 Chun Chou
Chun Chou Michael T. Chin
Michael T. Chin- Molecular Cardiology Research Institute, Tufts Medical Center, Boston, MA, United States
Heart failure (HF) with preserved ejection fraction (HFpEF) is a clinical syndrome characterized by signs and symptoms of HF in the presence of a normal left ventricular systolic function. Over the past decade, HFpEF has become increasingly prevalent, accounting for greater than 50% of all clinical HF presentations. HFpEF is a complex disease with heterogeneous clinical presentations and multiple non-cardiac comorbidities, which frequently co-exist and contribute to its pathophysiology. To date, only a handful of therapies have been proven to improve, albeit marginally, the outcomes in HFpEF. The development of effective therapeutic agents is in part hampered by the lack of animal models that adequately recapitulate human HFpEF. Although numerous pre-clinical models developed over the years have been labeled as “HFpEF” specific, there has not been a consensus on the appropriate standards for pre-clinical HFpEF models. Thus, the extent to which they truly mirror human HFpEF cannot be systematically validated. Recently, a new algorithm (H2FPEF) was developed to standardize the clinical diagnosis of HFpEF. In this review, with the aid of the clinical H2FPEF scoring system, we evaluate the clinical applicability and translational values of various murine models of HFpEF.
1 Introduction
Heart failure (HF) is a clinical syndrome characterized by a constellation of symptoms including dyspnea, fluid retention and fatigue caused by cardiac dysfunction (Klainer et al., 1965; McKee et al., 1971). Traditionally, the extent of pump dysfunction is approximated by the percentage of blood volume ejected per beat or ejection fraction (EF) (Sharma and Kass, 2014). Over the past decades, extensive basic science studies and clinical research have focused on HF associated with impaired left ventricular (LV) systolic function, characterized by a reduced EF, also known as HF with reduced EF (HFrEF). These efforts led to the development of several effective therapeutic regiments for HFrEF patients, culminating in the birth of guideline-directed medical therapy (GDMT) (Heidenreich et al., 2022). However, nearly 50% of patients with HF symptoms have a normal or preserved EF (HFpEF, defined as EF ≥ 50%) (Gok et al., 2020; Nair, 2020) and the majority do not benefit from GDMT (Borlaug, 2020). These observations therefore suggest HFrEF and HFpEF are in fact fundamentally distinct with respect to their pathophysiology (Ferrari et al., 2015; Borlaug, 2016) and animal models of HFrEF may not provide clinically applicable insights into the pathogenesis of HFpEF.
1.1 Challenges in modeling HFpEF
Unlike HFrEF, HF in HFpEF patients usually does not trace back to a primary injury of cardiomyocytes but rather a plethora of extra-cardiac perturbations (Ho et al., 2016), including obesity (Savji et al., 2018), diabetes (Lejeune et al., 2021), aging and hypertension (Eaton et al., 2016) as well as cardiac abnormalities, such as atrial fibrillation (Packer et al., 2020). Because of the predominantly cardiac origin of HFrEF pathogenesis, treatments aiming at optimizing cardio-hemodynamic imbalance often bestow disease modifying benefits for HFrEF patients. In contrast, the ill-defined etiology of HFpEF has hampered the development of HFpEF-specific drugs. Consequently, treatment of HFpEF still mostly focuses on symptom reduction. The block in bench-to-bedside translation may in part be due to a lack of pre-clinical HFpEF models that adequately recapitulate heterogeneity of the human condition.
1.2 Approach to validating HFpEF models
Although the complex etiologic and pathophysiologic paths of HFpEF pathogenesis pose a great challenge to the development of pre-clinical models, this process can be aided by a consensus on what HFpEF features the animals should manifest. First, the ejection fraction should be preserved. Second, clinical symptoms of HFpEF should be present. While assessment of shortness of breath and exercise intolerance in animal models may be less straightforward than in humans, various measurable parameters can provide a global impression if clinical symptoms of HFpEF are present. These include assessing exercise capacity by voluntary or forced exercise and pulmonary congestion by post-mortem lung weights or pulmonary vasculature. Lastly, a new diagnostic algorithm [H2FPEF score(Reddy et al., 2018)] that defines the most relevant clinical features (obesity, hypertension, atrial fibrillation, pulmonary hypertension, aging and diastolic dysfunction) of HFpEF has been recently developed (Figure 1). Such an approach simplifies and standardizes HFpEF diagnosis, but it is not without controversy, garnering criticisms ranging from oversimplification of disease heterogeneity to misclassification particularly in patients with low H2FPEF score(Ho et al., 2020; Churchill et al., 2021; Sanders-van Wijk et al., 2021). Nevertheless, the H2FPEF scoring algorithm has been validated in various patient cohorts(Packer et al., 2020; Churchill et al., 2021; Faxen et al., 2021; Tada et al., 2021) and its proven prognostic utility in human(Sueta et al., 2019; Parcha et al., 2021) further suggests that it sufficiently identifies key pathophysiologic determinants of HFpEF outcomes. Thus, the H2FPEF scoring system may be adapted as a novel standard to assess the clinical applicability and translational value of various pre-clinical HFpEF models. In the following sections, we include murine models that are presented as “HFpEF” in the literature or used to evaluate therapeutic options in the pre-clinical phase. All models are evaluated based on whether ejection fraction is preserved, whether clinical signs and symptoms of HFpEF are present, and scored in cardiac and extra-cardiac domains of the HF2PEF scoring system (Table 1).
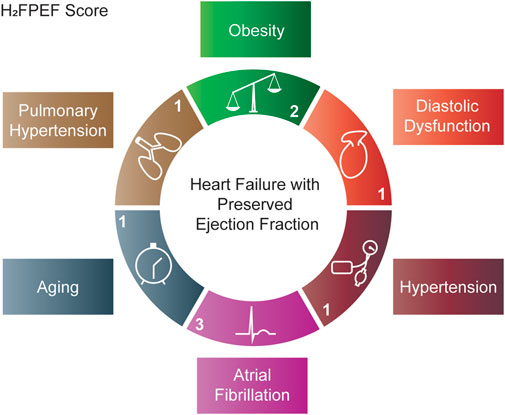
FIGURE 1. H2FPEF scoring system used to validate murine models of HFpEF. The H2FPEF score(Reddy et al., 2018) combines clinical and echocardiographic patient characteristics in the cardiac domain: diastolic dysfunction (E/E′) and atrial fibrillation as well as extra-cardiac domains: hypertension, aging (>60 years old), obesity (BMI >30 kg/m2) and pulmonary hypertension (pulmonary artery systolic pressure >35 mmHg). The score allocated to each domain is denoted. A total score ≥6 correlates with a >90% probability of HFpEF.
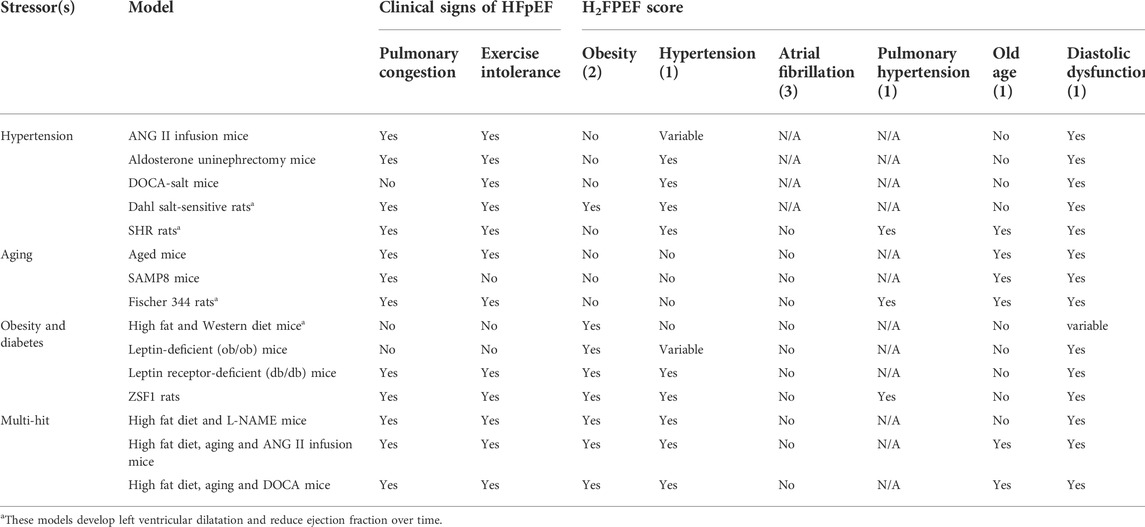
TABLE 1. Evaluation of murine HFpEF models by H2FPEF score.
2 Single stressor models
For the past decades, pre-clinical models of HFpEF have relied on single perturbations. Because of their limited applicability to human HFpEF, it is perhaps not surprising that therapeutic agents which were effective in these pre-clinical models often failed in clinical trials. A few unsuccessful attempts include angiotensin-converting enzyme inhibitors, angiotensin receptor one blockers and mineralocorticoid receptor antagonists (Khan et al., 2017; Lin et al., 2022) as well as nitric oxide donors and cyclic guanosine monophosphate stimulating therapies (Redfield et al., 2013; Komajda et al., 2017; Borlaug et al., 2018), and angiotensin receptor–neprilysin inhibitiors (Solomon et al., 2020; Wachter et al., 2020). Thus, pre-clinical models with single stressor may not represent a reliable platform for bench-to-bedside transition. However, they should not be dismissed as artifactual as such models often allow for detailed mechanistic studies on how each stressor singly contributes to cardiac manifestation of human HFpEF and may pave the way to the development of combination therapies. In the section below, we discuss the strengths and weaknesses of single-hit models of HFpEF with special focus on the three most significant risk factors: hypertension, aging and obesity.
