 Lucas A. Machado
Lucas A. Machado Eduardo Krempser
Eduardo Krempser Ana Carolina Ramos Guimarães
Ana Carolina Ramos Guimarães
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Drug Discov., 21 October 2022
Sec. Anti-Infective Agents
Volume 2 - 2022 | https://doi.org/10.3389/fddsv.2022.954911
HIV-1 integrase is an essential enzyme for the HIV-1 replication cycle, and currently, integrase inhibitors are in the first line of treatment in many guidelines. Despite the discovery of new inhibitors, including a new class of molecules with different mechanisms of action, resistance is still a relevant problem, and adding new options to the therapeutic arsenal to fight viral resistance is a Sisyphean task. Because of the difficulty and cost of in vitro screenings, machine learning-driven ligand-based virtual screenings are an alternative that can not only cut costs but also use valuable information about active compounds with yet unknown mechanisms of action. In this work, we describe a thorough model exploration and hyperparameter tuning procedure in a dataset with class imbalance and show several models capable of distinguishing between compounds that are active or inactive against the HIV-1 integrase. The best of the models was then used to screen the natural product atlas for active compounds, resulting in a myriad of molecules that share features with known integrase inhibitors. Here we also explore the strengths and shortcomings of our models and discuss the use of the applicability domain to guide in vitro screenings and differentiate between the “predictable” and “unknown” regions of the chemical space.
Over the past few years, the development of new antiretroviral therapies (ARTs) has significantly increased the life expectancy of people living with HIV (Human Immunodeficiency Virus), the causative agent of acquired immunodeficiency syndrome (AIDS) (Moore and Chaisson, 1999). The most recent compounds used in ART formulations are integrase strand transfer inhibitors (INSTIs). The HIV integrase (IN) is a major HIV-1 enzyme, and its inhibitors are being used in several first-line HIV treatments (el Bouzidi et al., 2020). The IN is an interesting study case because it has two sites that may be exploited for inhibition: the active site—in which the inhibition impairs the strand-transfer reaction -, and the so-called allosteric site—in which the drug binding impairs the binding of the co-factor lens epithelium-derived growth factor (LEDGF) (Christ and Debyser, 2013).
Despite the discovery of a class of drugs for each of two different sites of IN, resistance mutations against both types of inhibitors have been reported, causing disruption of binding sites and, in some cases, therapeutic failure (Feng et al., 2013; Machado and Guimarães, 2020; Mbhele et al., 2021), generating a demand for new molecules that could potentially translate into therapies. In silico screenings (or virtual screening—VS.) approaches are a set of methods used to screen extensive collections of molecules before the in vitro tests of drug candidates, reducing the number of molecules to be tested. Currently, several groups are exploiting the potential of Machine Learning (ML) in drug discovery (Li et al., 2017; Stephenson et al., 2019; Zhou et al., 2021). The models applied range from the use of fingerprints and molecular descriptors such as BCUTs to the use of connectivity graphs in graph neural networks (Cheung and Moura, 2020).
In 2015, an ML VS. method was used to search for new active ligands capable of inhibiting HIV-1 IN, using active and assumed-inactive compounds (Kurczyk et al., 2015). ML was also used to specifically find allosteric inhibitors in 2017 (Li et al., 2017). However, since 2015, more than 100 new compounds tested against the IN were added to the BindingDB alone (Wassermann and Bajorath, 2011). Here, we built models based on molecular descriptors that are able of discerning between compounds that are active and inactive against the IN regardless of the mechanism of action, using data from the BindingDB, and thoroughly screening a combination of resampling and modeling methods. The models were efficient in discerning between active and inactive compounds even in a set of molecules with different mechanisms of action. The best model was used to screen the Natural Product Atlas (NPA) database (van Santen et al., 2019) for new inhibitor candidates, resulting in a set of compounds that were predicted as positives by the models.
A dataset of 7,165 compounds tested against the IN was obtained from the BindingDB. In addition, INSTIs (Raltegravir, Elvitegravir, Bictegravir, Dolutegravir, and Cabotegravir) and allosteric integrase inhibitors (ALLINI-1 and ALLINI-2) were added (Kessl et al., 2012; Feng et al., 2013). The compounds with IC50 higher than 1 μM were considered inactive, and the ones with IC50 below or equal to 1 μM were considered active. The pairwise Tanimoto coefficient was calculated for all the compounds using the maccs fingerprint implemented in the RDkit package (Landrum, 2013), and duplicates of the compounds (Tanimoto coefficient equals 1) were removed and the average IC50 of all the copies was used. For the screening, we used all compounds in the Natural Product Atlas.
For each compound, all the molecular descriptors implemented in the Python library MORDRED (Moriwaki et al., 2018) were calculated using as input the SMILES representation of each compound. From this final dataset, 30% of the compounds were sampled and set aside to form a test set, and the remaining 70% were used as the training set. To guarantee the representativity of the test set, its entries were divided into clusters based on their distances in feature space using the k-means algorithm. After that, proportional samples were drawn from each cluster to form the test set; the ideal number of clusters was determined using the elbow method, and—in order to avoid any distortion caused by outliers–features were normalized using z-scores for the clustering procedure. The choice of standardization can considerably affect the quality of the final model, especially with the presence of outliers, being the investigation of alternative methods of an opportunity for future analyses To further test the models, a set of HIV-1 integrase decoys was retrieved from DUDE-E (Mysinger et al., 2012) to assess how the models behave when predicting the activity of compounds that were not deliberately tested or designed as inhibitors. The z-score of each feature was calculated again, this time just for the training set, and the mean and standard deviation values obtained for each feature were used to standardize all the other sets of compounds.
We built three different groups of models: using 10 features, 30 features, and 50 features, the feature selection process was carried out by calculating the mutual information between variables in the training set and the response variable, and the top 10, 30, and 50 features were used in each of the cases. The different feature selection procedures were combined with three different sampling strategies: undersampling, Synthetic Minority Oversampling Technique (SMOTE), and undersampling+SMOTE. For the SMOTE procedures, we used three nearest neighbors to generate the synthetic entries. The combinations of different numbers of features and sampling strategies were used to train a logistic regression (LR) model, a Random Forest (RF) model (Breiman, 2001), a Support Vector Machine (SVM) model (Shmilovici, 2009), and a Multilayer perceptron (MLP) (Castro et al., 2017).
