
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Drug Discov. , 30 October 2023
Sec. Technologies and Strategies to Enable Drug Discovery
Volume 3 - 2023 | https://doi.org/10.3389/fddsv.2023.1289314
 Manisha Padmakumar†
Manisha Padmakumar† Steven Biesmans*†
Steven Biesmans*† Jorge S. Valadas†
Jorge S. Valadas† Jan R. Detrez
Jan R. Detrez Gaëlle GilletPriscillia Bresler
Gaëlle GilletPriscillia Bresler Marie-Laure Clénet
Marie-Laure Clénet Irena Kadiu*
Irena Kadiu*Neuroscience drug discovery has faced significant challenges due to restricted access to relevant human cell models and limited translatability of existing preclinical findings to human pathophysiology. Induced pluripotent stem cells (iPSCs) have emerged as a promising solution, offering the potential to generate patient-specific cell types, including in the recent years, iPSC-derived microglia (iMGL). Current methods rely on complex and time-consuming differentiation procedures, leading to considerable batch-to-batch variability consequently hindering the establishment of standardized and reproducible high-throughput functional screening approaches. Addressing these challenges is critical in ensuring the generation of homogenous iMGL populations with consistent functional properties. In this study we describe an improved high-yield protocol for generating iMGL, which allows for increased reproducibility and flexibility in the execution of high-throughput functional screens. We introduce a two-step process in embryoid bodie (EB) maintenance and a stop point allowing for cryopreservation at the hematopoietic progenitor cell (iHPC) stages. Furthermore, we demonstrate inter-operator robustness of this modified protocol in a range of high-throughput functional assays including phagocytosis, lysosomal acidification, chemotaxis, and cytokine release. Our study underscores the importance of quality control checks at various stages of iPSC-differentiation and functional assay set up, highlighting novel workarounds to the existing challenges such as limited yield, flexibility, and reproducibility, all critical in drug discovery.
The high attrition rate in neuroscience drug discovery is multifactorial and can be attributed in part to lack of access to human primary cellular material or tissues at onset or during disease progression and limited translatability of transgenic rodent models. A lack of insight into human disease pathobiology beyond molecular signatures identified in end stage post-mortem tissues is a limiting step in understanding mechanistic and temporal drivers of human disease. Contrary to systemic inflammatory conditions, where blood or inflamed peripheral tissue can be sampled in a relatively noninvasive manner during disease progression, the lack of tissue access at various stages of neurological disease progression has been a limiting factor in preclinical de-risking of therapeutic targets in neuroscience. Transgenic in vivo models have been utilized to obtain temporal mechanistic insights into neurological disease; however, while useful as pharmacokinetic/pharmacodynamic tools, they do not display the full range of pathology and behavioral phenotypes as human disease. Furthermore, the pathological mutations these models harbor may restrict biological insights and translatability only to familial patient populations. As such, integration of more translational tools such as pluripotent stem cell technologies at all levels of drug discovery from target identification and validation to screening and biological characterization of clinical candidates has become a necessity for drug discovery.
The idea of de-differentiation and reprograming a human mature somatic cell into any other cell type, including neurons and glial cells, which were previously inaccessible in living patients has truly revolutionized scientific research and drug discovery. Reprograming of dermal fibroblasts into induced iPSCs through retroviral transduction of the Yamanaka factors: cellular myelomatosis (c-MYC), Kruppel-like factor 4 (KLF4), octamer-binding transcription factors 3 and 4 (OCT3/4), and (sex-determining region Y)-box 2 (SOX2) in 2007 opened up exciting possibilities for human cellular and tissue modeling and consequently drug discovery (Takahashi et al., 2007).
While differentiation protocols for neuronal, astro- and oligodendro-glia quickly followed, the generation of iPSC-derived microglia proved to be technically challenging, lagging by a decade from the first publication of the Yamanaka study, with Abud et al. (2017) reporting the first method in 2017. Microglia are highly contextual environmental sensors. They possess exquisite capabilities in distinguishing and responding to a plethora of environmental cues such as alterations in neuronal firing patterns with active synaptic architecture modifications (dendritic spine formation or pruning) (Thion et al., 2018; Angelova and Brown, 2019). In contexts such as tissue injury or pathogen invasion, microglia prioritize phagocytic/lysosomal clearance to restore homeostasis, and cytokine release to recruit other cell types to the sites of injury (DiSabato et al., 2016; Salter and Stevens, 2017; Thion et al., 2018). Mimicking these brain tissue specific and contextual cues are critical for generation of iPSC-derived microglia-like cells as opposed to tissue macrophage-like cells.
Several methods for generation of mature and functional microglia from human induced pluripotent stem cells (iPSCs) have been described in the recent years (Abud et al., 2017; Douvaras et al., 2017; Pocock and Piers, 2018; Xu et al., 2020; McQuade and Blurton-Jones, 2021). These protocols follow the developmental lineage of innate microglia, from iPSCs to hematopoietic precursor cells to mature microglia, in some instances requiring co-culturing conditions with neurons or astrocytes to provide more complex environmental cues (Ginhoux et al., 2013). While successful, these protocols are lengthy, labor-intensive and provide little technical detail on potential stopping points that can shorten the experimental timelines or provide flexibility in study planning. Furthermore, variability in differentiation efficiency, yields, cell quality between different iPSC lines, batches, and operators pose a challenge to culture generation scale-up and/or high-throughput experimentation in a drug discovery setting (Banerjee et al., 2020).
In this study, we report a novel method to scale-up microglia generation from iPSCs. We introduce a stopping point consisting of banking/cryopreservation of cells at iHPC stage to enable much needed flexibility with both scale and timing of high-throughput functional screening. Additionally, we describe experimental conditions for assay miniaturization and orthogonal functional characterization of iMGL that address the existing reproducibility challenges associated with use of iPSC systems for drug discovery.
The iPSC line was obtained from Applied StemCell. Peripheral blood mononuclear cells (PBMCs) form healthy donors were isolated from whole blood by leukapheresis and shipped overnight in culture medium from University of Nebraska Medical Center (Omaha, Nebraska). Cell culture reagents used in this study are listed in Supplementary Table S1.
All procedures for generation, cryopreservation of iHPCs, subsequent differentiation to iMGL, and functional assays were performed under sterile conditions (biological safety cabinet). iPSCs were maintained in 6-well plates in feeder free conditions on growth factor-reduced Matrigel (Corning) in complete mTeSR™ plus medium (STEMCELL Technologies) for at least 2 passages. Cell culture was maintained through daily full medium exchange in a humidified incubator (20% O2, 5% CO2, 37°C) and passaged every 4–5 days. Two days prior to the differentiation process, iPSCs were switched to complete Essential 8™ Flex medium (Thermo Fisher Scientific). For medium exchanges throughout the iMGL generation protocol fresh medium was prepared for each feeding with single-use aliquoted growth factors added to the warm (37°C) culture medium just before use.
