 Alex Pu
Alex Pu Gautam Ramani
Gautam Ramani Yi-Ju Chen
Yi-Ju Chen James A. Perry
James A. Perry Charles C. Hong
Charles C. Hong- Department of Medicine, University of Maryland School of Medicine, Baltimore, MD, United States
Pulmonary arterial hypertension (PAH) is characterized by remodeling and narrowing of the pulmonary vasculature which results in elevations of pulmonary arterial pressures. Here, we conducted a genome-wide association study (GWAS) using the UK Biobank, analyzing the genomes of 493 individuals diagnosed with primary pulmonary hypertension, based on ICD-10 coding, compared to 24,650 age, sex, and ancestry-matched controls in a 1:50 case-control design. Genetic variants were analyzed by Plink’s firth logistic regression and assessed for association with primary pulmonary hypertension. We identified three linked variants in the PIM1 gene, which encodes a protooncogene that has been garnering interest as a potential therapeutic target for PAH, that were associated with PAH with genome wide significance, one (rs192449585) of which lies in the promoter region of the gene. We also identified 15 linked variants in the LINC01491 gene. These results provide genetic evidence supporting the role of PIM1 inhibitors as a potential therapeutic option for PAH.
Introduction
Pulmonary hypertension (PH) describes a heterogeneous group of systemic, heart, and lung disorders characterized by elevations in pulmonary arterial pressures. Pulmonary arterial hypertension (PAH) represents a subgroup of PH, characterized by remodeling and narrowing of the pulmonary vasculature. Idiopathic pulmonary arterial hypertension (IPAH), which was previously coined primary pulmonary hypertension (PPH), describes PAH when no alternative etiology can be identified. Much of our understanding of the pathogenesis of PAH is shaped by the seminal human genetic studies which revealed that mutations in BMPR2, a gene encoding bone morphogenetic protein receptor type 2, account for ∼80% of patients with hereditary PAH and ∼20% of sporadic cases (Southgate et al., 2020; Ruopp and Cockrill, 2022). Subsequent exome sequencing and genome-wide association studies (GWAS) have found associations of PAH with additional genes, including those involved in the SMAD signaling pathway (Nasim et al., 2011) as well as in KCNK3 (Ma et al., 2013), SOX17 and HLAD-DPA1/DPB1 (Gräf et al., 2018; Southgate et al., 2020). Nevertheless, there is an incomplete understanding of PAH pathogenesis and heterogeneity, including disease penetrance, onset, clinical course, and prognosis. Despite improved diagnosis and effective pharmacotherapy, PAH mortality remains high, ∼10% per year (Southgate et al., 2020), highlighting a large unmet medical need.
Using the UK Biobank cohort, we have previously carried out ICD-10 (International Classification of Diseases 10)-based GWAS for a variety of cardiovascular diseases (Ashvetiya et al., 2021; Alkhalfan et al., 2022; Gyftopoulos et al., 2022). Here, using the UK Biobank cohort, we carried out a GWAS of loci associated with the ICD-10 diagnostic code for primary pulmonary hypertension (I27.0), which encompasses IPAH and hereditary PAH but not other forms of associated PAH, and found novel genetic variants, including those in the PIM1 and LINC01491 genes, associated with it.
Methods
Ethical approval
The present study, which involved deidentified data obtained from the UK Biobank Resource under Application Number 49852, received the proper ethical oversight, including the determination by the University of Maryland, Baltimore Institutional Review Board that the study is not human research (IRB #: HF-00088022).
Study population
This study utilized data from the UK Biobank, a large ongoing prospective cohort study involving 506,682 participants between 2006 and 2010. Participants were aged 40–69 at the time of recruitment. Our study population was identified using the ICD-10 diagnostic code for primary pulmonary hypertension (I27.0) as either the participant’s main diagnosis (indicating reason for hospital admission) or secondary diagnosis code. This ICD-10 diagnostic code encompasses (1) IPAH, (2) hereditary PAH, (3) primary PAH, and (4) primary group 1 PH and excludes PAH due to secondary causes. This I27.0 code is most similar, but not identical, to Group 1 pulmonary arterial hypertension (PAH) based on the newer World Health Organization (WHO) grouping (Simonneau et al., 2019). Given the ambiguity of the definitions of “primary pulmonary hypertension,” we have henceforth classified each case as “pulmonary arterial hypertension (PAH).”
Genome-wide association study
The UK Biobank contains genetic data with over 820,000 genotyped SNPs and up to 90 million imputed variants available. Using data from the UK Biobank Resource on 487,310 subjects with imputed genotypes, we performed quality control by removing those with genetic relatedness exclusions (Data-Field 22018—UKB, https://biobank.ctsu.ox.ac.uk/crystal/field.cgi?id=22018; 1532 subjects), sex chromosome aneuploidy (Data-Field 22019 –UKB, https://biobank.ctsu.ox.ac.uk/crystal/field.cgi?id=22019; 651 subjects), mismatch between self-reported sex and genetically determined sex (Data-Field 31 –UKB, https://biobank.ctsu.ox.ac.uk/crystal/field.cgi?id=31; Data-Field 22001 –UKB, https://biobank.ctsu.ox.ac.uk/crystal/field.cgi?id=22001; 372 subjects), recommended genomic analysis exclusions (Data-Field 22010—UKB, https://biobank.ctsu.ox.ac.uk/crystal/field.cgi?id=22010; 480 subjects), and outliers for heterozygosity or missing rate (Data-Field 22017 –UKB, https://biobank.ctsu.ox.ac.uk/crystal/field.cgi?id=22077; 968 subjects).
Additionally, we chose to limit subject selection to the UK Biobank’s genetic ethnic grouping described as “White British” with very similar genetic ancestry based on principal components analysis and made available as part of the UK Biobank Resource (Data-Field 2,206—UKB, https://biobank.ctsu.ox.ac.uk/crystal/field.cgi?id=22006).
