
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Drug Discov., 25 April 2022
Sec. Anti-Infective Agents
Volume 2 - 2022 | https://doi.org/10.3389/fddsv.2022.898035
This article is part of the Research TopicDevelopment of COVID-19 Therapies: Lessons Learnt and Ongoing EffortsView all 13 articles
 Peter Buchwald1,2*
Peter Buchwald1,2*Blocking protein-protein interactions (PPIs) involved in the initiation of the cell attachment and entry of viruses is an important antiviral mechanism of action including for neutralizing antibodies. Doing it with small-molecule inhibitors (SMIs) is challenging, as it is for all other PPIs, and might require the exploration of chemical space beyond that of typical drug-like structures. However, it could lead to new antiviral agents suitable for oral administration and acting on alternative targets, considerations that are essential for the development of widely acceptable and broad-spectrum preventive or curative therapeutics. Fostemsavir, an antiretroviral that acts via blocking of the gp120–CD4 PPI, supports the feasibility of the concept. Here, a brief review of relevant drug design considerations is presented together with a summary of the progress made toward the identification of SMIs targeting the PPI between the SARS-CoV-2 spike protein and ACE2 that initiates the viral attachment and cellular entry of this coronavirus causing the COVID-19 pandemic. SMIs identified in various screening assays that were also confirmed to have antiviral activity in a live virus or pseudovirus assay with an IC50 < 30 µM so far include several organic dyes (methylene blue, Evans blue, Congo red, direct violet 1), verteporfin, DRI-C23041, and cannabigerolic and cannabidiolic acids. While specificity and activity profiles still need improvement, results so far already provide proof-of-principle evidence for the feasibility of SMIs targeting the SARS-CoV-2-S–hACE2 PPI. Methylene blue, which is approved for clinical use, is orally bioactive, and could act by multiple mechanisms of action, might have potential for repurposing for COVID-19 prevention and treatment.
New drugs introduced during the past century, such as antibacterials (penicillin, 1943) anti-inflammatories (cortisol, 1952), antipsychotics (chlorpromazine, 1953), contraceptives (norethindrone, 1960), anxiolytics (diazepam, 1963), immunosuppressant (cyclosporin A, 1983), antidepressants (fluoxetine, 1987), TNFα-inhibitors (infliximab, 1998), and PD-1–PD-L1 inhibitors (pembrolizumab, nivolumab, 2014)—all shown with their first year of US market approval, are responsible for most of the unprecedented medical progress that happened since then and have completely altered the way life is conducted in industrialized nations. However, truly effective antivirals are still lacking, as the recent coronavirus-inflicted COVID-19 pandemic made painfully clear. The search for antivirals has its own particular challenges, as viruses hijack the reproduction machinery of their host organisms, but progress in drug discovery and development as a whole has been frustratingly slow due to a variety of problems (Proudfoot, 2002; Munos, 2009; Paul et al., 2010; Scannell et al., 2012).
Despite enormous increases in research and development (R&D) investments, the number of newly introduced drugs in the United States remained stubbornly stagnant since the 1960s staying around 20–30 per year (Figure 1) and ∼85% of them represented no or only modest improvements (Wolff, 1995) demonstrating a pervasive need for innovation. This is probably best illustrated by the fact that the number of new drugs approved by the United States Food and Drug Administration (FDA) that were developed per $1 billion of R&D spending in the drug industry (inflation-adjusted) has been decreasing exponentially since 1950, being steadily halved about every 9 years (Scannell et al., 2012). This is mainly due to the highly increased regulatory burden, the unrealistic public expectation of no side effects, the need to outperform existing old drugs, and the depletion of effective new targets for traditional drug design approaches (Walters et al., 2011; Bodor and Buchwald, 2012; Scannell et al., 2012). Regarding the last, it is commonly estimated that there are only about 500 to 1,500 human protein targets that are both “druggable” and “disease modifying”, i.e., only about 2–7% of the ∼20,000 canonical (nonmodified) human proteins encoded by individual genes (Hopkins and Groom, 2002; Russ and Lampel, 2005). In general agreement with this, a survey of small-molecule drug targets counted ∼550 human proteins (plus another ∼180 non-human ones) (Santos et al., 2017). Thus, we are probably beginning to run out of traditional protein targets, at least human ones, and quite likely most low-hanging fruits among such targets that can provide therapeutic benefits have already been picked.
