 Lindsay J. Marshall
Lindsay J. Marshall Kathleen M. Conlee
Kathleen M. Conlee- Animal Research Issues, The Humane Society of the United States, Gaithersburg, MD, United States
Creating and developing new drugs can take decades, costs millions of dollars, requires untold human effort and usually, takes thousands of animal lives. Despite regulators professing confidence in non-animal approaches and guidance documents that permit submission of non-animal data, toxicity testing is routinely carried out in animals, employing rodents (invariably mice) and non-rodents. However, extensive preclinical testing in animals is still no guarantee that drugs will be safe and/or effective. In fact, more than nine out of every ten drugs that appear safe from animal trials will fail when tested in people, often due to unexplained toxicity or a lack of efficacy. This paper will describe recent advances in drug development where non-animal approaches have been used, to explore how and where these could be applied more widely to revolutionize the drug development pipeline and accelerate the creation of safe and effective medicines. As one case study, we look at the small molecule channel modifiers developed to address the consequences of the mutated chloride channel in the fatal genetic condition, cystic fibrosis. We then take a closer look at where drug development could be accelerated by focusing on innovative, human biology-based testing methods. Finally, we put forward recommendations, targeting all stakeholders, including the public, that will be needed to put this into practice and enable drug development to become more efficient - focusing on human-biology based testing and cutting out the middle-mouse.
1 Introduction
“The child with the salty brow shall die.” This was the prognosis for people born with cystic fibrosis (CF), before the condition had even been named. There was terrible recognition that tasting salt when you kissed your baby was a harbinger of doom. Decades of tireless research by (among others) Dorothy Andersen and Paul di Sant’Agnese provided the key observations that children dying of this condition were not producing digestive enzymes from their pancreas (Andersen, 1938); that the levels of salt (sodium chloride) in their sweat was much higher than healthy children (Di Sant’Agnese et al., 1953) and this led to the condition being named as cystic fibrosis. In 1989, an important breakthrough occurred when the gene responsible for CF disease was identified (Rommens et al., 1989). This study revealed that the “CF gene,” encoded a protein called the cystic fibrosis transmembrane conductance regulator (CFTR), responsible for regulating chloride ion movement out of cells.
1.1 Understanding cystic fibrosis
Identification of the gene was a crucial advance that was hailed as the last piece of the puzzle in the search for a cure for CF. However, as is customary for modelling human genetic diseases, the identification of a specific, causative gene leads to the creation of multiple “artificial” animal models, since animals do not naturally have CF. Identification of the gene associated with CF was confirmed in 1989 and the first animal model was created a couple of years later (Snouwaert et al., 1992). Animal models of CF initially used mice, but since these manipulated mice failed to fully recapitulate the human condition, and the animals did not have symptoms of the lung disease, this expanded to other animals including rats, ferrets, sheep and pigs. There are now more than 750 different genetic animal models of CF, of which more than 690 use mice (Leenaars et al., 2020). Since animals do not have CF naturally, there has to be some manipulation of the animal in order to generate symptoms of the disease and this requires either genetic mutation techniques, used for the genetic models, or non-genetic approaches, including the use of drugs, deliberate infection with pathogens, or grafting human tissues into the animals’ lungs. There are more than 220 non-genetic animal models for CF, again the majority of these use mice, but there are also rats, pigs, monkeys, and rabbits (Leenaars et al., 2021).
The Canadian database which logs the different mutations in CFTR that give rise to CF currently stands at 2,114 (http://www.genet.sickkids.on.ca/StatisticsPage.html). Even before considering the ethics and scientific relevance of this exercise, the costs and time required to develop an animal model for each of these mutations would be exorbitant. One commercial service offering development of genetically modified animals charges a basic fee of around $4,000 for a single mutation and this does not include technical support (such as animal care and surgery) or consumables (e.g., surgical equipment) (https://brcf.medicine.umich.edu/cores/transgenic-animal-model/fees/; https://www.umassmed.edu/globalassets/transgenic-animal-modeling-core/documents/whycostsomuch2015.pdf). To create one animal version of each CF mutation found in people would therefore cost more than 8 million US dollars. This is a very conservative estimate since costs would be greater for larger animals (rabbits costs at least twenty times more than mice and mini pigs are over one hundred times more costly than mice https://minipigs.dk/products-services/enquiries) and it is more expensive and time consuming to create the complex models which would more accurately represent the patient population (almost half of the UK CF population have two different mutations https://www.cysticfibrosis.org.uk/sites/default/files/2020-12/2019%20Registry%20Annual%20Data%20report_Sep%202020.pdf). Developing pig models of the spectrum of CF mutations found in humans would cost upwards of 190 million US dollars.
