 Ida Lin1†‡
Ida Lin1†‡ Peter B. Rupert2‡Kristina Pilat1†
Peter B. Rupert2‡Kristina Pilat1† Raymond O. Ruff1†
Raymond O. Ruff1† Della J. Friend2Man Kid Chan1†Midori Clarke1†
Della J. Friend2Man Kid Chan1†Midori Clarke1† Benjamin G. Hoffstrom3†Jane Carter1†Soheil Meshinchi1Ashok D. Bandaranayake1†
Benjamin G. Hoffstrom3†Jane Carter1†Soheil Meshinchi1Ashok D. Bandaranayake1† Christopher Mehlin1†James M. Olson1*†
Christopher Mehlin1†James M. Olson1*† Roland K. Strong2*
Roland K. Strong2* Colin E. Correnti1*†
Colin E. Correnti1*†- 1Clinical Research Division, Fred Hutchinson Cancer Center, Seattle, WA, United States
- 2Division of Basic Sciences, Fred Hutchinson Cancer Center, Seattle, WA, United States
- 3Antibody Technology Resource, Fred Hutchinson Cancer Center, Seattle, WA, United States
Mesothelin is a glypiated, cell-surface glycoprotein expressed at low levels on normal mesothelium but overexpressed by many cancers. Implicated in cell adhesion and multiple signaling pathways, mesothelin’s precise biological function and overall structure remain undefined. Antibodies targeting mesothelin have been engineered into immunotoxins, antibody-drug conjugates, CAR-T cells, or bispecific T cell engagers as candidate therapeutics but most face challenges, including binding epitopes that are not optimal for selected modalities. Here we describe the isolation and characterization of a novel anti-mesothelin antibody, 1A12, including crystallographic mapping of the 1A12 epitope in relation to other antibodies (amatuximab, anetumab). 1A12 possesses uniquely favorable properties, including a membrane-proximal epitope, and enabled structure determination of the complete mesothelin ectodomain. We incorporated 1A12 into two different bispecific T cell engaging architectures with various anti-CD3 co-targeting elements as candidate therapeutics, demonstrating in vitro functionality and potency.
Introduction
The glycophosphatidylinositol-linked, cell surface glycoprotein mesothelin (MSLN) is normally limited in expression to mesothelial cells lining the pleura, peritoneum, and pericardium, but is highly overexpressed by many cancers, including ovarian, pancreatic, lung, cholangiocarcinoma, and mesothelioma: a highly aggressive lung cancer associated with asbestos exposure, with few effective treatment options, and very poor prognoses (Tang et al., 2013b; Hilliard, 2018; Cinausero et al., 2019; Davis et al., 2021). The biological function and full three-dimensional structure of MSLN remain undefined, but MSLN is functionally associated with cell survival, proliferation, adherence, and tumor progression, possibly signaling through the MAPK/ERK, JNK, and/or PI3K/AKT pathways; MSLN overexpression is correlated with poorer patient outcomes across cancer types. Confoundingly, however, MSLN knockout mice lack any clear phenotype (Bera and Pastan, 2000). MSLN is expressed as a precursor fusion protein with megakaryocyte potentiating factor (MPF); subsequent cleavage by furin during export releases MPF (∼30 kDa) as a soluble protein leaving MSLN proper (∼35 kDa) anchored on the cell surface. The only characterized ligand for MSLN is the cancer antigen/biomarker mucin 16 (MUC16 or CA125).
MSLN was first identified using the monoclonal antibody K1, which was isolated by immunizing mice with the ovarian adenocarcinoma cell line OVCAR-3 (Chang et al., 1992a; Chang et al., 1992b; Chang and Pastan, 1996). Though unsuitable for clinical development itself, many subsequent anti-MSLN antibodies have been isolated and evaluated for therapeutic applications and are summarized in Supplementary Tables S1, S2. The Fab fragment of one such antibody, the chimeric antibody amatuximab/MORAb-009, has been crystallized alone and used as a co-crystallization chaperone in complex with an immunodominant, 64-residue N-terminal fragment (N-terminal domain: NTD) of MSLN, revealing a compact, right-handed α-superhelical fold comprising five short helices and connecting loops (Ma et al., 2012). Soluble forms of MSLN, collectively referred to as “soluble mesothelin-related peptides” (SMRPs), are defined by a secreted isoform (UniProt: Q13421-2), as well as shed forms of the ectodomain that arise from lipase- and protease-mediated cleavage of the membrane anchored receptor, that can be detected in the serum of healthy and diseased humans (Scholler et al., 1999; Hassan et al., 2006; Hellstrom et al., 2006; Ho et al., 2006; Sapede et al., 2008). The presence of SMRPs in circulation serves as a clinically-useful biomarker for cancer detection and monitoring tumor progression, but also reduces the efficacy of antibody-based therapies by acting as an antigen sink, particularly in the tumor microenvironment (Pastan and Zhang, 2012; Awuah et al., 2016; Liu et al., 2020).
Since many of the available antibodies bind similar or overlapping epitopes on the immunodominant N-terminal half of MSLN, we employed immunization strategies focusing responses to distinct, membrane proximal MSLN epitopes to generate antibodies with potentially improved clinical performance, maintaining cross-reactivity across human and cynomolgus MSLN orthologs to enable future surrogate in vivo toxicity evaluation. We used the TRIANNI humanized mouse platform to generate antibodies with fully human variable domain (VH/VL) cassettes (TRIANNI, 2016; Cameron et al., 2020; Peter et al., 2021). Isolated antibodies were biochemically evaluated, with univalent equilibrium dissociation constants (KDs) ranging down to the tens-of-nanomolar range. Using combinations of the lead TRIANNI antibody, amatuximab, and anetumab as either Fab or single-chain Fv (scFv) fragments, three co-crystal structures of MSLN fragments spanning up to the full ectodomain were determined, mapping the distinct epitopes of these three antibodies. The structures also showed that the right-handed α-superhelix structure of the N-terminal domain extends through the entire ectodomain except for an extended, variable, disordered loop. Agreement between the experimentally determined MSLN crystal structures and state-of-the-art predictions by RoseTTAFold and AlphaFold was remarkable (Kim et al., 2004; Jumper et al., 2021; Varadi et al., 2022). The lead TRIANNI antibody was engineered into several bispecific, T cell-redirecting formats with anti-CD3 antibodies which showed high potencies in in vitro tumor cell killing assays and resistance to SMRP competition.
Materials and methods
Immunization, hybridoma generation, and antibody isolation
TRIANNI humanized mice (TRIANNI, 2016) were immunized over a 12-week period using Adjuplex™ adjuvant and 3T3 cells engineered to express full length mesothelin. Splenocytes were isolated, and hybridomas generated by electrofusion (Greenfield, 2014). Hybridomas were cultured in microtiter plates and secreted antibodies were identified by flow cytometry using both HEK 293 and MV-4-11 cells expressing the full length MSLN and counter-screening with parental controls. Polyclonal positives were subcloned using a ClonePix II™ and validated mesothelin binding antibody secretion by flow cytometry.
Protein expression and purification
We employed the Daedalus human cell line expression platform for the production and purification of all reported proteins, using methods described previously (Bandaranayake et al., 2011). The expression system utilizes suspension adapted HEK293 FreeStyle™ cells (ThermoFisher catalog #R79007) and lentiviral transduction to generate cell lines that secrete proteins at high levels. The lentiviral vector contains a cis-linked fluorescent protein reporter driven by an internal ribosome entry site (IRES) that allows for tracking of relative protein expression levels. All mammalian proteins described in Supplementary Table S5 were purified directly from conditioned media using HisTrap FF Crude columns (GE catalog #17528601) and subsequently polished on a Superose 6 10/300 GL SEC column (GE #17517201) using an AKTA pure 25 instrument. For crystallization, MSLN fragments were produced from HEK293 FreeStyle™ cells treated with kifunensine (Chang et al., 2007) to reduce N-glycan heterogeneity and overall amount. Antibody Fab fragments were produced by papain cleavage of IgGs and the Fab purified on a MabSelect protein A column; antibody scFvs of 1A12 and anetumab were engineered with a flexible (GGGS)4 linker. The amatuximab scFv was produced with a thrombin cleavable linker incorporating an internal HisTag for purification.
