 Virgilio Romanelli
Virgilio Romanelli Carmen Cerchia
Carmen Cerchia Antonio Lavecchia
Antonio Lavecchia- “Drug Discovery” Laboratory, Department of Pharmacy, University of Naples “Federico II”, Napoli, Italy
Drug discovery is a costly and time-consuming process, especially because of the significant expenses connected with the high percentage of clinical trial failures. As such, there is a need for new paradigms enabling the optimization of the various stages, from hit identification to market approval. The upsurge in the use of artificial intelligence (AI) technologies and the advent of deep learning (DL) demonstrated a lot of promise in rethinking and redesigning the traditional pipelines in drug discovery, including de novo molecular design. In this regard, generative models have greatly impacted the de novo design of molecules with desired properties and are being increasingly integrated into real world drug discovery campaigns. Herein, we will briefly appraise recent case studies utilizing generative models for chemical structure generation in the area of anticancer drug discovery. Finally, we will analyze current challenges and limitations as well as the possible strategies to overcome them, outlining potential future directions to advance this exciting field.
1 Introduction
The path from the identification to successful market approval of a new medicine is lengthy and complicated, often lasting more than 10 years and requiring impressive financial resources (Kiriiri et al., 2020). This process depends upon many steps, from target discovery and validation, hit identification, lead optimization, rigorous validation and testing in both preclinical and clinical context, regulatory approval, and post-marketing surveillance. However, due to intrinsic safety concerns, unsatisfactory efficacy against specific diseases, and complex regulatory procedures, the attrition rate of therapeutic candidates remains substantial (Waring et al., 2015). Therefore, exploring new technologies is pivotal for the industry to improve the efficiency of the drug discovery process. Computational approaches have emerged as valuable tools to model, simulate, and evaluate molecular interactions and processes or to predict physicochemical properties relevant for pharmacokinetics (PK) and toxicity. For instance, virtual screening enables the quick evaluation of compound libraries, to facilitate the discovery of prospective drug candidates with the desired interactions and characteristics (Lavecchia and Di Giovanni, 2013).
The increasing amount of biomedical data and the advancements in computing capabilities laid the foundations for the use of machine learning (ML) or artificial intelligence (AI) techniques for mining such data, retrieve useful patterns and support decision making (Lavecchia, 2015; 2019; Cerchia and Lavecchia, 2023). In addition, deep learning (DL) has demonstrated significant potential in tackling challenging tasks related to drug discovery, such as de novo molecular design, that is the process of (computationally) creating novel potential ligands from scratch (Lecun et al., 2015). Deep generative models have evolved as a fascinating area of research, with implications across a multitude of domains, delivering exceptional outcomes in the realm of image generation (Radford et al., 2016), textual synthesis (Bowman et al., 2016), speech reproduction (Oord et al., 2016), and even music composition (Engel et al., 2017). In the case of generative models for molecule generation, a DL-based model is trained using chemical structures (typically obtained from databases) to identify patterns and learn the basics of chemistry; then, the extracted knowledge is employed to construct novel molecules, that is, the de novo design (Cerchia and Lavecchia, 2023). In this mini-review, we focus on the most recent case studies on the application of generative models in the field of anticancer drug discovery; rather than delving into the technical details about the models used, which have been extensively reviewed elsewhere (Cheng et al., 2021; Meyers et al., 2021; Tong et al., 2021; Bilodeau et al., 2022; Wang et al., 2022; Cerchia and Lavecchia, 2023; Pang et al., 2023), we decided to put more emphasis on the preclinical evaluation of the generated candidates.
2 Generative models in brief
The rise of generative models prompted fresh ideas and new perspectives in the challenging field of drug design and development. While conventional approaches mostly depend on human expertise, generative models rely on the recent advancements in deep learning to address molecular design: identifying a set of compounds that possess the desired characteristics for a specific target by establishing a mapping function between properties and molecular structures (Kusner et al., 2017; Segler et al., 2018; Jin et al., 2021). In the last years, the application of generative models with varying molecular representations, architectures, and target design problems had an exponential growth (Cheng et al., 2021; Meyers et al., 2021; Tong et al., 2021; Bilodeau et al., 2022; Wang et al., 2022; Cerchia and Lavecchia, 2023).
The process of molecule generation with generative models can be divided into main steps (Figure 1A): first, a reference database or dataset is selected and converted into a computer-readable representation. In most cases, such representation is string-based, such as SMILES format, or graph-based, in the form of molecular graphs. These latter are subsequently converted into a numeric feature matrix, in order to train the generative model to learn chemical patterns from existing molecules. On the basis of the different model’s architecture, the matrices feed the model, and the molecular generation process is reiterated until the desired properties scores are reached.