2.1 Hypertension
Systemic hypertension is one of the most significant comorbidities in HFpEF patients (Heidenreich et al., 2022). In the past decades, animal studies have revealed a causal relationship between elevated blood pressure and LV structural alterations. Mechanistically, systemic hypertension induces arterial stiffness, which disproportionately increases afterload on the heart, leading to accelerated concentric LV hypertrophy and fibrosis (Samson et al., 2016). These changes may ultimately result in decreased diastolic as well as systolic dysfunction (Valero-Munoz et al., 2017). However, despite high prevalence of systemic hypertension in patients with HFpEF, intensive blood pressure control only reduces HFpEF events but does not alter disease outcomes (Upadhya et al., 2021). Conceivably, elevated blood pressure may initially contribute to the pathogenesis of HFpEF but subsequently play a less significant role during disease propagation.
Small animal models of hypertension have many features of human HFpEF and may help elucidate some aspects of HFpEF pathogenesis. Although these models commonly do not reproduce the slow onset of hypertension seen in patients with HFpEF, the relatively rapid development of cardiac phenotypes accelerates experiment turn-around. Below, we evaluate the clinical applicability of various hypertension induced HFpEF murine models using the H2FPEF scoring system.
2.1.1 Angiotensin II infusion (H2FPEF score 1–2)
Chronic stimulation of angiotensin II (ANG II) receptor one through an osmotic mini-pump has been a reliable model to induce HF. Although the relevance of supraphysiologic levels of ANGII in human diseases has been questioned, studies using this model have revealed some unexpected functions of ANGII in promoting HF. Notably, ANGII-induced left ventricular hypertrophy and remodeling occurred both in the presence (Ichihara et al., 2001) or absence (Matsumoto et al., 2013) of hypertension, suggesting that hypertension may not be the primary mechanism by which ANGII induces cardiac remodeling. Supporting the involvement of hypertension-independent factors, ANGII-induced cardiac hypertrophy is strain dependent. Whereas C57BL/6 mice develop compensatory concentric hypertrophy with diastolic dysfunction and fibrosis, BALB/c mice show severe LV dilation and systolic dysfunction (Peng et al., 2011), which is uncommon in patients with HFpEF and more resembles dilated cardiomyopathy. Nonetheless, pulmonary congestion developed in both strains (Peng et al., 2011), as a likely consequence of cardiac dysfunction but pulmonary hypertension has not been reported in this model. Exercise intolerance is evident and appears to be secondary to ANG II-induced skeletal muscle atrophy and mitochondrial dysfunction (Kadoguchi et al., 2015). Thus, the ANGII-infusion model recapitulates some aspects of human HFpEF, but other critical risk factors and comorbidities such as old age and obesity are neglected.
2.1.2 Aldosterone and salt infusion after unilateral nephrectomy (H2FPEF score 2)
Initially described in rats, the combination of aldosterone infusion, unilateral nephrectomy and 1% sodium chloride (NaCl) loading induces blood pressure elevation, concentric LV hypertrophy with diastolic dysfunction and pulmonary congestion in both C57BL/6 and FB/N mice (Tanaka et al., 2016; Valero-Munoz et al., 2016), although pulmonary hypertension was not assessed. These mice also exhibited exercise impairment (Wilson et al., 2009).Notably, the circulating aldosterone levels (∼6.5 ng/ml) in this model (Tanaka et al., 2014) appear to be within the same order of magnitude as human patients with acute HF (∼18 ng/ml) (Girerd et al., 2013). However, the effect of aging and obesity could not be assessed in this model.
2.1.3 Deoxycorticosterone acetate (DOCA)-salt administration (H2FPEF score 2)
This model was originally developed in 1969 to study hypertension in rats (Willard, 1969) and involves administration of DOCA for 4 weeks by intraperitoneal injection or subcutaneous implant, followed by unilateral nephrectomy and salt loading with 1% NaCl in drinking water. While DOCA salt-treated mice develop cardiac hypertrophy and fibrosis (Lovelock et al., 2012; Jeong et al., 2013), blood pressure appears to be either unaffected or only mildly elevated (Mohammed et al., 2010; Lovelock et al., 2012), consistent with the idea that mineralocorticoid may promote LV hypertrophy independent of increased afterload. Nevertheless, diastolic dysfunction and fibrosis can be markedly exacerbated with additional pressure overload via transverse-aortic constriction (TAC) (Mohammed et al., 2010), suggesting that mineralocorticoid and elevated afterload may synergistically contribute to the pathophysiology. Furthermore, reduction of nitric oxide production has also been demonstrated as a pathogenic mechanism in this model (Silberman et al., 2010). Exercise intolerance is evident in DOCA-salt mice after TAC (Methawasin et al., 2016). Pulmonary congestion, however, is not seen in DOCA-salt-treated mice and the effect of aging and obesity could not be assessed either.
2.1.4 Dahl salt-sensitive rats (H2FPEF score 4)
This strain of salt-sensitive Sprague-Dawley rats is only genetically distinguishable from the salt-resistant Dahl rat strain by a polymorphism in the renin gene (Rapp et al., 1989). When placed on high salt diet, Dahl salt-sensitive rats develop marked hypertension, concentric LV hypertrophy with diastolic dysfunction, exercise intolerance (Guazzi et al., 2001) and postmortem evidence of pulmonary congestion (Doi et al., 2000; Omori et al., 2012), but pulmonary hypertension has not been reported in this model. Notably, continued high salt diet results in transition to LV systolic dysfunction and HFrEF at old age (Doi et al., 2000), a phenotype infrequently observed in human HFpEF patients. Additionally, the clinical applicability of this model is further limited as markedly elevated (>170 mmHg) systolic blood pressure often develops (Doi et al., 2000) compared to moderate hypertension seen in HFpEF patients.
2.1.5 Spontaneous hypertensive rats (H2FPEF score 4)
Originally used to model essential hypertension in human, this inbred strain of rats develops concentric LV hypertrophy with preserved systolic function (Pfeffer et al., 1979; Heyen et al., 2002) and impaired exercise capacity (Campos et al., 2020) in the first year of age. However, between 18 and 24 months of age, LVEF starts to decrease in some colonies with signs of diastolic dysfunction and pulmonary congestion (Pfeffer et al., 1979; Heyen et al., 2002) with evidence of pulmonary hypertension (Aharinejad et al., 1996). Thus, although the lengthy duration required for HF development may mimic some aspects of human HFpEF which is prevalent in the aging population, the transition to HFrEF (which is rarely observed in HFpEF patients) significantly reduces the clinical applicability of the spontaneous hypertensive rats (SHR) model.
2.2 Aging
According to the Framingham Heart Study and Baltimore Longitudinal Study on Aging, aging positively correlates with prevalence of LV hypertrophy, even in the absence of hypertension. Furthermore, diastolic function worsens with age, likely due to increased stiffness of myocardium while the LV systolic function remains intact at rest (Upadhya and Kitzman, 2017). Additionally, endothelial dysfunction may also lead to vascular-ventricular uncoupling and afterload mismatch, collectively implying a causal role of aging in HFpEF pathogenesis. The short life span of rodents makes them more cost-effective to study than long-lived large animals. Below, we summarize the strengths and weaknesses of various aging rodent models with respect to their clinical applicability in modeling human HFpEF.
2.2.1 Aged mice (H2FPEF score 2)
Natural aging in mice is associated with the development of maladaptive cardiac changes, as is observed in humans. Starting at an age of 24 months, C57BL/6 mice display several of the HFpEF pathologies, including LV hypertrophy, diastolic dysfunction (Dai et al., 2009; Roh et al., 2019), pulmonary congestion and exercise intolerance (Roh et al., 2020). However, pulmonary hypertension has not been reported. Interestingly, blood pressure is not elevated in this model (Roh et al., 2020), supporting the notion that aging may be an independent risk factor for HFpEF. Notably, these phenotypes of cardiac aging can be partially reversed with caloric restriction, rapamycin treatment and exercise (Dai et al., 2014; Roh et al., 2020). Other comorbidities such as obesity have not been described in this model.
2.2.2 Accelerated senescence model (SAMP, H2FPEF score 2)
The senescence accelerated prone (SAMP) strain was generated by selective inbreeding of AKR/J mice (Takeda et al., 1997). Starting at the age of 10 months, these mice begin to display accelerated age-associated phenotypes seen in aging disorders in humans (Karuppagounder et al., 2017). Mutations in the DNA repair gene and alteration in oxidative stress response appear to be the underlying mechanism of accelerated senescence (Tanisawa et al., 2013). Male SAMP8 mice develop LV hypertrophy with diastolic dysfunction as early as 6 months of age (Reed et al., 2011). Intriguingly, female mice retain normal diastolic function compared to the control senescence-resistant strain (Gevaert et al., 2017), a pattern contrary to the female-skewed HFpEF prevalence in human (Scantlebury and Borlaug, 2011). However, when fed with a high fat and high salt diet, female SAMP8 mice also display features of HFpEF with hypertension and pulmonary edema (Gevaert et al., 2017), suggesting a potential synergy between metabolic disturbance and female sex in driving HFpEF pathology. Pulmonary hyptertension and exercise intolerance have not been reported in these mice. Obesity has not been reported in this model even with high fat diet. Thus, untreated SAMP8 mice may represent a better model for studying diastolic dysfunction than HFpEF.