SPR quantitative interaction analyses
Surface plasmon resonance (SPR) experiments were performed at 25°C on a Biacore T100 instrument (Cytiva) using a running buffer of 10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.05% surfactant P20 with 0.1 mg/mL bovine serum albumin. Goat anti-human IgG, Fcγ fragment specific antibody (Jackson ImmunoResearch: 109005098) was amine coupled to 2 flow cells of a Series S CM4 chip (∼2,800 RUs). Each antibody (1 μg/mL 1A12 or 0.3 μg/mL Amatuximab) was injected at 10 μL/min over 1 flow cell of immobilized anti-human IgG Fcγ for 35, 105, 35 or 30 s to capture 57.1 ± 0.8, 96 ± 1, 82 ± 2 or 81.9 ± 0.6 RUs of MDT846/984, MDT846/985, MDT1119/1121 or Amatuximab, respectively. MSLN-Avi was prepared by cleaving MDT536 with tobacco etch virus protease. Purified MSLN-Avi was run as a concentration series at 50 μL/min over both the captured antibody and anti-human IgG Fcγ alone surfaces. MSLN-Avi concentrations (serial 2-fold dilutions starting at 1 μM for the 1A12 constructs and 25 nM for Amatuximab) were run in duplicate, randomized, and included a buffer blank every 4th injection. MSLN-Avi was injected for 5 min and allowed to dissociate for 7 min from 1A12 to 3 min from Amatuximab. In addition, triplicate 50 nM samples of MSLN-Avi alternated with buffer blanks were injected over Amatuximab and allowed to dissociate for 2 h. The CM4 chip was regenerated with 10 mM glycine, pH 1.5 at 50 μL/min for 30 s, or 45 s for MDT1119/1121, and antibody recaptured prior to each MSLN-Avi injection. Data was double referenced and analyzed in BiaEval 2.0.4 with a 1:1 kinetic binding model.
T-cell cytotoxicity and cytokine release assays
Cancer cell lines OVCAR-3 and SKOV-3 (ATCC #HTB-161 and #HTB-77) were lentivirally transduced to express a near-infrared fluorescent reporter. Similarly, the NOMO-1 line (DSMZ #ACC-542) was marked with a gene encoding GFP. Cryopreserved healthy, unstimulated human donor PBMCs were obtained from Bloodworks NW. On the day of assay preparation, T-cells were isolated using a CD3+ magnetic negative selection kit (Stemcell Technologies #17951) according to the manufacturer’s instructions. All further assay preparation and sample collection was automated using a Microlab Starlet liquid handler (Hamilton). Target cells were co-cultured with purified T-cells at a 5:1 E:T ratio. Test molecules were added with all conditions performed in triplicate. Cells were incubated at 37°C for 48 H.
At 24 H, supernatant was collected to screen for cytokine release. At 48 H, cytotoxicity was measured by obtaining target cell counts. For the adherent lines, OVCAR-3 and SKOV-3, cells were imaged using an ImageXpress Nano and positive fluorescence was analyzed using the accompanying MetaXpress software (Molecular Devices). For the suspension line, NOMO-1, cells were collected, mixed with 1x DAPI, and run on an iQue Screener Plus (Sartorius) and live target cells were identified by forward/side scatter distribution, negativity for DAPI, and positive fluorescence. Raw cell counts were exported to Microsoft Excel and percent cytotoxicity was calculated utilizing the following formula:
Fitted curves were generated by applying a Sigmoidal, 4PL, x is log (concentration) model using Prism (GraphPad).
Cytokine quantification was performed using the iQue Qbeads® PlexScreen kit (Sartorius) as per the manufacturer’s protocol. Briefly, supernatants and assay kit cytokine standard were incubated with capture beads specific to IFNγ and IL-2. The samples were incubated for 2 H at room temperature with shaking (1,250 rpm). The beads were washed and incubated with prepared detection reagents to detect bound cytokines. Supernatant and reagent addition was performed on the Microlab Starlet liquid handler. The data was collected on the iQue Screen Plus, measuring fluorescence intensity in the BL2 channel. Cytokine concentrations were interpolated from cytokine standards and the resultant data was fitted using the Sigmoidal, 4PL, x is log (concentration) model in Prism.
Crystallography
Crystallization of free MSLN fragments proved infeasible, even after extensive screening. Alternatively, MSLN fragment complexes with combinations of Fabs and/or scFvs derived from amatuximab, anetumab, or 1A12 were isolated by SEC in 12 mM PIPES (pH = 7.2), 150 mM NaCl, 1 mM EDTA and concentrated to ∼10 mg/mL. Diffraction-quality crystals (dmin = 2.6 Å) of the ternary complex of MSLN fragment #1 with the scFvs of anetumab and 1A12 were obtained by vapor diffusion over a well solution of 1.2–1.4 M ammonium sulfate, 100 mM bicine (pH = 9.0), 3% v/v glycerol after multiple rounds of macro-seeding. Diffraction-quality crystals (dmin = 3.3 Å) of the binary complex of kifunensine-treated MSLN fragment #2 with 1A12 Fab were obtained by vapor diffusion over a well solution of 19%–22% w/w polyethylene glycol (Mr = 3,350), 100 mM TRIS (pH = 8.0), plus 250 mM sodium citrate. The highest quality crystals were obtained after multiple rounds of macro-seeding into drops of 1 mL of 3–6 mg/mL protein solution mixed with 1 mL of well solution. Crystals were cryopreserved by the addition of 15%–20% v/v glycerol to the well solution. Diffraction-quality crystals (dmin = 4.3 Å) of the ternary complex of kifunensine-treated MSLN fragment #2, the amatuximab scFv, and the 1A12 Fab were obtained by vapor diffusion over a well solution of 1.3–1.5 M ammonium sulfate, 50 mM TRIS (pH = 8.0), plus 14 mM ZnCl2. Diffraction data were collected at the Advanced Light Source (Berkeley, CA) beamlines 5.0.1 or 5.0.2 and integrated and scaled with HKL-2000 (Otwinowski and Minor, 1997). Initial phases for the MSLN fragment #2/1A12 Fab complex structure were determined by molecular replacement using Phaser (McCoy et al., 2007) as implemented in the CCP4 software suite (Winn et al., 2011) with 4F3F.pdb (Ma et al., 2012) (MSLN NTD/amatuximab Fab complex) as the search model, though preliminary electron density maps only supported building of a partial model. This partial model was used as a search model to generate initial phases by molecular replacement for the ternary MSLN fragment #2/amatuximab scFv/1A12 Fab and MSLN fragment #1/anetumab scFv/1A12 scFv complex structures. To complete model building, the RoseTTAFold MSLN model was used as a guide template iteratively across the three models. Model building and positional refinement were performed with refmac (Murshudov et al., 1997) and COOT (Emsley and Cowtan, 2004), including placing ordered solvent molecules, followed by a final round of TLS refinement (Winn et al., 2001). Residues or side chains that did not exhibit clear 2Fobs-Fcalc electron density when contoured at 0.7σ were removed or truncated to the Cβ atom. The quality of the final models was assessed using ProCheck and Molprobity (Supplementary Table S3).
