
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Drug Discov. , 09 May 2023
Sec. Anti-Cancer Drugs
Volume 3 - 2023 | https://doi.org/10.3389/fddsv.2023.1182146
 Irene Tang1
Irene Tang1 Lauren Schwimmer1†
Lauren Schwimmer1† Shenda Gu1
Shenda Gu1 Wei Wei Prior1†
Wei Wei Prior1† Hieu Van Tran1Allan Chan1†Anna McClain1†
Hieu Van Tran1Allan Chan1†Anna McClain1† Christopher C. Fraser1*Chunyan Sun2Meimei Si3
Christopher C. Fraser1*Chunyan Sun2Meimei Si3 Guijiang Wang2Yunxia Zhao2Ning Zhang2Jiayu Fu2Mengxin Liu2
Guijiang Wang2Yunxia Zhao2Ning Zhang2Jiayu Fu2Mengxin Liu2 Chuanzeng Cao2Shihao Chen1*
Chuanzeng Cao2Shihao Chen1*Cell surface molecules PD-L1 and CD47 are potent inhibitors of adaptive and innate anti-cancer immunity. We sought to generate a safe, therapeutic, bispecific antibody specifically targeting, and blocking both PD-L1 and CD47 inhibitory activity. Novel anti-PDL-1 and anti-CD47 antibodies with favorable inhibitory activity, were humanized and constructed into a unique bi-specific antibody intended for clinical use. Previous pre-clinical and clinical studies using anti-CD47 antibodies indicated anemia and thrombocytopenia as potential risks. QL401 is a PD-L1 x CD47 bispecific antibody engineered to reduce effect on red blood cells while retaining potent phagocytic activation of macrophages in vitro and delayed tumor growth in vivo. QL401 comprises three functional components: a PD-L1 binding Fab arm, a CD47 binding scFv arm, and a human IgG4 backbone. The PD-L1 binding arm provides both tumor targeting and blocking of PD-1 for reactivating T cells. The CD47 arm blocks the binding of SIRPα, while the IgG4 Fc retains Fc gamma receptor binding to provide a phagocytic signal. In preclinical efficacy studies, QL401 potently blocked SIRPα to promote phagocytosis of tumor cells with sub-nanomolar potency. In vivo efficacy studies in mouse xenograft tumor models showed QL401 to be comparable or superior to PD-L1 or CD47 monoclonal antibodies alone or in combination. In vitro safety evaluation of QL401 showed significantly reduced binding and phagocytosis of red blood cells, in contrast to CD47 monoclonal antibodies. In addition, QL401 did not induce hemagglutination. In non-human primates, QL401 was well tolerated up to 100 mg/kg without reduction of red blood cells or platelets below the normal range. QL401 is presently in a human phase I safety study.
Ideally, an immunotherapeutic drug would take advantage of triggering both innate and adaptive immunity in an anti-tumor response, activating tumor antigen uptake, processing and presentation, in parallel initiating or enhancing an anti-tumor T cell cytotoxic response. As such, a significant number of clinical trials are underway with this mechanistic approach in mind (Tang et al., 2018; Upadhaya et al., 2021). At the forefront of most cancer immunotherapy combinations are blocking antibodies that prevent the inhibitory checkpoint receptor PD-1 on T cells from interacting with its ligand PD-L1 on tumor microenvironment macrophages and/or tumor cells. Cancer checkpoint inhibitor therapy using anti-PD-1 or anti-PD-L1 antibodies is effective, but only in a certain patient population, and a significant number of patients relapse or become resistant to therapy (Vesely et al., 2022). The clinical success of the treatment varies according to the cancer type (Siu et al., 2017), and the relative immunological status of the tumor (Cindy et al., 2021). Evidence suggests augmented tumor antigen presentation plays a role in successful checkpoint inhibitor therapy (Mpakali and Stratikos, 2021). Clinical trial results with anti-PD-1/PD-L1 therapy can be improved when given in combination with other drugs that increase tumor antigen uptake and presentation, leading to a larger pool of T cell clones targeting tumor neoantigens (Patel and Minn, 2018).
The recent advancement in understanding the contribution of the innate immune system, as it relates to efficiency of tumor antigen uptake and presentation, has been highlighted by the success in pre-clinical studies using approaches to increase phagocytosis of tumor cells, resulting in an enhanced adaptive immune response (Nam et al., 2018). One mechanism by which cancer cells evade phagocytosis is via expression of CD47, which binds to the inhibitory SIRPα transmembrane protein expressed by APCs (Maute et al., 2022). Approaches to inhibit the SIRPα-CD47 interaction using monoclonal antibodies showed enhanced phagocytosis of tumor cells in vitro, and efficacy in mouse tumor xenograft models in vivo (Willingham et al., 2012). A role for the adaptive immune response has been demonstrated in pre-clinical mouse models, where treatment of tumor-bearing mice with anti-CD47 antibodies is effective, CD8 T cell dependent, and initiates cross-priming by dendritic cells (Tseng et al., 2013; Liu J et al., 2015). Interestingly a combination of irradiation and CD47 blockade was shown to significantly enhance efficacy and with a pronounced abscopal effect due to inflammatory macrophages, independent of T cells (Nishiga et al., 2022). Pre-clinical efficacy in mouse tumor models has proven to be variable in that CD47 or PD-L1 blockade are not always effective as single agents or in combination (Sockolosky et al., 2016; Nishiga et al., 2022). In a B16F10 model neither anti-CD47 nor anti–PD-L1 administered as monotherapy were effective at controlling tumor growth or extending survival, and only moderate efficacy was observed when administered in combination. In a CT26 syngeneic mouse model neither monotherapies nor a combination resulted in increased survival (Sockolosky et al., 2016). One proposed mechanism for limited efficacy is anti-CD47 antibody binding to CD47 expressed on red blood cells and other cells in the body, thereby inducing an “antigen sink” (Nishiga et al., 2022).
Despite some variable efficacy data in mouse models, successful pre-clinical studies have encouraged transition of CD47-SIRPα blockers into the clinic. Multiple CD47-SIRPα blocking-based immunotherapy clinical trials have been initiated, alone or in combination with other therapies (Bouwstra et al., 2022; Maute et al., 2022). Several obstacles have been encountered in the clinic including poor therapeutic effect and toxicity. Toxicology profiles, and severity, have been wide ranging, depending on the antibody used. Monotherapy, in particular, has shown poor therapeutic efficacy in patients with solid tumors (ORRs less than 5%) (Son et al., 2022), with significant dose-limiting toxicity (DLT), primarily anemia and thrombocytopenia (Patnaik et al., 2020; Ansell et al., 2021). Overcoming anemia and thrombocytopenia observed in early clinical trials with anti-CD47 antibodies has been a major focus of research, and various approaches have been taken, such as altering RBC binding characteristics of antibodies, or targeting SIRPα directly (Ring et al., 2017; Puro et al., 2020; Andrejeva et al., 2021; Qu et al., 2022).
We sought to generate a safe therapeutic molecule with enhanced anti-tumor activity, capable of blocking the inhibitory activity of adaptive and innate checkpoint molecules PD-L1 and CD47. Novel antibodies to human PD-L1 and CD47 were generated that have high affinity binding and blocking kinetics in vitro and showed efficacy in xenograft tumor models. A bispecific antibody (QL401) was constructed with an anti-PD-L1 Fab linked to an anti-CD47 scFv in a 1 + 1 format with a human IgG4 Fc region and Knobs-into-Holes mutations. QL401 induces phagocytosis of tumor cells by human macrophages and demonstrates potent anti-tumor activity in human PBMC xenograft mouse models. In vitro, QL401 shows reduced human RBC binding with significantly attenuated RBC phagocytosis by human macrophages, and lacks RBC hemagglutination activity compared to an anti-CD47 monoclonal antibody. A GLP non-human primate multi-dose toxicology study mimicked in vitro results with minor changes in hematologic parameters including RBC and platelet counts. As a result of efficacy and safety studies QL401 has entered a Phase I clinical trial.
All mouse experiments were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) and carried out according to protocol. The following xenograft models, human PBMC-NOG-Raji and human PBMC-NOG-A375 were performed by inoculating 60 female NOG mice with 3 × 106 human PBMCs in 0.1 mL/mouse by intravenous tail vein injection as well as subcutaneous inoculation of tumor cells in the right forelimb (3 × 106 in 0.1 mL/mouse with 1:1 Matrigel). When the average tumor volume reached 100 mm3, mice were randomly divided into 6 groups and dosed intraperitoneally twice a week, for 3 consecutive weeks as indicated. For the human PBMC-NPG-Raji xenograft model, 3 × 106 Raji cells in 0.1 mL saline (1:1 Matrigel) were inoculated subcutaneously in the right axillary of 70 NPG mice. When the average tumor volume reached 150 mm3, 28 mice were selected and randomly divided into 4 groups as indicated. Mice were dosed twice a week for 2 consecutive weeks. Tumor volume was measured twice a week. In a humanized syngeneic mouse model, MC38 cells (5 × 105 cells in 0.1 mL) expressing human PD-L1 were inoculated subcutaneously in the right axilla of 66 transgenic mice expressing human PD-1. When the average tumor volume reached 70 mm3, 40 mice were selected and randomly divided into 5 groups as indicated. Mice were dosed by intraperitoneal injection 3 times a week for 3 weeks. Tumor size and body weight were measured 3 times a week.
The cynomolgus monkey toxicology study was carried out according to Institutional Animal Care and Use Committee (IACUC) guidelines after review and approval. 40 cynomolgus monkeys, 20 male and 20 female, were randomly divided into 4 groups: the excipient control group, QL401 10 mg/kg, 30 mg/kg and 100 mg/kg groups. Dosing was administered one time per week for 4 weeks, followed by a 4-week recovery period. Once the cynomolgus monkeys were accustomed to wearing a telemetry vest, the EMKA01 system was used to carry out cardiovascular, respiratory and nervous system safety pharmacological tests before and after the first drug administration, and data collection, analysis and summary were performed on the 3 animals from each sex. The collection time was before the first administration (day-9), 2 h, 24 h, 48 h, 96 h and 168 h after the first administration. Evaluation indicators included cardiovascular system ECG (heart rate HR, PR interval, QRS Interval, QT interval, QTcf), blood pressure (mean arterial pressure, systolic blood pressure SP, diastolic blood pressure DP) and respiratory system (respiratory frequency Frequ, tidal volume TV) and nervous system (alertness, posture, gait, climbing, convulsions, tremors, myoclonus, general motility, grooming, lacrimation, piloerection, ptosis, retching/vomiting, salivation, abnormal/stereotyped behavior, urination and defecation, vocalizations, respiration, pupillary light response, blink reflex, eye position/symmetry, pupil size, balance and coordination, auditory startle response).