2.2.3 Fischer 344 rats (H2FPEF score 3)
The aged Fischer 344 rats develop pathophysiology of aging in various organs, including the heart (Horgan et al., 2014). Starting at 22 months of age, Fischer 344 rats begin to develop progressive LV diastolic function (Walker et al., 2006) with evident pulmonary congestion and exercise intolerance at 25 months of age (Choi et al., 2009). While severe pulmonary hypertension is evident (Suen et al., 2019) by 28 months of age, these animals also develop LV systolic dysfunction and chamber dilation, which is infrequently seen in human HFpEF (Walker et al., 2006), thus limiting the use of Fischer 344 rats as an HFpEF model.
2.3 Obesity and diabetes
Being overweight has been identified as a major risk factor for HFpEF in large outcome trials and registries (Haass et al., 2011; Shah et al., 2016). Indeed, obesity induces LV hypertrophy (Turkbey et al., 2010) and is associated with worsened diastolic function (Bartkowiak et al., 2021). Although increased adiposity appears to promote inflammation, endothelial dysfunction and insulin resistance, all of which are frequently seen in HFpEF patients, the precise molecular mechanism by which obesity leads to HFpEF remains unclear. Our understanding of the role obesity plays in HFpEF pathogenesis is further complicated by the paradoxical observation that among symptomatic HFpEF patients, mortality in fact gradually increases with both low and high BMI (Kenchaiah et al., 2007), with the nadir of risk for all-cause mortality at a BMI of 32–33 kg/m2 (Haass et al., 2011; Zhang et al., 2019). Conceivably, unintentional weight loss, reflecting progressive HF, may explain the increased mortality in patients with low BMI (Huang et al., 2021). In contrast, intentional weight loss, such as bariatric surgery, has been shown to decrease cardiac hypertrophy and LV filling pressure and to improve diastolic function, implying an active role of obesity in propagating HFpEF (Ikonomidis et al., 2007). Thus, these data suggest that obesity may play a causal and a less clearly defined perpetuating role in HFpEF pathogenesis.
Diabetes is also frequently seen in HFpEF patients and is associated with increased all-cause mortality (MacDonald et al., 2008; Aguilar et al., 2010; Kristensen et al., 2017). Various mechanisms of how diabetes promotes HFpEF have been proposed, ranging from systemic effects, such as neurohormonal, sympathetic and cytokine imbalance to cardiac-specific effects, including interstitial fibrosis, cardiomyocyte hypertrophy secondary to titin dysfunction (Boudina and Abel, 2007; von Bibra et al., 2016). Although diabetes appears to have a well-established role in HFpEF development, less is known regarding its impact on established HFpEF. Notably, while metformin has been reported to improve HFpEF outcomes (Wang et al., 2021), none of the other anti-diabetes medications have been shown to confer survival benefits (Mgbemena et al., 2021) except for SGLT2 inhibitors (whose cardioprotective effect is likely independent of its anti-glycemic function(Anker et al., 2021; Ebell, 2022)). Thus, these observations support diabetes as a risk factor for HFpEF development but challenge the notion that diabetes continues to play an active role in established HFpEF.
Although murine models of obesity and diabetes-induced HFpEF closely resemble human phenotypes, they share several limitations. First, these pre-clinical models often present with fulminant hyperglycemia and insulin-resistance at young age, which contrasts with the gradual disease progression seen in human. Additionally, the early onset of morbid obesity and severe hyperglyemia does not allow studies exploring the effect of mild obesity in established HFpEF, which represents most patient scenarios. Lastly, most murine models of obesity also inevitably develop diabetes and thus limit the possibility to investigate the exclusive role of obesity in HFpEF pathogenesis. Below, we highlight the strengths and weaknesses of each obesity/diabetes-induced HFpEF murine model.
2.3.1 High fat/western diet (H2FPEF score 2–3)
Obesity is one the most significant comorbidities in HFpEF patients and has been implicated in driving HFpEF (Sorimachi et al., 2021; Withaar et al., 2021a). In pre-clinical models, unhealthy food consumption is most commonly modeled by a high fat diet with >60% of daily caloric intake derived from fat, or a western diet consisted of daily intake of 36% fat and 36% sucrose. Animals fed with either diet consistently develop obesity with glucose intolerance at a young age. These animals frequently display cardiac phenotypes, including hypertrophy, fibrosis and diastolic dysfunction (Christopher et al., 2010; Leopoldo et al., 2010; Manrique et al., 2013; Carbone et al., 2015; Huang et al., 2016), but may also develop systolic dysfunction and reduced LVEF (Cheng et al., 2011). Pulmonary congestion and exercise intolerance have not been described in these models. Notably, some groups report no cardiac phenotypes in mice fed with high-fat diet (Brainard et al., 2013). The discrepancy between these studies may be attributed to the difference in study duration, caloric intake and the exact composition of the diet. Thus, when fed with select diets, the diet-induced obesity model may approximate human HFpEF.
2.3.2 ob/ob mice (H2FPEF score 3–4)
The leptin-deficient ob/ob mice develop obesity and type II diabetes secondary to hyperphagia, hyperglycemia and insulin resistance (Ingalls et al., 1950; Lindstrom, 2007). ob/ob mice display rapid weight gain within 2 weeks after birth and by 4 weeks of age, they develop marked hyperglycemia (Westman, 1968; Lindstrom, 2007). Ensuing cardiac dysfunction is characterized by LV hypertrophy with diastolic dysfunction (Christoffersen et al., 2003) and cardiac fibrosis (Manolescu et al., 2014). Notably, systolic function remains unchanged (Christoffersen et al., 2003). Hypertension has not been described. Although these mice develop exercise intolerance, this is likely attributed to excess body weight (Pelleymounter et al., 1995) rather than cardiac dysfunction as ob/ob mice do not display other overt HF symptoms such as pulmonary congestion before their premature death. While the ob/ob model appears to phenotypically recapitulate several aspects of human HFpEF, their clinical applicability is limited in two ways. First, obese patients with leptin mutations are rare. Second, the cardiomyocyte dysfunction in ob/ob mice is partially attributed to the lack of leptin signaling in cardiomyocytes, as administration of leptin to cardiomyocytes ex-vivo rescues their pro-apoptotic phenotype (Trivedi et al., 2008) and restores their contractility (Dong et al., 2006).
2.3.3 db/db mice (H2FPEF score 5)
The leptin receptor-deficient, or diabetic (db/db) mice harbor a point mutation in the leptin receptor gene, altering its sensitivity to leptin(Chen et al., 1996). These mice spontaneously develop morbid obesity accompanied by severe hyperglycemia secondary to type II diabetes (Chen et al., 1996). Although db/db mice display insulin resistance at younger age compared to humans, the late-stage manifestation of insulin secretion defect well approximates the pathogenesis of type II diabetes in human, making db/db mice a valuable model in exploring the combined contribution of obesity and type II diabetes to HFpEF. Starting at 16 weeks of age, db/db mice begin to develop LV hypertrophy (Barouch et al., 2003; Van den Bergh et al., 2006) with histological evidence of cardiomyocyte hypertrophy (Barouch et al., 2003) and fibrosis (Plante et al., 2014). Notably, there is diastolic dysfunction and pulmonary congestion with aging (Papinska et al., 2016), paralleled by severely reduced exercise capacity by 12 weeks (Ostler et al., 2014), while LV systolic function remains preserved (Mori et al., 2014). Spontaneous hypertension has also been reported (Habibi et al., 2017; Sukumaran et al., 2017). Pulmonary hypertension has not been described in this model. Interestingly, the brain natriuretic peptide levels are reduced, similar to obese HF patients where circulating brain natriuretic peptide levels remain non-elevated given the degree of HF (Wang et al., 2004). Together, these features make the db/db mice an attractive model for studying pathogenic mechanisms of HFpEF. One major deficiency of this model is that obesity, type II diabetes and the ensuing cardiac phenotypes develop rather early in life compared to human patients.
2.3.4 Zucker fatty and spontaneously hypertensive heart failure rats (H2FPEF score 5)
The Zucker fatty and spontaneously hypertensive heart failure F1 hybrid (ZSF1) was developed from crossing rat strains with two different leptin receptor mutations, of which the male was derived from the SHR background. Between 10 and 20 weeks of age, ZSF1 rats develop HFpEF characterized by hypertension, concentric LV hypertrophy, diastolic dysfunction, pulmonary congestion, pulmonary hypertension (Morales-Cano et al., 2019) and exercise intolerance (Schauer et al., 2020) as well as obesity and insulin resistance (Griffin et al., 2007; Mohanan et al., 2011; van Dijk et al., 2016). In addition to cardiac phenotypes, the ZSF1 rats also develop chronic kidney disease (Griffin et al., 2007), which is also frequently seen in HFpEF patients. While the renal involvement makes it challenging to assess the contribution of obesity and metabolic disturbances per se to HFpEF pathogenesis, ZSF1 nevertheless represents an attractive model of HFpEF.
3 Multi-hit models
The models described above are mostly unifactorial disease models that predominantly rely on one perturbation to induce HFpEF. Over the past few years, multifactorial pre-clinical models that consider two or more perturbations to mimic human HFpEF have been developed. Below we evaluate the strengths and weaknesses of each multi-hit model, again using the H2FPEF scoring system.
