
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Drug Discov. , 20 October 2022
Sec. Anti-Infective Agents
Volume 2 - 2022 | https://doi.org/10.3389/fddsv.2022.969983
 Gustavo Schottlender1Juan Manuel Prieto2
Gustavo Schottlender1Juan Manuel Prieto2 Miranda Clara Palumbo1
Miranda Clara Palumbo1 Florencia A. Castello1
Florencia A. Castello1 Federico Serral1
Federico Serral1 Ezequiel J. Sosa2
Ezequiel J. Sosa2 Adrián G. Turjanski2
Adrián G. Turjanski2 Marcelo A. Martì2*
Marcelo A. Martì2* Darío Fernández Do Porto1,2*
Darío Fernández Do Porto1,2*Phenotypic screening is a powerful technique that allowed the discovery of antimicrobials to fight infectious diseases considered deadly less than a century ago. In high throughput phenotypic screening assays, thousands of compounds are tested for their capacity to inhibit microbial growth in-vitro. After an active compound is found, identifying the molecular target is the next step. Knowing the specific target is key for understanding its mechanism of action, and essential for future drug development. Moreover, this knowledge allows drug developers to design new generations of drugs with increased efficacy and reduced side effects. However, target identification for a known active compound is usually a very difficult task. In the present work, we present a powerful reverse virtual screening strategy, that can help researchers working in the drug discovery field, to predict a set of putative targets for a compound known to exhibit antimicrobial effects. The strategy combines chemical similarity methods, with target prioritization based on essentiality data, and molecular-docking. These steps can be tailored according to the researchers’ needs and pathogen’s available information. Our results show that using only the chemical similarity approach, this method is capable of retrieving potential targets for half of tested compounds. The results show that even for a low chemical similarity threshold whenever domains are retrieved, the correct domain is among those retrieved in more than 80% of the queries. Prioritizing targets by an essentiality criteria allows us to further reduce, up to 3–4 times, the number of putative targets. Lastly, docking is able to identify the correct domain ranked in the top two in about two thirds of cases. Bias docking improves predictive capacity only slightly in this scenario. We expect to integrate the presented strategy in the context of Target Pathogen database to make it available for the wide community of researchers working in antimicrobials discovery.
Antimicrobial resistance is nowadays one of the most challenging public health issues. Strikingly, the World Health Organization (WHO) recently revealed that there has been little progress in the development of new antibiotics to tackle drug-resistant infections. In this critical situation, the design of novel antimicrobial agents or therapies is pressingly required. Currently, it is accepted that the identification of appropriate targets is a critical step in the design of new drugs. In the postgenomic era, integrative computational approaches facilitate the identification and prioritization of candidate targets (Serral et al., 2021). Target-Pathogen (http://target.sbg.qb.fcen.uba.ar) (Sosa et al., 2018) is a unique resource that helps to achieve this task, combining structural druggability datasets, essentiality analysis, metabolic context, genomic, and expression data to rank gene/proteins according to their potential as novel antimicrobial targets. Previous reports, from our and other groups, used this tool to select and prioritize molecular targets of several relevant pathogens such as M. tuberculosis (Defelipe et al., 2016), K. pneumoniae (Ramos et al., 2018; Serral et al., 2022), Bartonella bacilliformis (Farfán-López et al., 2020; Serral et al., 2021), Trypanosoma cruzi (Osorio-Méndez et al., 2016; Coutinho et al., 2021) and Schistosoma mansoni (Lobo-Silva et al., 2020). Once the target is selected, the challenge moves to the identification of a small molecule that can inhibit the protein target’s function, allows further pharmacological validation of the target, and ultimately drives the development of a new antibiotic.
Phenotypic screening for antimicrobial compounds is a powerful technique that yielded over the last century many molecules that became first-line drugs to treat infectious diseases (Ribeiro da Cunha et al., 2019). For example, Daptomycin, a fermentation product from Streptomyces roseosporus now used to treat Gram-positive infections, was discovered by a screening of compounds for antibiotic activity in the 1980s and approved by the FDA in 2003 (Katz and Baltz, 2016). In high throughput phenotypic screening assays, usually thousands (or even more) of compounds are tested for their capacity to inhibit microbial growth in-vitro (Landeta and Mejia-Santana, 2021). Most promising compounds are further studied in different growth conditions and even in animal models. Moreover, in the field of natural products, crude extracts are routinely screened for their potential antimicrobial activities, and positive results lead to active compound molecular purification and characterization (Harvey, 2007). Indeed, this is how penicillin, possibly the world’s most important antimicrobial agent, was discovered (Ligon, 2004).
Once an active compound is found, the next problem is to identify its molecular target, the biological molecule of the host to which the compound binds to perform its effect. Knowing a compound’s molecular target is essential for understanding its mechanism of action, and key for further medicinal chemistry development, or in other words, for the active to become a lead compound for therapeutic development (Davis, 2020). Possibly, the most common antimicrobial drug targets are essential proteins. These proteins usually perform a vital function for the microorganism, such as an enzymatic reaction that, when inhibited by the drug, leads to the pathogen’s death (Gerdes et al., 2006).
Target identification for an active compound is however a very difficult task. In the pre-genomic era, biological studies of the compound effect on the host narrowed down their action mechanism over the years, until the definitive target survived scrutiny. For example, for Isoniazid, the first front-line drug against TB, early studies in the fifties showed that it inhibited the synthesis of cell division (Barclay and Ebert, 1953), and in the following 20 years many hypotheses were put forward until Winder et al. showed it inhibits mycolic acid biosynthesis, by analyzing the lipid extracts using thin layer chromatography (Winder and Collins, 1970). Consequently, several lipid-metabolizing enzymes were proposed as targets. Davidson and Takayama (1979) later showed that isoniazid inhibited a desaturase involved in the synthesis of C24 and C26 monounsaturated fatty acids. The final identification came, however, only after the genetic characterization of resistant strains (Vilchèze and Jacobs, 2007). Presently, a powerful way to identify an active compound target at the genomic scale is to induce the development of resistance, either naturally or with some mutagen, and then compare the parent and resistant strain genomes. Those genes with relevant mutations are the candidate targets. This technique has been, for example, successfully used to identify bedaquiline’s target in mycobacteria, which to everybody’s surprise turned out to be the ubiquitous ATP synthase (Andries et al., 2005; Kundu et al., 2016). The process, however, is costly and time-consuming and can fail for several reasons, such as the impossibility of obtaining a resistance strain, or the difficulty in correctly identifying the relevant resistance causing mutation (Farha and Brown, 2016). In this context, several bioinformatic approaches have been proposed and applied in order to aid researchers in the identification of an active compound’s target (Hasan et al., 2006; Shanmugam and Natarajan, 2010; Lee et al., 2011; Rahman et al., 2014; Mondal et al., 2015; Neelapu et al., 2015; Cloete et al., 2016; Defelipe et al., 2016; Kaur et al., 2017; Mohana and Venugopal, 2017; Wadood et al., 2017; Oany et al., 2018; Ramos et al., 2018; Uddin and Jamil, 2018; Shuvo et al., 2019; Farfán-López et al., 2020; Karim et al., 2020; Lobo-Silva et al., 2020; Aslam et al., 2021; Chakkyarath et al., 2021; Serral et al., 2022).
Virtual Screening is usually referred to as the process by which bioinformatic and chemoinformatic tools are used to select compounds that potentially bind (and inhibit) the desired protein target (Gimeno et al., 2019). In the first steps, compounds can be selected based on chemical similarity to known binders of the desired target, or to similar proteins (Bajusz et al., 2015; Radusky et al., 2017); or filtered according to the presence of specific functional groups known to bind to the target known pharmacophore. In a second, more precise step, selected compounds (either from a preselected, as mentioned above, or chemical diverse set) are docked to the target, and those predicted to have binding energy below a selected threshold are passed for experimental testing (Kumar et al., 2019). Many docking programs include options to encourage the formation of specific molecular interactions (Jones et al., 1997; Friesner et al., 2004; Corbeil et al., 2012; Ruiz-Carmona et al., 2014). In previous works from our group, we developed and thoroughly tested such a bias strategy in the context of the AutoDock4 program (Morris et al., 2009), which can be used for pose prediction and Virtual Screening and is freely available as AutoDock Bias (Arcon et al., 2019). The method is a powerful and easy to use tool to improve docking performance, which can be also applied in a high-throughput fashion for VS. It allows guided docking towards pharmacophoric interactions and precise localization of atoms or groups in a defined 3D region relative to the target structure.
