 Ana L. Chávez-Hernández
Ana L. Chávez-Hernández Edgar López-López
Edgar López-López José L. Medina-Franco
José L. Medina-Franco
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Drug Discov., 21 June 2023
Sec. In silico Methods and Artificial Intelligence for Drug Discovery
Volume 3 - 2023 | https://doi.org/10.3389/fddsv.2023.1222655
This article is part of the Research TopicDrug Discovery and Development Explained: Introductory Notes for the General PublicView all 11 articles
Chemical and biological data are the cornerstone of modern drug discovery programs. Finding qualitative yet better quantitative relationships between chemical structures and biological activity has been long pursued in medicinal chemistry and drug discovery. With the rapid increase and deployment of the predictive machine and deep learning methods, as well as the renewed interest in the de novo design of compound libraries to enlarge the medicinally relevant chemical space, the balance between quantity and quality of data are becoming a central point in the discussion of the type of data sets needed. Although there is a general notion that the more data, the better, it is also true that its quality is crucial despite the size of the data itself. Furthermore, the active versus inactive compounds ratio balance is also a major consideration. This review discusses the most common public data sets currently used as benchmarks to develop predictive and classification models used in de novo design. We point out the need to continue disclosing inactive compounds and negative data in peer-reviewed publications and public repositories and promote the balance between the positive (Yang) and negative (Yin) bioactivity data. We emphasize the importance of reconsidering drug discovery initiatives regarding both the utilization and classification of data.
Data and the increasing role of predictive models, including machine and deep learning (Mouchlis et al., 2021; Bajorath et al., 2022), are the cornerstone of modern drug discovery programs (Zhang et al., 2022). The increasing use of computational methods that recently included deep learning is reducing the time and financial costs of finding drug candidates (Zhang et al., 2022). For instance, computer-aided drug design (CADD) has led to the discovery of more than seventy approved drugs (Sabe et al., 2021) including remdesivir as an emergency treatment against SARS-CoV-2 in 2021 (Dos Santos Nascimento et al., 2021).
CADD methods are typically divided into two main categories, structure-based drug design (SBDD) and ligand-based drug design (LBDD) that rely on the three-dimensional (3D) structure data available for one or more molecular targets, or the structure-activity data of ligands, respectively. Examples of deep learning applications in SBDD include AlphaFold to assist in homology modeling, and DiffDock in molecular docking. AlphaFold predicts 3D protein structures according to their amino acid sequences (Jumper et al., 2021), and DiffDock predicts the binding mode between the ligand and specific protein target (Corso et al., 2022). One of the most notable approaches in LBDD are quantitative structure-activity relationships (QSAR) (Dos Santos Nascimento et al., 2021). Current QSAR methods use machine learning and deep learning (Soares et al., 2022) that can be divided into linear methods and nonlinear methods (Patel et al., 2014; Greener et al., 2022). Linear methods include linear regression, multiple linear regression, partial least squares, and principal component analysis (Patel et al., 2014). Nonlinear methods include artificial neural networks, k-nearest neighbors, and Bayesian neural nets, to name a few examples (Patel et al., 2014; Greener et al., 2022).
Advances in deep learning models have a significant progress in molecule generation, representing a big step forward in bridging the gap between chemical entities and drug-like properties (Krishnan et al., 2021). Deep learning algorithms are currently used in the renewed interest in the de novo design of chemical libraries. In 2020, the successful application of deep learning in drug discovery, that included the de novo design using deep learning, was selected by the Massachusetts Institute of Technology Technology Review as one of the top ten breakthrough technologies (Juskalian et al., 2023).
De novo design is aimed at generating new chemical entities (NCE) with desired properties (Palazzesi and Pozzan, 2022). De novo design based on deep learning algorithms (Palazzesi and Pozzan, 2022) requires a large number of compounds that may demand significant computational resources. However, bioactivity data for a biological endpoint is not always sufficient. The lack of data has led to the development of new methods for compound selection and applications for deep learning algorithms are being developed (Guo M et al., 2021).
Knowledge-based drug design frequently involves quality data (Perron et al., 2022b) to develop models with useful predictions (Schneider et al., 2020). To this end, rethinking the methodologies used for drug discovery and development campaigns is crucial. The quality of data sets, decoy data sets and inactive compounds used in predictive models, and de novo design models need to be reviewed and discussed.
The main purpose of this manuscript is discussing the importance of quality data, decoy data sets, and the balance needed between inactive (i.e., “Yin”) and active (“Yang”) compounds currently employed in de novo design and developing predictive models of biological activity to generate NCE. Following up on previous studies (Schneider et al., 2020; Bajorath et al., 2022; Cherkasov, 2023), we comment on the need to rethink the way to drug design and develop campaigns. The manuscript is organized into four main sections. After this Introduction, Section 2 presents an overview of de novo design. Section 3 discusses the main public data sources used to develop predictive models. Section 4 discusses criteria to generate quality data sets. The last section presents a summary of conclusions and perspectives.
De novo design aims to generate new chemical structures from scratch with desired predicted properties, e.g., absorption, distribution, metabolism, excretion, toxicity (ADMET), other drug-likeness properties, and biological activities (Palazzesi and Pozzan, 2022). The two main strategies for de novo design can be classified into SBDD and LBDD (vide supra) (Zhang et al., 2022). A recent example of a structured-based de novo design is the RELATION model that learns from the desired geometric features of protein-ligand complexes to generate new molecules (Wang et al., 2022). The generation process applies a fragment-based strategy given an initial chemical scaffold embedded in the binding site of the target protein. The pre-trained model generates molecules iteratively by sequentially adding, deleting, inserting, or replacing and linking fragments (Zhang et al., 2022).
In contrast, ligand-oriented de novo design focuses on the ligands themselves, thereby generating compounds with new chemical structures with novel scaffolds from active compounds while optimizing the desired properties (Xie et al., 2022). A general workflow is schematically summarized in Figure 1 which has seven main steps (Krishnan et al., 2021; Zhang et al., 2022): 1) Selecting compound data sets from public or in-house sources (further discussed in Section 3); 2) Filtering molecular data sets with desired properties such as drug-likeness. In the example of Figure 1 a data set with three subsets of compounds is represented with a star, triangle, and circle, respectively. The compounds represented with a star have drug-like properties (Lipinski et al., 2001; Veber et al., 2002); those represented with triangles comply with some of the drug-likeness properties, and those represented with circles are not compliant. Other approaches to select compounds from the data sets use molecular fingerprints (Kadurin et al., 2017) or filter compounds directly via similarity-based virtual screening instead of designing NCE from scratch (Tong et al., 2021). 3) Selecting the molecular representation as a basis to learn and represent the structures and properties of molecules, e.g., SMILES (Weininger, 1988), SELFIES (Krenn et al., 2020) or molecular graphs (Simonovsky and Komodakis, 2018). 4) Developing and validating the model for molecule generation using metrics such as the operating characteristic curve. 5) Optimizing the model by combining reinforcement learning and property prediction (Olivecrona et al., 2017). 6) Generating molecules de novo, 7) Assessing the biological activity of the compounds designed in relevant in vitro or in vivo models.
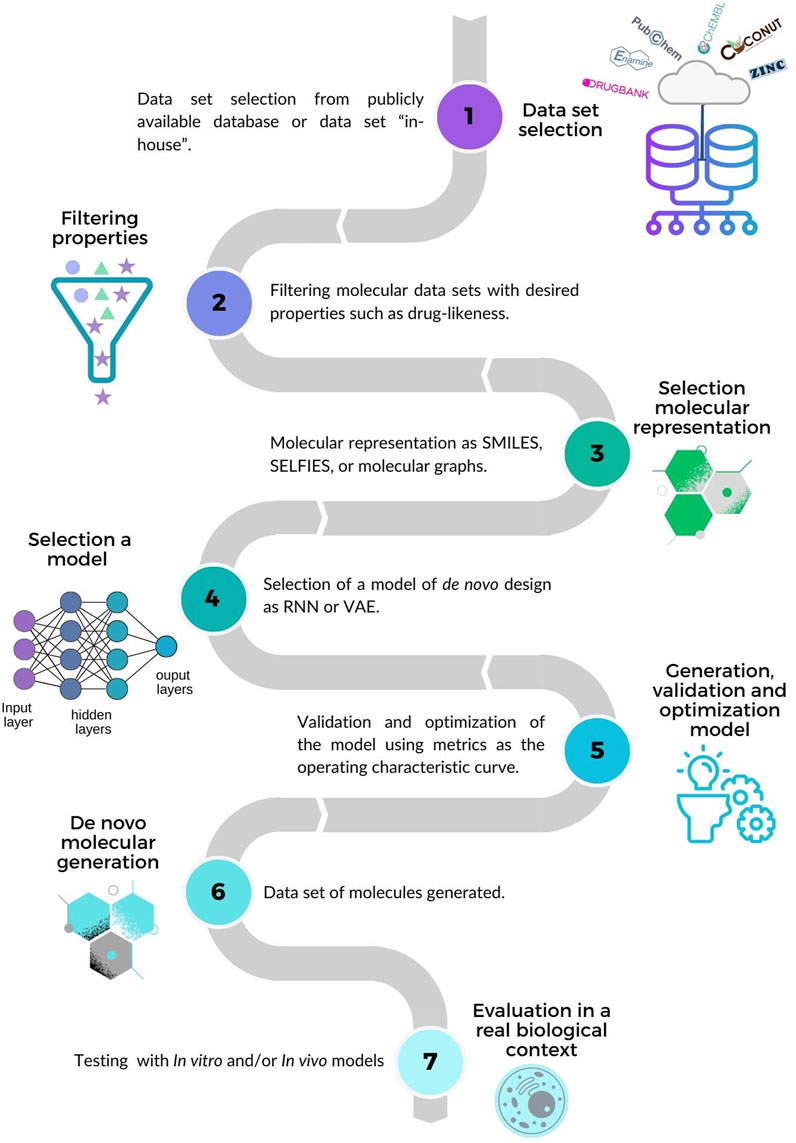
FIGURE 1. Overview of ligand-based de novo design. 1) Selecting data sets. 2) Filtering molecular data sets with desired properties such as drug-likeness. In this example, compounds represented with stars comply with drug-likeness properties (Lipinski et al., 2001; Veber et al., 2002). 3) Choosing a molecular representation. 4) Selecting a de novo design model. 5) Developing, validating and optimizing the model. 6) Generating molecules de novo. 7) Testing the compounds in a relevant biological experiment.