However, CF is a great example of how human-centred interventions (as opposed to animal-based research) have played a great role in improving understanding of CF such that the bleak prognosis for the child with the salty brow has improved over time. In the 1960s and ‘70s, increased survival was associated with the creation of dedicated CF centers, these were crucial for sharing best practices for nutrition and physiotherapy and for fast, aggressive treatment of infections. With these interventions and the knowledge sharing offered by the dedicated networks of CF physicians, parents and families, the median survival age for babies born with CF reached 11 years in the 70s and is still on the increase (Matthews et al., 1964; Doershuk et al., 1965) (Figure 1).
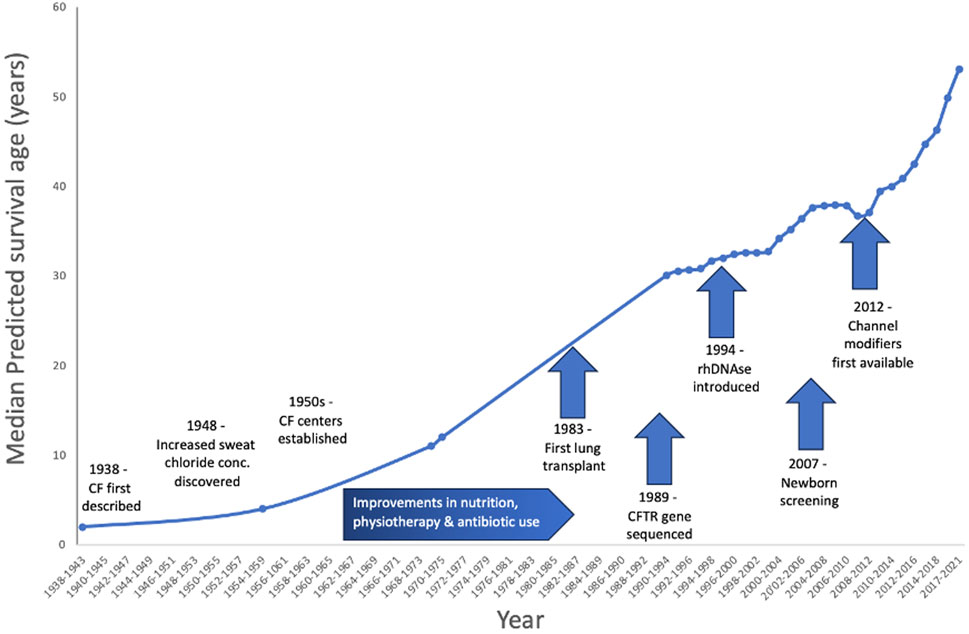
FIGURE 1. Increase in predicted survival over time for people born with CF. Improvements and advances in physiotherapy, antibiotic use and even lung transplantation have all contributed to this increase. However, aside from drugs to reduce the thick, sticky mucus and improvements in antibiotics, there were no specific treatments for CF - until 2012, when the first CFTR channel modifiers were approved for clinical use–note the steep increase in life expectancy since that time.