A pharmacokinetic study was performed after a single intravenous infusion in cynomolgus monkeys. Eighteen cynomolgus monkeys (9 male and 9 female) were randomly divided into 3 groups according to their body weight, and given a 5 mg/kg, 15 mg/kg, or 45 mg/kg dose of QL401. Blood was collected before administration of dosing (0 h) and then post dosing collections done 5 min, 1.0, 2.0, 6.0, 24, 48, 72, 120, 168, 240, 336, and 504 h post dose. The concentration of QL401 in the serum of each group was detected by ELISA, and the pharmacokinetic parameters calculated using Phoenix WinNonlin 8.1 software. A drug-binding receptor assay was used to detect the occupancy of CD47-bound QL401 on the surface of erythrocytes after administration. Whole blood was also collected on day 8, 15, and 22 after administration, serum was separated, and the anti-drug antibodies in serum samples was determined by ECL.
For generation of monoclonal antibodies specific for human PD-L1, Balb/c or SJL mice were immunized subcutaneously with recombinant His-tagged extracellular domain of human PD-L1 protein (Gene ID CD274, GenBank Accession #Q9NZQ7, Amino Acids 19-238). CD47 monoclonal antibodies were generated by immunizing with recombinant His-tagged extracellular domain of human CD47 protein (Gene ID CD47, GenBank Accession #Q08722, amino acids 19–141, C33S mutation). 6–8 week old female SJL/J mice (Jackson Labs, or Balb/c mice (Charles River) were immunized. Adjuvants used for immunization were RIBI (Sigma) and CPG ODN 1826 (Invivogen). For immunizations, RIBI 200 μg (0.5 μg/μL × 400 μL) was warmed to 37C, and diluted into 1.57 mL PBS. 200 μg of human CD47 or PD-L1 protein were added to 100 μg (1 μg/μL) CPG, mixed and added to RIBI/PBS for a total volume of 2 mL. Mice were immunized by injection of 25 μL in the rear foot pad, and 75 μL proximal to the inguinal lymph-node. Mice were boosted weekly with ½ dose for 8 weeks. Lymphocytes isolated from bone marrow and spleen of immunized mice were subsequently fused with mouse myeloma cells to generate hybridomas. Briefly, spleen and lymph nodes were collected from immunized mice, RBC lysed and single cell suspensions made. Lymph node cells 1.5 × 107 were mixed with 1.5 × 107 SP2 cells or spleen cells 1.0 × 108 were mixed with SP2 cells (2 × 107), and fused by slow addition of 35%–50% PEG1500 (Sigma). The fusion mixture was then diluted by slow addition of serum free DMEM medium, pelleted, and resuspended in fusion medium (DMEM + 10% FBS + 10% medium NCTC-109) supplemented with 10% medium E at a density of 2.5 × 104/100 uL and plated in 96-well plates. 2X HAT was added the next day for selection. After 6–7 days, half of the remaining medium was replaced with fusion medium + 2% medium E and HT. The hybridoma culture supernatant was collected on days 10–14 for screening.
The VH and VL sequences of hybridomas were obtained by RT-PCR and cloned into the mammalian expression vector pcDNA3.4TOPO (Invitrogen) modified with additional restriction sites. Variable heavy chains were assembled by Gibson assembly, using the prepared vector, VH PCR fragments, and a gBlock DNA fragment (Integrated DNA Technologies) encoding the human IgG2 constant region (Gene ID IGHG2, GenBank Accession #P01859). Variable light regions were cloned using the same method via assembly of VL PCR fragments with a gBlock DNA fragment encoding the human constant kappa region (Gene ID IGKC, GenBank Accession #P01834). The assembled vectors were used to transform DH5α competent E. coli (New England Bio, C2987). Single colonies were picked and sequenced. Mouse anti-PD-L1 and anti-CD47 antibodies were humanized by grafting mouse CDRs into the closest human framework acceptor (Jones et al., 1986) and backmutations were introduced to minimize impact on binding affinity (Foote and Winte., 1992). The human frameworks used are:
Clone D39.5: IGHV1-2*06 and IGKV1-NL1*01.
Clone C44.1: IGHV3-11*01 and IGKV3-11*01.
Clone E15.1: IGVH1-2*02 and IGKV2-30*02.
Clone E24.6: IGHV1-3*01 and IGKV4-1*01.
Humanized antibodies were cloned for mammalian expression by Gibson assembly of VH and VL DNA fragments with the human IgG1 constant region (Gene ID IGHG1, GenBank Accession #P01857) and the human constant kappa region. Humanized antibodies were cloned for mammalian expression by Gibson assembly of VH and VL DNA fragments with the human IgG2 constant region (Gene ID IGHG2, GenBank Accession #P01859) and human constant kappa region (Gene ID IGKC, GenBank Accession #P01834) as above. Proteins were produced by transfecting plasmids into Expi293 or ExpiCHO cells using the ExpiFectamine transient expression system (ThermoFisher). Six days following transfection, supernatants were filtered, and protein titers were determined by an IgG quantitation protocol using the Octet Red96 (ForteBio). IgG was purified by Mab Select Sure Protein-A column purification on an ACTA PURE system and dialyzed overnight in PBS.
Humanized anti-PD-L1 clones huD39.5.2.3 and anti-CD47 clones huE15.1 and E24.6 were reformatted into bispecific antibodies. CD47 monoclonal antibodies were converted into scFvs as described previously (Huston et al., 1988) by linking the variable domains via a (GGGGS) 3 linker. The scFvs were further stabilized by incorporating an extra disulfide bond using VH44C and VL100C mutations (Reiter et al., 1994). Bispecific antibodies were constructed in a 1 + 1 format using Knob-into-Hole mutations (Hole, S354C, T366S L368A Y407V. Knob, Y349C, T366W) (Merchant et al., 1998), in a human IgG4 Fc. Additional protein A binding mutations (H435R, Y436F) (Jendeberg et al., 1997) were introduced on the knob Fc to facilitate purification. Antibodies were cloned into a pcDNA3.4TOPO vector (ThermoFisher) and transiently transfected into Expi293 or ExpiCHO cells for expression. For non-GMP research use, antibodies were purified on Protein A column and further purified by size exclusion chromatography. Purity was verified via HPLC and SDS-PAGE.
Purified antibodies were characterized for affinity to the antigen using Octet Red96 (ForteBio, Sartorius) by loading purified antibodies (25–50 nM) onto anti-human Heavy Chain (AHC) capture sensors and measuring rates of association and dissociation of histidine tagged recombinant human PD-L1 or CD47 protein (ForteBio Data Analysis Software).
Immulon HBX plates (VWR) were coated with 2 μg/mL hPDL1-Fc (R&D Systems) overnight at 4°C. Plates were washed 3 times with PBST and blocked for 1 h at room temperature with 4% NFDM/PBS. Blocking was removed and antibody dilutions in 4% NFDM/PBS were added and incubated at room temperature for 1 h. Plates were washed 3 times with PBST. 1ug/mL huCD47-C33S_His was added to each well and incubated for 1 h at RT, followed by washing 3 times with PBST. Anti-His-HRP (AbCam) was added to each well and incubated at RT for 45 min. After washing 6 times with PBST, the assay was developed with TBM, followed by 2N sulfuric acid.
To evaluate anti-PD-L1 antibodies, 2 days prior to flow cytometry analysis, A431 cells were seeded at 30%–60% confluence in the absence or the presence of 1,000 U/mL of recombinant human IFN-γ to stimulate PD-L1 expression. In preparation for flow cytometry, cells were detached with Trypsin/EDTA, washed, resuspended in FACS buffer (PBS, 2% FBS) and aliquoted into 96 well plates at 1 × 105 cells/well. Cells were stained with 5 μg/mL anti-PD-L1 antibodies, positive control antibody “C1-IgG1” or isotype IgG1 control on ice for 45 min followed by washing and secondary staining with 1:500 diluted Goat anti-human IgG-AF647. Cells were analyzed by flow cytometry (Canto, or LSRFortessa, BD Biosciences) following a final wash and addition of 7-AAD. Human and cynomolgus monkey CD47 transfected CHO cells, Raji (ATCC CCL-86) and MM.1S cells (ATCC CRL-2974) were aliquoted into 96 well plates at 1 × 105 cells/well and incubated with serial diluted anti-CD47 antibodies in FACS buffer, followed by AF647-labeled F (ab’) 2 goat anti-human IgG and 7-AAD. After flow cytometry, data was analyzed using FlowJo and CD47 mean fluorescence intensity (MFI) calculated after gating on FSC/SSC, and viability.
For evaluation of cell binding by bispecific antibodies, HEK 293-PD-L1, or CHO-KI-CD47 cells were harvested and washed with FACS buffer. 1 × 105 cells per well were distributed in 96-well v-bottom plates. Serial dilutions of test antibodies were added and incubated on ice for 20 min. Cells were washed then resuspended in 50 μL of secondary antibody, AF647 F (ab’)2 Goat anti-hu IgG, Fc specific at 1:500 dilution (Jackson ImmunoResearch) and incubated on ice for 15 min. Finally, cells were resuspended in 100 μL of FACS buffer containing 7-AAD.
To compare blocking ability of humanized anti-PD-L1 antibodies, HEK293 cells expressing PD-L1 were harvested by Accutase and resuspended at 4 × 106 cells/mL in FACS buffer. In a 96 well plate 30 μL of cells (120,000), 30 μL of 6 μg/mL rhPD-1 His tagged (2 μg/mL final concentration), and 30 μL of serially titrated anti-PDL1 antibodies were combined and incubated for 20 min on ice. Following incubation, cells were washed, and bound PD-1 was detected with anti-His Tag-APC Mouse IgG1 (R&D Cat# IC050A). Cells were analyzed by flow cytometry following a final wash and addition of 7-AAD. Blocking by humanized antibodies were compared using the same assay with rhPD-1 Fc-biotin at 0.5 μg/mL final concentration and detected with Streptavidin-APC (R&D).
For evaluating SIRPα blocking of anti-CD47 monoclonal antibodies, Raji cells (ATCC CCL-86) were counted, washed with FACS buffer, and blocked with human FcR blocking reagent (Miltenyi Biotech). Cells were mixed with 1 μg/mL rh-SIRPα/Fc chimeric protein, then with serially diluted anti-CD47 antibodies (20 μg/mL, 1:4 dilutions). Cells were incubated on ice for 20 min, washed twice with FACS buffer, and incubated 25 min in 1:500 diluted anti-human IgG1 Fc PE secondary antibody in FACS buffer. Samples were stained with 7-AAD prior to flow cytometry analysis (BD FACS Canto II or LSR Fortessa cell analyzer).
To test PD-1 and SIRPα blocking by bispecific antibodies, HEK 293-PD-L1, or CHO-KI-CD47 cells were harvested and washed one time with FACS buffer. 1.2 × 105 cells per well were distributed in 96-well v-bottom plates. 0.5 μg/mL PD-1-biotin or 1.25 ug/mL SIRPα -biotin were added to the cells and incubated for 5 min on ice. Serial dilutions of test antibodies were added and incubated on ice for 20 min. The cells were washed 2 times with 200 uL FACS buffer. Cells were resuspended in 50 μL of secondary antibody, Streptavidin-APC (R&D) at 10µL/10^6 cells and incubated on ice for 15 min. Finally, the cells were washed 2 times with 200 uL FACS buffer and resuspended in 120 uL FACS buffer containing 7-AAD prior to analysis.