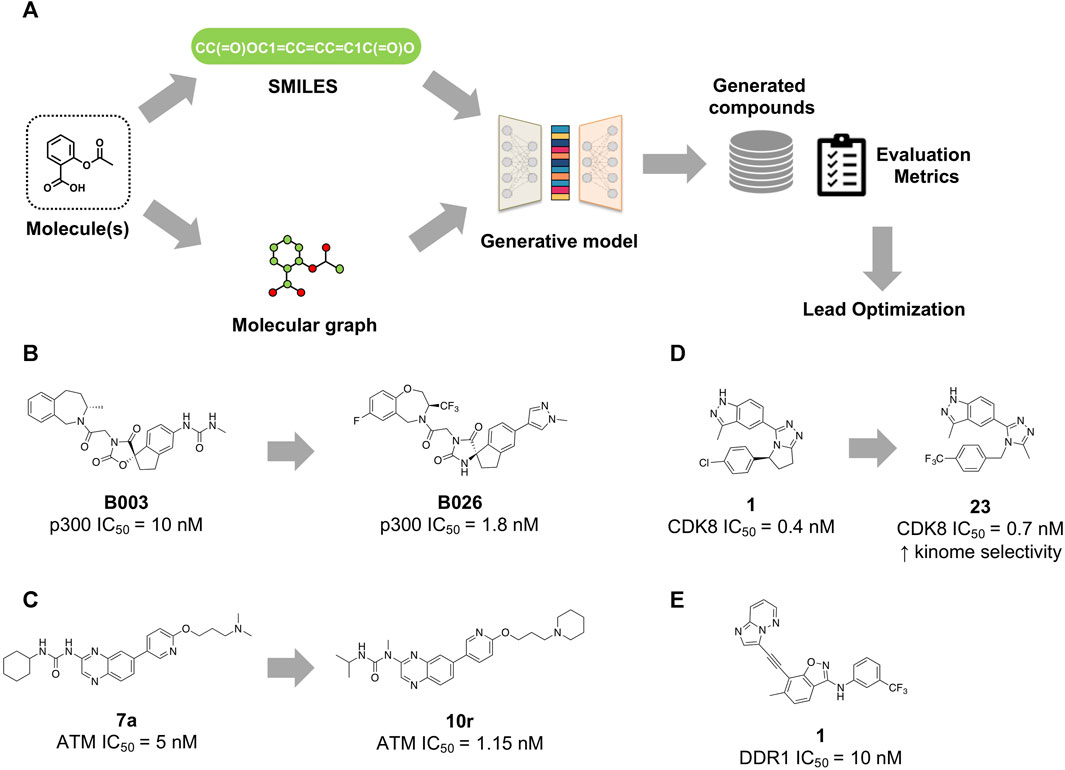
FIGURE 1. (A) Schematic overview of generative model workflow. Molecular structures are encoded either as SMILES strings or molecular graphs. The deep learning (DL)-based model is first trained with data from molecular databases and then generates new chemical structures with similar properties. Evaluation metrics are then used to validate the compounds’ structure quality. Compounds designed with the aid of generative models (B) Compound B003, identified by using a LSTM-based generative model, and its optimized derivative B026 as inhibitors of p300 histone acetyltransferase. (C) Compound 7a, designed by a goal-directed molecular generation approach, and its derivative 10r, as ATM kinase inhibitors. (D) Compound 1, identified by the AI-based platform Chemistry42, and its optimized derivative 23 as CDK8 inhibitors. (E) Compound 1, generated by means of the GENTRL approach as DDR1 kinase inhibitor.
Most generative models rely on deep neural networks. These latter usually sample new compounds from a latent representation of molecular structure learned by the neural network during model training; thus, they basically behave, as “statistical structure generators” (Tropsha et al., 2023). The most critical and prone to error step in the design process is the evaluation of the target property of the generated molecules, and multiple approaches to accomplish such evaluation procedure have been proposed, including quantitative structure-activity relationship (QSAR) models, or systematically adjusting the parameters of the generative model to prioritize the construction of molecules with the required properties (for example, by one-shot or few-shot learning, or reinforcement learning), or setting the evaluation criterion to the probabilities that the generative model learned. On the basis of the ML algorithm used for the adaptive model optimization, we can refer to “reward” or “fitness” functions. The encoding of chemical structures and the subsequent learning phase are key aspects of the model optimization, which could be considered similar to chemical descriptor calculation in traditional QSAR modelling (Tropsha et al., 2023). QSAR methods have progressively evolved with regard to both the complexity of statistical or ML-based approaches to build models and the size of the data sets used. While in QSAR modelling the molecular descriptors are conceptualized to numerically characterize molecular structures at different levels of structure representation, DL-based models require molecular descriptors to be properly engineered, because molecules are represented by vectors in artificially constructed high-dimensional spaces that are used in learning tasks using neural network architectures (Tropsha et al., 2023). Moreover, DL-models are more computationally demanding, require higher amounts of data for training and suffer of a “black box problem,” that is, tracing how and why the DL model makes a certain decision. Traditional ML approaches depend more on domain knowledge, such as statistical theories and mathematical models, whereas DL models rather extract patterns that are not necessarily explainable.
Among the first and most prominent contributions, it is worth mentioning the work from Gomez-Bombarelli et al. (Gómez-Bombarelli et al., 2018), and the REINVENT model developed within AstraZeneca (Olivecrona et al., 2017; Blaschke et al., 2020; Loeffler et al., 2023). In order to enable insightful comparisons between the growing number of generative models with different flavors (outlined in Table 1), a variety of benchmarks and metrics have been proposed. These benchmarks evaluate models on the basis of factors such as distribution learning, chemical diversity, validity and novelty (Brown et al., 2019; Polykovskiy et al., 2020).