On day-1, iPSCs cultured in 6-well culture plates were dissociated to single cell suspension (to enable uniform EB formation) by incubation with 1 mL of warm (37°C) Accutase® (Sigma-Aldrich) for 10 min at 37°C followed by gentle scraping. Dissociation time may vary for each iPSC line and preliminary testing to assess optimal dissociation time without affecting cell quality/viability is advised. Cells were pooled and centrifuged at 300 g at RT for 5 min followed by resuspension in Essential 8™ Flex Medium (Thermofisher) supplemented with 10 µM Y-27632 dihydrochloride (Abcam). Any residual clumps in the pooled cells were dissociated by gentle trituration (3-4 times) with a 1 mL micropipette. Cell viability and numbers were assessed using a Countess II automated cell counter (Thermofisher). Cells were supplemented with additional complete Essential 8™ Flex medium with 10 μM Y-27632 dihydrochloride to achieve a density range of 0.5–1.25 × 106 cells per well. Optimal seeding density varies by cell line and may be adjusted to obtain a small not dense EB (size should not exceed 300 µm). For the iPSC line used in this study, cells were seeded at a density of 1 × 106 cells/well (∼200 cells/microwell). A volume of 2 mL of cell suspension was added to each well of 6-well AggreWell™ 400 plates (STEMCELL Technologies) pretreated with anti-adherence rinsing solution (STEMCELL Technologies). To ensure cells remain confined to the microwells, plates were centrifuged at 100 g for 3 min at RT. Following centrifugation, cells were maintained in a humidified incubator under normoxic conditions (20% O2, 5% CO2, 37°C).
On day 0, the embryoid bodies were captured using a 37 µm reversible cell strainer and resuspended in iPSC-derived iHPCs. Differentiation Base Medium as previously described (Abud et al., 2017), supplemented with fibroblast growth factor 2 (FGF2; 50 ng/mL), bone morphogenetic protein 4 (BMP4; 50 ng/mL), Activin-A (12.5 ng/mL), Y-27632 dihydrochloride (1 μM) and lithium chloride (2 mM). EBs (in 60 mL of medium) were transferred to T225 flasks pre-treated with anti-adherence solution (STEMCELL Technologies) for 10 min at RT. Flasks were then transferred to a humidified incubator with hypoxic cell culture conditions (5% O2, 5% CO2, 37°C).
On day 2, the EBs were captured on a 37 µm reversible cell strainer and resuspended in iHPC differentiation base medium. Prior to use, the culture media was pre-warmed and pre-equilibrated in a hypoxic condition (5% O2, 5% CO2, 37°C) for 1 h and supplemented with FGF2 (50 ng/mL) and vascular endothelial growth factor (VEGF; 50 ng/mL). The EBs were then cultured in 60 mL medium per flask in T225 culture flasks (pre-treated with anti-adherence solution) in a humidified incubator under hypoxic cell culture conditions (5% O2, 5% CO2, 37°C) for 2 days.
On day 4 the EBs were captured using a 37 µm reversible cell strainer, resuspended in iHPC Differentiation Base Medium containing FGF2 (50 ng/mL), VEGF (50 ng/mL), thrombopoietin (TPO; 50 ng/mL), stem cell factor (SCF; 10 ng/mL), interleukin 3 (IL-3; 10 ng/mL) and interleukin 6 (IL-6; 50 ng/mL) (Day 4 iHPC differentiation medium) and cultured in T225 culture flasks (pre-treated with Anti-Adherence Solution) in a humidified incubator under normoxic conditions (20% O2, 5% CO2, 37°C). Every 2 days, cells were supplemented with 30 mL of fresh Day 4 iHPC differentiation medium.
To ensure a high yield of iHPC, EBs were maintained in culture for 10–18 days, supplemented with day 4 medium every 2–3 days (30 mL per T225 flask).
Between Day 10 and Day 18, iHPC morphology was assessed and cells were harvested. EBs were collected from the flasks using a 25 mL serological pipette and transferred to a conical tube through filtration into a 37 µm reversible cell strainer, whereby the embryoid bodies were captured and optionally re-cultured in 60 ml Day 4 iHPC differentiation medium for a second harvest. The flowthrough containing iHPCs was transferred to new tubes and analyzed by flow cytometry to assess viability, yield, and hematopoietic lineage marker expression (Abud et al., 2017; CD34+ CD43+ iHPC; Supplementary Figure S1). Cells were then centrifuged at 300 g for 5 min and supernatant was carefully removed to not disturb the cell pellet. Cells were gently resuspended in the appropriate volume of CryoStor® CS10 (STEMCELL Technologies) to obtain a suspension containing 2 × 106 cells/mL. Cells were maintained in cryo-storage isopropanol containers at −80°C for 24 h, and then transfered to a liquid nitrogen storage system for long term cryopreservation.
Cryopreserved iHPC were quickly thawed by gentle swirling at 37°C in a water bath. Following testing for viability, cells were cultured in Day 4 iHPC Differentiation Medium (see above) supplemented with 10 μM Y-27632 dihydrochloride and 1 μg/mL laminin (Merck) at 3 × 105 cells per well in a 6-well plate pre-coated with poly-D-lysine (PDL; Corning) and 20 μg/mL laminin. Cells were maintained in a humidified incubator (20% O2, 5% CO2, 37°C). On the next day, medium was changed to iMGL differentiation medium supplemented with macrophage colony-stimulating factor (M-CSF; 25 ng/mL), transforming growth factor beta (TGF-ß1; 50 ng/mL) and interleukin 34 (IL-34; 100 ng/mL). Cells were maintained for 14 days in 6-well plates through supplementation of 1 mL medium per well every 2–3 days, until used for various in vitro assays. Floating cells were collected first, and adherent ones were detached by a gentle wash in warm 1X phosphate-buffered saline (PBS; Thermo Fisher Scientific) followed by gentle scraping. Cells were centrifuged at 300 g for 5 min, resuspended in complete iMGL differentiation medium and counted to obtain the necessary cell density for functional assays.