Fresh PBMCs were diluted to 2 × 106 cells/mL in X-Vivo 15 medium (Lonza). SEB (Millipore) was added to the PBMCs at 200 ng/mL 100 ul of serially diluted antibodies and 100 ul of the PBMC/SEB antigen mixture were added into 96-well flat-bottom plates. Following incubation at 37°C for 48 h, supernatants were analyzed for IL-2 by ELISA according to manufacturer’s instructions (R&D Systems).
PBMCs were isolated from human buffy coat (Stanford) using density gradient centrifugation (Miltenyi Biotec) and washed 4 times with PBS. CD4+ T cells were isolated (Miltenyi Biotec) from PBMCs according to manufacturer’s protocol, and resuspended in X-Vivo 15 medium (Fisher Scientific) at 4 × 106 cells/mL. Monocyte-derived dendritic cells (DCs) were generated from positively selected CD14+ monocytes (Miltenyi Biotec). Purified monocytes were seeded at 5 × 105 cells/mL in complete RPMI-1640 media supplemented with 10% FBS for 7 days. Cultures were supplemented with recombinant human (rh-) IL-4 (1000 U/mL) (R&D) and rh granulocyte-macrophage colony-stimulating factor (rh-GMCSF) (500 U/mL) (R&D) on days 0, 2, and 5. Immature DCs were harvested on day 7 and resuspended in 5 mL RPMI-1640, 10% FBS medium. Cells were incubated with occasional mixing with 20 μg/mL mitomycin C for 1 h at 37°C followed by washing and resuspension in X-Vivo 15 media to 4 × 105 cells/mL.
In 96 well flat bottom plates, 50 ul DCs (2 × 104/well), 100 μL serially diluted anti-PD-L1 antibodies, and 50 μL purified CD4+ T cells (2 × 105 CD4+ T cells/per well) were mixed at a 1:10 ratio (DCs: T cells). Plates were incubated for 5 days, and the supernatants analyzed by ELISA for IFN-γ release (R&D).
Human PBMC were isolated by Ficoll gradient. To isolate monocytes, 1 × 108 cells were incubated for 2 h in a T75 flask with RPMI-1640 media 10% FBS to allow monocytic cells to settle and attach. Non-adherent cells were removed, and the flask was incubated with media containing 20 ng/mL recombinant human-M-CSF (Miltenyi Biotec, cat# 130-096-492) for 7 days, then with 20 ng/mL rh-M-CSF and 10 ng/mL rh IL-10 (Miltenyi Biotec, cat# 130-098-448) for an additional 2 days. The monocyte-derived macrophages were washed, harvested by Trypsin-EDTA, resuspended in media and quantified. Macrophages were combined at 1:4 ratio with CFSE-labeled MM.1S cells, and test antibodies in a round bottom ultra-low attachment 96 well plates (Costar, cat# 7007)., and incubated at 37°C for 2 h. The αCD47 antibodies were titrated in RPMI-1640 + 10% heat inactivated FBS media. Cells were resuspended and transferred to a V-bottom polypropylene plate for FACS staining with anti-CD36 APC (Thermo Fisher), and 7-AAD. Samples were analyzed by flow cytometry using BD LSR Fortessa and further analyzed using FlowJo gating on Live CFSE/APC double positives indicating phagocytosis of target cells with macrophages.
Fresh whole blood was obtained from Stanford Blood Center. Human red blood cells (RBC) were isolated following purification of mononuclear cells from whole blood by density gradient centrifugation. RBCs were washed 3 times with DPBS and resuspended at 2 × 107 cells/mL in DPBS. Titrated anti-hCD47 antibodies, and isotype controls were mixed with RBCs in a round-bottom 96-well plate and incubated at room temperature for 2 h. Hemagglutination was evaluated by image analysis.
Human red blood cells obtained from buffy coat after isolation of mononuclear cells by density gradient centrifugation were transferred into 50 mL conical tubes. RBC was washed 3 times and resuspended in DPBS to make 10% solution. 1 × 106 cells per well were distributed into 96-well v-bottom plates. Serial dilutions of monoclonal or bispecific antibodies were added and incubated at 4C for 30 min. RBC were then washed 2 times with FACS buffer. RBC were resuspended in 100 μL of secondary antibody AF647 F (ab’) 2 goat anti-hu IgG, Fc specific at 1:500 dilution (Jackson ImmunoResearch) and incubated at 4C for 15 min. RBC were then washed and resuspended in FACS buffer for flow cytometric analysis.
Recombinant human M-CSF and recombinant human IL-10 monocyte-derived macrophages were generated as described. Purified RBC were labelled with 1 μM of CellTrace CFSE (ThermoFisher). CFSE labelled RBC (1 × 105 cells/well) and macrophages (2.5 × 104 cells/well) were incubated for 2 h in 5% CO2 at 37C with serial dilutions of test antibodies in 96 well ultra-low attachment u-bottom plates (Costar). Cells were then transferred to 96-well v-bottom polypropylene plates, spun down, and washed one time with DPBS with 20% heat-inactivated FBS. Cells were resuspended with DPBS with 20% HI FBS and APC-conjugated anti-human CD36 antibody (ThermoFisher) was added to wells containing testing antibodies and incubated on ice for 20 min. Cells were washed 2 times with 200 uL DPBS with 20% HI FBS. Finally, cells were resuspended with buffer containing 7-AAD. Samples were analyzed by flow cytometry using BD LSR Fortessa and further analyzed using FlowJo gating on Live CFSE/APC double positives indicating phagocytosis of RBC with macrophages induced by anti-CD47 antibodies.
To generate anti-human PD-L1 antibodies, mice were immunized with a recombinant extracellular domain of human PD-L1. Hybridomas were produced and screened for anti-hPD-L1 antibodies by both Elisa and binding to hPD-L1 expressing cells. The variable heavy (VH) and variable light (VL) sequences were obtained and mouse anti-hPD-L1 antibodies humanized by grafting mouse complementarity-determining regions (CDRs) into the closest human framework acceptor, with the human IgG1 constant region and the human constant kappa region. Purified antibodies were characterized for affinity to the antigen, measuring rates of association and dissociation of histidine tagged recombinant human PD-L1 protein. Four humanized anti-hPD-L1 antibodies with the best monovalent binding kinetics are shown in Table 1, with KD’s ranging from 1.12 nM (hC69.1) to 0.268 nM (hA75.1).
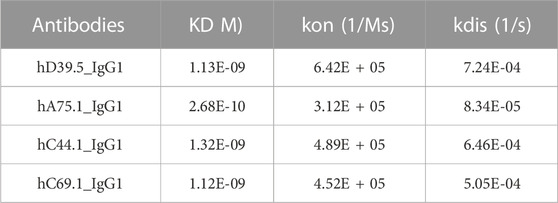
TABLE 1. Monovalent binding kinetics of humanized anti-hPD-L1 Antibodies.
To evaluate cellular binding capacity of anti-hPD-L1 antibodies, A431 cancer cells were cultured with human IFN-γ to stimulate PD-L1 expression. Flow cytometric analysis was performed and binding curves calculated for EC50 values (Figure 1A; Table 2). Similarly, binding EC50 values were calculated for HEK293 cells expressing human (Figure 1B) or cynomolgus monkey (Figure 1C) PD-L1. All four humanized anti-hPD-L1 antibodies bound both human and cynomolgus monkey PD-L1, with D39.5 showing the overall lowest EC50 of cell binding (Table 2). In order to check for specificity, each of the four humanized anti-hPD-L1 antibodies were tested for binding to hPD-L1/HEK293 cells in the presence of rhPD-1 His tag, or rhPD-1 Fc-biotin protein. (Figure 1D; Table 2). The four humanized anti-hPD-L1 antibodies tested could out-compete hPD-1 protein for binding, confirming specificity and ability to block ligand–receptor interaction.
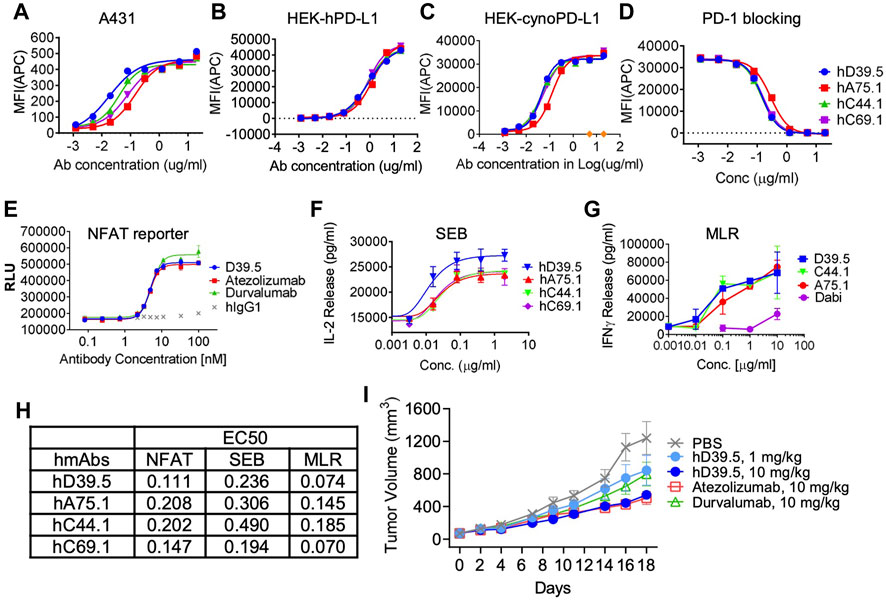
FIGURE 1. Characterization of humanized anti-hPD-L1 antibodies. Binding kinetics of four selected humanized antibodies (hD39.5, hA75.1, hC44.1, hCD69.1) to A431 cells (A) or HEK cells engineered to overexpress human (B), or cynomolgus monkey (cyno) PD-L1 (C). Cells were stained with 5 ug/mL anti-PD-L1 antibodies, followed by washing and secondary staining with Goat anti-human IgG-AF647. Cells were then analyzed by flow cytometry. (D) rhPD-1-His tagged protein blocking of anti-hPD-L1 antibodies to hPDL1/HEK293 cells. Bound hPD-1 was detected with anti-His Tag-APC Mouse IgG1. Results were confirmed using rhPD-1 Fc-biotin at and detection with Streptavidin-APC. Anti-hPD-L1 antibodies were evaluated using three in vitro cell based assays: PD-L1/PD-1 TCR blocking NFAT reporter assay (E), potentiation of IL-2 expression from human PBMC T cells after SEB stimulation (F) and in an MLR assay combining human allogeneic monocyte-derived dendritic cells with purified CD4+ T cells and analyzed for IFN-γ expression (G). Summary of EC50s for each assay is shown in (H,I) hD39.5 was dosed i.p into human PD-1 expressing transgenic mice harboring MC38 tumors expressing human PD-L1 (8 mice per group). Anti-PDL1 antibodies Atezolizumab and Durvalumab (10 mg/kg) were used for comparison.