Deep learning, currently used in ligand-based de novo design, learns the probability distribution of molecular data and generates continuous or discrete latent representations for molecules with property optimization (Gómez-Bombarelli et al., 2018). The algorithms map the learned probability distribution and molecule representation into novel molecules while optimizing molecular properties (Bilodeau et al., 2022) through the tuning of hyperparameters (Perron et al., 2022a; Bender et al., 2022). Advances in deep learning are significantly advancing molecule generation, representing a big step forward in bridging the gap between chemical entities and drug-like properties (Krishnan et al., 2021).
Ligand´s properties can be optimized in two steps: 1) property-based generation, wherein models would learn the chemical space of molecules with desirable properties; and 2) novel molecules are generated within a desired property space (Bilodeau et al., 2022). Examples of ligand-based de novo design are deep neural networks (DNN), recurrent neural networks (RNNs) (Olivecrona et al., 2017), and variational autoencoders (VAE) (Gómez-Bombarelli et al., 2018). Olivercroma et al. (Olivecrona et al., 2017) proposed the REIVENT model that uses RNN for de novo design. They introduced a reinforcement learning method to fine-tune the pre-trained RNN so the model could generate structures with desirable properties. Recently, Blaschke et al. released REINVENT 2.0 (Blaschke et al., 2020) making the code freely accessible in Github.
Ligand-based de novo design using DNN (Palazzesi and Pozzan, 2022) requires a large number of compounds that demand more computational resources. The DNN architecture is prone to problems because of fitting numerous parameters. For this reason, a large training data set is needed to reduce the risk of overfitting. However, sufficient bioactivity data for a biological endpoint is not always available (Wu et al., 2018). The lack of sufficient data has led to using methods for compound selection or the development of new methods for compound selection. Altae-Tran et al. (Altae-Tran et al., 2017) demonstrated how the one-shot learning paradigm can be used to address the overfitting problem; they used DNN to transform small molecules into embedding vectors in a continuous feature space whose similarity measures are then iteratively learned. They showed that this DNN architecture offers convincing performance in many activity prediction tasks given limited amounts of training. On the other hand, computer scientists advise using algorithms that can detect meaningful patterns in small data sets, which is a typical case in the early stage of drug discovery (Schneider and Clark, 2019). For instance, an initial approach to de novo design is to start from small data sets of compounds with diverse structures and diverse properties of pharmaceutical relevance (Chávez-Hernández and Medina-Franco, 2023).
The availability of gold standard datasets as well as independently generated data sets are valuable in generating well-performing models (Vamathevan et al., 2019). Dissimilarity-based compound selection could be improved if one focused the selection on a structural diverse dataset (for instance derived from natural products). Some approaches proposed suggest using quality data sets using a dissimilarity-based compound selection method such as the MaxMin or MaxSum algorithms (Leach and Gilleteds, 2007). Recently, we reported the use of the MaxMin algorithm for the selection of natural product subsets (Chávez-Hernández and Medina-Franco, 2023) using the Universal Natural Product Database (UNPD) (Gu et al., 2013). In that study, the natural product subsets generated had the most diverse chemical structures with physicochemical properties of pharmaceutical interest similar to the original data set. Chemical structures in the natural product subsets were represented with SMILES encoding chirality, an important feature of natural products.
The first step in de novo design is to select, from the vast chemical space, the appropriate subset of all possible molecules for a desired biological activity (Schneider et al., 2000). To have an idea, the size of the chemical space has been estimated at around 1060 small molecules and between 1020–1024 for all molecules up to 30 atoms that comply with Lipinski’s rule-of-five (Reymond, 2015). According to Yang et al. compound data sets can be classified into on-demand databases, collections containing bioactivity data, compounds databases commercially available, and natural products databases (Yang et al., 2019). Herein, we include benchmark, decoy and inactive compounds data sets as others categories as illustrated in Figure 2. In this figure, on-demand databases are further divided into commercially available (e.g., Enamine-REAL, CHEMriya and Freedom Space) (Chemspace, 2023) and in-house (e.g., Pfizer and AstraZeneca). The figure shows examples of compound databases in other categories which are discussed in the remainder of this section.

FIGURE 2. Classification of compound databases and representative examples of each one. For the discussion of this manuscript, databases are split into six main categories: on-demand, commercial availability, bioactivity, natural products, benchmark and decoys.
Among the different types of chemical databases, de novo design employs libraries from different categories outlined in Figure 2. Specific examples are ChEMBL (Davies et al., 2015; Mendez et al., 2019), PubChem (Kim et al., 2023), DrugBank (Wishart et al., 2006; Wishart et al., 2008; Wishart et al., 2018), Enamine´s REadily AccessibLe (REAL) (Enamine, 2023), CHEMriya (CHEMriya, 2023), Freedom Space (Chemspace, 2023), ZINC-22 (Tingle et al., 2023), and MoleculeNet (Wu et al., 2018) which more details for each one are provided in Table 1 and further commented in the next sections.
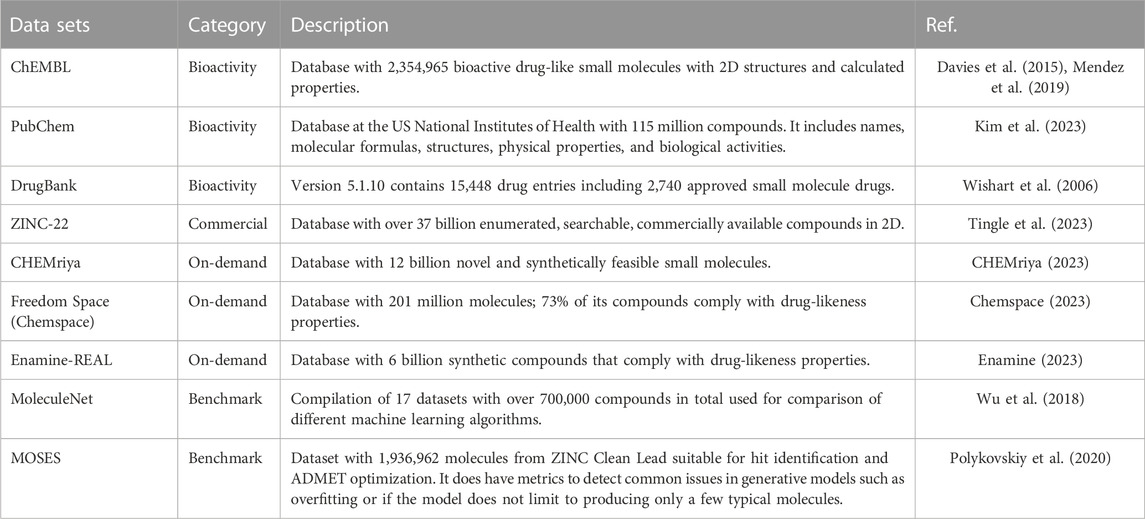
TABLE 1. Main sources of public molecular data sets used in de novo design.
Early approaches to ligand-based de novo design involved fragment compounds into unique building blocks which could be recombined to make new molecules. A number of commercial suppliers of chemical samples offer large make-on-demand collections that can be reliably synthesized because the building blocks are available as well as the synthetic routes and methods (Warr et al., 2022; Korn et al., 2023). There are also large collections of fragments or building blocks commercially available. Examples of on-demand compound databases and suppliers are REAL (Enamine) (Enamine, 2023), CHEMriya (OTAVA) (CHEMriya, 2023), and Freedom Space (Chemspace) (Chemspace, 2023) (Table 1). REAL database (Enamine, 2023) comprises over 6 billion molecules that comply with the traditional drug-likeness criteria. CHEMriya (CHEMriya, 2023) contains 12 billion novel and synthetically feasible small molecules whose molecules are not explicitly listed in the public domain. Freedom Space (Chemspace, 2023) contains 201 million molecules and 73% of its compounds are drug-like (as assessed with the “rule of five”). Examples of on-demand in-house databases from the pharmaceutical industry are 1015 compounds of AZ Space (AstraZeneca) (Grebner, 2022), 1019 compounds of JFS (Johnson & Johnson) (Warr, 2021), 1018 compounds of PGVL (Pfizer) (Hu et al., 2012), 1017 compounds BICLAIM (Boehringer Ingelheim) (Korn et al., 2023), and 1020 compounds MASSIV (Merck/EMD) (Korn et al., 2023).
One of the largest and long-standing compendiums of commercially available compounds in ZINC. The most recent version, ZINC-22 (Tingle et al., 2023) contains over 37 billion enumerated, searchable, commercially available compounds in 2D. Over 4.5 billion have been built in biologically relevant ready-to-dock 3D formats (Tingle et al., 2023). Some examples of de novo design using ZINC include the design of inhibitors of DDR1 (discoidin domain receptor 1, a kinase target implicated in fibrosis and other diseases) (Zhavoronkov et al., 2019) and compounds with activity towards the dopamine receptor D2 (Liu et al., 2019; Maziarka et al., 2020).
De novo design based on deep learning algorithms frequently use PubChem, ChEMBL, and DrugBank to select subsets of compounds focused on a biological target or biological endpoint as the design of ligands (Li et al., 2018; Li et al., 2022; Liu et al., 2019). PubChem (Kim et al., 2023) is a freely accessible database from the US National Institutes of Health (NIH) with over 115 million compounds. At the time of writing, the most recent version release of ChEMBL is 32 (Davies et al., 2015; Mendez et al., 2019) and contains 2,354,965 compounds bioactive drug-like small molecules with 2D structures and calculated properties. DrugBank (Wishart et al., 2006; Wishart et al., 2008; Wishart et al., 2018) version 5.1.10 (released 2023-01-04) contains 15,448 drug entries including 2,740 approved small molecule drugs, 1,577 approved biologics (proteins, peptides, vaccines, and allergens), 134 nutraceuticals and over 6,717 experimental (discovery-phase) drugs. Some applications include the de novo design of SARS-CoV-2 Mpro inhibitors (Li et al., 2022), the design of ligands against the adenosine receptor (A2AR) (Liu et al., 2019), and the generation of compounds analogs to celecoxib (used to manage symptoms of various types of arthritis pain and reduce precancerous polyps in the colon) (Li et al., 2018; DRUGBANK, 2023).