TABLE 1. Exemplary deep-learning models for molecule generation.
The molecule “validity” refers to the generated molecules’ adherence to chemical rules, including valence criteria and acceptable atom and bond types. Validity confirms the chemical feasibility of the structures generated. In order to gauge the model’s ability to produce novel chemical matter, “uniqueness” assesses the proportion of non-redundant chemical structures among the valid ones, while “novelty” refers to the portion of generated molecules that are absent from the training set or other reference datasets. The drug-likeness of the generated molecules is quantified in terms of “quantitative estimation of drug-likeness” (QED) (Bickerton et al., 2012), and mostly refers to the adherence of physicochemical descriptors to marketed oral drugs. A higher QED would be desirable, as it indicates the attractiveness of compounds as “hits.” The “synthetic accessibility score” (SAS) gives an estimation on the difficulty to synthesize the generated compounds (Ertl and Schuffenhauer, 2009). A SAS value higher than 5 denotes a compound that is more challenging to synthesize. The publicly available platforms MOSES (Polykovskiy et al., 2020) and GuacaMol (Brown et al., 2019) allow the evaluation of the quality of generated molecules as well as the performances of the model against other “baseline” models. Most of the works published on generative models built upon 2D ligand information (Tong et al., 2021; Bilodeau et al., 2022), thus possibly missing information about protein-ligand binding sites; however, some exemplary applications of “3D generative models” are reviewed elsewhere (Xie et al., 2022; Cerchia and Lavecchia, 2023). In the following section, we report recent case studies regarding the application of generative models to discover potential anticancer agents and corroborated by extensive experimental validation.
3 Applications of generative models in anticancer drug discovery
The evolution of deep-learning and generative models has also opened new perspectives in anticancer drug discovery. The first applications reported in literature mostly described theoretical work, focusing on the model’s performances rather than on the experimental validation of the generated compounds (Cerchia and Lavecchia, 2023).
In the work proposed by Zhou et al. (Yang et al., 2020), a neural network model based on LSTM (long short-term memory) was used to design small molecules able to inhibit the histone acetyltransferase (HAT) class of enzymes. These enzymes, which include p300 and CREB-binding protein (CBP), play crucial roles in many biological processes, including cell development, cell differentiation, environmental stress response, and, in pathological contexts, cancer progression (Iyer et al., 2004). The model was first trained with about 200,000 bioactive molecules taken from ChEMBL and then fine-tuned with a set containing 135 p300 inhibitors and 576 macrocycle molecules with proved activity against challenging targets, e.g., protein−protein interactions (PPI). Among the 500,000 molecules generated, 670 were selected by using specific filters: (a) they do not violate more than one of Lipinski’s rules of five, (b) they possess less complicated structures (synthetic accessibility score ≤4.5), and (c) they show favorable molecular docking scores. Through these filters, the selection was reduced to 20 molecules and after visual analysis compound B003 was proposed for synthesis and bioactivity assays (Figure 1B). This compound showed an IC50 value of 10 nM against p300 using radioactive acetyltransferase activity assay. Compound B003 underwent optimization cycles to improve potency, drug metabolism and pharmacokinetic properties, thus leading to the identification of compound B026 (IC50 = 1.8 nM). This latter exhibited very high (2000-fold) selectivity within the HAT family, as well as significant inhibition of growth of androgen-receptor-positive (AR+) prostate cancer cell lines (IC50 values of 9.8 and 4.4 nM in LnCaP-FGC and 22Rv1 cell lines, respectively). In Balb/c female mice bearing MV-4-11 tumor cells, orally administered B026 significantly inhibited tumor growth by 75% at 50 mg/kg and 85.7% at 100 mg/kg.