We defined cases with the ICD-10 code I27.0 as specified above. The set of cases was purged of relatedness by removing one of each related pair in an iterative fashion until no related subjects remained. Relatedness was defined as a kinship value of 0.0442 or greater in the data provided from the UK Biobank Resource. For each identified case, 50 controls were selected from the remaining pool of unrelated participants, matching for age, sex, and ancestry. This was done with incremental tolerance expansion for age and ancestry. For age, the tolerance ranged from 0 (exact match) to a max of 7 years. For ancestry, principal components (PCs) supplied by the UK Biobank were used. We assessed the similarity in ancestry by calculating the mathematical distance in a graph plotting the PC1 x PC2. The tolerance for ancestry ranged from a calculated distance of 2 PC units to a maximum of 80 PC units, with PC1 ranging from 0 to +400 and PC2 ranging from −300 to +100.
To perform this GWAS, Plink’s firth logistic regression was used, adjusting for age, sex, and 5 PCs using data supplied by the UK Biobank Resource. Firth regression was used for analysis as it has been shown to yield the best combination of control for type I error and power for detecting low-frequency variants (Wang, 2014; Chang et al., 2015). The PAH cases were analyzed with the 40 million imputed genetic variants provided by the UK Biobank with imputation quality scores greater than 0.80. This analysis incorporated covariates of sex, age, and principal components 1 through 5 to adjust for ancestry. Precalculated PC data for the first 40 principal components were supplied by the UK Biobank. Only the first 5 PCs had significance with p-values <0.05 in our preliminary analysis. As a result, we only included the first 5 PCs for our analysis.
Identification of pulmonary arterial hypertension variants
A minor allele frequency (MAF) of at least 0.5% and a significance value of < 5E-08 was used to identify variants of interest.
Comorbidity and biomarker analysis
The UK Biobank also contains participant baseline clinical information, including comorbidities, height, weight, body mass index, and basic laboratory values. The ICD-10 code diagnoses, ABO blood type, 5 biometric markers, and 61 biomarkers in the identified PAH cases and the matched controls were examined. The significance of these conditions and variables with PAH patients in comparison to age, sex, and ancestry-matched controls were determined by two-sample t-test.
Results
A total of 493 (243 males, 250 females) individuals were identified to have a diagnosis of PAH from a total of 472,124 participants, representing a prevalence of 1 in 1,000. Among these individuals, the mean (SD) age of diagnosis was 63.4 (8.21) years. Of the 490 cases with available BMI data, 355 (72%) had a BMI of 25 and above.
Genome-wide association study
Our GWAS of variants with MAF of at least 0.5% identified a total of 24 variants in 7 distinct loci (Figure 1). We identified 3 variants associated with PAH related to the PIM1 gene, which encodes the Pim-1serine/threonine-protein kinase, which is involved in cytokine signaling, cell cycle progression and oncogenesis (Holder and Abdulkadir, 2014). One variant was rs192449585 (MAF 0.76%; OR 3.808; p = 4.78E-09), located in the first amino acid of the exonic region of the PIM1 isoform 1 gene in a region of high density Dnase I hypersensitivity signals and predicted transcription start site for several tissues (Figure 2). Further, this variant lies in the transcription factor binding region of 28 transcription factors, including POLR2A, IRF1, CTCR, TAF1, and TBP (Figure 2, Supplementary Figure S1), and alters 13 regulatory motifs, including TATA, YY1, and SIN3A (Supplementary Table S1). Additionally, this variant was found to have an eigenPC score of 5.046, which are functional prediction scores ranging from −3.2 to +72 based on conservation, allele frequencies, deleteriousness prediction and epigenomic signals using an unsupervised learning method (Ionita-Laza et al., 2016). An eigenPC score of 5.046 places rs192449585 at the top 99.66th percentile of all variants. The prevalence of PAH in heterozygotes with the rs192449585 variant was 340 per 100,000, compared to 98 per 100,000 in the wild types (Figure 3A). Interestingly, two individuals were found to be homozygous for rs192449585, both of whom are not diagnosed with PAH and are females 49 years of age and older. The second variant was rs183926708 (MAF 0.75%; OR 4.002; p = 1.85E-10), located in the intergenic region −6,549 bases from the PIM1 gene. The third variant was rs187386578 (MAF 0.75%; OR 3.626; p = 5.28E-09), located in the intergenic region 56,310 bases from the PIM1 gene. The three variants were found to be linked in our linkage disequilibrium analysis. A locus zoom plot of rs183926708, the top variant, can be found in Supplementary Figure S2.
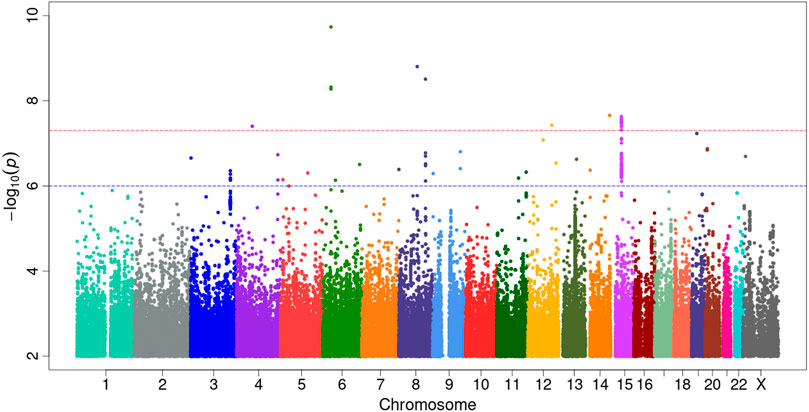
FIGURE 1. Genome-wide association study analysis for variants associated with pulmonary arterial hypertension. Manhattan plot of GWAS results with minor allele frequency >0.5% for PAH. The red dotted line denotes genome wide significance at p = 5E-8. The significance is displayed on the y-axis as -log10 of the p-value, and the results lie according to their chromosomal location on the x-axis.