Natural product databases (Gómez-García and Medina-Franco, 2022; Saldívar-González et al., 2022) are important in drug discovery. From drugs approved by 2020 about 23% are natural products or derivatives (Newman and Cragg, 2020). Natural products have a diversity of privileged scaffolds (Atanasov et al., 2021; Grigalunas et al., 2022) and molecular fragments (Chávez-Hernández et al., 2020a; Chávez-Hernández et al., 2020b) that depend on the particular source (Medina-Franco et al., 2022b); a diversity of chiral centers; and a larger fraction of sp3 carbon atoms and functional groups (Atanasov et al., 2021; Grigalunas et al., 2022).
Privileged structures were defined by Evans et al. (Evans et al., 1988) as chemical structures capable of providing useful ligands for more than one receptor judicious modification of such structures could be a viable alternative in the search for new receptor agonists and antagonists. Schneider and Schneider (2017) define a privileged structure as a chemical structure that may be considered to possess geometries suitable for decoration with side chains, such that the resulting products bind to different target proteins or a ligand that potently interacts with one (selective binder) or many target receptors (promiscuous binder). To this end, natural products are used in the development of pseudo-natural products, compounds that are generated through a de novo combination of natural product fragments, allowing the exploration of uncharted areas of biologically relevant chemical space that are different from the chemical space covered by the compounds from which they are derived (Grigalunas et al., 2022).
Representative natural product datasets that can be used in de novo design are Collection of Open NatUral ProdUcTs (COCONUT) (Sorokina et al., 2021), SuperNatural 3.0 (Gallo et al., 2023), UNPD (Gu et al., 2013), NuBBEDB (Pilon et al., 2017; Saldívar-González et al., 2019), SistematX (Scotti et al., 2018; Costa et al., 2021), CIFPMA (Olmedo et al., 2017; Olmedo and Medina-Franco, 2020), PeruNPDB (Barazorda-Ccahuana et al., 2023), BIOFACQUIM (Pilón-Jiménez et al., 2019; Sánchez-Cruz et al., 2019), UNIIQUIM(UNIIQUIM, 2015), and are summarized in Table 2.
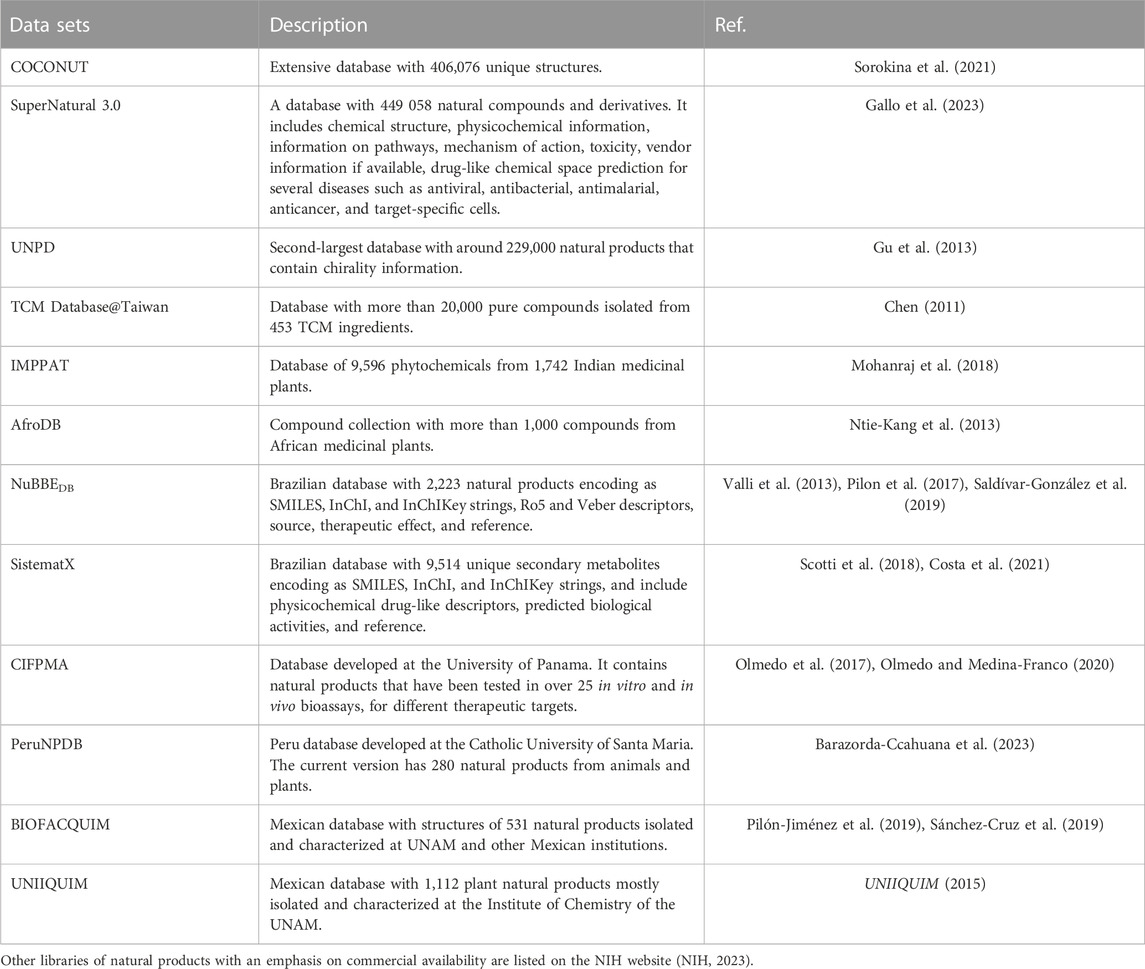
TABLE 2. Examples of natural product databases in the public domain.
SuperNatural 3.0, COCONUT and UNPD are the most extensive natural product databases. SuperNatural 3.0 (Gallo et al., 2023) is arguably the most extensive natural product database with 449,058 natural compounds and derivatives; followed by COCONUT (Sorokina et al., 2021) with 406,076 unique structures (no encoding stereochemistry) and UNPD (Gu et al., 2013) with 197,201 natural products that contain chirality information.
Several public natural products databases compile the compounds isolated and characterized from a geographical region or the country of origin as China, India and Africa. For instance, Chinese Traditional Medicine (TCM) Database@Taiwan (Chen, 2011) is a non-commercial TCM database with more than 20,000 pure compounds isolated from 453 TCM ingredients; A curated database of Indian Medicinal Plants, Phytochemistry And Therapeutics (IMPPAT) (Mohanraj et al., 2018) is a manually curated database of 9,596 phytochemicals from 1,742 Indian medicinal plants; and AfroDB (Ntie-Kang et al., 2013) with more than 1,000 small and structural diversity compounds from African medicinal plants.
Representative Latin American databases (Gómez-García and Medina-Franco, 2022) are NuBBEDB (Pilon et al., 2017; Saldívar-González et al., 2019), SistematX (Scotti et al., 2018; Costa et al., 2021) from Brazil; CIFPMA (Olmedo et al., 2017; Olmedo and Medina-Franco, 2020) from Panama; PeruNPDB (Barazorda-Ccahuana et al., 2023) from Peru; BIOFACQUIM (Pilón-Jiménez et al., 2019; Sánchez-Cruz et al., 2019) and UNIIQUIM (UNIIQUIM, 2015) from Mexico. The current version of NuBBEDB (Pilon et al., 2017; Saldívar-González et al., 2019) contains 2,223 natural products encoding as linear notations as SMILES. SistematX (Scotti et al., 2018; Costa et al., 2021) has 9,514 unique secondary metabolites arising from 20,934 botanical occurrences across five families. Other natural product collections from Latin America are CIFPMA, the Natural Products Database from the University of Panama, Republic of Panama (Olmedo et al., 2017; Olmedo and Medina-Franco, 2020)with 354 compounds. CIFPMA molecules have the potential to show target selectivity in biochemical assays and are useful molecules to identify reference compounds for virtual screening campaigns (Olmedo et al., 2017; Olmedo and Medina-Franco, 2020). The first version of the Peruvian Natural Products Database (PeruNPDB) had 280 natural products isolated from plants and animal sources (Barazorda-Ccahuana et al., 2023). BIOFACQUIM (Pilón-Jiménez et al., 2019; Sánchez-Cruz et al., 2019) contains 531 natural products isolated and characterized at the School of Chemistry of the National Autonomous University of Mexico (UNAM) and other Mexican institutions. UNIIQUIM (UNIIQUIM, 2015) with 1,112 plant natural products mostly isolated and characterized at the Institute of Chemistry of the UNAM.
The development of reliable machine learning algorithms has been limited due to the lack of standard benchmark datasets to compare the efficacy of the methods proposed (Jain and Nicholls, 2008). Furthermore, machine learning in chemistry compared with other areas such as computer speech and vision has a main disadvantage, the data recovery (Wu et al., 2018; Guo et al., 2022), because of measuring chemical properties often requires specialized instruments; as a result, datasets with experimentally determined results are small and often not sufficiently large to cover the high-demanding needs of machine-learning tasks (Wu et al., 2018). Another challenge is data splitting (the way in which datasets are split into training data and testing data). Some are random selection and rational selection. The former is randomly extracting a compound’s fraction from the data set. In contrast to rational selection, training and testing are selected from the same clusters of compounds. Random selection is common in machine learning but is often not correct for chemical data (Sheridan, 2013). In response to these challenges, standard benchmark data sets are being developed to evaluate de novo design protocols [(Wu et al., 2018; Brown et al., 2019; Polykovskiy et al., 2020). One example is MoleculeNet (Wu et al., 2018), a large-scale data set built upon multiple public databases. MoleculeNet is organized into regression and classification datasets and has over 700,000 compounds tested on a range of different properties subdivided into four categories (quantum mechanics, physical chemistry, biophysics, and physiology). Another example is the Molecular Sets (MOSES) (Polykovskiy et al., 2020) that contains 1,936,962 molecules (split into training, testing and scaffold datasets) and a set of metrics to evaluate the quality and diversity of generated structures. Metrics detect common issues in generative models such as overfitting or if the de novo design model just generates fairly common (not novel) structures (Brown et al., 2019; Polykovskiy et al., 2020). The developers of MOSES implemented and compared several molecular generation models and suggested using the results as reference points for further advancements in generative chemistry research.