Deng et al. (2023) proposed a combination of goal-oriented molecule generation and virtual screening to discover new inhibitors of the ATM (ataxia telangiectasia mutated) enzyme. This latter is a member of the Phosphoinositide 3-kinase (PI3K)-related kinases (PIKKs) and is activated in response to DNA damage, particularly DNA double-strand breaks. When DNA damage occurs, as a result of exposure to ionizing radiation or other damaging agents, ATM is activated and initiates a signaling cascade that helps coordinate DNA repair, regulate cell cycle checkpoints, and activate cellular defense mechanisms such as apoptosis (Lee and Paull, 2007). As ATM represents a feasible target for radiotherapy and chemotherapy sensitization, specific ATM inhibitors have been studied to increase the efficacy of anticancer therapies. However, no ATM inhibitor has been approved to date, while there are only few inhibitors in clinical trials. Since PI3K and ATM kinases share a good degree of structural homology, they might display affinity for similar chemical moieties. Some well-known PI3K inhibitors possess peculiar scaffolds, such as quinolines and quinoxalines, that could be repositioned for ATM inhibitors. To this aim, the authors employed a scaffold-constrained goal-directed generative model. They first obtained a representative conformation of ATM in complex with the inhibitor M4076 (Stakyte et al., 2021) by molecular dynamics. Then, they established the target specificity and ligand electrostatic similarity as target optimization properties. The ligand similarity part involved obtaining scores by calculating the Tanimoto similarity of the generated molecules to the electrostatic fingerprint of the inhibitor molecule template. The target specificity, on the other hand, involved the structure-based binding affinity prediction. The authors used an advanced reinforcement learning algorithm, the hill climbing algorithm, to fine-tune the RNN generation model to maximize the generation of the highest-scoring molecules. This procedure generated 20,194 compounds; according to Lipinski’s rules, 6,543 potential leads were obtained, which were then further reduced to 1,868 on the basis of synthetic feasibility and drug-like characteristics. Through molecular docking simulations, 30 molecules were selected. Following ADME analysis and experimental evaluations, compound 7a (Figure 1C) was chosen as the starting compound demonstrating strong inhibitory activity on ATM (IC50 = 5 nM). Extensive SAR studies eventually led to compound 10r (IC50 = 1.15 nM), with potent inhibitory activity on ATM and improved metabolic stability. Compound 10r showed synergistic antiproliferative effects with irinotecan against MCF-7 and SW620 cells. Further experiments showed that 10r combined with irinotecan suppressed HCT116 cells proliferation by inducing apoptosis and cell cycle arrest in the G2/M phase. The oral administration of compound 10r combined with irinotecan (40 mg/kg) induced a significant and dose-dependent tumor volume and tumor weight inhibition (up to 85.35%) in a human colon cancer SW620 xenograft model, suggesting its potential as a promising candidate drug combined with chemotherapy for cancer treatment.
Zhavoronkov et al. (Li et al., 2023) reported the discovery of cyclin-dependent kinase 8 (CDK8) inhibitors for the treatment of cancer by using a suite of computational tools, including also generative models. CDK8 is an enzyme belonging to the cyclin-dependent kinase (CDK) family. This kinase regulates the cell cycle and is active in the regulation of gene transcription through phosphorylation of specific proteins involved in this process (Szilagyi and Gustafsson, 2013). The overexpression of CDK8 has been associated with the development of various cancers, including colorectal cancer (Xi et al., 2019). Starting from the crystal structure of CDK8 in complex with a reference inhibitor (PDB: 5IDN), the authors first highlighted the important interactions between the residues composing the binding pocket and the co-crystallized ligand. Then, they decided to keep fixed the chemical groups forming interactions with the “hinge” motif and the P-loop of the kinase while modifying the linker, a pyrrolidine group, by using a combination of generative chemistry and structure-based approaches. Compound 1 (Figure 1D), with a dihydropyrrolotriazole fragment as a linker was thus identified, showing a strong affinity for CDK8 with an IC50 of 0.4 ± 0.1 nM as well as antiproliferative effects on the acute myeloid leukemia (AML) cell line MV4-11 with an IC50 of 2.4 ± 0.4 nM. Although compound 1 qualified as a good starting point, it displayed low selectivity for CDK8. In fact, compound 1 and its first-generation derivatives showed strong binding affinity to GSK3B (glycogen synthase kinase-3 beta), which is involved in some cellular toxicity processes. Further modifications to the chlorophenyl group led to the discovery of compound 23, which displayed strong affinity with CDK8 (IC50 = 0.70 ± 0.4 nM) and promising anti-proliferative activity against MV4-11 (IC50 = 11.8 nM). It also showed good microsome stability and low clearance in vivo, with increased CDK8 selectivity. Compound 23 also showed significant tumor growth inhibition in the AML CDX model as monotherapy and in solid tumor syngeneic models as a combination with the PDL1 antibody. The results of further preclinical profiling for compound 23 are being awaited. The generative chemistry approach employed in the above-described work has been incorporated into Chemistry42, an AI-based commercial platform (Ivanenkov et al., 2023), integrating different, interconnected generative and predictive models (about 40) together with a reinforcement learning system to evaluate and select optimal molecules and make comparisons. The platform allows to generate a huge number of molecules that pass within a reward pipeline. This latter prioritizes high-scoring molecules, those that achieve specific objectives such as metabolic stability, safety, potency, and synthetic feasibility. Then, the models “learn” from both highly- and poorly-scoring molecules and thus they are re-trained to generate better molecules.
Before the release of this platform, Zhavoronkov et al. published several studies about generative models for de novo design (Putin et al., 2018b; 2018a; Polykovskiy et al., 2018; Zhavoronkov et al., 2019), also establishing a network of collaborations among research groups involved in this field. One of the most prominent works was related to GENTRL (generative tensorial reinforcement learning) (Zhavoronkov et al., 2019), a deep generative model used to discover potent discoidin domain receptor 1 (DDR1) inhibitors in a very short time. DDR1 is a collagen-activated kinase whose overexpression has been associated with many pathological phenomena such as fibrosis and cancer (Moll et al., 2019). GENTRL model combined reinforcement learning, variational inference, tensor decomposition (to capture the relationships between the compounds and their properties) and self-organizing maps (SOMs) as reward functions. The model was trained using first ZINC datasets, and then DDR1 kinase inhibitors. Among the identified compounds, 1 and 2 strongly inhibited DDR1 with IC50 of 10 nM and 21 nM, respectively. Then, compounds 1 and 2 were tested in human cells to evaluate the inhibition of DDR1 and the effect on fibrosis. Both molecules demonstrated high inhibitory activity, reducing the induction of fibrosis markers such as α-actin, CCN2 and collagen production. Finally, 1 (Figure 1E) showed good efficacy and pharmacokinetic properties also in mice, confirming good oral bioavailability, with half-life of about 3.5 h and significant plasma concentrations after intravenous and oral administration. The results presented in the work (Zhavoronkov et al., 2019) raised some critiques, in particular that the selectivity profile towards different kinases might be questionable; moreover, compound 1 was very similar to ponatinib (Walters and Murcko, 2020). The authors acknowledged that selectivity of compound 1 should have been assessed more in depth and that it requires further optimization; however, the compound was still novel, and therefore, patentable (Zhavoronkov and Aspuru-Guzik, 2020). This debate stimulated the reflection that the scientific community should adopt some guidelines to better assess the molecules obtained by generative models, similarly to those generated by medicinal chemists.