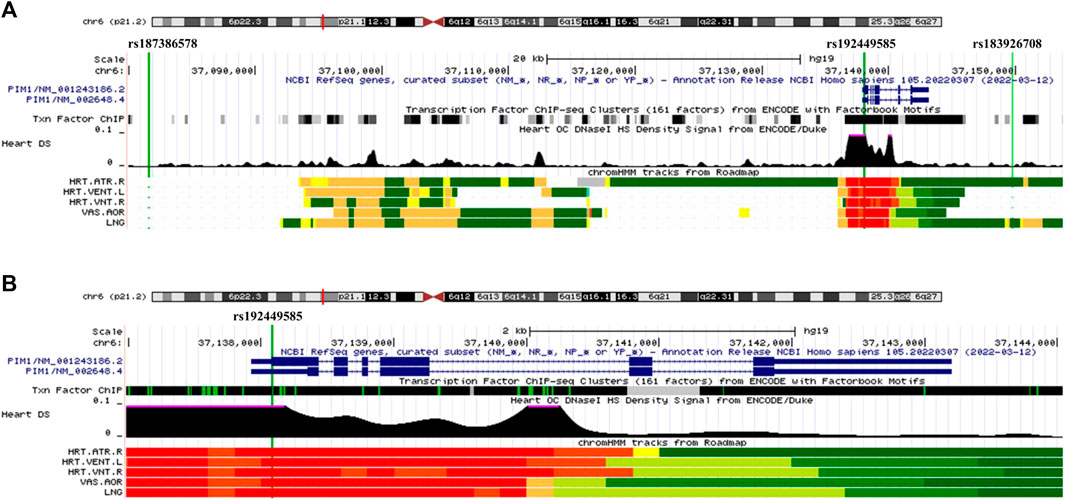
FIGURE 2. UCSC Genome Browser view of genomic region for PIM1 variants. (A) UCSC Genome Browser for all three variants. Green vertical line depicts location of variant, labeled on top. Dark blue regions indicate the 6 exons for PIM1 plus untranslated regions (5′ and 3′ UTRs are narrower dark blue sections). Black region of “Txn Factor ChIP” indicates density of transcription factor ChIP-seq clusters from ENCODE. Black graphic for “Heart DS” indicates the Dnase I hypersensitivity density signal from ENCODE/Duke. Bottom five tracks indicate the predicted chromatin state from Roadmap. Red indicates a transcription start site region for five tissues, HRT.ATR.R (Right Atrium), HTR.VENT.L (Left Ventricle), HRT.VNT.R (Right Ventricle), VAS.AOR (Aorta), LNG (Lung). (B) UCSC Genome Browser focused on exonic variant rs192449585 and PIM1 gene.
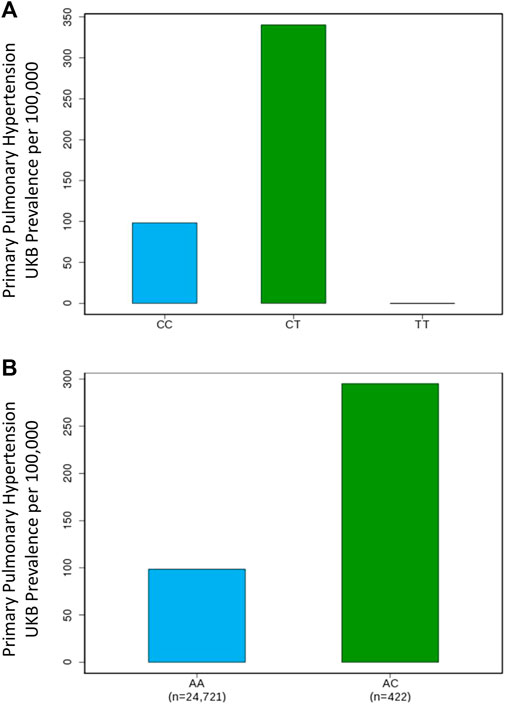
FIGURE 3. Genome-wide association study results for PIM1 and LINC01491 variants associated with pulmonary arterial hypertension. (A) Prevalence of PAH in the UK Biobank increases with PIM1 variant rs192449585 status. CC denotes wildtype, CT denotes heterozygous carrier, TT denotes homozygous carrier. (B) Prevalence of PAH in the UK Biobank increases with LINC01491 variant rs145733648 status. AA denotes wildtype, AC denotes heterozygous carrier.
Our GWAS identified 15 variants related to LINC01491. All of the identified variants lie in the intergenic region of LINC01491. The top variant within this locus was rs145733648 (MAF 0.85%; OR 3.303; p = 2.34E-0.8). The prevalence of PAH in heterozygotes with the rs145733648 variant was 295 per 100,000, compared to 98 per 100,000 in the wild types (Figure 3B). Information regarding the other LINC01491 variants can be found in Table 1. A locus zoom plot of rs145733648 can be found in Supplementary Figure S3.