The coordinates for the final models have been deposited in the RCSB PDB (Berman et al., 2000), accession codes: 8CZ8 (MSLN fragment #1/anetumab scFv/1A12 scFv complex), 8CYH (MSLN fragment #2/1A12 Fab complex), and 8CXC (MSLN fragment #1/anetumab scFv/1A12 scFv complex).
Results
Isolating anti-MSLN antibodies targeting novel epitopes
In order to target distinct regions of the MSLN ectodomain with novel antibody-based reagents, murine 3T3 cells were engineered to surface express a chimeric murine/human MSLN molecule, where the membrane-proximal ectodomain sequence of murine MSLN (residues 464-625, based on UniProt: Q61468) was swapped with the corresponding region of human MSLN (residues 470-630, based on UniProt: Q13421-1). The logic was that murine protein sequences would not be immunogenic in mice focusing induced humoral responses on the human membrane-proximal region (Supplementary Figure S1A). TRIANNI mice, which produce chimeric antibodies from a full repertoire of human heavy and light chain variable domains, while maintaining mouse constant domains, were immunized with the engineered murine 3T3 cells and hybridomas were isolated. Sequencing mesothelin-positive monoclonal hybridomas yielded 22 paired heavy (VH) and light (VL) chain variable domain sequences of which 18 belonged to a single sequence family (Supplementary Figure S1B), where the VH domain was derived from the IGHV3-23*01 germline, defining a single clonotype: sequence identities across these 18 CDRH3 and CDRL3 segments were >80% and >78%, respectively. There were few predicted deamidation or isomerization liabilities (Yan et al., 2018) and no non-standard cysteines or glycosylation sites, particularly in the CDRs. The clonotype archetype, clone 1A12, as well as other related clones, were reformatted by replacing the murine constant regions with human constant regions (aglycosylated IgG1) and recombinantly expressed using FreeStyle 293F cells (Bandaranayake et al., 2011). The binding of 1A12, as an IgG1, to MSLN+ and MSLN-knockout cell lines was evaluated by flow cytometry in head-to-head comparisons with amatuximab, showing comparable sensitivity and specificity (Figure 1A). The interaction parameters of 1A12 and amatuximab for recombinant MSLN were quantitated by surface plasmon resonance (SPR) biosensor analyses, showing univalent KDs of 64.4 ± 0.1 nM (ligand: captured 1A12 IgG, analyte: MSLN ectodomain; Figure 1B) and 112 ± 1 pM (ligand: captured amatuximab IgG, analyte: MSLN ectodomain; Figure 1C). The reported univalent KD of amatuximab for MSLN is 1.5 nM (Hassan et al., 2007), and of anetumab for MSLN is 10 nM (Golfier et al., 2014).
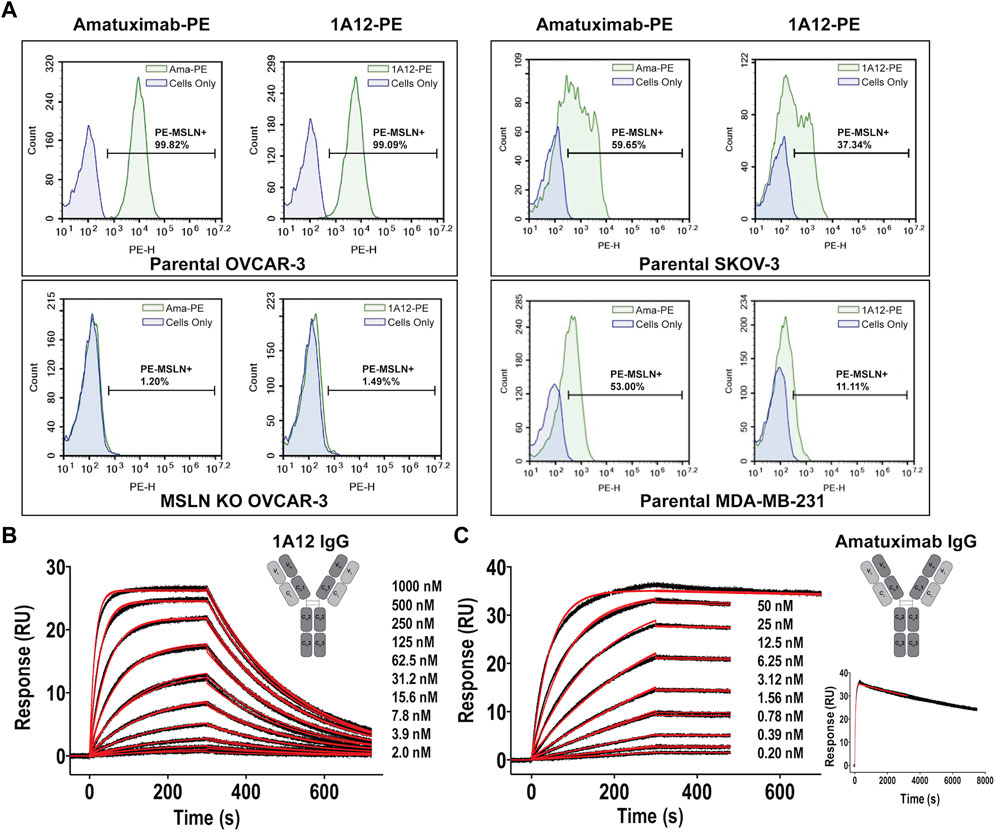
FIGURE 1. Binding characterization of antibody clone 1A12 to MSLN. (A) Comparative staining of MSLN expressing cell lines OVCAR-3 (top, left panels) and SKOV-3 and MDA-MB-231 (right panels) with amatuximab and clone 1A12 by flow cytometry. Staining of OVCAR-3 cells where MSLN was genetically knocked-out confirms target specificity (bottom, left panels). SPR sensorgrams showing the interaction analyses of (B) the 1A12 Fab (formatted as a human IgG1 antibody) and (C), the amatuximab Fab (formatted as a human IgG1 antibody). Black lines represent the duplicate experimental response curves; red lines represent the modeled kinetic curves. Insets show (B,C) schematic representations of the engineered antibody constructs, analyte concentrations, and (C) an extended analysis at highest analyte concentration.
Antibody co-crystal structures of MSLN ectodomain fragments
In order to confirm that the desired targeting of 1A12 was achieved and to compare epitopes, three co-crystal structures with recombinant MSLN ectodomain fragments [residues 434-590 (#1), and 296-605 (#2)] were determined at dmins ranging from 2.6 to 4.3 Å (Figure 2; Supplementary Table S3). MSLN fragments were produced in human cell lines using the Daedalus protein expression system (Bandaranayake et al., 2011). Using combinations of either Fab or scFv fragments of 1A12, amatuximab, and anetumab as co-crystallization “chaperones” enabled these structure determinations, including a fragment of MSLN (#2) spanning the entire ectodomain, revealing its overall fold. Antibody Fab fragments were produced by papain cleavage of IgGs and purified on a MabSelect™ protein A column; antibody scFvs were engineered with a flexible (GGGS)4 linker and either purified separately or as co-expressed MSLN ectodomain complexes. Even in the lowest resolution structure, the final electron density maps were cleanly resolvable, ultimately allowing modeling of the entire structure, including N-acetylglucosamine residues at the two N-glycan sites in MSLN (N388, N523), except for a prominent disordered loop (residues 405-414). This disordered loop spans an eight amino acid insertion (QAPRRPLP) only present in human MSLN isoform 1 (UniProt: Q13421-1) among human isoforms and Pan troglodytes isoform 4 (A0A2J8J880) among primate MSLN sequences, though a similar insert of matching length does appear in a few more distantly related primate orthologs.