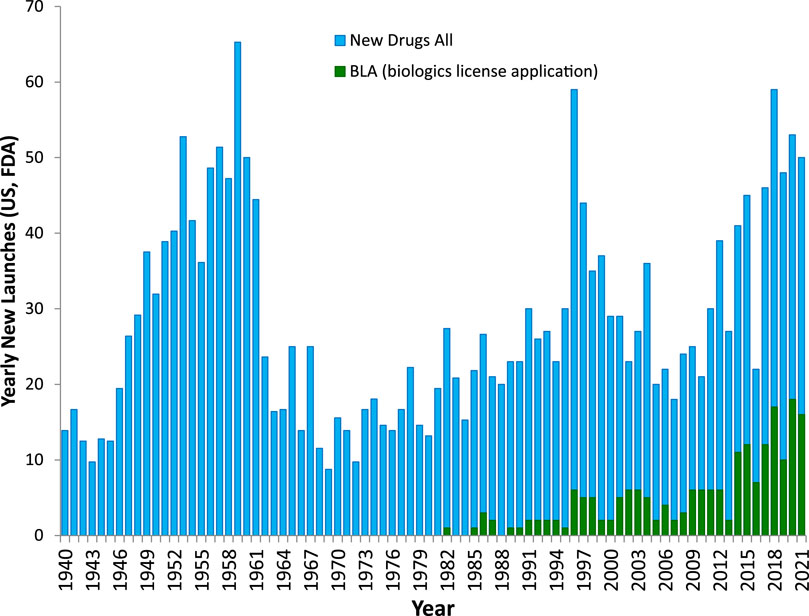
FIGURE 1. The number of all new drugs launched annually in the United States with FDA approval. The number of all new drugs are shown as blue columns with that of new biologics (approved biologic license applications, BLAs) as superimposed green columns. Except for a few peaks in the 1950s, 1990s, and the last decade, it has been quite steady in the 20–30 per year range. Graphic prepared based on data from (Reuben, 1996; Mullard, 2016a; 2020).
Protein-protein interactions (PPIs), the focus of the present review, represent possible additional, alternate targets as evidenced by the increasing number of clinically approved biologics targeting them (Figure 1). For example, one of the latest such successes was the development of cancer immunotherapies targeting immune checkpoint PPIs such as CD80–CTLA4 and PD-1–PD-L1, which has been named Science Breakthrough of the Year in 2013 (Couzin-Frankel, 2013). Unfortunately, PPIs are difficult to modulate with small molecules as the corresponding protein interfaces tend to lack well-defined ligand-binding sites where sufficiently strong interactions can take place to ensure the energy of interaction needed for high affinity binding. Nevertheless, the sheer number of such PPIs, estimated to be in the 300,000 (Zhang et al., 2012; Cheng et al., 2018) to 650,000 (Stumpf et al., 2008) range for humans, implies that a considerable number should still be druggable. Drugs need to be quite potent to be able to compete with naturally present ligands, to be sufficiently specific for their intended target, and to not need unacceptably high doses. Typically, they need to have affinities in the mid-nanomolar range. For example, the median value for all approved drugs has been estimated to be around 20 nM (Overington et al., 2006), which corresponds to a free energy of binding of ΔG0 = –RT·lnKD = –5.94·log10KD [kJ/mol] = 45.7 kJ/mol. To achieve such high energy, small-molecule endogenous agonists and drugs of classic targets such as G-protein coupled receptors (GPCRs) typically bind at binding sites that are fully buried and allow interactions along the entire ligand surface (Figure 2A) (Buchwald, 2019). Since PPI interfaces tend to be relatively large and flat surfaces that lack such well-defined deep pockets, strong binding is difficult to achieve here with small molecules, as interactions are limited to only parts of the total ligand surface. This is illustrated in Figure 2, which compares the 3D structure of a typical fully buried small-molecule agonist at a classic GPCR target (purinergic P2Y12 receptor) with that of a surface-bound small-molecule inhibitor (SMI) of a PPI (venetoclax bound to BCL-2).