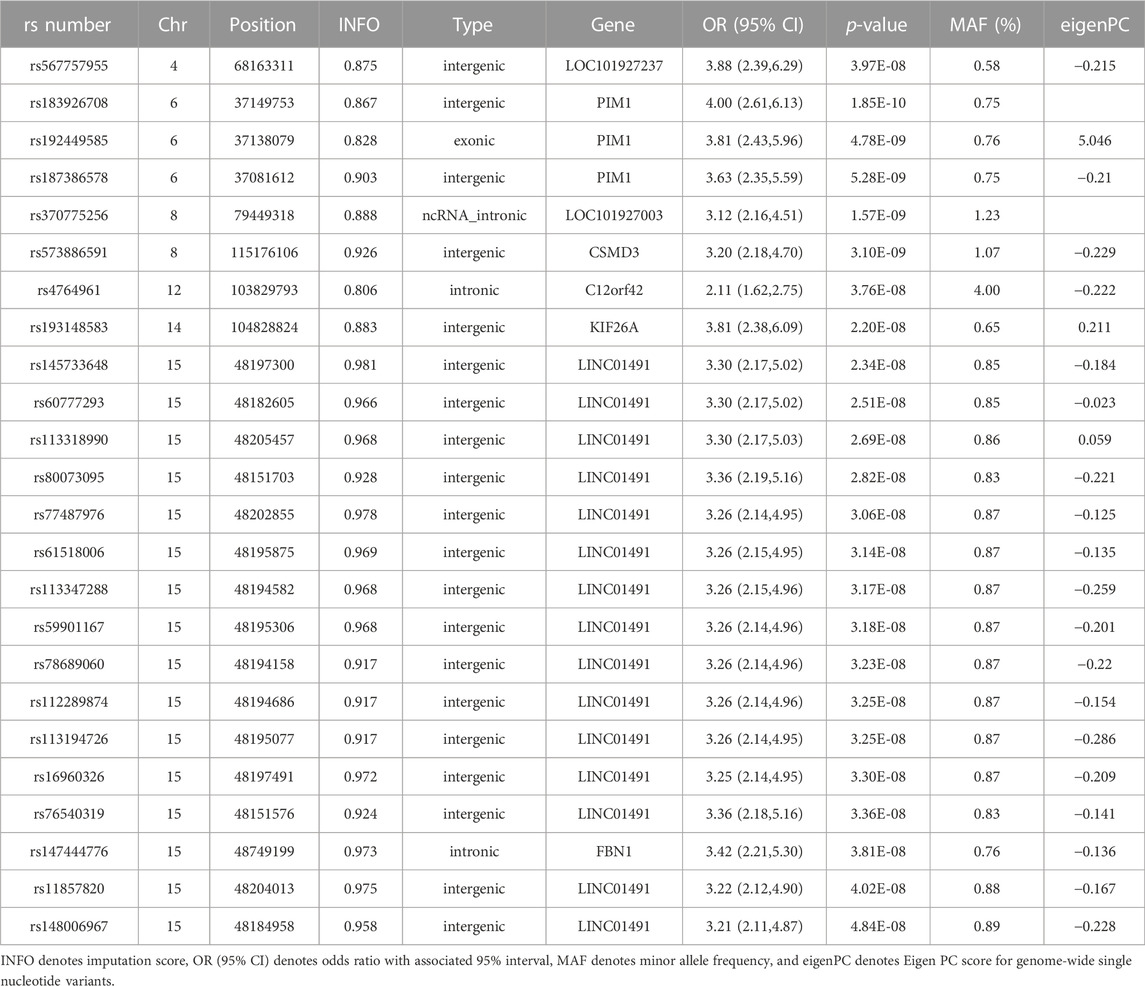
TABLE 1. Detailed genome wide association study results.
In addition, 6 other variants at 5 other loci with MAF over 0.5% were found to be associated with PAH. Information regarding these can be found in Table 1, with information regarding the further breakdown of cases and controls for each variant in Supplementary Table S2. Of note, variant rs147444776 was identified in the intronic region of FBN1 gene, which encodes fibrillin-1, a structural protein involved in elastic and non-elastic connective tissue. Quantile-quantile plots (QQ Plots) are provided in Supplementary Figure S4 to illustrate that the GWAS quality was well controlled.
Comorbidities and biomarkers
The cases of PAH were more likely to be diagnosed with dyspnea (R06.0; 24% vs. 2%, p < 0.0001), chest pain (R07.4; 25% vs. 7%, p < 0.0001), syncope and collapse (R55; 14% vs. 3%, p < 0.0001), all of which are common symptoms of PAH. PAH cases were also more likely to be diagnosed with congestive heart failure (I500; 29% vs. 1%, p < 0.0001), chronic kidney disease (N18.9; 15% vs. 1%, p < 0.0001), and atrial fibrillation/flutter (I48; 53% vs. 6%, p < 0.0001). Additionally, PAH cases were more likely to report a family history of heart disease (Z82.4; 20% vs. 4%, p < 0.0001) than controls.
Interestingly, individuals with a diagnosis of PAH were also more likely to be diagnosed with left ventricular failure (I50.1; 25% vs. 1%, p < 0.0001), chronic obstructive pulmonary disease (J44.9; 20% vs. 3%, p < 0.0001), and pulmonary embolism (I26.9; 12% vs. 1%, p < 0.0001). Additionally, PAH cases were also more likely to have a co-diagnosis of hypothyroidism (E03.9; 13% vs. 5%, p < 0.0001), iron deficiency anemia (D50.9; 10% vs. 2%, p < 0.0001), sleep apnea (G47.3; 5% vs. 1%, p < 0.0001), non-insulin-dependent diabetes mellitus (E11.9; 21% vs. 6%, p < 0.0001), pure hypercholesterolemia (E78.0; 38% vs. 13%, p < 0.0001), and essential hypertension (I10; 65% vs. 29%, p < 0.0001). A full list of co-diagnoses can be found in Table 2.