4 Challenges and potential future directions
Undoubtedly, generative models provided proof-of-concept of their significance within drug discovery projects. However, there is room for improvements, in order to overcome some limitations. Alternative molecular representations should be more extensively explored, to address the problem of molecule’s validity often encountered using SMILES. While few studies have implemented three-dimensional representation and featurization (Xie et al., 2022), SELF-referencIng Embedded Strings (SELFIES) (Krenn et al., 2020), or DeepSMILES (O’Boyle and Dalke, 2018) have been also proposed. However, their performances highly depend on the model’s settings.
There is a compelling need of robust benchmarks and metrics towards more standardized procedures as well as uncertainty quantification in model’s predictions, to improve decision making (Mervin et al., 2021; Ballester, 2023). The existing metrics are inherently limited and provide only an incomplete view of the effectiveness of the generative models for real-world applications. Given the increasingly complex DL architectures that are currently being reported in literature, suitable methods and best practices to fairly judge model’s performances are absolutely required. Further crucial elements for expanding the use of DL-based models in the biological sciences and reducing their black box nature are the model’s interpretability and rationality of the predictions. In fact, there is increased interest in explainable artificial intelligence and a rise in the utilization of feature attribution methods (such as SHAP values) (Rodríguez-Pérez and Bajorath, 2020). Still, the most reliable and rigorous standard for evaluating the generated molecules is the experimental validation; moreover, assessing whether or not the candidate will make it to the clinic requires further investigation, well outside the model’s predictive ability. It is worth noting that, in the above-described case studies, the role of the generative models was mainly to generate new initial molecular design, which then underwent several optimization cycles. Optimization was mostly guided by more classical approaches, such as structure-based design, docking, and, above all, human expertise.
5 Conclusion and outlook
The realm of drug discovery is currently facing exciting new opportunities and challenges as a result of the constant growth and implementation of AI technologies. These latter made significant strides in the field of small molecule design, particularly in the areas of molecular generation and protein structure prediction, with a growing number of AI-derived medications undergoing clinical testing (Pun et al., 2023). Herein, we focused on the most recent advances and exemplary applications of molecular generative models in the field of anticancer drug discovery. However, there are differing views. On the one hand, there is a generalized dissatisfaction over the fact that the AI-promised revolution has yet to happen, at least in the field of chemistry (Author Anonymous, 2023). One main concern regards the lack of high-quality data available in an appropriate amount to feed the hungry AI systems. Undoubtedly, more data are required, both from simulations and experiments, as well as the cryptic information from failed experiments. In addition, such data should be more readily accessible to the public domain. It has been suggested that a possible solution lies in pre-training models using abundant unlabeled data for self-supervised learning (Ballester, 2023). The majority of AI successes were accomplished in text, speech, and images processing, whereas dealing with the more complex biology data requires considerably more work and time. Therefore, at least for the time being, the knowledge and skills of human experts remain still crucial. Future significant developments in this area are anticipated, with the long-term goal of creating an autonomous design-make-test-analyze cycle with the integration of generative models into automated laboratory systems capable of synthesizing the generated compounds and conducting their experimental evaluation (Schneider et al., 2020; Mervin et al., 2021).
