. (2013) 35:129-33. doi: 10.1007/s10529-012-0992-6
74. Huynh NC, Everts V, Pavasant P, Ampornaramveth RS. Inhibition of histone deacetylases enhances the osteogenic differentiation of human periodontal ligament cells. J Cell Biochem. (2016) 117:1384-95. doi: 10.1002/jcb.25429
75. Offenbacher S, Odle BM, Van Dyke TE. The use of crevicular fluid prostaglandin E2 levels as a predictor of periodontal attachment loss. J Periodontal Res. (1986) 21:101-12. doi: 10.1111/j.1600-0765.1986.tb01443.x
76. Hannas AR, Pereira JC, Granjeiro JM, Tjaderhane L. The role of matrix metalloproteinases in the oral environment. Acta Odontol Scand. (2007) 65:1-13. doi: 10.1080/00016350600963640
77. Ryan ME, Ramamurthy S, Golub LM. Matrix metalloproteinases and their inhibition in periodontal treatment. Curr Opin Periodontol. (1996) 3:85-96.
78. Kiili M, Cox SW, Chen HY, Wahlgren J, Maisi P, Eley BM, et al. Collagenase-2 (Mmp-8) and Collagenase-3 (Mmp-13) in adult periodontitis: molecular forms and levels in gingival crevicular fluid and immunolocalisation in gingival tissue. J Clin Periodontol. (2002) 29:224-32. doi: 10.1034/j.1600-051x.2002.290308.x
79. Sorsa T, Tjaderhane L, Konttinen YT, Lauhio A, Salo T, Lee HM, et al. Matrix metalloproteinases: contribution to pathogenesis, diagnosis and treatment of periodontal inflammation. Ann Med. (2006) 38:306-21. doi: 10.1080/07853890600800103
80. Golub LM, Lee HM, Ryan ME, Giannobile WV, Payne J, Sorsa T. Tetracyclines inhibit connective tissue breakdown by multiple non-antimicrobial mechanisms. Adv Dent Res. (1998) 12:12-26. doi: 10.1177/08959374980120010501
81. Smith GN Jr., Mickler EA, Hasty KA, Brandt KD. Specificity of inhibition of matrix metalloproteinase activity by doxycycline: relationship to structure of the enzyme. Arthritis Rheum. (1999) 42:1140-6.
82. Walker C, Preshaw PM, Novak J, Hefti AF, Bradshaw M, Powala C. Long-term treatment with sub-antimicrobial dose doxycycline has no antibacterial effect on intestinal flora. J Clin Periodontol. (2005) 32:1163-9. doi: 10.1111/j.1600-051X.2005.00840.x
83. Zhang Y, Gu Y, Lee HM, Hambardjieva E, Vrankova K, Golub LM, et al. Design, synthesis and biological activity of new polyenolic inhibitors of matrix metalloproteinases: a focus on chemically-modified curcumins. Curr Med Chem. (2012) 19:4348-58. doi: 10.2174/092986712802884295
84. Kobayashi T, Yoshie H. Host responses in the link between periodontitis and rheumatoid arthritis. Curr Oral Health Rep. (2015) 2:1-8. doi: 10.1007/s40496-014-0039-2
85. Chen HH, Huang N, Chen YM, Chen TJ, Chou P, Lee YL, et al. Association between a history of periodontitis and the risk of rheumatoid arthritis: a nationwide, population-based, case-control study. Ann Rheum Dis. (2013) 72:1206-11. doi: 10.1136/annrheumdis-2012-201593
86. de Molon RS, Rossa C Jr., Thurlings RM, Cirelli JA, Koenders MI. Linkage of periodontitis and rheumatoid arthritis: current evidence and potential biological interactions. Int J Mol Sci. (2019) 20. doi: 10.3390/ijms20184541
87. Han JY, Reynolds MA. Effect of anti-rheumatic agents on periodontal parameters and biomarkers of inflammation: a systematic review and meta-analysis. J Periodontal Implant Sci. (2012) 42:3-12. doi: 10.5051/jpis.2012.42.1.3
88. Zhang J, Xu C, Gao L, Zhang D, Li C, Liu J. Influence of anti-rheumatic agents on the periodontal condition of patients with rheumatoid arthritis and periodontitis: a systematic review and meta-analysis. J Periodontal Res. (2021) 56:1099-115. doi: 10.1111/jre.12925
89. Benjamin O, Bansal P, Goyal A, Lappin SL. Disease Modifying Anti-Rheumatic Drugs (Dmard). Treasure Island, FL: Statpearls (2022).