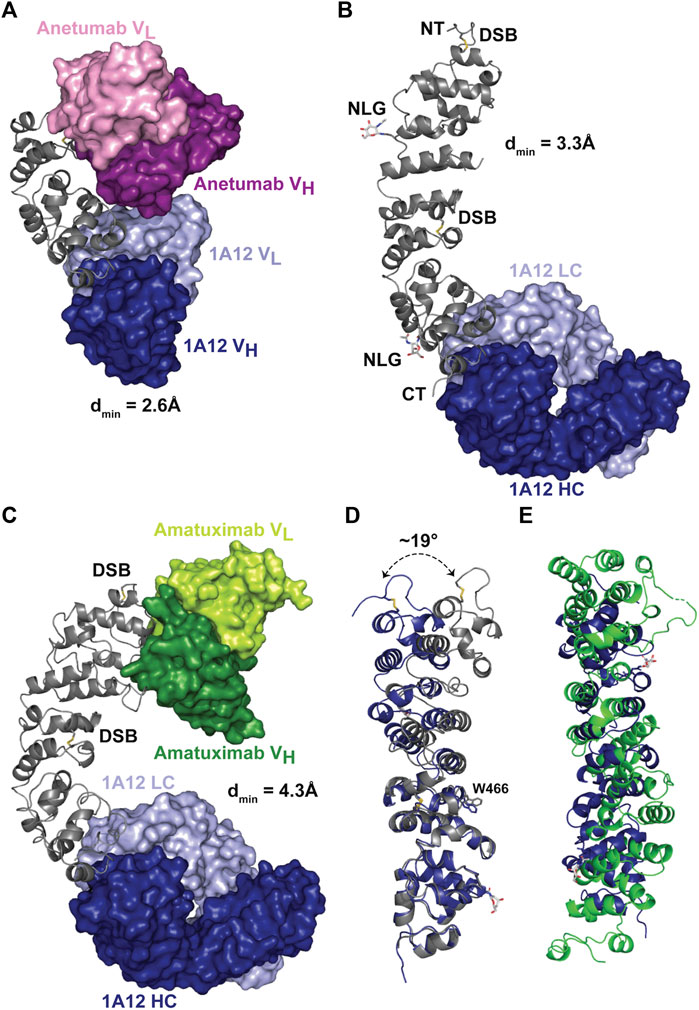
FIGURE 2. X-ray crystal structures of MSLN bound to 1A12, amatuximab, and anetumab. (A) Crystal structure of 1A12 and anetumab scFvs in complex with a minimized fragment of the MSLN ectodomain, spanning residues 434-590. The MSLN domain is shown in a ribbon representation, colored gray, while he variable domains of the scFvs are shown in molecular surface representations, colored as indicated. (B) Crystal structure of 1A12 Fab in complex with the MSLN ectodomain, spanning residues 296-605. The resolvable N/C termini (“NT”, “CT”), disulfide bonds (yellow connectors) and glycosylation sites (“NLG”, with resolvable sugar group shown in a stick representation) are denoted. (C) Crystal structure of 1A12 Fab/amatuximab scFv in complex with the MSLN ectodomain, spanning residues 296-605, colored as indicated. (D) Structural alignment of the MSLN ectodomains from the 1A12 Fab/amatuximab scFv/MSLN296-605 (gray) and 1A12 Fab/MSLN296-605 (blue) complex structures, shown in ribbon representations, highlighting the flexibility of the ectodomain (superposition on C⍺ 524-589). (E) Superposition of the Rif1 NTD (green) and the MSLN ectodomain (blue) based on the Dali alignment.
The MSLN ectodomain is composed of a series of short amphipathic helices that wind in a coiled fashion to produce a slightly bent, elongated structure (Figures 2A–C), extending the compact, right-handed α-superhelical fold observed in the preceding structure of the MSLN NTD (Ma et al., 2012). Each helix is oriented to position its non-polar face towards the central axis resulting in the packing of an extended hydrophobic core. The ectodomain contains two disulfide bonds, with the first (C302-C326) in the NTD securing the initial helical packing and the second (C450-C476) bridging between superhelical turns near the middle of the ectodomain. This disulfide constrains the packing of several surrounding helices resulting in the solvent-exposure of W466 and defining a hinge point that is the largest element of overall flexibility observed in the MSLN ectodomain (∼19°) when the two full length MSLN ectodomain were superimposed on their C-terminal ends (residues 476-587; Figure 2D).
One goal of MSLN structural analyses was to advance its functional annotation by identifying structurally related proteins with potentially informative, related functions. However, the overall fold of the extracellular domain of MSLN is fairly distinct, based on structural similarity searches using the DALI server (Holm, 2020). The closest match was the NTD of Saccharomyces cerevisiae Rif1 [5NW5.pdb (Mattarocci et al., 2017)], but the structural similarity was weak, with a Z-score of 4.9, obvious also by visual inspection (Figure 2E), and there was no obvious sequence similarity. Functionally, Rif1 is an intracellular DNA binding protein involved in double-strand break processing, telomere capping, replication initiation, silencing, and rDNA stability, providing no useful information about MSLN extracellular domain function/s. However, despite the relative uniqueness of the MSLN structure as determined experimentally, state-of-the-art structure prediction algorithms performed extremely well, even in detail. We compared the MSLN crystal structures with its predicted structure, available either through the AlphaFold protein structure database (Jumper et al., 2021; Varadi et al., 2022) (rmsd = 0.80 Å, residues 449-589) or as a one-side blinded prediction using RoseTTAFold (Baek et al., 2021) (rmsd = 1.63 Å; residues 448-589; Supplementary Figure S2). While slight deviations from experiment were noted throughout both predictions, both algorithms correctly identified the 405-414 disordered loop. Indeed, the accuracy of these results was such that at least one of the crystallographer co-authors decided to check the status of their retirement plan.
These three antibody/MSLN co-crystal structures enabled high-resolution mapping of the epitopes of anetumab and 1A12 and the relationship between these and that of amatuximab (Supplementary Figure S3A; Supplementary Tables S3, S4). The amatuximab scFv was used in combination with the 1A12 Fab to generate one of the two full length MSLN co-crystal structures which recapitulated all the salient details of the previously reported co-crystal structure with the isolated MSLN NTD. Anetumab binds near the middle of the ectodomain and 1A12, as intended, binds near the C-terminus. All three antibodies have distinct epitopes which do not cross-block, though all three preferentially bind to relatively negatively-charged patches on MSLN, which overall displays a patchwork of positive/negative surface charge (Supplementary Figure S3B). At the antibody/MSLN interfaces, amatuximab buries ∼890 Å2 of solvent-accessible surface and makes 6-8 hydrogen bonds and two salt bridges; anetumab buries ∼825 Å2 making 6-8 hydrogen bonds and 2-3 salt bridges; and 1A12 buries ∼600 Å2 and interacts primarily through hydrophobic contacts plus 4-6 hydrogen bonds. The reduced footprint of 1A12 likely contributes to its reduced affinity for MSLN: ∼6-fold weaker than anetumab and ∼40-fold weaker than amatuximab.