Accuracy of predictive models depends on data quality and quantity. Also, the balance between active and inactive compounds is important, which remains an issue to resolve. Historically, the publication of active compounds in a given assay or with a particular endpoint has been prioritized over inactive molecules. For example, a recent comprehensive analysis of published screening bioactivity data shows that in ChEMBL V.29 (release in 2022) there is a large number of active compounds (ca. 71%) with respect to the inactive ones (ca. 31%); contrary to what it would be expected (López-López et al., 2022). These results highlight the relevance of changing the mindset about the importance and utility of inactive or negative data (keeping in mind that the definition of “inactive” is subjective as it depends on the particular biological assay and the predefined threshold to deem a compound inactive).
Decoy data sets have been developed in an attempt to reduce the gap between inactive (or negative) and active compounds. Decoy molecules are assumed non-active but have high physicochemical property similarity (but not topologically) to reference compounds (Réau et al., 2018). Decoys are useful to evaluate benchmark models that were assembled in the absence of inactive compounds experimentally measured (Irwin, 2008) and can be used to enrich de novo design models. Table 3 summarizes examples of large databases of experimentally tested active or inactive compounds, decoy datasets, and tools to generate decoys for specific projects.
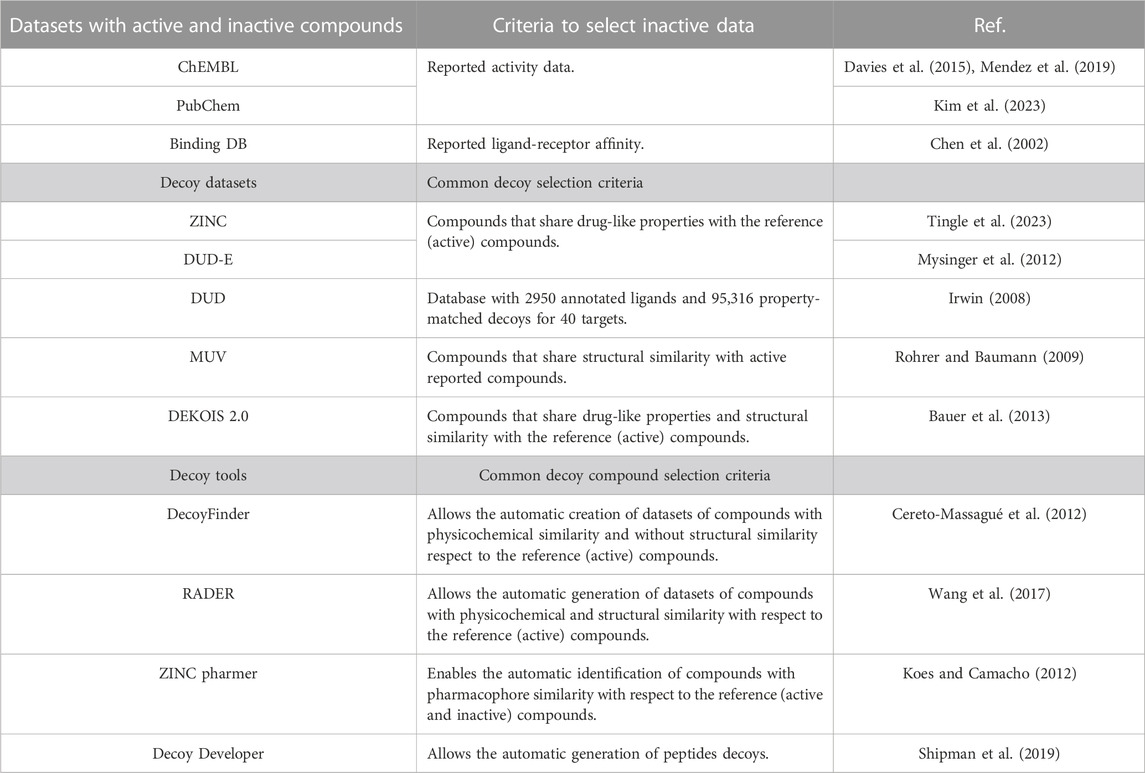
TABLE 3. Examples of potential inactive and decoy resources for enriching de novo design models.
Decoy compounds have been used to describe, explore, and expand the knowledge of active molecules. For example, rationalizing the physicochemical, chemical, biological, and clinical data of active compounds (López-López et al., 2021a). Recently, decoys can be employed in several de novo protocols based on ligand or structure as summarized in Table 4.
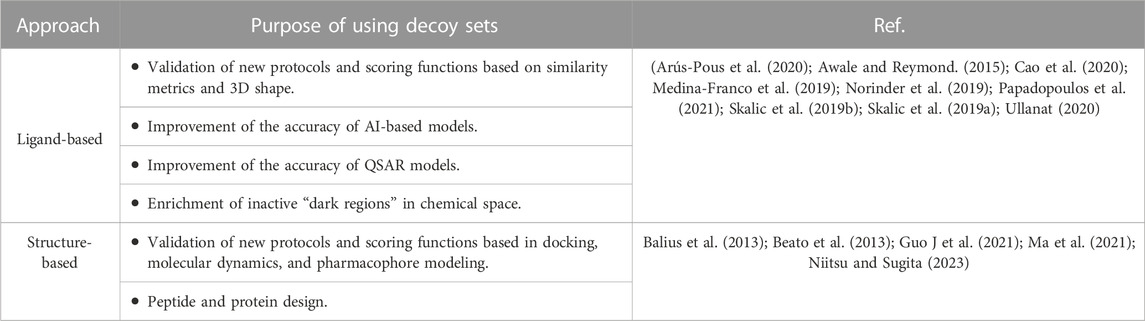
TABLE 4. Examples of applications of decoys in de novo design.
The quality of a data set is multifaceted. Commonly, it is associated with the experimental reproducibility of each data point and the experimental similarities between the protocols used to derive such data. Another important aspect of data quality is the balance between active and inactive compound. The latter is specially a challenge in public data sets due to the overall lack of published negative data. Finding qualitative yet better quantitative relationships between chemical structures and biological activity has been long pursued in medicinal chemistry and drug discovery. With the rapid increase and deployment of the predictive machine and deep learning methods, as well as the increased interest in the de novo design of chemical libraries (Mouchlis et al., 2021), the quantity and quality of data are becoming a central point in the discussion of the type of data sets needed (Schneider et al., 2020). While the more data (Cherkasov, 2023), the better, it is also true that the quality of the data available (that might not be quite large) is also crucial. Furthermore, the balance between active and inactive compounds is also a major consideration (López-López et al., 2022). Table 5 summarizes criteria for generating quality data sets. The list is not exhaustive but covers what the authors consider key points based on experience and what has been discussed extensively in the literature. Each point is supported by the references indicated in the table and further commented in the next subsections.
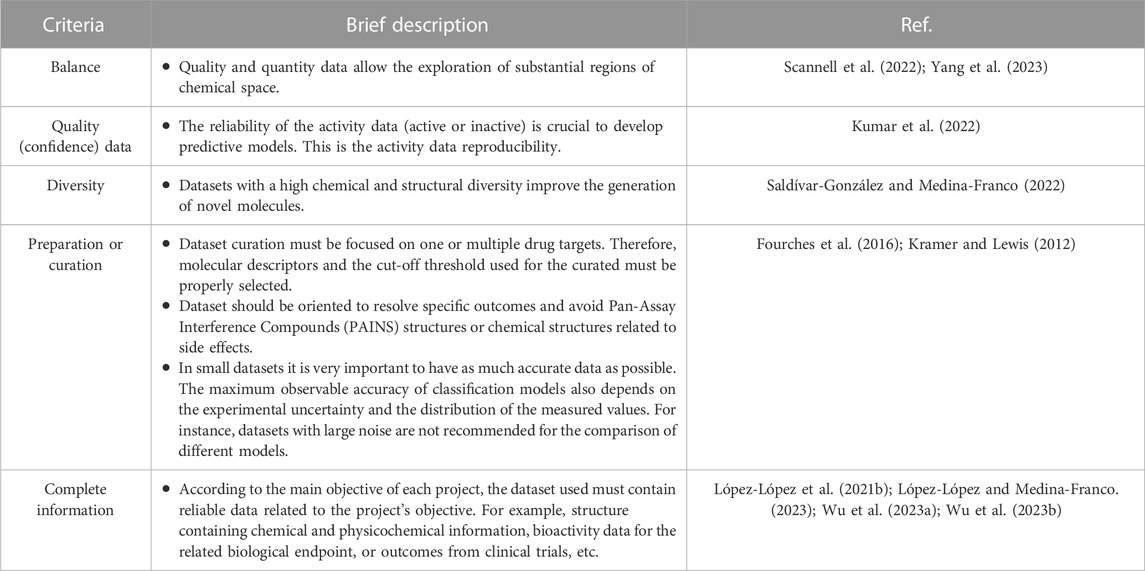
TABLE 5. Overview of suggested general criteria to generate quality datasets useful in de novo design.
As discussed previously, several current data sets in the public domain are unbalanced due to the infrequent practice of reporting inactive compounds and negative data in general. Historically, the negative and inactive data of preclinical compounds has been ignored by most journals that favor the publication of most active compounds and positive results (Medina-Franco and López-López, 2022). However, inactive and negative data are essential in drug design and development. For example, the analysis of high-quality inactive and negative data improves clinical success rate, reduces costs associated with drug development, and reduces the side effects rates (Hayes and Hunter, 2012; López-López and Medina-Franco, 2023). Moreover, data mining and AI approaches are largely benefitted from inactive compounds (Yu, 2021; López-López et al., 2022). The use of inactive and negative data allows real data augmentation to develop AI models, improve their accuracy, and reduce the rate of false-positive cases (Korkmaz, 2020; IBM, 2022). Also, the inactive and negative data facilitates the generation of QSPRs models that allows the rationalization of basically any property (Kramer and Lewis, 2012; Norinder et al., 2019).
An unwritten rule on AI and computational projects in general is "garbage in, garbage out". This perspective has direct implications in drug design (Bajorath et al., 2022). Recent studies have demonstrated that the use of quality data allows generating of AI models with higher accuracy than the AI models generated from larger datasets but with low-quality.