90. Serhan CN. Systems approach to inflammation resolution: identification of novel anti-inflammatory and pro-resolving mediators. J Thromb Haemost. (2009) 7(Suppl 1):44-8. doi: 10.1111/j.1538-7836.2009.03396.x
91. Serhan CN. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol. (2007) 25:101-37. doi: 10.1146/annurev.immunol.25.022106.141647
92. Bannenberg G, Serhan CN. Specialized pro-resolving lipid mediators in the inflammatory response: an update. Biochim Biophys Acta. (2010) 1801:1260-73. doi: 10.1016/j.bbalip.2010.08.002
93. Arita M, Bianchini F, Aliberti J, Sher A, Chiang N, Hong S, et al. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J Exp Med. (2005) 201:713-22. doi: 10.1084/jem.20042031
94. Arita M, Yoshida M, Hong S, Tjonahen E, Glickman JN, Petasis NA, et al. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc Natl Acad Sci USA. (2005) 102:7671-6. doi: 10.1073/pnas.0409271102
95. Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J Exp Med. (2002) 196:1025-37. doi: 10.1084/jem.20020760
96. Bannenberg GL, Chiang N, Ariel A, Arita M, Tjonahen E, Gotlinger KH, et al. Molecular circuits of resolution: formation and actions of resolvins and protectins. J Immunol. (2005) 174:4345-55. doi: 10.4049/jimmunol.174.7.4345
97. Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and Protectin D1 activate inflammation-resolution programmes. Nature. (2007) 447:869-74. doi: 10.1038/nature05877
98. Chen C. Cox-2's new role in inflammation. Nat Chem Biol. (2010) 6:401-2. doi: 10.1038/nchembio.375
99. El-Sharkawy H, Aboelsaad N, Eliwa M, Darweesh M, Alshahat M, Kantarci A, et al. Adjunctive treatment of chronic periodontitis with daily dietary supplementation with omega-3 fatty acids and low-dose aspirin. J Periodontol. (2010) 81:1635-43. doi: 10.1902/jop.2010.090628
100. Naqvi AZ, Hasturk H, Mu L, Phillips RS, Davis RB, Halem S, et al. Docosahexaenoic acid and periodontitis in adults: a randomized controlled trial. J Dent Res. (2014) 93:767-73. doi: 10.1177/0022034514541125
101. Castro Dos Santos NC, Andere N, Araujo CF, de Marco AC, Kantarci A, Van Dyke TE, et al. Omega-3 pufa and aspirin as adjuncts to periodontal debridement in patients with periodontitis and type 2 diabetes mellitus: randomized clinical trial. J Periodontol. (2020) 91:1318-27. doi: 10.1002/JPER.19-0613
102. Elwakeel NM, Hazaa HH. Effect of Omega 3 fatty acids plus low-dose aspirin on both clinical and biochemical profiles of patients with chronic periodontitis and type 2 diabetes: a randomized double blind placebo-controlled study. J Periodontal Res. (2015) 50:721-9. doi: 10.1111/jre.12257
103. Hajishengallis G, Reis ES, Mastellos DC, Ricklin D, Lambris JD. Novel mechanisms and functions of complement. Nat Immunol. (2017) 18:1288-98. doi: 10.1038/ni.3858
104. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. (2010) 11:785-97. doi: 10.1038/ni.1923
105. Patters MR, Niekrash CE, Lang NP. Assessment of complement cleavage in gingival fluid during experimental gingivitis in man. J Clin Periodontol. (1989) 16:33-7. doi: 10.1111/j.1600-051X.1989.tb01609.x
106. Niekrash CE, Patters MR. Simultaneous assessment of complement components C3, C4, and B and their cleavage products in human gingival fluid. Ii. Longitudinal changes during periodontal therapy. J Periodontal Res. (1985) 20:268-75. doi: 10.1111/j.1600-0765.1985.tb00434.x
107. Grande MA, Belstrom D, Damgaard C, Holmstrup P, Thangaraj SS, Nielsen CH, et al. Complement split product C3c in saliva as biomarker for periodontitis and response to periodontal treatment. J Periodontal Res. (2021) 56:27-33. doi: 10.1111/jre.12788