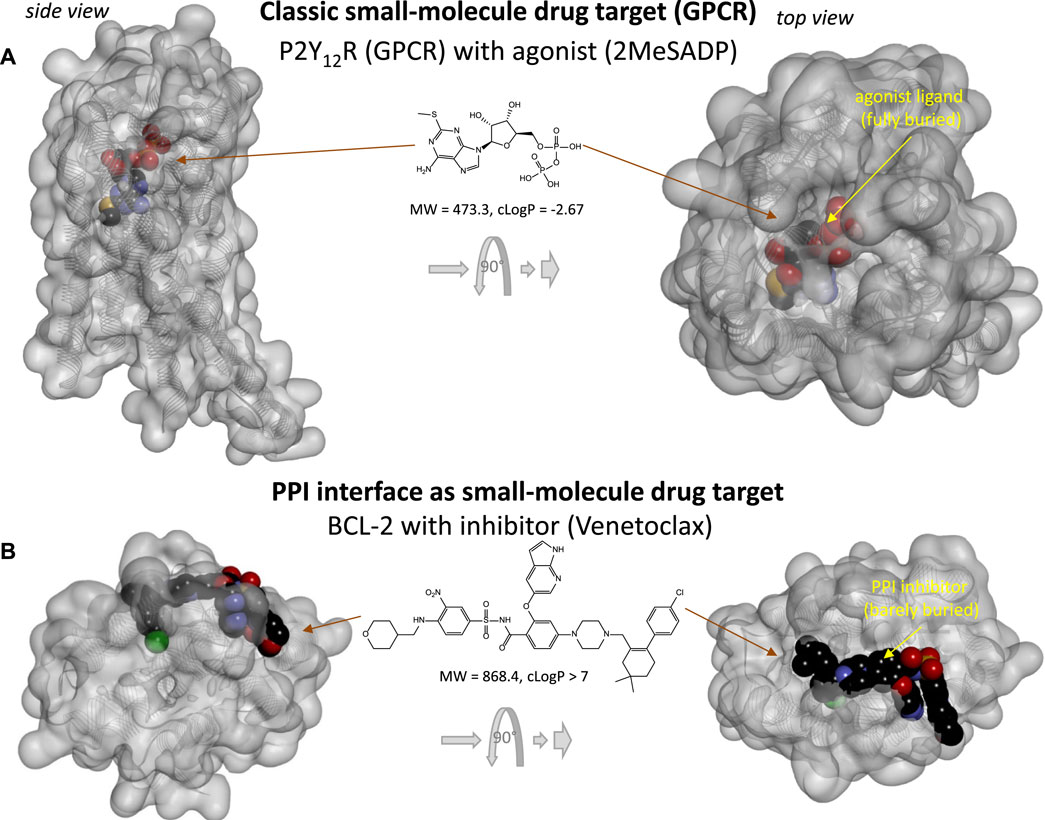
FIGURE 2. Comparing the binding of small-molecule drugs at PPI interface to that at classic targets such as G-protein coupled receptors (GPCRs). (A) 3D structure of the purinergic P2Y12 receptor (a type Aδ GPCR) with an agonist (2MeSADP). Protein structure (PDB ID# 4PXZ (Zhang et al., 2014)) is shown covered with a semi-transparent gray surface; the ligand is shown as darker CPK structure. Two different perspectives are included with the one on the right being a 90° rotated and somewhat enlarged view. Here, the receptor-bound ligand is faded as it fully buried inside the receptor and obscured by the covering surface. (B) 3D structure of BCL-2 (a pro-survival protein targeted in cancer therapeutics) with an FDA-approved SMI of PPI (venetoclax). Structure (PDB ID# 6O0K (Birkinshaw et al., 2019)) is shown as before; however, here the bound PPI inhibitor is barely buried leaving large parts of its surface exposed as indicated by the more vivid colors where directly visible.
Not surprisingly, binding pockets on protein-protein interfaces that are suitable to accommodate small molecules are indeed considerably smaller than those of traditional protein-ligand interactions (Fuller et al., 2009). Typically, existing drugs target a single binding pocket with an average volume of ∼300 Å3, whereas SMIs of PPIs target multiple (3–5) smaller pockets (∼100 Å3) (Fuller et al., 2009). As the achievable maximum energy is limited by the pocket size (Buchwald, 2008), adequate binding affinity at protein interfaces can only be achieved by molecules large enough to reach a sufficient number of such smaller pockets (as illustrated in Figure 2B). The need for larger size can also be seen from the perspective of ligand efficiency, LE, defined as the binding energy per unit size–typically the binding free energy per non-hydrogen atom (Na), LE = ΔG0/Na (Hopkins et al., 2004). Typical ligand-receptor protein interactions have LE of ∼1.5 kJ/atom (Hopkins et al., 2004; Hajduk, 2006; Reynolds et al., 2007; Buchwald, 2008), which corresponds to an about two-fold increase in affinity (decrease in KD or IC50) with the addition of each (non-hydrogen) atom. Such high LE is almost impossible to achieve at PPI interfaces where the bound SMI ligand can interact only along part of its surface (Figure 2B). Thus, to achieve the free energy needed for 20 nM binding (45.7 kJ/mol) with an LE of 1 kJ/atom, structures with more than 45 non-hydrogen atoms are needed, which is already larger than desired for typical “druggability”. SMIs of PPIs were indeed found to be larger than classic drugs including receptor ligands, ion channel modulators, and enzyme inhibitors (Neugebauer et al., 2007).
