 O. A. Arosarena
O. A. Arosarena A. S. Saribas4†
A. S. Saribas4†
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Drug Discov. , 11 April 2024
Sec. Anti-Cancer Drugs
Volume 4 - 2024 | https://doi.org/10.3389/fddsv.2024.1278163
Reversible phosphorylation is the basis for signal transduction in eukaryotic cells, and this is tightly controlled by the complex interplay of kinases and phosphatases. Many malignancies are characterized by dysregulation of the delicate protein phosphorylation balance. The targeting of protein phosphatases has been gaining attention as their role in cancer development and progression has been elucidated. The protein phosphatase-2A (PP2A) family of phosphatases are the primary cellular serine/threonine phosphatases. PP2A heterotrimers containing the B55α (PR55α) regulatory subunit have been associated with oncogenic signaling, and B55 subunits are found exclusively in forms of PP2A in which the carboxyl terminus of the catalytic subunit (PP2Ac) is methylated. Methylation of PP2Ac is primarily mediated by leucine carboxyl methyltransferase-1 (LCMT-1). Demethylation is controlled by an esterase, PP2A methylesterase (PME-1). We tested two potential LCMT-1 small molecule inhibitors and found that methyl 4-methyl-2-[(2-methylbenzoyl)amino]-5-[[(3-methylphenyl)amino]carbonyl]-3-thiophenecarboxylate (henceforth referred to as Compound 2) significantly inhibited PP2Ac methylation in vitro (p = 0.0024), and in the MDA-MB-231 breast carcinoma (p = 0.0431) and Rosi melanoma (p = 0.0335) cell lines. Compound 2 significantly reduced survival in HEK-293, HS-5, MDA-MB-231 and Rosi cells; and constrained clonogenic colony formation in MCF7, MDA-MB-231 and Rosi cells. The LCMT-1inhibitor induced G0/G1 cell cycle arrest in Rosi cells (p = 0.0193) and induced apoptosis in MDA-MB-231 cells (p < 0.0001). Increased phosphorylation of the receptor-interacting serine/threonine protein kinase-1 (RIPK1) was detected in MDA-MB-231 (p = 0.0273) and Rosi cells (p = 0.0179) in response to treatment with Compound 2. These data add to the body of evidence pointing to LCMT-1 as an oncogenic target.
Reversible phosphorylation is the basis for signal transduction in eukaryotic cells, and this is tightly controlled by the complex interplay of kinases and phosphatases. Protein phosphorylation is one of the most common post-translational modifications, altering structural conformation and function. It is necessary for a variety of cellular functions including mitosis, cell death, metabolism, organelle trafficking, differentiation and migration (Heo et al., 2022; Cervone et al., 2018; OConnor et al., 2018). Aberrant phosphorylation is a mark of disease states including cancer, diabetes and neurodegenerative diseases.
It is estimated that two-thirds of proteins encoded by the human genome undergo phosphorylation (Ardito et al., 2017), and there are 538 known human kinases (Zhang et al., 2021). More than two-thirds of phosphorylation events occur on serine, threonine or tyrosine residues. Serine phosphorylation is the most common event, followed by threonine phosphorylation, with tyrosine phosphorylation being the rarest. In contrast to the kinases, there are approximately 200 known phosphatases, the majority of which are tyrosine phosphatases (Sacco et al., 2012). Of the two families of serine/threonine phosphatases, the protein phosphatase-2A family (PP2A) is known to regulate development, apoptosis, transcription, translation, growth and cell division (Moura and Conde, 2019; Vaneynde et al., 2022).
Many malignancies are characterized by dysregulation of the delicate protein phosphorylation balance due to mutations, chromosomal rearrangements or epigenetic modifications resulting in constitutive kinase activation (Cicenas et al., 2018). Much more is known about the kinases than the phosphatases, and several kinase inhibitors have been successful in the treatment of some malignancies. However, these successes have been limited by the development of drug resistance. The targeting of protein phosphatases has been gaining attention as their role in cancer development and progression has been elucidated (Haesen et al., 2014; Turdo et al., 2021; Remmerie and Janssens, 2019; Xiao et al., 2018; Vainonen et al., 2021; Dai et al., 2017; Sun et al., 2021; Uddin et al., 2020; DArcy et al., 2019). LB-100, a PP2A inhibitor, is currently in clinical trials (Chung et al., 2017; Ronk et al., 2022).
PP2A enzymes have a heterotrimeric structure consisting of scaffolding (A), catalytic (C) and regulatory (B) subunits. While there are two isoforms each of the A and C subunits, substrate specificity is mediated primarily by the 23 isoforms of the B subunits (Haesen et al., 2014; Vaneynde et al., 2022). PP2A heterotrimers containing the B55α (PR55α) regulatory subunit have been associated with oncogenic signaling (Smits et al., 1992; Hein et al., 2016; Di et al., 2017; Hein et al., 2019), and B55 subunits are found exclusively in forms of PP2A in which the carboxyl terminus of the catalytic subunit (PP2Ac) is methylated at leucine 309 (Longin et al., 2007).
Methylation of PP2Ac is controlled by two enzymes, a methyl transferase (leucine carboxyl methyltransferase-1 (LCMT-1)), which adds the methyl group with S-adenosylmethionine (SAM) as the methyl donor; and an esterase (PP2A methyl esterase (PME-1)), which removes the methyl group (Figures 1A, B) (Lee and Pallas, 2007; Stanevich et al., 2011; Sangodkar et al., 2016; Lee et al., 2018). Methylation and demethylation of PP2Ac modulates PP2A activity. LCMT-1 is necessary for normal progression through mitosis and its overexpression has been associated with poor prognosis in hepatocellular carcinoma (Lee and Pallas, 2007; Zhang et al., 2023). We hypothesized that a small molecule inhibitor of LCMT-1 would inhibit cancer cell survival in a manner similar to LB-100. While LB-100 inhibits the catalytic subunit of PP2A, the LCMT-1 inhibitor would indirectly inhibit PP2A heterotrimers containing the B55α regulatory subunit by preventing PP2Ac methylation.
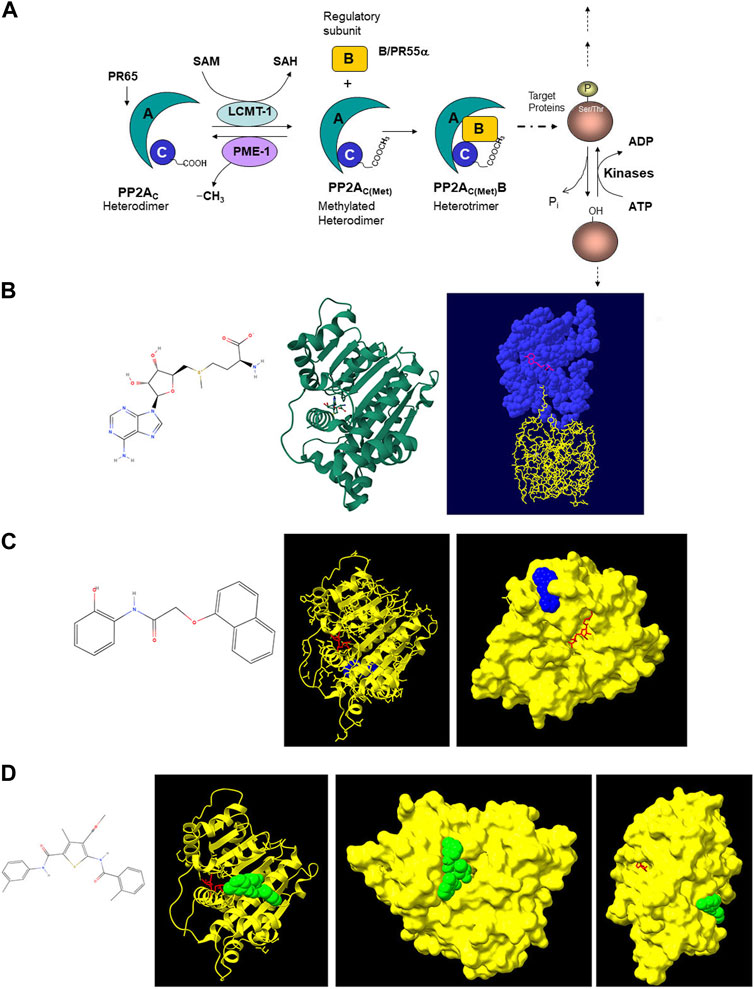
Figure 1. (A) Schematic diagram of PP2A assembly and activity. PP2A enzymes have a heterotrimeric structure consisting of one scaffolding (A) subunit (PR65), one catalytic (C) subunit and one regulatory (B) subunit. Owing to various isoforms of each of these subunits (2 A, 2 C and 23 B isoforms), 92 different heterotrimeric complexes can be assembled, each with its own specific substrate. Methylation of the catalytic subunit of PP2A near its carboxyl terminus at leucine-309 facilitates docking of certain regulatory subunits, including the B55α subunit (PR55α). This methylation is catalyzed by LCMT-1, the primary cellular PP2A methyltransferase. In this reaction, SAM is the methyl donor. PP2Ac is demethylated by PME-1. (B) Left: structure of SAM (generated with MolView software). Center: X-ray crystallographic model of LCMT-1 in complex with SAM (Image from RCSB Protein Data Bank (PDB, RCSB.org) of PDB ID 3IEI (Stanevich V, Jiang L, Satyshur KA, Li Y, Jeffrey PD, Li Z, Semmelhack MF, Xing Y. Structural insights into novel functions of a pro-survival PP2A-specific methyltransferase. To be published). Right: X-ray crystallographic model of LCMT-1 (blue) in complex with SAM (pink) and PP2Ac (yellow). (Stanevich V, Jiang L, Satyshur KA, Li Y, Jeffrey PD, Li Z, Menden P, Semmelhack MF, Xing Y. The structural basis for tight control of PP2A methylation and function by LCMT-1 (2011) Mol Cell 41:331–342) created with Swiss-PdbViewer (Guex and Peitsch, 1997). (C) Left: structure of Compound 1 (N-(2-Hydroxyphenyl)-2-(1-naphthal enyloxy)acetamide, generated with MolView software). Center and Right: X-ray crystallographic models of LCMT-1 (yellow) in complex with SAM (red) and Compound 1 (blue). Note that Compound 1 does not interact with LCMT-1 at the SAM binding site. (Tsai ML, Cronin N, Djordjevic S. The structure of human leucine carboxyl methyltransferase one that regulates protein phosphatase PP2A (2011) Acta Crystallogr D Biol Crystallogr 67:14-24) created with AutoDock Vina (Trott and Olson, 2010; Eberhardt et al., 2021). (D) Left: Structure of the Compound 2 (methyl 4-methyl-2-[(2-methylbenzoyl)amino]-5-[[(3-methylphenyl)amino]carbonyl]-3-thiophenecarboxylate, generated with MolView software). Right X-ray crystallographic models of LCMT-1 (yellow) in complex with Compound 2 (green) and SAM (red). Note that Compound 2 does not interact with LCMT-1 at the SAM binding site. (Tsai ML, Cronin N, Djordjevic S. The structure of human leucine carboxyl methyltransferase one that regulates protein phosphatase PP2A (2011) Acta Crystallogr D Biol Crystallogr 67:14–24) created with AutoDock Vina (Trott and Olson, 2010; Eberhardt et al., 2021).
This study was granted exempt status from the Temple University Institutional Review Board (protocol #13653) because commercially available cell lines were used.
Library screening was performed by Dr. Saribas while he was employed by Tetralogic Pharmaceuticals, which is no longer in business. Small molecule libraries (ASINEX and CHEM DIV03) comprising a total of 36,550 compounds that were not SAM analogues were screened using a radioactive LCMT-1 activity assay (Supplementary Figure S1). This screen identified two potential LCMT-1 inhibitors, N-(2-Hydroxyphenyl)-2-(1-naphthal enyloxy)acetamide (henceforth referred to as Compound 1) and (3-Thiophenecarboxylic acid, 4-methyl-2-[(2-methylbenzoyl)amino]-5-[[(3-methylphenyl)amino]carbonyl]-, methyl ester (also known as Methyl 4-methyl-2-[(2-methylbenzoyl)amino]-5-[[(3-methylphenyl)amino]carbonyl]-3-thiophenecarboxylate), henceforth referred to as Compound 2, Figures 1C, D and e, Supplementary Figure S2).