To optimize the hyperparameters of the RF model, a random search with three-fold cross-validation was performed, exploring the number of trees (integers from 200 to 2000), the maximum tree depth (integers from 10 to 110), the minimum number of samples required to split an internal node (2, 5, 10,15,20), the minimum number of samples required to be at a leaf node (1, 2, 4, 8), and whether or not to use bootstrap.
For the hyperparameter optimization of the SVM, a grid search with three-fold cross-validation was carried out, to explore the values of C (0.001, 0.01, 0.1, 1, 10,100) and values of Gamma (0.001, 0.01, 0.1, 1, 10) in an RBF kernel.
Finally, for the MLP, we built a model with two hidden layers, varying the number of nodes from: half the input size, the input size, and twice the input size. Lambda values for L2 regularization (0, 0.01, 0.001, 0.0001), batch size (32, 64 and 128), and learning rates (0.1, 0.01, 0.001, 0.0001) were also explored. The optimization was carried out through three-fold cross-validation using 10 epochs, and the best architecture was used for the optimization of the number of epochs also using cross-validation.
After hyperparameter optimization of LR, RF, SVM, and MLP models, the optimum probability thresholds were calculated by averaging the Youden’s J index over 500 cross-validation rounds adjusting the models to 80% of the training set observations, and validating against the remaining 20%.
The performance of the models was calculated by testing them against the test set, and further validation was done by testing against a set of 6,650 decoys; the model with the highest precision against the test set was used for the predictions in the NPA dataset. To add information on possibility of assay interference, the molecules predicted as active were submitted to the prediction of PAINS (Pan-Assay INterference compounds) using filters of functional groups from the ChEMBL database, as well as several filters of properties calculated with the RDKit. They were also clustered by a hierarchical clustering algorithm using the Tanimoto distance matrix, and the best-scoring molecule from each cluster was chosen as representative.
To assure that the predictions were within the applicability domain of the models, we merged the training and NPA set, carried out a principal component analysis (PCA), and using the first three principal components we drew a convex hull (Netzeva et al., 2005) around the training set, leaving out the NPA compounds that were outside the boundaries of the hull.
After applying the IC50 cutoff and excluding duplicated molecules based on Tanimoto coefficients, we obtained 1,439 active compounds, 5,598 inactive compounds, and 28902 NPA compounds. The test set comprised 408 active compounds and 1704 inactive compounds sampled from the 50 clusters. The number of rule of five violations (Pollastri, 2010) of each compound within the training and test set was assessed in order to guarantee that the models were not just discerning between drug-like and non-drug-like compounds (Figure 1); 74% of the active compounds and 79% of the inactive compounds showed zero violations of the rule of five, and therefore are all highly suitable as orally bioavailable drugs (Giménez et al., 2010). Both datasets are also well distributed in relation to the number of rule-of-five violations of the compounds, with the inactive set showing only a slightly increased number of compounds with no violations, while the active ones have a slightly increased number of compounds with one violation.
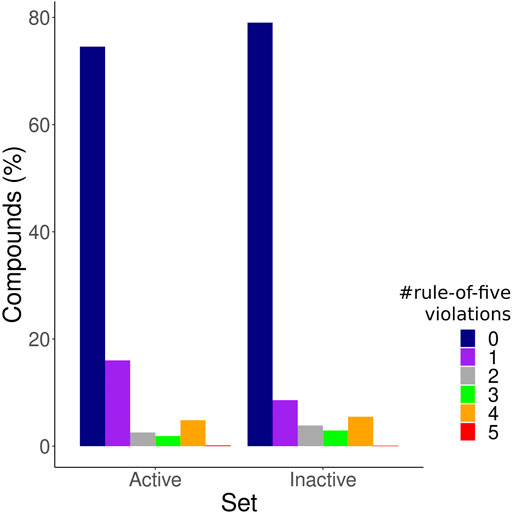
FIGURE 1. Druglikeness of molecules within the training set. The bar plot depicts the frequency of compounds with 0, 1, 2, 3, four and five rule of five violations among the active compounds set, and the inactive compounds set.
Given the number of active compounds in the dataset, resampling strategies (SMOTE and SMOTE+undersampling) were implemented. The training set using SMOTE was comprised of 3,894 inactive compounds and 3,093 active compounds and synthetic active samples. The SMOTE+undersampling set contained 1947 inactive samples and 2062 active and synthetic active samples. The pairwise Tanimoto coefficient of the training/test set molecules shows rare entries with values greater than 0.7, confirming that the dataset is quite diverse (Figure 2A). Figure 2B displays the first three principal components of the training and test set. We emphasize that the test set was sampled from 50 clusters of compounds (proportional to cluster size), and it is possible to see that the compounds used for the test of the models are distributed throughout all the regions of the feature space where compounds of the training set can also be found. It suggests that sampling the entries of the test set from different clusters helped achieve a representative test set, spanning even some of the less populated regions of the relevant feature space.

FIGURE 2. Variability of the training and test set. (A) displays the pairwise Tanimoto coefficient of the training and test set; while (B) displays the three first PCs of the training and test set and the percentage of variance explained by each one of them, where the training set is shown in blue, and the test set in lime green.
The evaluation of the models showed that RF using 30 features and the SMOTE strategy was the highest precision model, and when considering the F1 score, the two best models have the same performance. All the top five models are RF or SVM models using SMOTE, followed by RF models using undersampling+SMOTE. Suggesting that just undersampling generates a loss of information, and therefore, precision values lower than 0.8 (Table 1). LR models had the least promising performances, suggesting that there are essential non-linear relationships that must be considered to correctly classify the compounds. MLP also underperformed, even being theoretically capable of capturing the non-linearities, possibly because the reduced number of data points hindered the optimization of the model. RF and SVM models also performed better against the decoy set (Table 2), with the top five models correctly classifying almost all the decoys, even when including the ones outside the convex hull. LR and MLP models underperformed against the decoy set too, and the combination of SMOTE and MLP had the worst performance regardless of the number of features used. RF+SMOTE using 30 features had an accuracy of 0.97 when tested against the decoy set, both when considering the decoys within the hull or all of them.