Author contributions
VR: Writing–original draft. CC: Writing–original draft, Conceptualization, Data curation, Supervision, Writing–review and editing. AL: Supervision, Writing–review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
AL acknowledges funding from the Italian Ministry of Education Progetti di Rilevante Interesse Nazionale (PRIN) grant 2022P5LPHS. CC acknowledges PON R&I 2014–2020 Asse IV “Istruzione e Ricerca per il recupero—REACT-EU” Azione IV.4 “Contratti di Ricerca su tematiche dell’Innovazione” and support from the University of Naples “Federico II” (Grant FRA 2022, Line C, INITIATIVE). VR acknowledges the PhD scholarship funded by DM351/2022 PNRR, CUP: E66E22000360006.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Author Anonymous (2023). For chemists, the AI revolution has yet to happen. Nature 617, 438. doi:10.1038/d41586-023-01612-x
Ballester, P. J. (2023). The AI revolution in chemistry is not that far away. Nature 624, 252. doi:10.1038/d41586-023-03948-w
Bickerton, G. R., Paolini, G. V., Besnard, J., Muresan, S., and Hopkins, A. L. (2012). Quantifying the chemical beauty of drugs. Nat. Chem. 4, 90–98. doi:10.1038/nchem.1243
Bilodeau, C., Jin, W., Jaakkola, T., Barzilay, R., and Jensen, K. F. (2022). Generative models for molecular discovery: recent advances and challenges. Wiley Interdiscip. Rev. Comput. Mol. Sci. 12, e1608. doi:10.1002/wcms.1608
Blaschke, T., Arús-Pous, J., Chen, H., Margreitter, C., Tyrchan, C., Engkvist, O., et al. (2020). REINVENT 2.0: an AI tool for de novo drug design. J. Chem. Inf. Model. 60, 5918–5922. doi:10.1021/acs.jcim.0c00915
Bowman, S. R., Vilnis, L., Vinyals, O., Dai, A. M., Jozefowicz, R., and Bengio, S. (2016). Generating sentences from a continuous space. CoNLL 2016 - 20th SIGNLL Conf. Comput. Nat. Lang. Learn. Proc., 10–21. doi:10.18653/v1/k16-1002
Brown, N., Fiscato, M., Segler, M. H. S., and Vaucher, A. C. (2019). GuacaMol: benchmarking models for de novo molecular design. J. Chem. Inf. Model. 59, 1096–1108. doi:10.1021/acs.jcim.8b00839
Cerchia, C., and Lavecchia, A. (2023). New avenues in artificial-intelligence-assisted drug discovery. Drug Discov. Today 28, 103516. doi:10.1016/j.drudis.2023.103516
Cheng, Y., Gong, Y., Liu, Y., Song, B., and Zou, Q. (2021). Molecular design in drug discovery: a comprehensive review of deep generative models. Brief. Bioinform. 22, bbab344. doi:10.1093/bib/bbab344
Deng, D., Yang, Y., Zou, Y., Liu, K., Zhang, C., Tang, M., et al. (2023). Discovery and evaluation of 3-quinoxalin urea derivatives as potent, selective, and orally available ATM inhibitors combined with chemotherapy for the treatment of cancer via goal-oriented molecule generation and virtual screening. J. Med. Chem. 66, 9495–9518. doi:10.1021/acs.jmedchem.3c00082
Engel, J., Resnick, C., Roberts, A., Dieleman, S., Norouzi, M., Eck, D., et al. (2017). “Neural audio synthesis of musical notes with WaveNet autoencoders,” in 34th International Conference on Machine Learning, ICML 2017, Sydney, Australia (PMLR 70), 1771–1780.
Ertl, P., and Schuffenhauer, A. (2009). Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 1, 8–11. doi:10.1186/1758-2946-1-8
Gómez-Bombarelli, R., Wei, J. N., Duvenaud, D., Hernández-Lobato, J. M., Sánchez-Lengeling, B., Sheberla, D., et al. (2018). Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent. Sci. 4, 268–276. doi:10.1021/acscentsci.7b00572
Goodfellow, I., Pouget-Abadie, J., Mirza, M., Xu, B., Warde-Farley, D., Ozair, S., et al. (2020). Generative adversarial networks. Commun. ACM 63, 139–144. doi:10.1145/3422622
Hochreiter, S., and Schmidhuber, J. (1997). Long short-term memory. Neural comput. 9, 1735–1780. doi:10.1162/neco.1997.9.8.1735
Ivanenkov, Y. A., Polykovskiy, D., Bezrukov, D., Zagribelnyy, B., Aladinskiy, V., Kamya, P., et al. (2023). Chemistry42: an AI-driven platform for molecular design and optimization. J. Chem. Inf. Model. 63, 695–701. doi:10.1021/acs.jcim.2c01191
Iyer, N. G., Özdag, H., and Caldas, C. (2004). p300/CBP and cancer. Oncogene 23, 4225–4231. doi:10.1038/sj.onc.1207118
Jin, W., Barzilay, R., and Jaakkola, T. (2021). Chapter 11: junction tree variational autoencoder for molecular graph generation. RSC Drug Discov. Ser. (PMLR), 228–249. doi:10.1039/9781788016841-00228
Kingma, D. P., and Welling, M. (2014). “Auto-encoding variational bayes,” in 2nd Int. Conf. Learn. Represent. ICLR 2014 - Conf. Track Proc.
Kiriiri, G. K., Njogu, P. M., and Mwangi, A. N. (2020). Exploring different approaches to improve the success of drug discovery and development projects: a review. Futur. J. Pharm. Sci. 6, 27–12. doi:10.1186/s43094-020-00047-9
Krenn, M., Häse, F., Nigam, A. K., Friederich, P., and Aspuru-Guzik, A. (2020). Self-referencing embedded strings (SELFIES): a 100% robust molecular string representation. Mach. Learn. Sci. Technol. 1, 045024. doi:10.1088/2632-2153/aba947
Kusner, M. J., Paige, B., and Hemández-Lobato, J. M. (2017). “Grammar variational autoencoder,” in 34th International Conference on Machine Learning, ICML 2017, Sydney, Australia (PMLR 70), 3072–3084.