For mass cytometry, 1 × 106 iMGL were harvested and washed once with iMGL Differentiation medium. PBMCs were thawed and washed once with DMEM Glutamax containing 10% fetal bovine serum (FBS) were washed with Maxpar® Cell Staining buffer (Fluidigm) followed by immunostaining with a cocktail of metal tagged antibodies (Supplementary Table S2) for 45 min at 4°C and 15 min at room temperature (RT). Cells were washed thrice using the Maxpar® Cell Staining buffer and fixed using 4% paraformaldehyde (PFA) in 1X PBS for 10 min at RT then stored at 4°C until data acquisition. Cells were treated with Maxpar® 191/193Ir DNA Intercalator (50 nM, Fluidigm) diluted in 4% PFA in 1X PBS and incubated at 4°C overnight. Cells were washed twice with Maxpar® water (Fluidigm) and centrifuged at 800 g. Cells were resuspended in EQ™ Four Element Calibration beads (Fluidigm) diluted in Maxpar® Cell Acquisition Solution (Fluidigm) at 1:4 ratio, filtered through a 40 μm cell strainer and acquired on a Helios™ mass cytometer (Fluidigm). Mass cytometry data were analyzed using Cytobank (Beckman Coulter). Normalization beads were gated out and single cells were then gated based on DNA content and cell size to exclude debris and doublets. The monocyte subset in PBMCs was gated based on CD11b and CD172a surface markers. iPSC microglia were gated based on CD45 and CD11b expression. For dimensionality reduction and visualization, viSNE maps were generated according to the expression levels of all markers using the t-distributed stochastic linear embedding (t-SNE) algorithm available on Cytobank. All samples were included within the same viSNE analysis for direct comparison.
iMGL were seeded at a density of 1 × 104 cells/well in a 384-well plate and allowed to attach for 24 h. Cells were washed with 1X PBS at RT and fixed with 4% PFA in 1X PBS for 10 min at RT. Cells were washed once with 1X PBS followed by blocking and permeabilization with Odyssey™ Blocking Buffer supplemented with 0.2% Triton X (Sigma Aldrich, LI-COR Biosciences™). Cells were incubated overnight at 4°C with the primary antibody diluted in the blocking buffer. Cells were then washed thrice for 15 min in 1X PBS followed by 1 h incubation with secondary antibodies (Supplementary Table S3) diluted in the blocking buffer. After three washes with 1X PBS, cells were incubated with a nuclear stain (Hoechst 33342, 1:1000) for 20 min at RT and washed with 1X PBS. Images were acquired with Zeiss LSM880 confocal microscope using Zeiss ZEN black imaging software.
Day 25 iMGL were plated at 5 × 103 cells/well on PDL- and laminin-coated 384-well plates (Corning) with three technical replicates per condition. Cells were allowed to attach for 1 h and then the positive control wells were pre-treated with Cytochalasin D (CytoD, Merck) at concentrations ranging from 1.25 to 10 µM. 1 h post-treatment, pHrodo™ Red Zymosan Bioparticles™ (Invitrogen) were added to all the remaining wells at increasing concentrations (0.125–0.5 µg/well). Phagocytic uptake of bioparticles by iMGL was captured through time lapse live imaging every 30 min for 24 h on an IncuCyte® S3 Live-Cell Analysis System (Sartorius), at 10x magnification. All images were automatically processed by Incucyte® analysis software and the total integrated intensity of phagocytic beads corrected for confluence at baseline was reported.
Day 25 iMGL were plated on a PDL- and laminin-coated plate at 5 × 103 iMGL/well and allowed to adhere for 1 h prior to functional assessment. Bafilomycin A1 (a V-ATPase inhibitor) was added at concentrations ranging from 0.01 to 1 μM for 1 h followed by addition of LysoTracker™ Red at 50 nM and Hoechst 33342 at 10 μg/mL in iMGL medium. Live iMGL were imaged using a 40x objective in an Opera Phenix™ High-Content Screening System (Perkin Elmer). Lysosomes were identified as LysoTracker™ Red positive structures. Analysis of lysosomal characteristics was performed on the Harmony® High-Content Imaging and Analysis Software (Perkin Elmer) and normalized on cellular density.
Day 25 iMGL were plated at a density of 2.5 × 103 per well on the top of the insert of a IncuCyte® ClearView 96-Well Chemotaxis Plate (Essen BioScience). Chemoattractants including monocyte chemoattractant protein-1 (MCP-1; 0.1 μg/mL), complement component 5a (C5a; 0.25 μg/mL), adenosine diphosphate (ADP; 100 µM) were added to the bottom wells. The top porous insert (8 µm diameter pores) was carefully placed to match the corresponding chemoattractant wells and incubated at 37°C, 5% CO2 for 15 min to allow the cells to settle. iMGL migration towards the chemoattractant was measured for 24 h on the IncuCyte® S3 Live-Cell Analysis System, utilizing the IncuCyte® Chemotaxis Cell Migration Assay module (10x imaging), per manufacturer’s instructions. All images of cells present on both sides of the porous membrane were automatically processed by IncuCyte® software. Cell migration was reported as the decrease in cellular density on the top of the porous membrane.
Day 25 iMGL were seeded on PDL- and laminin-coated 384 well plates at a density of 1 × 104 cells per well and allowed to attach for 1 h. Cells were then treated with increasing concentrations of liposaccharide (LPS; 0.11–1 μg/mL, Sigma), peptidoglycan from Gram-positive Bacillus subtilis (PGN-BS; 3.33–30 μg/mL, Invivogen) or pHrodo™ Red Zymosan Bioparticles™ (0.25–1 µg/well, Invitrogen). Following a 24 h stimulation, cell culture fluids were collected and centrifuged at 300 g for 5 min at 37°C to remove cellular debris. Supernatants were collected and analyzed using Meso Scale Discovery V-Plex Proinflammatory Panel 1 Human Kit (Meso Scale Diagnostics) per manufacturer’s instructions.
All data are presented as mean values ± standard deviation (S.D.). Data and statistical analysis were performed in GraphPad Prism. Two-way ANOVA followed by Tukey’s multiple comparison was used to compare different conditions with a p < 0.05 considered statistically significant.
While published methods for iPSC-differentiation to iMGL provide a good starting point, we identified several challenges that precluded their use for drug discovery for two primary reasons: First, low yield represents a limiting factor in execution of high-content immune phenotyping and functional readouts. Second, lengthy differentiation protocols with no stopping points increase the likelihood of cell culture contaminations and more importantly require months of planning, making them incompatible with dynamic and flexible drug discovery needs. To address these challenges, we modified the existing methods to improve cellular yield through scale-up of EB cultures using AggreWell™ plates and T225 flasks. We introduced a stopping point at iHPCs stage to improve experimental planning flexibility and miniaturized functional readouts to 384-well format to reduce cell number requirements. To scale-up the generation of iMGL, iPSCs were allowed to form uniform EBs in AggreWell™ plates. These EBs were then differentiated for 10 days in T225 flasks in varying growth factor cocktails in normoxic and hypoxic conditions to generate minimum 1 × 108 iHPCs. These cells were either cryopreserved for future differentiation into iMGL or maintained in culture under normoxic conditions for 15 days in medium supplemented with M-CSF, TGF-β1, IL-34 and insulin for use in functional assays (Figure 1A). Several quality control assessments were performed during the differentiation process starting with confirmation of pluripotency characteristics through immunostaining of the iPSCs for stem cell markers OCT4, SOX2, and TRA-1-8. Immunocyctochemistry confirmed that the iPSC line used in this study expresses pluripotency markers (Figure 1B). Uniformity of the EBs was assessed prior to their seeding to ensure optimal yield and viability of iHPCs. Bright field images show optimal and uniform growth of EBs in AggreWell™ and T225 flasks (Figure 1C). A change in culture conditions from 6-well plates to T225 flasks and lengthening maintenance time of EBs resulted in a yield of 15–20 iMGL per iPSC (Supplementary Figure S1). Yield is cell line-dependent and amenable to additional scale-up based on cell number needs with similar culture handling effort. Since the optimal stopping point in the differentiation protocol was determined to be at the iHPC stage, morphological and phenotypical assessment were performed of microglia differentiated from cryopreserved iHPCs after 2 weeks in culture. Quality control (presence of hematopoietic lineage markers CD34 and CD43) and ability to differentiate into iMGL of cryopreserved iHPC and those generated following a non-cession protocol was assessed. No visible differences in viability (95% post-thaw-data not shown) and differentiation capacity were observed between the two protocols suggesting cryopreservation at iHPC stage does not disrupt iMGL essential functions (Supplementary Figure S1). iMGL displayed a typical ramified morphology and expressed classical microglial markers such as P2RY12, CD11b, and IBA1 (Figure 1D). Since one of the challenges in establishing iPSC-derived microglial differentiation methods has been the propensity of iHPCs to differentiate into monocytic populations, iMGL generated with our protocol were compared to primary human monocytes at single cell resolution immune phenotyping by mass cytometry. Data show iMGL express canonical markers including CD45, CD11b, TMEM 119, TREM2 and C3aR. Mass cytometry data demonstrate higher expression levels of microglia classical markers in iMGL compared to primary human monocytes, indicating these cells are microglia-like (Figure 1E). These data confirmed the suitability of the modified scale-up and a stop point approach for generation of high yield microglia-like cells.