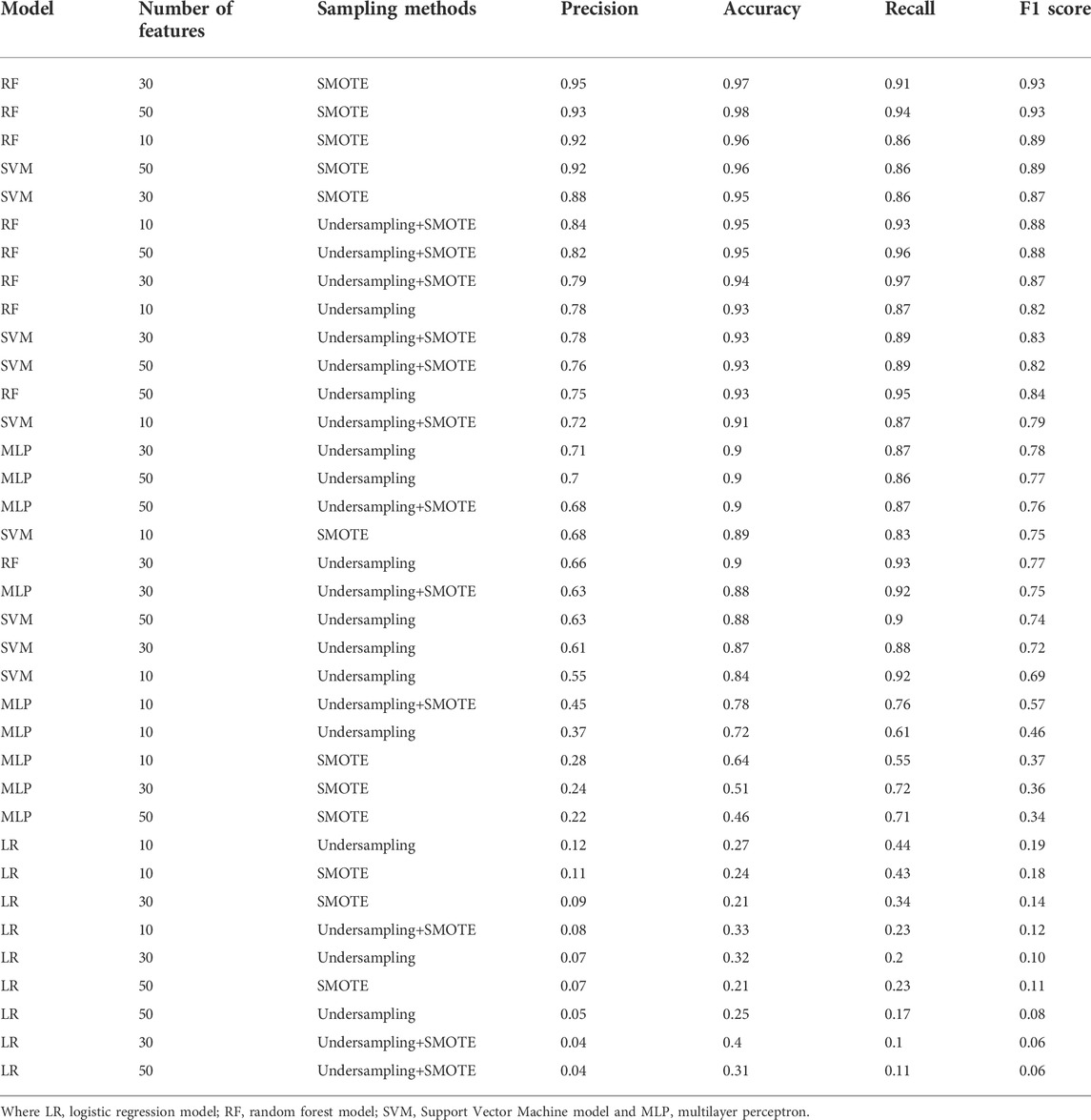
TABLE 1. Model performance. The model, number of features, sampling strategy and performance is shown for each of the models.
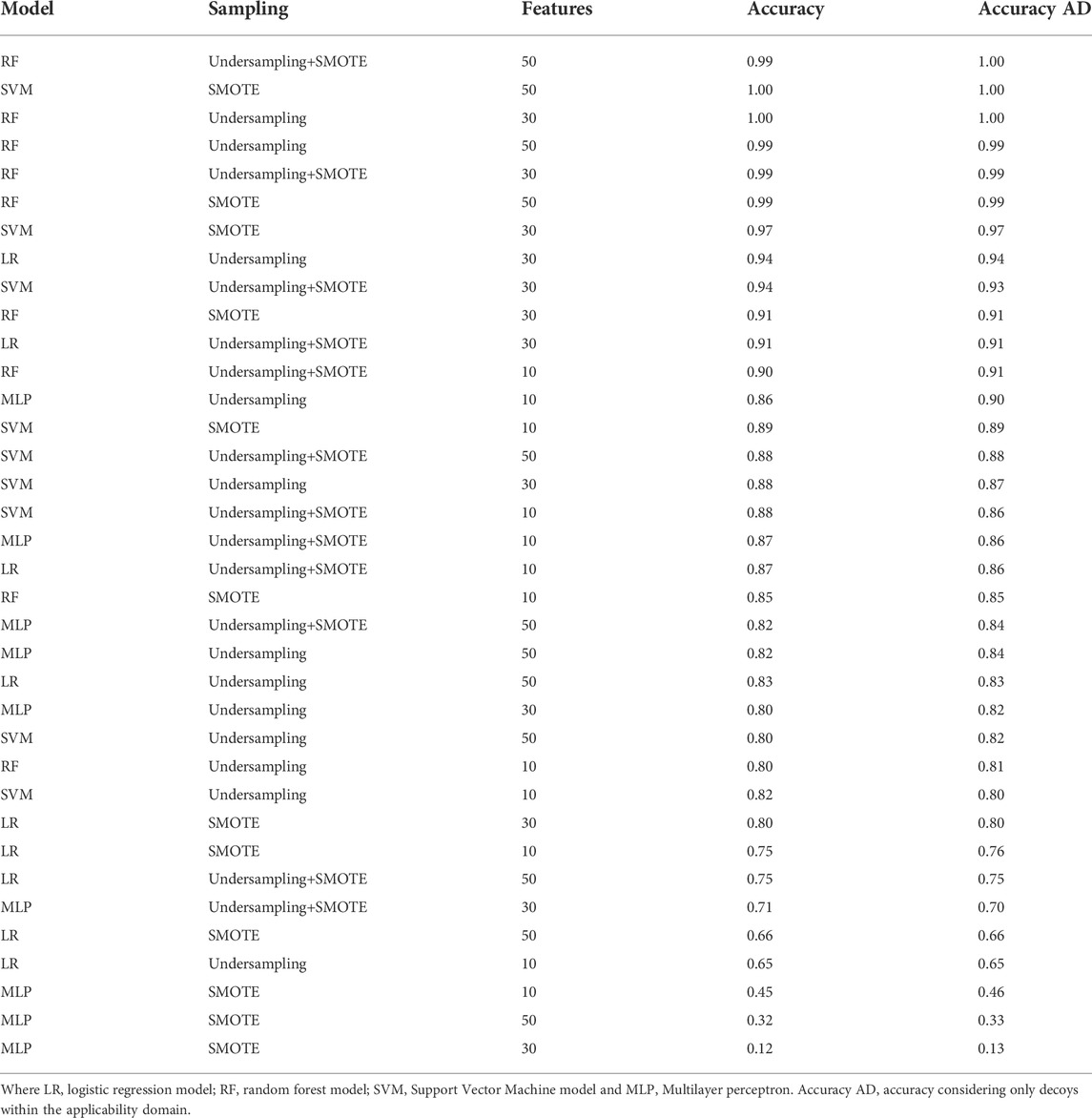
TABLE 2. Model performance on decoy set. The model, number of features, sampling strategy and performance is shown for each of the models.