In general, a compound dataset with a large or broad applicability domain, as captured by the diversity of the contents, can give rise to predictive models with a large coverage. This is, molecules from diverse chemical structures could be conveniently interpolated in those models. As a comparison in an experimental setting, high-throughput screening of chemical diverse libraries increases the chances to find hit compounds for targets for which no hit compounds have been previously identified.
Due to the rapid expansion of the chemical universe, recently called the ‘Big Bang’ of the chemical universe (Cherkasov, 2023) it is relatively easy to have access to large and diverse regions of the chemical space. However, a practical challenge is to manage such large compound data sets computationally while developing and testing new models. A similar practical problem emerged when combinatorial chemistry was at its peak: it was challenging to design rationally novel large and diverse combinatorial libraries. To tackle this problem numerous diversity selection algorithms have been developed (Leach and Gillet, 2007). We recently applied a dissimilarity-based compound selection method to obtain three diverse subsets of natural products (with 14,994, 7,497, and 4,998 compounds, respectively) from the UNP. The subsets, that are freely available, can be readily used for the novo design applications and as benchmarks for similarity/diversity analysis (Chávez-Hernández and Medina-Franco, 2023).
A general curation protocol used on drug discovery datasets is to eliminate duplicate structures, canonize their SMILES representation, eliminate salts, and metals. However, according to the main goal of the de novo design model, additional steps to prepare a dataset could be taking into account, for example: 1) eliminating compounds with structural PAINS to reduce the rate of false-positive compounds prediction; 2) deleting compounds reported with side effects and/or ADMET deficiencies, to prioritize the generation of safe and optimization compounds.; or 3) making sure to keep in the dataset compounds with high activity confidence to improve the quality of predicted outputs. This list must be adapted according to the main goal of the de novo design model. It is also noted the need to develop robust and consistent protocols that take into scout metal-containing compounds as they have a major role in medicinal inorganic chemistry (Medina-Franco et al., 2022a).
Chemical structures should contain the required or relevant information for the goals of the study. For instance, compounds should be annotated with stereochemistry information if the 3D structure and conformation is critical; electronic density and quantum chemical data if the reactivity is key point to predict; the type of the biological activity data such as biochemical, cell-based or functional assays; drug-drug interaction data, pharmacogenomics, or post-marketing annotations; should be aligned with the type of outcome to be predicted and later validated experimentally.
One of the major perspectives of the de novo design is using balanced data sets (as much as experimental data is available) to build reliable models. Similar to QSAR predictive models, it is also crucial the validation of de novo protocols using standard and well-curated benchmark datasets (discussed in Section 3.6). With the increasing data availability to generate and train new models, it is becoming increasingly easy to explore regions of chemical space previously uncharted and continue contributing to the so-called “big bang” expansion of the chemical space. A major perspective in this direction is to explore biologically relevant compounds but outside the traditional small molecule chemical space (Medina-Franco et al., 2014). For instance, exploring metallodrugs (Medina-Franco et al., 2022a), macrocycles (Liang et al., 2022), peptides, or the combination of commonly explored chemical spaces, e.g., pseudo-natural products (discussed in Section 3.5).
Among the main types of datasets used in the novo design are on-demand collections, compounds annotated with biological activity, commercially available libraries, and natural products. More recently, a large benchmark data set was developed for machine learning applications. Although there is a general agreement in machine learning that the more data, the better, it is becoming more and more evident to consider the reliability and the quality of the data sets as critical features of the data. Part of the quality is associated with the balance between inactive and active compounds (in a rough analogy with the Yin-Yang concept), tasks that are not always feasible due to the general scarcity of negative (inactive compounds). The later point further emphasizes the continued need to publish and disclose negative results. Due to the fact that the experimental data of inactive compounds are not common, the community is using decoy data sets that by themselves are subject to design and refining using rational approaches. Decoy data sets try to fill the void of experimentally determined inactive molecules. Major criteria to take into account to generate compound data sets with high quality include balanced data sets in terms of active and inactive compounds (when the experimental information is available), structural and chemical diversity, curation or preparation according to the goals of the project, and complete information. All these together contribute to the perspectives of de novo design that foresees a continued and rapid expansion of molecules with the potential to become drugs.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Authors are grateful to DGAPA, UNAM, Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (PAPIIT), grant no. IN201321. We also thank the Dirección General de Cómputo y de Tecnologías de Información y Comunicación (DGTIC), UNAM, for the computational resources to use Miztli supercomputer at UNAM under project LANCAD-UNAM-DGTIC-335; and the innovation space UNAM-HUAWEI the computational resources to use their supercomputer under project-7 “Desarrollo y aplicación de algoritmos de inteligencia artificial para el diseño de fármacos aplicables al tratamiento de diabetes mellitus y cáncer”.
AC-H and EL-L are thankful to CONACyT, Mexico, for the Ph.D. scholarships number 847870 and 894234, respectively.
The author JLM-F declared that he was an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
2D/3D, two-dimensional/three-dimensional; ADMET, absorption, distribution, metabolism, excretion, and toxicity; AI, artificial intelligence; CADD, computer-aided drug design; COCONUT, Collection of Open NatUral ProdUcTs; DNN, deep neural networks; HBA, hydrogen bond acceptors; HBD, hydrogen bond donors; IMPPAT, A curated database of Indian Medicinal Plants, Phytochemistry And Therapeutics; MW, molecular weight; LBDD, ligand-based drug design; log P, octanol-water partition coefficient; NCE, new chemical entities; NIH(US), National Institutes of Health; PAINS, pan-assay interference compounds; Peru NPDB, Peruvian Natural Products Database; QSAR, quantitative structure-activity relationships; REAL, Enamine’s REadily AccessibLe; RNNs, recurrent neural networks; SBDD, structure-based drug design; TCM, Traditional Chinese Medicine; TPSA, topological surface area; UNPD, Universal Natural Product Database.
Altae-Tran, H., Ramsundar, B., Pappu, A. S., and Pande, V. (2017). Low data drug discovery with one-shot learning. ACS central Sci. 3 (4), 283–293. doi:10.1021/acscentsci.6b00367
Arús-Pous, J., Patronov, A., Bjerrum, E. J., Tyrchan, C., Reymond, J. L., Chen, H., et al. (2020). SMILES-based deep generative scaffold decorator for de-novo drug design. J. cheminformatics 12 (1), 38. doi:10.1186/s13321-020-00441-8
Atanasov, A. G., Zotchev, S. B., Dirsch, V. M., and Supuran, C. T.International Natural Product Sciences Taskforce (2021). Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 20 (3), 200–216. doi:10.1038/s41573-020-00114-z
Awale, M., and Reymond, J-L. (2015). Similarity mapplet: Interactive visualization of the directory of useful decoys and ChEMBL in high dimensional chemical spaces. J. Chem. Inf. Model. 55 (8), 1509–1516. doi:10.1021/acs.jcim.5b00182
Bajorath, J., Chávez-Hernández, A. L., Duran-Frigola, M., Fernández-de Gortari, E., Gasteiger, J., López-López, E., et al. (2022). Chemoinformatics and artificial intelligence colloquium: Progress and challenges in developing bioactive compounds. J. cheminformatics 14 (1), 82. doi:10.1186/s13321-022-00661-0
Balius, T. E., Allen, W. J., Mukherjee, S., and Rizzo, R. C. (2013). Grid-based molecular footprint comparison method for docking and de novo design: Application to HIVgp41. J. Comput. Chem. 34 (14), 1226–1240. doi:10.1002/jcc.23245
Barazorda-Ccahuana, H. L., Ranilla, L. G., Candia-Puma, M. A., Cárcamo-Rodriguez, E. G., Centeno-Lopez, A. E., Davila-Del-Carpio, G., et al. (2023). PeruNPDB: The Peruvian natural products database for in silico drug screening. Sci. Rep. 13 (1), 7577. doi:10.1038/s41598-023-34729-0
Bauer, M. R., Ibrahim, T. M., Vogel, S. M., and Boeckler, F. M. (2013). Evaluation and optimization of virtual screening workflows with DEKOIS 2.0-a public library of challenging docking benchmark sets. J. Chem. Inf. Model. 53 (6), 1447–1462. doi:10.1021/ci400115b
Beato, C., Beccari, A. R., Cavazzoni, C., Lorenzi, S., and Costantino, G. (2013). Use of experimental design to optimize docking performance: The case of LiGenDock, the docking module of LiGen, a new de novo design program. J. Chem. Inf. Model. 53 (6), 1503–1517. doi:10.1021/ci400079k
Bender, A., Schneider, N., Segler, M., Patrick Walters, W., Engkvist, O., and Rodrigues, T. (2022). Evaluation guidelines for machine learning tools in the chemical sciences. Nat. Rev. Chem. 6 (6), 428–442. doi:10.1038/s41570-022-00391-9
Bilodeau, C., Jin, W., Jaakkola, T., Barzilay, R., and Jensen, K. F. (2022). Generative models for molecular discovery: Recent advances and challenges. Comput. Mol. Sci. 12 (5), e1608. doi:10.1002/wcms.1608
Blaschke, T., Arús-Pous, J., Chen, H., Margreitter, C., Tyrchan, C., Engkvist, O., et al. (2020). Reinvent 2.0: An AI tool for de novo drug design. J. Chem. Inf. Model. 60 (12), 5918–5922. doi:10.1021/acs.jcim.0c00915
Brown, N., Fiscato, M., Segler, M. H. S., and Vaucher, A. C. (2019). GuacaMol: Benchmarking models for de Novo molecular design. J. Chem. Inf. Model. 59 (3), 1096–1108. doi:10.1021/acs.jcim.8b00839
Cao, L., Goreshnik, I., Coventry, B., Case, J. B., Miller, L., Kozodoy, L., et al. (2020). De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 370 (6515), 426–431. doi:10.1126/science.abd9909
Cereto-Massagué, A., Guasch, L., Valls, C., Mulero, M., Pujadas, G., and Garcia-Vallvé, S. (2012). DecoyFinder: An easy-to-use python GUI application for building target-specific decoy sets. Bioinformatics 28 (12), 1661–1662. doi:10.1093/bioinformatics/bts249
Chávez-Hernández, A. L., and Medina-Franco, J. L. (2023). Natural products subsets: Generation and characterization. Artif. Intell. Life Sci. 3, 100066. doi:10.1016/j.ailsci.2023.100066
Chávez-Hernández, A. L., Sánchez-Cruz, N., and Medina-Franco, J. L. (2020a). A fragment library of natural products and its comparative chemoinformatic characterization. Mol. Inf. 39 (11), e2000050. doi:10.1002/minf.202000050
Chávez-Hernández, A. L., Sánchez-Cruz, N., and Medina-Franco, J. L. (2020b). Fragment library of natural products and compound databases for drug discovery. Biomolecules 10 (11), 1518. doi:10.3390/biom10111518
Chemriya, (2023). CHEMriya. Available at: https://chemriya.com/ (accessed May 13, 2023).