The x-ray crystallographic structures of human LCMT-1 (https://doi.org/10.2210/pdb3O7W/pdb), the SET (catalytic) domain of human histone-lysine-N-methyltransferase 2E (MLL5 methyltransferase, https://doi.org/10.2210/pdb5HT6/pdb), and both chains of the heterodimeric acidic residue methyltransferase 1 (ARMT1, https://doi.org/10.2210/pdb6UMQ/pdb) were downloaded from the RCSB Protein Data Bank (RCSB.org) (Berman et al., 2000). The SET domain is conserved in class II and III methyltransferases (Katz et al., 2003). Like LCMT-1, both chains of ARMT1 contain a Rossman fold catalytic domain characteristic of class I methyltransferases (Katz et al., 2003; Dennis et al., 2020). Protein-ligand interactions between the SET domain of MLL5 and Compound 2, the SET domain of MLL5 and SAM, both chains of ARMT1 and Compound 2, and both chains of ARMT1 and SAM were modelled with SwissDock software (Grosdidier et al., 2011). Because interactions between LCMT-1, and Compounds one and two could not be analyzed with SwissDock due to the size of LCMT-1, these interactions were analyzed with AutoDock Vina software (Trott and Olson, 2010; Eberhardt et al., 2021). Docking parameters and outputs can be viewed at https://mynotebook.labarchives.com/share/ Arosarena%2520Lab/NjMuN3w5NTAwNzAvNDkv VHJlZU5vZGUvMTEzNTA4NzM5MXwxNjEuNw==. Outputs from SwissDock and AutoDock Vina were converted to Protein Data Bank file format with OpenBabel software (OBoyle et al., 2011). Outputs were visualized with University of California San Francisco (UCSF) ChimeraX software (Goddard et al., 2018; Pettersen et al., 2021; Meng et al., 2023).
Recombinant human LCMT-1 (rhLCMT-1) and rhPP2Ac were obtained from SignalChem (Richmond, British Columbia, Canada). Methyltransferase assays were performed with 100 ng rhLCMT-1 and 91.62 ng rhPP2Ac (2:1 M ratio) (Stanevich et al., 2011) in reaction buffer containing 20 mM Tris (pH 8.0), 50 mM NaCl, 1 mM EDTA. rhLCMT-1 was incubated with Compound 1 (Chemspace, San Jose, California), Compound 2 (Akos Consulting and Solutions, Amtsgericht, Freiburg, Germany) or sinefungin, a SAM analogue, (Active Motif, Carlsbad, California), each at 50 µM final concentration, for 10 min at room temperature. rhPP2Ac was added to the reactions with 20 µM (final concentration) SAM (New England Biolabs, Ipswich, Massachusetts, Supplementary Figure S1B). The negative control reaction did not contain SAM. Reactions were incubated at 37°C for 30 min (Supplementary Figure S1C). Because the inhibitors gave false positive results with the secondary reactions used in colorimetric and bioluminescent methyltansferase assays, LCMT-1 activity was assessed using dot immunoblots. Reactions were applied to nitrocellulose membranes (Cytiva, Marlborough, Massachusetts). The membranes were blocked with tris-buffered saline (TBS) with 5% nonfat dry milk. Membranes were probed with an antibody directed to demethylated PP2Ac (Santa Cruz Biotechnology, Dallas, Texas, catalog #sc-13601, 1:1,000 dilution). Membranes were treated with infrared (IR)-dye anti-mouse antibody (Li-Cor, Lincoln, Nebraska, catalog #926–32210, 1:20,000 dilution) and densities were measured with Li-Cor Image Studio (version 5.2) software. rhLCMT-1 inhibition assays were performed three times, each time with five replicates for each condition.
The human MDA-MB-231 and MCF7 breast carcinoma cell lines were obtained from the Fox Chase Cancer Center cell culture core facility. These cell lines have been previously shown to be sensitive to glucose starvation-mediated PR55α depletion (Di et al., 2017). Additional cell lines (501-MEL, Rosi, UM-SCC-12, UM-SCC-14a, UM-SCC-49, UM-SCC-69, SCC-15, SCC-25, A549) were screened for sensitivity to PR55α depletion with glucose starvation assays. 501-MEL and Rosi cells were gifted by Dr. Rafal Kaminski (Center for Neurovirology and Gene Editing, Lewis Katz School of Medicine at Temple University (LKSOM)). The human embryonic kidney (HEK-293) and A549 cells were gifted by Dr. Mary Barbe (Center for Translational Medicine, LKSOM). SCC-15 cells were gifted by Dr. M. Alexandra Monroy (Diagenode, Philadelphia, Pennsylvania). HS-5 and SCC-25 cells were obtained from the American Type Culture Collection. UM-SCC-12, UM-SCC-14a, UM-SCC-49 and UM-SCC-69 cells were obtained from the Department of Otolaryngology at the University of Michigan. We determined that Rosi melanoma cells were susceptible to glucose starvation (Supplementary Figure S3). SCC-15 and SCC-25 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) with Ham’s F-12 (DMEM/F-12, Corning®, Glendale, Arizona, catalog #90–090-PB) with 10% fetal bovine serum (FBS, Corning® catalog #35–015-CV). The remaining cells were grown in DMEM (Corning® catalog #50–003-PC) with 10% FBS. Cells were maintained in a 5% carbon dioxide atmosphere at 37°C and tested free of mycoplasma contamination. Cells were not maintained in culture for more than 6 months without cryopreservation.
MCF7, MDA-MB-231 and Rosi cells were treated with Compound 1 or Compound 2 at a concentration of 20 µM in complete medium, and lysates were harvested after 2, 4, 6, 8 and 12 h in radioimmunoprecipitation assay buffer containing protease inhibitors (Boster Bio, Pleasanton, California, catalog #AR1182-1) and phosphatase inhibitors (Santa Cruz Biotechnology catalog #sc-45044 and #sc-45045). Protein concentrations were measured with a bicinchoninic acid assay (G-Biosciences, Saint Louis, Missouri). Fifty micrograms of each lysate were used for bis-tris polyacrylamide gel electrophoresis. Resolved lysates were transferred to nitrocellulose membranes. Membranes were blocked with TBS with 5% bovine serum albumin before incubation with primary antibodies.
Antibodies obtained from Cell Signaling Technology (Danvers Massachusetts) were, anti-PP2Ac (catalog #2038, 1:1,000 dilution), anti-phospho-adenosine monophosphate-activated protein kinase (pAMPKα2 threonine 172, catalog #2535, 1:1,000 dilution). Antibodies obtained from Proteintech (Rosemont, Illinois) include anti-phospho-receptor-interacting serine/threonine protein kinase-1 antibodies (pRIPK1 serine 161 (catalog #66854-1-Ig, 1:1,000 dilution) and pRIPK1 serine 166 (catalog #28252-1-AP, 1:10,000 dilution), anti-mixed lineage kinase domain like pseudokinase (MLKL, catalog #66675-1-Ig, 1:5,000 dilution), anti-AMPKα2 (catalog #18167-1-AP, 1:500 dilution), anti-phospho-acetyl-coenzyme A carboxylase (pACC1 serine 79, catalog #29119-1-AP, 1:1,000 dilution), anti-ACC1 (catalog #21923-1-AP, 1:2,000 dilution) and anti-β-actin (catalog #66009-1-Ig, 1:50,000 dilution). The anti-RIPKI antibody was obtained from AbClonal (Woburn, Massachusetts, catalog #A7414, 1:750 dilution). The anti-phospho-MLKL antibody was obtained from R&D Systems (T357, catalog #MAB9187, Minneapolis, Minnesota, 1:1,000 dilution). The LCMT-1 antibody was obtained from Origene (Rockville, Maryland, catalog #TA503127S, 1:2,000 dilution). Appropriate IR-dye secondary antibodies used (Alexa Fluor® 790-conjugated anti-mouse (catalog #115–655–146, 1:50,000 dilution), Alexa Fluor® 680-conjugated anti-mouse (catalog #115–625–146, 1:50,000 dilution), and Alexa Fluor® 680-conjugated anti-rabbit (catalog #115–625–144, 1:50,000 dilution) were obtained from Jackson ImmunoResearch Laboratories (West Grove, Pennsylvania). Band densities were normalized to β-actin, the endogenous control. At least five replicates were used for statistical analyses.
HS-5, HEK-293, MCF7, MDA-MB-231 and Rosi cells were seeded in 96-well optical bottom tissue culture plates (Corning, New York) at a density of 2.5 × 103 cells/well. After 24 h, the media were changed to media containing LB-100 (Cayman Chemical Company, Ann Arbor, Michigan) or Compound 2 at various concentrations (50–0.31 µM). Control cells were given fresh media. After 48 h the media were aspirated and plates were washed with warmed Dulbecco’s phosphate-buffered saline (DPBS) with calcium and magnesium (Sigma-Aldrich, catalog #D1283). Cells were stained with 3 µM calcein-AM (AAT Bioquest, Pleasanton, California, catalog #22002) in DPBS for 30 min in the dark at 37°C. Plates were immediately read with a Biotek fluorescent plate reader (excitation 485 nm, emission 520 nm). The cytotoxicity assays were performed at least three times, each time with six replicates per condition.
A mammalian LCMT-1 plasmid expression vector was obtained from Origene (pCMV6-LCMT1, catalog #RC200018). HEK-293 cells were transfected with the plasmid using PolyJet™ transfection reagent (SignaGen Laboratories, Frederick, Maryland) according to manufacturer’s directions. After 5 h, the medium was changed. The next day, the transfected cells were seeded in 96-well optical bottom tissue culture plates for cytotoxicity assays as previously described. After 24 h, the medium was changed to medium containing Compound 2 at various concentrations (0.78–50 µM), or fresh medium with the carrier (dimethyl sulfoxide) in the case of the control. After 48 h, cells were stained with calcein-AM and fluorescence was measured.
MDA-MB-231 cells were also transfected with the LCMT-1 expression vector using Lipofectamine 3,000 transfection reagent (Thermo Fisher Scientific, Waltham, Massachusetts) according to the manufacturer’s transfection protocol for MDA-MB-231 cells. After 5 h the medium was changed. The cells were split 1:100 the following day, and 2 days after transfection, selection with G418 sulfate (Mediatech, Herndon, Virginia) at 700 ng/mL for 2 weeks commenced to develop stably transfected cells. After the selection process, cells were used in Compound 2 cytotoxicity assays as previously described.
MCF7, MDA-MB-231 and Rosi cells were seeded in 96-well tissue culture plates (Celltreat, Pepperell, Massachusetts) at a density of 90 cells per well (Mayr et al., 2018). After 24 h, the media were changed to media containing LB-100 or Compound 2 at various concentrations (50–3.25 µM). Control cells were given fresh media. After 1 week the media were aspirated and the wells were washed with PBS. Cells were stained with crystal violet solution (0.25 g crystal violet in 100 mL 20% methanol) for 10 min. The wells were washed 6 times with PBS to remove excess crystal violet. Colonies were counted with an ECHO Revolve hybrid microscope at ×1.5 magnification. There were ten replicates for each condition.
MCF7, MDA-MB-231 and Rosi cells were seeded in 6-well tissue culture plates (Celltreat) at a density of 5 × 105 cells per well. After 24 h, the DMEM was changed to media containing 20 µM LB-100 or Compound 2. Control cells were given fresh media. After 24 h, cells were labeled with 50 µM bromodeoxyuridine (BrdU, BioGems, Westlake Village, California, catalog #5911439). Cells were lifted with 0.25% trypsin, 2.21 mM ethylenediaminetetraacetic acid (Corning catalog #25–053-CI) after 4 h and fixed with 70% ethanol overnight at 4°C. Cells were pelleted and cellular DNA was denatured with HCl. After neutralization with 1 M Tris, pH 8.5, cells were labeled with FITC-anti-BrdU (Biolegend, San Diego, California, catalog #364104, 5 µL/100 μL cell suspension) and 7-aminoactinomycin-D (7-AAD, Biolegend, catalog #420404, 1 µL/100 μL cell suspension). Cells were analyzed with a Becton-Dickinson FACSymphony™ flow cytometer. Cell cycle analyses were performed three times.