In what concerns the features selected on the highest-precision model, 22 of them are BCUT descriptors, five are Barysz Matrix descriptors, and the three remaining are an E-state descriptor, a molar refractivity descriptor, and a molecularID descriptor. BCUT descriptors—which are eigenvalue-based descriptors - have been described before in studies with antimalarials as the most influential features in activity (Roy et al., 2002; Hou et al., 2016; Sarkar et al., 2016; Danishuddin et al., 2019), and here too these were the dominant features, corroborating the observations that were made in other studies, and suggesting the importance of BCUTs in ligand-based strategies.
The first step was to determine the applicability domain of the models. Figure 3 shows the three first PCs of the NPA dataset and the training/test set. It is possible to see the regions in which the datasets overlap, and consequently, the regions in which the models can be applied to discern between active and inactive compounds. Since only the NPA compounds inside the convex hull drawn with the training set were used, 1,283 compounds were left out of the prediction. Here we used a convex hull with only three dimensions, but the hull can be expanded to higher dimensions at an increased computational cost, and the use of more dimensions could enhance the resolutions of the boundaries. The convex hull presents itself as an interesting tool for two antagonistic approaches: one could use it either to understand the boundaries within which the model works or to determine which regions of the chemical space are sufficiently “unknown” and should be prioritized during in vitro screenings to find new compounds. From the remaining 27619 molecules from the NPA, 246 were predicted as active by the best-performing model, from which 106 presented no rule-of-five violations. From all the 106 active compounds, 63 passed the PAINS filter. The molecules predicted as active were quite diverse, with an average Tanimoto coefficient of 0.4 (Figure 4), meaning that the molecules were still remarkably different among themselves, which could be explained by the fact that models were trained in a heterogeneous and mechanistically diverse dataset. In Figure 4 it is also possible to observe how the molecules predicted as active cluster together in feature space, with few exceptions.

FIGURE 3. PCA of the training set and NPA set. The graph shows the three first PCs of the training set/test set (purple), and the NPA set (dark green), as well as the percentage of variance explained by each one of them, to show the regions in which they overlap.
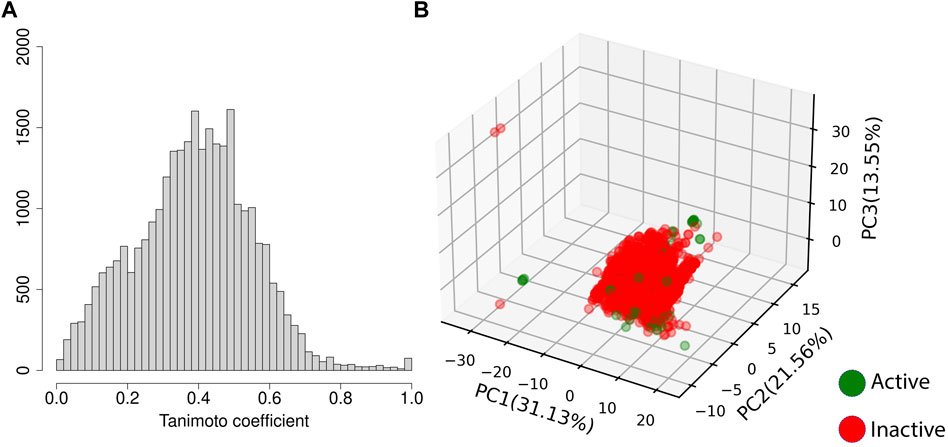
FIGURE 4. Profile of the molecules predicted as active. (A) shows the pairwise Tanimoto coefficients of the molecules predicted as active; while (B) shows the first three PCs of the NPA set, and the percentage of variance explained by each one of them. The compounds predicted as inactive are shown in red, and the ones predicted as actives are shown in green.
Most of the top 15 molecules (Figure 5) were derived from fungi and bacteria, with Streptomyces sp. being the most predominant source of molecules. Among the set of compounds predicted as active (Figure 6), the most frequent Murcko scaffold (Bemis & Murcko, 1996) (considering continuous scaffolds with a minimum of six heavy atoms) was 7,8,9,10-tetrahydro-5H-benzo[b]carbazole, followed by xanthene. Most of the scaffolds shown are large and complex structures that are ubiquitous among molecules from the Natural Product Atlas.