1.2 The drug development pipeline today
Traditionally, animals are used in drug development (Figure 2) to demonstrate the safety of the preparation (for a comprehensive review of the drug development process, see the paper by Singh et al. in this edition (Singh et al., 2023)). As stated by Dr van Norman in her 2019 article about animal use for drug safety testing, “[t]here is no doubt that the use of animals in science and medicine has significantly benefitted human beings.” However, despite an historical reliance on animals for medical advances, drug attrition rates of more than 90% (Sun et al., 2022) indicate that animals are not predictive for humans and a drug that is safe for animals is not always safe (or effective) in people (Van Norman, 2019; Van Norman, 2020). Unfortunately, it is not possible to carry out a stringent comparison of the possible failure rates if drugs were tested using the non-animal approaches. The data submitted by the pharmaceutical companies as part of the regulatory approval are not publicly available and whilst the current legislative requirements do not demand animal data, many of the drug safety guidelines to which pharmaceutical companies refer make reference to submission of preclinical pharmacology data from animals. It is therefore not possible to calculate the likely “failure rate” of a drug tested solely on human based methods or using human data. Despite a mounting body of evidence that the human based tools are (and will be) more predictive than animals, we cannot simply assume that the non-animal tests will always accurately predict human responses. However, as data from human relevant tools accumulate, this may change. Indeed, the case study of CF presented here indicates that patient-derived samples and developing tests using human cells offer insight into the biological effects of potential drugs and can revolutionise treatments.
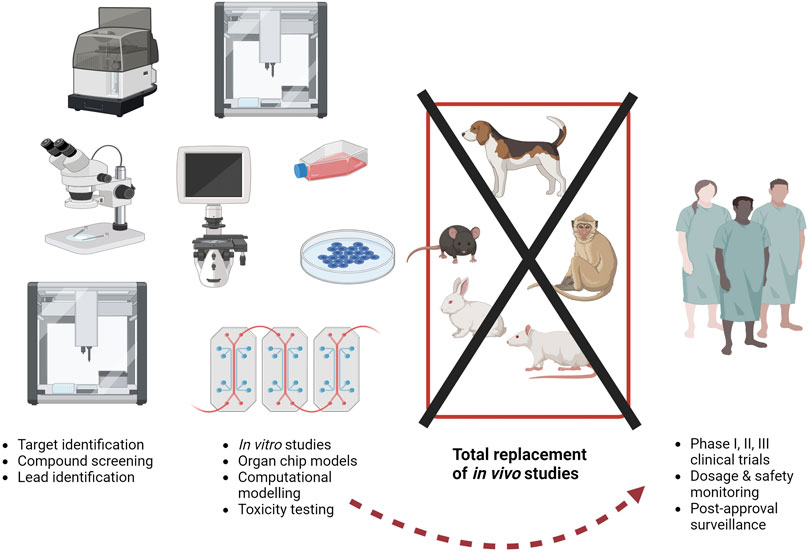
FIGURE 2. The current drug development paradigm is time consuming and costly, partly due to a reliance on animals. Estimates suggest around 10 to 5 years and 518 million to tens of billions of US dollars are required to progress from target identification (at the left of the figure) to regulatory approval (at the right pf the figure). As of today, animals may be used throughout this process, but the use of animal models is no guarantee of success, as even after approval, severe adverse effects in the extended patient population can result in drug withdrawal. More sustained, dedicated funding to the methods described on the left here (in vitro studies etc.) could help to bypass animal studies completely. Image created with BioRender.com.
The new approach methodologies (NAMs) are innovative methods that no longer rely on the use of live animals, these include human cells, tissues and organs, organ-chips or microphysiological systems (MPS), use of human data or human volunteers, and computer modelling. When considering the uniquely human nature of a disease, these methods offer a more relevant and physiologically accurate approach to understanding the disease features and therefore developing an effective treatment. They also provide more confidence in the predictivity of the data obtained—recent research has shown that MPS can recreate species-specific effects (Jang et al., 2019) and that MPS using human liver cells are better able to predict toxic drugs than animal testing (Ewart et al., 2022). In fact, analysis of possible savings that could be realised if human liver MPS were included for safety testing within the drug development pipeline revealed that these might reach around 3 billion USD in drug development costs (Ewart et al., 2022).