MCF7, MDA-MB-231 and Rosi cells were treated with LB-100 and Compound 2 at 5–20 µM concentrations. After 48 h, cells were lifted with Accutase and washed twice with PBS (pH 7.4) with 10% FBS. Cells were resuspended in annexin V binding buffer (10 mM HEPES (pH 7.4), 140 mM NaCl and 2.5 mM CaCl2) and labeled with FITC-anti-Annexin V and 7-AAD (Biolegend, FITC Annexin V Apoptosis Detection Kit with 7-AAD, catalog #640922). Cell surface annexin V expression was assessed with flow cytometry.
Were performed with Prism Graphpad version 10.0.1.218 software. All data were subjected to normality and lognormality tests (Shapiro-Wilk and Kolmogorov-Smirnov tests) prior to comparison of means. One-way analyses of variance (ANOVA) with Dunnett’s multiple comparisons test were used for normally distributed data, while Brown-Forsythe and Welch ANOVAs with Dunnett’s T3 multiple comparisons tests were used for data that were not normally distributed. A two-way ANOVA was used to compare cytotoxicity between Compound 2 and LB-100 treated cells, and between cells overexpressing LCMT-1 and controls.
Molecular modeling of Compound 1 and 2 interactions with LCMT-1 using AutoDock Vina yielded one binding site for each compound, with nine different conformations at the binding site for the ligands. The conformations with the lowest binding free energies were visualized with UCSF ChimeraX software (Figures 1C, D). Molecular modeling revealed that neither Compound 1 nor Compound 2 occupied the SAM binding site of LCMT-1, or the binding site of the PP2Ac carboxyl terminus. Compound 1 appeared to occupy a cleft in the LCMT-1 protein near the SAM binding site, while Compound 2 occupied a cleft near the PP2Ac carboxyl terminus binding site.
Molecular modeling of Compound 2 binding with the SET domain of MLL5 methyltransferase using SwissDock yielded three possible binding sites with 10 ligand conformations for each binding site. Like the modeling results for MLL5 methyltransferase, SwissDock yielded three possible binding sites for each chain of ARMT1 with 10 ligand conformations for each binding site. The conformation/binding site with the lowest free energy and highest SwissDock Full Fitness rank was visualized with UCSF ChimeraX software. The modelling revealed that Compound 2 may dock at the SAM binding site in the SET domain of MLL5, and partially so in the α chain of ARMT1. Compound 2 may also partially dock in the β chain of ARMT1 (Supplementary Figure S4).
rhLCMT-1 activity assays revealed that Compound 2, but not Compound 1, inhibited methylation of rhPP2Ac to the same degree as the SAM analogue sinefungin, a natural compound used as a global inhibitor of all methyltransferases (p = 0.0024, Figure 2A). rhLCMT-1 activity in Compound 2- and sinefungin-containing reactions did not differ from the negative control.
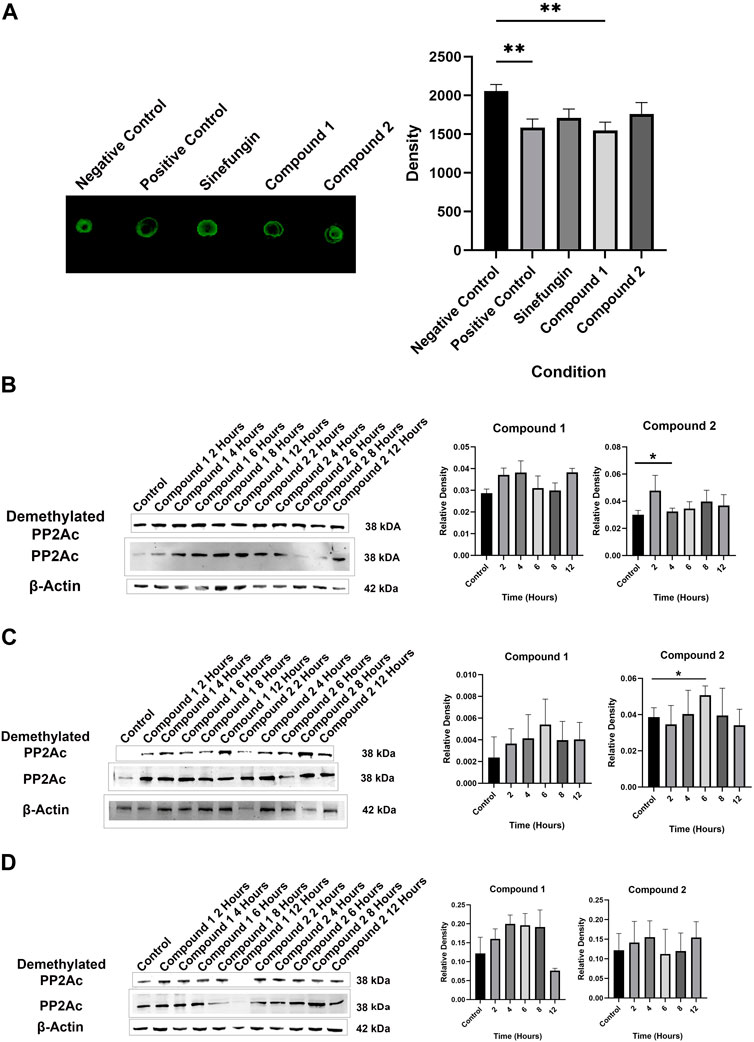
Figure 2. (A) Representative dot immunoblots demonstrating decreased methylation of rhPP2Ac by rhLCMT-1 in the presence of sinefungin and Compound 2. Bars represent means ± standard errors of the mean (SEM). (B) Representative Western blot demonstrating demethylation of PP2Ac in MDA-MB-231 cells in the presence of Compound 2, but not in the presence of Compound 1 (means ± SEM). (C) Representative Western blot demonstrating demethylation of PP2Ac in Rosi cells in the presence of Compound 2, but not in the presence of Compound 1 (means ± SEM). (D) Neither Compound 1 nor Compound 2 significantly decreased PP2Ac methylation in MCF7 cells (means ± SEM).
Compound 1 did not decrease PP2Ac methylation in MCF7 (p = 0.0716), MDA-MB-231 (p = 0.0730) or Rosi cells (p = 0.5517) in a statistically significant manner. Treatment with Compound 2 resulted in decreased PP2Ac methylation as compared to control in MDA-MB-231 cells which was significant at 4 h (p = 0.0431, Figure 2B), and in Rosi cells at 6 h (p = 0.0335, Figure 2C). Compound 2 did not significantly decrease methylation of PP2Ac in MCF7 cells (p = 0.7768, Figure 2D). Because Compound 1 did not significantly inhibit PP2Ac methylation in the LCMT-1 activity assays nor in the cell lines, Compound 2 was used for further analyses.
Because LCMT-1 knock-out gene editing is associated with lethality in embryos (MacKay et al., 2013) and LCMT-1 targeting with RNA interference results in apoptotic cell death in cancer cell lines (Longin et al., 2007; Lee and Pallas, 2007), we sought to determine if LCMT-1 inhibition with Compound 2 would affect cell survival. Cytotoxicity assays revealed that Compound 2 inhibited survival of HEK-293 and Rosi cells at concentrations between 5 and 20 µM (p = 0.0192 at 5 μM, p = 0.0014 at 10 μM, and p = 0.0003 at 20 µM for HEK-293 cells; p = 0.0003 at 5 μM, p < 0.0001 at 10 μM and 20 µM for Rosi cells), while LB-100 inhibited survival at 20 µM (p = 0.0036 for HEK-293 cells, p = 0.0035 for Rosi cells, Figures 3A, B). In HS-5 cells, Compound 2 cytotoxicity at 20 µM (p = 0.0004) was comparable to that of LB-100 at 20 µM (p = 0.0087, Figure 3C). In MDA-MB-231 cells, LB-100 inhibited survival in concentrations between 25 and 50 µM (p = 0.0117 at 25 µM and p = 0.0023 at 50 µM), while Compound 2 significantly inhibited survival at 25 µM (p = 0.0425, Figure 3D). Neither LB-100 nor Compound 2 significantly inhibited survival in MCF7 cells at the concentrations tested (p = 0.3204, Figure 3E). However, all cultures treated with Compound 2 contained necrotic figures not seen in cultures treated with LB-100. Half maximal effective concentrations of Compound 2 were lower than that of LB-100 for all cell lines tested (Table 1).
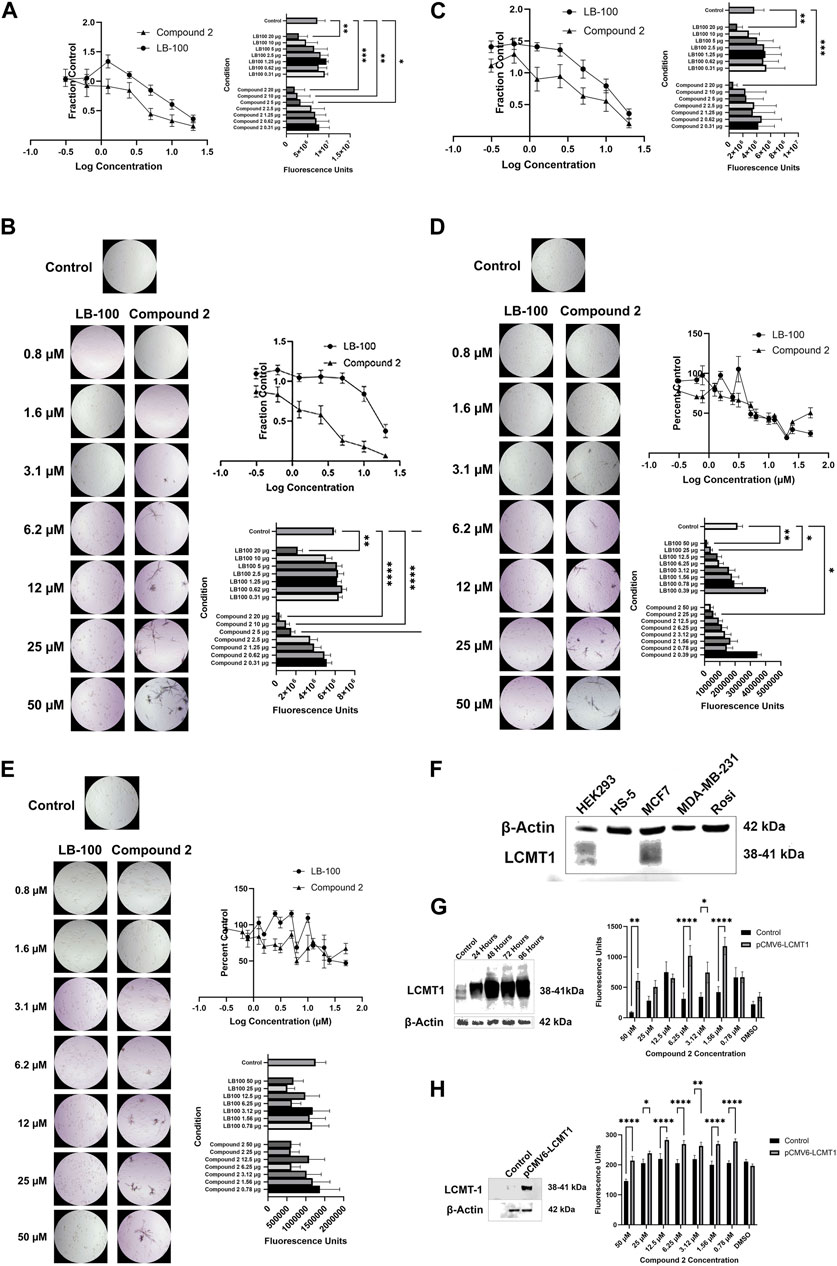
Figure 3. (A) Compound 2 inhibited survival of HEK-293 cells at concentrations between 5 and 20 µM (p = 0.0192 at 5 μM, p = 0.0014 at 10 μM, and p = 0.0003 at 20 µM), while LB-100 inhibited survival at 20 µM (p = 0.0036). Data represent means ± SEM. (B) Representative photomicrographs demonstrating cell death in cultures treated with LB-100 and necrotic figures in cultures treated with Compound 2. Compound 2 inhibited survival of Rosi cells at concentrations between 5 and 20 µM (p = 0.0003 at 5 μM, p < 0.0001 at 10 μM, and p < 0.0001 at 20 µM), while LB-100 inhibited survival at 20 µM (p = 0.0035). Note necrotic figures in cultures treated with Compound 2. Data represent means ± SEM. (C) In HS-5 cells, Compound 2 cytotoxicity at 20 µM (p = 0.0004) was comparable to that of LB-100 at 20 µM (p = 0.0087). Data represent means ± SEM. (D) In MDA-MB-231 cells, LB-100 inhibited survival in concentrations between 25 and 50 µM (p = 0.0117 at 25 µM and p = 0.0023 at 50 µM), while Compound 2 significantly inhibited survival at 25 µM (p = 0.0425). Data represent means ± SEM. (E) Neither LB-100 nor Compound 2 significantly inhibited survival in MCF7 cells at the concentrations tested (p = 0.3204). Data represent means ± SEM. (F) Baseline expression of LCMT-1 in cell lines. Relative overexpression of LCMT-1 is demonstrated in HEK-293 and MCF7 cells. (G) HEK-293 cells were transfected with a LCMT-1 mammalian expression vector (left). After 48 h, cells were treated with Compound 2. Overexpression of LCMT-1 in HEK-293 cells enhanced resistance to Compound 2-mediated cell death (right). (H) MDA-MB-231 cells were transfected with a LCMT-1 mammalian expression vector (left). After selection of stable transfectants, cells were treated with Compound 2. Overexpression of LCMT-1 in MDA-MB-231 induced resistance to Compound 2-mediated cell death (right).