3.1 High fat diet and L-NAME (H2FPEF score 4)
In a recent report, Schiattarella et al. presented the first two-hit mouse model that resembles human HFpEF (Schiattarella et al., 2019). Briefly, C57BL/6N male mice were fed a high-fat diet (60% of daily caloric intake from fat) and a constitutive nitric oxide synthase inhibitor, L-NAME for 15 weeks. The combination of treatments results in LV hypertrophy with diastolic dysfunction, pulmonary congestion, exercise intolerance, hypertension and obesity. However, pulmonary hypertension was not reported. Importantly, high-fat diet or L-NAME alone was insufficient to induce such phenotypes. Mechanistically, the combination treatment appears to promote the expression of inducible nitric oxide synthase (iNOS) in cardiomyocytes, which in turn alters the misfolded protein response cascade in a cardiomyocyte-intrinsic manner. Notably, genetic or pharmacological inhibition of iNOS ameliorates cardiac phenotypes without reducing hyperglycemia or body weight. These findings seemingly contradict recent randomized trials showing an improvement in exercise tolerance in HFpEF patients following administration of inorganic nitric oxide (Borlaug et al., 2016; Zamani et al., 2017; Borlaug et al., 2018). However, these clinical studies have small sample sizes with skewed patient ethnicity. Furthermore, the rather immediate improvement following nitric oxide treatment likely results from transient afterload reduction secondary to vasodilation rather than reversal of cardiac remodeling. Overall, the two-hit mouse model well approximates human HFpEF and provides a unique perspective on its pathogenesis.
3.2 High fat diet, aging and ANG II infusion (H2FPEF score 5)
Recently, Withaar et al. developed a multifactorial HFpEF mouse model, which involves aging C57BL/6J female mice to 18–24 months of age, followed by 12 weeks of high fat diet and 4 weeks of ANG II infusion through an osmotic mini pump (Withaar et al., 2021b). These mice subsequently develop many HFpEF-like phenotypes, including concentric LV hypertrophy with diastolic dysfunction, cardiac fibrosis, reduced exercise tolerance and pulmonary congestion, although pulmonary hypertension has not been assessed. The LV systolic function is preserved. Notably, in contrast to recent findings in the EMPEROR-Preserved trial (Anker et al., 2021), an SGLT2 inhibitor, dapagliflozin, did not show significant cardioprotective effects in this triple-hit HFpEF mouse model (Withaar et al., 2021b). Rather, a GLP-1 agonist, liraglutide, markedly improves cardiac function, despite similarly effective glycemic control achieved by both medications. Conceivably, these mice may model a subgroup of HFpEF patients who may benefit more from a GLP-1 agonist than a SGLT2 inhibitor. Alternatively, dapagliflozin may not be therapeutically identical to empagliflozin, which may be addressed by the ongoing DELIVER trial (Solomon et al., 2022). Another drawback of the study by Withaar et al. is the exclusive use of female mice. Although HFpEF phenotype is indeed more prevalent in aged female population, similar sets of risk factors and comorbidities are present in males. Future studies are necessary to address sex-specific effects in this model.
3.3 High fat diet, aging and DOC (H2FPEF score 5)
A recent study by Deng et al. utilized a combinatorial model, which involves feeding C57BL/6J mice high fat diet from 3 to 16 months of age followed by intraperitoneal injection of DOC during the last month (Deng et al., 2021). This model encompasses three most significant risk factors and comorbidities in HFpEF: hypertension, obesity and aging. Treated mice develop LV hypertrophy with diastolic dysfunction, fibrosis and exercise intolerance and pulmonary edema, although pulmonary hypertension was not formally evaluated. Notably, treatment with empagliflozin improves diastolic relaxation, pulmonary edema and exercise tolerance, but intriguingly did not result in weight reduction. Although both sexes are included, further sub-group analyses were not performed.
4 Concluding remarks
In this review, we assessed the clinical applicability and translational value of various murine HFpEF models. While the H2FPEF scoring system defines the most clinically relevant features that HFpEF animal models should manifest, whether these comorbidities are mechanistically relevant to HFpEF pathogenesis remains unclear. Although treating comorbidities has not materially altered outcome, it remains possible that these comorbidities contribute to the development of HFpEF rather than propagation of disease. Thus, modeling HFpEF comorbidities represents a reasonable first step in understanding the precise pathogenesis of the disease.
It is evident that multifactorial models appear to better approximate human HFpEF than those with a single perturbation, making them attractive tools for future HFpEF research. However, we also note that some multi-hit models differ from humans with regards to their response to therapy. Therefore, we advocate that future HFpEF pre-clinical studies designed to test the efficacy of therapeutic agents should consider using multiple models. Furthermore, it remains unclear if the HFpEF phenotypes observed in high scoring multifactorial models are strain-specific, as is seen in several single-hit models. In addition to validating these experimental findings in different strains of mice, we also propose further exploration of multi-hit models in outbred colonies as they may more faithfully reflect the heterogeneous phenotypes and responses to therapy that are often seen in a diverse human population. Because HFpEF-associated comorbid conditions are not present in equal severity in all HFpEF patients, uni-factor models may in fact approximate subsets of HFpEF patients. In addition, these pre-clinical models allow examination of contribution of each risk factor in isolation without additional comorbidities confounding the findings. Thus, although single stressor models generally have lower H2FPEF scores, they should not be disregarded as artifactual.
As the highest scoring parameter in the H2FPEF algorithm, atrial fibrillation is highly associated with increased risk of HF (Packer et al., 2020). Mechanistically, atrial fibrillation in HFpEF patients developed from left atrial remodeling induced by a pro-inflammatory state secondary to metabolic disorders and hypertension (Kuo et al., 2022). Yet, none of the HFpEF murine models, whether encompassing single or multiple factors, showed atrial fibrillation. The discrepancy is likely due to the intrinsic resistance to arrhythmia in mice as they lack the critical cardiac mass (Byrd et al., 2005; Qu, 2006), highlighting just one of the many major differences between rodents and humans. Anatomical discrepancies between the organisms often complicate imaging, electrophysiological studies and surgical interventions. Physiological disparities, such as markedly higher heart rate in rodents and correlating action potentials, changes in metabolism, as well as difference in oxygen and calcium usage may in fact represent an unsurmountable obstacle to accurately modeling human HFpEF. Thus, a “perfect” murine HFpEF model that can generate immediately translatable findings may not exist. Rather, we envision the “imperfect” murine models to instead serve as screening platforms for novel targetable molecules or pathways, the validation of which should ultimately be carried out in large mammal models with physiology closer to that of humans.
Author contributions
MC conceived of the article and did background literature review. CC did further literature review and wrote the initial draft of the manuscript. Both authors reviewed and edited the final version.
Funding
This work was supported by the National Heart, Lung and Blood Institute, National Institutes of Health, Award Number 1R61HL154137-01A1 to MC.
Acknowledgments
We thank all members of the Chin lab for valuable discussion and critical reading of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Aguilar, D., Deswal, A., Ramasubbu, K., Mann, D. L., and Bozkurt, B. (2010). Comparison of patients with heart failure and preserved left ventricular ejection fraction among those with versus without diabetes mellitus. Am. J. Cardiol. 105, 373–377. doi:10.1016/j.amjcard.2009.09.041
Aharinejad, S., Schraufnagel, D. E., Bock, P., Mackay, C. A., Larson, E. K., Miksovsky, A., et al. (1996). Spontaneously hypertensive rats develop pulmonary hypertension and hypertrophy of pulmonary venous sphincters. Am. J. Pathol. 148, 281–290.