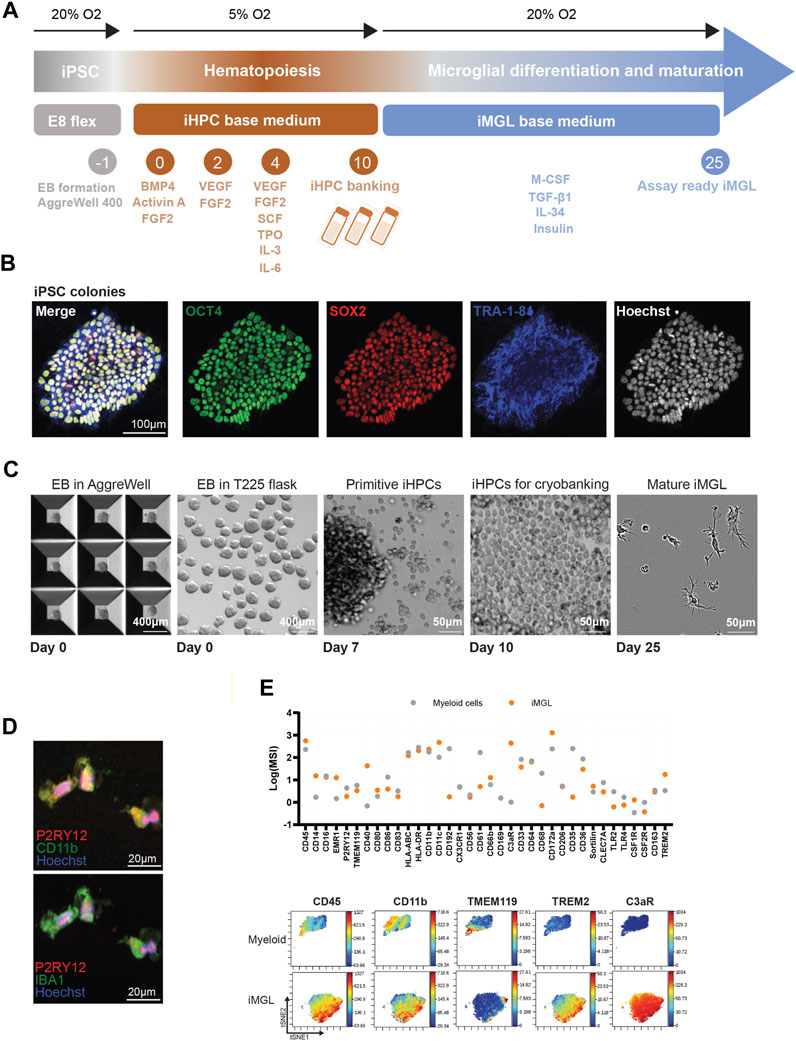
FIGURE 1. iMGL generation and phenotypic characterization. (A) Outline of the large scale iMGL generation protocol and nomenclature (B) Immunofluorescence images demonstrating expression of pluripotency markers OCT4, SOX2, and TRA-1-8 in the iPSC cells. (C) Images showing generation and maintenance of uniform (similar shape and size) EBs in AggreWell™ microwell plates, transfer to T225 flasks, iHPCs generation and cryopreservation, as well as ramified morphology of iMGL 15 days in culture from cryo-preserved iHPCs. (D) Immunocytochemistry of iMGL demonstrating expression of microglia markers including P2RY12, CD11b, and Iba1. (E) Mass cytometry comparative analysis of immune marker expression in iMGL and primary human monocytes. Data was captured as mean signal intensity (MSI) and representative two-dimensional projections of single-cell data generated by t-SNE (n = 15,000 cells/population, randomly sampled).
Next, we established several functional assays in miniaturized 384-well format to enable a higher throughput functional screening allowing for conservation of cells and reagents, while promoting more uniform and controlled cell culture environments (minimizing experimental variability). To ascertain the reproducibility of our methods the same iPSC line was independently differentiated into iMGL by three operators with variable degrees of experience in handling iPSCs. Cells were assessed in a range of functional readouts including phagocytic uptake, lysosomal function, and cytokine release. These were deemed as relevant readouts for microglial homeostatic roles including their ability to survey the central nervous system (CNS) environment, clear cellular debris and modify the synaptic architecture.
To assess phagocytic function, iMGL were exposed to increasing concentrations of pHrodo™ Red Zymosan Bioparticles™ which fluoresce in lysosomal acidic pH upon uptake by iMGL (Figures 2A, B). Time-lapse imaging over 24 h revealed a robust and concentration-dependent uptake of particles, demonstrating that iMGL generated from our modified protocol are capable of phagocytosis (Figure 2C). Data show similar levels of phagocytic capacity in iMGL generated by different operators. Pre-treatment with CytoD, a cell-permeable inhibitor of actin polymerization disrupting endocytosis was used to demonstrate assay response (Figure 2D).