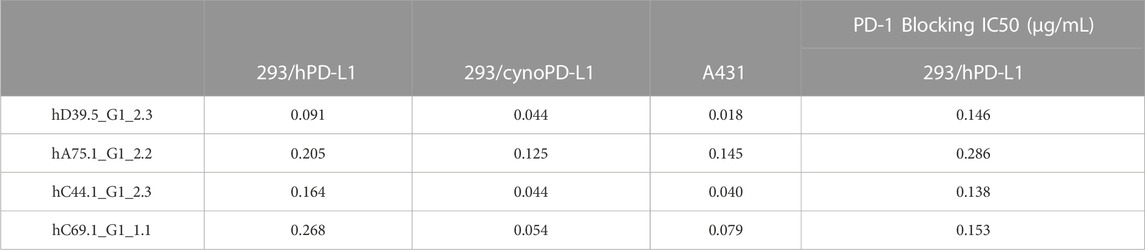
TABLE 2. Binding EC50 (μg/mL) of humanized anti-hPD-L1 Antibodies.
Next, experiments were carried out to test the potency of blocking PD-1 inhibition by the four humanized anti-hPD-L1 antibodies in 3 cell based functional assays (Figures 1E–G). First, antibody blocking efficiency was evaluated in a TCR - NFAT reporter assay sensitive to PD-1 mediated suppression (Figure 1E). Second, we tested antibodies for the enhanced IL-2 production in human peripheral blood mononuclear cells (PBMC) stimulated with Staphyolococcus enterotoxin B (SEB) T cell super antigen (Figure 1F). Third, we looked for enhanced interferon-γ release in a mixed lymphocyte reaction (MLR), where human T cells were stimulated with PBMC-derived allogeneic dendritic cells (Figure 1G). Comparison of EC50’s for the three functional assays are summarized in Figure 1H. Based on binding and in vitro functional data we opted to proceed with humanized anti-hPD-L1 antibody D39.5 for further characterization. D39.5 was tested in a surrogate syngeneic mouse model where murine colon adenocarcinoma MC38 tumors expressing human PD-L1 were established in hPD-1+ C57BL/6 mice, and efficacy compared to clinically available anti-hPD-L1 antibodies atezolizumab and durvalumab (Figure 1I). Efficacy was obtained with both hD39.5 and atezolizumab at equivalent doses.
Initial discovery, humanization and further in vitro and in vivo validation of novel set of humanized anti-hPD-L1 antibodies with high affinity binding and blocking led to selection of hD39.5 for in vivo validation, and as a lead candidate for development of an anti-hPD-L1/anti-CD47 bispecific antibody.
Similar to the generation of novel anti-human PD-L1 antibodies, mice were immunized with protein (recombinant His-tagged extracellular domain of human CD47 protein), and hybridomas screened for anti-hCD47 antibodies by both Elisa and binding to hCD47 expressing cells. Mouse anti-hCD47 antibodies were humanized, and purified antibodies characterized for affinity to histidine tagged recombinant human CD47 protein. Monovalent binding kinetics of the three most potent mouse and humanized anti-hCD47 lead antibodies (B26.2, E15.1, and E24.6) were compared as shown in Table 3. The rank-order between mouse and humanized antibodies were different, with huE15.1.4.2 having the highest affinity for CD47 among the humanized antibodies, with a KD of 6.7 nM.
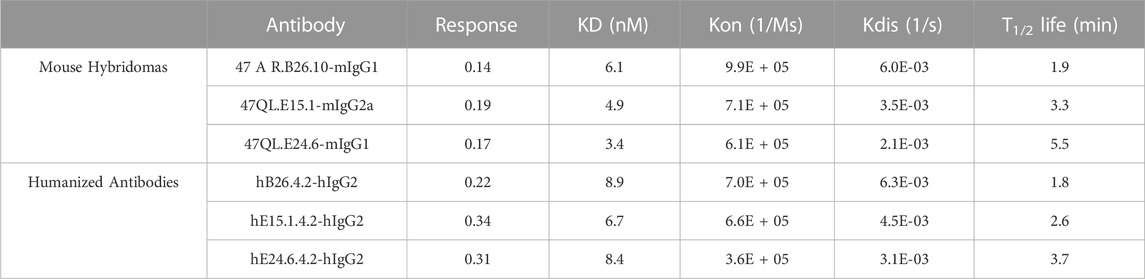
TABLE 3. Mouse anti-hCD47 and humanized antibody binding kinetics.
Binding affinity of the three mouse anti-hCD47, and derivative humanized anti-hCD47 antibodies was determined using CHO cells expressing either human or cynomolgus monkey CD47, as well as human Raji and MM1S cell lines. (Table 4). All three mouse and humanized anti-hCD47 antibodies showed variable high affinity binding to hCD47, depending on the cell lines tested (0.057–0.363 μg/mL), as well as CHO cells expressing cynomolgus monkey CD47 (0.130–0.490 μg/mL).
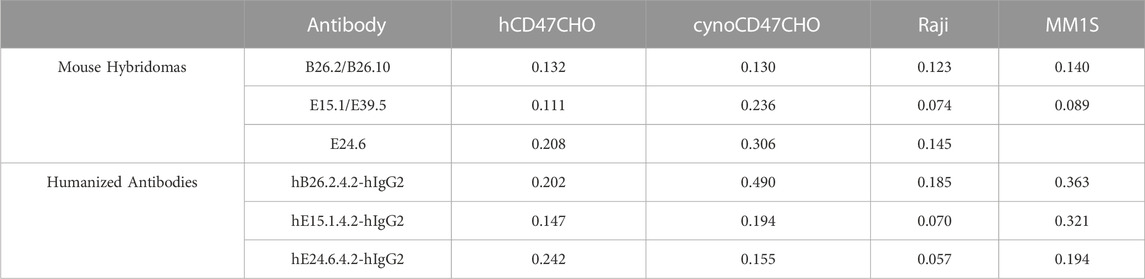
TABLE 4. Mouse anti-hCD47 and humanized antibody cell binding (EC50 μg/mL).
An assay was performed to ensure specificity and ability of anti-hCD47 antibodies to inhibit binding of hCD47 to its ligand, SIRPα (Figure 2A). The results demonstrated the anti-hCD47 antibodies effectively block association of rh-SIRPα/Fc chimeric protein with hCD47 on Raji cells, and CHO cells transfected with hCD47. Humanized antibody clone hE15.1 was the most potent SIRPα blocker, with an IC50 of 0.034 μg/mL (Figure 2B).
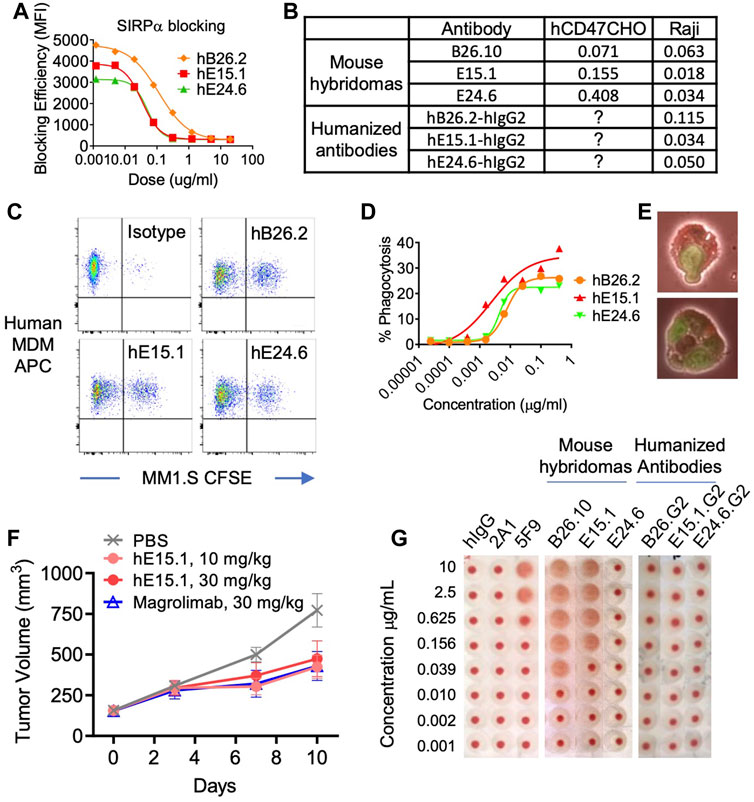
FIGURE 2. Characterization of humanized anti-hCD47 antibodies. (A) Blocking of anti-hCD47 antibodies by rh-SIRPα/Fc chimeric protein. Raji cells were mixed with rh-SIRPα/Fc chimeric protein prior to adding anti-CD47 antibodies. After incubation and washing cells were stained with anti-hIgG1 Fc-PE secondary antibody then analyzed by flow cytometry. IC50s are summarized in (B). Phagocytic potentiation by humanized anti-hCD47 antibodies (C–E). Human monocyte-derived macrophages (hMDM) were combined with CFSE-labeled MM.1S cells and anti-hCD47 antibodies. After incubation cells were stained with macrophage specific anti-CD36 APC and analyzed by flow cytometry. CFSE/APC double positives indicating phagocytosis of target cells with macrophages were enhanced with antibodies hB26.2, hE15.1, and hE24.6 compared to an isotype control antibody (C), dose response curve (D) representative microscopy demonstrating hMDM phagocytosis of MM1. S-CFSE (E). (F) In a human PBMC-NPG-Raji xenograft model, Raji-tumor bearing NPG mice (7 per group) were transplanted with human PBMC and dosed twice a week for 2 consecutive weeks with hE15.1 (10 and 30 mg/kg) or magrolimab (30 mg/kg). Tumor volume was measured twice a week until end of study. (G) Anti-hCD47 antibody induction of human RBC hemagglutination. hRBC were isolated and incubated in the presence of a titrated human isotype antibody (hIgG), anti-CD47 antibodies 2A1, or 5F9 (magrolimab), mouse anti-hCD47 antibodies B26.10, hE15.1 and hE24.6, or their humanized counterparts. Hemagglutination was evaluated by image analysis.
Tumor associated CD47 binding to SIRPα on human myeloid cells inhibits phagocytosis. We therefor tested the three-lead humanized anti-hCD47 antibodies (hB26.2, hE15.1, and hE24.6) for modulation of human PBMC monocyte derived macrophages phagocytic capability (Figures 2C, D). Analysis of anti-CD36 APC stained macrophages after co-culture with CFSE-labeled target MM.1S cells and humanized anti-hCD47 antibodies demonstrated a dose dependent increase in CFSE/APC double positive clusters, indicative of macrophage engulfed target cells (Figure 2C). Figure 2D shows the macrophage phagocytosis of MM.1S in presence of increasing concentrations of humanized αhCD47. Humanized clone hE15.1 was the most potent with the largest increase in percent of macrophages engulfing MM1S cells (30%). Flow cytometry-based phagocytosis detection was confirmed by microscopy (Figure 2E).
