 Bruno O. Villoutreix
Bruno O. Villoutreix Iker Badiola
Iker Badiola Abdel-Majid Khatib
Abdel-Majid Khatib- 1Université Paris Cité, Inserm UMR 1141 NeuroDiderot, Robert-Debré Hospital, Paris, France
- 2Akttyva Therapeutics, Inc., Mansfield, MA, United States
- 3Department of Cell Biology and Histology, Faculty of Medicine and Nursing, University of Basque Country (UPV/EHU), Leioa, Spain
- 4RYTME, Bordeaux Institute of Oncology (BRIC)- UMR1312 Inserm, University of Bordeaux, Pessac, France
- 5Institut Bergonié, Bordeaux, France
Furin is involved in the endoproteolytic processing of various protein precursors implicated in many diseases such as diabetes, obesity, atherosclerosis, cancer, Alzheimer’s disease and viral infection including COVID-19. Recently, cell entry of SARS-CoV-2 was found to require sequential cleavage of the viral spike glycoprotein (S protein) at the S1/S2 and the S2ʹ cleavage sites. The S1/S2 site (PRRAR) can be cleaved by the proprotein convertase furin that facilitates membrane fusion and viral spread. Here we summarized the recent findings on furin and S protein structures, the role of S protein cleavage by furin during SARS-CoV-2 infection. We analyzed 12 diverse representative inhibitors of furin using a chemoinformatic approach starting from a list of 628 compounds downloaded from the ChEMBL database. Among those, only 76 survived a soft rule of five filtering step. Structural alerts are present on most of these molecules while some compounds are also predicted to act on toxicity targets. No clinical trials are presently listed at the ClinicalTrials.gov website regarding small molecule inhibitors of furin.
SARS-CoV-2 Infection and Furin
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), responsible for the pandemic disease COVID-19, is a vigorous infection that has affected millions of people worldwide. This contagious respiratory infection provokes flu-like symptoms such as cough with or without fever that spreads more easily than flu. Of the infected people, up to 20% seemed to develop respiratory difficulties including pneumonia (Hu et al., 2021).
Like with other coronaviruses (CoVs), SARS-CoV-2 presents spike glycoproteins (S protein) on their viral envelope used by the virus to bind host receptors and fuse with cell membrane, a molecular event crucial for the viral infection. The S proteins are type I transmembrane proteins composed of two subunits S1 and S2. The S1 subunit is responsible for receptor binding while the S2 subunit is involved in membrane fusion (Figure 1). The S1 domain contains an N-terminal domain (NTD) and a receptor‐binding domain (RBD) used for binding the SARS-CoV-2 receptor: the angiotensin‐converting enzyme 2 (ACE2) and heparan sulfate (Clausen et al., 2020; Walls et al., 2020). To facilitate fusion, the S protein that is synthesized as immature protein precursor, is proteolyticaly cleaved at the S1/S2 boundary and at the S2ʹ site located close to the S2 fusion peptide (FP) (Figure 1). Indeed, upon cleavage at the S1/S2 site, and engagement of ACE2 with the spike RBD domain, the S2ʹ site is also cleaved by serine proteases, leading to S2 fusion peptide generation that allows viral–host membrane fusion initiation (Figure 2).
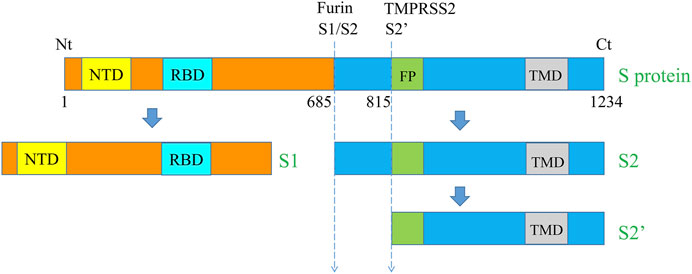
FIGURE 1. Schematic representation of the major SARS‐CoV‐2 S protein domains and the cleavage sites. The S protein of SARS‐CoV‐2 is a transmembrane protein that contains various domains including NTD, RBD, FP, and TMD. The total aa sequence length (1,273 aa) and the position of the cleavage sites S1/S2 (685/686) and S2′ (815/816) are also indicated. NTD, N-terminal domain, RBD, receptor-binding domain, FP, Fusion peptide, TMD, transmembrane domain, Nt, N-terminal and Ct, C-terminal.
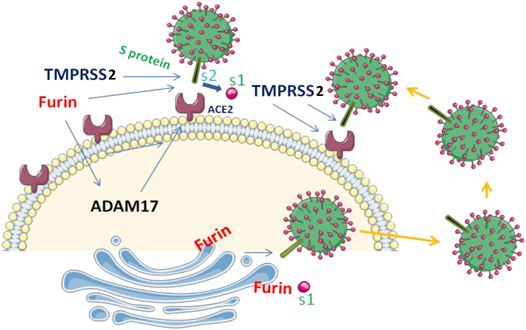
FIGURE 2. S protein cleavage and SARS‐CoV‐2 infection. SARS-CoV-2 entry into the cells is mediated by the binding of the viral spike (S) protein to ACE2 and the cleavage of S protein by furin and TMPRSS2 at the S1/S2 site. ACE2 can also be cleaved by TMPRSS2 and ADAM17 and ADAM17 precursor involves furin for its cleavage and activation.