For drug safety testing, there are national and international guidance documents that have to be adhered to, and these indicate that animal studies should be undertaken (e.g., the International Council on Harmonisation guidance M3 (R2) for “Non-clinical safety studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals” states that “The development of a pharmaceutical is a stepwise process involving an evaluation of both animal and human efficacy and safety information” and goes on to detail what number and species of animals should be used throughout the process U. S. Food and Drug Administration). However, many of the regulatory agencies claim that they do not require data from animals and that they would be willing to examine data from the non-animal methods and the UK government recently declared that [“t]here is no United Kingdom legislation that mandates animal testing” (UK Parliament, 2023). Indeed, the Food and Drug Administration Modernization Act 2.0, which was signed into US law in 2022, permits drug developers to make use of “certain alternatives to animal testing, including cell-based assays and computer models, to obtain an exemption from the Food and Drug Administration to investigate the safety and effectiveness of a drug” (One hundred and seventeenth Congress of the United States, 2023). Likewise, for drug efficacy testing, (these are studies undertaken to evaluate whether a treatment will be effective), the regulatory agencies do not insist on animal data although animal models of disease are often used here, and so it seems that the door is open for non-animal data to be submitted to support product registration.
1.3 In vitro breakthroughs for CF show the way forwards
In 2006, researchers at Vertex first used human cell cultures with bronchial epithelial cells (taken from the upper airways of people) to develop a method to measure the activity of the CFTR channel (Van Goor et al., 2006). They used this human-relevant tool to screen hundreds of thousands of different chemicals and identify compounds that would increase CFTR activity. This assay revealed many hundreds of compounds with the desired activity, and the researchers then got rid of the ones that were unlikely to be successful drugs based on chemistry (through comparing structures to drugs that have been recognised as toxic). Fifty-three compounds were selected for further investigation using intestinal organoid models—note that organoids are tiny, cell-based models that retain the structure and function of the “parent” organ (Dekkers et al., 2013). Accessing bronchial epithelial cells is quite difficult and can require sedation, making this a complicated procedure to carry out in very young children (the intended patient population), but the cells to create intestinal organoids are more easily accessible and patients have reported limited discomfort with this technique (Servidoni et al., 2013). Thus, intestinal organoids offer a robust method for screening hundreds (or even thousands) of potential drugs. Using this approach, several likely compounds can be rapidly and efficiently assessed using the intestinal organoids, and the activity can be verified with the airways model so that the most promising candidate(s) can quickly move to the clinic.
1.4 Personalising medicine with personalised tools
Another advantage to using intestinal organoids from people with CF is that this offers a simple system with which to reveal patient specific treatments. Organoids are three dimensional structures, so that when CFTR is active, fluid influx leads to swelling of the organoid and measuring the volume of the organoid is a simple way to know whether the drug applied to the organoid is activating CFTR (where increased volume indicates active CFTR). Using this approach, patient-specific theratyping is possible, where organoids from patients can be screened against multiple compounds, or combinations of compounds, to find the optimal dose and identity of drugs to give each individual patient (Clancy et al., 2019; Conti et al., 2022). The major advantage is that the organoid retains the exact same genetic mutation in CFTR as the person, and so “treating” the organoid reflects what will happen in that individual. This testing is just not possible using animal models, or at least it would require that an animal model was available for every single individual with CF, which would take time and money and thousands of animal lives (as described above), and still does not guarantee success, given that animals do not exhibit the lung disease that we are trying to treat! Thus, adoption of the human cell-based intestinal organoid models as a screening platform makes this a very efficient way of screening individual people with CF, to ensure that the medication given will be effective, and therefore can be used to personalise treatment.
1.5 Non-animal methods could indicate where drugs won’t work for patients
The example of curcumin demonstrates another way in which in vitro, human cell-based tools can be used to help advance drug discovery, or at least to prevent false hope. Curcumin is a derivative of the spice turmeric; this was force-fed to genetically modified CF mice (who bore the mutation most frequently found in people with CF) for 3 days, before the electrical activity of their airways and intestines was measured, as an indicator of CFTR activity. The study also used cultures of hamster kidney cells with mutated CFTR inserted in them and tracked the processing of the CFTR protein, showing that curcumin enhanced the insertion of CFTR in the appropriate cellular localisation (Egan et al., 2004). These results suggested that curcumin could correct the CFTR-dependent deficit but unfortunately, this was not recapitulated for people with CF. When the studies were repeated with airways epithelial cells isolated from someone with CF, curcumin did not increase the electrical activity (Song et al., 2004). These data are evidence of the issue with translational failures–where data in animals are not recapitulated in people and indicate that caution is needed in interpreting data from non-human model systems. We suggest that the incorporation of more human-relevant tools, such as the airways models or intestinal organoids, in the drug discovery pipeline, would help reduce these failures.