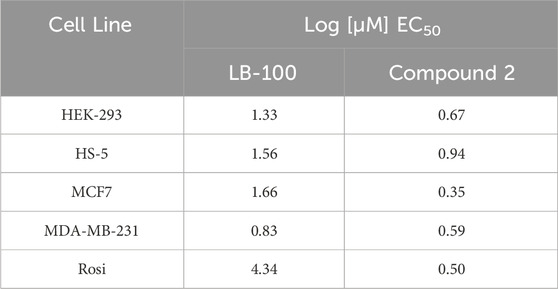
Table 1. Logarithms of half maximal effective concentrations for LB-100 and Compound 2 in cell lines tested.
Baseline expression of LCMT-1 was assessed in the HEK-293, HS-5, MCF7, MDA-MB-231 and Rosi cell lines. This revealed relative LCMT-1 overexpression in HEK-293 and MCF7 cells (Figure 3F). LCMT-1 was overexpressed in HEK-293 and MDA-MB-231 cells with the pCMV6-LCMT1 mammalian plasmid vector (Figures 3F, H). For HEK-293 cells, Only cells treated with 50 uM Compound 2 had decreased survival as compared to cells treated with other concentrations of Compound 2 (p = 0.0001 for HEK-293 parent cells and p = 0.0291 for HEK-293pCMV6-LCMT1 cells). LCMT-1 overexpression significantly increased survival of HEK-293 cells in the presence of Compound 2 (p = 0.0006).
For MDA-MB-231 cells parent cells treated with 50 uM Compound 2 had decreased survival as compared to cells treated with other concentrations of Compound 2 (p < 0.0001). In cells overexpressing LCMT-1, there was no difference in survival between cells treated with Compound 2 and untreated cells (p = 0.8629). There was increased survival of MDA-MB-231pCMV6-LCMT1 cells in the presence of Compound 2 at 25 uM (p = 0.002), 12.5 uM (p < 0.0001), 6.25 uM (p = 0.0004), 3.12 uM (p = 0.0013), 1.56 uM (p < 0.0001) and 0.78 uM (p < 0.0001).
Silencing of PR55α inhibits clonogenic colony formation in pancreatic cancer cells (Hein et al., 2016; Hein et al., 2019) and breast cancer cells (Di et al., 2017). We sought to determine whether prevention of PR55α binding to the PP2A holoenzyme through LCMT-1 inhibition would have similar effects. Both LB-100 and Compound 2 inhibited colony formation in MDA-MB-231 (p < 0.0001 for both compounds) and Rosi (p < 0.0001 for both compounds) cells in a concentration-dependent manner (Figures 4A, B). Colony formation in MCF7 cells was inhibited to a lesser extent (p = 0.0012 for LB-100, and p = 0.0035 for Compound 2, Figure 4C).
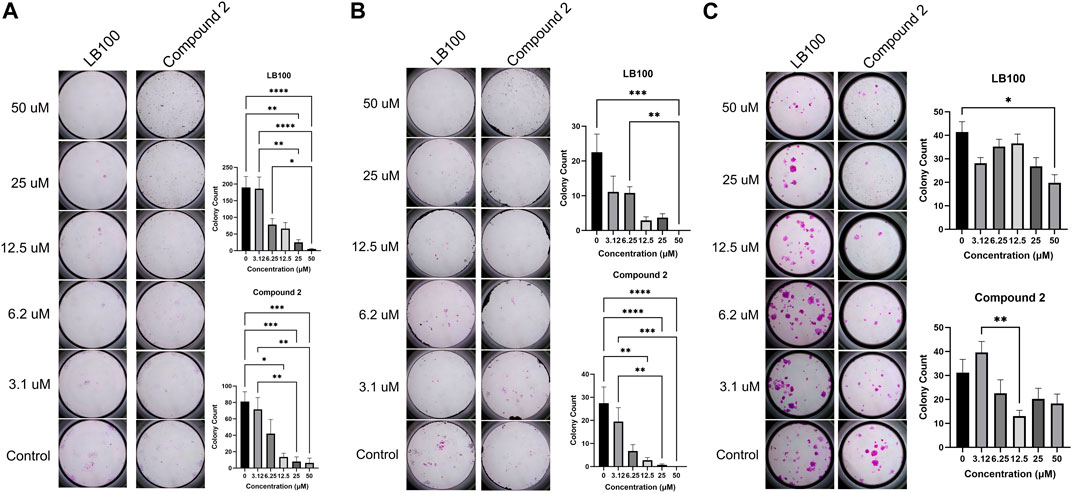
Figure 4. (A) LB-100 and Compound 2 inhibit colony formation in MDA-MB-231 cells in a concentration-dependent manner. Data represent means ± SEM. (B) LB-100 and Compound 2 inhibit colony formation in Rosi cells. Data represent means ± SEM. (C) LB-100 and Compound 2 inhibit colony formation in MCF7 cells. Data represent means ± SEM.
LCMT-1 plays an essential role in normal progression through mitosis (Lee and Pallas, 2007; Stanevich et al., 2011; Zhang et al., 2023). LB-100 has been shown to induce cell cycle arrest in colorectal cancer cells (Dai et al., 2017). To determine whether Compound 2 effects on cell survival and colony formation might be due to mitotic interference, cell cycle analysis was performed with BrdU flow cytometry. While LB-100 induced G2/M-phase arrest in MDA-MB-231 and Rosi cells (p < 0.0001 for both comparisons), Compound 2 induced G0/G1 arrest only in Rosi cells (p = 0.0193, Figures 5A, B). LB-100 induced G0/G1 arrest in MCF7 cells (p = 0.0063 for G0/G1 and p < 0.0001 for S-phase).
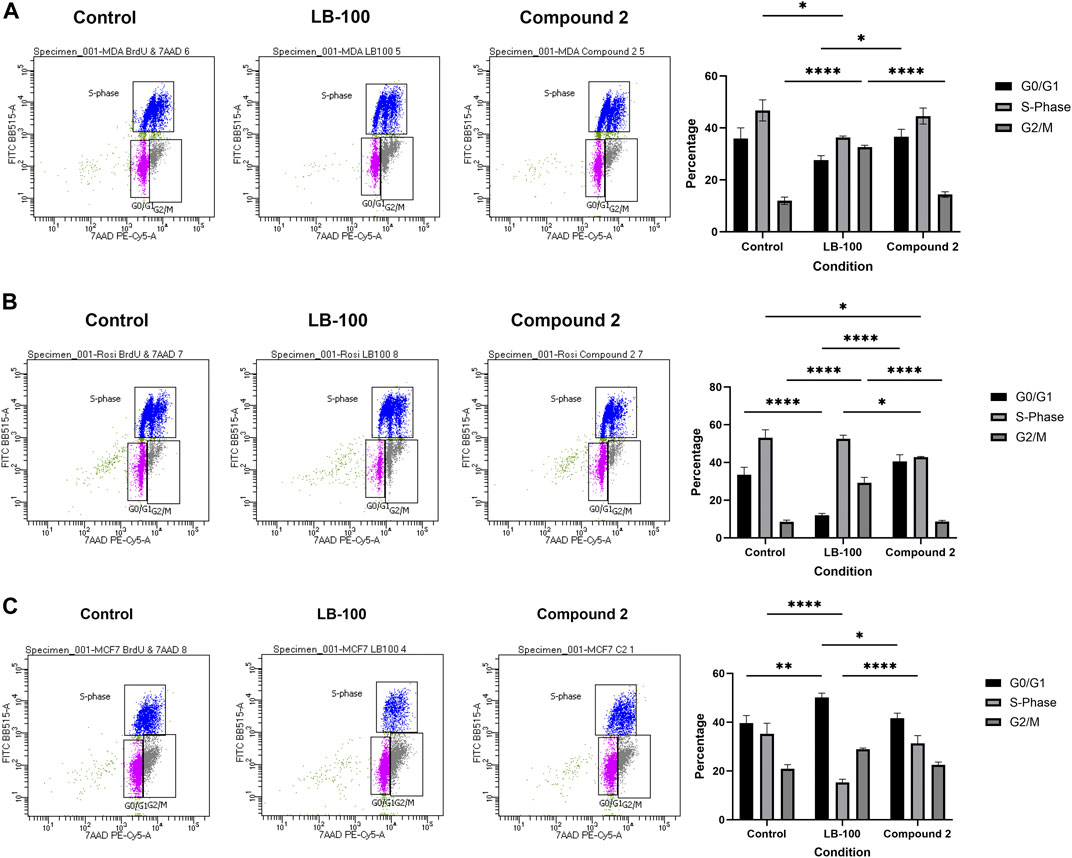
Figure 5. (A) Representative BrdU cell cycle analyses demonstrating that LB-100 induces G2/M-phase arrest in MDA-MB-231 cells. Compound 2 did not change cell cycle dynamics in this cell line. Data represent means ± SEM. (B) LB-100 induces G2/M-phase arrest in Rosi cells, while Compound 2 induced G0/G1 arrest. Data represent means ± SEM. (C) LB-100 induces G0/G1 arrest in MCF7 cells. Compound 2 did not change cell cycle dynamics in this cell line. Data represent means ± SEM.
Silencing of LCMT-1 (Lee and Pallas, 2007; Longin et al., 2007) and PR55α (Lee and Pallas, 2007; Di et al., 2017) result in apoptosis, and LB-100 has been shown to induce apoptosis in colorectal cancer cells (Dai et al., 2017). To determine whether the results of the cell survival and clonogenic assays represented apoptosis induced by LB-100 and Compound 2, annexin V assays were performed. Treatment with Compound 2 resulted in increased cell surface annexin V expression in MDA-MB-231 cells (p < 0.0001 at 20 µM), and in decreased cell surface annexin V in MCF7 cells (p = 0.0013 at 10 µM and p = 0.0397 at 20 µM). Compound 2 did not induce apopotosis in Rosi cells (p > 0.9999 at 5 μM, p = 0.6764 at 10 µM and p = 0.3002 at 20 µM). LB-100 induced apoptosis in MDA-MB-231 (p = 0.0258 at 20 µM) and Rosi cells (p = 0.0019 at 5 μM, p = 0.0126 at 10 μM, and p = 0.0007 at 20 μM, Figure 6).