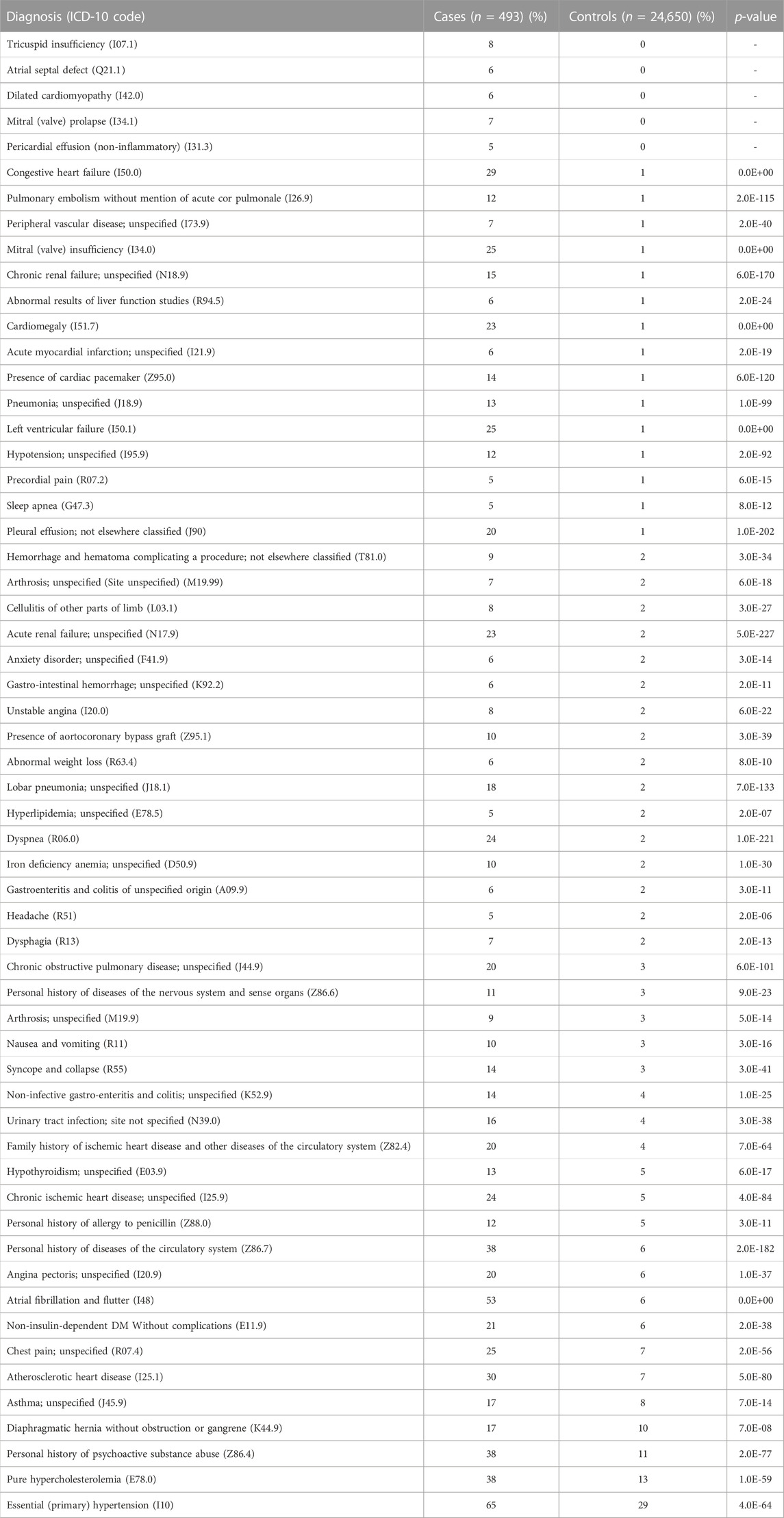
TABLE 2. ICD-10 diagnoses associated with PAH.
On average, PAH cases had higher BMIs (28.68 vs. 27.51, p < 0.0001) than controls. Individuals with PAH had a higher mean serum urea (39.3 vs. 33.9 mg/dL, p < 0.0001), creatinine (0.824 vs. 0.735 mg/dL, p = 0.0001), uric acid (5.93 vs. 5.32 mg/dL, p < 0.0001) and cystatin C (1.11 vs. 0.939 mg/dL, p < 0.0001) levels, consistent with the higher rate of diagnosis of chronic kidney disease in that population when compared to controls. While individuals with PAH were more likely to be diagnosed with hypercholesterolemia, they had a lower mean cholesterol (201 vs. 221 mg/dL, p < 0.0001) compared to the control population. Consistent with findings regarding diabetes mellitus type 2, the PAH cohort had a higher HbA1c (5.7% vs. 5.5%, p < 0.0001) and fasting glucose level (97.3 vs. 93.3 mg/dL, p = 0.01) when compared to the control cohort. These results are shown in Table 3.
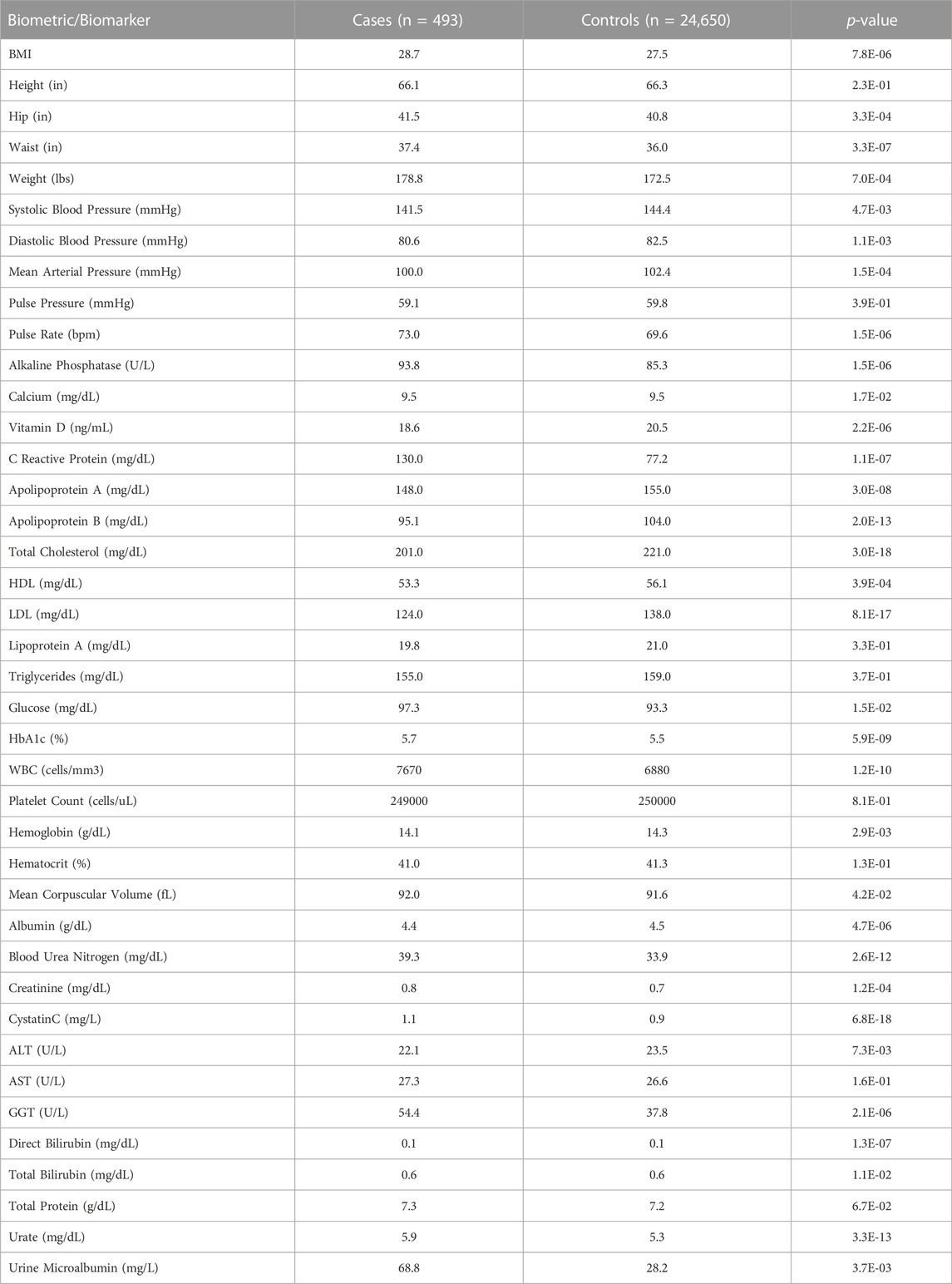
TABLE 3. Biometrics and biomarkers in PAH cases and controls.
Discussion
In our GWAS using data available from the UK Biobank, we discovered 3 linked variants in the PIM1 gene associated with PAH. One of these variants (rs192449585) is particularly noteworthy because, while it does not alter the resulting amino acid sequence, it lies in the promoter region of the gene and overlaps with the binding sites of transcription factors such as RNA Polymerase as well as regulatory motifs such as TATA, suggesting it may impact PIM1 expression. This is supported by the variant’s high eigenPC score of 5.046, an aggregate predictive score for functional importance, which helps to prioritize likely causal variants in a given genomic region (Jeffcott and Whitwell, 1973).
While PIM1 is more commonly viewed as a key player in the role of tumorigenesis, Paulin and colleagues have provided evidence for its potential role in PAH (Paulin et al., 2011; Renard et al., 2013; Lampron et al., 2020). PIM1 has been hypothesized to amplify the growth signals from STAT3, an isoform of the signal transducers and activators of transcription (STAT) protein family, to activate transcription factor nuclear factor of activated T cells (NFAT), which in turn promotes the pro-proliferative and anti-apoptotic phenotype of PAH (Paulin et al., 2011). While STAT3 and NFAT1 are constitutively expressed in normal pulmonary arteries, PIM1 was found to only be present in PAH, making it a therapeutic target of interest. Further studies have corroborated this, showing that inhibition of PIM1 leads to decreased KU70 (lupus Ku autoantigen protein p70) recruitment to damaged DNA sites, effectively inhibiting DNA non-homologous end-joining, which allows for increased apoptosis in PAH pulmonary artery smooth muscle cells. In the same