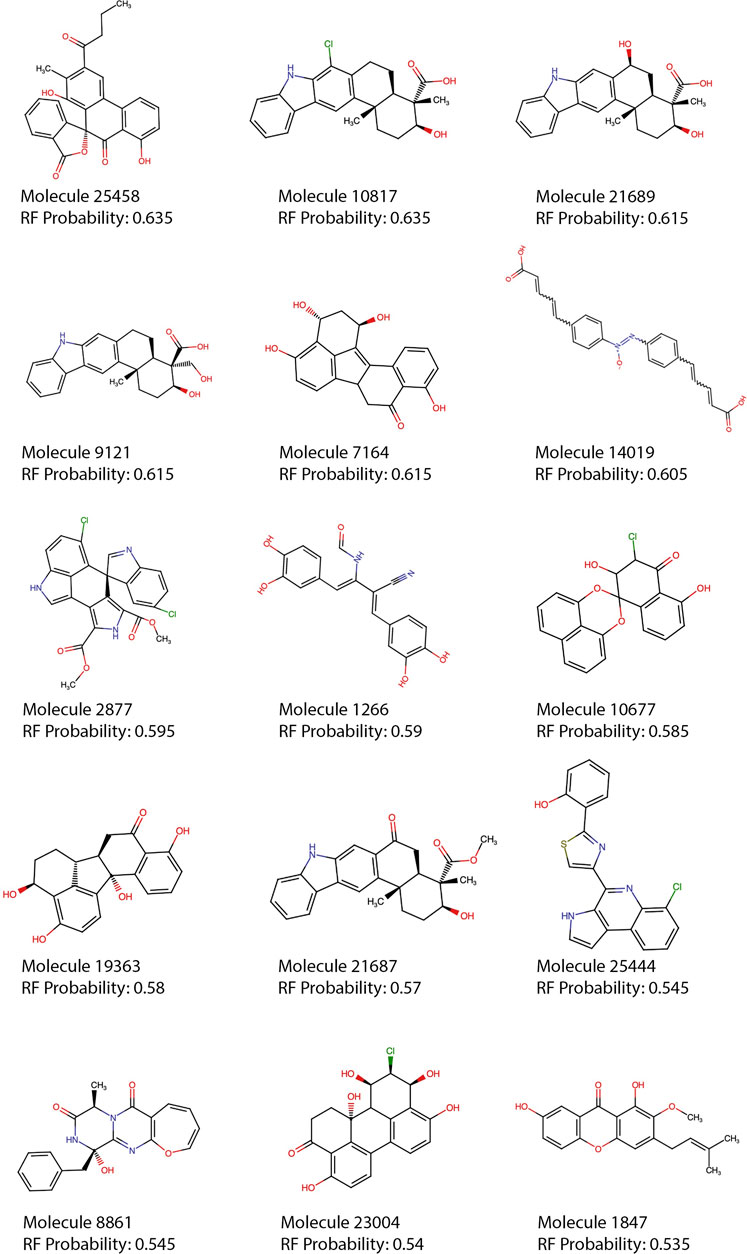
FIGURE 5. Top 15 RF predictions. Each of the top 15 molecules predicted as active by the best model (RF, using 30 features and SMOTE) are shown. Molecules with rule of five violations were filtered out of the top 15 and can be seen in the supplementary material (Supplementary Table S2).
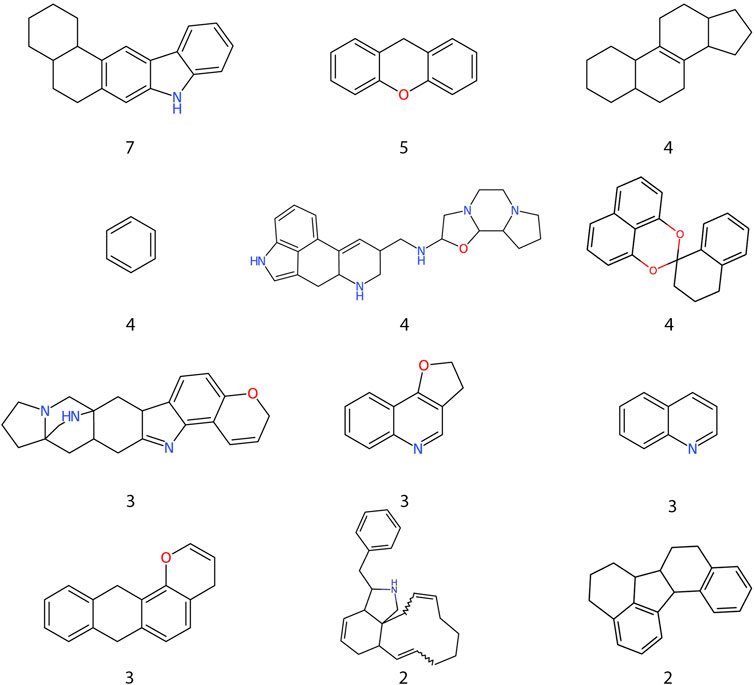
FIGURE 6. Frequent scaffolds in compounds predicted as active. Murcko scaffolds extracted from the RF highest probability compounds and their frequency inside the dataset.
Most of the top 15 molecules shown in Figure 5, present groups resembling the diketoacid moieties found in INSTIs, pointing out that the model seems to be selecting molecules containing hydroxylated aromatic compounds. However, the exact pattern found in diketoacids is rarely complete in the selected natural compounds or at least is slightly different, which could be caused by the lack of available compounds containing exact matches. It is also possible, however, that only two coplanar oxygens could also interact with Mg2+ ions in the active site of the enzyme. For instance, in molecules 7,164 and 10677 the third oxygen of the triad is offset by one position, making the configuration slightly different from the known INSTIs. Candidate 25458 shows protruding oxygens like dolutegravir, but one of them is not free to interact with Mg2+ ions, and instead is linked to another ring in the molecule scaffold; in the other segment of the molecule, the coplanar oxygens are separated by a methyl group. Molecule 1847 also resembles dolutegravir diketoacid moiety, except for the fact that one of the three coplanar oxygens protruding from the ring is linked to a methyl group, which could difficult strand-transfer inhibition, but could still be an interesting subject to test; mainly because its closest correspondence within the set of active compounds (Figure 7) is an experimental INSTI-like diketoacid inhibitor with an IC50 of 600 nM. A similar case is 8,861, which contains a methyl between the coplanar oxygens protruding from the rings. Its closest correspondent in the dataset of active compounds is an experimental integrase inhibitor which also lacks the third coplanar oxygens, but contains the halogenated ring characteristic of the commercial INSTIs and displays an IC50 of 7.6 nM. It suggests that perhaps the absence of the third Mg-contacting oxygen does not completely impair activity. The presence of only two coplanar oxygens in the rings, however, can be reminiscent of IN “cinnamoyl inhibitors” (Buolamwini and Assefa, 2002) and therefore can still be a sign of inhibitory activity. Molecules 7,164 and 1,266 share the same “cinnamoyl inhibitor” as their closest neighbor in the dataset of active compounds.