1.6 Comparing patients and mutations allows wider drug use
The non-animal, human relevant tools can be used to reveal insights into the association between physiology and disease symptoms, that are not possible in animals, and that allow researchers to estimate the likely clinical effect of a drug. For example, clinical data comparing the electrical potential difference across the airways in people with or without CF indicates that increasing the amount of CFTR in the cell membrane by around 15%–30% would help to “normalise” this electrical activity (which is associated with ion movement, and therefore CFTR activity) in CF. Researchers can then test potential drugs, existing drugs and combinations of these, in human airways cell models to find those compounds that increase membrane CFTR and enhance CFTR activity. An additional advantage of using human cell-based systems is the ability to carry out direct comparisons. Models developed using CF cells can be compared with non-CF cultures, but importantly models from people with CF who have different mutations in their CFTR can be compared. As we have already mentioned above, research with genetically engineered animal models is almost prohibitively expensive, time consuming, and cannot accurately model human responses, whereas the human cell-based approach allows researchers to clarify the relationship between the mutation in CFTR and its activity, and therefore to make intelligent decisions regarding which CFTR modifiers may be helpful for individual people with CF. Although there is one specific mutation that affects the majority of people with CF (Kerem et al., 1989), as described above, there are over 2000 different mutations. In vitro, cell-based methods offer a more efficient manner with which to test the effects of drugs on different mutations and combinations of mutations (since people with CF may have two different mutations). It is very gratifying to see this potential utility of non-animal models reflected at some level of regulations, as the FDA granted a “label extension” for one of the CFTR modifiers, based purely on data from human cell models (Ratner, 2017), enabling the broader use of this drug for many more people with CF.
1.7 Drug repurposing with cell-based tools
The realisation that patient-derived intestinal organoids could be used to filter hundreds or compounds to detect possible CFTR modifiers for treating CF led to another important advance - that of the application of human relevant tools in drug repurposing, an approach suitable for applications beyond CF. Drug repurposing is an efficient way of finding new treatments as it employs existing, approved drugs for a purpose other than that for which they were approved, and so this bypasses the lengthy, expensive safety testing that is needed for entirely new compounds.
This extends beyond CF: there are other examples of drug repurposing where the efficacy data were obtained using human cell-based approaches. For example, for SARS-CoV2, researchers tested seven clinically approved drugs on airways models made of human cells, measuring the ability of these drugs to prevent the virus getting inside the cells (Si et al., 2020). When the drugs were tested at concentrations and flow rate equivalent to those found in human blood, they found that only two of them showed great promise in terms of preventing infection and, soon after, a clinical trial was set up to assess the effects of one of these drugs in people with COVID. Additionally, a human stem cell-based system showed that a biological therapeutic could be effective in a rare neuropathy disease (Rumsey et al., 2022) and data from an organ chip demonstrated that a medication used in Type 2 diabetes could prevent chemotherapy-induced kidney toxicity (Cohen et al., 2021). This is hopefully the start of a shift in the regulatory paradigm and is indicative of enhancing flexibility to enable accelerated access to safe and effective drugs for all patients.
2 Discussion
This paper uses CF as an example to show how the incorporation of non-animal tools into the drug development can be transformational. Two examples of human cell-based systems presented here -namely the use of intestinal organoids and airways epithelial cell cultures—revealed the CFTR modifying activity of small molecule therapeutics that have gone on to revolutionise life for people with CF. Given the current regulatory requirements (detailed in Figure 2) the CFTR modifier drugs were tested (for toxicity) in animals, but there was no way that animal models of CF, given the lack of respiratory involvement in mice, for example, could show the efficacy that was needed to give the confidence that these drugs would be disruptive for people living with CF and for parents of babies born with CF.