Earlier studies revealed that SARS-CoV-2 has superior affinity for the ACE2 receptor compared to SARS-CoV (Shang et al., 2020; Lemmin et al., 2020) and was associated to S protein cleavage by furin, a proprotein convertase member (also known as dibasic-processing enzyme) that is ubiquitously expressed and involved in the proteolytic activation of various protein precursors (Siegfried et al., 2020; Soulet et al., 2020; Scamuffa et al., 2014). Indeed, cell entry of SARS-CoV-2 depends on binding of the viral S protein to the cellular receptor ACE2 and on S protein priming by furin and TMPRSS2. TMPRSS2 also cleaves ACE2 that augment viral infectivity (Figure 2) (Heurich et al., 2014; Hoffmann et al., 2020). In addition, TMPRSS2 competes with the ADAM17 for ACE2 cleavage (Haga et al., 2008; Haga et al., 2010; Wong et al., 2015). Like various MMPs, ADAM17 precursor also requires furin for its activation (Wong et al., 2015). Thereby, the high expression of furin as found in lung could be exploited by the SARS-CoV-2 for the cleavage of S protein and ACE2 leading to enhanced spread of the virus. However, although furin is the main protease responsible for the cleavage at the site S1/S2, this proteolytic cleavage may also happen in the absence of furin, suggesting the involvement of other proprotein convertases in these processes (Jaimes et al., 2020; Wrobel et al., 2020).
In comparison to all known SARS-CoVs, the protein of SARS-CoV-2 possesses a unique four-residue PRRA insertion at the cleavage site S1/S2 (Figure 1). Studies analyzing the effect of the mutation of this site in various cell lines and animal models revealed that this deletion significantly reduced viral infection. On the other hand, the cleavage at the S1/S2 site was found not to be required for infection and replication in Vero E6 cells (Lau et al., 2020; Johnson et al., 2021; Peacock et al., 2021). Lately, using infected hamster models, Lau et al. reported that compared to the wild-type, SARS-CoV-2 with mutated S1/S2 site replicated weakly in tracheal and lung tissues (Lau et al., 2020). Accordingly, deletion of this site was confirmed to reduce by up to 10-fold the replication in human cell lines, and reduce the severity of COVID-19 in hamsters and K18-hACE2 transgenic mice (Johnson et al., 2021). Taken together, these findings indicate the potential use of furin inhibitors to modulate SARS-CoV-2 infection.
Furin 3D Structure, Substrates and Inhibitors
Furin 3D Structure
Twenty-four crystal structures of furin are available at the Protein Data Bank (Burley et al., 2021) at the time of writing. Furin is a 794 amino acid calcium-dependent multidomain enzyme. The structure of furin in complex with meta-guanidinomethyl-Phac-RVR-Amba (PDB file 5JXH) at 2.0 Angstrom resolution (Dahms et al., 2014) is shown as an example (Figure 3A). Only the catalytic and P domains are present in these experimental 3D structures (i.e., the other domains: the cysteine-rich domain, the transmembrane domain and the C-terminal cytoplasmic domain are not visible). On the N-terminal side, the protein also contains a so-called prodomain that acts as an intramolecular chaperone, and seems important to assist proper folding of the enzyme. This prodomain was reported to regulate the furin activity in vitro and in vivo (López et al., 2021; Descarpentrie et al., 2022). The catalytic domain is similar to the one of subtilisin, it consists of a twisted beta-sheet composed of seven parallel and one antiparallel beta-stands flanked by five adjacent and two peripheral helices (Henrich et al., 2003). Yet, the active site cleft of furin differs from that of subtilisins with respect to depth, shape and charges (i.e., the furin active site is mainly electronegative with at least six negatively charged residues directly surrounding the Ser368-His194-Asp153 active site triad). Furin preferentially recognizes/cleaves the sequence motif RXR/KR-X (the “-” indicates the scissile peptide bond) found in various protein precursors (Siegfried et al., 2005; Scamuffa et al., 2008; He et al., 2020a). While the catalytic activity of furin is strictly calcium-dependent, the three calcium ions found in the catalytic domain are not directly involved in the catalytic reaction, they are indirectly important by forming the right geometry of the active site. The adjacent P domain is organized as a separate eight-stranded β-sandwich and is thought to be important for the catalytic activity of the enzyme.