study, they found PIM1 inhibition resulted in improved vascular remodeling and pulmonary hemodynamics in mice models (Lampron et al., 2020). To our knowledge, our study is the first to show a human genetic association of PIM1 with PAH. Since pipeline drug targets with human genetic evidence of disease association are more likely to lead to approved drugs than those without such evidence (Nelson et al., 2015; Hong, 2022), our study lends important support for PIM1 inhibition as a promising therapeutic approach for PAH. Moreover, as plasma PIM1 levels have been associated with PAH disease severity, our findings further corroborate the role of PIM1 in the pathogenesis of PAH and as a potential therapeutic target (Renard et al., 2013; Zhu et al., 2019).
In addition to our findings regarding PIM1, we also identified 15 linked variants in LINC01491, which encodes long intergenic non-coding RNA (lncRNA) 1491. While LINC01491 has not been well characterized in the literature, other lncRNAs have been found to play a role in the pathogenesis of PAH (Han et al., 2021), with lncRNA H19 being a specific example showing promise as a biomarker for severity of PAH as well as a potential therapeutic target (Omura et al., 2020). Studies to verify our findings regarding LINC01491 may be warranted to confirm its role in PAH pathogenesis and explore its utility as a biomarker or therapeutic target.
Our study also provides information corroborating our current understanding of PAH and associated comorbidities. For example, recent studies have found that iron deficiency is correlated with PAH disease severity, with a prevalence ranging from 30% to 65% in patients with IPAH (Rosenkranz et al., 2020). We found that our PAH cases were more likely to be diagnosed with iron deficiency and had slightly lower levels of hemoglobin compared to matched controls, although our findings showed a smaller prevalence of iron deficiency. Additionally, our study found that urine microalbumin levels were higher in PAH (Table 2), in comparison to controls, consistent with recent findings that low grade albuminuria is prevalent in PAH patients (Nickel et al., 2019). Moreover, we observed a higher prevalence of non-insulin-dependent diabetes and higher mean hemoglobin A1c in our PAH cases, consistent with prior studies linking insulin resistance to PAH (Zamanian et al., 2009; Pugh et al., 2011; Hemnes et al., 2019). Interestingly, our study found that patients with PAH generally had lower levels of Vitamin D and were more likely to have comorbid hypothyroidism. While the mechanism behind these findings is still unclear, our results are consistent with findings from prior epidemiological studies and further suggest that studies to elucidate these relationships may be warranted (Callejo et al., 2020; Rosenkranz et al., 2020).