Finally, in some cases, a third step is performed, where a smaller set of compounds is evaluated using Molecular Dynamics methods in order to obtain a more precise estimation of their binding free energy, and only those with higher affinity are tested (Sabe et al., 2021). In recent years, informatics approaches based on Machine Learning (ML) have gained strength for drug discovery (Lau et al., 2021), including the field of antimicrobial discovery (Vamathevan et al., 2019; Lau et al., 2021). For example, Halicin, an effective drug against many multidrug resistant microbes, is one of the most notable recently discovered antimicrobials using ML techniques (Stokes et al., 2020). Furthermore, the increasing performance of computers allows combining of molecular docking with more computational-demanding approaches such as MD, machine learning, and quantum mechanics QM calculations to further improve overall project performance (Caballero, 2021).
Reverse virtual screening can thus be described as the process where an already known active compound is screened against a set of potential targets, using the same (or similar) tools as those used for the typical case. For antimicrobial compounds, found using phenotypic assays, except for cases of compounds targeting DNA or the cell membrane, the universe of potential targets can be circumscribed to all the organism proteins and the aim is thus to find a small group of potential protein targets to study further and validate experimentally.
In the present work, we combined chemical similarity methods, with target prioritization based on essentiality and expression data, and knowledge-based docking to analyze the potential of reverse virtual screening. We provide a comparison for different organisms, with different proteome sizes and available information, and show the potential and drawbacks of each reverse screening step.
The selected pathogens are Mycobacterium tuberculosis (Mtb), Bartonella bacilliformis (Bb), Klebsiella pneumoniae (Kp), and Trypanosoma cruzi (Tc), all corresponding to important challenges for health care systems, particularly in developing countries. Mtb is the causative agent of tuberculosis (TB), an infectious disease that despite being preventable and treatable, continues to be a global health threat (World Health Organization 2021). It is estimated that 25% of the world population is infected with latent Mtb. These individuals constitute a great reservoir of mycobacteria, presenting between a 5%–10% probability of developing active TB throughout life. TB is positioned as the 13th cause of death worldwide, and until the pandemic caused by COVID19, the first cause of death by a single infectious agent (World Health Organization 2021). Bb is responsible for Carrion’s disease, an ancient vector-borne biphasic illness dating from the pre-Columbian era, restricted to the South American Andes, including Ecuador, Peru, and Colombia (Gomes and Ruiz, 2018). Bb infectious causes two well-defined clinical phases. The early stage, named Oroya fever, causes severe acute hemolytic anemia. High fatality rates (40%–88%) have been described in patients without any antibiotic treatment. Even more, with adequate antibiotic treatment, the fatality rate is around 11% (Farfán-López et al., 2020). The development of dermal eruptions, known as Peruvian warts, and commonly present on the extremities and the head is typical of the chronic phase of Carrion’s disease. Although this phase is hardly fatal, dermal eruptions can be accompanied by acute pains in joints and bones, headache, fever, and lymphadenopathy (Minnick et al., 2014). Kp is a Gram-negative, non-motile, rod-shaped enterobacterium. From a clinical perspective, it represents one of the most important pathogenic bacterium (Podschun and Ullmann, 1998; Podschun et al., 2001). It is commonly reported as an etiologic agent of either bacterial pneumonia or community-acquired urinary tract infections. However, it can cause any type of infection in hospitals, including breaks in patients under intensive care and newborns, which is likely associated with its ability to spread fast in hospital settings. Finally, Tc is a kinetoplastid protozoan that causes Chagas disease. This infection affects 6–8 million people worldwide (Schofield et al., 2006), most of them neglected populations (Schijman, 2018). Tc traverses an acute phase, evolving to an asymptomatic or symptomatic chronic phase, with different degrees of progression and severity (Rassi et al., 2010). It is important to remark that all these pathogens present antimicrobial resistance for first and second-line treatments, and thus, new drugs are urgently required (Mazzeti et al., 2021); (Van den Kerkhof et al., 2020).
Based on the available genomic, proteomic, and ligand binding information for the selected pathogens, our results show that reverse virtual screening is a powerful tool for predicting (or narrowing the number of) potential targets, for new compounds showing antimicrobial activity. We also provide a detailed step-by-step pipeline that allows implementation of the described strategy in other pathogens. Finally, further integration of the proposed methodology in the context of the Target Pathogen database is discussed and provided.
All proteomes were downloaded from Target Pathogen (http://target.sbg.qb.fcen.uba.ar/patho/). Each protein was blasted against Uniprot (The UniProt Consortium, 2018) to obtain the corresponding Uniprot ID. Duplicated and erroneous sequences were deleted.
We assigned each protein sequence domain using HMMER (http://hmmer.org/) and the PFAM domain database (Mistry et al., 2021). Domains were assigned whenever the e-value was below 1x10−5 (in order to obtain a reasonable and confident number of domains for each protein to keep only highly reliable domains), they score higher than the corresponding gathering cutoff, and the protein sequence harbored over 60% of the domain length.
For each analyzed organism, we built an internal database. This internal protein (domain)-ligand database consists mainly of two connected tables, one containing PFAM domains and their respective proteins, and the other, the ligands and their associated PFAM domains. Therefore, for each ligand, the set of known binding domains can be retrieved. The ligand-protein (domain) connections are derived from either the PDB (Berman et al., 2003) or ChEMBL (Gaulton et al., 2017). In the case of the PDB, there are lots of complex structures harboring a protein and its ligand. For each structure, the Pfam domains present in the corresponding protein are annotated in the PDB database. We, therefore, extracted from the PDB all annotated domain-ligand pairs, and corresponding PDB ID, using a customized in-house script. PDB database annotates as ligands not only drug-like compounds but any non-protein molecule, such as salts, metals, and cosolvents. Since we are interested only in drug-like molecules, we filtered PDB-derived ligands using MOAD (Hu et al., 2005), to keep only those that are classified as drug-like, i.e., peptides of 10 amino acids or less, oligonucleotides of four nucleotides or less, small organic molecules and cofactors.
Concerning ChEMBL, we first assigned PFAM domains to each ChEMBL target using HMMER. Secondly, we retrieved all ligands which are annotated as binding to a single protein (not protein complexes) that contain at least one domain from the pathogens proteome and have a pchembl value >6 (pchembl is a measure of protein activity inhibition) (Bento et al., 2014). Finally, since many ChEMBL targets harbored more than one domain, we attempted to address which of these domains actually binds the ligand. For this sake, we compared each of the multidomain-target binding ligands to those retrieved from the PDB using Chemical Similarity (see below). If one of these multidomain-protein-binding ligands shares chemical similarity (TI > 0.4) with a PDB ligand that binds a given domain of the multidomain proteins, we assign this as the corresponding ligand-binding domain in the ChEMBL table.
It is interesting to note, that contrary to what we expected, although ChEMBL allows us to retrieve more ligands compared to PDB, they show a relatively smaller number of targets. In other words, ChEMBL has many ligands but fewer targets. Therefore, the coverage or diversity of domains obtained from PDB is larger than those from ChEMBL.
Chemical similarity between compound pairs was quantified using the Tanimoto Index (T.I.), as previously described (Bajusz et al., 2015). Briefly, starting from each compound SMILES we obtained the corresponding Morgan Fingerprints (Morgan, 1965; Rogers and Hahn, 2010) (a series of binary digits that represent the absence or presence of particular chemical substructures in the ligands) using RDkit (Landrum, 2016). This index is computed as
Metabolic information (betweenness-centrality and choke-points) for each target was downloaded from Target Pathogen (Sosa et al., 2018). From the metabolic perspective betweenness-centrality, reflects the participation of a given reaction as an intermediary in many transformations. It is assumed that the blockage of proteins associated with high betweenness-centrality would generate disequilibrium in many different pathways, and thus be of high impact. Choke-points are reactions that uniquely produce or consume a given product or substrate (Yeh et al., 2004). Thereby, choke-point blockage may lead to the lack of essential compounds or the accumulation of a potentially toxic metabolite in the cell. For this reason, proteins associated with these reactions are supposed to be relevant for drug discovery.
To consider whether a protein was essential, we downloaded available large-scale information from Target Pathogen. Since genomic scale essentiality experiments were available only for Mtb, we predicted which proteins of the other three organisms are essential blasting each organism’s proteome using the Database of Essential Genes (Zhang, 2004) (coverage> 80%, e-value<1.10–10). If homologs proteins are found, it is possible that the queried proteins are also essential, since functions encoded by essential genes are broadly conserved in microorganisms.