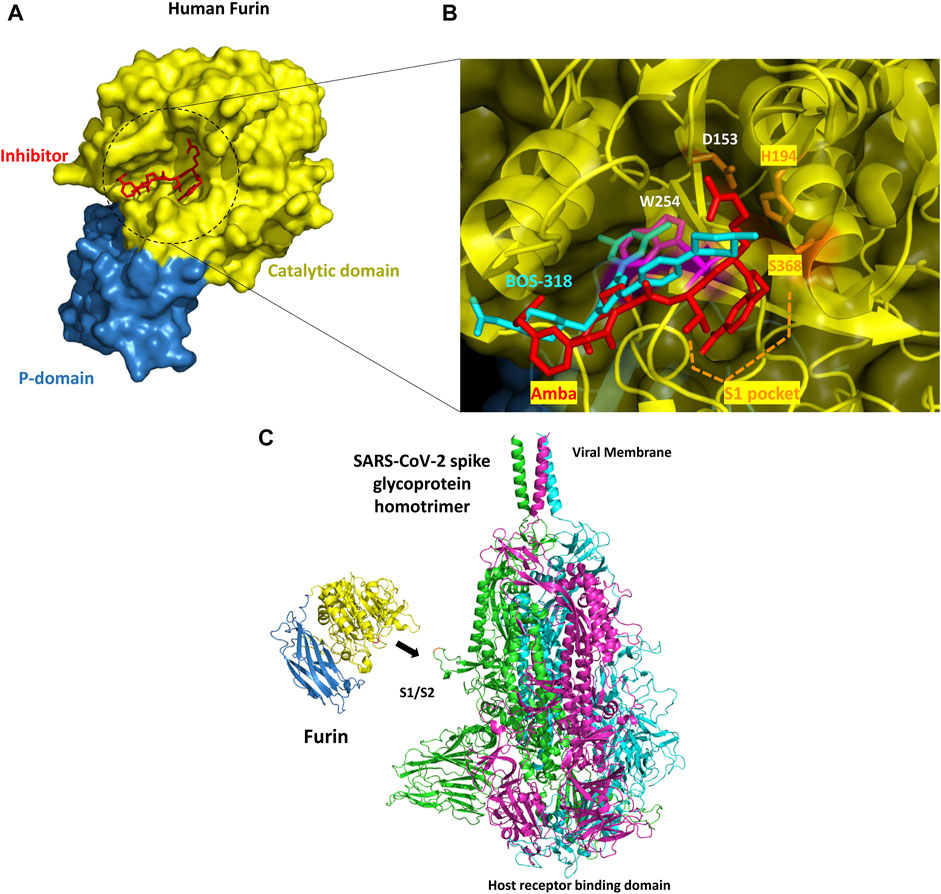
FIGURE 3. (A) Overall structure of human furin. The molecular surface is shown (the catalytic domain and the P-domain are in yellow and blue, respectively) with a view down the active site (PDB file 5JXH). A small inhibitor (meta-guanidinomethyl-Phac-RVR-Amba or Amba compound) co-crystallized in the catalytic site is shown in red (B) Zoom into the catalytic site. The experimental structure of BOS-318 in complex with furin (PDB file 7LCU) was superimposed onto furin co-crystallized with an Amba compound (PDB file 5JXH). Amba compounds and in general all known furin inhibitors have a P1 residue inserted into the S1 pocket and they make contacts with the catalytic triad residues. By contrast, BOS-318 does not directly engage with the catalytic triad, does not interact with the S1 pocket and seems to induce the flip of furin W254 (magenta, side chain present in 5JXH), thereby creating novel binding subpocket that allows the binding of the dichlorophenyl moiety of BOS-318 (without such change in the orientation of the W side chain, major steric clashes would occur and the binding of BOS-318 would not be possible) (C) Overview of the SARS-CoV-2 spike glycoprotein homotrimer - human furin complex. Furin was manually positioned nearby the S1/S2 region cleavage site. The three monomers of the SARS-CoV-2 S protein are colored green, magenta, and cyan while furin is showed in blue and yellow. The catalytic site of the enzyme is next to the S protein PRRAR cleavage site loop (in red). There seems to be enough room to accommodate three furin molecules on the S protein trimer at the same time.
New information about the catalytic site have been recently reported with the experimental structure of furin in complex with a small inhibitor named BOS-318 (Douglas et al., 2022). Furin inhibitors usually make significant contacts with the catalytic triad residues and have a P1 residue inserted into the S1 binding pocket (Figure 3B). This is the case for instance with the Amba family of compounds. But the selective BOS-318 inhibitor does not make such contacts (PDB file 7LCU). Surprisingly, BOS-318 seems to induce a flip of furin W254 which in turn creates a novel binding pocket that is filled by the dichlorophenyl moiety of BOS-318. This novel mode of binding found in that study provides novel insights to rationally design novel inhibitors of furin.
Spike Substrate
Furin have been linked to various pathologies including Alzheimer’s disease, tumorigenesis, and infections (Thomas, 2002). In all these diseases, furin activity was found to mediate the proteolytic activation of various protein precursors directly linked to the initiation and/or progression of these pathologies. The cleavage of the furin substrates can occurs inside the cell at the Golgi apparatus and on the cell surface membrane where furin is located (Molloy et al., 1998). Indeed, furin is predominantly localized to the TGN can reach the plasma membrane and internalized back to the TGN (Molloy et al., 1998). In Alzheimer’s disease, furin mediates the cleavage of α- and β-secretase precursors, involved in the accumulation of amyloid-β (Aβ), the principal constituent of senile plaques (He et al., 2020a). The implication of furin in tumor progression was revealed based on changes in several biological functions that directly affect the malignant and the metastatic potential of tumor cells after furin inhibition (Basak et al., 2010; He et al., 2020a; Siegfried et al., 2020). This through repressed processing and activation of many molecules involved in cell proliferation, adhesion, invasion and survival (Scamuffa et al., 2008; Basak et al., 2009; López et al., 2021). The protein precursors include metalloproteases, adhesion molecules, growth factors, and growth factor receptors (He et al., 2020b; Siegfried et al., 2020). The role of furin in various viral infection is also well established ((Jaimes et al., 2020)– (Burley et al., 2021)). The acquisition of the infectious capacity and/or cell-cell spread of various viruses require the maturation of the viral cell surface glycoproteins by furin. These include HIV-1, Ebola virus, Hong Kong influenza virus, the severe acute respiratory syndrome coronavirus (Anderson et al., 1993; Basak et al., 2001; Thomas, 2002). Inhibition of processing of some of these viral surface glycoproteins by furin inhibitors was found to abrogate the virus-induced cellular cytopathicity (Anderson et al., 1993; Basak et al., 2001; Thomas, 2002).