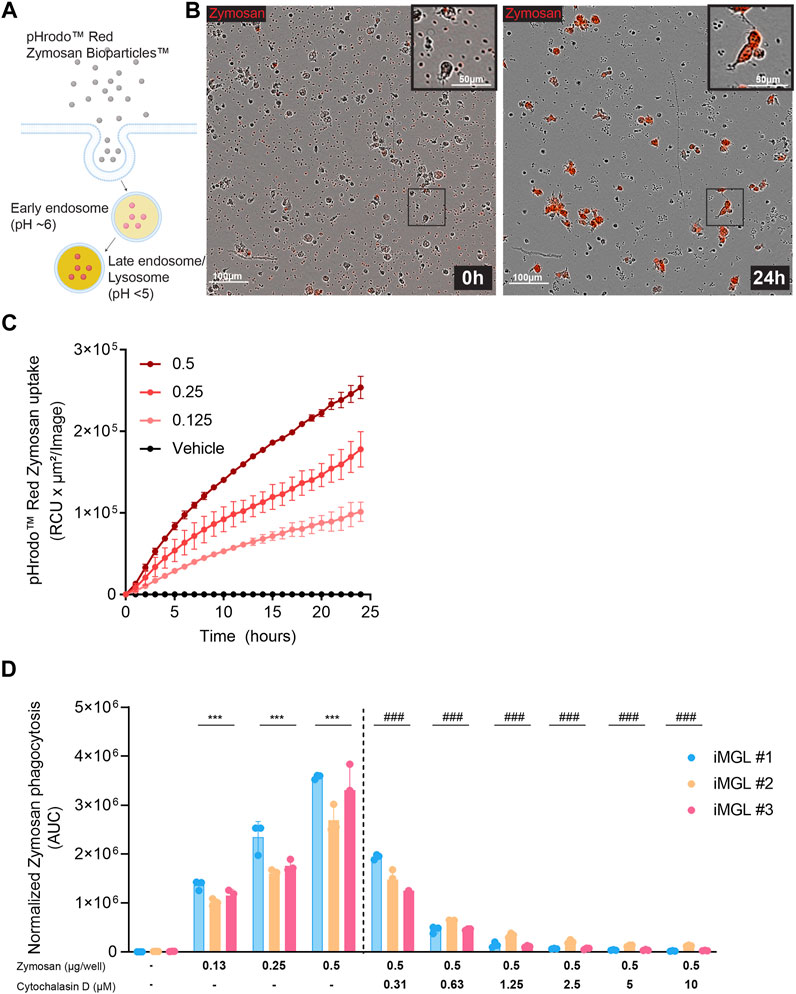
FIGURE 2. Phagocytic uptake in iMGL. (A) A schematic depicting pHrodoRed™ zymosan bioparticles™ internalization into acidified endo-lysosomal compartments through phagocytosis. (B) Representative time lapse frames of iMGL uptake of zymosan particles at 0 and 24 h. (C) Dose- and time-dependent increase of pHrodamine signal as a measure of phagocytic activity in iMGL over 24 h. (D) Phagocytic activity across the iMGL line differentiated independently and assessed for phagocytic activity by three operators. Disruption of actin polymerization by CytoD prevents zymosan uptake in a dose dependent manner. Data were generated from three technical replicates per condition. Data are shown as mean ± S.D. Data were analyzed using two-way ANOVA followed by Tukey’s post hoc test; ***p < 0.001 (comparing to untreated condition), and ###p < 0.001 (comparing to zymosan at 0.5 μg/well condition).
Lysosomal degradation of cellular debris, autophagic or misfolded protein products is critical for microglial clearance functions and maintenance of tissue homeostasis (Pomilio et al., 2020). Genetic variants impacting aspects of lysosomal function have been broadly implicated in neurodegenerative disorders including Alzheimer’s, Parkinson’s disease, and Frontotemporal dementia genome wide association (GWAS) studies, and human disease pathobiology (Root et al., 2021). Therefore, high-throughput assays evaluating various aspects of lysosomal function such as lysosomal acidification, deemed critical in load degradation, would be useful in identifying modifiers of this biology. To assess lysosomal acidification, iMGLs were cultured in 384-well plates and exposed to Lysotracker™ dye which fluoresces when protonated in acidic pH, followed by high resolution high-content confocal imaging. Bafilomycin A1, a V-ATPase inhibitor blocking lysosomal acidification as well as an inhibitor of autophagosome-lysosomal fusion, was used to assess assay sensitivity to perturbation of these lysosomal functions (Maharjan et al., 2022) (Figure 3A). Treatment with increasing concentrations of Bafilomycin A1 showed a significant and dose-dependent decrease in Lysotracker™ dye fluorescence intensity suggesting interference with lysosomal acidification. Similar outcomes were observed in iMGL generated by 3 different operators but with varying amplitudes in signal intensity (Figure 3B). These findings were paralleled by a dose-dependent decrease in numbers of lysosomal puncta in iMGL exposed to the highest doses if Bafilomycin A1 across operators (Figure 3C).
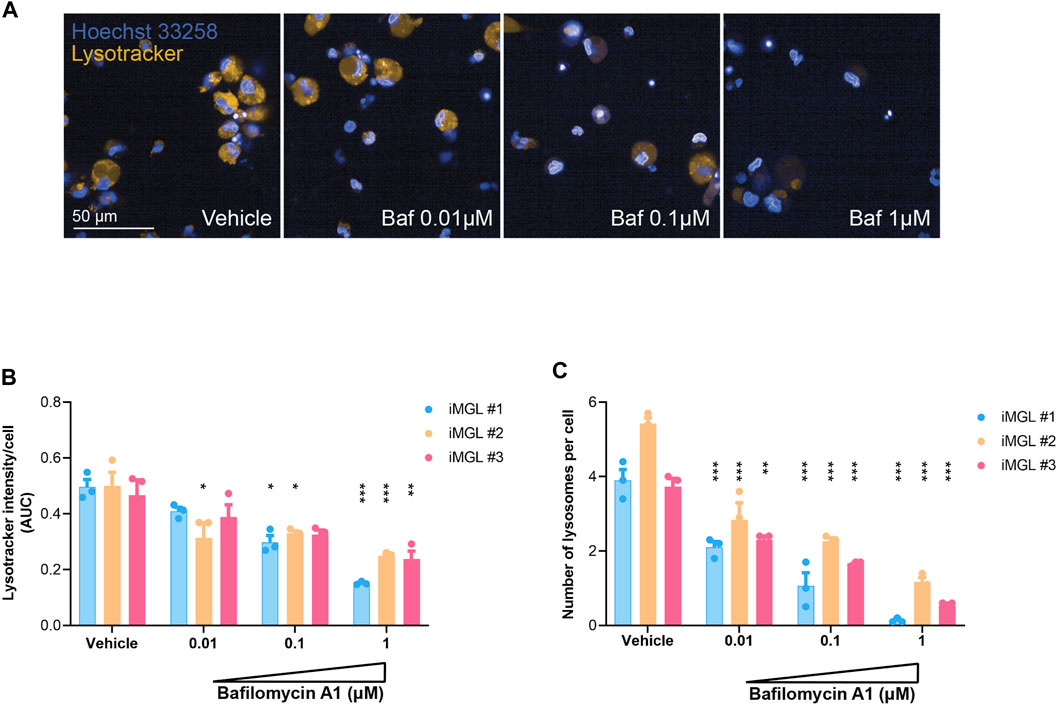
FIGURE 3. Lysosomal acidification assay. (A) Representative images of iMGL cultured in 384-well plates and exposed to LysoTracker™ dye in pH-neutral medium or post-treatment with incremental doses of Bafilomycin A1 (a V-ATPase inhibitor/acidification blocker). Hoechst 33342 was used to visualize nuclei in blue. (B) LysoTracker™ signal intensity in iMGL generated from three independent differentiation campaigns (three different operators). Cells were maintained in pH-neutral medium or exposed to incremental doses of Bafilomycin A1 and overall signal from high resolution/content imaging was normalized to cell numbers (nuclei). (C) Effect of incremental doses of Bafilomycin A1 on lysosomal biogenesis captured as number of LysoTracker™ positive puncta (lysosomes) normalized to numbers of iMGL (Hoechst nuclear stain). Data were generated from three technical replicates per condition. Data are shown as mean ± S.D. Data were analyzed using two-way ANOVA followed by Tukey’s post hoc test; *p < 0.05, **p < 0.01 and ***p < 0.001 (Bafilomycin compared to vehicle-treated groups compared).