Molecular docking calculations were performed for each randomly selected query ligand against the PDB structures that correspond to the first 10 domains that bind the most chemically similar compounds to the query (similar domains are ranked according to ligand T.I. as described), excluding those that also bind organic cofactors. From the selected PDB crystals, all hetatoms (solvent molecules and/or ligands) were removed, except in those domains that harbor ionic cofactors that display a key role in protein-ligand interactions. After that, we prepared the PDB files for docking, adding hydrogen atoms and charges information in the corresponding pdbqt files using AutoDockTools version 4.2.6 (Morris et al., 2009).
The potential ligand binding site of the selected target protein, was the one where the ligand (similar to the query) binds to its corresponding domain. All docking calculations were performed with AutoDockTools. The grid size was set to 15 × 15 × 15 Å, with a grid spacing of 0.375 Å, and the grid center was designated at the position where the corresponding ligand binds the respective PDB structure. The following docking parameters were adopted: ga_run = 100, rmstol = 2 Å.
Molecular docking calculations were also performed using the Bias docking protocol, a script-based method that allows the introduction of different types of biases for fine-tuning AutoDock calculations, developed in our group. Biases are introduced as energy rewards for each ligand atom that participates in previously identified protein-ligand relevant molecular interactions, usually hydrogen bonds or hydrophobic (Arcon et al., 2019). In the present case, key protein-ligand interactions for the bias were derived from the protein-ligand interactions observed in the complex structure of the protein (harboring the same target domain) with its corresponding ligand which is similar to the query compound.
The results are organized as follows. First, we explore the druggable genomes of four distinct microorganisms with significantly different genome sizes, and available ligand and target information. Secondly, we analyze the predictive capacity of the proposed strategy using only chemical similarity and domain information. Finally, we analyzed whether the use of biological properties of each microorganism’s proteins (i. e, metabolic context and essentiality) and molecular docking allow us to select (or narrow) the most suitable target candidates.
The first step of our reverse virtual screening pipeline uses chemical similarity to find potential targets of a given compound (Figure 1). We used the same idea described in our previous work, where we showed that using chemical similarity it is possible to find a set of potential binders to a given target protein by looking at compounds that are similar to those known to bind to homolog proteins (Radusky et al., 2017). In the present pipeline, we first search for compounds that are effectively known to bind proteins (binder compounds), that are chemically similar to the query inhibitor up to a user-defined threshold (we will use the term query to refer to the phenotypic active compound for which we are seeking the target). Therefore, those proteins from the target organisms that are homologs to the known targets of the binder compounds (i.e., those similar to the query), are the query’s potential targets. In this scheme, the success will depend on: 1) the number of potential different targets, 2) the number and diversity of known binders to the potential targets’ homologs, 3) the chemical similarity method and threshold used, and 4) the method used to define the protein homologs.
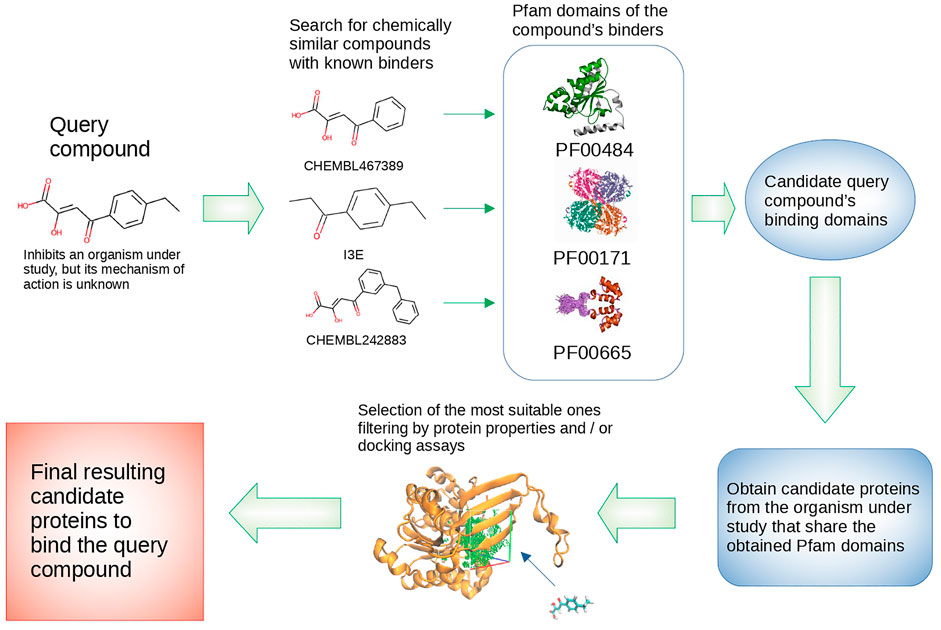
FIGURE 1. Reverse virtual screening pipeline.
To analyze the impact of these parameters on the pipeline predictive capacity we selected four pathogens, namely Mtb, Bb, Kp, and Tc, with significantly different proteome sizes and available ligand and target data (Table 1). To define homolog proteins in our pipeline, we will work not with the whole proteins (i.e., the full protein sequence), but with individual domains as defined in the PFAM database (Mistry et al., 2021). As shown in Table 1, although proteome sizes differ significantly, the total number of unique domains is similar. We will use four categories to define a homolog binder protein. The first (g1), most stringent, group corresponds to proteins with a co-crystallized ligand (as found in the PDB), and with over 95% identity to the potential target (i.e., almost the same protein domain but from the same, or another organism). The second (g2) corresponds to co-crystallized ligands found in proteins from any organism, binding to the same PFAM domain of the potential target. In this group, thus, we are assuming that two proteins sharing the same domain according to PFAM are homologs. Group 3 (g3) and 4 (g4) correspond to the same identity thresholds but define the known targets of the ligands using the Chembl database (Gaulton et al., 2017).
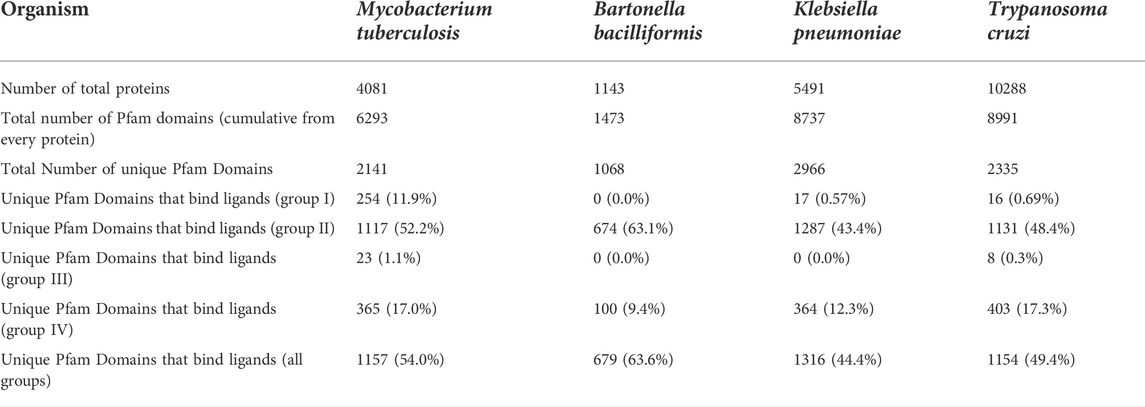
TABLE 1. Total count of ligands-target pairs that can be associated in M tuberculosis, B bacilliformis, K pneumoniae and T cruzi, using PFAM and Chembl data.
Table 1 shows the number of ligands-target pairs that can be associated with each pathogen using the four mentioned group categories. If we look at specific organisms domains (g1 and g3), only Mtb displays a significant number of known ligands. This is not unexpected since there is a long tradition in the study of Mtb ligand-target pairs. For the other organisms, the number of known ligands is virtually null. However, when the analysis is extended to homolog domains (G2 and G4), now between 20% and 60% of the domains can be assigned a tentative ligand. This moderately wide coverage is the key to a successful reverse screening pipeline.
We now turn our attention to the ligand chemical-similarity threshold and analyze the resulting predictive capacity of the pipeline first step. Figure 2A shows the number of different domains that are retrieved on average as a function of their chemical similarity threshold to the query. Ligand queries are obtained from a random subset of 1000 PDB and Chembl ligands respectively, and the compounds from our datasets (from which we retrieve their respective binding domains) that present T.I. > 0.9 with the queries were excluded in the evaluations. The plot shows, for example, that if we retrieve all targets whose ligands are between 0.6–0.7 similar to the query compound, on average we find only 2-3 different domains. As expected the smaller the threshold (i.e., more dissimilar compounds) the more different domains are retrieved, and it grows exponentially. The data shows that on average the threshold should be no smaller than 0.5 which results in up to about 10 different domains. Again, and consistent with previous observations for the PDB dataset, more domains are found.