In the context of COVID-19, it was suggested that furin could cleave the SARS-CoV-2 S protein. The experimental structure of furin is seen oriented toward a cleavage site, located between the S1/S2 subunits of the S protein and present on a solvent exposed loop structure (Figure 3C). This loop should accommodate into the deep groove located in the catalytic domain of the protease and should make numerous hydrophobic, polar and electrostatic interactions with the enzyme. The S proteins assemble as a trimer and are in part inserted into the viral membrane. The molecule system is formed of three identical protomers that strongly binds to ACE2 receptor upon activation, a receptor found on the surface of many cell types. The S protein is here a theoretical 3D model structure derived from several cryo-EM structures (Fertig et al., 2022) including PDB files 6VSB (Wrapp et al., 2020) and 6XR8 (Mendez et al., 2019). The model was developed because some loops were missing in the experimental structures. The binding of the S protein to the ACE2 receptor requires several proteolytic cleavages and conformational changes. One such cleavage on the S protein (cleavage motif PRRAR) is performed by furin, at least on one site located at the S1/S2 boundary. This cleavage is expected to promote disordering of the S protein and then exposure of a domain (the RBD domain) critical for ACE2 binding. Yet, as mentioned above, S protein processing can occur independently of furin although the presence of this protease significantly increases cleavage.
Small Molecules Inhibitors
It has been suggested that furin inhibitors would be very valuable to reduce but not completely abolish viral spread. Several small molecules have been shown to inhibit furin. Yet, most of the time, these involve peptides and peptide-like structures that are often not well suited for human administration due to short half-life, instability, degradation by proteases or difficulties to administrate via oral route. In fact, it seems that most furin preclinical candidates have been stopped thus far due to various absorption, distribution, metabolism, excretion and toxicity problems (ADME-Tox, e.g., cellular toxicity, low cell permeability, chemical instability, etc.) (see below). A search with furin as keyword at the https://www.clinicaltrials.gov/ website did not output ongoing clinical trials involving direct small molecule inhibitors. To investigate previously reported furin inhibitors, we mined the ChEMBL database (Mendez et al., 2019). We downloaded 628 compounds that have been tested on furin and removed molecules that are too far away from the rule of 5. This rule was introduced in 1997, and suggests that to administrate a small molecule by oral route, the molecular weight (MW) of the compound should be less than 500 daltons, the lipophilicity (evaluated via computed logP) cLogP should not exceed 5, the H-bond donor count (sum of OH + NH groups) should be less than 5 and the H-bond acceptor count (sum of O + N atoms) should be less than 10 (Lipinski, 2000). As some of the compounds available in ChEMBL for furin have not been extensively optimized toward clinical candidates, we initially only applied a soft filter on MW and selected molecules with MW ∼600 or less. This simple filtering step led to the selection of 76 molecules. Then we focused on activity characterized by the so-called pChEMBL value. As defined at the ChEMBL website, the pChEMBL value allows a number of roughly comparable measures of half-maximal response concentration/potency/affinity to be compared on a negative logarithmic scale. For example, an IC50 measurement of 1 nM would have a pChEMBL value of 9. This value is defined as: -log(molar IC50, XC50, EC50, AC50, Ki, Kd or Potency). It was possible to obtain a pChEMBL value for only 42 molecules. Then, removing duplicate compounds and clustering the molecules by structural similarities, we ended up with 11 drug-like molecules that inhibit human furin (Figure 4). To these compounds we manually added a molecule missing in the ChEMBL database named BOS-318 (Figure 3B), this compound has been recently co-crystallized with furin and was shown to be selective and cell-permeable (Douglas et al., 2022).