Next, Raji cells were engrafted into immunodeficient NPG mice and treated with humanized anti-hCD47 antibody hE15.1, or the clinically available anti-hCD47 antibody, magrolimab (Figure 2F). Both hE15.1 (10 and 30 mg/kg) and magrolimab treatment reduced tumor growth.
Red blood cells (RBC) express CD47 and are prone to hemagglutination when exposed to anti-hCD47 antibodies, leading to safety concerns in clinical trials (Son et al., 2022). Human RBCs were exposed to mouse hybridoma derived antibodies as well as the derivative humanized αhCD47 antibodies (Figure 2G). While 5F9 (Magrolimab) and mouse B26.2 and hE15.1 antibodies induced dose dependent RBC hemagglutination, exposure to the humanized versions did not show increased hemagglutination when compared to a hIgG control.
In summary in vitro and in vivo validation of humanized αhCD47 antibodies led to selection of hE15.1 and hE24.6 as lead candidates for development of an anti-hPD-L1/anti-hCD47 bispecific antibodies.
Humanized anti-hPD-L1 antibody hD39.5 and anti-hCD47 antibodies hE15.1 and hE24.6 were engineered into a bispecific PD-L1-CD47 format (Figure 3A). hD39.5 was modified to be the Fab region only and hE15.1 and hE24.6 hCD47 monoclonal antibodies converted into scFv’s by linking the variable domains, and further stabilized by incorporating an extra disulfide bond. Bispecific PD-L1-CD47 antibodies were constructed in a 1 + 1 format using Knobs-into-Holes mutations in the human IgG4 Fc region. Additional protein A binding mutations were introduced on the knob Fc region to facilitate purification. Two PD-L1-CD47 bispecific antibodies were made for evaluation: hD39.5- hE15.1 and hD39.5- hE24.6.
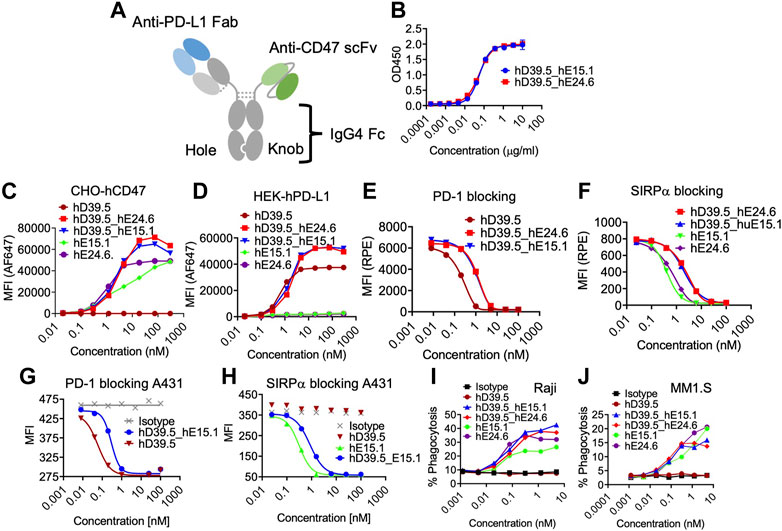
FIGURE3. Characterization of human PD-L1/CD47 bispecific antibodies. (A) Format of bi-specific PD-L1-CD47 antibody using hD39.5 Fab region only and hE15.1 or hE24.6 hCD47 monoclonal antibody scFv. Additional modifications include, knob-in-hole mutations in the human IgG4 Fc region, and protein A binding mutations in the knob Fc region to facilitate purification. Two PD-L1-CD47 bispecific antibodies were made for evaluation: hD39.5- hE15.1 and hD39.5- hE24.6. (B) A PD-L1-CD47 bispecific ELISA was performed by coating plates with hPDL1-Fc washing and then adding a titration of hD39.5- hE15.1 or hD39.5- hE24.6. After incubation, huCD47-C33S_his was added followed by incubation, washing and addition of anti-His-HRP. The assay was developed with TBM. (C,D) Binding affinity to CHO cells expressing hCD47 (C) or HEK cells expressing hPD-L1 (D) was performed by adding bispecific antibodies hD39.5- hE15.1 and hD39.5- hE24.6 and compared to hD39.5, hE15.1 or hE24.6 monoclonal antibodies alone. (E–H) Blocking of bispecific antibodies and individual monoclonals were performed with hPD-1-biotin protein on HEK cells expressing hPD-L1 and A431 cells (E,G respectively), or hSIRPα -biotin proteins on CHO cells expressing hCD47 and A431 cells (F, H respectively). (I,J) Phagocytosis of Raji (I) and MM1. S cell lines (J) in the presence of bispecific hD39.5- hE15.1, hD39.5- hE24.6 or monoclonal hD39.5, hE15.1 or hE24.6 antibodies by human PBMC monocyte derived macrophages.
To compare relative hPD-L1 and hCD47 binding affinity of the two bispecific antibodies, a bispecific elisa and binding to transfected cell lines were tested (Figures 3B–D). Affinity of the two antibodies were equivalent in both Elisa and cellular binding assays. In the cell based assay, bispecific antibodies showed superior binding compared to the individual anti-hPD-L1 and anti-hCD47 antibodies alone (Figures 3C, D respectively). Counter to the binding data, humanized antibodies were more potent than bispecific antibodies at out-competing binding of hPD-1 and hSIRPα to their respective ligands on transfected cells and on the A431 cell line (Figures 3E–H). Next, we compared the two bispecific antibodies and anti-hCD47 monoclonal antibodies for modulation of human PBMC monocyte derived macrophages phagocytic capability of Raji and MM1S cell lines (Figures 3I, J respectively). The bispecific antibodies showed similar increasing dose dependent phagocytic activity equal to or better than each anti-hCD47 antibody alone.
We further investigated the lead bispecific molecule hD39.5- hE15.1 (QL401) for in vivo efficacy and for RBC hemagglutination. For in vivo efficacy, a human PBMC transplanted xenograft model in immunodeficient NOG mice was used with engraftment of A375 or Raji human tumorigenic cell lines (Figures 4A, B). In the NOG A375 PBMC model, mice were dosed with QL401 (1, 3, 10 mg/kg), or clinically available anti hCD47 or αhPD-L1 antibodies (magrolimab and durvalumab respectively) alone or in combination at 10 mg/kg (Figure 4A). Similar anti-tumor efficacy was obtained with QL401 (3 and 10 mg/kg), as well as magrolimab or durvalumab alone or in combination. In the NOG Raji PBMC model (Figure 4B), mice were dosed with QL401 (1,10,30 mg/kg), magrolimab or anti-hPD-L1 antibody atezolizumab (10 mg/kg). While treatment with atezolizumab alone did not show an anti-tumor response, both QL401 and magrolimab demonstrated significant inhibition of tumor growth.
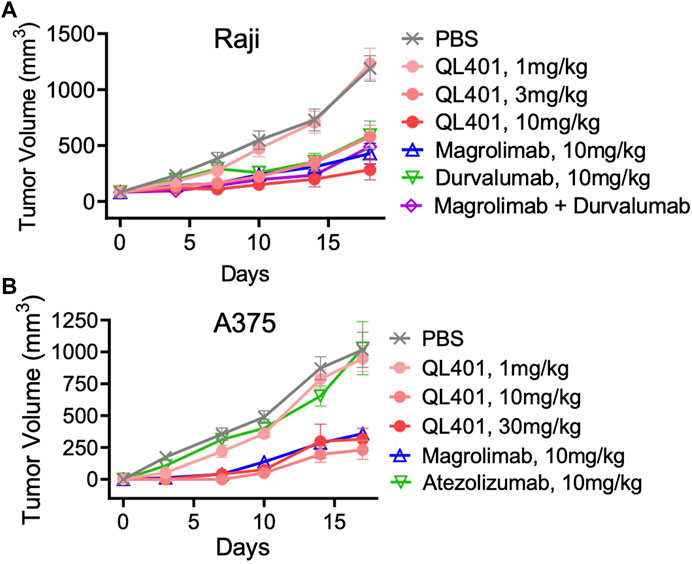
FIGURE 4. In vivo validation of bispecific antibody lead candidate QL401 (hD39.5- hE15.1) in human PBMC-NOG-Raji (A) and human PBMC-NOG-A375 (B) xenograft models. Female NOG mice were randomized and inoculated with human PBMC iv, and tumor cells subQ (5 mice per group) and dosed intraperitoneally twice a week with antibodies as indicated.
Because hCD47 is expressed on human RBC, and anemia was observed in anti-hCD47 antibody clinical trials, we initially evaluated QL401 for potential effects on human RBC in vitro (Figures 5A–C). QL401 was less potent than the parental humanized hE15.1 monoclonal antibody in binding to RBC derived from two different blood donors at an EC50 of 1.99–1.79 and 0.69–0.93 nM respectively (Figure 5A). Human RBC from two different blood donors were exposed to increasing concentrations of QL401, the parental humanized hE15.1 monoclonal antibody or an isotype control antibody (Figure 5B). While hemagglutination occurred in a concentration dependent manner with hE15.1, no effect was seen with an isotype control antibody or QL401 in RBC from either donor. Next, human monocyte derived macrophages were tested for enhanced capacity to phagocytose RBC in vitro in the presence or absence of QL401 or hE15.1 (Figure 5C). 50%–80% of macrophages were capable of phagocytosing RBC in the presence of hE15.1, whereas RBC phagocytosis was significantly attenuated in response to presence of QL401. In vitro results validated further pharmacology studies in non-human primates.