Chemspace (2023). Freedom space. Available at: https://chem-space.com/compounds/freedom-space (accessed May 13, 2023).
Chen, C. Y-C. (2011). TCM Database@Taiwan: The world’s largest traditional Chinese medicine database for drug screening in silico. PloS one 6 (1), e15939. doi:10.1371/journal.pone.0015939
Chen, X., Lin, Y., Liu, M., and Gilson, M. K. (2002). The binding database: Data management and interface design. Bioinformatics 18 (1), 130–139. doi:10.1093/bioinformatics/18.1.130
Cherkasov, A. (2023). The ‘Big Bang’ of the chemical universe. Nat. Chem. Biol. 19, 667–668. doi:10.1038/s41589-022-01233-x
Corso, G., Stärk, H., Jing, B., et al. (2022). DiffDock: Diffusion steps, twists, and turns for molecular docking. arXiv [q-bio.BM]. Available at: http://arxiv.org/abs/2210.01776.
Costa, R. P. O., Lucena, L. F., Silva, L. M. A., Zocolo, G. J., Herrera-Acevedo, C., Scotti, L., et al. (2021). The SistematX web portal of natural products: An update. J. Chem. Inf. Model. 61 (6), 2516–2522. doi:10.1021/acs.jcim.1c00083
Davies, M., Nowotka, M., Papadatos, G., Dedman, N., Gaulton, A., Atkinson, F., et al. (2015). ChEMBL web services: Streamlining access to drug discovery data and utilities. Nucleic acids Res. 43 (W1), W612–W620. doi:10.1093/nar/gkv352
Dos Santos Nascimento, I. J., de Aquino, T. M., and da Silva-Júnior, E. F. (2021). Drug repurposing: A strategy for discovering inhibitors against emerging viral infections. Curr. Med. Chem. 28 (15), 2887–2942. doi:10.2174/0929867327666200812215852
DRUGBANK (2023). Celecoxib. Available at: https://go.drugbank.com/drugs/DB00482 (accessed May 13, 2023).
Enamine (2023). Real database. Available at: https://enamine.net/compound-collections/real-compounds/real-database (accessed May 13, 2023).
Evans, B. E., Rittle, K. E., Bock, M. G., DiPardo, R. M., Freidinger, R. M., Whitter, W. L., et al. (1988). Methods for drug discovery: Development of potent, selective, orally effective cholecystokinin antagonists. J. Med. Chem. 31 (12), 2235–2246. doi:10.1021/jm00120a002
Fourches, D., Muratov, E., and Tropsha, A. (2016). Trust, but verify II: A practical guide to chemogenomics data curation. J. Chem. Inf. Model. 56 (7), 1243–1252. doi:10.1021/acs.jcim.6b00129
Gallo, K., Kemmler, E., Goede, A., Becker, F., Dunkel, M., Preissner, R., et al. (2023). SuperNatural 3.0-a database of natural products and natural product-based derivatives. Nucleic acids Res. 51 (D1), D654–D659. doi:10.1093/nar/gkac1008
Gómez-Bombarelli, R., Wei, J. N., Duvenaud, D., Hernández-Lobato, J. M., Sánchez-Lengeling, B., Sheberla, D., et al. (2018). Automatic chemical design using a data-driven continuous representation of molecules. ACS central Sci. 4 (2), 268–276. doi:10.1021/acscentsci.7b00572
Gómez-García, A., and Medina-Franco, J. L. (2022). Progress and impact of Latin American natural product databases. Biomolecules 12 (9), 1202. doi:10.3390/biom12091202
Grebner, C. (2022). Webinar: "exploration and mining of large virtual chemical spaces. Available at: https://youtu.be/fMrI11SXwpU (accessed May 13, 2023).
Greener, J. G., Kandathil, S. M., Moffat, L., and Jones, D. T. (2022). A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 23 (1), 40–55. doi:10.1038/s41580-021-00407-0
Grigalunas, M., Brakmann, S., and Waldmann, H. (2022). Chemical evolution of natural product structure. J. Am. Chem. Soc. 144 (8), 3314–3329. doi:10.1021/jacs.1c11270
Gu, J., Gui, Y., Chen, L., Yuan, G., Lu, H. Z., and Xu, X. (2013). Use of natural products as chemical library for drug discovery and network pharmacology. PloS one 8 (4), e62839. doi:10.1371/journal.pone.0062839
Guo J, J., Janet, J. P., Bauer, M. R., Nittinger, E., Giblin, K. A., Papadopoulos, K., et al. (2021). DockStream: A docking wrapper to enhance de novo molecular design. J. cheminformatics 13 (1), 89. doi:10.1186/s13321-021-00563-7
Guo M, M., Thost, V., Li, B., et al. (2021). “Data-efficient graph grammar learning for molecular generation,” in International conference on learning representations, 9. February 2021. Available at: https://research.ibm.com/publications/data-efficient-graph-grammar-learning-for-molecular-generation (accessed May 13, 2023).
Guo, M., Thost, V., Li, B., et al. (2022). Data-efficient graph grammar learning for molecular generation. arXiv [cs.LG]. Available at: http://arxiv.org/abs/2203.08031.
Hayes, A., and Hunter, J. (2012). Why is publication of negative clinical trial data important? Br. J. Pharmacol. 167 (7), 1395–1397. doi:10.1111/j.1476-5381.2012.02215.x
Hu, Q., Peng, Z., Sutton, S. C., Na, J., Kostrowicki, J., Yang, B., et al. (2012). Pfizer global virtual library (PGVL): A chemistry design tool powered by experimentally validated parallel synthesis information. ACS Comb. Sci. 14 (11), 579–589. doi:10.1021/co300096q
IBM (2022). How to use AI to discover new drugs and materials with limited data. Available at: https://research.ibm.com/blog/ai-discovery-with-limited-data#fnref-1 (accessed April 16, 2023).
Irwin, J. J. (2008). Community benchmarks for virtual screening. J. computer-aided Mol. Des. 22 (3-4), 193–199. doi:10.1007/s10822-008-9189-4
Jain, A. N., and Nicholls, A. (2008). Recommendations for evaluation of computational methods. J. computer-aided Mol. Des. 22 (3-4), 133–139. doi:10.1007/s10822-008-9196-5
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596 (7873), 583–589. doi:10.1038/s41586-021-03819-2
Juskalian, R., Regalado, A., Orcutt, M., et al. (2023). 10 breakthrough technologies 2020. Available at: https://www.technologyreview.com/10-breakthrough-technologies/2020/ (accessed February 26, 2020).
Kadurin, A., Aliper, A., Kazennov, A., Mamoshina, P., Vanhaelen, Q., Khrabrov, K., et al. (2017). The cornucopia of meaningful leads: Applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget 8 (7), 10883–10890. doi:10.18632/oncotarget.14073
Kim, S., Chen, J., Cheng, T., Gindulyte, A., et al. (2023). PubChem 2023 update. Nucleic acids Res. 51 (D1), D1373–D1380. doi:10.1093/nar/gkac956
Koes, D. R., and Camacho, C. J. (2012). ZINCPharmer: Pharmacophore search of the ZINC database. Nucleic acids Res. 40, W409–W414. Web Server issue). doi:10.1093/nar/gks378
Korkmaz, S. (2020). Deep learning-based imbalanced data classification for drug discovery. J. Chem. Inf. Model. 60 (9), 4180–4190. doi:10.1021/acs.jcim.9b01162
Korn, M., Ehrt, C., Ruggiu, F., Gastreich, M., and Rarey, M. (2023). Navigating large chemical spaces in early-phase drug discovery. Curr. Opin. Struct. Biol. 80, 102578. doi:10.1016/j.sbi.2023.102578
Kramer, C., and Lewis, R. (2012). QSARs, data and error in the modern age of drug discovery. Curr. Top. Med. Chem. 12 (17), 1896–1902. doi:10.2174/156802612804547380
Krenn, M., Häse, F., Nigam, A., Friederich, P., and Aspuru-Guzik, A. (2020). Self-referencing embedded strings (SELFIES): A 100% robust molecular string representation. Mach. Learn. Sci. Technol. 1 (4), 045024. doi:10.1088/2632-2153/aba947
Krishnan, S. R., Bung, N., Bulusu, G., and Roy, A. (2021). Accelerating de novo drug design against novel proteins using deep learning. J. Chem. Inf. Model. 61 (2), 621–630. doi:10.1021/acs.jcim.0c01060
Kumar, S. A., Ananda Kumar, T. D., Beeraka, N. M., Pujar, G. V., Singh, M., Narayana Akshatha, H. S., et al. (2022). Machine learning and deep learning in data-driven decision making of drug discovery and challenges in high-quality data acquisition in the pharmaceutical industry. Future Med. Chem. 14 (4), 245–270. doi:10.4155/fmc-2021-0243
Leach, A. R., and Gillet, V. J. (2007). “Selecting diverse dets of compounds,” in An introduction to chemoinformatics (Dordrecht: Springer Netherlands), 119–139. doi:10.1007/978-1-4020-6291-9_6
Li, S., Wang, L., Meng, J., Zhao, Q., Zhang, L., and Liu, H. (2022). De Novo design of potential inhibitors against SARS-CoV-2 Mpro. Comput. Biol. Med. 147, 105728. doi:10.1016/j.compbiomed.2022.105728
Li, Y., Zhang, L., and Liu, Z. (2018). Multi-objective de novo drug design with conditional graph generative model. J. cheminformatics 10 (1), 33. doi:10.1186/s13321-018-0287-6
Liang, Y., Fang, R., and Rao, Q. (2022). An insight into the medicinal chemistry perspective of macrocyclic derivatives with antitumor activity: A systematic review. Molecules 27 (9), 2837. doi:10.3390/molecules27092837
Lipinski, C. A., Lombardo, F., Dominy, B. W., and Feeney, P. J. (2001). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. drug Deliv. Rev. 46 (1-3), 3–26. doi:10.1016/s0169-409x(00)00129-0