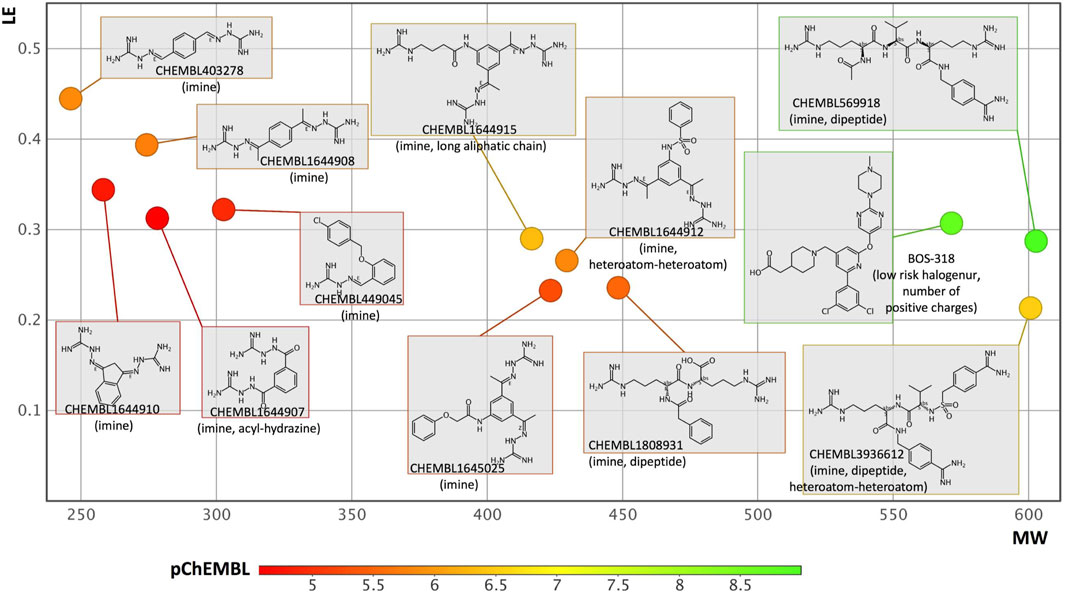
FIGURE 4. Analysis of furin inhibitors present in the ChEMBL database. After applying simple filtering rules (see text), removing duplicates and clustering the molecules based upon structural similarities, 11 molecules were selected. To these compounds, a recently co-crystallized selective furin inhibitor named BOS-318 was added. The plot considers MW and LE values while the compounds are color coded according to the their pChEMBL values (i.e., green means more potent). Next to each compound, the ChEMBL-ID (or name) is written. Some structural alerts and physico-chemical problems that may impede development are noted between parentheses. These data were generated with the FAF-Drugs server. More specific endpoints were computed with the interpretable-ADMET server, for some compounds no information could be obtained as outside the applicability domain of the method, for the others, the main warnings are as follow: compound CHEMBL403278, putative inhibition of hERG (cardiotoxicity), and inhibition of OATP1B3 organic anion transporting polypeptides (a critical transporter); CHEMBL1644908 or CHEMBL1644915, putative inhibitor of hERG; CHEMBL1644910 or CHEMBL449045, putative inhibitor of hERG and OATP1B3; CHEMBL1645025, putative hERG and Pgp-inhibitor and possible respiratory toxicity; CHEMBL1808931, putative inhibitor of OATP1B3, modulator of nuclear receptors and possible respiratory toxicity; CHEMBL1644912 putative inhibitor of hERG and modulator of nuclear receptors; CHEMBL569918, putative inhibitor of OATP1B3; BOS-318, CHEMBL3936612 and CHEMBL1644907, tend to be outside the applicability domain of the method and further computations will be required.
Analysis of the molecules was performed with our software FAF-Drugs (Lagorce et al., 2017) and the visualization carried out with DataWarrior (Sander et al., 2015). While potency is an important parameter for drug discovery, other metrics can be used to evaluate small molecules and help to select compounds for further developments. One such metric is called ligand efficiency or LE. This value normalizes the experimental activity to molecular weight, heavy atom count, or other molecular size quantifier (i.e., an acceptable level for LE is around ≥ 0.3) (Hopkins et al., 2004). LE metrics are thus considered as valuable indicator of compound quality in the early stage of drug discovery as they help focus on molecules that are relatively small and yet that have a significant target activity, suggesting that optimization of the molecules should be easier. Indeed, in most cases, compound optimization usually deteriorates parameters such as MW and log P (e.g., increasing these two parameters often lead to increased interaction with off-targets and anti-targets, render oral administration more difficult…). In Figure 4, we plotted LE as a function of MW (e.g., to for instance see if compounds could grow in size) while color coding the molecules with the computed pChEMBL value. We note for instance a peptide-like molecule (CHEMBL569918) with a high MW that shows high potency with a reasonable starting LE, yet such molecule would require significant optimization before becoming a clinical candidate. Some molecules with a relatively small MW could be more interesting to optimize as they have a favorable LE but the starting chemistry is eventually not optimal. Indeed, some structural alerts (i.e., toxicophores, simple means to flag potential chemical hazards and problematic compounds) are often noticed in these molecules (please see details about major structural alerts at the FAF-Drugs website). Such chemical groups often impede further developments (e.g., many reactive warheads react with off-targets) and/or have been found to interfere with the reading of high-throughput screening experiments and/or can be promiscuous aggregators. Next to more global physical-chemistry properties and structural alerts mentioned above, more specific ADMET endpoints can also be predicted. We used the recently reported Interpretable-ADMET web service (Wei et al., 2022) and found that several compounds could be inhibitors of important toxicity targets such as hERG (cardiotoxicity) (see legend of Figure 4). Overall, this analysis suggests that important medicinal chemistry efforts will be needed to develop furin inhibitors that could enter clinical trials. Yet, the available compounds together with several experimental structures are highly valuable and should definitively facilitate the design of novel, more ADME-Tox friendly inhibitors exploiting novel furin subpockets.