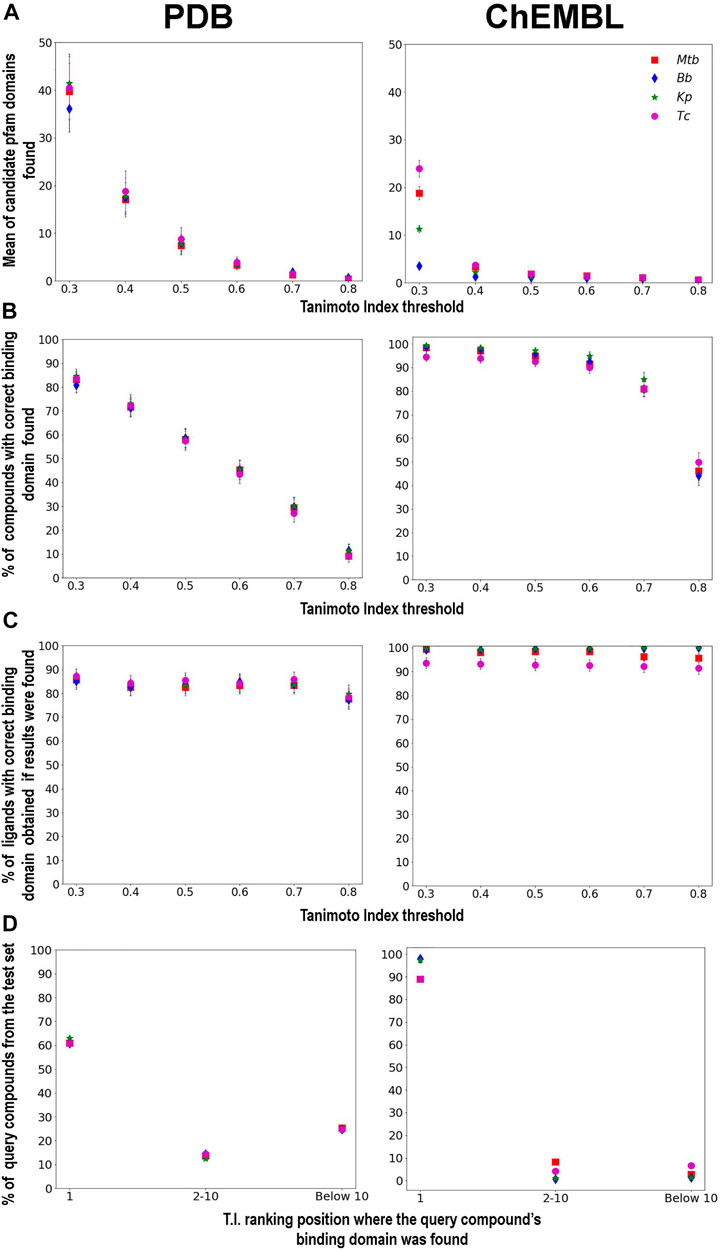
FIGURE 2. Mean of Pfam domains found per T.I. threshold (A). Percentage of query ligands with the correct Pfam domain obtained by each T.I. threshold globally (B), and only when at least one candidate binding domain was retrieved (C). Histogram of T.I. similarity ranking positions where the ligand that binds the correct query’s interaction domain is obtained (D). Plots are built based on 1000 queries extracted either from the PDB (left) and ChEMBL (right) datasets.
Figure 2B shows a first glimpse of the pipeline performance. Here we evaluate for each chemical similarity threshold, the percentage of query ligands that result in a positive identification (or retrieval) of the correct target domain. Of course, the number of retrieved domains increases as the threshold diminishes. For the PDB set, the plot shows a linear relationship and that using a threshold of T.I. = 0.5, the target is found for more than half of the query compounds. Interestingly, for the Chembl set, the plot shows that plateau/saturation is reached at a threshold of 0.6 of chemical similarity, with about 80% of queries compound finding the potential target domain.
Figure 2C is similar to 2B, but now we determine the percentage of correct domain retrieval, considering only those cases where for the given chemical similarity threshold, at least one domain is retrieved. In other words, it measures the method’s predictive value whenever a result is obtained. The results show that even for a low chemical similarity threshold whenever domains are retrieved, the correct domain is among those retrieved in more than 80% of the queries for the PDB set and 90% for the ChEMBL set.
Finally, Figure 2D shows the ranking capability of the chemical similarity index regarding correct domain identification. The results show that in around 60% of the cases in the PDB datasets (and above 90% in the ChEMBL ones), the most similar ligand (i.e., first ranked) results in correct domain retrieval. This is consistent with our working hypothesis, i.e., that more chemically similar ligands tend to bind the same domains. It also shows that ranking the retrieved domains by chemical similarity is a good first approach to identify the correct domain.
We now turn our attention to the second step of our pipeline which is related to each potential target’s biological properties. We selected 10 drugs for each species whose binding target is known and also known to cause growth inhibition of (or kill) the respective bacteria. As we could not find 10 drugs with a known active binder for Tc, this organism was discarded for further analysis. For each drug, we determined the group of potential binding domains (using the previously described chemical similarity approach) and analyzed whether maintaining only essential proteins or those associated with high centrality or choke-point reactions could allow us to reduce the number of potential targets while keeping the true target. The results, displayed in Figure 3A, indicate that in almost every case the correct target could still be found after filtering by essentiality. This is not the case for centrality or choke points as filters, showing that these two characteristics are not adequate for describing current drug targets. The effect of filtering by essentiality is shown in Figure 3B, where for each species the number of potential targets before and after essentiality filtering are displayed. For both Mtb and Kp, a significant (3–4 times) reduction in the number of potential targets results after filtering. For Bb reduction is negligible, possibly due to lack of knowledge of essential proteins, i.e., filter is useless. In conclusion, essentiality was the only biological property that allowed us to narrow the candidate target list without ruling out the putative valid binders, so essential candidate genes/proteinas should be given priority as the potential true targets.
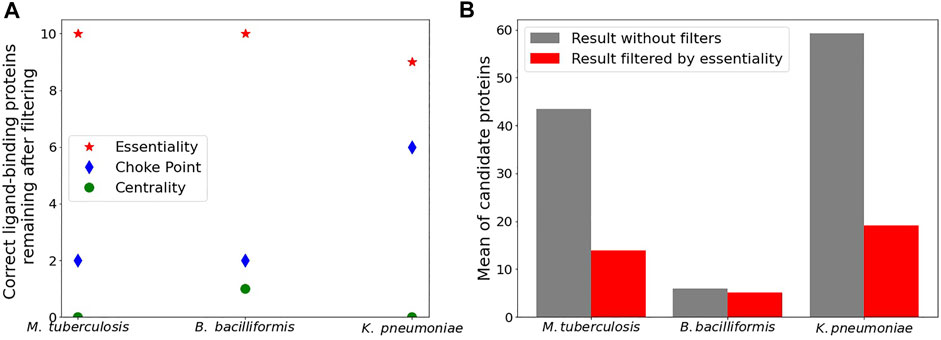
FIGURE 3. Ligand - target pairs in which the correct binding domain remains, after filtering by corresponding protein features: Essentiality or involving a choke point or a central reaction in a metabolic pathway (A). Mean number of potential protein domains after and before filtering by essentiality (B). Data obtained from sets of 10 known drugs each, used to treat respectively M. tuberculosis, B. bacilliformis and K. pneumoniae.
The final step of the proposed pipeline aims to: 1) discard domains that are not likely to bind the query, and 2) confirm that one (or more) of the potential targets is (are) able to bind the query, using Molecular Docking. After performing the above-mentioned steps, for each query, we have a group of usually less than 10 potential targets with their corresponding known ligands. For those derived from PDB, we can take advantage of the known protein-ligand complex structure and the similarity between the query and bound ligand, to define key protein-ligand interactions, which can be subsequently used in the Bias Docking Scheme. An example of the idea is presented in Figure 4.