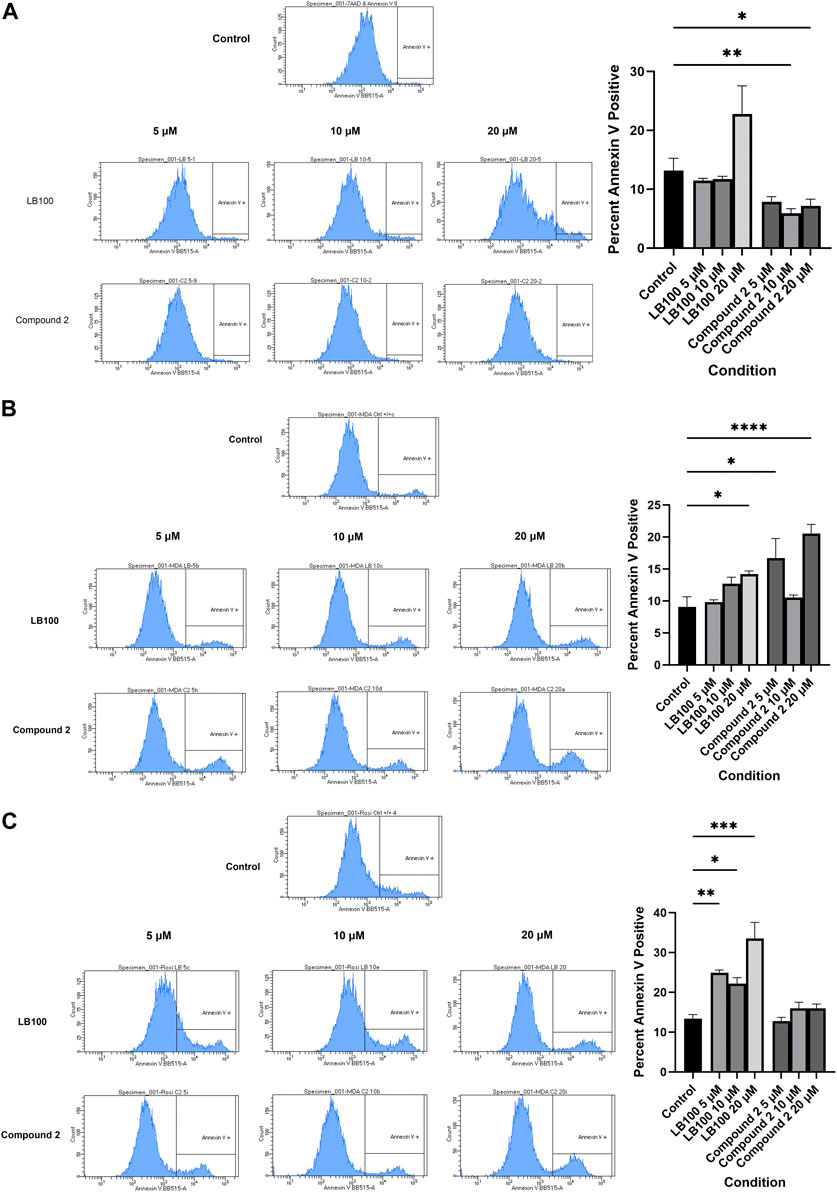
Figure 6. (A) Representative histogram demonstrating that Compound 2 decreases cell surface annexin V expression in MCF7 cells. Data represent means ± SEM. (B) Both LB-100 and Compound 2 induced apoptosis in MDA-MB-231 cells. Data represent means ± SEM. (C) LB-100, but not Compound 2, induces apoptosis in Rosi cells. Data represent means ± SEM.
Compound 2 decreased cytosolic expression of PP2Ab in MCF7 and Rosi cells between 4 and 6 hours after treatment. Decreased PP2Ab cytosolic expression has been reported in response to PP2Ac demethylation (Lee et al., 2018; Guffens et al., 2023). Glucose deprivation has been shown to induce cell death in some cell lines through calcium-dependent PP2Ac demethylation, and this form of cell death is mediated by RIPK1 phosphorylation (Lee et al., 2018). We found that treatment with compound 2 resulted in increased RIPK1 phosphorylation at serine 161 in the cancer cell lines, but this was statistically significant only for MDA-MB-231 (p = 0.0273) and Rosi cells (p = 0.0179, Figure 7). Phosphorylation of RIPK1 in the presence of Compound 2 did not statistically increase in MCF7 cells (p = 0.0598). None of the cell lines demonstrated RIPK1 phosphorylation at serine 166. There was increased phosphorylation of MLKL in all of the cell lines at 2–4 hours after treatment with Compound 2.
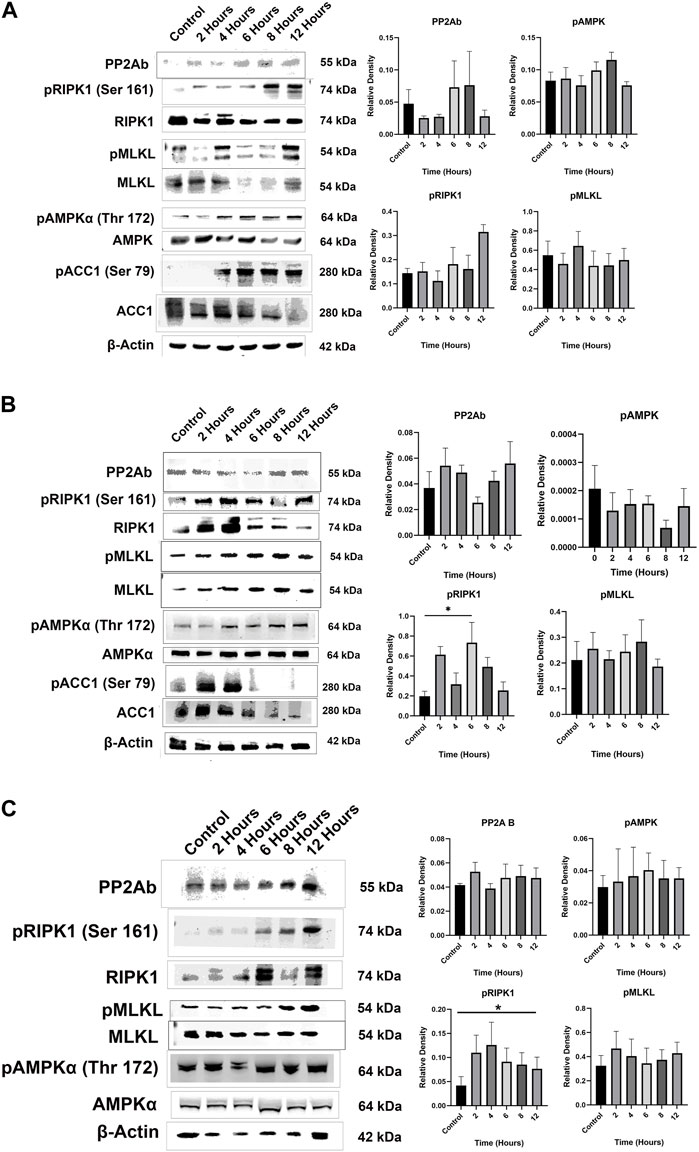
Figure 7. (A) Representative Western blots demonstrate that treatment with Compound 2 resulted in decreased cytosolic expression of PP2Ab in MCF7 cells beginning at 4 h. Cytosolic PP2Ab expression recovered after 6 h. Treatment with Compound 2 resulted in increased phosphorylation of RIPK1, MLKL, AMPKα and ACC1 in MCF7 cells. Data represent means ± SEM. (B) Representative Western blots demonstrate that treatment with Compound 2 decreased cytosolic expression of PP2Ab at 6 h, with recovery of cytosolic PP2Ab at 8 h. Compound 2 increased phosphorylation of RIPK1 in Rosi cells (p = 0.0179) and MLKL. Increased phosphorylation of AMPKα and ACC1 was not statistically significant. Data represent means ± SEM. (C) Representative Western blots demonstrate that treatment with Compound 2 did not change cytosolic expression PP2Ab or phosphorylation of pAMK in MDA-MB-231 cells. Compound 2 increased phosphorylation of RIPK1 (p = 0.0273) and MLKL. Data represent means ± SEM.
Dai et al. reported that treatment of colorectal cancer cells with LB-100 induced phosphorylation of AMPK (Dai et al., 2017). We did not find increased phosphorylation of AMPK in our cell lines in response to treatment with Compound 2 (p = 0.0906 for MCF7 cells, p = 0.0660 for MDA-MB-231 cells, and p = 0.6775 for Rosi cells). However, treatment with Compound 2 increased phosphorylation of ACC1 in MCF7 and Rosi cells.
Dysregulation of the delicate balance between kinases and phosphatases characteristic of most cancers is complex, and can be mediated by gain of function mutations in kinases, loss of function mutations in phosphatases, overexpression of microRNAs that target phosphatases, deletions or mutations in genes that activate phosphatases, or overexpression of oncogenes that inactivate phosphatases (Remmerie and Janssens, 2019; Ivovic et al., 2023; Johnson et al., 2024). Most therapies aimed at restoring this balance have targeted kinases, in part because they were discovered before the phosphatases, and because the importance of the phosphatases has been discovered relatively recently (Turdo et al., 2021). PP2A is a major serine/threonine phosphatase, and several direct and indirect PP2A activators have shown therapeutic efficacy for a variety of malignancies.
Direct activators of PP2A include phenothiazines and their analogues, metformin, sodium selenate, forskolin and NSC49L (Johnson et al., 2024). These molecules are thought to exert their effects by stabilizing the PP2A heterotrimer (Farrington et al., 2020; Leonard et al., 2020). Indirect activators of PP2A target endogenous PP2A inhibitors such as PME-1, Suvar/Enhancer of zeste/Trithorax (SET), 2 A inhibitory protein (TIPRL1), α-endosulfine, ARPP-16, ARPP-19 and cancerous inhibitor of PP2A (CIP2A) (Haesen et al., 2014; Johnson et al., 2024). These include FTY720, CM-1231, OSU-25, OP449, Bortezomib, ethoxysanguarine, TD52, actigenin, celastrol, niclosamide, polyphyllin I and Lapatinib (Johnson et al., 2024).
However, few PP2A activators have advanced to clinical trials due to their side effect profiles and also because PP2A plays a complicated role in oncogenic signaling, behaving as both a tumor suppressor and promoter. PP2A downstream effects are mediated by substrate specificity, which is in turn dependent upon the heterotrimeric composition. While deletions of the gene for the B55α subunit have been detected in breast (Beca et al., 2015) and prostate (Cheng et al., 2011; Zhao et al., 2019) cancers, PP2A heterotrimers with the B55α subunit have been shown to promote the transcription of Wnt-responsive genes such as cyclin D1 and c-Myc through dephosphorylation of β-catenin (Wlodarchak and Xing, 2016; Pippa and Odero, 2020). PP2A-B55α also activates mitogen activated protein kinase signaling through dephosphorylation of the kinase suppressor of Ras one and Raf-1 (Ory et al., 2003). The B55α subunit activates the human papilloma virus strain 16 long control region in cells with a deletion in the short arm of chromosome 11 (Smits et al., 1992), sustains oncogenic signaling in pancreatic cancer cells (Hein et al., 2016; Hein et al., 2019), and mediates resistance to glucose-starvation-mediated cell death in breast cancer cells (Di et al., 2017). Thus, different mechanisms of PP2A dysregulation are dependent upon the cellular genetic, epigenetic and metabolic environment.
The rationale for PP2A inhibition is based on the role that PP2A, including PP2A-B55α, plays in the DNA damage response (Dohoney et al., 2004; Chowdhury et al., 2005; Li et al., 2007; Kalev et al., 2012; Li et al., 2015; Mazhar et al., 2019). PP2A inhibitors allow cells with damaged DNA to enter the cell cycle, resulting in cytotoxicity due to unstable chromatin (Li et al., 2015; Mazhar et al., 2019; Johnson et al., 2024). Several PP2A inhibitors (okadaic acid, calyculin A, tautomycin, tautomycetin, fostriecin, cantharidin, noncantharidein, and LB-100) have shown efficacy against a variety of cancer cells, but only LB-100, a cantharidin derivative, has advanced to clinical trials (Chung et al., 2017) due to toxicities with other PP2A inhibitors (Johnson et al., 2024). LB-100 restricts tumor growth in melanoma (Hu et al., 2022), colorectal (Dai et al., 2017), hepatocellular (Sun et al., 2021) and breast (Uddin et al., 2020) cancer cells. LB-100 has shown promise as a sensitizer for DNA-damaging therapies (chemotherapy and radiotherapy) (Wei et al., 2013; Bai et al., 2014; Bai et al., 2014; Lv et al., 2014; Chang et al., 2015; Gordon et al., 2015; Hong et al., 2015; Zhang et al., 2015; Ho et al., 2016; Hu et al., 2017; Ho et al., 2018a; Hao et al., 2018; Song et al., 2021; Gao et al., 2022; Ronk et al., 2022), as well as an enhancer of chimeric antigen receptor T-cell therapy (Cui et al., 2020) and immune checkpoint inhibition (Ho et al., 2018b; Maggio et al., 2020; Mirzapoiazova et al., 2021).