Engineering novel anti-MSLN T cell-redirecting biologics
One widely employed approach for developing antibodies targeting tumor antigens into therapeutics is to engineer bispecific reagents, where one antibody specificity is directed against the tumor antigen and the other antibody specificity is directed against a T cell-associated antigen, often the CD3 signaling moiety of the T cell holoreceptor (Arvedson et al., 2022). When infused, these reagents recruit and activate otherwise non-tumor specific, bulk T cells, generating potent anti-tumor responses. 1A12 was engineered into two different bispecific architectures: “IgG-scFv” (Figure 3A) and “scFv-Fc” (Figure 3B). Engineered protein and antibody sequences are appended in Supplementary Table S5 and were produced using the Daedalus protein expression system in human cell lines (Bandaranayake et al., 2011). The 1A12 IgG-scFv was initially tested with four anti-CD3 scFv moieties derived from the parental antibodies OKT3 (Kjer-Nielsen et al., 2004), UCHT1 (Arnett et al., 2004), MICRO194 (Kischel et al., 2009), and ADI26906 (Walker Laura et al., 2020; Liu et al., 2023). All four expressed well using the FreeStyle™ 293F mammalian tissue culture system and could be purified to homogenous, monodisperse species stable even after a freeze-thaw cycle (Supplementary Figure S4). 1A12 was also used to engineer an scFv-Fc construct with OKT3. The 1A12/OKT3 IgG-scFv and scFv-Fc constructs retained binding to MSLN, with univalent KDs for soluble MSLN of 63.0 ± 0.1 nM (ligand: captured 1A12 IgG-scFv, analyte: MSLN ectodomain) and 103 ± 1 nM (ligand: captured 1A12 scFv-Fc, analyte: MSLN ectodomain), respectively (Figures 3C, D), within two-fold of the parent, un-engineered 1A12 IgG.
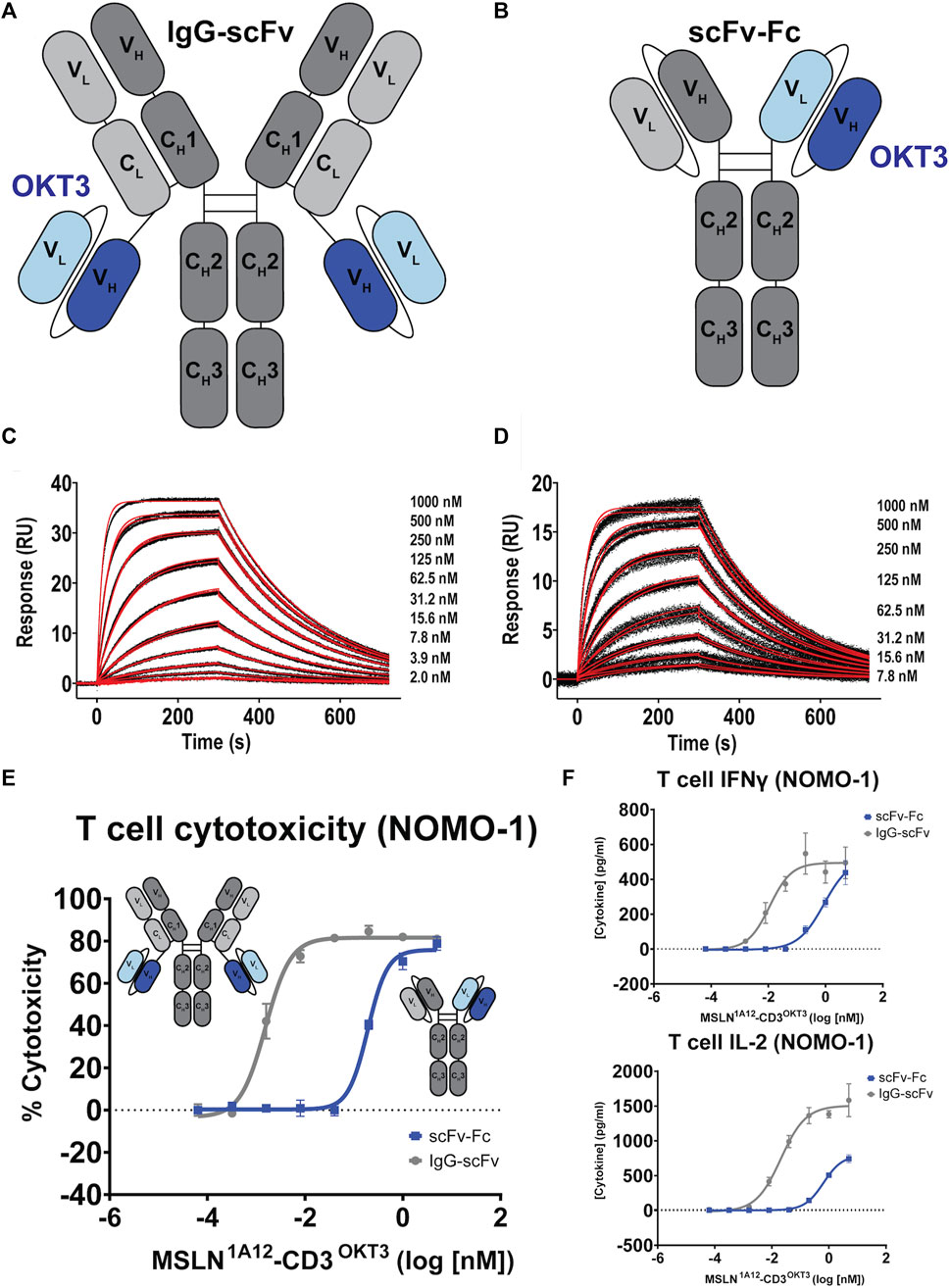
FIGURE 3. Functional comparison of 1A12-derived IgG-scFv and scFv-Fc bispecific T cell engagers. (A,B) Schematics of aglycosylated IgG-scFv and scFv-Fc bispecific constructs incorporating OKT3 scFvs. (C,D) SPR sensorgrams showing binding of the 1A12/OKT3 IgG-scFv and scFv-Fc to recombinant MSLN. Black lines represent the duplicate experimental response curves; red lines represent the modeled kinetic curves. Insets show analyte concentrations. (E) T cell-mediated cytotoxicity comparing the potency of the two bispecific formats on MSLN-expressing NOMO-1 cells, colored as indicated. Insets show construct schematics. (F) Resulting IFNγ and IL-2 production as a function of bispecific antibody concentration colored as indicated.
Focusing on the OKT3 fusions because of its long history and superior product quality in the IgG-scFv format, both IgG-scFv (“IgG1A12-scFvOKT3”) and scFv-Fc (“scFv-Fc1A12/OKT3”) constructs demonstrated in vitro functional T cell engagement and activation in the nanomolar-to-picomolar range against the MSLN+, human acute monocytic leukemia cell line NOMO-1 in redirected cytotoxicity and cytokine interferon-γ (IFN-γ) and interleukin 2 (IL-2) release assays (Figures 3E, F). Because of its greater potency, reasonably a product of the greater inherent valency of a bi-bivalent IgG-scFv over that of a bi-monovalent scFv-Fc, IgG1A12-scFvOKT3 was advanced for head-to-head testing with the analogous constructs incorporating amatuximab (alternate anti-MSLN moiety) or ADI26906 (alternate anti-CD3 moiety): IgGAMA-scFvOKT3, IgG1A12-scFvADI, and IgGAMA-scFvADI. IgG-scFv constructs with 1A12-based MSLN-targeting moieties (IgG1A12-scFv) were roughly a half-log more potent than IgGAMA-scFv constructs, with either OKT3 anti-CD3 moieties (IgG-scFvOKT3; Figure 4A) or ADI26906 moieties (IgG-scFvADI; Figure 4B) in in vitro redirected cytotoxicity assays against OVCAR-3 cells. The improved potency of both IgG1A12-scFv constructs over amatuximab-based bispecifics was reasonably the result of targeting an epitope closer to the tumor cell surface, tightening the T cell synapse, as has been previously observed (Bluemel et al., 2010; Tang et al., 2013a).