There are several limitations in this study. First, the identification of genetic variants in the GWAS does not prove functional causality, and further studies should be done to confirm these findings. Additionally, these findings should be validated in a patient population with a more diverse genetic origin. The use of ICD-10 codes to select cases is also a limitation as many diseases may be misdiagnosed in the real world. In particular, PAH is often misdiagnosed, which is one reason referral to specialty centers is recommended. ICD-10 based studies offer an opportunity to study a wide array of diseases and medical conditions, especially rare ones like PAH, in a large, “real world” setting. The prevalence of PAH in the UK Biobank is much higher than the previously reported prevalence of 10–15 cases per million people (Ruopp and Cockrill, 2022), reflecting issues with the diagnosis of “primary” PH in the real world. This is further corroborated by the fact that, even though PAH by definition is a diagnosis that excludes left heart disease, pulmonary disease, and thromboembolic disease, there was a higher rate of co-diagnosis of these three conditions in our PAH cases. Additionally, the ICD-10 diagnosis code for “primary pulmonary hypertension” does not provide information about rare subtypes of Group 1 PH, including pulmonary veno-occlusive disease/pulmonary capillary hemangiomatosis or hereditary hemorrhagic telangiectasia associated PAH.
Our study also found markedly higher rates of chronic and acute renal failure, atherosclerotic heart disease, essential hypertension, and peripheral vascular disease as well as higher mean CRP levels in PAH patients. Given that these co-morbid conditions can be considered vascular diseases, one wonders whether PAH involves a more general vascular pathology. Regardless of these limitations and confounding factors, our findings identify novel genetic variants associated with PAH and provide further support for the notion of repositioning PIM1 inhibitors like TP-3654, which was originally developed for advanced solid tumors (Lampron et al., 2020), for the treatment of PAH, possibly in conjunction with PIM1 detection in pulmonary vasculature or elevated plasma PIM1 levels.
Conclusion
Using ICD-10 diagnostic codes for PAH, we identified 3 linked variants in the PIM1 gene in our GWAS of the UK Biobank, further suggesting the role of PIM1 in the pathogenesis of PAH in humans and providing key human genetic evidence in support of the potential of PIM1 inhibitors as a therapeutic option for PAH. We also identified 15 linked variants in the LINC01491 gene, contributing to the growing literature of lncRNAs contributing to the pathogenesis of PAH.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
Author contributions
AP developed and suggested the hypothesis of this manuscript and wrote the majority of the draft. Y-JC provided data analysis. GR provided subject matter expertise in pulmonary hypertension to identify strengths and weaknesses of the methods of this study. JP and CH developed the methods, provided subject matter expertise in GWAS and omics and provided guidance to authors on the manuscript.
Funding
This work was supported by the National Institute of General Medical Sciences, R01GM118557 and National Heart, Lung, and Blood Institute, R01HL1351291 to CCH, and National Heart, Lung, and Blood Institute, 1U01HL137181 to JP. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fddsv.2023.1127736/full#supplementary-material
References
Alkhalfan, F., Gyftopoulos, A., Chen, Y.-J., Williams, C. H., Perry, J. A., and Hong, C. C. (2022). Identifying genetic variants associated with the ICD10 (International Classification of Diseases10)-based diagnosis of cerebrovascular disease using a large-scale biomedical database. PloS One 17, e0273217. doi:10.1371/journal.pone.0273217
Ashvetiya, T., Fan, S. X., Chen, Y.-J., Williams, C. H., O’Connell, J. R., Perry, J. A., et al. (2021). Identification of novel genetic susceptibility loci for thoracic and abdominal aortic aneurysms via genome-wide association study using the UK Biobank Cohort. PloS One 16, e0247287. doi:10.1371/journal.pone.0247287
Callejo, M., Barberá, J. A., Duarte, J., and Perez-Vizcaino, F. (2020). Impact of nutrition on pulmonary arterial hypertension. Nutrients 12, 169. doi:10.3390/nu12010169
Chang, C. C., Chow, C. C., Tellier, L. C., Vattikuti, S., Purcell, S. M., and Lee, J. J. (2015). Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 4, 7. doi:10.1186/s13742-015-0047-8
Gräf, S., Haimel, M., Bleda, M., Hadinnapola, C., Southgate, L., Li, W., et al. (2018). Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat. Commun. 9, 1416. doi:10.1038/s41467-018-03672-4
Gyftopoulos, A., Chen, Y.-J., Wang, L., Williams, C. H., Chun, Y. W., O’Connell, J. R., et al. (2022). Identification of novel genetic variants and comorbidities associated with ICD-10-based diagnosis of hypertrophic cardiomyopathy using the UK Biobank cohort. Front. Genet. 13, 866042. doi:10.3389/fgene.2022.866042
Han, Y., Ali, M. K., Dua, K., Spiekerkoetter, E., and Mao, Y. (2021). Role of long non-coding RNAs in pulmonary arterial hypertension. Cells 10, 1892. doi:10.3390/cells10081892