3 Recommendations
Finally, by using cystic fibrosis as a case study, we offer a few brief recommendations for some of the points where we see implementation or action is needed to enable wider use of the non-animal methods for drug development. For the purpose of this review, we have focused on the development and application of the non-animal approaches. We appreciate the historical advances that occurred as a result of animal use, including, for example, the discovery of insulin in 1921 and the advent of the polio vaccine in 1953. However, we also believe that the successful, historical use of animals, particularly in the face of rapidly evolving non-animal technologies, does not scientifically justify their continued use. This reflects the viewpoints reported in notable reviews such as the report on the use of dogs as subjects of biomedical research (The National Academies of Sciences, Engineering, and Medicine, 2020) and the general public, who are invested in the use of non-animal technologies to replace animals (Savanta, 2022). Therefore, the recommendations below are directed at accelerating the replacement of animals rather than considering how and where animal use should be continued.
Regulatory transparency is key-the agencies should be encouraged to publicly publish where they have accepted data form non-animal methods, to ensure that animal use is not duplicated.
Biobanking (where patient samples or volunteer biological material is curated and stored for widespread use by researchers) should be incentivized. This is particularly important for rare disease like CF, where clinical trials are limited by the number of patients with the same mutation in a specific geographic location. Providing global access to the tissue, and developing methods for developing assays based on these tissues, would help to address this disparity. It is also important to ensure that the biological material deposited in the biobanks fully reflects the differences in the human population (Ghosh et al., 2022).
Funding agencies need to analyse where their funding is not providing the expected, or acceptable, return on investment. This may require retrospective reviews of projects but would be invaluable in identifying where projects or topics are failing to deliver and therefore could inform future funding strategies. Presently, over 90% of drugs fail in people (Sun et al., 2022) and a proportion of this failure is directly related to the use of animals as models (Van Norman, 2019). Understanding where the animal models continually fail enables the decision to no longer fund this sort of research, and could allow diversion of the award money to in vitro, epidemiological, or computer modelling based research that could help to advance the field. This is an issue that should resonate with the public, given their role in research. In the UK in 2018, £1.2billion funding originated from medical charities, representing around 14% of all health related research funding in the UK (Fraser of Allander Institute, 2021). In the US, the largest funder of biomedical research is the National Institutes of Health (NIH), which relies on taxpayer dollars. The NIH awards an estimated half of its total budget of over 47 billion dollars to animal-based research (U.S. Government Accountability Office, 2019), yet our analysis has revealed that less than 1% of this is dedicated to organ chips, for example. The public are therefore heavily invested in research, either voluntarily through charitable donations, or through their taxes and the same public is vocal in its desire to move away from using animals in medical research (IPSOS MORI, 2018; Savanta, 2022), although this depends on several factors, including the type of animal and purpose of research (Brunt and Weary, 2021).
At the Humane Society of the United States, we are keen to prioritise funding to development and use of the non-animal methods. In 2023 we introduced a bill in Maryland that requires that laboratories using animals have to contribute to a research fund which is available for non-animal method developers. This legislation creates a precedent for the transition toward the non-animal methods of the future. The subsequent funding shift will help to accelerate scientific discovery by allowing for early adoption of promising non-animal methods. However, this shift impacts just one state of the US and we need more. If the NIH and other government funding agencies could make the commitment to shift an annual 5%–10% of their budgets to non-animal research, we might see more advances, like the CFTR modifier drugs, more quickly.