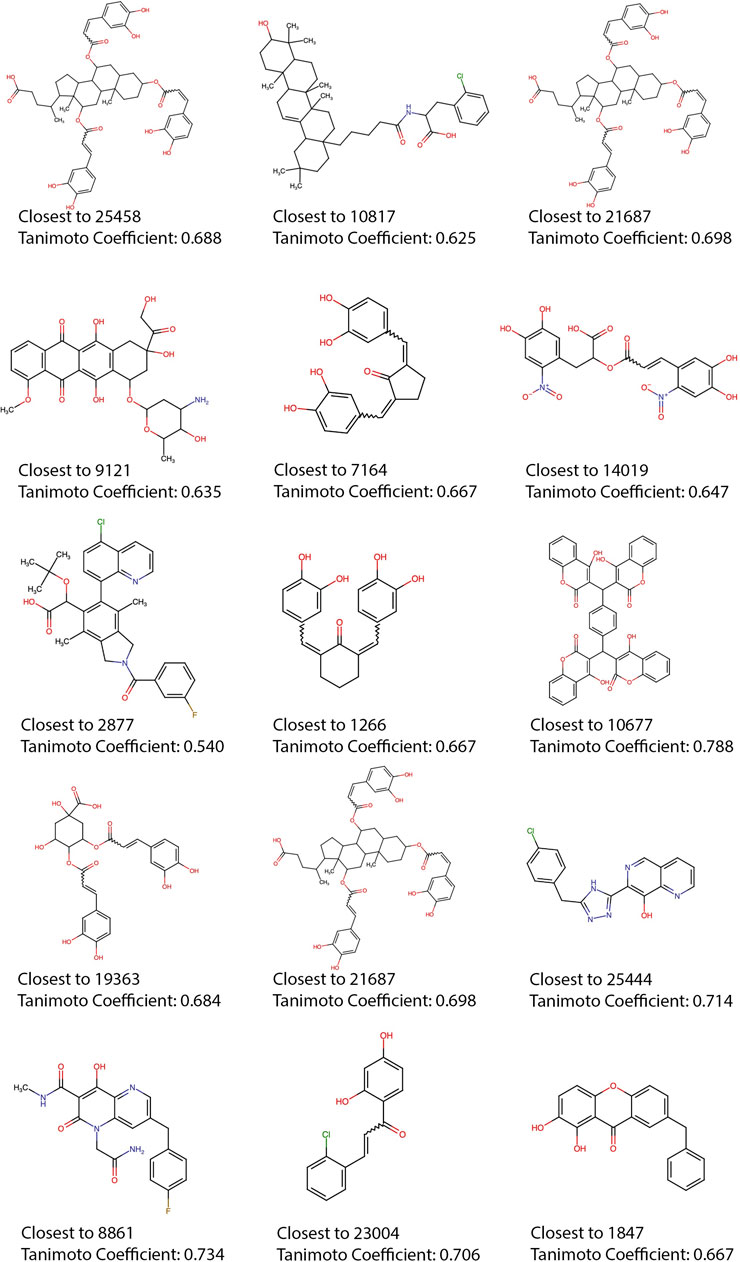
FIGURE 7. Training set compounds closest to the top 15. Each of the compounds with higher Tanimoto coefficients in relation to the top 15 predicted active compounds, the Tanimoto coefficients are shown, as well as the name of the neighbors (shown in Figure 5).
Compounds 7,164, 10677, 1963, and 23004 seem to display a recurring theme of 8-hydroxy-1-tetralone regions and halogenated rings. Albeit having features present in known highly active inhibitors, in these molecules, the moieties are reoriented in the structures in ways that could impair functionality. The nearest neighbor of 23004 is a compound that was investigated as ALLINI, but despite the similarity concerning the presence of the halogenated and hydroxylated rings, both molecules are topologically very distinct. Compound 9,121–oridamycin B –, except for two small groups is very similar to xiamycin, a compound that was shown before as active against HIV-1 (Ding et al., 2010), the same similarity is observed with compound 21687–xiamycin E–and 19-methoxyl-xiamycin. These three compounds are interesting cases because xiamycin was only shown to be anti-HIV-1 in culture assays and is believed to block viral entry.
The model selected for the predictions has a clear preference for polyhydroxylated aromatic compounds, which were the most investigated types of compounds in the early days of IN drug development (Buolamwini and Assefa, 2002), and therefore very frequent within the dataset. The active compounds were divided into 30 clusters, and the most populated clusters—which comprise more than 50% of the active molecules - were explored here (Figure 8), the cluster information for all molecules can be found in Supplementary Table S1. Molecules 25458, 10817, 10677, 8,861, and 7,164 are representative of five of the major clusters, which are populated by compounds with similar structural features that were also classified as active. The average pairwise Tanimoto coefficient of cluster 4, which has molecule 10817 as representative was 0.83, showing that the molecules within the cluster are highly similar and most of them also have similarities with xiamycin. Other molecules that share the same scaffold as 10817 are 9,121 and 21686—both with high scores according to the best model. The intracluster pairwise similarities highlight the capability of the model to make consistent predictions and capture scaffold information despite the use of features that do not explicitly represent the connectivity maps. Despite the consistency, some compounds show structural features of active compounds but in different spatial configurations, highlighting the possible shortcomings of the method.