Anker, S. D., Butler, J., Filippatos, G., Ferreira, J. P., Bocchi, E., Bohm, M., et al. (2021). Empagliflozin in heart failure with a preserved ejection fraction. N. Engl. J. Med. 385, 1451–1461. doi:10.1056/NEJMoa2107038
Barouch, L. A., Berkowitz, D. E., Harrison, R. W., O'donnell, C. P., and Hare, J. M. (2003). Disruption of leptin signaling contributes to cardiac hypertrophy independently of body weight in mice. Circulation 108, 754–759. doi:10.1161/01.CIR.0000083716.82622.FD
Bartkowiak, J., Spitzer, E., Kurmann, R., Zurcher, F., Krahenmann, P., Garcia-Ruiz, V., et al. (2021). The impact of obesity on left ventricular hypertrophy and diastolic dysfunction in children and adolescents. Sci. Rep. 11, 13022. doi:10.1038/s41598-021-92463-x
Borlaug, B. A., Melenovsky, V., and Koepp, K. E. (2016). Inhaled sodium nitrite improves rest and exercise hemodynamics in heart failure with preserved ejection fraction. Circ. Res. 119, 880–886. doi:10.1161/CIRCRESAHA.116.309184
Borlaug, B. A., Anstrom, K. J., Lewis, G. D., Shah, S. J., Levine, J. A., Koepp, G. A., et al. (2018). National heart, L., and blood institute heart failure clinical research, NEffect of inorganic nitrite vs placebo on exercise capacity among patients with heart failure with preserved ejection fraction: The INDIE-HFpEF randomized clinical trial. JAMA 320, 1764–1773. doi:10.1001/jama.2018.14852
Borlaug, B. A. (2016). Defining HFpEF: where do we draw the line? Eur. Heart J. 37, 463–465. doi:10.1093/eurheartj/ehv561
Borlaug, B. A. (2020). Evaluation and management of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 17, 559–573. doi:10.1038/s41569-020-0363-2
Boudina, S., and Abel, E. D. (2007). Diabetic cardiomyopathy revisited. Circulation 115, 3213–3223. doi:10.1161/CIRCULATIONAHA.106.679597
Brainard, R. E., Watson, L. J., Demartino, A. M., Brittian, K. R., Readnower, R. D., Boakye, A. A., et al. (2013). High fat feeding in mice is insufficient to induce cardiac dysfunction and does not exacerbate heart failure. PLoS One 8, e83174. doi:10.1371/journal.pone.0083174
Byrd, G. D., Prasad, S. M., Ripplinger, C. M., Cassilly, T. R., Schuessler, R. B., Boineau, J. P., et al. (2005). Importance of geometry and refractory period in sustaining atrial fibrillation: testing the critical mass hypothesis. Circulation 112, I7–I13. doi:10.1161/CIRCULATIONAHA.104.526210
Campos, H. O., Drummond, L. R., Rodrigues, Q. T., Lima, P. M., Primola-Gomes, T. N., and Coimbra, C. C. (2020). Exercise capacity in different stages of hypertension in spontaneously hypertensive rats. J. Sports Med. Phys. Fit. 60, 800–805. doi:10.23736/S0022-4707.20.10369-4
Carbone, S., Mauro, A. G., Mezzaroma, E., Kraskauskas, D., Marchetti, C., Buzzetti, R., et al. (2015). A high-sugar and high-fat diet impairs cardiac systolic and diastolic function in mice. Int. J. Cardiol. 198, 66–69. doi:10.1016/j.ijcard.2015.06.136
Chen, H., Charlat, O., Tartaglia, L. A., Woolf, E. A., Weng, X., Ellis, S. J., et al. (1996). Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell 84, 491–495. doi:10.1016/s0092-8674(00)81294-5
Cheng, Y., Li, W., Mcelfresh, T. A., Chen, X., Berthiaume, J. M., Castel, L., et al. (2011). Changes in myofilament proteins, but not Ca(2)(+) regulation, are associated with a high-fat diet-induced improvement in contractile function in heart failure. Am. J. Physiol. Heart Circ. Physiol. 301, H1438–H1446. doi:10.1152/ajpheart.00440.2011
Choi, S. Y., Chang, H. J., Choi, S. I., Kim, K. I., Cho, Y. S., Youn, T. J., et al. (2009). Long-term exercise training attenuates age-related diastolic dysfunction: association of myocardial collagen cross-linking. J. Korean Med. Sci. 24, 32–39. doi:10.3346/jkms.2009.24.1.32
Christoffersen, C., Bollano, E., Lindegaard, M. L., Bartels, E. D., Goetze, J. P., Andersen, C. B., et al. (2003). Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology 144, 3483–3490. doi:10.1210/en.2003-0242
Christopher, B. A., Huang, H. M., Berthiaume, J. M., Mcelfresh, T. A., Chen, X., Croniger, C. M., et al. (2010). Myocardial insulin resistance induced by high fat feeding in heart failure is associated with preserved contractile function. Am. J. Physiol. Heart Circ. Physiol. 299, H1917–H1927. doi:10.1152/ajpheart.00687.2010
Churchill, T. W., Li, S. X., Curreri, L., Zern, E. K., Lau, E. S., Liu, E. E., et al. (2021). Evaluation of 2 existing diagnostic scores for heart failure with preserved ejection fraction against a comprehensively phenotyped cohort. Circulation 143, 289–291. doi:10.1161/CIRCULATIONAHA.120.050757
Dai, D. F., Santana, L. F., Vermulst, M., Tomazela, D. M., Emond, M. J., Maccoss, M. J., et al. (2009). Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 119, 2789–2797. doi:10.1161/CIRCULATIONAHA.108.822403
Dai, D. F., Karunadharma, P. P., Chiao, Y. A., Basisty, N., Crispin, D., Hsieh, E. J., et al. (2014). Altered proteome turnover and remodeling by short-term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell 13, 529–539. doi:10.1111/acel.12203
Deng, Y., Xie, M., Li, Q., Xu, X., Ou, W., Zhang, Y., et al. (2021). Targeting mitochondria-inflammation circuit by beta-hydroxybutyrate mitigates HFpEF. Circ. Res. 128, 232–245. doi:10.1161/CIRCRESAHA.120.317933
Doi, R., Masuyama, T., Yamamoto, K., Doi, Y., Mano, T., Sakata, Y., et al. (2000). Development of different phenotypes of hypertensive heart failure: systolic versus diastolic failure in Dahl salt-sensitive rats. J. Hypertens. 18, 111–120. doi:10.1097/00004872-200018010-00016
Dong, F., Zhang, X., Yang, X., Esberg, L. B., Yang, H., Zhang, Z., et al. (2006). Impaired cardiac contractile function in ventricular myocytes from leptin-deficient ob/ob obese mice. J. Endocrinol. 188, 25–36. doi:10.1677/joe.1.06241
Eaton, C. B., Pettinger, M., Rossouw, J., Martin, L. W., Foraker, R., Quddus, A., et al. (2016). Risk factors for incident hospitalized heart failure with preserved versus reduced ejection fraction in a multiracial cohort of postmenopausal women. Circ. Heart Fail. 9 (10), e002883. doi:10.1161/CIRCHEARTFAILURE.115.002883
Ebell, M. H. (2022). Empagliflozin reduces hospitalization for heart failure with preserved ejection fraction, but not mortality outcomes. Am. Fam. Physician. 105. Online.
Faxen, U. L., Venkateshvaran, A., Shah, S. J., Lam, C. S. P., Svedlund, S., Saraste, A., et al. (2021). Generalizability of HFA-PEFF and H2FPEF diagnostic algorithms and associations with heart failure indices and proteomic biomarkers: Insights from PROMIS-HFpEF. J. Card. Fail. 27, 756–765. doi:10.1016/j.cardfail.2021.02.005
Ferrari, R., Bohm, M., Cleland, J. G., Paulus, W. J., Pieske, B., Rapezzi, C., et al. (2015). Heart failure with preserved ejection fraction: uncertainties and dilemmas. Eur. J. Heart Fail. 17, 665–671. doi:10.1002/ejhf.304
Gevaert, A. B., Shakeri, H., Leloup, A. J., Van Hove, C. E., De Meyer, G. R. Y., Vrints, C. J., et al. (2017). Endothelial senescence contributes to heart failure with preserved ejection fraction in an aging mouse model. Circ. Heart Fail. 10 (6), e003806. doi:10.1161/CIRCHEARTFAILURE.116.003806
Girerd, N., Pang, P. S., Swedberg, K., Fought, A., Kwasny, M. J., Subacius, H., et al. (2013). Serum aldosterone is associated with mortality and re-hospitalization in patients with reduced ejection fraction hospitalized for acute heart failure: analysis from the EVEREST trial. Eur. J. Heart Fail. 15, 1228–1235. doi:10.1093/eurjhf/hft100
Gok, G., Kilic, S., Sinan, U. Y., Turkoglu, E., Kemal, H., and Zoghi, M. (2020). Epidemiology and clinical characteristics of hospitalized elderly patients for heart failure with reduced, mid-range and preserved ejection fraction. Heart Lung 49, 495–500. doi:10.1016/j.hrtlng.2020.03.023
Griffin, K. A., Abu-Naser, M., Abu-Amarah, I., Picken, M., Williamson, G. A., and Bidani, A. K. (2007). Dynamic blood pressure load and nephropathy in the ZSF1 (fa/fa cp) model of type 2 diabetes. Am. J. Physiol. Ren. Physiol. 293, F1605–F1613. doi:10.1152/ajprenal.00511.2006
Guazzi, M., Brenner, D. A., Apstein, C. S., and Saupe, K. W. (2001). Exercise intolerance in rats with hypertensive heart disease is associated with impaired diastolic relaxation. Hypertension 37, 204–208. doi:10.1161/01.hyp.37.2.204