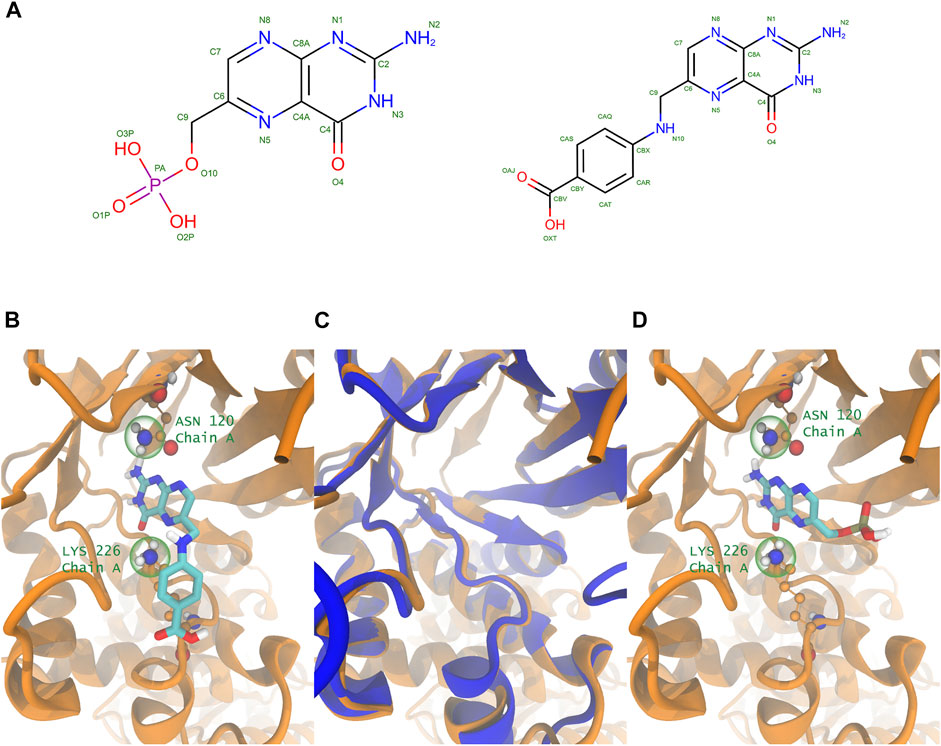
FIGURE 4. Using known protein-ligand structure and query-ligand similarity to define potential ligand interactions for bias-docking. Comparison between PMM (query ligand example) and 22D (similar compound retrieved, that interacts with the correct binding domain) (A). Key atoms from residues that interact with the ligand 22D in the crystal 6OFW, which are used to define the bias sites for the query (B). Structural alignment of both structures that share the same Pfam domain containing respectively 22D (6OFW, in orange) and PMM (1EYE in blue), where the structural similarity between both proteins is shown (C). Query ligand (PMM) correctly docked in the structure 6OFW, with coincident interactions with the bias sites (D).
Key to the present step is to be able to discern which of the potential targets actually binds the query and which do not. In order to compare all the possible binding domains evaluated for a ligand, we transformed the Autodock results (Population and estimated binding energy) to a Z-score which allows comparing the results for different domains and ranking each pose. Examples of the results are presented in Figure 5 A and B for the ligands 9OR, which binds PF01070 (present in the structure 6A21) and 89U, which interacts with PF00091 (in the crystal 5XLZ). The obtained representing crystal for PF01070 is 2ZRV (that has the compound FNR, similar to 9OR by a T.I. of 0.63) and 5LYJ (that contains the ligand 7BA, that shares a T.I. of 0.49 with 89U) for PF00091.
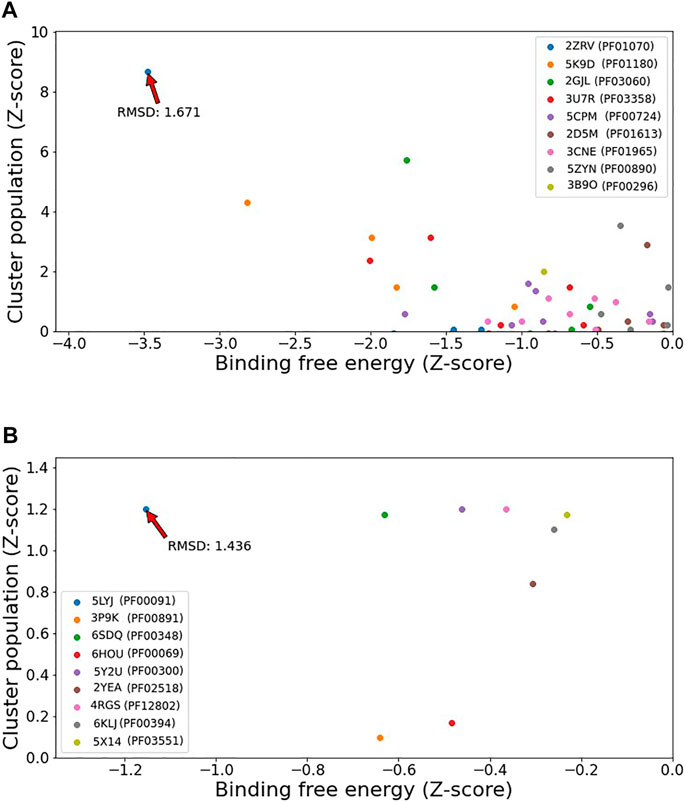
FIGURE 5. Z-normalized plot of binding free energy (x-axis) and cluster population of similar docked ligand’s poses (y-axis) comparing all the candidate binding domains (represented by a structure for each) with the correct one indicated for the compounds 9OR (A) and 89U (B).
The results from Figure 5 clearly show how the docking allows selection of the correct domain, and a corresponding good quality model of the ligand bound structure. Proper ligand pose is evidenced as an upper-left outlier dot in the Z-score normalized population vs. binding energy plot, which consistently displays low RMSD values of the query ligand heavy atoms with respect to the corresponding reference structure. Wrong domains, on the contrary, lack or have less significant (in terms of Z-score) outliers. The summary of the results presented in Figure 6, shows that in both cases (either with queries derived from PDB or ChEMBL) docking is able to identify the correct domain in the first ranks (by Z-score) in most, about two thirds, of cases. Bias docking improves predictive capacity only slightly in this scenario (more detailed information in Supplementary Table S1).
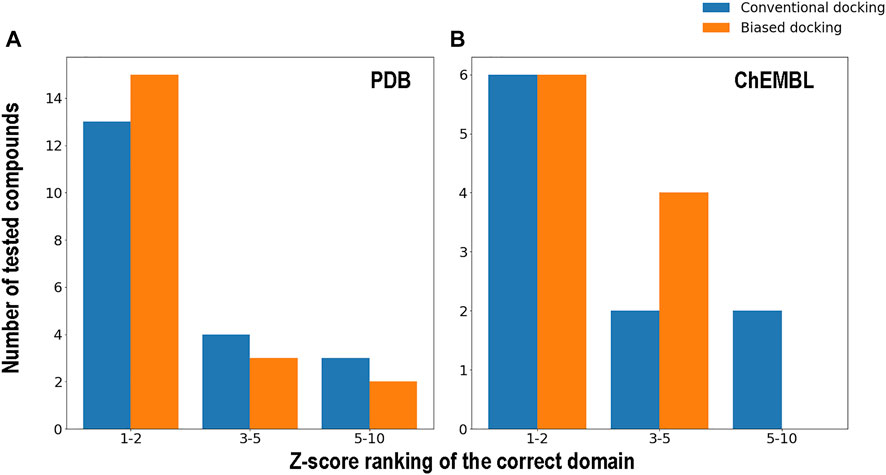
FIGURE 6. Histogram showing the positions where the correct binding domain was found in the biased and conventional docking z-score rankings made for the 20 compounds evaluated from the PDB dataset (A) and the 10 tested ligands from the ChEMBL dataset (B).
Taken the results altogether, we can discuss the whole pipeline and how each step contributes and can be used to reach the desired target. It is clear from the first proteome coverage and database comparison, that present knowledge allows the identification of a known ligand at the domain scale for a significant percentage (more than half) of a microbial proteome. As more ligands and their targets are identified these numbers can only increase. The first step of the proposed strategy, using ligand chemical similarity to infer potential targets, is already quite powerful. On average 50% of query compounds result in positive target identification for a chemical similarity index of 0.5. Moreover, the first ranked, and thus most similar ligand, is actually a ligand of the correct target domain in 80% of cases. Higher chemical similarities (> 0.7) result in a close to a unique and positive identification of the binding domain but are expected for about 10%–20% of tested ligands. On the other end, for those queries where retrieved domains result from lower chemical similarity (0.2–0.4 range), tens of domains are retrieved and thus a high rate of false positives is expected. This scenario is where the following steps of the strategy are relevant. The second step is just a filter due to potential target characteristics. Since we are expected to look for targets that actually kill the microbial, it is expected that an essentiality filter allows reducing the number of potential targets. For this filter to be effective it is key to be able to determine which proteins are essential for the microbe, either through direct observation or by homology with essential gene lists.