Intravital microscopy studies have provided insights on the dynamic states of microglia under homeostatic and acute injury contexts (Hefendehl et al., 2014). Microglia are highly contextual sensors and dynamic responders to neuronal and environmental cues. These surveillance functions are enabled through cytoskeletal rearrangements that enable filopodial movement along the neuronal soma and dendrites for regulation of neuronal excitability. Migratory functions allow for microglial chemotaxis towards sites of injury for tissue debris clearance and trophic support (Salter and Stevens, 2017; Li and Barres, 2018). Since ability to migrate towards a chemotactic gradient is a critical microglial function with potential implications in human disease pathobiology, we established a trans-well migration assay towards chemotactic factors including MCP-1, ADP, and C5a (Figure 4A). Random migratory activity towards wells containing culture medium alone was set as a baseline. MCP-1 was selected as a negative control since microglia do not express C-C chemokine receptor 2 (CCR2). Time-lapse imaging of the top surface area of the porous membrane over 24 h showed a significant migratory activity towards C5a measured as disappearance of cells from the top well. This outcome was consistent across the iMGL batches generated by the three operators. Some migratory activity against the ADP gradient was observed for operators 1 and 2 but not for operator 3 (Figure 4B). iMGL showed differential responses to the various chemoattractants. C5a induced a rapid and robust chemotaxis, ADP mildly increased movement, and as expected since iMGLs (similarly to primary microglia) lack the monocyte marker CCR2, MCP-1 had no effect on cell migration (Figure 4C). These data show iMGL display migratory functions; however, choice of chemoattractant and signal window should be carefully considered as they are sensitive to batch-to-batch differentiation.
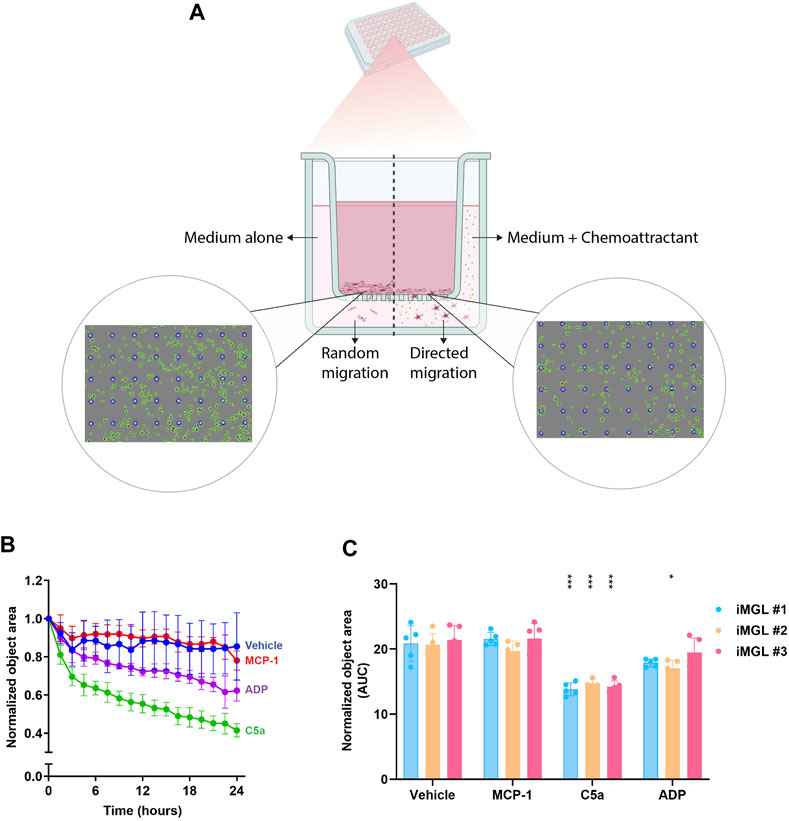
FIGURE 4. Migratory functions of iMGL. (A) A schematic depicting trans well chemotactic migration of iMGL seeded on optically clear porous membranes separating the top and bottom wells (containing either medium or chemotactic factors) of a 96-well chemotaxis plate. Representative images in the schematic, were captured in phase/contrast. A mask was applied to visualize cells (green) and pores (blue). Images show cells disappearing from the top well when exposed to cell culture medium (random undirected migration) or a chemoattractant (directed migration). (B) Timelapse data capturing iMGL chemotaxis over 24 h in presence of medium or chemoattractant including MCP-1 0.1 μg/mL, ADP 100 μM, and C5a 0.25 μg/mL. Values were normalized to number of cells on the top wells at 0 h. (C) Chemotactic activity in iMGLs from the same line differentiated by 3 independent operators. Data were generated through five technical replicates and presented as mean ± S.D. Analysis was performed using two-way ANOVA followed by Tukey’s post hoc test; *p < 0.05, and ***p < 0.001 (chemoattractant-exposed groups were compared to vehicle/medium controls).
One of the critical functions of microglia is immune surveillance and defense against CNS-invading pathogens. In the context of neurodegenerative proteinopathies misfolded proteins such as amyloid beta and synuclein activate similar pattern/pathogen recognition receptors such as toll-like receptors 2 (TLR2) and 4 (TLR4) through molecular mimicry (Liu et al., 2012, 2; Hughes and Minee, 2020, 4; Gorecki et al., 2021). Upon activation, microglia respond by increasing pro-inflammatory and chemotactic factors necessary for recruitment of local microglia or brain infiltrating/surveilling peripheral immune cells at the sites of injury (Salter and Stevens, 2017; Li and Barres, 2018). To establish optimal cytokine release assay conditions iMGL were exposed to increasing concentrations of PGN-BS (TLR2 agonist), LPS (TL4 agonist), or zymosan (TLR2 and TLR6 agonist). A significant dose-dependent cytokine release was observed across stimuli (Figures 5A–C). Stimulant doses with a maximal assay window were used in subsequent experiments to assess assay variability across three independent differentiations (operators). Data demonstrated that cells from all operators were responsive to inflammatory stimuli, with the line differentiated by operator 1 (iMGL#1) displaying higher cytokine levels compared to iMGL#2 and #3. At the highest concentration iMGL responded similarly to all three stimuli as shown by similar levels of the various pro-inflammatory factors (Figures 5D–F). These data overall demonstrate reproducible pro-inflammatory responses in iMGL generated by different operators.