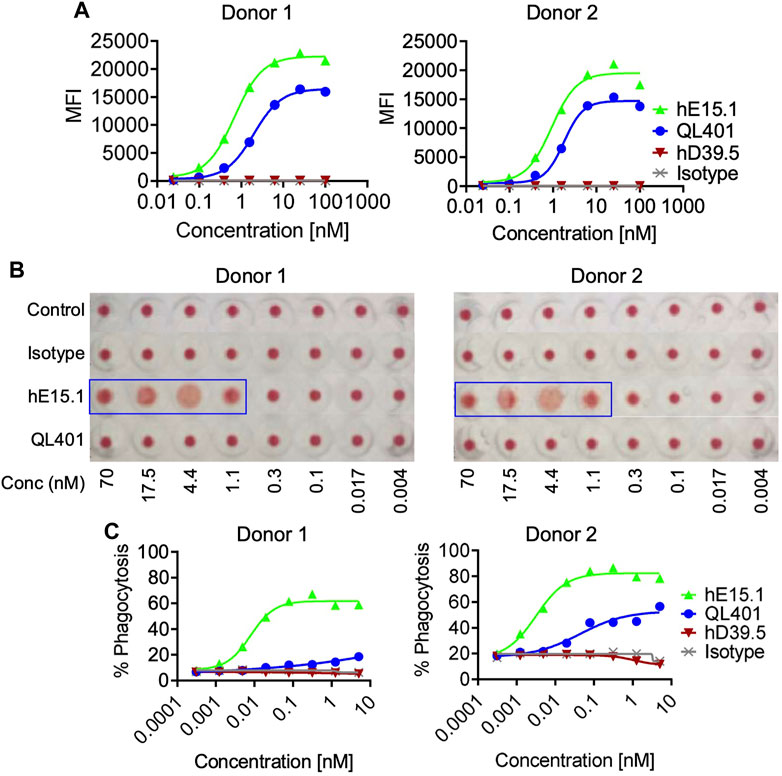
FIGURE 5. Evaluation of lead candidate QL401 (hD39.5- hE15.1) or parental anti-CD47 clone hE15.1 impact on human RBCs in vitro. (A) Titrated antibodies were added and incubated with purified human RBC from 2 separate whole blood cell donors, followed by secondary antibody AF647 F (ab’) 2 goat anti-hu IgG. RBC were then washed and resuspended in FACS buffer for flow cytometric analysis. (B) Antibody induction of human RBC hemagglutination. hRBC from 2 separate donors were isolated and incubated in the presence of a titrated human isotype antibody (hIgG), QL401, or hE15.1 in round-bottom 96-well plated and incubated at room temperature for 2 h. Hemagglutination was evaluated by image analysis. (C) Human monocyte-derived macrophages (hMDM) from 2 separate PBMC donors were combined with CFSE-labeled MM.1S cells and antibodies in round bottom ultra-low attachment 96 well plates and incubated at 37°C for 2 h, then stained with macrophage specific anti-CD36 APC and analyzed by flow cytometry. CFSE/APC double positives indicating phagocytosis of target cells by macrophages.
In preparation for a non-human primate GLP toxicology study, a scalable production of QL401 was established to meet dosing requirements, with the final yield of pure drug substance between 1.0–1.4 g/L. Testing of a GMP batch showed a favorable production and stability profile (Table 5). QL401 maintained greater than 95% monomer by HPLC-SEC after a 4-week incubation period at 42°C (Figures 6A, B). QL401 also showed a favorable serum stability profile, being stable in human serum for at least 7 days without loss of binding in a bispecific sandwich ELISA (Figure 6C).
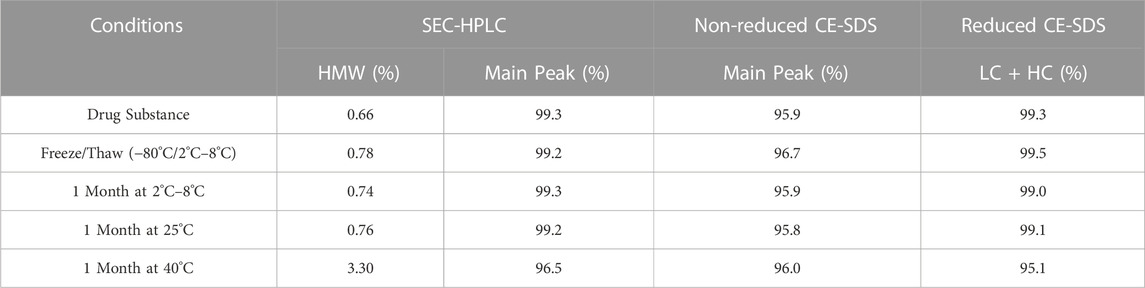
TABLE 5. PDL1 CD47 GMP production for GLP toxicology study.
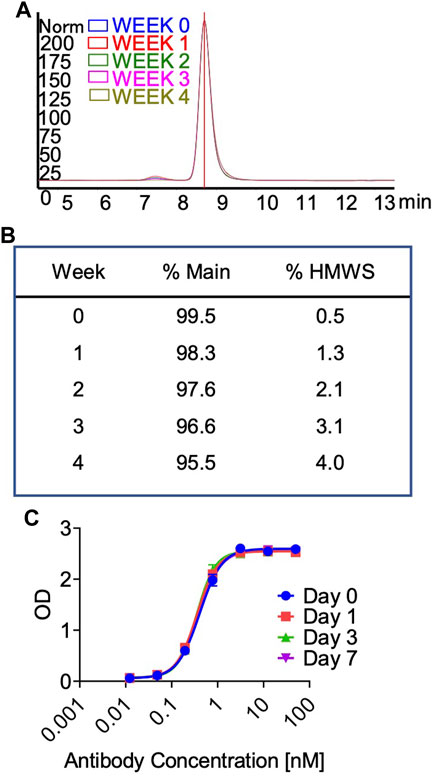
FIGURE 6. High temperature stress test and serum stability of QL401. (A,B) Percent monomer of QL401 after a 1–4 weeks incubation period at 42°C determined by HPLC-SEC. (C) Stability of QL401 after incubation in human serum for 7 days and assayed in a bi-specific sandwich ELISA.
In vivo pharmacokinetics was evaluated after the first dose of four groups (5 male and 5 female per group) at 0, 10, 30, or 100 mg/kg in a GLP cynomolgus monkey toxicology study (Figure 7A). The pharmacokinetic profile of QL401 was promising, with the range of the average half life (T ½) in the 3 doses at 24.7–120 h) (Table 6). Cmax was dose proportional, ranging from 270 g/mL average at a 10 mg/kg dose to 3,700 g/mL average at the 100 mg/kg dose. The AUC0-T, volume of distribution (Vd), and clearance (Cl) were also dose proportional (2,765–206,975 H*pg/mL, 274–52.4 mL/kg, and 3.74–0.31 mL/h*kg respectively.
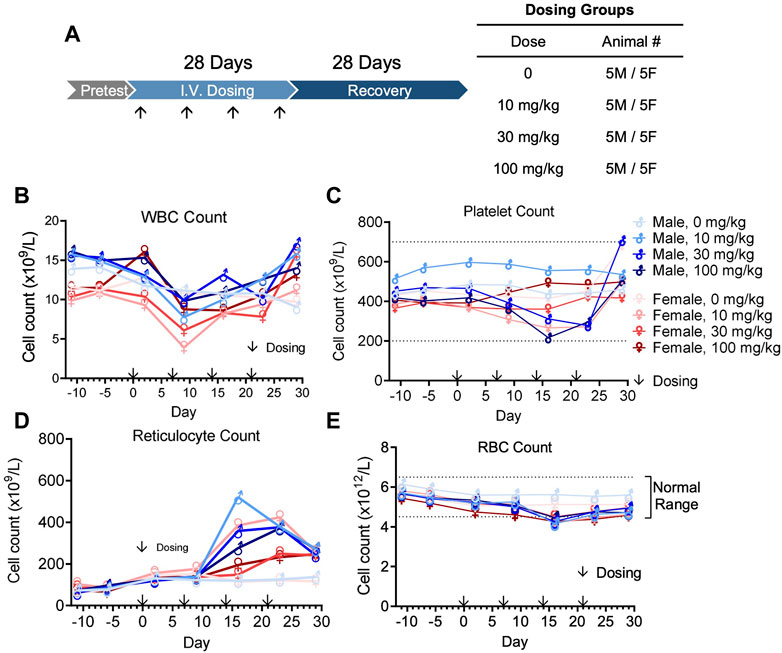
FIGURE 7. Cynomolgus monkey GLP toxicology study with bispecific antibody QL401. (A) Schematic of dosing and recovery schedule. 40 cynomolgus monkeys, 20 male and 20 female, were randomly divided into 4 groups: the excipient control group, QL401 10 mg/kg, 30 mg/kg and 100 mg/kg groups. Dosing was administered one time per week for 4 weeks, followed by a 4-week recovery period. Peripheral blood was collected at the indicated timepoints for WBC (B), platelet (C), reticulocyte (D) and RBC counts (E) in response to the treatment.
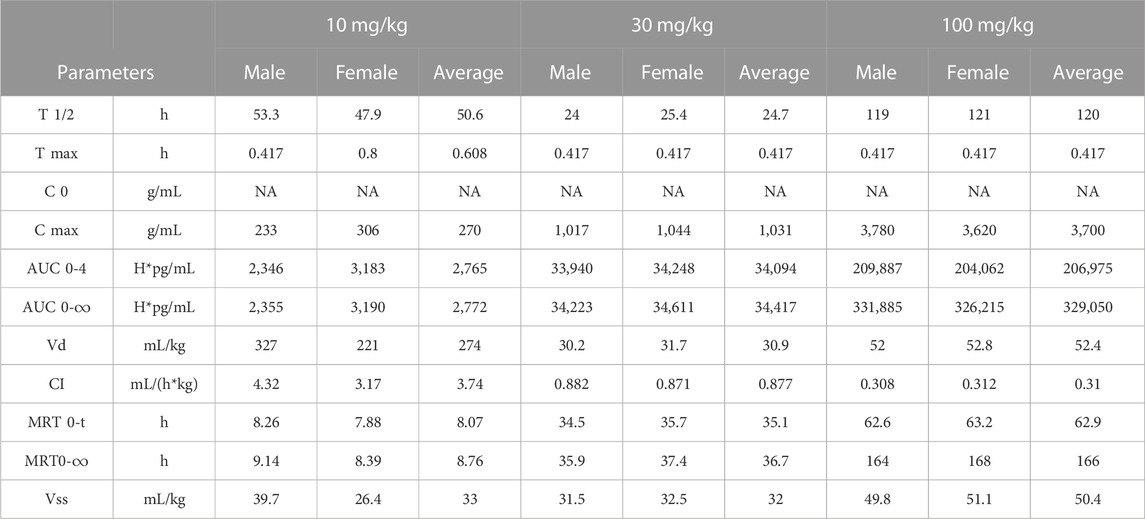
TABLE 6. Pharmacokinetics in cynomolgus monkeys.
The GLP toxicology study was performed with weekly dosing for 4 weeks followed by a 4-week recovery and observation period (Figure 7A). No severe adverse side effects were observed up to 100 mg/kg. WBC counts remained in a normal range except for a minimal decline after the second dose, returning to normal levels within 1 week (Figure 7B). Platelet counts transiently declined minimally after the third dose of QL401, but remained within a normal range (Figure 4C). Reticulocyte counts were elevated after the third dose (highest value 520.4 × 109/L max in a male at 10 mg/kg) above non-treated control values (Figure 7D). RBC counts transiently declined minimally (J. Vet. Med. Sci. 79 (5): 881–888, 2017) after the third dose of QL401 (lowest value 4.15 × 1012/L in a male at 10 mg/kg) (Figure 7E). The cynomolgus GMP toxicology study indicated that QL401 has a very low risk of hematological toxicities.