Haass, M., Kitzman, D. W., Anand, I. S., Miller, A., Zile, M. R., Massie, B. M., et al. (2011). Body mass index and adverse cardiovascular outcomes in heart failure patients with preserved ejection fraction: results from the irbesartan in heart failure with preserved ejection fraction (I-preserve) trial. Circ. Heart Fail. 4, 324–331. doi:10.1161/CIRCHEARTFAILURE.110.959890
Habibi, J., Aroor, A. R., Sowers, J. R., Jia, G., Hayden, M. R., Garro, M., et al. (2017). Sodium glucose transporter 2 (SGLT2) inhibition with empagliflozin improves cardiac diastolic function in a female rodent model of diabetes. Cardiovasc. Diabetol. 16, 9. doi:10.1186/s12933-016-0489-z
Heidenreich, P. A., Bozkurt, B., Aguilar, D., Allen, L. A., Byun, J. J., Colvin, M. M., et al. (2022). 2022 AHA/ACC/HFSA guideline for the management of heart failure: executive summary: a report of the American college of cardiology/american heart association joint committee on clinical practice guidelines. Circulation. 145 (18), e876–e894. doi:10.1161/CIR.0000000000001062
Heyen, J. R., Blasi, E. R., Nikula, K., Rocha, R., Daust, H. A., Frierdich, G., et al. (2002). Structural, functional, and molecular characterization of the SHHF model of heart failure. Am. J. Physiol. Heart Circ. Physiol. 283, H1775–H1784. doi:10.1152/ajpheart.00305.2002
Ho, J. E., Enserro, D., Brouwers, F. P., Kizer, J. R., Shah, S. J., Psaty, B. M., et al. (2016). Predicting heart failure with preserved and reduced ejection fraction: the international collaboration on heart failure subtypes. Circ. Heart Fail. 9, e003116. doi:10.1161/CIRCHEARTFAILURE.115.003116
Ho, J. E., Redfield, M. M., Lewis, G. D., Paulus, W. J., and Lam, C. S. P. (2020). Deliberating the diagnostic dilemma of heart failure with preserved ejection fraction. Circulation 142, 1770–1780. doi:10.1161/CIRCULATIONAHA.119.041818
Horgan, S., Watson, C., Glezeva, N., and Baugh, J. (2014). Murine models of diastolic dysfunction and heart failure with preserved ejection fraction. J. Card. Fail. 20, 984–995. doi:10.1016/j.cardfail.2014.09.001
Huang, J. P., Cheng, M. L., Wang, C. H., Shiao, M. S., Chen, J. K., and Hung, L. M. (2016). High-fructose and high-fat feeding correspondingly lead to the development of lysoPC-associated apoptotic cardiomyopathy and adrenergic signaling-related cardiac hypertrophy. Int. J. Cardiol. 215, 65–76. doi:10.1016/j.ijcard.2016.03.239
Huang, P., Guo, Z., Liang, W., Wu, Y., Zhao, J., He, X., et al. (2021). Weight change and mortality risk in heart failure with preserved ejection fraction. Front. Cardiovasc. Med. 8, 681726. doi:10.3389/fcvm.2021.681726
Ichihara, S., Senbonmatsu, T., Price, E., Ichiki, T., Gaffney, F. A., and Inagami, T. (2001). Angiotensin II type 2 receptor is essential for left ventricular hypertrophy and cardiac fibrosis in chronic angiotensin II-induced hypertension. Circulation 104, 346–351. doi:10.1161/01.cir.104.3.346
Ikonomidis, I., Mazarakis, A., Papadopoulos, C., Patsouras, N., Kalfarentzos, F., Lekakis, J., et al. (2007). Weight loss after bariatric surgery improves aortic elastic properties and left ventricular function in individuals with morbid obesity: a 3-year follow-up study. J. Hypertens. 25, 439–447. doi:10.1097/HJH.0b013e3280115bfb
Ingalls, A. M., Dickie, M. M., and Snell, G. D. (1950). Obese, a new mutation in the house mouse. J. Hered. 41, 317–318. doi:10.1093/oxfordjournals.jhered.a106073
Jeong, E. M., Monasky, M. M., Gu, L., Taglieri, D. M., Patel, B. G., Liu, H., et al. (2013). Tetrahydrobiopterin improves diastolic dysfunction by reversing changes in myofilament properties. J. Mol. Cell. Cardiol. 56, 44–54. doi:10.1016/j.yjmcc.2012.12.003
Kadoguchi, T., Kinugawa, S., Takada, S., Fukushima, A., Furihata, T., Homma, T., et al. (2015). Angiotensin II can directly induce mitochondrial dysfunction, decrease oxidative fibre number and induce atrophy in mouse hindlimb skeletal muscle. Exp. Physiol. 100, 312–322. doi:10.1113/expphysiol.2014.084095
Karuppagounder, V., Arumugam, S., Babu, S. S., Palaniyandi, S. S., Watanabe, K., Cooke, J. P., et al. (2017). The senescence accelerated mouse prone 8 (SAMP8): a novel murine model for cardiac aging. Ageing Res. Rev. 35, 291–296. doi:10.1016/j.arr.2016.10.006
Kenchaiah, S., Pocock, S. J., Wang, D., Finn, P. V., Zornoff, L. A., Skali, H., et al. (2007). Body mass index and prognosis in patients with chronic heart failure: insights from the candesartan in heart failure: assessment of reduction in mortality and morbidity (CHARM) program. Circulation 116, 627–636. doi:10.1161/CIRCULATIONAHA.106.679779
Khan, M. S., Fonarow, G. C., Khan, H., Greene, S. J., Anker, S. D., Gheorghiade, M., et al. (2017). Renin-angiotensin blockade in heart failure with preserved ejection fraction: a systematic review and meta-analysis. ESC Heart Fail. 4, 402–408. doi:10.1002/ehf2.12204
Klainer, L. M., Gibson, T. C., and White, K. L. (1965). The epidemiology of cardiac failure. J. Chronic Dis. 18, 797–814. doi:10.1016/0021-9681(65)90019-6
Komajda, M., Isnard, R., Cohen-Solal, A., Metra, M., Pieske, B., Ponikowski, P., et al. (2017). Effect of ivabradine in patients with heart failure with preserved ejection fraction: the EDIFY randomized placebo-controlled trial. Eur. J. Heart Fail. 19, 1495–1503. doi:10.1002/ejhf.876
Kristensen, S. L., Mogensen, U. M., Jhund, P. S., Petrie, M. C., Preiss, D., Win, S., et al. (2017). Clinical and echocardiographic characteristics and cardiovascular outcomes according to diabetes status in patients with heart failure and preserved ejection fraction: A report from the I-preserve trial (irbesartan in heart failure with preserved ejection fraction). Circulation 135, 724–735. doi:10.1161/CIRCULATIONAHA.116.024593
Kuo, J. Y., Jin, X., Sun, J. Y., Chang, S. H., Chi, P. C., Sung, K. T., et al. (2022). Insights on distinct left atrial remodeling between atrial fibrillation and heart failure with preserved ejection fraction. Front. Cardiovasc. Med. 9, 857360. doi:10.3389/fcvm.2022.857360
Lejeune, S., Roy, C., Slimani, A., Pasquet, A., Vancraeynest, D., Vanoverschelde, J. L., et al. (2021). Diabetic phenotype and prognosis of patients with heart failure and preserved ejection fraction in a real life cohort. Cardiovasc. Diabetol. 20, 48. doi:10.1186/s12933-021-01242-5
Leopoldo, A. S., Sugizaki, M. M., Lima-Leopoldo, A. P., Do Nascimento, A. F., Luvizotto Rde, A., De Campos, D. H., et al. (2010). Cardiac remodeling in a rat model of diet-induced obesity. Can. J. Cardiol. 26, 423–429. doi:10.1016/s0828-282x(10)70440-2
Lin, Y., Zhong, X., Liu, M., Zhang, S., Xiong, Z., Huang, Y., et al. (2022). Risk stratification and efficacy of spironolactone in patients with heart failure with preserved ejection fraction: secondary analysis of the TOPCAT randomized clinical trial. Cardiovasc. Drugs Ther. 36, 323–331. doi:10.1007/s10557-021-07178-y
Lindstrom, P. (2007). The physiology of obese-hyperglycemic mice [ob/ob mice]. ScientificWorldJournal. 7, 666–685. doi:10.1100/tsw.2007.117
Lovelock, J. D., Monasky, M. M., Jeong, E. M., Lardin, H. A., Liu, H., Patel, B. G., et al. (2012). Ranolazine improves cardiac diastolic dysfunction through modulation of myofilament calcium sensitivity. Circ. Res. 110, 841–850. doi:10.1161/CIRCRESAHA.111.258251
MacDonald, M. R., Petrie, M. C., Varyani, F., Ostergren, J., Michelson, E. L., Young, J. B., et al. (2008). Impact of diabetes on outcomes in patients with low and preserved ejection fraction heart failure: an analysis of the candesartan in heart failure: assessment of reduction in mortality and morbidity (CHARM) programme. Eur. Heart J. 29, 1377–1385. doi:10.1093/eurheartj/ehn153
Manolescu, D. C., Jankowski, M., Danalache, B. A., Wang, D., Broderick, T. L., Chiasson, J. L., et al. (2014). All-trans retinoic acid stimulates gene expression of the cardioprotective natriuretic peptide system and prevents fibrosis and apoptosis in cardiomyocytes of obese ob/ob mice. Appl. Physiol. Nutr. Metab. 39, 1127–1136. doi:10.1139/apnm-2014-0005
Manrique, C., Demarco, V. G., Aroor, A. R., Mugerfeld, I., Garro, M., Habibi, J., et al. (2013). Obesity and insulin resistance induce early development of diastolic dysfunction in young female mice fed a western diet. Endocrinology 154, 3632–3642. doi:10.1210/en.2013-1256
Matsumoto, E., Sasaki, S., Kinoshita, H., Kito, T., Ohta, H., Konishi, M., et al. (2013). Angiotensin II-induced cardiac hypertrophy and fibrosis are promoted in mice lacking Fgf16. Genes Cells 18, 544–553. doi:10.1111/gtc.12055