Liu, X., Ye, K., van Vlijmen, H. W. T., Ijzerman, A. P., and van Westen, G. J. P. (2019). An exploration strategy improves the diversity of de novo ligands using deep reinforcement learning: A case for the adenosine A2A receptor. J. cheminformatics 11 (1), 35. doi:10.1186/s13321-019-0355-6
López-López, E., Bajorath, J., and Medina-Franco, J. L. (2021a). Informatics for chemistry, biology, and biomedical sciences. J. Chem. Inf. Model. 61 (1), 26–35. doi:10.1021/acs.jcim.0c01301
López-López, E., Cerda-García-Rojas, C. M., and Medina-Franco, J. L. (2021b). Tubulin inhibitors: A chemoinformatic analysis using cell-based data. Molecules 26 (9), 2483. doi:10.3390/molecules26092483
López-López, E., Fernández-de Gortari, E., and Medina-Franco, J. L. (2022). Yes SIR! On the structure-inactivity relationships in drug discovery. Drug Discov. today 27 (8), 2353–2362. doi:10.1016/j.drudis.2022.05.005
López-López, E., and Medina-Franco, J. L. (2023). Towards decoding hepatotoxicity of approved drugs through navigation of multiverse and consensus chemical spaces. Biomolecules 13 (1), 176. doi:10.3390/biom13010176
Ma, B., Terayama, K., Matsumoto, S., Isaka, Y., Sasakura, Y., Iwata, H., et al. (2021). Structure-based de novo molecular generator combined with artificial intelligence and docking simulations. J. Chem. Inf. Model. 61 (7), 3304–3313. doi:10.1021/acs.jcim.1c00679
Maziarka, L., Pocha, A., Kaczmarczyk, J., Rataj, K., Danel, T., and Warchoł, M. (2020). Mol-CycleGAN: A generative model for molecular optimization. J. cheminformatics 12 (1), 2. doi:10.1186/s13321-019-0404-1
Medina-Franco, J. L., Flores-Padilla, E. A., and Chávez-Hernández, A. L. (2022b). “Chapter 23 - discovery and development of lead compounds from natural sources using computational approaches,” in Evidence-based validation of herbal medicine. Editor P. K. Mukherjee Second Edition (Elsevier), 539–560. doi:10.1016/B978-0-323-85542-6.00009-3
Medina-Franco, J. L., López-López, E., Andrade, E., Ruiz-Azuara, L., Frei, A., Guan, D., et al. (2022a). Bridging informatics and medicinal inorganic chemistry: Toward a database of metallodrugs and metallodrug candidates. Drug Discov. today 27 (5), 1420–1430. doi:10.1016/j.drudis.2022.02.021
Medina-Franco, J. L., and López-López, E. (2022). The essence and transcendence of scientific publishing. Front. Res. metrics Anal. 7, 822453. doi:10.3389/frma.2022.822453
Medina-Franco, J. L., Martinez-Mayorga, K., and Meurice, N. (2014). Balancing novelty with confined chemical space in modern drug discovery. Expert Opin. drug Discov. 9 (2), 151–165. doi:10.1517/17460441.2014.872624
Medina-Franco, J. L., Naveja, J. J., and López-López, E. (2019). Reaching for the bright StARs in chemical space. Drug Discov. today 24 (11), 2162–2169. doi:10.1016/j.drudis.2019.09.013
Mendez, D., Gaulton, A., Bento, A. P., Chambers, J., De Veij, M., Félix, E., et al. (2019). ChEMBL: Towards direct deposition of bioassay data. Nucleic acids Res. 47 (D1), D930–D940. doi:10.1093/nar/gky1075
Mohanraj, K., Karthikeyan, B. S., Vivek-Ananth, R. P., Chand, R. P. B., Aparna, S. R., Mangalapandi, P., et al. (2018). Imppat: A curated database of indian medicinal plants, phytochemistry and therapeutics. Sci. Rep. 8 (1), 4329. doi:10.1038/s41598-018-22631-z
Mouchlis, V. D., Afantitis, A., Serra, A., Fratello, M., Papadiamantis, A. G., Aidinis, V., et al. (2021). Advances in de novo drug design: From conventional to machine learning methods. Int. J. Mol. Sci. 22 (4), 1676. doi:10.3390/ijms22041676
Mysinger, M. M., Carchia, M., Irwin, J. J., and Shoichet, B. K. (2012). Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 55 (14), 6582–6594. doi:10.1021/jm300687e
Newman, D. J., and Cragg, G. M. (2020). Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 83 (3), 770–803. doi:10.1021/acs.jnatprod.9b01285
NIH (2023). Natural product libraries. Available at: https://www.nccih.nih.gov/grants/natural-product-libraries.
Niitsu, A., and Sugita, Y. (2023). Towards de novo design of transmembrane α-helical assemblies using structural modelling and molecular dynamics simulation. Phys. Chem. Chem. Phys. PCCP 25 (5), 3595–3606. doi:10.1039/d2cp03972a
Norinder, U., Naveja, J. J., López-López, E., Mucs, D., and Medina-Franco, J. L. (2019). Conformal prediction of HDAC inhibitors. SAR QSAR Environ. Res. 30 (4), 265–277. doi:10.1080/1062936X.2019.1591503
Ntie-Kang, F., Zofou, D., Babiaka, S. B., Meudom, R., Scharfe, M., Lifongo, L. L., et al. (2013). AfroDb: A select highly potent and diverse natural product library from african medicinal plants. PloS one 8 (10), e78085. doi:10.1371/journal.pone.0078085
Olivecrona, M., Blaschke, T., Engkvist, O., and Chen, H. (2017). Molecular de-novo design through deep reinforcement learning. J. cheminformatics 9 (1), 48. doi:10.1186/s13321-017-0235-x
Olmedo, A. D., and Medina-Franco, J. L. (2020). “Chemoinformatic approach: The case of natural products of Panama,” in Cheminformatics and its applications (IntechOpen). doi:10.5772/intechopen.87779
Olmedo, D. A., González-Medina, M., Gupta, M. P., and Medina-Franco, J. L. (2017). Cheminformatic characterization of natural products from Panama. Mol. Divers. 21 (4), 779–789. doi:10.1007/s11030-017-9781-4
Palazzesi, F., and Pozzan, A. (2022). “Deep learning applied to ligand-based de novo drug DesignDe novo drug design,” in Artificial intelligence in drug design. Editor A. Heifetz (New York, NY: Springer US), 273–299. doi:10.1007/978-1-0716-1787-8_12
Papadopoulos, K., Giblin, K. A., Janet, J. P., Patronov, A., and Engkvist, O. (2021). De novo design with deep generative models based on 3D similarity scoring. Bioorg. Med. Chem. 44, 116308. doi:10.1016/j.bmc.2021.116308
Patel, H. M., Noolvi, M. N., Sharma, P., Jaiswal, V., Bansal, S., Lohan, S., et al. (2014). Quantitative structure–activity relationship (QSAR) studies as strategic approach in drug discovery. Med. Chem. Res. Int. J. rapid Commun. Des. Mech. action Biol. Act. agents 23 (12), 4991–5007. doi:10.1007/s00044-014-1072-3
Perron, Q., da Silva, V. B. R., Atwood, B., and Gaston-Mathé, Y. (2022b). Key points to succeed in Artificial Intelligence drug discovery projects. Chem. Int. 44 (1), 19–21. doi:10.1515/ci-2022-0106
Perron, Q., Mirguet, O., Tajmouati, H., Skiredj, A., Rojas, A., Gohier, A., et al. (2022a). Deep generative models for ligand-based de novo design applied to multi-parametric optimization. J. Comput. Chem. 43 (10), 692–703. doi:10.1002/jcc.26826
Pilon, A. C., Valli, M., Dametto, A. C., Pinto, M. E. F., Freire, R. T., Castro-Gamboa, I., et al. (2017). NuBBEDB: An updated database to uncover chemical and biological information from Brazilian biodiversity. Sci. Rep. 7 (1), 7215. doi:10.1038/s41598-017-07451-x
Pilón-Jiménez, B. A., Saldívar-González, F. I., Díaz-Eufracio, B. I., and Medina-Franco, J. L. (2019). Biofacquim: A Mexican compound database of natural products. Biomolecules 9 (1), 31. doi:10.3390/biom9010031
Polykovskiy, D., Zhebrak, A., Sanchez-Lengeling, B., Golovanov, S., Tatanov, O., Belyaev, S., et al. (2020). Molecular sets (MOSES): A benchmarking platform for molecular generation models. Front. Pharmacol. 11, 565644. doi:10.3389/fphar.2020.565644
Réau, M., Langenfeld, F., Zagury, J-F., Lagarde, N., and Montes, M. (2018). Decoys selection in benchmarking datasets: Overview and perspectives. Front. Pharmacol. 9, 11. doi:10.3389/fphar.2018.00011
Reymond, J-L. (2015). The chemical space project. Accounts Chem. Res. 48 (3), 722–730. doi:10.1021/ar500432k
Rohrer, S. G., and Baumann, K. (2009). Maximum unbiased validation (MUV) data sets for virtual screening based on PubChem bioactivity data. J. Chem. Inf. Model. 49 (2), 169–184. doi:10.1021/ci8002649
Sabe, V. T., Ntombela, T., Jhamba, L. A., Maguire, G. E. M., Govender, T., Naicker, T., et al. (2021). Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. Eur. J. Med. Chem. 224, 113705. doi:10.1016/j.ejmech.2021.113705
Saldívar-González, F. I., Aldas-Bulos, V. D., Medina-Franco, J. L., and Plisson, F. (2022). Natural product drug discovery in the artificial intelligence era. Chem. Sci. 13 (6), 1526–1546. doi:10.1039/d1sc04471k
Saldívar-González, F. I., and Medina-Franco, J. L. (2022). Approaches for enhancing the analysis of chemical space for drug discovery. Expert Opin. drug Discov. 17 (7), 789–798. doi:10.1080/17460441.2022.2084608
Saldívar-González, F. I., Valli, M., Andricopulo, A. D., da Silva Bolzani, V., and Medina-Franco, J. L. (2019). Chemical space and diversity of the NuBBE database: A chemoinformatic characterization. J. Chem. Inf. Model. 59 (1), 74–85. doi:10.1021/acs.jcim.8b00619