Lavecchia, A. (2015). Machine-learning approaches in drug discovery: methods and applications. Drug Discov. Today 20, 318–331. doi:10.1016/j.drudis.2014.10.012
Lavecchia, A. (2019). Deep learning in drug discovery: opportunities, challenges and future prospects. Drug Discov. Today 24, 2017–2032. doi:10.1016/j.drudis.2019.07.006
Lavecchia, A., and Di Giovanni, C. (2013). Virtual screening strategies in drug discovery: a critical review. Curr. Med. Chem. 20, 2839–2860. doi:10.2174/09298673113209990001
Lecun, Y., Bengio, Y., and Hinton, G. (2015). Deep learning. Nature 521, 436–444. doi:10.1038/nature14539
Lee, J. H., and Paull, T. T. (2007). Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 26, 7741–7748. doi:10.1038/sj.onc.1210872
Li, Y., Liu, Y., Wu, J., Liu, X., Wang, L., Wang, J., et al. (2023). Discovery of potent, selective, and orally bioavailable small-molecule inhibitors of CDK8 for the treatment of cancer. J. Med. Chem. 66, 5439–5452. doi:10.1021/acs.jmedchem.2c01718
Loeffler, H. H., He, J., Tibo, A., Janet, J. P., Voronov, A., Mervin, L., et al. (2023). REINVENT4: modern AI–driven generative molecule design. chemRxiv. doi:10.26434/chemrxiv-2023-xt65x
Mervin, L. H., Johansson, S., Semenova, E., Giblin, K. A., and Engkvist, O. (2021). Uncertainty quantification in drug design. Drug Discov. Today 26, 474–489. doi:10.1016/j.drudis.2020.11.027
Meyers, J., Fabian, B., and Brown, N. (2021). De novo molecular design and generative models. Drug Discov. Today 26, 2707–2715. doi:10.1016/j.drudis.2021.05.019
Mikolov, T., Karafiát, M., Burget, L., Jan, C., and Khudanpur, S. (2010). “Recurrent neural network based language model,” in Proceedings of the 11th Annual Conference of the International Speech Communication Association, INTERSPEECH 2010 (Makuhari), 1045–1048. doi:10.21437/interspeech.2010-343
Moll, S., Desmoulière, A., Moeller, M. J., Pache, J. C., Badi, L., Arcadu, F., et al. (2019). DDR1 role in fibrosis and its pharmacological targeting. Biochim. Biophys. Acta - Mol. Cell Res. 1866, 118474. doi:10.1016/j.bbamcr.2019.04.004
O’Boyle, N. M., and Dalke, A. (2018). DeepSMILES: an adaptation of SMILES for use in machine-learning of chemical structures. ChemRxiv 1–9. doi:10.26434/chemrxiv.7097960.v1
Olivecrona, M., Blaschke, T., Engkvist, O., and Chen, H. (2017). Molecular de-novo design through deep reinforcement learning. J. Cheminform. 9, 48–14. doi:10.1186/s13321-017-0235-x
Oord, A. van den, Dieleman, S., Zen, H., Simonyan, K., Vinyals, O., Graves, A., et al. (2016). WaveNet: a generative model for raw audio. arXiv Prepr. arXiv1609.03499. Available at: http://arxiv.org/abs/1609.03499.
Pang, C., Qiao, J., Zeng, X., Zou, Q., and Wei, L. (2023). Deep generative models in de novo drug molecule generation. J. Chem. Inf. Model. doi:10.1021/acs.jcim.3c01496
Polykovskiy, D., Zhebrak, A., Sanchez-Lengeling, B., Golovanov, S., Tatanov, O., Belyaev, S., et al. (2020). Molecular sets (MOSES): a benchmarking platform for molecular generation models. Front. Pharmacol. 11, 565644. doi:10.3389/fphar.2020.565644
Polykovskiy, D., Zhebrak, A., Vetrov, D., Ivanenkov, Y., Aladinskiy, V., Mamoshina, P., et al. (2018). Entangled conditional adversarial autoencoder for de novo drug discovery. Mol. Pharm. 15, 4398–4405. doi:10.1021/acs.molpharmaceut.8b00839
Popova, M., Isayev, O., and Tropsha, A. (2018). Deep reinforcement learning for de novo drug design. Sci. Adv. 4, eaap7885. doi:10.1126/sciadv.aap7885
Pun, F. W., Ozerov, I. V., and Zhavoronkov, A. (2023). AI-powered therapeutic target discovery. Trends Pharmacol. Sci. 44, 561–572. doi:10.1016/j.tips.2023.06.010
Putin, E., Asadulaev, A., Ivanenkov, Y., Aladinskiy, V., Sanchez-Lengeling, B., Aspuru-Guzik, A., et al. (2018a). Reinforced adversarial neural computer for de novo molecular design. J. Chem. Inf. Model. 58, 1194–1204. doi:10.1021/acs.jcim.7b00690
Putin, E., Asadulaev, A., Vanhaelen, Q., Ivanenkov, Y., Aladinskaya, A. V., Aliper, A., et al. (2018b). Adversarial threshold neural computer for molecular de novo design. Mol. Pharm. 15, 4386–4397. doi:10.1021/acs.molpharmaceut.7b01137