The last, the molecular docking proposed step, is relevant when several potential domains are retrieved by chemical similarity. Our results show that docking is capable of selecting the true binding domain among others. Indeed, the presence of a clear outlier in the z-score population vs. binding energy plot not only highlights the true binding domain, but also yields a model of the corresponding protein domain-ligand structure. This model structure can be used, for example, to design single point mutants that would prevent binding of the compound, yielding a valuable tool for experimental confirmation. Also, the lack of a proper outlier possibly hits to the fact that none of the retrieved domains is the true target, and therefore that the ligand possibly binds to one which lacks a known similar binder.
To perform an application of the proposed strategy, we run through the pipeline a set of 776 compounds developed by GSK that were shown to kill Mtb in a phenotypic screening assay (Hersey, 2013), and whose targets are largely unknown. Our results show that 69% of the drugs are similar to at least one ligand from our binders database with a TI greater than 0.4. The predicted target domains are presented in SI Supplementary Table S2.
Previous work used in-silico methods to predict potential binding domains (Martínez-Jiménez et al., 2013), for 31 of the mentioned GSK compounds. In 19 cases our prediction has coincidences with that reported previously. More importantly, for 42 GSK set drugs, targets were experimentally validated (Mugumbate et al., 2015; Trofimov et al., 2018). Most of them (30) were shown to bind Dihydrofolate reductase (DHFR). Our methodology was able to correctly predict the binding domain for 18 of these 30 ligands. DHFR’s domain (PF00186) was retrieved as the top result (the most similar binder compound in the database) for nine of these drugs, among the top 10 results for another three compounds, and between the best 20 candidates for the last six ligands. For the remaining 11 molecules, those that do not bind DHFR, we predicted the corresponding binding-domains for only two of them, that bind to KasA (Pfam PF00109 and PF02801), and MmpL3 (Pfam PF03176) respectively. In both cases, the closest binder compound corresponds to the true target (Supplementary Table S3).
Finally we applied our chemical-similarity approach to a recently discovered antibiotic (Fabimycin), whose mechanism of action was also determined (Parker et al., 2020, 2022). This is an interesting example, because Fabymycin is not present in either ChEMBL or PDB at the moment of the present work, which makes it eligible as proof of concept for our strategy. This drug is active against Gram negative bacteria (including Klebsiella pneumoniae) and acts as an inhibitor of the enzyme FabI, that corresponds to the domain Enoyl-Acyl carrier protein reductase (Pfam PF13561). According to our results, the most similar ligand of Fabimycin present in chemical databases is CHEMBL1652621 (with TI of 0.78) which is bound to FabI protein of Staphylococcus aureus and Burkholderia pseudomallei. This protein, as expected, belongs to the PF13561 domain (Enoyl-(Acyl carrier protein) reductase), present in FabI protein of Klebsiella pneumoniae. In conclusion our methodology was able to correctly retrieve the FabI as the target in Klebsiella pneumoniae.
Our bioinformatic strategy allows, starting from a compound (the query) which is known to kill (or show any other clear phenotypic effect) against a given target organism, to determine potential domains and corresponding target organism’s proteins that are likely to be molecular targets of the query. The pipeline consists of three steps (chemical similarity, essentiality filter, and molecular docking) that can be tailored according to the researchers’ needs and available information. Overall, we show that potential targets can be retrieved for half of the tested compounds with a 0.5 chemical similarity index. Whenever a potential target is retrieved there is more than an 80% chance that the actual target is among the handful of retrieved domains. We expect to integrate the presented strategy in the context of Target Pathogen database (Sosa et al., 2018) to make it available to the wide community of researchers pursuing the development of novel antimicrobials.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
AT, MM, and DF Conceived the study design. GS, JP, MP, FC, FS, and ES contributed tools and performed data analysis. MM, DF, and GS drafted the manuscript with input from the other authors. All authors read and approved the final version of the manuscript.
Fellowship supported from CONICET to MP, FC, and FS. Fellowship supported from Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT) to GS and JP. ES, AT, MM, and DF are members of CONICET. This work was supported by Agencia Nacional de Promoción Científica y Tecnológica (ANPCyT, PICT-2018-04663 to DF) and Universidad de Buenos Aires (20020190200275BA to DF).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fddsv.2022.969983/full#supplementary-material
Aslam, M., Shehroz, M., Ali, F., Zia, A., Pervaiz, S., and Shah, M. (2021). Chlamydia trachomatis core genome data mining for promising novel drug targets and chimeric vaccine candidates identification (2021). Comput. Biol. Med. 136, 104701.
Andries, K., Verhasselt, P., Guillemont, J., Göhlmann, H. W. H., Neefs, J.-M., Winkler, H., et al. (2005). A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307, 223–227. doi:10.1126/science.1106753
Arcon, J. P., Modenutti, C. P., Avendaño, D., Lopez, E. D., Defelipe, L. A., Ambrosio, F. A., et al. (2019). AutoDock bias: Improving binding mode prediction and virtual screening using known protein-ligand interactions. Bioinformatics 35, 3836–3838. doi:10.1093/bioinformatics/btz152
Bajusz, D., Rácz, A., and Héberger, K. (2015). Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 7, 20. doi:10.1186/s13321-015-0069-3
Barclay, W. R., and Ebert, R. H. (1953). The effect of cortisone on the vascular reactions to serum sickness and tuberculosis. Ann. N. Y. Acad. Sci. 56, 634–636. doi:10.1111/j.1749-6632.1953.tb27384.x
Bento, A. P., Gaulton, A., Hersey, A., Bellis, L. J., Chambers, J., Davies, M., et al. (2014). The ChEMBL bioactivity database: An update. Nucleic Acids Res. 42, D1083–D1090. doi:10.1093/nar/gkt1031
Berman, J., Bourne, P., Westbrook, J., and Zardecki, C. (2003). The protein data bank. Protein Struct., 389–405. doi:10.1201/9780203911327.ch14
Butina, D. (1999). Unsupervised data base clustering based on daylight’s fingerprint and Tanimoto similarity: A fast and automated way to cluster small and large data sets. J. Chem. Inf. Comput. Sci. 39, 747–750. doi:10.1021/ci9803381
Caballero, J. (2021). The latest automated docking technologies for novel drug discovery. Expert Opin. Drug Discov. 16, 625–645. doi:10.1080/17460441.2021.1858793
Chakkyarath, V., Shanmugam, A., and Natarajan, J. (2021). Prioritization of potential drug targets and antigenic vaccine candidates against Klebsiella aerogenes using the computational subtractive proteome-driven approach. J. Proteins Proteom. 12, 201–211. doi:10.1007/s42485-021-00068-9
Cloete, R., Oppon, E., Murungi, E., Schubert, W.-D., and Christoffels, A. (2016). Resistance related metabolic pathways for drug target identification in Mycobacterium tuberculosis. BMC Bioinforma. 17, 75. doi:10.1186/s12859-016-0898-8
Corbeil, C. R., Williams, C. I., and Labute, P. (2012). Variability in docking success rates due to dataset preparation. J. Comput. Aided. Mol. Des. 26, 775–786. doi:10.1007/s10822-012-9570-1