Sánchez-Cruz, N., Pilón-Jiménez, B. A., and Medina-Franco, J. L. (2019) Functional group and diversity analysis of BIOFACQUIM: A Mexican natural product database. F1000Research 8, Chem Inf Sci-2071. doi:10.12688/f1000research.21540.2
Scannell, J. W., Bosley, J., Hickman, J. A., Dawson, G. R., Truebel, H., Ferreira, G. S., et al. (2022). Predictive validity in drug discovery: What it is, why it matters and how to improve it. Nat. Rev. Drug Discov. 21 (12), 915–931. doi:10.1038/s41573-022-00552-x
Schneider, G., and Clark, D. E. (2019). Automated de novo drug design: Are we nearly there yet? Angew. Chem. 58 (32), 10792–10803. doi:10.1002/anie.201814681
Schneider, G., Clément-Chomienne, O., Hilfiger, L., Schneider, , Kirsch, , Böhm, , et al. (2000). Virtual screening for bioactive molecules by evolutionary de novo design. Angew. Chem. 39 (22), 4130–4133. doi:10.1002/1521-3773(20001117)39:22<4130:aid-anie4130>3.0.co;2-e
Schneider, P., and Schneider, G. (2017). Privileged structures revisited. Angew. Chem. 56 (27), 7971–7974. doi:10.1002/anie.201702816
Schneider, P., Walters, W. P., Plowright, A. T., Sieroka, N., Listgarten, J., Goodnow, R. A., et al. (2020). Rethinking drug design in the artificial intelligence era. Nat. Rev. Drug Discov. 19 (5), 353–364. doi:10.1038/s41573-019-0050-3
Scotti, M. T., Herrera-Acevedo, C., Oliveira, T. B., Costa, R. P. O., Santos, S. Y. K. d. O., Rodrigues, R. P., et al. (2018). SistematX, an online web-based cheminformatics tool for data management of secondary metabolites. Molecules 23 (1), 103. doi:10.3390/molecules23010103
Sheridan, R. P. (2013). Time-split cross-validation as a method for estimating the goodness of prospective prediction. J. Chem. Inf. Model. 53 (4), 783–790. doi:10.1021/ci400084k
Shipman, J. T., Su, X., Hua, D., and Desaire, H. (2019). DecoyDeveloper: An on-demand, de novo decoy glycopeptide generator. J. proteome Res. 18 (7), 2896–2902. doi:10.1021/acs.jproteome.9b00203
Simonovsky, M., and Komodakis, N. (2018). “GraphVAE: Towards generation of small graphs using variational autoencoders,” in Artificial neural networks and machine learning – icann 2018 (Springer International Publishing), 2018, 412–422. doi:10.1007/978-3-030-01418-6_41
Skalic, M., Jiménez, J., Sabbadin, D., and De Fabritiis, G. (2019b). Shape-based generative modeling for de novo drug design. J. Chem. Inf. Model. 59 (3), 1205–1214. doi:10.1021/acs.jcim.8b00706
Skalic, M., Sabbadin, D., Sattarov, B., Sciabola, S., and De Fabritiis, G. (2019a). From target to drug: Generative modeling for the multimodal structure-based ligand design. Mol. Pharm. 16 (10), 4282–4291. doi:10.1021/acs.molpharmaceut.9b00634
Soares, T. A., Nunes-Alves, A., Mazzolari, A., Ruggiu, F., Wei, G. W., and Merz, K. (2022). The (Re)-evolution of quantitative structure-activity relationship (qsar) studies propelled by the surge of machine learning methods. J. Chem. Inf. Model. 62 (22), 5317–5320. doi:10.1021/acs.jcim.2c01422
Sorokina, M., Merseburger, P., Rajan, K., Yirik, M. A., and Steinbeck, C. (2021). COCONUT online: Collection of open natural products database. J. cheminformatics 13 (1), 2. doi:10.1186/s13321-020-00478-9
Tingle, B. I., Tang, K. G., Castanon, M., Gutierrez, J. J., Khurelbaatar, M., Dandarchuluun, C., et al. (2023). ZINC-22─A free multi-billion-scale database of tangible compounds for ligand discovery. J. Chem. Inf. Model. 63 (4), 1166–1176. doi:10.1021/acs.jcim.2c01253
Tong, X., Liu, X., Tan, X., Jiang, J., Xiong, Z., et al. (2021). Generative models for de novo drug design. J. Med. Chem. 64 (19), 14011–14027. doi:10.1021/acs.jmedchem.1c00927
Ullanat, V. (2020). “Variational autoencoder as a generative tool to produce de-novo lead compounds for biological targets,” in 2020 14th international conference on innovations in information Technology (IIT), 102–107. doi:10.1109/IIT50501.2020.9299078
UNIIQUIM (2015). Uniiquim. Available at: https://uniiquim.iquimica.unam.mx/ (accessed May 13, 2023).
Valli, M., dos Santos, R. N., Figueira, L. D., Nakajima, C. H., Castro-Gamboa, I., Andricopulo, A. D., et al. (2013). Development of a natural products database from the biodiversity of Brazil. J. Nat. Prod. 76 (3), 439–444. doi:10.1021/np3006875
Vamathevan, J., Clark, D., Czodrowski, P., Dunham, I., Ferran, E., Lee, G., et al. (2019). Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 18 (6), 463–477. doi:10.1038/s41573-019-0024-5
Veber, D. F., Johnson, S. R., Cheng, H-Y., Smith, B. R., Ward, K. W., and Kopple, K. D. (2002). Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 45 (12), 2615–2623. doi:10.1021/jm020017n
Wang, L., Pang, X., Li, Y., Zhang, Z., and Tan, W. (2017). Rader: A RApid DEcoy retriever to facilitate decoy based assessment of virtual screening. Bioinformatics 33 (8), 1235–1237. doi:10.1093/bioinformatics/btw783
Wang, M., Hsieh, C-Y., Wang, J., Wang, D., Weng, G., Shen, C., et al. (2022). Relation: A deep generative model for structure-based de novo drug design. J. Med. Chem. 65 (13), 9478–9492. doi:10.1021/acs.jmedchem.2c00732
Warr, W. A., Nicklaus, M. C., Nicolaou, C. A., and Rarey, M. (2022). Exploration of ultralarge compound collections for drug discovery. J. Chem. Inf. Model. 62 (9), 2021–2034. doi:10.1021/acs.jcim.2c00224
Warr, W. (2021). Report on an NIH workshop on ultralarge chemistry databases. Chemrxiv: 43. Available at: https://chemrxiv.org/engage/chemrxiv/article-details/60c75883bdbb89984ea3ada5.
Weininger, D. (1988). SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 28 (1), 31–36. doi:10.1021/ci00057a005
Wishart, D. S., Feunang, Y. D., Guo, A. C., Lo, E. J., Marcu, A., Grant, J. R., et al. (2018). DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic acids Res. 46 (D1), D1074–D1082. doi:10.1093/nar/gkx1037
Wishart, D. S., Knox, C., Guo, A. C., Cheng, D., Shrivastava, S., Tzur, D., et al. (2008). DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic acids Res. 36, D901–D906. doi:10.1093/nar/gkm958
Wishart, D. S., Knox, C., Guo, A. C., Shrivastava, S., Hassanali, M., Stothard, P., et al. (2006). DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic acids Res. 34, D668–D672. doi:10.1093/nar/gkj067
Wu, A., Ye, Q., Zhuang, X., Chen, Q., Zhang, J., Wu, J., et al. (2023a). Elucidating structures of complex organic compounds using a machine learning model based on the 13C NMR chemical shifts. Precis. Chem. 1 (1), 57–68. doi:10.1021/prechem.3c00005
Wu, J., Xiao, Y., Cai, H., Zhao, D., Li, Y., et al. (2023b). DeepCancerMap: A versatile deep learning platform for target- and cell-based anticancer drug discovery. Eur. J. Med. Chem. 255, 115401. doi:10.1016/j.ejmech.2023.115401
Wu, Z., Ramsundar, B., Feinberg, E. N., Gomes, J., Geniesse, C., Pappu, A. S., et al. (2018). MoleculeNet: A benchmark for molecular machine learning. Chem. Sci. 9 (2), 513–530. doi:10.1039/c7sc02664a
Xie, W., Wang, F., Li, Y., Lai, L., and Pei, J. (2022). Advances and challenges in de novo drug design using three-dimensional deep generative models. J. Chem. Inf. Model. 62 (10), 2269–2279. doi:10.1021/acs.jcim.2c00042
Yang, J., Wang, D., Jia, C., Wang, M., Hao, G., and Yang, G. (2019). Freely accessible chemical database resources of compounds for in silico drug discovery. Curr. Med. Chem. 26 (42), 7581–7597. doi:10.2174/0929867325666180508100436
Yang, X., Yang, G., and Chu, J. (2023). The balanced matrix factorization for computational drug repositioning. arXiv [cs.CE]. Available at: http://arxiv.org/abs/2301.06448.
Yu, H. (2021). Responsible use of negative research outcomes-accelerating the discovery and development of new antibiotics. J. antibiotics 74 (9), 543–546. doi:10.1038/s41429-021-00439-w
Zhang, Y., Luo, M., Wu, P., Wu, S., Lee, T. Y., and Bai, C. (2022). Application of computational biology and artificial intelligence in drug design. Int. J. Mol. Sci. 23 (21), 13568. doi:10.3390/ijms232113568
Keywords: big data, chemoinformatics, chemical libraries, data quality, de novo design, drug discovery, machine learning, negative results
Citation: Chávez-Hernández AL, López-López E and Medina-Franco JL (2023) Yin-yang in drug discovery: rethinking de novo design and development of predictive models. Front. Drug Discov. 3:1222655. doi: 10.3389/fddsv.2023.1222655
Received: 15 May 2023; Accepted: 12 June 2023;
Published: 21 June 2023.
Edited by:
Ho Leung Ng, Atomwise Inc., United StatesReviewed by:
Rodolpho C. Braga, InsilicAll, BrazilCopyright © 2023 Chávez-Hernández, López-López and Medina-Franco. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José L. Medina-Franco, bWVkaW5hamxAdW5hbS5teA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.