Hemnes, A. R., Luther, J. M., Rhodes, C. J., Burgess, J. P., Carlson, J., Fan, R., et al. (2019). Human PAH is characterized by a pattern of lipid-related insulin resistance. JCI Insight 4, e123611. doi:10.1172/jci.insight.123611
Holder, S. L., and Abdulkadir, S. A. (2014). PIM1 kinase as a target in prostate cancer: Roles in tumorigenesis, castration resistance, and docetaxel resistance. Curr. Cancer Drug Targets 14, 105–114. doi:10.2174/1568009613666131126113854
Hong, C. C. (2022). The grand challenge of discovering new cardiovascular drugs. Front. Drug Discov. 2, 1027401. doi:10.3389/fddsv.2022.1027401
Ionita-Laza, I., McCallum, K., Xu, B., and Buxbaum, J. D. (2016). A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat. Genet. 48, 214–220. doi:10.1038/ng.3477
Jeffcott, L. B., and Whitwell, K. E. (1973). Twinning as a cause of foetal and neonatal loss in the thoroughbred mare. J. Comp. Pathol. 83, 91–106. doi:10.1016/0021-9975(73)90032-7
Lampron, M.-C., Vitry, G., Nadeau, V., Grobs, Y., Paradis, R., Samson, N., et al. (2020). PIM1 (moloney murine leukemia provirus integration site) inhibition decreases the nonhomologous end-joining DNA damage repair signaling pathway in pulmonary hypertension. Arterioscler. Thromb. Vasc. Biol. 40, 783–801. doi:10.1161/ATVBAHA.119.313763
Ma, L., Roman-Campos, D., Austin, E. D., Eyries, M., Sampson, K. S., Soubrier, F., et al. (2013). A novel channelopathy in pulmonary arterial hypertension. N. Engl. J. Med. 369, 351–361. doi:10.1056/NEJMoa1211097
Nasim, M. T., Ogo, T., Ahmed, M., Randall, R., Chowdhury, H. M., Snape, K. M., et al. (2011). Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum. Mutat. 32, 1385–1389. doi:10.1002/humu.21605
Nelson, M. R., Tipney, H., Painter, J. L., Shen, J., Nicoletti, P., Shen, Y., et al. (2015). The support of human genetic evidence for approved drug indications. Nat. Genet. 47, 856–860. doi:10.1038/ng.3314
Nickel, N. P., de Jesus Perez, V. A., Zamanian, R. T., Fessel, J. P., Cogan, J. D., Hamid, R., et al. (2019). Low-grade albuminuria in pulmonary arterial hypertension. Pulm. Circ. 9, 2045894018824564. doi:10.1177/2045894018824564
Omura, J., Habbout, K., Shimauchi, T., Wu, W.-H., Breuils-Bonnet, S., Tremblay, E., et al. (2020). Identification of long noncoding RNA H19 as a new biomarker and therapeutic target in right ventricular failure in pulmonary arterial hypertension. Circulation 142, 1464–1484. doi:10.1161/CIRCULATIONAHA.120.047626
Paulin, R., Courboulin, A., Meloche, J., Mainguy, V., Dumas de la Roque, E., Saksouk, N., et al. (2011). Signal transducers and activators of transcription-3/pim1 axis plays a critical role in the pathogenesis of human pulmonary arterial hypertension. Circulation 123, 1205–1215. doi:10.1161/CIRCULATIONAHA.110.963314
Pugh, M. E., Robbins, I. M., Rice, T. W., West, J., Newman, J. H., and Hemnes, A. R. (2011). Unrecognized glucose intolerance is common in pulmonary arterial hypertension. J. Heart Lung Transpl. Off. Publ. Int. Soc. Heart Transpl. 30, 904–911. doi:10.1016/j.healun.2011.02.016
Renard, S., Paulin, R., Breuils-Bonnet, S., Simard, S., Pibarot, P., Bonnet, S., et al. (2013). Pim-1: A new biomarker in pulmonary arterial hypertension. Pulm. Circ. 3, 74–81. doi:10.4103/2045-8932.109917
Rosenkranz, S., Howard, L. S., Gomberg-Maitland, M., and Hoeper, M. M. (2020). Systemic consequences of pulmonary hypertension and right-sided heart failure. Circulation 141, 678–693. doi:10.1161/CIRCULATIONAHA.116.022362
Ruopp, N. F., and Cockrill, B. A. (2022). Diagnosis and treatment of pulmonary arterial hypertension: A Review. JAMA 327, 1379–1391. doi:10.1001/jama.2022.4402
Simonneau, G., Montani, D., Celermajer, D. S., Denton, C. P., Gatzoulis, M. A., Krowka, M., et al. (2019). Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 53, 1801913. doi:10.1183/13993003.01913-2018
Southgate, L., Machado, R. D., Gräf, S., and Morrell, N. W. (2020). Molecular genetic framework underlying pulmonary arterial hypertension. Nat. Rev. Cardiol. 17, 85–95. doi:10.1038/s41569-019-0242-x
Wang, X. (2014). Firth logistic regression for rare variant association tests. Front. Genet. 5, 187. doi:10.3389/fgene.2014.00187
Zamanian, R. T., Hansmann, G., Snook, S., Lilienfeld, D., Rappaport, K. M., Reaven, G. M., et al. (2009). Insulin resistance in pulmonary arterial hypertension. Eur. Respir. J. 33, 318–324. doi:10.1183/09031936.00000508
Keywords: pulmonary arterial hypertension, genome wide association study, PIM1, ICD-10, UK biobank
Citation: Pu A, Ramani G, Chen Y-J, Perry JA and Hong CC (2023) Identification of novel genetic variants, including PIM1 and LINC01491, with ICD-10 based diagnosis of pulmonary arterial hypertension in the UK Biobank cohort. Front. Drug Discov. 3:1127736. doi: 10.3389/fddsv.2023.1127736
Received: 19 December 2022; Accepted: 24 January 2023;
Published: 08 February 2023.
Edited by:
David Milan, Leducq Foundation, FranceReviewed by:
Carrie Welch, Columbia University, United StatesEmmanouil Tampakakis, Johns Hopkins University, United States
Copyright © 2023 Pu, Ramani, Chen, Perry and Hong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James A. Perry, anBlcnJ5QHNvbS51bWFyeWxhbmQuZWR1; Charles C. Hong, Y2hhcmxlcy5ob25nQHNvbS51bWFyeWxhbmQuZWR1