Coutinho, J. V. P., Rosa-Fernandes, L., Mule, S. N., de Oliveira, G. S., Manchola, N. C., Santiago, V. F., et al. (2021). The thermal proteome stability profile of Trypanosoma cruzi in epimastigote and trypomastigote life stages. J. Proteomics 248, 104339. doi:10.1016/j.jprot.2021.104339
Davidson, L. A., and Takayama, K. (1979). Isoniazid inhibition of the synthesis of monounsaturated long-chain fatty acids in Mycobacterium tuberculosis H37Ra. Antimicrob. Agents Chemother. 16, 104–105. doi:10.1128/aac.16.1.104
Davis, R. L. (2020). Mechanism of action and target identification: A matter of timing in drug discovery. iScience 23, 101487. doi:10.1016/j.isci.2020.101487
Defelipe, L. A., Do Porto, D. F., Pereira Ramos, P. I., Nicolás, M. F., Sosa, E., Radusky, L., et al. (2016). A whole genome bioinformatic approach to determine potential latent phase specific targets in Mycobacterium tuberculosis. Tuberculosis 97, 181–192. doi:10.1016/j.tube.2015.11.009
Farfán-López, M., Espinoza-Culupú, A., García-de-la-Guarda, R., Serral, F., Sosa, E., Palomino, M. M., et al. (2020). Prioritisation of potential drug targets against Bartonella bacilliformis by an integrative in-silico approach. Mem. Inst. Oswaldo Cruz 115, e200184. doi:10.1590/0074-02760200184
Farha, M. A., and Brown, E. D. (2016). Strategies for target identification of antimicrobial natural products. Nat. Prod. Rep. 33, 668–680. doi:10.1039/c5np00127g
Friesner, R. A., Banks, J. L., Murphy, R. B., Halgren, T. A., Klicic, J. J., Mainz, D. T., et al. (2004). Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749. doi:10.1021/jm0306430
Gaulton, A., Hersey, A., Nowotka, M., Bento, A. P., Chambers, J., Mendez, D., et al. (2017). The ChEMBL database in 2017. Nucleic Acids Res. 45, D945–D954. doi:10.1093/nar/gkw1074
Gerdes, S., Edwards, R., Kubal, M., Fonstein, M., Stevens, R., and Osterman, A. (2006). Essential genes on metabolic maps. Curr. Opin. Biotechnol. 17, 448–456. doi:10.1016/j.copbio.2006.08.006
Gimeno, A., Ojeda-Montes, M. J., Tomás-Hernández, S., Cereto-Massagué, A., Beltrán-Debón, R., Mulero, M., et al. (2019). The light and dark sides of virtual screening: What is there to know? Int. J. Mol. Sci. 20, E1375. doi:10.3390/ijms20061375
Gomes, C., and Ruiz, J. (2018). Carrion’s disease: The sound of silence. Clin. Microbiol. Rev. 31, 000566-e117. doi:10.1128/cmr.00056-17
Harvey, A. L. (2007). Natural products as a screening resource. Curr. Opin. Chem. Biol. 11, 480–484. doi:10.1016/j.cbpa.2007.08.012
Hasan, S., Daugelat, S., Rao, P. S. S., and Schreiber, M. (2006). Prioritizing genomic drug targets in pathogens: Application to Mycobacterium tuberculosis. PLoS Comput. Biol. 2, e61. doi:10.1371/journal.pcbi.0020061
Hu, L., Benson, M. L., Smith, R. D., Lerner, M. G., and Carlson, H. A. (2005). Binding MOAD (mother of all databases). Proteins 60, 333–340. doi:10.1002/prot.20512
Jones, G., Willett, P., Glen, R. C., Leach, A. R., and Taylor, R. (1997). Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 267, 727–748. doi:10.1006/jmbi.1996.0897
Karim, M., Islam, M. D. N., and Nurnabi Azad Jewel, G. (2020). In Silicoidentification of potential drug targets by subtractive genome analysis of Enterococcus faecium DO. doi:10.1101/2020.02.14.948232
Katz, L., and Baltz, R. H. (2016). Natural product discovery: Past, present, and future. J. Ind. Microbiol. Biotechnol. 43, 155–176. doi:10.1007/s10295-015-1723-5
Kaur, D., Kutum, R., Dash, D., and Brahmachari, S. K. (2017). Erratum: Data intensive genome level analysis for identifying novel, non-toxic drug targets for multi drug resistant Mycobacterium tuberculosis. Sci. Rep. 7, 46825. doi:10.1038/srep46825
Kumar, N., Sood, D., Tomar, R., and Chandra, R. (2019). Antimicrobial peptide designing and optimization employing large-scale flexibility analysis of protein-peptide fragments. ACS Omega 4, 21370–21380. doi:10.1021/acsomega.9b03035
Kundu, S., Biukovic, G., Grüber, G., and Dick, T. (2016). Bedaquiline targets the ε subunit of mycobacterial F-ATP synthase. Antimicrob. Agents Chemother. 60, 6977–6979. doi:10.1128/AAC.01291-16
Landeta, C., and Mejia-Santana, A. (2021). Union is strength: Target-based and whole-cell high-throughput screens in antibacterial discovery. J. Bacteriol. 204, e0047721. doi:10.1128/JB.00477-21
Landrum, G. (2016). RDkit; Open-source cheminformatics. Available at: http://www.rdkit.org.
Lau, H. J., Lim, C. H., Foo, S. C., and Tan, H. S. (2021). The role of artificial intelligence in the battle against antimicrobial-resistant bacteria. Curr. Genet. 67, 421–429. doi:10.1007/s00294-021-01156-5
Lee, D.-Y., Chung, B. K. S., Yusufi, F. N. K., and Selvarasu, S. (2011). In silico genome-scale modeling and analysis for identifying anti-tubercular drug targets. Drug Dev. Res. 72, 121–129. doi:10.1002/ddr.20408
Ligon, B. L. (2004). Penicillin: Its discovery and early development. Semin. Pediatr. Infect. Dis. 15, 52–57. doi:10.1053/j.spid.2004.02.001
Lobo-Silva, J., Cabral, F. J., Amaral, M. S., Miyasato, P. A., de Freitas, R. P., Pereira, A. S. A., et al. (2020). The antischistosomal potential of GSK-J4, an H3K27 demethylase inhibitor: Insights from molecular modeling, transcriptomics and in vitro assays. Parasit. Vectors 13, 140. doi:10.1186/s13071-020-4000-z
Mazzeti, A. L., Capelari-Oliveira, P., Bahia, M. T., and Mosqueira, V. C. F. (2021). Review on experimental treatment strategies against trypanosoma cruzi. J. Exp. Pharmacol. 13, 409–432. doi:10.2147/jep.s267378
Minnick, M. F., Anderson, B. E., Lima, A., Battisti, J. M., Lawyer, P. G., and Birtles, R. J. (2014). Oroya fever and verruga peruana: Bartonelloses unique to South America. PLoS Negl. Trop. Dis. 8, e2919. doi:10.1371/journal.pntd.0002919
Mistry, J., Chuguransky, S., Williams, L., Qureshi, M., Salazar, G. A., Sonnhammer, E. L. L., et al. (2021). Pfam: The protein families database in 2021. Nucleic Acids Res. 49, D412–D419. doi:10.1093/nar/gkaa913
Mohana, R., and Venugopal, S. (2017). In silico analysis of toxins of Staphylococcus aureus for validating putative drug targets. Infect. Disord. Drug Targets 17, 130–142. doi:10.2174/1871526516666161230150219
Mondal, S. I., Ferdous, S., Akter, A., Mahmud, Z., Karim, N., Islam, M. M., et al. (2015). Identification of potential drug targets by subtractive genome analysis of Escherichia coli O157:H7: An in silico approach. Adv. Appl. Bioinform. Chem. 49, 49–63. doi:10.2147/aabc.s88522
Morgan, H. L. (1965). The generation of a unique machine description for chemical structures-A technique developed at chemical abstracts service. J. Chem. Doc. 5, 107–113. doi:10.1021/c160017a018
Morris, G. M., Huey, R., Lindstrom, W., Sanner, M. F., Belew, R. K., Goodsell, D. S., et al. (2009). AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791. doi:10.1002/jcc.21256
Mugumbate, G., Abrahams, K. A., Cox, J. A. G., Papadatos, G., van Westen, G., Lelièvre, J., et al. (2015). Mycobacterial dihydrofolate reductase inhibitors identified using chemogenomic methods and in vitro validation. PLoS One 10, e0121492. doi:10.1371/journal.pone.0121492
Neelapu, N., Mutha, N., and Akula, S. (2015). Identification of potential drug targets in Helicobacter pylori strain HPAG1 by in silico genome analysis. Infect. Disord. Drug Targets 15, 106–117. doi:10.2174/1871526515666150724111528
Oany, A. R., Mia, M., Pervin, T., Hasan, M. N., and Hirashima, A. (2018). Identification of potential drug targets and inhibitor of the pathogenic bacteria Shigella flexneri 2a through the subtractive genomic approach. Silico Pharmacol. 6, 11. doi:10.1007/s40203-018-0048-2