Radford, A., Metz, L., and Chintala, S. (2016). Unsupervised representation learning with deep convolutional generative adversarial networks. arXiv 2015. arXiv.1511.06434. doi:10.48550/arXiv.1511.06434
Rodríguez-Pérez, R., and Bajorath, J. (2020). Interpretation of machine learning models using shapley values: application to compound potency and multi-target activity predictions. J. Comput. Aided. Mol. Des. 34, 1013–1026. doi:10.1007/s10822-020-00314-0
Schneider, P., Walters, W. P., Plowright, A. T., Sieroka, N., Listgarten, J., Goodnow, R. A., et al. (2020). Rethinking drug design in the artificial intelligence era. Nat. Rev. Drug Discov. 19, 353–364. doi:10.1038/s41573-019-0050-3
Segler, M. H. S., Kogej, T., Tyrchan, C., and Waller, M. P. (2018). Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci. 4, 120–131. doi:10.1021/acscentsci.7b00512
Silver, D., Schrittwieser, J., Simonyan, K., Antonoglou, I., Huang, A., Guez, A., et al. (2017). Mastering the game of go without human knowledge. Nature 550, 354–359. doi:10.1038/nature24270
Stakyte, K., Rotheneder, M., Lammens, K., Bartho, J. D., Grädler, U., Fuchß, T., et al. (2021). Molecular basis of human ATM kinase inhibition. Nat. Struct. Mol. Biol. 28, 789–798. doi:10.1038/s41594-021-00654-x
Szilagyi, Z., and Gustafsson, C. M. (2013). Emerging roles of Cdk8 in cell cycle control. Biochim. Biophys. Acta - Gene Regul. Mech. 1829, 916–920. doi:10.1016/j.bbagrm.2013.04.010
Tong, X., Liu, X., Tan, X., Li, X., Jiang, J., Xiong, Z., et al. (2021). Generative models for de novo drug design. J. Med. Chem. 64, 14011–14027. doi:10.1021/acs.jmedchem.1c00927
Tropsha, A., Isayev, O., Varnek, A., Schneider, G., and Cherkasov, A. (2023). Integrating QSAR modelling and deep learning in drug discovery: the emergence of deep QSAR. Nat. Rev. Drug Discov., 1–15. doi:10.1038/s41573-023-00832-0
Walters, W. P., and Murcko, M. (2020). Assessing the impact of generative AI on medicinal chemistry. Nat. Biotechnol. 38, 143–145. doi:10.1038/s41587-020-0418-2
Wang, M., Wang, Z., Sun, H., Wang, J., Shen, C., Weng, G., et al. (2022). Deep learning approaches for de novo drug design: an overview. Curr. Opin. Struct. Biol. 72, 135–144. doi:10.1016/j.sbi.2021.10.001
Waring, M. J., Arrowsmith, J., Leach, A. R., Leeson, P. D., Mandrell, S., Owen, R. M., et al. (2015). An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 14, 475–486. doi:10.1038/nrd4609
Xi, M., Chen, T., Wu, C., Gao, X., Wu, Y., Luo, X., et al. (2019). CDK8 as a therapeutic target for cancers and recent developments in discovery of CDK8 inhibitors. Eur. J. Med. Chem. 164, 77–91. doi:10.1016/j.ejmech.2018.11.076
Xie, W., Wang, F., Li, Y., Lai, L., and Pei, J. (2022). Advances and challenges in de novo drug design using three-dimensional deep generative models. J. Chem. Inf. Model. 62, 2269–2279. doi:10.1021/acs.jcim.2c00042
Yang, Y., Zhang, R., Li, Z., Mei, L., Wan, S., Ding, H., et al. (2020). Discovery of highly potent, selective, and orally efficacious p300/CBP histone acetyltransferases inhibitors. J. Med. Chem. 63, 1337–1360. doi:10.1021/acs.jmedchem.9b01721
Zhavoronkov, A., and Aspuru-Guzik, A. (2020). Reply to ‘Assessing the impact of generative AI on medicinal chemistry. Nat. Biotechnol. 38, 146. doi:10.1038/s41587-020-0417-3
Keywords: drug discovery, artificial intelligence, machine-learning, deep-learning, generative models, anticancer agents
Citation: Romanelli V, Cerchia C and Lavecchia A (2024) Deep generative models in the quest for anticancer drugs: ways forward. Front. Drug Discov. 4:1362956. doi: 10.3389/fddsv.2024.1362956
Received: 29 December 2023; Accepted: 22 January 2024;
Published: 02 February 2024.
Edited by:
Giuseppe Felice Mangiatordi, National Research Council (CNR), ItalyReviewed by:
David Ryan Koes, University of Pittsburgh, United StatesDomenico Gadaleta, Mario Negri Institute for Pharmacological Research (IRCCS), Italy
Copyright © 2024 Romanelli, Cerchia and Lavecchia. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carmen Cerchia, Y2FybWVuLmNlcmNoaWFAdW5pbmEuaXQ=; Antonio Lavecchia, YW50b25pby5sYXZlY2NoaWFAdW5pbmEuaXQ=