Conclusion
Since its discovery in the 1980s, furin has acquired the status of therapeutic target for the treatment of various diseases including cancer (Siegfried et al., 2020; He et al., 2022). The discovery of a furin cleavage site in the S protein of SARS-CoV-2 virus further stimulates research on this enzyme for the treatment of infectious diseases. The availability of several high-resolution experimental 3D structures in complex with different types of inhibitors (peptide-like and non-peptide like) should facilitate the design of novel high-quality inhibitors (Osman et al., 2022). However, furin is ubiquitous and mediates the activation of various constitutively secreted substrates involved in numerous physiological processes such as angiogenesis, extracellular matrix remodeling and others (Scamuffa et al., 2006; Lahlil et al., 2009). Thereby the use of furin inhibitors as therapeutic approach may affect these biological functions and induces several side effects. In addition, the ability of furin to control the expression and/or activity of TMPRSS2, and ADAM17 (Haga et al., 2008; Haga et al., 2010; Heurich et al., 2014; Wong et al., 2015; Hoffmann et al., 2020), suggests that furin inhibitors can also interfere with the biological function mediated by these proteases. Thereby, prior any application in clinical setting, the side effect of the developed furin inhibitors should be evaluated and more effort should be oriented toward the development of reversible furin inhibitors, of which the action could be stopped when necessary to prevent any potential side effects. Taken together, while it is clear that furin is a challenging therapeutic target, it definitively deserves further investigation.
Author Contributions
BV, IB and A-MK wrote and drafted the manuscript. All authors contributed to the article and approved the final version.
Funding
This work was supported by SPARK 2020 (University of Bordeaux) to A-MK and grants from INCA and ARC (BV). This work was supported by the INSERM institute.
Conflict of Interest
Author BV was employed by the company Akttyva Therapeutics, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Anderson, E. D., Thomas, L., Hayflick, J. S., and Thomas, G. (1993). Inhibition of HIV-1 Gp160-dependent Membrane Fusion by a Furin-Directed Alpha 1-antitrypsin Variant. J. Biol. Chem. 268, 24887. doi:10.1016/s0021-9258(19)74548-7
Basak, A., Chen, A., Scamuffa, N., Mohottalage, D., Basak, S., and Khatib, A.-M. (2010). Blockade of Furin Activity and Furin-Induced Tumor Cells Malignant Phenotypes by the Chemically Synthesized Human Furin Prodomain. Curr. Med. Chem. 17, 2214–2221. doi:10.2174/092986710791331040
Basak, A., Khatib, A.-M., Mohottalage, D., Basak, S., Kolajova, M., Bag, S. S., et al. (2009). A Novel Enediynyl Peptide Inhibitor of Furin that Blocks Processing of proPDGF-A, B and proVEGF-C. PLoS ONE 4, e7700. doi:10.1371/journal.pone.0007700
Basak, A., Zhong, M., Munzer, J. S., Chrétien, M., and Seidah, N. G. (2001). Implication of the Proprotein Convertases Furin, PC5 and PC7 in the Cleavage of Surface Glycoproteins of Hong Kong, Ebola and Respiratory Syncytial Viruses: a Comparative Analysis with Fluorogenic Peptides. Biochem. J. 353, 537–545. doi:10.1042/0264-6021:3530537
Burley, S. K., Bhikadiya, C., Bi, C., Bittrich, S., Chen, L., Crichlow, G. V., et al. (2021). RCSB Protein Data Bank: Powerful New Tools for Exploring 3D Structures of Biological Macromolecules for Basic and Applied Research and Education in Fundamental Biology, Biomedicine, Biotechnology, Bioengineering and Energy Sciences. Nucleic Acids Res. 49, D437–D451. doi:10.1093/nar/gkaa1038
Clausen, T. M., Sandoval, D. R., Spliid, C. B., Pihl, J., Perrett, H. R., Painter, C. D., et al. (2020). SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 183, 1043–1057. doi:10.1016/j.cell.2020.09.033
Dahms, S. O., Hardes, K., Becker, G. L., Steinmetzer, T., Brandstetter, H., and Than, M. E. (2014). X-ray Structures of Human Furin in Complex with Competitive Inhibitors. ACS Chem. Biol. 9, 1113–1118. doi:10.1021/cb500087x
Descarpentrie, J., Araúzo-Bravo, M. J., He, Z., François, A., González, Á., Garcia-Gallastegi, P., et al. (2022). Role of Furin in Colon Cancer Stem Cells Malignant Phenotype and Expression of LGR5 and NANOG in KRAS and BRAF-Mutated Colon Tumors. Cancers 14, 1195. doi:10.3390/cancers14051195