Over the last decade there have been major shifts in concepts regarding immunity and cancer. Inhibitory molecules such as PD-L1 and CD47, among others, have been recognized as key determinants in tumor immune evasion (Robert, 2020). The recognition of blocking PD-L1/PD-1 interactions in activating anti-tumor T-cell responses has translated to the clinic with significant therapeutic results for the treatment of cancer (Siu et al., 2017). Therapeutic success has been particularly beneficial tor the treatment of tumors that are thought to be more immunogenic such as melanoma (Huang and Zappasodi, 2022), with overall response rate of 33% to anti-PD-1 (pembrolizumab) therapy (Ribas et al., 2016). However, long lasting benefit in melanoma is only seen in approximately half of patients even with an optimal combination treatment of anti- PD-L1 (nivolumab) and anti- CTLA-4 (ipilimumab) (Wolchok et al., 2022). Response rates for other cancers, such as pancreatic cancer are poor (Isaacsson and Antonarakis, 2018), and thought to be related to the limited immune cell presence in the tumor. In some cancers anti-PD-L1 or anti-PD-1 response can be improved with synergistic combination therapies, such as chemotherapies, radiation therapy and immunotherapeutics (Upadhaya et al., 2021). A potential mechanism for improved efficacy is enhanced tumor antigen presentation, resulting in generation of tumor reactive cytotoxic T cells (Mpakali and Stratikos, 2021).
CD47 is another immunosuppressive molecule that inhibits anti-tumor immunity through a different mechanism than the PD-L1/PD-1 interaction. CD47 is expressed on multiple cell types including cancer cells, and suppresses macrophage phagocytosis of tumor cells by binding to its inhibitory receptor SIRPα (Zhao et al., 2022). SIRPα mediated suppression of phagocytosis has the potential to limit the adaptive immune response by attenuating the abundance of tumor antigens. A logical step to take advantage of blocking CD47-SIRPα interaction for the treatment of cancer has been translated into pre-clinical and clinical studies with anti-CD47 antibodies (Maute et al., 2022; Son et al., 2022), SIRPα fusion proteins and anti-SIRPα antibodies (Zhang et al., 2020). Unfortunately CD47 blocking therapies have had limited success in clinical trials and have encountered dose limiting toxicities, particularly anemia and thrombocytopenia (Bouwstra et al., 2022). Early phase clinical trials testing the combination of anti-PD-1/PD-L1 with anti-CD47 antibodies indicated mostly mild to moderate toxicities, favoring further clinical investigation (Lakhani et al., 2021).
Because the rationale for combining blocking antibodies to PD-L1 and CD47 was compelling for both improving efficacy and safety, we, and others (Wang et al., 2020; Chen et al., 2021; Wang et al., 2023), have taken advantage of conventional antibody engineering to develop human anti-PD-L1/anti-CD47 therapeutic bispecific antibodies. Reported molecular structures vary, but have in common IgG1 or IgG4 backbones using knobs-in-holes technologies. The source, and molecular characteristics of the CD47 and PD-L1 binding domains are distinct, being derived from standard mouse hybridoma approaches (Wang et al., 2023) or common light chain phage display libraries (Chen et al., 2021) in Fab, or single domain (VHH) format (Wang et al., 2020). QL401 is in a 1 + 1 format on a human IgG4 backbone, with Knob-in-Hole and Protein A binding mutations.
We generated novel antibodies against human PD-L1 or CD47 by immunizing mice, and screening hybridoma supernatants for binding and blocking activity, then humanized for further characterization. Humanized anti-PD-L1 antibodies were further validated in functional in vitro immune cell assays, and a final candidate selected, hD39.5, which proved to be efficacious in a MC-38/hPD-L1 - hPD-1/C57BL/6 mouse tumor model. hD39.5 was converted to a Fab for bi-specific antibody engineering. Humanized anti-human CD47 antibodies were also screened in a validation process including evaluation of human monocyte phagocytosis of tumor cells and for efficacy in a mouse xenograft model. Human RBC express CD47, and anemia has been a limiting factor in clinical trials. Therefor hemagglutination was assayed as one of the criteria for selecting humanized anti-CD47 antibodies for further evaluation. Two anti-hCD47 antibodies, hE15.1 and hE24.6 were selected and converted into scFv for testing in a bispecific anti-PD-L1-CD47 format.
The bispecific platform for the clinical candidate QL401 was constructed in a 1 + 1 format on a human IgG4 backbone, which retained Fc gamma receptor binding to provide a phagocytic signal, and with Knob-in-Hole mutations. Protein A binding mutations were introduced on the knob Fc region to facilitate purification. Two additional functional components of QL401 were the anti-PD-L1 binding Fab arm derived from hD39.5, and the anti-CD47 binding scFv arm from hE15.1. The resulting bispecific construct provided sufficient yield, recovery, purity and stability of GMP grade protein to proceed with a QLP toxicology study.
Overall QL401 has potent in vitro activity in binding and functional assays. QL401 demonstrated high affinity binding and blocking (PD-1 and SIRPα) for both arms (anti-PD-L1 Fab and anti-CD47 scFv) in cell based assays. However, blocking of PD1 by anti-PD-L1 arm and of SIRPα by anti-CD47 arm was less efficient than the parental antibody clones. Phagocytosis capacity of QL401 of human macrophages engulfing tumor cell line was potent and equivalent to the parental antibody. In vivo in mouse/human-PBMC-tumor xenograft models, QL401 showed similar efficacy to clinical benchmarks Magrolimab and Durvalumab alone or in combination. Early clinical studies with anti-CD47 antibodies pointed to anemia and thrombocytopenia as potential dose limiting toxicities. Previous reports demonstrated lower binding of an affinity engineered anti-CD47 IgG4 antibody resulted in a better RBC toxicology profile (Thaker et al., 2022). Anti-PD-L1/anti-CD47 bispecific antibodies with significantly lower binding affinity of the anti-CD47 arm compared to the higher affinity anti-PD-L1 arm have been described (Chen et al., 2021; Wang et al., 2023) Such an approach has the potential to provide a better safety profile by preferentially targeting PD-L1/CD47 double positive tumors rather than single positive circulating RBC. QL401 has similar binding affinities and blocking capability for both anti-PD-L1 and anti-CD47 arms. QL401 maintained potent in vitro binding, blocking and enhanced functional responses for both PD-L1 and CD47, but demonstrated a clearly reduced effect on human RBC. QL401 showed lower binding affinity to RBC compared to CD47 expressing tumor cells, and remarkably no hemagglutination activity, and little activity in a human monocyte/RBC phagocytosis assay. In a QL401 cynomolgus GMP multi-dose toxicology (10, 30, 100 mg/kg) minor fluctuations in RBC and platelet counts were observed but remained within the normal range. In contrast, Magrolimab was reported to induce hemagglutination and phagocytosis of RBCs in vitro (Liu X et al., 2015), suggesting potential toxicities. A non-human primate pharmacokinetic and toxicology study of Magrolimab did result in dose-dependent anemia (Liu et al., 2022).
In summary, novel potent anti-human PD-L1 and anti-human CD47 monoclonal antibodies were isolated, humanized and formatted into a bi-specific antibody for clinical use. QL401 maintained potent macrophage phagocytic activity against tumor cells in vitro, and delayed tumor growth in vivo. QL401 demonstrated reduced human RBC binding, hemagglutination activity and RBC phagocytosis in vitro, which was reflected in a non-human primate toxicology safety profile, consistent with moving forward into a clinical Phase I safety study in humans.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by Institutional Animal Care and Use Committee: QLSF Biotherapeutics.
IT, LS, WW, HV, AC, AM, SG, CF, CS, MS, GW, YZ, NZ, JF, and ML contributed experimental data to this manuscript. CF prepared the manuscript. All authors contributed to the article and approved the submitted version.