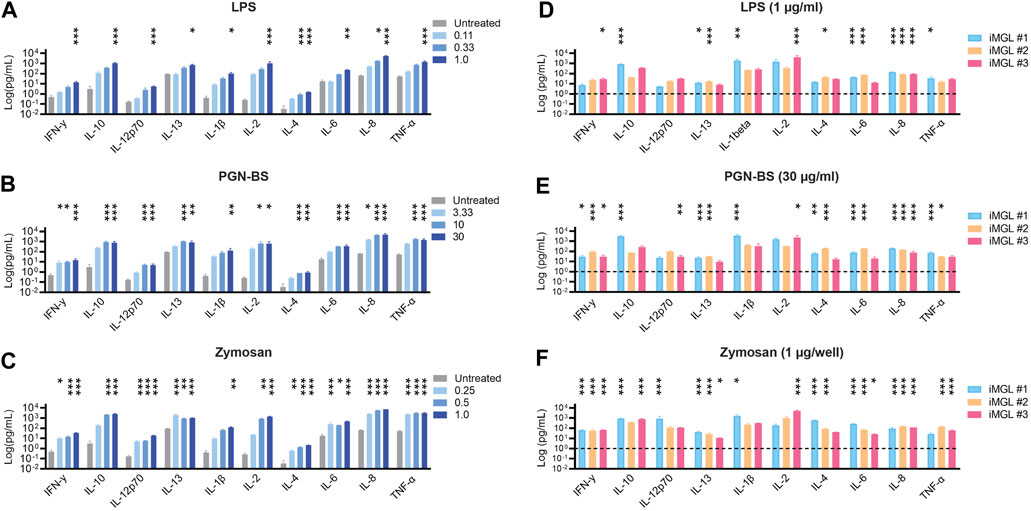
FIGURE 5. High-throughput cytokine release assay. (A) LPS (0.11–1 μg/mL), (B) PGN-BS (3.33–30 μg/mL) and (C) pHrodoRed™ zymosan (0.25–1 μg/well) dose-dependent induction of cytokine release by iMGL. iMGL from three independent differentiations/operators display similar levels of cytokine release in response to highest concentrations of (D) LPS (1 μg/mL) (E) PGN (30 μg/mL), and (F) pHrodoRed™ zymosan (1 μg/well). Data shown as mean ± S.D. were generated from three technical replicates. Analysis was performed using two-way ANOVA followed by Tukey’s post hoc test; ns. p > 0.05, *p < 0.05, **p < 0.01, ***p < 0.001 (stimulant-treated groups were compared to vehicle-treated controls).
In the recent years microglia have come into focus for drug discovery due to an increased understanding of their versatility in the maintenance of CNS homeostasis, and their genetic implication in the pathobiology of neurological disease. With technological advances in the field of iPSCs, it is now possible to reduce reliance solely on surgical/post-mortem human brain samples, or rodent models for investigation of mechanistic drivers of human disease pathobiology and validation of novel therapeutic approaches. However, when considering iPSC-based protocols, reproducibility among operators has not been addressed. Moreover, introduction of additional flexibility in cell generation and scale-up with negligible impact on reproducibility is critical, as multiple independent rounds of differentiations could introduce batch effects in functional screens.
Here, we present a scaled-up differentiation protocol for generating human microglia that behave consistently across differentiation batches/operators in high-throughput functional assays. By establishing optimal conditions for EB maintenance and harvesting in larger vessels, we were able to increase iHPC yields several folds while reducing culture maintenance effort and reagent costs. By introducing a cessation point at the microglial progenitor stage we were able to introduce flexibility in experimental planning and shorten time in culture reducing the risk of culture loss due to contamination.
Microglia generated from this protocol are distinct from human blood monocytes, expressing classical human microglial cell surface markers. Furthermore, they display a ramified morphology and are functionally active in phagocytosis, lysosomal clearance, chemotaxis, and cytokine release assays. These functional readouts were established in medium- and higher throughput formats to minimize cell and reagent use, while reducing well-to-well and over all experimental variability. These assays can be multiplexed with other cell types (i.e., neurons) or readouts such as cellular viability providing additional biological insights through monitoring of live cultures over time.
There are some challenges and considerations to our experimental set up that are worth noting. First, in this manuscript we report data generated on a single human iPSC line from a healthy donor, differentiated, and assessed functionally by three different operators. Due to genetic backgrounds or pathogenic mutations, iPSC lines vary in cellular health and differentiation times. We recommend thorough genetic characterization of iPSCs (especially of what are considered “healthy” lines) as well as an extended culture period of 10–18 days at the iHPC stage to evaluate differentiation rate and confirm the optimal yield times prior to any large scale differentiation campaigns. Secondly, although multiple harvests are possible with prolonged maintenance in culture of EBs, in our experience the yield and quality of iHPCs declines over time. Some minor differences in levels of iMGL activity in certain assays were observed in cells generated by different operators. Beyond minor differentiation-related variability, these findings can be explained by factors including insufficient assay window with a specific reagent/readout, differences in culture manipulation, or growth factor batch differences. Lastly, although monocultures of iPSC-derived microglia are being used routinely for target identification, validation, and drug discovery we need to interpret the output with caution. These cells are highly contextual surveyors of complex environmental cues, adjusting their functions dynamically based on intercellular crosstalk. Therefore, functional outcomes measured in mono-cultures may be divergent to those observed in more complex environments such as organoids or in vivo models. Although neuroimmunology drug discovery is now more translationally enabled through the access to iMGL from living patients, from a novel target validation or drug discovery perspective orthogonal testing in primary human cells, organoids, chimeric or transgenic mouse models may be necessary to derisk novel drug target ideas ahead of clinical trials.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Human iPSC lines and human PBMCs were obtained under informed consent. Cells were registered by the UCB biobank in accordance with the Belgian Federal law of 19 December 2008 (Doc 54 3262/005) on the Procurement and Use of Human Biological Material and the Belgian royal decree of January 9th, 2018, on Biobanks, and the Belgian Advisory Committee on Bioethics Opinion No. 45 of 19 January 2009 on human biological material banks intended for research.