McKee, P. A., Castelli, W. P., Mcnamara, P. M., and Kannel, W. B. (1971). The natural history of congestive heart failure: the Framingham study. N. Engl. J. Med. 285, 1441–1446. doi:10.1056/NEJM197112232852601
Methawasin, M., Strom, J. G., Slater, R. E., Fernandez, V., Saripalli, C., and Granzier, H. (2016). Experimentally increasing the compliance of titin through RNA binding motif-20 (RBM20) inhibition improves diastolic function in a mouse model of heart failure with preserved ejection fraction. Circulation 134, 1085–1099. doi:10.1161/CIRCULATIONAHA.116.023003
Mgbemena, O., Zhang, Y., and Velarde, G. (2021). Role of diabetes mellitus in heart failure with preserved ejection fraction: a review article. Cureus 13, e19398. doi:10.7759/cureus.19398
Mohammed, S. F., Ohtani, T., Korinek, J., Lam, C. S., Larsen, K., Simari, R. D., et al. (2010). Mineralocorticoid accelerates transition to heart failure with preserved ejection fraction via "nongenomic effects". Circulation 122, 370–378. doi:10.1161/CIRCULATIONAHA.109.915215
Mohanan, A., Gupta, R., Dubey, A., Jagtap, V., Mandhare, A., Gupta, R. C., et al. (2011). TRC120038, a novel dual AT(1)/ET(A) receptor blocker for control of hypertension, diabetic nephropathy, and cardiomyopathy in ob-ZSF1 rats. Int. J. Hypertens. 2011, 751513. doi:10.4061/2011/751513
Morales-Cano, D., Callejo, M., Barreira, B., Mondejar-Parreno, G., Esquivel-Ruiz, S., Ramos, S., et al. (2019). Elevated pulmonary arterial pressure in Zucker diabetic fatty rats. PLoS One 14, e0211281. doi:10.1371/journal.pone.0211281
Mori, J., Patel, V. B., Abo Alrob, O., Basu, R., Altamimi, T., Desaulniers, J., et al. (2014). Angiotensin 1-7 ameliorates diabetic cardiomyopathy and diastolic dysfunction in db/db mice by reducing lipotoxicity and inflammation. Circ. Heart Fail. 7, 327–339. doi:10.1161/CIRCHEARTFAILURE.113.000672
Nair, N. (2020). Epidemiology and pathogenesis of heart failure with preserved ejection fraction. Rev. Cardiovasc. Med. 21, 531–540. doi:10.31083/j.rcm.2020.04.154
Omori, Y., Ohtani, T., Sakata, Y., Mano, T., Takeda, Y., Tamaki, S., et al. (2012). L-Carnitine prevents the development of ventricular fibrosis and heart failure with preserved ejection fraction in hypertensive heart disease. J. Hypertens. 30, 1834–1844. doi:10.1097/HJH.0b013e3283569c5a
Ostler, J. E., Maurya, S. K., Dials, J., Roof, S. R., Devor, S. T., Ziolo, M. T., et al. (2014). Effects of insulin resistance on skeletal muscle growth and exercise capacity in type 2 diabetic mouse models. Am. J. Physiol. Endocrinol. Metab. 306, E592–E605. doi:10.1152/ajpendo.00277.2013
Packer, M., Lam, C. S. P., Lund, L. H., and Redfield, M. M. (2020). Interdependence of atrial fibrillation and heart failure with a preserved ejection fraction reflects a common underlying atrial and ventricular myopathy. Circulation 141, 4–6. doi:10.1161/CIRCULATIONAHA.119.042996
Papinska, A. M., Soto, M., Meeks, C. J., and Rodgers, K. E. (2016). Long-term administration of angiotensin (1-7) prevents heart and lung dysfunction in a mouse model of type 2 diabetes (db/db) by reducing oxidative stress, inflammation and pathological remodeling. Pharmacol. Res. 107, 372–380. doi:10.1016/j.phrs.2016.02.026
Parcha, V., Malla, G., Kalra, R., Patel, N., Sanders-Van Wijk, S., Pandey, A., et al. (2021). Diagnostic and prognostic implications of heart failure with preserved ejection fraction scoring systems. ESC Heart Fail. 8, 2089–2102. doi:10.1002/ehf2.13288
Pelleymounter, M. A., Cullen, M. J., Baker, M. B., Hecht, R., Winters, D., Boone, T., et al. (1995). Effects of the obese gene product on body weight regulation in ob/ob mice. Science 269, 540–543. doi:10.1126/science.7624776
Peng, H., Yang, X. P., Carretero, O. A., Nakagawa, P., D'ambrosio, M., Leung, P., et al. (2011). Angiotensin II-induced dilated cardiomyopathy in Balb/c but not C57BL/6J mice. Exp. Physiol. 96, 756–764. doi:10.1113/expphysiol.2011.057612
Pfeffer, J. M., Pfeffer, M. A., Fishbein, M. C., and Frohlich, E. D. (1979). Cardiac function and morphology with aging in the spontaneously hypertensive rat. Am. J. Physiol. 237, H461–H468. doi:10.1152/ajpheart.1979.237.4.H461
Plante, E., Menaouar, A., Danalache, B. A., Broderick, T. L., Jankowski, M., and Gutkowska, J. (2014). Treatment with brain natriuretic peptide prevents the development of cardiac dysfunction in obese diabetic db/db mice. Diabetologia 57, 1257–1267. doi:10.1007/s00125-014-3201-4
Qu, Z. (2006). Critical mass hypothesis revisited: role of dynamical wave stability in spontaneous termination of cardiac fibrillation. Am. J. Physiol. Heart Circ. Physiol. 290, H255–H263. doi:10.1152/ajpheart.00668.2005
Rapp, J. P., Wang, S. M., and Dene, H. (1989). A genetic polymorphism in the renin gene of dahl rats cosegregates with blood pressure. Science 243, 542–544. doi:10.1126/science.2563177
Reddy, Y. N. V., Carter, R. E., Obokata, M., Redfield, M. M., and Borlaug, B. A. (2018). A simple, evidence-based approach to help guide diagnosis of heart failure with preserved ejection fraction. Circulation 138, 861–870. doi:10.1161/CIRCULATIONAHA.118.034646
Redfield, M. M., Chen, H. H., Borlaug, B. A., Semigran, M. J., Lee, K. L., Lewis, G., et al. (2013). Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 309, 1268–1277. doi:10.1001/jama.2013.2024
Reed, A. L., Tanaka, A., Sorescu, D., Liu, H., Jeong, E. M., Sturdy, M., et al. (2011). Diastolic dysfunction is associated with cardiac fibrosis in the senescence-accelerated mouse. Am. J. Physiol. Heart Circ. Physiol. 301, H824–H831. doi:10.1152/ajpheart.00407.2010
Roh, J. D., Hobson, R., Chaudhari, V., Quintero, P., Yeri, A., Benson, M., et al. (2019). Activin type II receptor signaling in cardiac aging and heart failure. Sci. Transl. Med. 11, eaau8680. doi:10.1126/scitranslmed.aau8680
Roh, J. D., Houstis, N., Yu, A., Chang, B., Yeri, A., Li, H., et al. (2020). Exercise training reverses cardiac aging phenotypes associated with heart failure with preserved ejection fraction in male mice. Aging Cell 19, e13159. doi:10.1111/acel.13159
Samson, R., Jaiswal, A., Ennezat, P. V., Cassidy, M., and Le Jemtel, T. H. (2016). Clinical phenotypes in heart failure with preserved ejection fraction. J. Am. Heart Assoc. 5, e002477. doi:10.1161/JAHA.115.002477
Sanders-Van Wijk, S., Barandiaran Aizpurua, A., Brunner-La Rocca, H. P., Henkens, M., Weerts, J., Knackstedt, C., et al. (2021). The HFA-PEFF and H2 FPEF scores largely disagree in classifying patients with suspected heart failure with preserved ejection fraction. Eur. J. Heart Fail. 23, 838–840. doi:10.1002/ejhf.2019
Savji, N., Meijers, W. C., Bartz, T. M., Bhambhani, V., Cushman, M., Nayor, M., et al. (2018). The association of obesity and cardiometabolic traits with incident HFpEF and HFrEF. JACC Heart Fail. 6, 701–709. doi:10.1016/j.jchf.2018.05.018
Scantlebury, D. C., and Borlaug, B. A. (2011). Why are women more likely than men to develop heart failure with preserved ejection fraction? Curr. Opin. Cardiol. 26, 562–568. doi:10.1097/HCO.0b013e32834b7faf
Schauer, A., Draskowski, R., Jannasch, A., Kirchhoff, V., Goto, K., Mannel, A., et al. (2020). ZSF1 rat as animal model for HFpEF: Development of reduced diastolic function and skeletal muscle dysfunction. ESC Heart Fail. 7, 2123–2134. doi:10.1002/ehf2.12915
Schiattarella, G. G., Altamirano, F., Tong, D., French, K. M., Villalobos, E., Kim, S. Y., et al. (2019). Nitrosative stress drives heart failure with preserved ejection fraction. Nature 568, 351–356. doi:10.1038/s41586-019-1100-z
Shah, S. J., Kitzman, D. W., Borlaug, B. A., Van Heerebeek, L., Zile, M. R., Kass, D. A., et al. (2016). Phenotype-specific treatment of heart failure with preserved ejection fraction: a multiorgan roadmap. Circulation 134, 73–90. doi:10.1161/CIRCULATIONAHA.116.021884
Sharma, K., and Kass, D. A. (2014). Heart failure with preserved ejection fraction: mechanisms, clinical features, and therapies. Circ. Res. 115, 79–96. doi:10.1161/CIRCRESAHA.115.302922
Silberman, G. A., Fan, T. H., Liu, H., Jiao, Z., Xiao, H. D., Lovelock, J. D., et al. (2010). Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation 121, 519–528. doi:10.1161/CIRCULATIONAHA.109.883777
Solomon, S. D., Mcmurray, J. J. V., and Committee, P.-H. S. (2020). Angiotensin-neprilysin inhibition in heart failure with preserved ejection fraction. Reply. N. Engl. J. Med. 382, 1182–1183. doi:10.1056/NEJMc2000284