108. Liang S, Krauss JL, Domon H, McIntosh ML, Hosur KB, Qu H, et al. The C5a receptor impairs Il-12-dependent clearance of porphyromonas gingivalis and is required for induction of periodontal bone loss. J Immunol. (2011) 186:869-77. doi: 10.4049/jimmunol.1003252
109. Maekawa T, Abe T, Hajishengallis E, Hosur KB, DeAngelis RA, Ricklin D, et al. Genetic and intervention studies implicating complement C3 as a major target for the treatment of periodontitis. J Immunol. (2014) 192:6020-7. doi: 10.4049/jimmunol.1400569
110. Abe T, Hajishengallis G. Optimization of the ligature-induced periodontitis model in mice. J Immunol Methods. (2013) 394:49-54. doi: 10.1016/j.jim.2013.05.002
111. Abe T, Hosur KB, Hajishengallis E, Reis ES, Ricklin D, Lambris JD, et al. Local complement-targeted intervention in periodontitis: proof-of-concept using a C5a receptor (Cd88) antagonist. J Immunol. (2012) 189:5442-8. doi: 10.4049/jimmunol.1202339
112. Tsukasaki M, Komatsu N, Nagashima K, Nitta T, Pluemsakunthai W, Shukunami C, et al. Host defense against oral microbiota by bone-damaging T Cells. Nat Commun. (2018) 9:701. doi: 10.1038/s41467-018-03147-6
113. Reis ES, Berger N, Wang X, Koutsogiannaki S, Doot RK, Gumas JT, et al. Safety profile after prolonged C3 inhibition. Clin Immunol. (2018) 197:96-106. doi: 10.1016/j.clim.2018.09.004
114. Hajishengallis G, Hasturk H, Lambris JD, Contributing a. C3-targeted therapy in periodontal disease: moving closer to the clinic. Trends Immunol. (2021) 42:856-64. doi: 10.1016/j.it.2021.08.001
115. De Souza AP, Planello AC, Marques MR, De Carvalho DD, Line SR. High-throughput DNA analysis shows the importance of methylation in the control of immune inflammatory gene transcription in chronic periodontitis. Clin Epigenet. (2014) 6:15. doi: 10.1186/1868-7083-6-15
116. de Faria Amormino SA, Arao TC, Saraiva AM, Gomez RS, Dutra WO, da Costa JE, et al. Hypermethylation and low transcription of Tlr2 gene in chronic periodontitis. Hum Immunol. (2013) 74:1231-6. doi: 10.1016/j.humimm.2013.04.037
117. De Oliveira NF, Andia DC, Planello AC, Pasetto S, Marques MR, Nociti FH Jr., et al. Tlr2 and Tlr4 Gene promoter methylation status during chronic periodontitis. J Clin Periodontol. (2011) 38:975-83. doi: 10.1111/j.1600-051X.2011.01765.x
118. Zhang S, Barros SP, Moretti AJ, Yu N, Zhou J, Preisser JS, et al. Epigenetic regulation of Tnfa expression in periodontal disease. J Periodontol. (2013) 84:1606-16. doi: 10.1902/jop.2013.120294
119. Ivanov M, Barragan I, Ingelman-Sundberg M. Epigenetic Mechanisms of importance for drug treatment. Trends Pharmacol Sci. (2014) 35:384-96. doi: 10.1016/j.tips.2014.05.004
Keywords: periodontal disease, inflammation, inflammatory response, host modulation therapy, non-surgical therapy
Citation: Bezerra B, Monajemzadeh S, Silva D and Pirih FQ (2022) Modulating the Immune Response in Periodontitis. Front. Dent. Med. 3:879131. doi: 10.3389/fdmed.2022.879131
Received: 18 February 2022; Accepted: 26 April 2022;
Published: 13 May 2022.
Edited by:
Elena Calciolari, University of Parma, ItalyReviewed by:
Hatice Hasturk, The Forsyth Institute, United StatesJoice Dias Correa, Pontifícia Universidade Católica de Minas Gerais, Brazil
Copyright © 2022 Bezerra, Monajemzadeh, Silva and Pirih. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Beatriz Bezerra, YmJlemVycmFAdWNsYS5lZHU=; Flavia Q. Pirih, ZnBpcmloQGRlbnRpc3RyeS51Y2xhLmVkdQ==