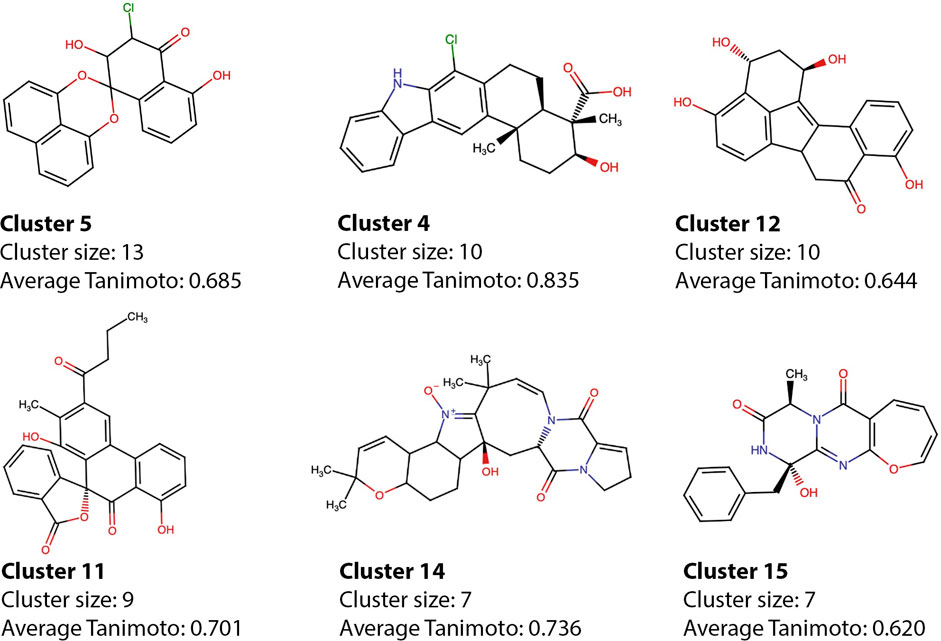
FIGURE 8. Cluster representatives from the active compounds. The representative of each of the main clusters is shown alongside with the cluster size and average intracluster Tanimoto coefficient. Representatives were chosen according to activity prediction.
It is important to emphasize the fact that for some of the molecules, the model could pinpoint fundamental substructures found in known inhibitors and select molecules with such features, while in others, the substructures were selected, but in spatial distributions that could hinder activity. Although not likely, it could be that because of the unknown mechanism of action of many molecules in the dataset, these selected compounds - that do not resemble structures of known actives but bear some of their substructures—could still be active, but act by different mechanisms. The use of features that do not explicitly consider the connectivity of the molecule could lead to some sort of “monkey’s paw effect” where molecules predicted as active bear similar moieties to known active compounds, but in spatial configurations that hinder activity. On the other hand, this kind of flexibility can be interesting to explore new areas of the chemical space while preserving some characteristics that are known to play a role in inhibition. More experiments are needed to further discuss if the molecules that lack resemblance to the known active compounds could act as inhibitors or are mere artifacts. The fact remains that the best-performing model could detect important structural features and select interesting compounds for screening, including some that are remarkably similar to highly active compounds. The complete list of molecules predicted as active can be found in Supplementary Table S2 and the ones that pass PAINS filters can be found in Supplementary Table S3. It is important to emphasize the fact that the molecules from the training set were not sorted by their mechanisms of action, and the mechanism is not even stated in the bindingDB. Therefore, the models could be selecting compounds that are either similar to INSTIs or ALLINIs, or act by alternative mechanisms.
Here we showed a thorough selection of models capable of discerning between active and inactive compounds from the BindingDB. RF model using SMOTE was shown to be the best strategy regardless of the number of features used. SMOTE was shown to be an interesting strategy both alone or in conjunction with undersampling; and can be very useful to deal with minority classes, which is usually a problem because of the lack of negative results available but can also be an issue when only a small group of active compounds is known. The performance of the models was also tested against IN decoys, to further validate their predictive capabilities.
We also implemented a convex hull strategy to limit the predictions to the boundaries of the applicability domain of the models, which is essential to state that a given observation cannot be interpolated using the training set. The hull can also be an interesting strategy for exploring compounds with features that were not screened before. For instance, the 1,283 compounds that were found to be outside the hull, probably share little chemical similarity with compounds already tested against the HIV-1 integrase. This information can be used to guide in vitro screenings, both to feed Machine Learning models with information about molecules with new properties, or to increase the chance of finding new classes of molecules in experimental procedures by exploring “unknown” regions of the feature space. Fifteen of the promising compounds were explored in the detail, being mostly bacteria-derived compounds with features that resemble some of the known inhibitors, and many others are listed in the supplementary tables. Some of the molecules, however, display inhibitor-like groups, but in different spatial configurations, which can arise from the choice of features used to train the model.
Machine learning-based approaches are a promising strategy for ligand-based virtual screenings. Here we make our contribution both to the suggestion of new inhibitor candidates against a key HIV-1 enzyme, and to expand on the model-building strategies for this kind of approach.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
LM designed the workflow, and carried out the model building and molecule screening; EK contributed with insights to the machine learning approach, model choice, and hyperparameter tunning; AG contributed with the choice of database to use for the screening, the idea of screening natural for compounds, and overall review.
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001 and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ)—Finance Code E-26/201.462/2021.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fddsv.2022.954911/full#supplementary-material
Bemis, G. W., and Murcko, M. A. (1996). The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 39 (15), 2887–2893. doi:10.1021/JM9602928
Bouzidi, K., Jose, S., Phillips, A. N., Pozniak, A., Ustianowski, A., Gompels, M., et al. (2020). First-line HIV treatment outcomes following the introduction of integrase inhibitors in UK guidelines. AIDS 34, 1823. doi:10.1097/QAD.0000000000002603
Buolamwini, J. K., and Assefa, H. (2002).,CoMFA and CoMSIA 3D QSAR and docking studies on conformationally-restrained cinnamoyl HIV-1 integrase inhibitors: Exploration of a binding mode at the active site, J. Med. Chem. 45, 841–852. doi:10.1021/JM010399H/ASSET/IMAGES/MEDIUM/JM010399HN00001
Castro, W., Oblitas, J., Santa-Cruz, R., and Avila-George, H. (2017). Multilayer perceptron architecture optimization using parallel computing techniques. PLOS ONE 12, e0189369. doi:10.1371/journal.pone.0189369