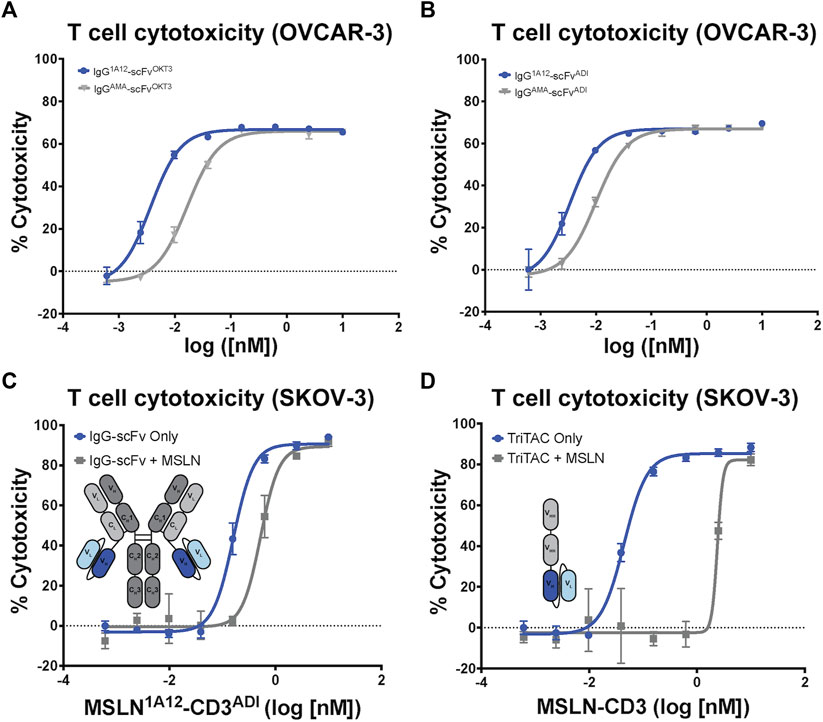
FIGURE 4. Comparison of amatuximab and 1A12 derived bispecific antibodies with different CD3 engagers highlights tolerance to shed MSLN. T cell-mediated cytotoxicity of IgG-scFv bispecific constructs utilizing either amatuximab or 1A12 and (A) OKT3 or (B) ADI as contrasting anti-CD3 scFvs. (C) T cell-mediated cytotoxicity of the MSLN1A12-CD3ADI IgG-scFv in the absence and presence of 30 nM soluble MSLN shows minimal impact on potency. (D) T cell-mediated cytotoxicity of the MSLN-CD3 TriTAC in the absence and presence of 30 nM soluble MSLN shows a profound impact on potency. Insets in (C,D) show schematic representations of the fusion constructs.
It is well established that SMRPs have the potential to confound antibody-based therapeutics by preventing the therapies from reaching the tumor cell surface (Zhang et al., 2007; Liu et al., 2022). To assess how soluble MSLN may affect the potency of T-cell engaging therapies we performed in vitro killing assays with SKOV-3 cells in the presence of a high concentration of recombinant MSLN. We compared the potency of the IgG1A12-scFvADI with that of a trispecific T-cell engager (known as HPN536) with a novel VHH-VHH-scFv format (referred to as a TriTAC). The TriTAC binds human MSLN, albumin and CD3 with monovalent affinities (KDs of .21 nM, 6.3 nM, and 6.6 nM respectively) and has reached clinical trials (Molloy et al., 2021). Both IgG1A12-scFv (Figure 4C) and the TriTAC molecule (Figure 4D) kill SKOV-3 cells with high potency (EC50s of 6.6 pM and 3.6 pM respectively). To mimic physiologic MSLN levels we performed the cytotoxicity experiment in the presence of 1ug/mL (∼30 nM) recombinant MSLN (Zhang et al., 2012). Both molecules were inhibited by the soluble ectodomain but the fold-change in potency was much improved (Figures 4C, D), with the bivalent molecule only showing a three-fold reduction in potency (compared to 60-fold for the VHH-VHH-scFv). This result suggests that bivalent binding of the IgG-scFv may have advantages over a monomeric T cell engager when soluble antigen sinks are a concern.
Discussion
Considerable effort over multiple modalities has been exerted to effectively target MSLN for anti-cancer therapy. Advances include broadening the panel of anti-MSLN antibodies available, developing approaches to overcome interference by cancer-associated SMRPs, and evaluating biologics from a structure-based perspective. Employing a novel immunization strategy using a chimeric form of MSLN, we have isolated a panel of fully human anti-MSLN antibodies targeting a more membrane-proximal epitope potentially improving the efficacy of engineered biologics. Using these antibodies, we have successfully determined the crystal structure of the intact MSLN ectodomain, revealing a formally novel fold and mapping the precise epitopes for two additional anti-MSLN antibodies. The structure of MSLN, comprising an extended superhelical bundle of α-helices, failed to provide useful insights into its precise molecular functions, partly due to its uniqueness, but does now provide the foundation for determining the details of interactions with its physiological partners. Building on our newly isolated antibodies, we have engineered multiple novel biologic reagents demonstrating uniquely advantageous in vitro properties as candidate therapeutics. Specifically, the bispecific T cell-redirecting IgG-scFv format, which binds avidly to cell-surface MSLN, was shown to be more potent than the scFv-Fc format and minimally sensitive to soluble MSLN. When comparing IgG-scFv molecules derived from 1A12 and amatuximab head-to-head, the 1A12-based molecules, which bind a membrane proximal epitope with lower affinity, demonstrated higher potency, highlighting the importance of epitope in the design of T cell-redirecting therapies. The next step in the clinical development of these reagents will be the evaluation of in vivo potency using mouse models and safety pharmacology using cynomolgus monkeys. While T cell-based therapies show great promise, the expression of MSLN in normal tissue requires that the potency and dosing of these modalities be carefully considered in order to minimize potential on-target, off-tumor toxicity (Haas et al., 2019; Molloy et al., 2021; Haas et al., 2023).
Data availability statement
Crystallographic structure factors and model coordinates have been deposited in the Protein Data Bank under accession codes: 8CXC, 8CYH, and 8CZ8. The sequences of all the protein constructs used in the study can be found in the Supplementary Material. All other data can be obtained by contacting the corresponding authors.
Author contributions
KP and RR conducted protein expression, purification, and biochemical characterization experiments. CC and PR designed and conducted all crystallization experiments. DF conducted surface plasmon resonance protein binding studies. CC designed all protein constructs. CC and BH conducted the antibody discovery campaign. AB, IL, and MaC conducted T-cell killing experiments. CC and MiC sequence verified all antibodies described in the study. SM, CM, JO, and RS facilitated all aspects of the research in their labs. JC provided logistical support for all efforts. CC, PR, and RS wrote the manuscript, which was edited by all authors. All authors contributed to the article and approved the submitted version.
Funding
This work was funded by the Washington Research Foundation, St. Baldrick’s Foundation, Children’s Oncology Group Foundation and by the Fred Hutchinson Cancer Research Center.