Author contributions
LJM: Conceptualization, Writing–original draft, Writing–review and editing. KMC: Writing–review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Andersen, D. H. (1938). CYSTIC FIBROSIS OF THE PANCREAS AND ITS RELATION TO CELIAC DISEASE: A CLINICAL AND PATHOLOGIC STUDY. Am. J. Dis. Child. 56, 344–399. doi:10.1001/archpedi.1938.01980140114013
Brunt, M. W., and Weary, D. M. (2021). Public consultation in the evaluation of animal research protocols. PLoS One 16, e0260114. doi:10.1371/journal.pone.0260114
Clancy, J. P., Cotton, C. U., Donaldson, S. H., Solomon, G. M., Vandevanter, D. R., Boyle, M. P., et al. (2019). CFTR modulator theratyping: current status, gaps and future directions. J. Cyst. Fibros. 18, 22–34. doi:10.1016/j.jcf.2018.05.004
Cohen, A., Ioannidis, K., Ehrlich, A., Regenbaum, S., Cohen, M., Ayyash, M., et al. (2021). Mechanism and reversal of drug-induced nephrotoxicity on a chip. Sci. Transl. Med. 13, eabd6299. doi:10.1126/scitranslmed.abd6299
Conti, J., Sorio, C., and Melotti, P. (2022). Organoid technology and its role for theratyping applications in cystic fibrosis. Child. (Basel) 10, 4. doi:10.3390/children10010004
Dekkers, J. F., Wiegerinck, C. L., De Jonge, H. R., Bronsveld, I., Janssens, H. M., De Winter-De Groot, K. M., et al. (2013). A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat. Med. 19, 939–945. doi:10.1038/nm.3201
Di Sant’Agnese, P. A., Darling, R. C., Perera, G., and Shea, E. (1953). Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas; clinical significance and relationship to the disease. Pediatrics 12, 549–563. doi:10.1542/peds.12.5.549
Doershuk, C., Matthews, L., and Tucker, A. (1965). Evaluation of a prophylactic and therapeutic program for patients with cystic fibrosis. Pediatrics 36, 675–688. doi:10.1542/peds.36.5.675
Egan, M. E., Pearson, M., Weiner, S. A., Rajendran, V., Rubin, D., Glockner-Pagel, J., et al. (2004). Curcumin, a major constituent of turmeric, corrects cystic fibrosis defects. Science 304, 600–602. doi:10.1126/science.1093941
Ewart, L., Apostolou, A., Briggs, S. A., Carman, C. V., Chaff, J. T., Heng, A. R., et al. (2022). Performance assessment and economic analysis of a human Liver-Chip for predictive toxicology. Commun. Med. 2, 154. doi:10.1038/s43856-022-00209-1
Fraser of Allander Institute (2021). The contribution of medical funding by charities to the UK economy. Strathclyde: Univerity of Strathclyde Business School.
Ghosh, S., Nehme, R., and Barrett, L. E. (2022). Greater genetic diversity is needed in human pluripotent stem cell models. Nat. Commun. 13, 7301. doi:10.1038/s41467-022-34940-z
Jang, K. J., Otieno, M., Ronxh, I. J., Lim, H., Ewart, L., Kodella, K., et al. (2019). Reproducing human and cross-species drug toxicities using a Liver-Chip. Sci. Transl. Med. 11, eaax5516. doi:10.1126/scitranslmed.aax5516
Kerem, B., Rommens, J. M., Buchanan, J. A., Markiewicz, D., Cox, T. K., Chakravarti, A., et al. (1989). Identification of the cystic fibrosis gene: genetic analysis. Science 245, 1073–1080. doi:10.1126/science.2570460
Leenaars, C. H., De Vries, R. B., Heming, A., Visser, D., Holthaus, D., Reijmer, J., et al. (2020). Animal models for cystic fibrosis: a systematic search and mapping review of the literature - Part 1: genetic models. Lab. Anim. 54, 330–340. doi:10.1177/0023677219868502
Leenaars, C. H., Vries, R. B., Reijmer, J., Holthaus, D., Visser, D., Heming, A., et al. (2021). Animal models for cystic fibrosis: a systematic search and mapping review of the literature. Part 2: nongenetic models. Lab. Anim. 55, 23677221990688. doi:10.1177/0023677221990688
Matthews, L. W., Doershuk, C. F., Wise, M., Eddy, G., and Nudelman, H. (1964). A therapeutic regimen for patients with cystic fibrosis. J. Pediatr. 65, 558–575. doi:10.1016/s0022-3476(64)80290-0
National Academies of Sciences, Engineering, and Medicine (2020). Necessity, use, and care of laboratory dogs at the U.S. Department of Veterans Affairs. Washington DC: National Academies Press.
One Hundred and Seventeenth Congress of the United States (2023). Consolidated appropriations Act, 2023. Avaliable at: https://www.congress.gov/117/plaws/publ328/PLAW-117publ328.pdf.