Methylation of the PP2Ac catalytic subunit at leucine 309 near its carboxyl terminus by LCMT-1 stabilizes binding of the B55α subunit to the PP2A heterotrimer (Longin et al., 2007). While LCMT-1 silencing promotes castration-resistant prostate cancer growth through increasing androgen receptor activity (Rasool et al., 2023), LCMT-1 overexpression carries a poor prognosis in hepatocellular carcinoma (Zhang et al., 2023). Because LCMT-1 is necessary for progression through mitosis, it has been identified as an oncologic target (Lee and Pallas, 2007; Longin et al., 2007; Zhang et al., 2023).
Neither of the compounds we studied as potential LCMT-1 inhibitors were analogues of SAM, nor were they PP2Ac peptidomimetics. While we recognize the limitations of molecular modelling (Limongelli, 2020), our models indicate that these compounds did not interact with LCMT-1 at the SAM or PP2Ac carboxyl terminus binding sites as we predicted (Figures 1B,C). Previous attempts at LCMT-1 inhibition using synthetic peptides based on the carboxyl terminus of PP2Ac revealed that these peptides were neither substrates nor inhibitors of LCMT-1, suggesting that LCMT-1 recognizes aspects of the tertiary and/or quaternary structure of PP2Ac (Xie and Clarke, 1994). We hypothesize that binding of Compound 2 to LCMT-1 may induce or prevent conformational changes in LCMT-1 that restrict binding or methylation of the PP2Ac carboxyl terminus, and this will have to be confirmed with x-ray crystallography. This could be consistent with the work of Stanevich et al., who determined that the conformation of the LCMT-1 active site pocket changes to allow binding of the PP2Ac carboxyl terminus (Stanevich et al., 2011).
Inhibition of LCMT-1 by Compound 2 had differential effects on the cell cycle (Figure 5), survival (Figures 3A–E) and induction of apoptosis (Figure 6) in the cell lines we tested, but Compound 2 inhibited clonogenic colony formation in all three of the cancer cell lines (Figure 4). The observed variability in response to Compound 2 among the cell lines may in part be explained by differences in baseline expression of LCMT-1, which was increased in HEK-293 and MCF7 cells (Figure 3F), and could explain their relative resistance to Compound 2. However, HS-5 cells demonstrated a similar response to Compound 2 as did HEK-293 cells without overexpression of LCMT-1. Though overexpression of LCMT-1 in HEK-293 and MDA-MB-231 cells increased resistance to Compound 2-mediated toxicity (Figures 3G, H), Compound 2 did not induce statistically significant PP2Ac demethylation in MCF7 cells (Figure 2D). This may be indicative of off-target effects of Compound 2 since our molecular modelling indicated that Compound 2 may dock at the SAM binding site of the SET domain of MLL5, and partially dock at the SAM binding site of the ARMT1 α chain. Compound 2 also partially docked with the ARMT1 β chain (Supplementary Figure S2). It is also possible that the effects were mediated by other protein phosphatases, as PP4 and PP6 are also LCMT-1 substrates (Lee et al, 2018). Relatively high concentrations of Compound 2 were needed to obtain cellular effects, which could also be indicative of off-target effects. However, this was also true of LB-100, and Compound 2 was more effective at inhibiting cell survival than was LB-100 in the cell lines tested (Figures 3A–D).
Compound 2 decreased cytosolic PP2Ab expression in MCF7 and Rosi cells after 4–6 h of exposure (Figure 7). This is consistent with a previous study demonstrating that PP2Ac demethylation induced by oxidative stress results in increased B55α subunit binding to PME-1 and translocation to the nucleus in glioblastoma cells after 4 h. PP2Ac demethylation resulted in increased phosphorylation of RIPK1 (Guffens et al., 2023). Our data indicate that cytosolic localization of the B55α subunit rebounded after 8 h.
We also demonstrated increased phosphorylation of RIPK1 at serine 161 in response to treatment with Compound 2 in our cell lines. While activation of both RIPK1 and AMPK pathways are linked to depletion of cellular adenosine triphosphate, activation of the AMPK pathway has been shown to prevent formation of the RIPK1-RIPK3 necroptosis complex (Lee et al., 2019; Zhang et al., 2023). Nevertheless, RIPK1 has been shown to communicate energetic stress to AMPK through the mechanistic target of rapamycin complex 1 (Najafov et al., 2021). Though we did not find statistically significant phosphorylation of AMPKα by Compound 2 in our cell lines, Compound 2 increased phosphorylation of AMPKα and ACC1 in MCF7 and Rosi cells. AMPK is the main kinase regulator of ACC1, inactivating ACC1 when cellular energy stores are low.
Increased phosphorylation of MLKL was noted in Rosi and MCF7 cells, but not in MDA-MB-231 cells. This suggests that while necroptosis may have been the mechanism of cell death in Rosi and MCF7 cells, a different mechanism occurred in MDA-MB-231 cells. This is consistent with Figure 6, which indicates that Compound 2 only induced apoptosis in MDA-MB-231 cells. Though increased RIPK1 phosphorylation was evident in MDA-MB-231 cells in response to Compound 2, RIPK1 can also contribute to apoptosis (Kaiser et al., 2014).
PP2A inhibition has been shown to impact other disease processes including nonalcoholic fatty liver disease (Chen et al., 2019), ultraviolet radiation-induced damage in retinal pigment epithelium (Li et al., 2018), depression (Lecca et al., 2016), renal and hepatic fibrosis (Nyamsuren et al., 2021), and cardiovascular disease (Zhang et al., 2019). The role that LCMT-1 may play in these disease processes has not been studied.
In conclusion, methyl 4-methyl-2-[(2-methylbenzoyl)amino]-5-[[(3-methylphenyl)amino]carbonyl]-3-thiophenecarboxylate (Compound 2) is an LCMT-1 inhibitor that may have anti-tumor effects including inhibition of survival, induction of cell cycle arrest and apoptosis in some cells. Compound 2 inhibits clonogenic colony formation, and these effects seem to be mediated by necroptosis through demethylation of PP2Ac in some cells, and apoptosis in others. Further studies are needed to confirm Compound 2 binding to LCMT-1 and potential off-target effects.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
OA: Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Visualization, Writing–original draft, Writing–review and editing. AS: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Writing–review and editing. EP: Investigation, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. OA’s work was supported by a Temple University Lewis Katz School of Medicine seed money grant and by the National Cancer Institute (1R21CA167126-01A1).
AS would like to acknowledge Dr. Stephen Condon, Ph.D., Vice President of Chemistry at Venatorx Pharmaceuticals, Incorporated (Malvern, Pennsylvania), formerly Head of Discovery Research at Tetralogic Pharmaceuticals, for releasing the identity of the compound structures and obtaining permission for use of the compounds in research. AS would also like to acknowledge Dr. Srinivas Chunduru, Ph.D., Chief Scientific Officer at EveryONE Medicines (Boston, Massachusetts and Malvern, Pennsylvania), formerly Director of Biology at Tetralogic Pharmaceuticals, for providing leadership and guidance during the library screening. Finally, AS would like to acknowledge Dr. Yigong Shi, Ph.D., President, Westlake University (Hangzhou, Zhejiang, China), formerly Professor of Biophysics at Princeton University. Dr. Shi’s laboratory at Princeton provided the recombinant proteins used in the radioactive LCMT-1 assays.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fddsv.2024.1278163/full#supplementary-material
Ardito, F., Giuliani, M., Perrone, D., Troiano, G., and Lo Muzio, L. (2017). The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy (Review). Int. J. Mol. Med. 40 (2), 271–280. doi:10.3892/ijmm.2017.3036
Bai, X., Zhi, X., Zhang, Q., Liang, F., Chen, W., Liang, C., et al. (2014a). Inhibition of protein phosphatase 2A sensitizes pancreatic cancer to chemotherapy by increasing drug perfusion via HIF-1α-VEGF mediated angiogenesis. Cancer Lett. 355 (2), 281–287. doi:10.1016/j.canlet.2014.09.048
Bai, X. L., Zhang, Q., Ye, L. Y., Hu, Q. D., Fu, Q. H., Zhi, X., et al. (2014b). Inhibition of protein phosphatase 2A enhances cytotoxicity and accessibility of chemotherapeutic drugs to hepatocellular carcinomas. Mol. cancer Ther. 13 (8), 2062–2072. doi:10.1158/1535-7163.MCT-13-0800
Beca, F., Pereira, M., Cameselle-Teijeiro, J. F., Martins, D., and Schmitt, F. (2015). Altered PPP2R2A and Cyclin D1 expression defines a subgroup of aggressive luminal-like breast cancer. BMC cancer 15, 285. doi:10.1186/s12885-015-1266-1
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The protein Data Bank. Nucleic acids Res. 28 (1), 235–242. doi:10.1093/nar/28.1.235
Cervone, N., Monica, R. D., Serpico, A. F., Vetrei, C., Scaraglio, M., Visconti, R., et al. (2018). Evidence that PP2A activity is dispensable for spindle assembly checkpoint-dependent control of Cdk1. Oncotarget 9 (7), 7312–7321. doi:10.18632/oncotarget.23329
Chang, K. E., Wei, B. R., Madigan, J. P., Hall, M. D., Simpson, R. M., Zhuang, Z., et al. (2015). The protein phosphatase 2A inhibitor LB100 sensitizes ovarian carcinoma cells to cisplatin-mediated cytotoxicity. Mol. cancer Ther. 14 (1), 90–100. doi:10.1158/1535-7163.MCT-14-0496
Chen, X. Y., Cai, C. Z., Yu, M. L., Feng, Z. M., Zhang, Y. W., Liu, P. H., et al. (2019). LB100 ameliorates nonalcoholic fatty liver disease via the AMPK/Sirt1 pathway. World J. Gastroenterol. 25 (45), 6607–6618. doi:10.3748/wjg.v25.i45.6607
Cheng, Y., Liu, W., Kim, S. T., Sun, J., Lu, L., Sun, J., et al. (2011). Evaluation of PPP2R2A as a prostate cancer susceptibility gene: a comprehensive germline and somatic study. Cancer Genet. 204 (7), 375–381. doi:10.1016/j.cancergen.2011.05.002
Chowdhury, D., Keogh, M. C., Ishii, H., Peterson, C. L., Buratowski, S., and Lieberman, J. (2005). gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol. Cell 20 (5), 801–809. doi:10.1016/j.molcel.2005.10.003