Cheung, M., and Moura, J. M. F. (2020). “Graph neural networks for COVID-19 drug discovery,” in Proceedings of the IEEE international conference on big data (big data), Atlanta, GA, USA, 10-13 December 2020 (IEEE), 5646–5648. doi:10.1109/BigData50022.2020.9378164
Christ, F., and Debyser, Z. (2013). The LEDGF/p75 integrase interaction, a novel target for anti-HIV therapy. Virology 435, 102–109. doi:10.1016/J.VIROL.2012.09.033
DanishuddinMadhukar, G., Malik, M. Z., and Subbarao, N. (2019). Development and rigorous validation of antimalarial predictive models using machine learning approaches. Sar. QSAR Environ. Res. 30, 543–560. doi:10.1080/1062936X.2019.1635526
Ding, L., Münch, J., Goerls, H., Maier, A., Fiebig, H. H., Lin, W. H., et al. (2010). Xiamycin, a pentacyclic indolosesquiterpene with selective anti-HIV activity from a bacterial mangrove endophyte. Bioorg. Med. Chem. Lett. 20, 6685–6687. doi:10.1016/J.BMCL.2010.09.010
Feng, L., Sharma, A., Slaughter, A., Jena, N., Koh, Y., Shkriabai, N., et al. (2013). The A128T resistance mutation reveals aberrant protein multimerization as the primary mechanism of action of allosteric HIV-1 integrase inhibitors. J. Biol. Chem. 288, 15813–15820. doi:10.1074/jbc.M112.443390
Giménez, B. G., Santos, M. S., Ferrarini, M., S Fernandes, J. P., and Paulo dos Santos Fernandes, J. (2010). Evaluation of blockbuster drugs under the rule-of-five. Ingentaconnect.Com. 65, 148–152. doi:10.1691/ph.2010.9733
Hou, X., Chen, X., Zhang, M., and Yan, A. (2016). QSAR study on the antimalarial activity of Plasmodium falciparum dihydroorotate dehydrogenase ( Pf DHODH) inhibitors. Sar. QSAR Environ. Res. 27, 101–124. doi:10.1080/1062936X.2015.1134652
Kessl, J. J., Jena, N., Koh, Y., Taskent-Sezgin, H., Slaughter, A., Feng, L., et al. (2012). Multimode, cooperative mechanism of action of allosteric HIV-1 integrase inhibitors. J. Biol. Chem. 287 (20), 16801–16811. doi:10.1074/jbc.M112.354373
Kurczyk, A., Warszycki, D., Musiol, R., Kafel, R., Bojarski, A. J., and Polanski, J. (2015). Ligand-based virtual screening in a search for novel anti-HIV-1 chemotypes. J. Chem. Inf. Model. 55, 2168–2177. doi:10.1021/acs.jcim.5b00295
Landrum, G. (2013). RDKit: A software suite for cheminformatics, computational chemistry, and predictive modeling. Available at: http://www.rdkit.org/RDKit_Overview.pdf.
Li, Y., Wu, Y., and Yan, A. (2017). Study of structure-active relationship for inhibitors of HIV-1 integrase LEDGF/p75 interaction by machine learning methods. Mol. Inf. 36, 1600127. doi:10.1002/minf.201600127
Machado, L. de A., and Guimarães, A. C. R. (2020). Evidence for disruption of Mg2+ pair as a resistance mechanism against HIV-1 integrase strand transfer inhibitors. Front. Mol. Biosci. 7, 170. doi:10.3389/fmolb.2020.00170
Mbhele, N., Chimukangara, B., and Gordon, M. (2021). HIV-1 integrase strand transfer inhibitors: A review of current drugs, recent advances and drug resistance. Int. J. Antimicrob. Agents 57, 106343. doi:10.1016/j.ijantimicag.2021.106343
Moore, R. D., and Chaisson, R. E. (1999). Natural history of HIV infection in the era of combination antiretroviral therapy. AIDS 13, 1933–1942. doi:10.1097/00002030-199910010-00017
Moriwaki, H., Tian, Y.-S., Kawashita, N., and Takagi, T. (2018). Mordred: A molecular descriptor calculator. J. Cheminform. 10, 4. doi:10.1186/s13321-018-0258-y
Mysinger, M. M., Carchia, M., Irwin, J. J., and Shoichet, B. K. (2012). Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 55, 6582–6594. doi:10.1021/jm300687e
Netzeva, T. I., Worth, A. P., Aldenberg, T., Benigni, R., Cronin, M. T. D., Gramatica, P., et al. (2005). Current status of methods for defining the applicability domain of (quantitative) structure-activity relationships. The report and recommendations of ECVAM Workshop 52 Altern. Lab. Anim. 33, 155–173. doi:10.1177/026119290503300209
Pollastri, M. P. (2010). Overview on the rule of five. Curr. Protoc. Pharmacol. 49, 12. doi:10.1002/0471141755.PH0912S49
Roy, K., De, A. U., and Sengupta, C. (2002). QSAR with electrotopological state atom index: Human factor xa inhibitor N 2 -aroylanthranilamides. Drug Des. Discov. 18, 33–43. doi:10.3109/10559610213502
Sarkar, S., Siddiqui, A. A., Saha, S. J., De, R., Mazumder, S., Banerjee, C., et al. (2016). Antimalarial activity of small-molecule benzothiazole hydrazones. Antimicrob. Agents Chemother. 60, 4217–4228. doi:10.1128/AAC.01575-15
Shmilovici, A. (2009). “Support vector machines,” in Data mining and knowledge discovery handbook (Boston, MA: Springer US), 231–247. doi:10.1007/978-0-387-09823-4_12
Stephenson, N., Shane, E., Chase, J., Rowland, J., Ries, D., Justice, N., et al. (2019). Survey of machine learning techniques in drug discovery. Curr. Drug Metab. 20, 185–193. doi:10.2174/1389200219666180820112457
van Santen, J. A., Jacob, G., Singh, A. L., Aniebok, V., Balunas, M. J., Bunsko, D., et al. (2019). The natural products atlas: An open access knowledge base for microbial natural products discovery. ACS Cent. Sci. 5, 1824–1833. doi:10.1021/acscentsci.9b00806
Wassermann, A. M., and Bajorath, J. (2011). BindingDB and ChEMBL: Online compound databases for drug discovery. Expert Opin. Drug Discov. 6, 683–687. doi:10.1517/17460441.2011.579100
Keywords: machine learning, HIV-1, integrase, natural compounds, inhibition
Citation: Machado LA, Krempser E and Guimarães ACR (2022) A machine learning-based virtual screening for natural compounds capable of inhibiting the HIV-1 integrase. Front. Drug. Discov. 2:954911. doi: 10.3389/fddsv.2022.954911
Received: 27 May 2022; Accepted: 26 September 2022;
Published: 21 October 2022.
Edited by:
Dario Fernández Do Porto, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaReviewed by:
Konda Reddy Karnati, Bowie State University, United StatesCopyright © 2022 Machado, Krempser and Guimarães. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lucas A. Machado, YmlvbHVjYXNtYWNoYWRvQGdtYWlsLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.