Conflict of interest
IL, MaC, MiC, AB, JC, CM, and CC were employees of the Fred Hutchinson Cancer Research Center but moved to Link Immunotherapeutics, Inc. during the drafting of the manuscript. AB, SM, CM, JO, and CC are named inventors on IP corresponding to the technology in this manuscript. AB, CM, JO, and CC are founders of Link Immunotherapeutics Inc., which recently licensed the technology for use in clinical applications.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fddsv.2023.1216516/full#supplementary-material
References
Arnett, K. L., Harrison, S. C., and Wiley, D. C. (2004). Crystal structure of a human CD3-epsilon/delta dimer in complex with a UCHT1 single-chain antibody fragment. Proc. Natl. Acad. Sci. U. S. A. 101, 16268–16273. doi:10.1073/pnas.0407359101
Arvedson, T., Bailis, J. M., Urbig, T., and Stevens, J. L. (2022). Considerations for design, manufacture, and delivery for effective and safe T-cell engager therapies. Curr. Opin. Biotechnol. 78, 102799. doi:10.1016/j.copbio.2022.102799
Awuah, P., Bera, T. K., Folivi, M., Chertov, O., and Pastan, I. (2016). Reduced shedding of surface mesothelin improves efficacy of mesothelin-targeting recombinant immunotoxins. Mol. Cancer Ther. 15, 1648–1655. doi:10.1158/1535-7163.MCT-15-0863
Baek, M., Dimaio, F., Anishchenko, I., Dauparas, J., Ovchinnikov, S., Lee, G. R., et al. (2021). Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876. doi:10.1126/science.abj8754
Bandaranayake, A. D., Correnti, C., Ryu, B. Y., Brault, M., Strong, R. K., and Rawlings, D. J. (2011). Daedalus: A robust, turnkey platform for rapid production of decigram quantities of active recombinant proteins in human cell lines using novel lentiviral vectors. Nucleic Acids Res. 31, e143. doi:10.1093/nar/gkr706
Bera, T. K., and Pastan, I. (2000). Mesothelin is not required for normal mouse development or reproduction. Mol. Cell Biol. 20, 2902–2906. doi:10.1128/MCB.20.8.2902-2906.2000
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The protein data bank. Nucleic Acids Res. 28, 235–242. doi:10.1093/nar/28.1.235
Bluemel, C., Hausmann, S., Fluhr, P., Sriskandarajah, M., Stallcup, W. B., Baeuerle, P. A., et al. (2010). Epitope distance to the target cell membrane and antigen size determine the potency of T cell-mediated lysis by BiTE antibodies specific for a large melanoma surface antigen. Cancer Immunol. Immunother. 59, 1197–1209. doi:10.1007/s00262-010-0844-y
Cameron, B., Dabdoubi, T., Berthou-Soulie, L., Gagnaire, M., Arnould, I., Severac, A., et al. (2020). Complementary epitopes and favorable developability of monoclonal anti-LAMP1 antibodies generated using two transgenic animal platforms. PLoS One 15, e0235815. doi:10.1371/journal.pone.0235815
Chang, K., Pai, L. H., Batra, J. K., Pastan, I., and Willingham, M. C. (1992a). Characterization of the antigen (CAK1) recognized by monoclonal antibody K1 present on ovarian cancers and normal mesothelium. Cancer Res. 52, 181–186.
Chang, K., and Pastan, I. (1996). Molecular cloning of mesothelin, a differentiation antigen present on mesothelium, mesotheliomas, and ovarian cancers. Proc. Natl. Acad. Sci. U. S. A. 93, 136–140. doi:10.1073/pnas.93.1.136
Chang, K., Pastan, I., and Willingham, M. C. (1992b). Isolation and characterization of a monoclonal antibody, K1, reactive with ovarian cancers and normal mesothelium. Int. J. Cancer 50, 373–381. doi:10.1002/ijc.2910500308
Chang, V. T., Crispin, M., Aricescu, A. R., Harvey, D. J., Nettleship, J. E., Fennelly, J. A., et al. (2007). Glycoprotein structural genomics: Solving the glycosylation problem. Structure 15, 267–273. doi:10.1016/j.str.2007.01.011
Cinausero, M., Rihawi, K., Cortiula, F., Follador, A., Fasola, G., and Ardizzoni, A. (2019). Emerging therapies in malignant pleural mesothelioma. Crit. Rev. Oncol. Hematol. 144, 102815. doi:10.1016/j.critrevonc.2019.102815
Davis, A. P., Kao, S. C., Clarke, S. J., Boyer, M., and Pavlakis, N. (2021). Emerging biological therapies for the treatment of malignant pleural mesothelioma. Expert Opin. Emerg. Drugs 26, 179–192. doi:10.1080/14728214.2021.1924670
Emsley, P., and Cowtan, K. (2004). Coot: Model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 60, 2126–2132. doi:10.1107/S0907444904019158
Golfier, S., Kopitz, C., Kahnert, A., Heisler, I., Schatz, C. A., Stelte-Ludwig, B., et al. (2014). Anetumab ravtansine: A novel mesothelin-targeting antibody-drug conjugate cures tumors with heterogeneous target expression favored by bystander effect. Mol. Cancer Ther. 13, 1537–1548. doi:10.1158/1535-7163.MCT-13-0926
Greenfield, E. A. (2014). Antibodies: A laboratory manual. New York: Cold Spring Harbor Laboratory Press.
Haas, A. R., Golden, R. J., Litzky, L. A., Engels, B., Zhao, L., Xu, F., et al. (2023). Two cases of severe pulmonary toxicity from highly active mesothelin-directed CAR T cells. Mol. Ther. 2023, 6. doi:10.1016/j.ymthe.2023.06.006
Haas, A. R., Tanyi, J. L., O'Hara, M. H., Gladney, W. L., Lacey, S. F., Torigian, D. A., et al. (2019). Phase I study of lentiviral-transduced chimeric antigen receptor-modified T cells recognizing mesothelin in advanced solid cancers. Mol. Ther. 27, 1919–1929. doi:10.1016/j.ymthe.2019.07.015
Hassan, R., Ebel, W., Routhier, E. L., Patel, R., Kline, J. B., Zhang, J., et al. (2007). Preclinical evaluation of MORAb-009, a chimeric antibody targeting tumor-associated mesothelin. Cancer Immun. 7, 20.
Hassan, R., Remaley, A. T., Sampson, M. L., Zhang, J., Cox, D. D., Pingpank, J., et al. (2006). Detection and quantitation of serum mesothelin, a tumor marker for patients with mesothelioma and ovarian cancer. Clin. Cancer Res. 12, 447–453. doi:10.1158/1078-0432.CCR-05-1477
Hellstrom, I., Raycraft, J., Kanan, S., Sardesai, N. Y., Verch, T., Yang, Y., et al. (2006). Mesothelin variant 1 is released from tumor cells as a diagnostic marker. Cancer Epidemiol. Biomarkers Prev. 15, 1014–1020. doi:10.1158/1055-9965.EPI-05-0334
Hilliard, T. S. (2018). The impact of mesothelin in the ovarian cancer tumor microenvironment. Cancers (Basel) 10, 277. doi:10.3390/cancers10090277
Ho, M., Onda, M., Wang, Q. C., Hassan, R., Pastan, I., and Lively, M. O. (2006). Mesothelin is shed from tumor cells. Cancer Epidemiol. Biomarkers Prev. 15, 1751. doi:10.1158/1055-9965.EPI-06-0479
Holm, L. (2020). Using Dali for protein structure comparison. Methods Mol. Biol. 2112, 29–42. doi:10.1007/978-1-0716-0270-6_3
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589. doi:10.1038/s41586-021-03819-2
Kim, D. E., Chivian, D., and Baker, D. (2004). Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 32, W526–W531. doi:10.1093/nar/gkh468
Kischel, R., Raum, T., Schlereth, B., Rau, D., Cierpka, R., and Kufer, P. (2009). Composition comprising cross-species-sepecific antibodies and uses thereof. United States: US Patent Application.