Chung, V., Mansfield, A. S., Braiteh, F., Richards, D., Durivage, H., Ungerleider, R. S., et al. (2017). Safety, tolerability, and preliminary activity of LB-100, an inhibitor of protein phosphatase 2A, in patients with relapsed solid tumors: an open-label, dose escalation, first-in-human, phase I trial. Clin. cancer Res. official J. Am. Assoc. Cancer Res. 23 (13), 3277–3284. doi:10.1158/1078-0432.CCR-16-2299
Cicenas, J., Zalyte, E., Bairoch, A., and Gaudet, P. (2018). Kinases and cancer. Cancer. Cancers (Basel) 10 (3), 63. doi:10.3390/cancers10030063
Cui, J., Wang, H., Medina, R., Zhang, Q., Xu, C., Indig, I. H., et al. (2020). Inhibition of PP2A with LB-100 enhances efficacy of CAR-T cell therapy against glioblastoma. Cancers (Basel) 12 (1), 139. doi:10.3390/cancers12010139
Dai, C., Zhang, X., Xie, D., Tang, P., Li, C., Zuo, Y., et al. (2017). Targeting PP2A activates AMPK signaling to inhibit colorectal cancer cells. Oncotarget 8 (56), 95810–95823. doi:10.18632/oncotarget.21336
DArcy, B. M., Prakash, A., and Honkanen, R. E. (2019). Targeting phosphatases in cancer: suppression of many versus the ablation of one. Oncotarget 10 (61), 6543–6545. doi:10.18632/oncotarget.27201
Dennis, T. N., Kenjic, N., Kang, A. S., Lowenson, J. D., Kirkwood, J. S., Clarke, S. G., et al. (2020). Human ARMT1 structure and substrate specificity indicates that it is a DUF89 family damage-control phosphatase. J. Struct. Biol. 212 (1), 107576. doi:10.1016/j.jsb.2020.107576
Di, C. G., Trusso, C. S., Zheng, X., Zhang, Q., and Mazzone, M. (2017). PHD2 targeting overcomes breast cancer cell death upon glucose starvation in a PP2A/B55α-mediated manner. Cell Rep. 18 (12), 2836–2844. doi:10.1016/j.celrep.2017.02.081
Dohoney, K. M., Guillerm, C., Whiteford, C., Elbi, C., Lambert, P. F., Hager, G. L., et al. (2004). Phosphorylation of p53 at serine 37 is important for transcriptional activity and regulation in response to DNA damage. Oncogene 23 (1), 49–57. doi:10.1038/sj.onc.1207005
Eberhardt, J., Santos-Martins, D., Tillack, A. F., and Forli, S. (2021). AutoDock Vina 1.2.0: new docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model 61 (8), 3891–3898. doi:10.1021/acs.jcim.1c00203
Farrington, C. C., Yuan, E., Mazhar, S., Izadmehr, S., Hurst, L., Allen-Petersen, B. L., et al. (2020). Protein phosphatase 2A activation as a therapeutic strategy for managing MYC-driven cancers. J. Biol. Chem. 295 (3), 757–770. doi:10.1074/jbc.RA119.011443
Gao, S., Shan, L., Zhang, M., Wang, Y., Zhan, X., Yin, Y., et al. (2022). Inhibition of PP2A by LB100 sensitizes bladder cancer cells to chemotherapy by inducing p21 degradation. Cell Oncol. (Dordr) 45 (6), 1203–1215. doi:10.1007/s13402-022-00710-8
Goddard, T. D., Huang, C. C., Meng, E. C., Pettersen, E. F., Couch, G. S., Morris, J. H., et al. (2018). UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 27 (1), 14–25. doi:10.1002/pro.3235
Gordon, I. K., Lu, J., Graves, C. A., Huntoon, K., Frerich, J. M., Hanson, R. H., et al. (2015). Protein phosphatase 2A inhibition with LB100 enhances radiation-induced mitotic catastrophe and tumor growth delay in glioblastoma. Mol. cancer Ther. 14 (7), 1540–1547. doi:10.1158/1535-7163.MCT-14-0614
Grosdidier, A., Zoete, V., and Michielin, O. (2011). SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic acids Res. 39 (Web Server issue), W270–W277. doi:10.1093/nar/gkr366
Guex, N., and Peitsch, M. C. (1997). SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18 (15), 2714–2723. doi:10.1002/elps.1150181505
Guffens, L., Derua, R., and Janssens, V. (2023). PME-1 sensitizes glioblastoma cells to oxidative stress-induced cell death by attenuating PP2A-B55α-mediated inactivation of MAPKAPK2-RIPK1 signaling. Cell Death Discov. 9 (1), 265. doi:10.1038/s41420-023-01572-1
Haesen, D., Sents, W., Lemaire, K., Hoorne, Y., and Janssens, V. (2014). The basic Biology of PP2A in hematologic cells and malignancies. Front. Oncol. 4, 347. doi:10.3389/fonc.2014.00347
Hao, S., Song, H., Zhang, W., Seldomridge, A., Jung, J., Giles, A. J., et al. (2018). Protein phosphatase 2A inhibition enhances radiation sensitivity and reduces tumor growth in chordoma. Neuro Oncol. 20 (6), 799–809. doi:10.1093/neuonc/nox241
Hein, A. L., Brandquist, N. D., Ouellette, C. Y., Seshacharyulu, P., Enke, C. A., Ouellette, M. M., et al. (2019). PR55α regulatory subunit of PP2A inhibits the MOB1/LATS cascade and activates YAP in pancreatic cancer cells. Oncogenesis 8 (11), 63. doi:10.1038/s41389-019-0172-9
Hein, A. L., Seshacharyulu, P., Rachagani, S., Sheinin, Y. M., Ouellette, M. M., Ponnusamy, M. P., et al. (2016). PR55α subunit of protein phosphatase 2A supports the tumorigenic and metastatic potential of pancreatic cancer cells by sustaining hyperactive oncogenic signaling. Cancer Res. 76 (8), 2243–2253. doi:10.1158/0008-5472.CAN-15-2119
Heo, K., Basu, H., Gutnick, A., Wei, W., Shlevkov, E., and Schwarz, T. L. (2022). Serine/threonine protein phosphatase 2A regulates the transport of axonal mitochondria. Front. Cell Neurosci. 16, 852245. doi:10.3389/fncel.2022.852245
Ho, W. S., Feldman, M. J., Maric, D., Amable, L., Hall, M. D., Feldman, G. M., et al. (2016). PP2A inhibition with LB100 enhances cisplatin cytotoxicity and overcomes cisplatin resistance in medulloblastoma cells. Oncotarget 7 (11), 12447–12463. doi:10.18632/oncotarget.6970
Ho, W. S., Sizdahkhani, S., Hao, S., Song, H., Seldomridge, A., Tandle, A., et al. (2018a). LB-100, a novel Protein Phosphatase 2A (PP2A) inhibitor, sensitizes malignant meningioma cells to the therapeutic effects of radiation. Cancer Lett. 415, 217–226. doi:10.1016/j.canlet.2017.11.035
Ho, W. S., Wang, H., Maggio, D., Kovach, J. S., Zhang, Q., Song, Q., et al. (2018b). Pharmacologic inhibition of protein phosphatase-2A achieves durable immune-mediated antitumor activity when combined with PD-1 blockade. Nat. Commun. 9 (1), 2126. doi:10.1038/s41467-018-04425-z
Hong, C. S., Ho, W., Zhang, C., Yang, C., Elder, J. B., and Zhuang, Z. (2015). LB100, a small molecule inhibitor of PP2A with potent chemo- and radio-sensitizing potential. Cancer Biol. Ther. 16 (6), 821–833. doi:10.1080/15384047.2015.1040961
Hu, B., Hao, S., Miao, Y., Deng, Y., Wang, J., Wan, H., et al. (2022). Inhibiting PP2A upregulates B7-H3 expression and potentially increases the sensitivity of malignant meningiomas to immunotherapy by proteomics. Pathology Oncol. Res. POR 28, 1610572. doi:10.3389/pore.2022.1610572
Hu, C., Yu, M., Ren, Y., Li, K., Maggio, D. M., Mei, C., et al. (2017). PP2A inhibition from LB100 therapy enhances daunorubicin cytotoxicity in secondary acute myeloid leukemia via miR-181b-1 upregulation. Sci. Rep. 7 (1), 2894. doi:10.1038/s41598-017-03058-4
Ivovic, D., Kabelikova, P., and Jurkovicova, D. (2023). Unraveling the complexity: a comprehensive analysis of the PP2A in cancer and its potential for novel targeted therapies. Neoplasma 70 (4), 485–499. doi:10.4149/neo_2023_230806N411
Johnson, H., Narayan, S., and Sharma, A. K. (2024). Altering phosphorylation in cancer through PP2A modifiers. Cancer Cell Int. 24 (1), 11. doi:10.1186/s12935-023-03193-1
Kaiser, W. J., Daley-Bauer, L. P., Thapa, R. J., Mandal, P., Berger, S. B., Huang, C., et al. (2014). RIP1 suppresses innate immune necrotic as well as apoptotic cell death during mammalian parturition. Proc. Natl. Acad. Sci. U. S. A. 111 (21), 7753–7758. doi:10.1073/pnas.1401857111
Kalev, P., Simicek, M., Vazquez, I., Munck, S., Chen, L., Soin, T., et al. (2012). Loss of PPP2R2A inhibits homologous recombination DNA repair and predicts tumor sensitivity to PARP inhibition. Cancer Res. 72 (24), 6414–6424. doi:10.1158/0008-5472.CAN-12-1667
Katz, J. E., Dlakic, M., and Clarke, S. (2003). Automated identification of putative methyltransferases from genomic open reading frames. Mol. Cell. proteomics MCP 2 (8), 525–540. doi:10.1074/mcp.M300037-MCP200
Lecca, S., Pelosi, A., Tchenio, A., Moutkine, I., Lujan, R., Herve, D., et al. (2016). Rescue of GABAB and GIRK function in the lateral habenula by protein phosphatase 2A inhibition ameliorates depression-like phenotypes in mice. Nat. Med. 22 (3), 254–261. doi:10.1038/nm.4037
Lee, H. Y., Itahana, Y., Schuechner, S., Fukuda, M., Je, H. S., Ogris, E., et al. (2018). Ca(2+)-dependent demethylation of phosphatase PP2Ac promotes glucose deprivation-induced cell death independently of inhibiting glycolysis. Sci. Signal. 11 (512), eaam7893. doi:10.1126/scisignal.aam7893
Lee, J. A., and Pallas, D. C. (2007). Leucine carboxyl methyltransferase-1 is necessary for normal progression through mitosis in mammalian cells. J. Biol. Chem. 282 (42), 30974–30984. doi:10.1074/jbc.M704861200
Lee, J. A., Wang, Z., Sambo, D., Bunting, K. D., and Pallas, D. C. (2018). Global loss of leucine carboxyl methyltransferase-1 causes severe defects in fetal liver hematopoiesis. J. Biol. Chem 293 (25), 9636–9650. doi:10.1074/jbc.RA118.002012
Lee, S. B., Kim, J. J., Han, S. A., Fan, Y., Guo, L. S., Aziz, K., et al. (2019). The AMPK-Parkin axis negatively regulates necroptosis and tumorigenesis by inhibiting the necrosome. Nat. Cell Biol. 21 (8), 940–951. doi:10.1038/s41556-019-0356-8
Leonard, D., Huang, W., Izadmehr, S., O'Connor, C. M., Wiredja, D. D., Wang, Z., et al. (2020). Selective PP2A enhancement through biased heterotrimer stabilization. Cell 181 (3), 688–701. doi:10.1016/j.cell.2020.03.038
Li, H. H., Cai, X., Shouse, G. P., Piluso, L. G., and Liu, X. (2007). A specific PP2A regulatory subunit, B56gamma, mediates DNA damage-induced dephosphorylation of p53 at Thr55. EMBO J. 26 (2), 402–411. doi:10.1038/sj.emboj.7601519
Li, X., Nan, A., Xiao, Y., Chen, Y., and Lai, Y. (2015). PP2A-B56ϵ complex is involved in dephosphorylation of gamma-H2AX in the repair process of CPT-induced DNA double-strand breaks. Toxicology 331, 57–65. doi:10.1016/j.tox.2015.03.007
Li, X. F., Li, S. Y., Dai, C. M., Li, J. C., Huang, D. R., and Wang, J. Y. (2018). PP2A inhibition by LB-100 protects retinal pigment epithelium cells from UV radiation via activation of AMPK signaling. Biochem. biophysical Res. Commun. 506 (1), 73–80. doi:10.1016/j.bbrc.2018.10.077