Douglas, L. E. J., Reihill, J. A., Ho, M. W. Y., Axten, J. M., Campobasso, N., Schneck, J. L., et al. (2022). A Highly Selective, Cell-Permeable Furin Inhibitor BOS-318 Rescues Key Features of Cystic Fibrosis Airway Disease. Cell Chem. Biol. (22), S2451. doi:10.1016/j.chembiol.2022.02.001
Fertig, T. E., Chitoiu, L., Terinte‐Balcan, G., Peteu, V. E., Marta, D., and Gherghiceanu, M. (2022). The Atomic Portrait of SARS‐CoV‐2 as Captured by Cryo‐electron Microscopy. J. Cell. Mol. Medi 26, 25–34. doi:10.1111/jcmm.17103
Haga, S., Nagata, N., Okamura, T., Yamamoto, N., Sata, T., Yamamoto, N., et al. (2010). TACE Antagonists Blocking ACE2 Shedding Caused by the Spike Protein of SARS-CoV Are Candidate Antiviral Compounds. Antivir. Res. 85, 551–555. doi:10.1016/j.antiviral.2009.12.001
Haga, S., Yamamoto, N., Nakai-Murakami, C., Osawa, Y., Tokunaga, K., Sata, T., et al. (2008). Modulation of TNF-α-Converting Enzyme by the Spike Protein of SARS-CoV and ACE2 Induces TNF-α Production and Facilitates Viral Entry. Proc. Natl. Acad. Sci. U.S.A. 105, 7809–7814. doi:10.1073/pnas.0711241105
He, Z., Khatib, A.-M., and Creemers, J. W. M. (2020). Loss of the Proprotein Convertase Furin in T Cells Represses Mammary Tumorigenesis in Oncogene-Driven Triple Negative Breast Cancer. Cancer Lett. 484, 40–49. doi:10.1016/j.canlet.2020.05.001
He, Z., Khatib, A.-M., and Creemers, J. W. M. (2022). The Proprotein Convertase Furin in Cancer: More Than an Oncogene. Oncogene 41, 1252–1262. doi:10.1038/s41388-021-02175-9
He, Z., Thorrez, L., Siegfried, G., Meulemans, S., Evrard, S., Tejpar, S., et al. (2020). The Proprotein Convertase Furin Is a Pro-oncogenic Driver in KRAS and BRAF Driven Colorectal Cancer. Oncogene 39, 3571–3587. doi:10.1038/s41388-020-1238-z
Henrich, S., Cameron, A., Bourenkov, G. P., Kiefersauer, R., Huber, R., Lindberg, I., et al. (2003). The Crystal Structure of the Proprotein Processing Proteinase Furin Explains its Stringent Specificity. Nat. Struct. Mol. Biol. 10, 520–526. doi:10.1038/nsb941
Heurich, A., Hofmann-Winkler, H., Gierer, S., Liepold, T., Jahn, O., and Pöhlmann, S. (2014). TMPRSS2 and ADAM17 Cleave ACE2 Differentially and Only Proteolysis by TMPRSS2 Augments Entry Driven by the Severe Acute Respiratory Syndrome Coronavirus Spike Protein. J. Virol. 88, 1293–1307. doi:10.1128/JVI.02202-13
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Krüger, N., Herrler, T., Erichsen, S., et al. (2020). SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 181, 271–280. doi:10.1016/j.cell.2020.02.052
Hopkins, A. L., Groom, C. R., and Alex, A. (2004). Ligand Efficiency: a Useful Metric for Lead Selection. Drug Discov. Today 9, 430–431. doi:10.1016/S1359-6446(04)03069-7
Hu, B., Guo, H., Zhou, P., and Shi, Z.-L. (2021). Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 19, 141–154. doi:10.1038/s41579-020-00459-7
Jaimes, J. A., Millet, J. K., and Whittaker, G. R. (2020). Proteolytic Cleavage of the SARS-CoV-2 Spike Protein and the Role of the Novel S1/S2 Site. iScience 23, 101212. doi:10.1016/j.isci.2020.101212
Johnson, B. A., Xie, X., Bailey, A. L., Kalveram, B., Lokugamage, K. G., Muruato, A., et al. (2021). Loss of Furin Cleavage Site Attenuates SARS-CoV-2 Pathogenesis. Nature 591, 293–299. doi:10.1038/s41586-021-03237-4
Lagorce, D., Bouslama, L., Becot, J., Miteva, M. A., and Villoutreix, B. O. (2017). FAF-Drugs4: Free ADME-Tox Filtering Computations for Chemical Biology and Early Stages Drug Discovery. Bioinformatics 33, 3658–3660. doi:10.1093/bioinformatics/btx491
Lahlil, R., Calvo, F., and Khatib, A.-M. (2009). The Potential Anti-tumorigenic and Anti-metastatic Side of the Proprotein Convertases Inhibitors. Recent. Pat Anticancer. Drug. Discov 4, 83–91. doi:10.2174/157489209787002470
Lau, S.-Y., Wang, P., Mok, B. W.-Y., Zhang, A. J., Chu, H., Lee, A. C.-Y., et al. (2020). Attenuated SARS-CoV-2 Variants with Deletions at the S1/S2 Junction. Emerg. Microbes Infect. 9, 837–842. doi:10.1080/22221751.2020.1756700
Lemmin, T., Kalbermatter, D., Harder, D., Plattet, P., and Fotiadis, D. (2020). Structures and Dynamics of the Novel S1/S2 Protease Cleavage Site Loop of the SARS-CoV-2 Spike Glycoprotein. J. Struct. Biol. X 4, 100038. doi:10.1016/j.yjsbx.2020.100038
Lipinski, C. A. (2000). Drug-like Properties and the Causes of Poor Solubility and Poor Permeability. J. Pharmacol. Toxicol. Methods 44, 235–249. doi:10.1016/s1056-8719(00)00107-6
López, J. J., Siegfried, G., Cantonero, C., Soulet, F., Descarpentrie, J., Smani, T., et al. (2021). Furin Prodomain ppFurin Enhances Ca2+ Entry through Orai and TRPC6 Channels' Activation in Breast Cancer Cells. Cancers 13, 1670. doi:10.3390/cancers13071670