Kjer-Nielsen, L., Dunstone, M. A., Kostenko, L., Ely, L. K., Beddoe, T., Mifsud, N. A., et al. (2004). Crystal structure of the human T cell receptor CD3 epsilon gamma heterodimer complexed to the therapeutic mAb OKT3. Proc. Natl. Acad. Sci. U. S. A. 101, 7675–7680. doi:10.1073/pnas.0402295101
Liu, C. Y., Ahonen, C. L., Brown, M. E., Zhou, L., Welin, M., Krauland, E. M., et al. (2023). Structure-based engineering of a novel CD3ε-targeting antibody for reduced polyreactivity. MAbs 15, 2189974. doi:10.1080/19420862.2023.2189974
Liu, X., Chan, A., Tai, C. H., Andresson, T., and Pastan, I. (2020). Multiple proteases are involved in mesothelin shedding by cancer cells. Commun. Biol. 3, 728. doi:10.1038/s42003-020-01464-5
Liu, X., Onda, M., Watson, N., Hassan, R., Ho, M., Bera, T. K., et al. (2022). Highly active CAR T cells that bind to a juxtamembrane region of mesothelin and are not blocked by shed mesothelin. Proc. Natl. Acad. Sci. U. S. A. 119, e2202439119. doi:10.1073/pnas.2202439119
Ma, J., Tang, W. K., Esser, L., Pastan, I., and Xia, D. (2012). Recognition of mesothelin by the therapeutic antibody MORAb-009: Structural and mechanistic insights. J. Biol. Chem. 287, 33123–33131. doi:10.1074/jbc.M112.381756
Mattarocci, S., Reinert, J. K., Bunker, R. D., Fontana, G. A., Shi, T., Klein, D., et al. (2017). Rif1 maintains telomeres and mediates DNA repair by encasing DNA ends. Nat. Struct. Mol. Biol. 24, 588–595. doi:10.1038/nsmb.3420
Mccoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C., and Read, R. J. (2007). Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. doi:10.1107/S0021889807021206
Molloy, M. E., Austin, R. J., Lemon, B. D., Aaron, W. H., Ganti, V., Jones, A., et al. (2021). Preclinical characterization of HPN536, a trispecific, T-cell-activating protein construct for the treatment of mesothelin-expressing solid tumors. Clin. Cancer Res. 27, 1452–1462. doi:10.1158/1078-0432.CCR-20-3392
Murshudov, G. N., Vagin, A. A., and Dodson, E. J. (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D. Biol. Crystallogr. 53, 240–255. doi:10.1107/S0907444996012255
Otwinowski, Z., and Minor, W. (1997). “Processing of X-ray diffraction data collected in oscillation mode,” in Meth. Enzymol. Editors C. W. Carter, and R. M. SWEET (United States: Academic Press).
Pastan, I., and Zhang, Y. (2012). Modulating mesothelin shedding to improve therapy. Oncotarget 3, 114–115. doi:10.18632/oncotarget.445
Peter, A. S., Roth, E., Schulz, S. R., Fraedrich, K., Steinmetz, T., Damm, D., et al. (2021). A pair of noncompeting neutralizing human monoclonal antibodies protecting from disease in a SARS-CoV-2 infection model. Eur. J. Immunol. 52, 770–783. doi:10.1002/eji.202149374
Sapede, C., Gauvrit, A., Barbieux, I., Padieu, M., Cellerin, L., Sagan, C., et al. (2008). Aberrant splicing and protease involvement in mesothelin release from epithelioid mesothelioma cells. Cancer Sci. 99, 590–594. doi:10.1111/j.1349-7006.2007.00715.x
Scholler, N., Fu, N., Yang, Y., Ye, Z., Goodman, G. E., Hellstrom, K. E., et al. (1999). Soluble member(s) of the mesothelin/megakaryocyte potentiating factor family are detectable in sera from patients with ovarian carcinoma. Proc. Natl. Acad. Sci. U. S. A. 96, 11531–11536. doi:10.1073/pnas.96.20.11531
Tang, Z., Feng, M., Gao, W., Phung, Y., Chen, W., Chaudhary, A., et al. (2013a). A human single-domain antibody elicits potent antitumor activity by targeting an epitope in mesothelin close to the cancer cell surface. Mol. Cancer Ther. 12, 416–426. doi:10.1158/1535-7163.MCT-12-0731
Tang, Z., Qian, M., and Ho, M. (2013b). The role of mesothelin in tumor progression and targeted therapy. Anticancer Agents Med. Chem. 13, 276–280. doi:10.2174/1871520611313020014
Trianni, I. (2016). Transgenic mice: Transforming targeted monoclonal antibody (mAb) therapeutics. Berlin, Germany: Springer.
Varadi, M., Anyango, S., Deshpande, M., Nair, S., Natassia, C., Yordanova, G., et al. (2022). AlphaFold protein structure database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439–D444. doi:10.1093/nar/gkab1061
Walker Laura, M., Pejchal, R., Krauland, E., Vasquez, M., and Leung Monica Wai, L. (2020). Anti-CD3-Binding domains and antibodies comprising them, and methods for their generation and use. United States: US patent application.
Winn, M. D., Ballard, C. C., Cowtan, K. D., Dodson, E. J., Emsley, P., Evans, P. R., et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D. Biol. Crystallogr. 67, 235–242. doi:10.1107/S0907444910045749
Winn, M. D., Isupov, M. N., and Murshudov, G. N. (2001). Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. D. Biol. Crystallogr. 57, 122–133. doi:10.1107/s0907444900014736
Yan, Q., Huang, M., Lewis, M. J., and Hu, P. (2018). Structure based prediction of asparagine deamidation propensity in monoclonal antibodies. MAbs 10, 901–912. doi:10.1080/19420862.2018.1478646
Zhang, J., Qiu, S., Zhang, Y., Merino, M., Fetsch, P., Avital, I., et al. (2012). Loss of mesothelin expression by mesothelioma cells grown in vitro determines sensitivity to anti-mesothelin immunotoxin SS1P. Anticancer Res. 32, 5151–5158.
Keywords: mesothelin, antibody, CD3, bispecific, T cell, cancer, X-ray crystallography
Citation: Lin I, Rupert PB, Pilat K, Ruff RO, Friend DJ, Chan MK, Clarke M, Hoffstrom BG, Carter J, Meshinchi S, Bandaranayake AD, Mehlin C, Olson JM, Strong RK and Correnti CE (2023) Novel mesothelin antibodies enable crystallography of the intact mesothelin ectodomain and engineering of potent, T cell-engaging bispecific therapeutics. Front. Drug Discov. 3:1216516. doi: 10.3389/fddsv.2023.1216516
Received: 04 May 2023; Accepted: 20 July 2023;
Published: 03 August 2023.
Edited by:
Yusuf Tutar, University of Health Sciences, TürkiyeReviewed by:
Sayed K. Goda, University of Derby, United KingdomChristian Klein, Roche Innovation Center Zurich, Switzerland
Copyright © 2023 Lin, Rupert, Pilat, Ruff, Friend, Chan, Clarke, Hoffstrom, Carter, Meshinchi, Bandaranayake, Mehlin, Olson, Strong and Correnti. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James M. Olson, amltLm9sc29uQHNlYXR0bGVjaGlsZHJlbnMub3Jn; Roland K. Strong, cnN0cm9uZ0BmcmVkaHV0Y2gub3Jn; Colin E. Correnti, Y29saW4uY29ycmVudGlAZ21haWwuY29t
†Present address: Ida Lin, Man Kid Chan, Midori Clarke, Jane Carter, Ashok D. Bandaranayake, Christopher Mehlin, Colin E. Correnti, Link Immunotherapeutics, Seattle, WA, United States; Kristina Pilat, Raymond O. Ruff, James M. Olson, Seattle Children’s Research Institute, Seattle, WA, United States; Benjamin G. Hoffstrom, University of California Los Angeles, Department of Medicine, Translational Oncology Research Lab, Santa Monica, CA, United States
‡These authors have contributed equally to this work and share first authorship