Solomon, S. D., Vaduganathan, M., Claggett, B. L., De Boer, R. A., Demets, D., Hernandez, A. F., et al. (2022). Baseline characteristics of patients with HF with mildly reduced and preserved ejection fraction: DELIVER trial. JACC Heart Fail. 10, 184–197. doi:10.1016/j.jchf.2021.11.006
Sorimachi, H., Obokata, M., Takahashi, N., Reddy, Y. N. V., Jain, C. C., Verbrugge, F. H., et al. (2021). Pathophysiologic importance of visceral adipose tissue in women with heart failure and preserved ejection fraction. Eur. Heart J. 42, 1595–1605. doi:10.1093/eurheartj/ehaa823
Suen, C. M., Chaudhary, K. R., Deng, Y., Jiang, B., and Stewart, D. J. (2019). Fischer rats exhibit maladaptive structural and molecular right ventricular remodelling in severe pulmonary hypertension: a genetically prone model for right heart failure. Cardiovasc. Res. 115, 788–799. doi:10.1093/cvr/cvy258
Sueta, D., Yamamoto, E., Nishihara, T., Tokitsu, T., Fujisue, K., Oike, F., et al. (2019). H2FPEF score as a prognostic value in HFpEF patients. Am. J. Hypertens. 32, 1082–1090. doi:10.1093/ajh/hpz108
Sukumaran, V., Tsuchimochi, H., Tatsumi, E., Shirai, M., and Pearson, J. T. (2017). Azilsartan ameliorates diabetic cardiomyopathy in young db/db mice through the modulation of ACE-2/ANG 1-7/Mas receptor cascade. Biochem. Pharmacol. 144, 90–99. doi:10.1016/j.bcp.2017.07.022
Tada, A., Nagai, T., Omote, K., Iwano, H., Tsujinaga, S., Kamiya, K., et al. (2021). Performance of the H2FPEF and the HFA-PEFF scores for the diagnosis of heart failure with preserved ejection fraction in Japanese patients: a report from the Japanese multicenter registry. Int. J. Cardiol. 342, 43–48. doi:10.1016/j.ijcard.2021.08.001
Takeda, T., Hosokawa, M., and Higuchi, K. (1997). Senescence-accelerated mouse (SAM): a novel murine model of senescence. Exp. Gerontol. 32, 105–109. doi:10.1016/s0531-5565(96)00036-8
Tanaka, K., Wilson, R. M., Essick, E. E., Duffen, J. L., Scherer, P. E., Ouchi, N., et al. (2014). Effects of adiponectin on calcium-handling proteins in heart failure with preserved ejection fraction. Circ. Heart Fail. 7, 976–985. doi:10.1161/CIRCHEARTFAILURE.114.001279
Tanaka, K., Valero-Munoz, M., Wilson, R. M., Essick, E. E., Fowler, C. T., Nakamura, K., et al. (2016). Follistatin like 1 regulates hypertrophy in heart failure with preserved ejection fraction. JACC Basic Transl. Sci. 1, 207–221. doi:10.1016/j.jacbts.2016.04.002
Tanisawa, K., Mikami, E., Fuku, N., Honda, Y., Honda, S., Ohsawa, I., et al. (2013). Exome sequencing of senescence-accelerated mice (SAM) reveals deleterious mutations in degenerative disease-causing genes. BMC Genomics 14, 248. doi:10.1186/1471-2164-14-248
Trivedi, P., Yang, R., and Barouch, L. A. (2008). Decreased p110alpha catalytic activity accompanies increased myocyte apoptosis and cardiac hypertrophy in leptin deficient ob/ob mice. Cell Cycle 7, 560–565. doi:10.4161/cc.7.5.5529
Turkbey, E. B., Mcclelland, R. L., Kronmal, R. A., Burke, G. L., Bild, D. E., Tracy, R. P., et al. (2010). The impact of obesity on the left ventricle: the multi-ethnic study of atherosclerosis (MESA). JACC Cardiovasc. Imaging 3, 266–274. doi:10.1016/j.jcmg.2009.10.012
Upadhya, B., and Kitzman, D. W. (2017). Heart failure with preserved ejection fraction in older adults. Heart Fail. Clin. 13, 485–502. doi:10.1016/j.hfc.2017.02.005
Upadhya, B., Willard, J. J., Lovato, L. C., Rocco, M. V., Lewis, C. E., Oparil, S., et al. (2021). Incidence and outcomes of acute heart failure with preserved versus reduced ejection fraction in SPRINT. Circ. Heart Fail. 14, e008322. doi:10.1161/CIRCHEARTFAILURE.121.008322
Valero-Munoz, M., Li, S., Wilson, R. M., Hulsmans, M., Aprahamian, T., Fuster, J. J., et al. (2016). Heart failure with preserved ejection fraction induces beiging in adipose tissue. Circ. Heart Fail. 9, e002724. doi:10.1161/CIRCHEARTFAILURE.115.002724
Valero-Munoz, M., Backman, W., and Sam, F. (2017). Murine models of heart failure with preserved ejection fraction: a "fishing expedition". JACC Basic Transl. Sci. 2, 770–789. doi:10.1016/j.jacbts.2017.07.013
Van den Bergh, A., Flameng, W., and Herijgers, P. (2006). Type II diabetic mice exhibit contractile dysfunction but maintain cardiac output by favourable loading conditions. Eur. J. Heart Fail. 8, 777–783. doi:10.1016/j.ejheart.2006.03.001
van Dijk, C. G., Oosterhuis, N. R., Xu, Y. J., Brandt, M., Paulus, W. J., Van Heerebeek, L., et al. (2016). Distinct endothelial cell responses in the heart and kidney microvasculature characterize the progression of heart failure with preserved ejection fraction in the obese ZSF1 rat with cardiorenal metabolic syndrome. Circ. Heart Fail. 9, e002760. doi:10.1161/CIRCHEARTFAILURE.115.002760
von Bibra, H., Paulus, W., and St John Sutton, M. (2016). Cardiometabolic syndrome and increased risk of heart failure. Curr. Heart Fail. Rep. 13, 219–229. doi:10.1007/s11897-016-0298-4
Wachter, R., Shah, S. J., Cowie, M. R., Szecsody, P., Shi, V., Ibram, G., et al. (2020). Angiotensin receptor neprilysin inhibition versus individualized RAAS blockade: design and rationale of the PARALLAX trial. ESC Heart Fail. 7, 856–864. doi:10.1002/ehf2.12694
Walker, E. M., Nillas, M. S., Mangiarua, E. I., Cansino, S., Morrison, R. G., Perdue, R. R., et al. (2006). Age-associated changes in hearts of male Fischer 344/Brown Norway F1 rats. Ann. Clin. Lab. Sci. 36, 427–438.
Wang, T. J., Larson, M. G., Levy, D., Benjamin, E. J., Leip, E. P., Wilson, P. W., et al. (2004). Impact of obesity on plasma natriuretic peptide levels. Circulation 109, 594–600. doi:10.1161/01.CIR.0000112582.16683.EA
Wang, J., Lu, Y., Min, X., Yuan, T., Wei, J., and Cai, Z. (2021). The association between metformin treatment and outcomes in type 2 diabetes mellitus patients with heart failure with preserved ejection fraction: a retrospective study. Front. Cardiovasc. Med. 8, 648212. doi:10.3389/fcvm.2021.648212
Westman, S. (1968). Development of the obese-hyperglycaemic syndrome in mice. Diabetologia 4, 141–149. doi:10.1007/BF01219435
Willard, P. W. (1969). A model for evaluation of thiazide-induced hypotension. J. Pharm. Pharmacol. 21, 406–408. doi:10.1111/j.2042-7158.1969.tb08280.x
Wilson, R. M., De Silva, D. S., Sato, K., Izumiya, Y., and Sam, F. (2009). Effects of fixed-dose isosorbide dinitrate/hydralazine on diastolic function and exercise capacity in hypertension-induced diastolic heart failure. Hypertension 54, 583–590. doi:10.1161/HYPERTENSIONAHA.109.134932
Withaar, C., Meems, L. M. G., and De Boer, R. A. (2021a). Fighting HFpEF in women: taking aim at belly fat. Eur. Heart J. 42, 1606–1608. doi:10.1093/eurheartj/ehaa952
Withaar, C., Meems, L. M. G., Markousis-Mavrogenis, G., Boogerd, C. J., Sillje, H. H. W., Schouten, E. M., et al. (2021b). The effects of liraglutide and dapagliflozin on cardiac function and structure in a multi-hit mouse model of heart failure with preserved ejection fraction. Cardiovasc. Res. 117, 2108–2124. doi:10.1093/cvr/cvaa256
Zamani, P., Tan, V., Soto-Calderon, H., Beraun, M., Brandimarto, J. A., Trieu, L., et al. (2017). Pharmacokinetics and pharmacodynamics of inorganic nitrate in heart failure with preserved ejection fraction. Circ. Res. 120, 1151–1161. doi:10.1161/CIRCRESAHA.116.309832
Zhang, J., Begley, A., Jackson, R., Harrison, M., Pellicori, P., Clark, A. L., et al. (2019). Body mass index and all-cause mortality in heart failure patients with normal and reduced ventricular ejection fraction: a dose-response meta-analysis. Clin. Res. Cardiol. 108, 119–132. doi:10.1007/s00392-018-1302-7
Keywords: heart failure preserved ejection fraction, mouse model, heart failure, cardiovascular disease, cardiac hypertrophy, diastolic dysfunction. (Min.5-Max. 8
Citation: Chou C and Chin MT (2022) Modeling heart failure with preserved ejection fraction in rodents: Where do we stand?. Front. Drug. Discov. 2:948407. doi: 10.3389/fddsv.2022.948407
Received: 19 May 2022; Accepted: 18 July 2022;
Published: 11 August 2022.
Edited by:
Leon Ptaszek, Harvard Medical School, United StatesReviewed by:
Nicholas Houstis, Harvard Medical School, United StatesCopyright © 2022 Chou and Chin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michael T. Chin, bWNoaW4zQHR1ZnRzbWVkaWNhbGNlbnRlci5vcmc=