MP: Data curation, Formal Analysis, Investigation, Methodology, Writing–original draft, Visualization. SB: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Project administration, Writing–review and editing. JV: Data curation, Formal Analysis, Methodology, Writing–original draft. JD: Methodology, Software, Writing–review and editing. GG: Data curation, Formal Analysis, Methodology, Visualization, Writing–original draft. PB and M-LC: Formal Analysis, Investigation, Methodology, Writing–review and editing. IK: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing–review and editing, Writing–original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors declare that this study was funded by a grant from the Walloon Region (Belgium)—DGO6—Convention 7790 from October 2019 and UCB Biopharma SRL. The funder (UCB) had the following involvement in the study: all authors were employees of UCB at the time of execution of the study. UCB was involved in study design, collection, analysis, interpretation of data, the writing of this article, and the decision to submit it for publication.
All workflow diagrams for iMGL generation and functional assay were generated in BioRender.
Authors MP, SB, JV, JD, GG, PB, M-LC, and IK were employed by UCB Biopharma SRL.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fddsv.2023.1289314/full#supplementary-material
Abud, E. M., Ramirez, R. N., Martinez, E. S., Healy, L. M., Nguyen, C. H. H., Newman, S. A., et al. (2017). IPSC-derived human microglia-like cells to study neurological diseases. Neuron 94 (2), 278–293. doi:10.1016/j.neuron.2017.03.042
Angelova, D. M., and Brown, D. R. (2019). Microglia and the aging brain: are senescent microglia the key to neurodegeneration? J. Neurochem. 151 (6), 676–688. doi:10.1111/jnc.14860
Banerjee, P., Paza, E., Perkins, E. M., James, O. G., Boyd, K., Lloyd, A. F., et al. (2020). Generation of pure monocultures of human microglia-like cells from induced pluripotent stem cells. Stem Cell Res. 49, 102046. doi:10.1016/j.scr.2020.102046
DiSabato, D., Quan, N., and Godbout, J. P. (2016). Neuroinflammation: the devil is in the details. J. Neurochem. 139 (2), 136–153. doi:10.1111/jnc.13607
Douvaras, P., Sun, B., Wang, M., Kruglikov, I., Lallos, G., Zimmer, M., et al. (2017). Directed differentiation of human pluripotent stem cells to microglia. Stem Cell Rep. 8 (6), 1516–1524. doi:10.1016/j.stemcr.2017.04.023
Ginhoux, F., Lim, S., Hoeffel, G., Low, D., and Huber, T. (2013). Origin and differentiation of microglia. Front. Cell. Neurosci. 7, 45. doi:10.3389/fncel.2013.00045
Gorecki, A. M., Anyaegbu, C. C., and Anderton, R. S. (2021). TLR2 and TLR4 in Parkinson’s disease pathogenesis: the environment takes a toll on the gut. Transl. Neurodegener. 10 (1), 47. doi:10.1186/s40035-021-00271-0
Hefendehl, J. K., Neher, J. J., Sühs, R. B., Kohsaka, S., Skodras, A., and Jucker, M. (2014). Homeostatic and injury-induced microglia behavior in the aging brain. Aging Cell 13 (1), 60–69. doi:10.1111/acel.12149
Hughes, C., Minee, L., Yi, J. H., Kim, S. C., Drews, A., George-Hyslop, P. S., et al. (2020). Beta amyloid aggregates induce sensitised TLR4 signalling causing long-term potentiation deficit and rat neuronal cell death. Commun. Biol. 3 (1), 79. doi:10.1038/s42003-020-0792-9
Li, Q., and Barres, B. A. (2018). Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 18 (4), 225–242. doi:10.1038/nri.2017.125
Liu, S., Liu, Y., Wenlin, H., Wolf, L., Kiliaan, A. J., Penke, B., et al. (2012). TLR2 is a primary receptor for Alzheimer’s amyloid β peptide to trigger neuroinflammatory activation. J. Immunol. 188 (3), 1098–1107. doi:10.4049/jimmunol.1101121
Maharjan, Y., Dutta, R. K., Son, J., Wei, X., Park, C., Kwon, H. M., et al. (2022). Intracellular cholesterol transport inhibition impairs autophagy flux by decreasing autophagosome-lysosome fusion. Cell Commun. Signal. CCS 20 (1), 189. doi:10.1186/s12964-022-00942-z
McQuade, A., and Blurton-Jones, M. (2021). Human induced pluripotent stem cell-derived microglia (HiPSC-Microglia). Methods Mol. Biol. 2454, 473–482. doi:10.1007/7651_2021_429
Pocock, J. M., and Piers, T. M. (2018). Modelling microglial function with induced pluripotent stem cells: an update. Nat. Rev. Neurosci. 19 (8), 445–452. doi:10.1038/s41583-018-0030-3
Pomilio, C., Gorojod, R. M., Riudavets, M., Vinuesa, A., Presa, J., Gregosa, A., et al. (2020). Microglial autophagy is impaired by prolonged exposure to β-amyloid peptides: evidence from experimental models and Alzheimer’s disease patients. GeroScience 42 (2), 613–632. doi:10.1007/s11357-020-00161-9
Root, J., Merino, P., Austin, N., Johnson, M., and Kukar, T. (2021). Lysosome dysfunction as a cause of neurodegenerative diseases: lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 154, 105360. doi:10.1016/j.nbd.2021.105360
Salter, M. W., and Stevens., B. (2017). Microglia emerge as central players in brain disease. Nat. Med. 23 (9), 1018–1027. doi:10.1038/nm.4397
Takahashi, K., Tanabe, K., Ohnuki, M., Narita, M., Ichisaka, T., Tomoda, K., et al. (2007). Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131 (5), 861–872. doi:10.1016/j.cell.2007.11.019
Thion, M. S., Ginhoux, F., and Garel., S. (2018). Microglia and early brain development: an intimate journey. Science 362 (6411), 185–189. doi:10.1126/science.aat0474
Xu, R., Li, X., Boreland, A. J., Anthony, P., Kwan, K., Hart, R. P., et al. (2020). Human IPSC-derived mature microglia retain their identity and functionally integrate in the chimeric mouse brain. Nat. Commun. 11 (1), 1577. doi:10.1038/s41467-020-15411-9

Keywords: iPSC-derived microglia, high-throughput functional screening, phagocytosis assay, lysosomal function, chemotaxis, cytokine release, drug discovery
Citation: Padmakumar M, Biesmans S, Valadas JS, Detrez JR, Gillet G, Bresler P, Clénet M-L and Kadiu I (2023) An improved method for large scale generation and high-throughput functional characterization of human iPSC-derived microglia. Front. Drug Discov. 3:1289314. doi: 10.3389/fddsv.2023.1289314
Received: 05 September 2023; Accepted: 11 October 2023;
Published: 30 October 2023.
Edited by:
Liang Xu, University of Kansas, United StatesReviewed by:
Emma Mead, University of Oxford, United KingdomCopyright © 2023 Padmakumar, Biesmans, Valadas, Detrez, Gillet, Bresler, Clénet and Kadiu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven Biesmans, c3RldmVuLmJpZXNtYW5zQHVjYi5jb20=; Irena Kadiu, aXJlbmEua2FkaXVAdWNiLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.