Ratner, M. (2017). FDA deems in vitro data on mutations sufficient to expand cystic fibrosis drug label. Nat. Biotechnol. 35, 606. doi:10.1038/nbt0717-606
Rommens, J. M., Iannuzzi, M. C., Kerem, B.-S., Drumm, M. L., Melmer, G., Dean, M., et al. (1989). Identification of the CysticFibrosis gene: chromosome walking and jumping. Science 245, 1059–1065. doi:10.1126/science.2772657
Rumsey, J. W., Lorance, C., Jackson, M., Sasserath, T., Mcaleer, C. W., Long, C. J., et al. (2022). Classical complement pathway inhibition in a “human-on-A-chip” model of autoimmune demyelinating neuropathies. Adv. Ther. 5, 2200030. doi:10.1002/adtp.202200030
Servidoni, M. F., Sousa, M., Vinagre, A. M., Cardoso, S. R., Ribeiro, M. A., Meirelles, L. R., et al. (2013). Rectal forceps biopsy procedure in cystic fibrosis: technical aspects and patients perspective for clinical trials feasibility. BMC Gastroenterol. 13, 91. doi:10.1186/1471-230X-13-91
Si, L., Bai, H., Rodas, M., Cao, W., Oh, C. Y., Jiang, A., et al. (2020). Human organs-on-chips as tools for repurposing approved drugs as potential influenza and COVID19 therapeutics in viral pandemics. BioRxv.
Singh, N., Vayer, P., Tanwar, S., Poyet, J.-L., Tsaioun, K., and Villoutreix, B. O. (2023). Drug discovery and development: introduction to the general public and patient groups. Front. Drug Discov. 3. doi:10.3389/fddsv.2023.1201419
Snouwaert, J. N., Brigman, K. K., Anne, M., Malouf, N. N., Boucher, R. C., Smithies, O., et al. (1992). An animal model for cystic fibrosis made by gene targeting. Science 257, 1083–1088. doi:10.1126/science.257.5073.1083
Song, Y., Sonawane, N. D., Salinas, D., Qian, L., Pedemonte, N., Galietta, L. J., et al. (2004). Evidence against the rescue of defective DeltaF508-CFTR cellular processing by curcumin in cell culture and mouse models. J. Biol. Chem. 279, 40629–40633. doi:10.1074/jbc.M407308200
Sun, D., Gao, W., Hu, H., and Zhou, S. (2022). Why 90% of clinical drug development fails and how to improve it? Acta Pharm. Sin. B 12, 3049–3062. doi:10.1016/j.apsb.2022.02.002
UK Parliament (2023). Question for department of health and social care. Avaliable at: https://questions-statements.parliament.uk/written-questions/detail/2023-09-19/200502.
U. S. Food and Drug Administration (2009). Guidance on non-clinical safety studies for the Conduct of human clinical trials and marketing authorization for pharmaceuticals M3. USA: FDA.
U.S. Government Accountability Office (2019). “Animal use in research: federal agencies should assess and report on their efforts to develop and promote alternatives,” in United States government accountability office. USA: GAO.
Van Goor, F., Straley, K. S., Cao, D., Gonzalez, J., Hadida, S., Hazlewood, A., et al. (2006). Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol. Lung Cell Mol. Physiol. 290, L1117–L1130. doi:10.1152/ajplung.00169.2005
Van Norman, G. A. (2019). Limitations of animal studies for predicting toxicity in clinical trials. JACC Basic Transl. Sci. 4, 845–854. doi:10.1016/j.jacbts.2019.10.008
Keywords: cystic fibrosis, in vitro, drug development, cystic fibrosis transmembrane conductance regulator, new approach methodologies
Citation: Marshall LJ and Conlee KM (2024) The case of the missing mouse—developing cystic fibrosis drugs without using animals. Front. Drug Discov. 4:1347246. doi: 10.3389/fddsv.2024.1347246
Received: 30 November 2023; Accepted: 15 January 2024;
Published: 31 January 2024.
Edited by:
Bruno Villoutreix, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Jeremy J. Yang, University of New Mexico, United StatesCopyright © 2024 Marshall and Conlee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lindsay Marshall, bG1hcnNoYWxsQGhzaS5vcmc=