Osorio-Méndez, J. F., Vizcaíno-Castillo, A., Manning-Cela, R., Hernández, R., and Cevallos, A. M. (2016). Expression of profilin in Trypanosoma cruzi and identification of some of its ligands. Biochem. Biophys. Res. Commun. 480, 709–714. doi:10.1016/j.bbrc.2016.10.044
Parker, E. N., Cain, B. N., Hajian, B., Ulrich, R. J., Geddes, E. J., Barkho, S., et al. (2022). An iterative approach guides discovery of the FabI inhibitor Fabimycin, a late-stage antibiotic candidate with in vivo efficacy against drug-resistant gram-negative infections. ACS Cent. Sci. 8, 1145–1158. doi:10.1021/acscentsci.2c00598
Parker, E. N., Drown, B. S., Geddes, E. J., Lee, H. Y., Ismail, N., Lau, G. W., et al. (2020). Implementation of permeation rules leads to a FabI inhibitor with activity against Gram-negative pathogens. Nat. Microbiol. 5, 67–75. doi:10.1038/s41564-019-0604-5
Podschun, R., Pietsch, S., Höller, C., and Ullmann, U. (2001). Incidence of Klebsiella species in surface waters and their expression of virulence factors. Appl. Environ. Microbiol. 67, 3325–3327. doi:10.1128/AEM.67.7.3325-3327.2001
Podschun, R., and Ullmann, U. (1998). Klebsiella spp. as nosocomial pathogens: Epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin. Microbiol. Rev. 11, 589–603. doi:10.1128/cmr.11.4.589
Radusky, L., Ruiz-Carmona, S., Modenutti, C., Barril, X., Turjanski, A. G., and Martí, M. A. (2017). LigQ: A webserver to select and prepare ligands for virtual screening. J. Chem. Inf. Model. 57, 1741–1746. doi:10.1021/acs.jcim.7b00241
Rahman, M. A., Noore, M. S., Hasan, M. A., Ullah, M. R., Rahman, M. H., Hossain, M. A., et al. (2014). Identification of potential drug targets by subtractive genome analysis of Bacillus anthracis A0248: An in silico approach. Comput. Biol. Chem. 52, 66–72. doi:10.1016/j.compbiolchem.2014.09.005
Ramos, P. I. P., Fernández Do Porto, D., Lanzarotti, E., Sosa, E. J., Burguener, G., Pardo, A. M., et al. (2018). An integrative, multi-omics approach towards the prioritization of Klebsiella pneumoniae drug targets. Sci. Rep. 8, 10755. doi:10.1038/s41598-018-28916-7
Rassi, A., Rassi, A., and Marin-Neto, J. A. (2010). Chagas disease. Lancet 375, 1388–1402. doi:10.1016/S0140-6736(10)60061-X
Ribeiro da Cunha, B., Fonseca, L. P., and Calado, C. R. C. (2019). Antibiotic discovery: Where have we come from, where do we go? Antibiot. (Basel) 8, E45. doi:10.3390/antibiotics8020045
Rogers, D., and Hahn, M. (2010). Extended-connectivity Fingerprints. J. Chem. Inf. Model. 50, 742–754. doi:10.1021/ci100050t
Ruiz-Carmona, S., Alvarez-Garcia, D., Foloppe, N., Garmendia-Doval, A. B., Juhos, S., Schmidtke, P., et al. (2014). rDock: a fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLoS Comput. Biol. 10, e1003571. doi:10.1371/journal.pcbi.1003571
Sabe, V. T., Ntombela, T., Jhamba, L. A., Maguire, G. E. M., Govender, T., Naicker, T., et al. (2021). Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. Eur. J. Med. Chem. 224, 113705. doi:10.1016/j.ejmech.2021.113705
Schijman, A. G. (2018). Molecular diagnosis of Trypanosoma cruzi. Acta Trop. 184, 59–66. doi:10.1016/j.actatropica.2018.02.019
Schofield, C. J., Jannin, J., and Salvatella, R. (2006). The future of Chagas disease control. Trends Parasitol. 22, 583–588. doi:10.1016/j.pt.2006.09.011
Serral, F., Castello, F. A., Sosa, E. J., Pardo, A. M., Palumbo, M. C., Modenutti, C., et al. (2021). From genome to drugs: New approaches in antimicrobial discovery. Front. Pharmacol. 12, 647060. doi:10.3389/fphar.2021.647060
Serral, F., Pardo, A. M., Sosa, E., Palomino, M. M., Nicolás, M. F., Turjanski, A. G., et al. (2022). Pathway driven target selection in Klebsiella pneumoniae: Insights into carbapenem exposure. Front. Cell. Infect. Microbiol. 12, 773405. doi:10.3389/fcimb.2022.773405
Shanmugam, A., and Natarajan, J. (2010). Computational genome analyses of metabolic enzymes in Mycobacterium leprae for drug target identification. Bioinformation 4, 392–395. doi:10.6026/97320630004392
Shuvo, M. S. R., Shakil, S. K., and Ahmed, F. (2019). Potential drug target identification of Legionella pneumophila by subtractive genome analysis: An in silico approach. Banglad. J. Microbiol. 35, 102–107. doi:10.3329/bjm.v35i2.42638
Sosa, E. J., Burguener, G., Lanzarotti, E., Defelipe, L., Radusky, L., Pardo, A. M., et al. (2018). Target-pathogen: A structural bioinformatic approach to prioritize drug targets in pathogens. Nucleic Acids Res. 46, D413–D418. doi:10.1093/nar/gkx1015
Stokes, J. M., Yang, K., Swanson, K., Jin, W., Cubillos-Ruiz, A., Donghia, N. M., et al. (2020). A deep learning approach to antibiotic discovery. Cell 181, 475–483. doi:10.1016/j.cell.2020.04.001
The UniProt Consortium (2018). UniProt: The universal protein knowledgebase. Nucleic Acids Res. 46, 2699. doi:10.1093/nar/gky092
Trofimov, V., Kicka, S., Mucaria, S., Hanna, N., Ramon-Olayo, F., Del Peral, L. V.-G., et al. (2018). Antimycobacterial drug discovery using Mycobacteria-infected amoebae identifies anti-infectives and new molecular targets. Sci. Rep. 8, 3939. doi:10.1038/s41598-018-22228-6
Uddin, R., and Jamil, F. (2018). Prioritization of potential drug targets against P. aeruginosa by core proteomic analysis using computational subtractive genomics and Protein-Protein interaction network. Comput. Biol. Chem. 74, 115–122. doi:10.1016/j.compbiolchem.2018.02.017
Vamathevan, J., Clark, D., Czodrowski, P., Dunham, I., Ferran, E., Lee, G., et al. (2019). Applications of machine learning in drug discovery and development. Nat. Rev. Drug Discov. 18, 463–477. doi:10.1038/s41573-019-0024-5
Van den Kerkhof, M., Sterckx, Y. G.-J., Leprohon, P., Maes, L., and Caljon, G. (2020). Experimental strategies to explore drug action and resistance in kinetoplastid parasites. Microorganisms 8, E950. doi:10.3390/microorganisms8060950
Vilchèze, C., and Jacobs, W. R. (2007). The mechanism of isoniazid killing: Clarity through the scope of genetics. Annu. Rev. Microbiol. 61, 35–50. doi:10.1146/annurev.micro.61.111606.122346
Wadood, A., Ghufran, M., Khan, A., Azam, S. S., Uddin, R., Waqas, M., et al. (2017). The methicillin-resistant S. epidermidis strain RP62A genome mining for potential novel drug targets identification. Gene Rep. 8, 88–93. doi:10.1016/j.genrep.2017.06.002
Winder, F. G., and Collins, P. B. (1970). Inhibition by isoniazid of synthesis of mycolic acids in Mycobacterium tuberculosis. J. Gen. Microbiol. 63, 41–48. doi:10.1099/00221287-63-1-41
Yeh, I., Hanekamp, T., Tsoka, S., Karp, P. D., and Altman, R. B. (2004). Computational analysis of Plasmodium falciparum metabolism: Organizing genomic information to facilitate drug discovery. Genome Res. 14, 917–924. doi:10.1101/gr.2050304
Keywords: virtual screeening, antimicrobials, drug disco/very, phenotypic screening, molecular docking, chemical similarity, neglected diseases
Citation: Schottlender G, Prieto JM, Palumbo MC, Castello FA, Serral F, Sosa EJ, Turjanski AG, Martì MA and Fernández Do Porto D (2022) From drugs to targets: Reverse engineering the virtual screening process on a proteomic scale. Front. Drug. Discov. 2:969983. doi: 10.3389/fddsv.2022.969983
Received: 15 June 2022; Accepted: 07 October 2022;
Published: 20 October 2022.
Edited by:
Leena Hanski, University of Helsinki, FinlandReviewed by:
Neeraj Kumar, Northwestern University, United StatesCopyright © 2022 Schottlender, Prieto, Palumbo, Castello, Serral, Sosa, Turjanski, Martì and Fernández Do Porto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcelo A. Martì, bWFydCYjeDAwMGVlOy5tYXJjZWxvQGdtYWlsLmNvbQ== Darîo Fernández Do Porto, ZGFyaW9mZEBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.