Mendez, D., Gaulton, A., Bento, A. P., Chambers, J., De Veij, M., Félix, E., et al. (2019). ChEMBL: towards Direct Deposition of Bioassay Data. Nucleic Acids Res. 47, D930–D940. doi:10.1093/nar/gky1075
Molloy, S. S., Thomas, L., Kamibayashi, C., Mumby, M. C., and Thomas, G. (1998). Regulation of Endosome Sorting by a Specific PP2A Isoform. J. Cell Biol. 142, 1399–1411. doi:10.1083/jcb.142.6.1399
Osman, E. E. A., Rehemtulla, A., and Neamati, N. (2022). Why All the Fury over Furin? J. Med. Chem. 65, 2747–2784. doi:10.1021/acs.jmedchem.1c00518
Peacock, T. P., Goldhill, D. H., Zhou, J., Baillon, L., Frise, R., Swann, O. C., et al. (2021). The Furin Cleavage Site in the SARS-CoV-2 Spike Protein Is Required for Transmission in Ferrets. Nat. Microbiol. 6, 899–909. doi:10.1038/s41564-021-00908-w
Sander, T., Freyss, J., von Korff, M., and Rufener, C. (2015). DataWarrior: an Open-Source Program for Chemistry Aware Data Visualization and Analysis. J. Chem. Inf. Model. 55, 460–473. doi:10.1021/ci500588j
Scamuffa, N., Calvo, F., Chrétien, M., Seidah, N. G., and Khatib, A. M. (2006). Proprotein Convertases: Lessons from Knockouts. FASEB J. 20, 1954–1963. doi:10.1096/fj.05-5491rev
Scamuffa, N., Sfaxi, F., Ma, J., Lalou, C., Seidah, N., Calvo, F., et al. (2014). Prodomain of the Proprotein Convertase Subtilisin/kexin Furin (ppFurin) Protects from Tumor Progression and Metastasis. Carcinogenesis 35, 528–536. doi:10.1093/carcin/bgt345
Scamuffa, N., Siegfried, G., Bontemps, Y., Ma, L., Basak, A., Cherel, G., et al. (2008). Selective Inhibition of Proprotein Convertases Represses the Metastatic Potential of Human Colorectal Tumor Cells. J. Clin. Invest. 118, 352–363. doi:10.1172/JCI32040
Shang, J., Ye, G., Shi, K., Wan, Y., Luo, C., Aihara, H., et al. (2020). Structural Basis of Receptor Recognition by SARS-CoV-2. Nature 581, 221–224. doi:10.1038/s41586-020-2179-y
Siegfried, G., Basak, A., Prichett-Pejic, W., Scamuffa, N., Ma, L., Benjannet, S., et al. (2005). Regulation of the Stepwise Proteolytic Cleavage and Secretion of PDGF-B by the Proprotein Convertases. Oncogene 24, 6925–6935. doi:10.1038/sj.onc.1208838
Siegfried, G., Descarpentrie, J., Evrard, S., and Khatib, A.-M. (2020). Proprotein Convertases: Key Players in Inflammation-Related Malignancies and Metastasis. Cancer Lett. 473, 50–61. doi:10.1016/j.canlet.2019.12.027
Soulet, F., Bodineau, C., Hooks, K. B., Descarpentrie, J., Alves, I., Dubreuil, M., et al. (2020). ELA/APELA Precursor Cleaved by Furin Displays Tumor Suppressor Function in Renal Cell Carcinoma through mTORC1 Activation. JCI Insight 5, 129070. doi:10.1172/jci.insight.129070
Thomas, G. (2002). Furin at the Cutting Edge: from Protein Traffic to Embryogenesis and Disease. Nat. Rev. Mol. Cell Biol. 3, 753–766. doi:10.1038/nrm934
Walls, A. C., Park, Y.-J., Tortorici, M. A., Wall, A., McGuire, A. T., and Veesler, D. (2020). Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 181, 281–292. doi:10.1016/j.cell.2020.02.058
Wei, Y., Li, S., Li, Z., Wan, Z., and Lin, J. (2022). Interpretable-ADMET: a Web Service for ADMET Prediction and Optimization Based on Deep Neural Representation. Bioinformatics. btac192. doi:10.1093/bioinformatics/btac192
Wong, E., Maretzky, T., Peleg, Y., Blobel, C. P., and Sagi, I. (2015). The Functional Maturation of A Disintegrin and Metalloproteinase (ADAM) 9, 10, and 17 Requires Processing at a Newly Identified Proprotein Convertase (PC) Cleavage Site. J. Biol. Chem. 290, 12135. doi:10.1074/jbc.M114.624072
Wrapp, D., Wang, N., Corbett, K. S., Goldsmith, J. A., Hsieh, C.-L., Abiona, O., et al. (2020). Cryo-EM Structure of the 2019-nCoV Spike in the Prefusion Conformation. Science 367, 1260–1263. doi:10.1126/science.abb2507
Keywords: furin, COVID-19, small molecules, drug discovery, S protein
Citation: Villoutreix BO, Badiola I and Khatib A-M (2022) Furin and COVID-19: Structure, Function and Chemoinformatic Analysis of Representative Active Site Inhibitors. Front. Drug. Discov. 2:899239. doi: 10.3389/fddsv.2022.899239
Received: 18 March 2022; Accepted: 25 April 2022;
Published: 10 May 2022.
Edited by:
Ho Leung Ng, Kansas State University, United StatesReviewed by:
José J. Naveja, Johannes Gutenberg University Mainz, GermanyCopyright © 2022 Villoutreix, Badiola and Khatib. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bruno O. Villoutreix, QnJ1bm8udmlsbG91dHJlaXhAaW5zZXJtLmZy; Abdel-Majid Khatib, bWFqaWQua2hhdGliQGluc2VybS5mcg==