This study received funding from Qilu Pharmacueticals. The funder's employees were involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication. The following authors who contributed to this manuscript were employees of QLSF Biotherapeutics: IT, LS, WWP, HVT, AC, AM, SG and CF, whereas CC, CS, MS, GW, YZ, NZ, JF, and ML were employees of Qilu Pharmaceutical Co.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Andrejeva, G., Capoccia, B. J., Hiebsch, R. R., Donio, M. J., Darwech, I. M., Puro, R. J., et al. (2021). Novel SIRPα antibodies that induce single-agent phagocytosis of tumor cells while preserving T cells. J. Immunol. 206 (4), 712–721. doi:10.4049/jimmunol.2001019
Ansell, S. M., Maris, M. B., Lesokhin, A. M., Chen, R. W., Flinn, I. W., Sawas, A., et al. (2021) Phase I study of the CD47 blocker TTI-621 in patients with relapsed or refractory hematologic malignancies. Clin. Cancer Res. 15;27(8), 2190–2199. doi:10.1158/1078-0432.CCR-20-3706
Bouwstra, R., van Meerten, T., and Bremer, E. (2022). CD47-SIRPα blocking-based immunotherapy: Current and prospective therapeutic strategies. Clin. Trans.l Med. 12 (8), e943. doi:10.1002/ctm2.943
Chen, S. H., Dominik, P. K., Stanfield, J., Ding, S., Yang, W., Kurd, N., et al. (2021). Dual checkpoint blockade of CD47 and PD-L1 using an affinity-tuned bispecific antibody maximizes antitumor immunity. J. Immunother. Cancer. 9 (10), e003464. doi:10.1136/jitc-2021-003464
Cindy, S., Yang, S. Y., Lien, S. C., Wang, B. X., Clouthier, D. L., Hanna, Y., et al. (2021). Pan-cancer analysis of longitudinal metastatic tumors reveals genomic alterations an,d immune landscape dynamics associated with pembrolizumab sensitivity. Nat. Commun. 12 (1), 5137. doi:10.1038/s41467-021-25432-7
Foote, J., and Winte, r. G. (1992). Antibody framework residues affecting the conformation of the hypervariable loops. J. Mo.l Biol. 224 (2), 487–499. doi:10.1016/0022-2836(92)91010-m
Huang, A. C., and Zappasodi, R. (2022). A decade of checkpoint blockade immunotherapy in melanoma: Understanding the molecular basis for immune sensitivity and resistance. Nat. Immunol. 23, 660–670. doi:10.1038/s41590-022-01141-1
Huston, J. S., Levinson, D., Mudgett-Hunter, M., Tai, M. S., Novotný, J., Margolies, M. N., et al. (1988). Protein engineering of antibody binding sites: Recovery of specific activity in an anti-digoxin single-chain fv analogue produced in Escherichia coli. Proc. Natl. Acad. .Sci. U. S. A. 85 (16), 5879–5883. doi:10.1073/pnas.85.16.5879
IsaacssonVelho, P., and Antonarakis, E. S. (2018). PD-1/PD-L1 pathway inhibitors in advanced prostate cancer. Expert. Rev. Clin. Pharmacol. 11 (5), 475–486. doi:10.1080/17512433.2018.1464388
Jendeberg, L., Nilsson, P., Larsson, A., Denker, P., Uhlén, M., Nilsson, B., et al. (1997) Engineering of Fc(1) and Fc(3) from human immunoglobulin G to analyze subclass specificity for staphylococcal protein A. J. Immunol. Methods. 14;201(1):25–34. doi:10.1016/s0022-1759(96)00215-3
Jones, P. T., Dear, P. H., Foote, J., Neuberger, M. S., and Winter, G. (1986). Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 321 (6069), 522–525. doi:10.1038/321522a0
Lakhani, N. J., Chow, L. Q. M., Gainor, J. F., LoRusso, P., Lee, K.-W., Chung, H. C., et al. (2021). Evorpacept alone and in combination with pembrolizumab or trastuzumab in patients with advanced solid tumours (ASPEN-01): A first-in-human, open-label, multicentre, phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 22, 1740–1751. doi:10.1016/S1470-2045(21)00584-2
Liu, B., Hu, X., Feng, K., Gao, R., Xue, Z., Zhang, S., et al. (2022). Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti-PD-1 therapy in lung cancer. Nat. Cancer 3 (1), 108–121. doi:10.1038/s43018-021-00292-8
Liu, J., Wang, L., Zhao, F., Tseng, S., Narayanan, C., Shura, L., et al. (2015) Pre-clinical development of a humanized anti-CD47 antibody with anti-Cancer therapeutic potential. PLoS One.;10(9):e0137345. doi:10.1371/journal.pone.0137345
Liu, X., Pu, Y., Cron, K., Deng, L., Kline, J., Frazier, W. A., et al. (2015). CD47 blockade triggers T cell-mediated destruction of immunogenic tumors. Nat. Med. 21(10):1209–1215. doi:10.1038/nm.3931
Maute, R., Xu, J., and Weissman, I. L. (2022). CD47-SIRPα-targeted therapeutics: Status and prospects. Immuno-oncol. Technol. 13, 100070. doi:10.1016/j.iotech.2022.100070
Merchant, A. M., Zhu, Z., Yuan, J. Q., Goddard, A., Adams, C. W., Presta, L. G., et al. (1998). An efficient route to human bispecific IgG. Nat. Biotechnol. 16 (7), 677–681. doi:10.1038/nbt0798-677
Mpakali, A., and Stratikos, E. (2021). The role of antigen processing and presentation in cancer and the efficacy of immune checkpoint inhibitor immunotherapy. Cancers (Basel) 13 (1), 134. doi:10.3390/cancers13010134
Nam, G. H., Lee, E. J., Kim, Y. K., Hong, Y., Choi, Y., Ryu, M. J., et al. (2018). Combined Rho-kinase inhibition and immunogenic cell death triggers and propagates immunity against cancer. Nat. Commun. 9 (1), 2165. doi:10.1038/s41467-018-04607-9
Nishiga, Y., Drainas, A. P., Baron, M., Bhattacharya, D., Barkal, A. A., Ahrari, Y., et al. (2022) Radiotherapy in combination with CD47 blockade elicits a macrophage-mediated abscopal effect. Nat. Cancer. 3(11):1351–1366. doi:10.1038/s43018-022-00456-0
Patel, S. A., and Minn, A. ,J. (2018). Combination cancer therapy with immune checkpoint blockade: Mechanisms and strategies. Immunity 48 (3), 417–433. doi:10.1016/j.immuni.2018.03.007
Patnaik, A., Spreafico, A., Paterson, A. M., Peluso, M., Chung, J.-K., Bowerset, B., et al. (2020). Results of a first-inhuman phase I study of SRF231, a fully human, high-affinity anti-CD47 antibody. J. Clin. Oncol. 38 (15), 3064. doi:10.1200/JCO.2020.38.15_suppl.3064
Puro, R. J., Bouchlaka, M. N., Hiebsch, R. R., Capoccia, B. J., Donio, M. J., Manning, P. T., et al. (2020). Development of AO-176, a next-generation humanized anti-CD47 antibody with novel anticancer properties and negligible red blood cell binding. Mol. Cancer. Ther. 19 (3), 835–846. doi:10.1158/1535-7163.MCT-19-1079
Qu, T., Li, B., and Wang, Y. (2022). Targeting CD47/SIRPα as a therapeutic strategy, where we are and where we are headed. Biomark. Res. 10 (1), 20. 13. doi:10.1186/s40364-022-00373-5
Reiter, Y., Brinkmann, U., Kreitman, R. J., Jung, S. H., Lee, B., and Pastan, I. (1994). Stabilization of the Fv fragments in recombinant immunotoxins by disulfide bonds engineered into conserved framework regions. Biochemistry 33 (18), 5451–5459. doi:10.1021/bi00184a014
Ribas, A., Hamid, O., Daud, A., Hodi, F. S., Wolchok, J. D., Kefford, R., et al. (2016). Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA 315 (15), 1600–1609. doi:10.1001/jama.2016.4059
Ring, N. G., Herndler-Brandstetter, D., Weiskopf, K., Shan, L., Volkmer, J. P., George, B. M., et al. (2017). Anti-SIRPα antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc. Natl. Acad. Sci. U. S. A. 114 (49), E10578–E10585. doi:10.1073/pnas.1710877114
Robert, C. (2020). A decade of immune-checkpoint inhibitors in cancer therapy. Nat. Commun. 11 (1), 3801. doi:10.1038/s41467-020-17670-y
Siu, L. L., Ivy, S. P., Dixon, E. L., Gravell, A. E., Reeves, S. A., and Rosner, G. L. (2017). Challenges and opportunities in adapting clinical trial design for immunotherapies. Clin. Cancer Res. 23 (17), 4950–4958. doi:10.1158/1078-0432.CCR-16-3079
Sockolosky, J. T., Dougan, M., Ingram, J. R., Ho, C. C., Kauke, M. J., Almo, S. C., et al. (2016). Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc. Natl. Acad. Sci. U.S. A. 113 (19), E2646–E2654. doi:10.1073/pnas.1604268113
Son, J., Hsieh, R. C., Lin, H. Y., Krause, K. J., Yuan, Y., Biter, A. B., et al. (2022). Inhibition of the CD47-sirpα axis for cancer therapy: A systematic review and meta-analysis of emerging clinical data. Front. Immunol. 13, 1027235. doi:10.3389/fimmu.2022.1027235
Tang, J., Yu, J. X., Hubbard-Lucey, V. M., Neftelinov, S. T., Hodge, J. P., and Lin, Y. (2018). Trial watch: The clinical trial landscape for PD1/PDL1 immune checkpoint inhibitors. Nat. Rev. Drug. Discov. 17 (12), 854–855. doi:10.1038/nrd.2018.210
Thaker, Y. R., Rivera, I., Pedros, C., Singh, A. R., Rivero-Nava, L., Zhou, H., et al. (2022). A novel affinity engineered anti-CD47 antibody with improved therapeutic index that preserves erythrocytes and normal immune cells. Front. Oncol. 12, 884196. doi:10.3389/fonc.2022.884196
Tseng, D., Volkmer, J. P., Willingham, S. B., Contreras-Trujillo, H., Fathman, J. W., Fernhoff, N. B., et al. (2013) Anti-CD47 antibody–mediated phagocytosis of cancer by macrophages primes an effective antitumor T-cell response. Proc. Natl. Acad. Sci. U. S. A.;110:11103–11108. doi:10.1073/pnas.1305569110
Upadhaya, S., Neftelino, S. T., Hodge, J. P., Oliva, C., Campbell, J. R., and Yu, J. X. (2021). Combinations take centre stage in PD1/PDL1 inhibitor clinical trials. Nat. Rev. Drug. Discov. 20 (3), 168–169. doi:10.1038/d41573-020-00204-y
Vesely, M. D., Zhang, T., and Chen, L. (2022). Resistance mechanisms to anti-PD cancer immunotherapy. Annu. Rev. Immunol. 40, 45–74. doi:10.1146/annurev-immunol-070621-030155
Wang, R., Zhang, C., Cao, Y., Wang, J., Jiao, S., Zhang, J., et al. (2023). Blockade of dual immune checkpoint inhibitory signals with a CD47/PD-L1 bispecific antibody for cancer treatment. Theranostics 13 (1), 148–160. doi:10.7150/thno.79367
Wang, Y., Ni, H., Zhou, S., He, K., Gao, Y., Wu, W., et al. (2020). Tumor-selective blockade of CD47 signaling with a CD47/PD-L1 bispecific antibody for enhanced anti-tumor activity and limited toxicity. Cancer Immunol. Immunother. 70, 365–376. doi:10.1007/s00262-020-02679-5
Willingham, S. B., Volkmer, J. P., Gentles, A. J., Sahoo, D., Dalerba, P., Mitra, S. S., et al. (2012). The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. U. S. A. 109 (17), 6662–6667. doi:10.1073/pnas.1121623109
Wolchok, J. D., Chiarion-Sileni, V., Gonzalez, R., Grob, J. J., Rutkowski, P., Lao, C. D., et al. (2022). Long-term outcomes with nivolumab plus ipilimumab or nivolumab alone versus ipilimumab in patients with advanced melanoma. J. Clin. Oncol. 40 (2), 127–137. doi:10.1200/JCO.21.02229
Zhang, W., Huang, Q., Xiao, W., Zhao, Y., Pi, J., Xu, H., et al. (2020). Advances in anti-tumor treatments targeting the CD47/sirpα Axis. Front. Immunol. 11, 18. doi:10.3389/fimmu.2020.00018
Keywords: checkpoint therapy, cd47, PD-L1, bispecific antibody, safety, efficacy
Citation: Tang I, Schwimmer L, Gu S, Wei Prior W, Tran HV, Chan A, McClain A, Fraser CC, Sun C, Si M, Wang G, Zhao Y, Zhang N, Fu J, Liu M, Cao C and Chen S (2023) Generation of a potent anti-PD-L1-CD47 bispecific antibody with a strong therapeutic and safety profile for cancer immunotherapy. Front. Drug Discov. 3:1182146. doi: 10.3389/fddsv.2023.1182146
Received: 08 March 2023; Accepted: 27 April 2023;
Published: 09 May 2023.
Edited by:
Werner J. Geldenhuys, West Virginia University, United StatesReviewed by:
Shang Su, University of Toledo, United StatesCopyright © 2023 Tang, Schwimmer, Gu, Wei Prior, Tran, Chan, McClain, Fraser, Sun, Si, Wang, Zhao, Zhang, Fu, Liu, Cao and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christopher C. Fraser, Y2ZyYXNlckBxbHNmYmlvLmNvbQ==; Shihao Chen, c2hpaGFvLmNoZW5AcWxzZmJpby5jb20=
†Present addresses: Lauren Schwimmer, Abalone Bio, Emeryville, CA, United States
Wei Wei Prior, Asher Bio, South San Francisco, CA, United States
Allan Chan, Bayer, Berkeley, CA, United States
Anna McClain, University of Texas, Austin, TX, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.