Limongelli, V. (2020). Ligand binding free energy and kinetics calculation in 2020. Wiley Interdiscip. Reviews-Computational Mol. Sci. 10 (4). doi:10.1002/wcms.1455
Longin, S., Zwaenepoel, K., Louis, J. V., Dilworth, S., Goris, J., and Janssens, V. (2007). Selection of protein phosphatase 2A regulatory subunits is mediated by the C terminus of the catalytic Subunit. J. Biol. Chem. 282 (37), 26971–26980. doi:10.1074/jbc.M704059200
Lv, P., Wang, Y., Ma, J., Wang, Z., Li, J. L., Hong, C. S., et al. (2014). Inhibition of protein phosphatase 2A with a small molecule LB100 radiosensitizes nasopharyngeal carcinoma xenografts by inducing mitotic catastrophe and blocking DNA damage repair. Oncotarget 5 (17), 7512–7524. doi:10.18632/oncotarget.2258
MacKay, K. B., Tu, Y., Young, S. G., and Clarke, S. G. (2013). Circumventing embryonic lethality with Lcmt1 deficiency: generation of hypomorphic Lcmt1 mice with reduced protein phosphatase 2A methyltransferase expression and defects in insulin signaling. PloS one 8 (6), e65967. doi:10.1371/journal.pone.0065967
Maggio, D., Ho, W. S., Breese, R., Walbridge, S., Wang, H., Cui, J., et al. (2020). Inhibition of protein phosphatase-2A with LB-100 enhances antitumor immunity against glioblastoma. J. Neurooncol 148 (2), 231–244. doi:10.1007/s11060-020-03517-5
Mayr, C., Beyreis, M., Dobias, H., Gaisberger, M., Pichler, M., Ritter, M., et al. (2018). Miniaturization of the clonogenic assay using confluence measurement. Int. J. Mol. Sci. 19 (3), 724. doi:10.3390/ijms19030724
Mazhar, S., Taylor, S. E., Sangodkar, J., and Narla, G. (2019). Targeting PP2A in cancer: combination therapies. Biochim. Biophys. Acta Mol. Cell Res. 1866 (1), 51–63. doi:10.1016/j.bbamcr.2018.08.020
Meng, E. C., Goddard, T. D., Pettersen, E. F., Couch, G. S., Pearson, Z. J., Morris, J. H., et al. (2023). UCSF ChimeraX: tools for structure building and analysis. Protein Sci. 32 (11), e4792. doi:10.1002/pro.4792
Mirzapoiazova, T., Xiao, G., Mambetsariev, B., Nasser, M. W., Miaou, E., Singhal, S. S., et al. (2021). Protein phosphatase 2A as a therapeutic target in small cell lung cancer. Mol. cancer Ther. 20 (10), 1820–1835. doi:10.1158/1535-7163.MCT-21-0013
Moura, M., and Conde, C. (2019). Phosphatases in mitosis: roles and regulation. Biomolecules 9 (2), 55. doi:10.3390/biom9020055
Najafov, A., Luu, H. S., Mookhtiar, A. K., Mifflin, L., Xia, H. G., Amin, P. P., et al. (2021). RIPK1 promotes energy sensing by the mTORC1 pathway. Mol. Cell 81 (2), 370–385.e7. doi:10.1016/j.molcel.2020.11.008
Nyamsuren, G., Rapp, G., Dihazi, H., Zeisberg, E. M., Tampe, D., Tampe, B., et al. (2021). PP2A phosphatase inhibition is anti-fibrotic through Ser77 phosphorylation-mediated ARNT/ARNT homodimer formation. Sci. Rep. 11 (1), 24075. doi:10.1038/s41598-021-03523-1
OBoyle, N. M., Banck, M., James, C. A., Morley, C., Vandermeersch, T., and Hutchison, G. R. (2011). Open Babel: an open chemical toolbox. J. Cheminform 3, 33. doi:10.1186/1758-2946-3-33
OConnor, C. M., Perl, A., Leonard, D., Sangodkar, J., and Narla, G. (2018). Therapeutic targeting of PP2A. Int. J. Biochem. Cell Biol. 96, 182–193. doi:10.1016/j.biocel.2017.10.008
Ory, S., Zhou, M., Conrads, T. P., Veenstra, T. D., and Morrison, D. K. (2003). Protein phosphatase 2A positively regulates Ras signaling by dephosphorylating KSR1 and Raf-1 on critical 14-3-3 binding sites. Curr. Biol. 13 (16), 1356–1364. doi:10.1016/s0960-9822(03)00535-9
Pettersen, E. F., Goddard, T. D., Huang, C. C., Meng, E. C., Couch, G. S., Croll, T. I., et al. (2021). UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30 (1), 70–82. doi:10.1002/pro.3943
Pippa, R., and Odero, M. D. (2020). The role of MYC and PP2A in the initiation and progression of myeloid leukemias. Cells 9 (3), 544. doi:10.3390/cells9030544
Rasool, R. U., O'Connor, C. M., Das, C. K., Alhusayan, M., Verma, B. K., Islam, S., et al. (2023). Loss of LCMT1 and biased protein phosphatase 2A heterotrimerization drive prostate cancer progression and therapy resistance. Nat. Commun. 14 (1), 5253. doi:10.1038/s41467-023-40760-6
Remmerie, M., and Janssens, V. (2019). PP2A: a promising biomarker and therapeutic target in endometrial cancer. Front. Oncol. 9, 462. doi:10.3389/fonc.2019.00462
Ronk, H., Rosenblum, J. S., Kung, T., and Zhuang, Z. (2022). Targeting PP2A for cancer therapeutic modulation. Cancer Biol. Med. 19 (10), 1428–1439. doi:10.20892/j.issn.2095-3941.2022.0330
Sacco, F., Perfetto, L., Castagnoli, L., and Cesareni, G. (2012). The human phosphatase interactome: an intricate family portrait. FEBS Lett. 586 (17), 2732–2739. doi:10.1016/j.febslet.2012.05.008
Sangodkar, J., Farrington, C. C., McClinch, K., Galsky, M. D., Kastrinsky, D. B., and Narla, G. (2016). All roads lead to PP2A: exploiting the therapeutic potential of this phosphatase. FEBS J. 283 (6), 1004–1024. doi:10.1111/febs.13573
Smits, P. H., Smits, H. L., Minnaar, R. P., Hemmings, B. A., Mayer-Jaekel, R. E., Schuurman, R., et al. (1992). The 55 kDa regulatory subunit of protein phosphatase 2A plays a role in the activation of the HPV16 long control region in human cells with a deletion in the short arm of chromosome 11. EMBO J. 11 (12), 4601–4606. doi:10.1002/j.1460-2075.1992.tb05562.x
Song, Q., Wang, H., Jiang, D., Xu, C., Cui, J., Zhang, Q., et al. (2021). Pharmacological inhibition of PP2A overcomes nab-paclitaxel resistance by downregulating MCL1 in esophageal squamous cell carcinoma (ESCC). Cancers (Basel) 13 (19), 4766. doi:10.3390/cancers13194766
Stanevich, V., Jiang, L., Satyshur, K. A., Li, Y., Jeffrey, P. D., Li, Z., et al. (2011). The structural basis for tight control of PP2A methylation and function by LCMT-1. Mol. Cell 41 (3), 331–342. doi:10.1016/j.molcel.2010.12.030
Sun, B., Zhong, F. J., Xu, C., Li, Y. M., Zhao, Y. R., Cao, M. M., et al. (2021). Programmed cell death 10 promotes metastasis and epithelial-mesenchymal transition of hepatocellular carcinoma via PP2Ac-mediated YAP activation. Cell Death Dis. 12 (9), 849. doi:10.1038/s41419-021-04139-z
Trott, O., and Olson, A. J. (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31 (2), 455–461. doi:10.1002/jcc.21334
Turdo, A., D'Accardo, C., Glaviano, A., Porcelli, G., Colarossi, C., Colarossi, L., et al. (2021). Targeting phosphatases and kinases: how to checkmate cancer. Front. Cell Dev. Biol. 9, 690306. doi:10.3389/fcell.2021.690306
Uddin, M. H., Pimentel, J. M., Chatterjee, M., Allen, J. E., Zhuang, Z., and Wu, G. S. (2020). Targeting PP2A inhibits the growth of triple-negative breast cancer cells. Cell Cycle 19 (5), 592–600. doi:10.1080/15384101.2020.1723195
Vainonen, J. P., Momeny, M., and Westermarck, J. (2021). Druggable cancer phosphatases. Sci. Transl. Med. 13 (588), eabe2967. doi:10.1126/scitranslmed.abe2967
Vaneynde, P., Verbinnen, I., and Janssens, V. (2022). The role of serine/threonine phosphatases in human development: evidence from congenital disorders. Front. Cell Dev. Biol. 10, 1030119. doi:10.3389/fcell.2022.1030119
Wei, D., Parsels, L. A., Karnak, D., Davis, M. A., Parsels, J. D., Marsh, A. C., et al. (2013). Inhibition of protein phosphatase 2A radiosensitizes pancreatic cancers by modulating CDC25C/CDK1 and homologous recombination repair. Clin. cancer Res. official J. Am. Assoc. Cancer Res. 19 (16), 4422–4432. doi:10.1158/1078-0432.CCR-13-0788
Wlodarchak, N., and Xing, Y. (2016). PP2A as a master regulator of the cell cycle. Crit. Rev. Biochem. Mol. Biol. 51 (3), 162–184. doi:10.3109/10409238.2016.1143913
Xiao, G., Chan, L. N., Klemm, L., Braas, D., Chen, Z., Geng, H., et al. (2018). B-Cell-Specific diversion of glucose carbon utilization reveals a unique vulnerability in B cell malignancies. Cell 173 (2), 470–484. doi:10.1016/j.cell.2018.02.048
Xie, H., and Clarke, S. (1994). Protein phosphatase 2A is reversibly modified by methyl esterification at its C-terminal leucine residue in bovine brain. J. Biol. Chem. 269 (3), 1981–1984. doi:10.1016/s0021-9258(17)42124-7
Zhang, C., Hong, C. S., Hu, X., Yang, C., Wang, H., Zhu, D., et al. (2015). Inhibition of protein phosphatase 2A with the small molecule LB100 overcomes cell cycle arrest in osteosarcoma after cisplatin treatment. Cell Cycle 14 (13), 2100–2108. doi:10.1080/15384101.2015.1041693
Zhang, H., Cao, X., Tang, M., Zhong, G., Si, Y., Li, H., et al. (2021). A subcellular map of the human kinome. Elife 10, e64943. doi:10.7554/eLife.64943
Zhang, L., Lu, L., Zhong, X., Yue, Y., Hong, Y., Li, Y., et al. (2019). Metformin reduced NLRP3 inflammasome activity in Ox-LDL stimulated macrophages through adenosine monophosphate activated protein kinase and protein phosphatase 2A. Eur. J. Pharmacol. 852, 99–106. doi:10.1016/j.ejphar.2019.03.006
Zhang, N., Lu, C., Mo, J., Wang, X., Liao, S., Liang, N., et al. (2023a). LCMT1 indicates poor prognosis and is essential for cell proliferation in hepatocellular carcinoma. Transl. Oncol. 27, 101572. doi:10.1016/j.tranon.2022.101572
Zhang, T., Xu, D., Trefts, E., Lv, M., Inuzuka, H., Song, G., et al. (2023b). Metabolic orchestration of cell death by AMPK-mediated phosphorylation of RIPK1. Science 380 (6652), 1372–1380. doi:10.1126/science.abn1725
Zhao, Z., Kurimchak, A., Nikonova, A. S., Feiser, F., Wasserman, J. S., Fowle, H., et al. (2019). PPP2R2A prostate cancer haploinsufficiency is associated with worse prognosis and a high vulnerability to B55α/PP2A reconstitution that triggers centrosome destabilization. Oncogenesis 8 (12), 72. doi:10.1038/s41389-019-0180-9
Keywords: humans, protein phosphatase, catalytic domain, methylation, eukaryotic cells, methyltransferases, inhibitor
Citation: Arosarena OA, Saribas AS and Papadopoulos EP (2024) A small molecule inhibitor of leucine carboxyl methyltransferase-1 inhibits cancer cell survival. Front. Drug Discov. 4:1278163. doi: 10.3389/fddsv.2024.1278163
Received: 15 August 2023; Accepted: 18 March 2024;
Published: 11 April 2024.
Edited by:
Zhe-Sheng Chen, St. John’s University, United StatesReviewed by:
Essa M. Saied, Humboldt University of Berlin, GermanyCopyright © 2024 Arosarena, Saribas and Papadopoulos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: O.A. Arosarena, b25laWRhQHRlbXBsZS5lZHU=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.