On the other hand, biologics, such as antibodies and fusion proteins, can interact with proteins along a broader surface and a variety of epitopes without having to rely solely on druggable pockets to achieve adequate affinity and specificity. An increasing number of biologics are being used clinically as they can be highly specific (Figure 1); however, they cannot cross cell membranes, thus cannot reach intracellular targets (Verdine and Walensky, 2007; Hughes et al., 2011; Neklesa et al., 2017), and their protein nature also causes solubility, stability, route of administration (i.e., no oral bioavailability), and biodistribution limitations. Further, since they are foreign proteins, they can act as antigens and elicit strong immune responses in some recipients (Suntharalingam et al., 2006; Wadman, 2006; Leader et al., 2008). All these problems are further exacerbated by their typically long elimination half-lives, which makes it difficult to rapidly eliminate unwanted effects when they occur (Huck et al., 2018). Not surprisingly, FDA-approved biologics encountered more post-market safety issues than did small-molecule drugs (Downing et al., 2017). SMIs of PPIs may represent viable alternatives lacking these problems, if the difficulties related to affinity/specificity can be overcome. While such SMIs were not pursued until relatively recently because they were considered unlikely to be successful due to the aforementioned challenges, during the last 2 decades, it has become clear that SMIs can be effective against at least some PPIs. Most small-molecule PPI modulators are SMIs (i.e., antagonists)—our sole focus here, as so far there are only a limited number of identified small-molecule PPI ‘agonists’ (i.e., enhancers or stabilizers) (Thiel et al., 2012; Milroy et al., 2014; Andrei et al., 2017). SMIs of PPIs, as antagonists in general, can be orthosteric, directly interfering with the interface and competing with the protein ligand, or allosteric, binding away from the interface but causing sufficient conformational change to block binding of the protein ligand.
Tens of PPI-targeting SMIs have reached preclinical development (Arkin and Wells, 2004; Wells and McClendon, 2007; Wilson, 2009; Buchwald, 2010; Arkin et al., 2014; Milroy et al., 2014; Song and Buchwald, 2015; Scott et al., 2016), and three are approved by the FDA for clinical use: lifitegrast (Gadek et al., 2002), venetoclax (Souers et al., 2013), and fostemsavir (Meanwell et al., 2018) (Figure 3). Lifitegrast (SAR 1118) is a LFA-1–ICAM-1 inhibitor developed first at Sunesis (Zhong et al., 2012) from a series originating at Genentech (Gadek et al., 2002) and then clinically by SARcode/Shire; it was approved by the FDA for the treatment of dry eye in 2016 (Xiidra) (Scott et al., 2016). Venetoclax (ABT-199) is part of a small-molecule series developed by Abbott and later AbbVie and designed to target PPIs in the B cell lymphoma 2 (BCL-2) family (Souers et al., 2013). It received FDA approval in 2015 for treatment of chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma (SLL), and later acute myeloid leukemia (AML) (Venclexta, Venclyxto) (Mullard, 2016b). Fostemsavir (BMS-663068) is a water soluble prodrug of temsavir developed by Bristol-Myers Squibb that acts by blocking gp120 binding to CD4 to limit HIV attachment and entry; it was approved by the FDA for clinical use in the US in 2020 as an antiretroviral for adults living with HIV/AIDS (Rukobia) (Meanwell et al., 2018). Finally, maraviroc (Selzentry) is an antiretroviral that can be considered an allosteric SMI of the gp120–CCR5 PPI as it targets CCR5 and stabilizes a conformation no longer recognized by the HIV envelope (Melby and Westby, 2009; Tan et al., 2013). These successes, and particularly that of fostemsavir reemphasize the feasibility of SMIs of PPIs as drug discovery strategy for antivirals. Such SMIs could yield novel therapies that are not only more patient friendly than antibodies (i.e., suitable for oral or inhaled administration), but also less immunogenic, more controllable (shorter half-life/better biodistribution), and possibly even less strain- and mutation-sensitive.
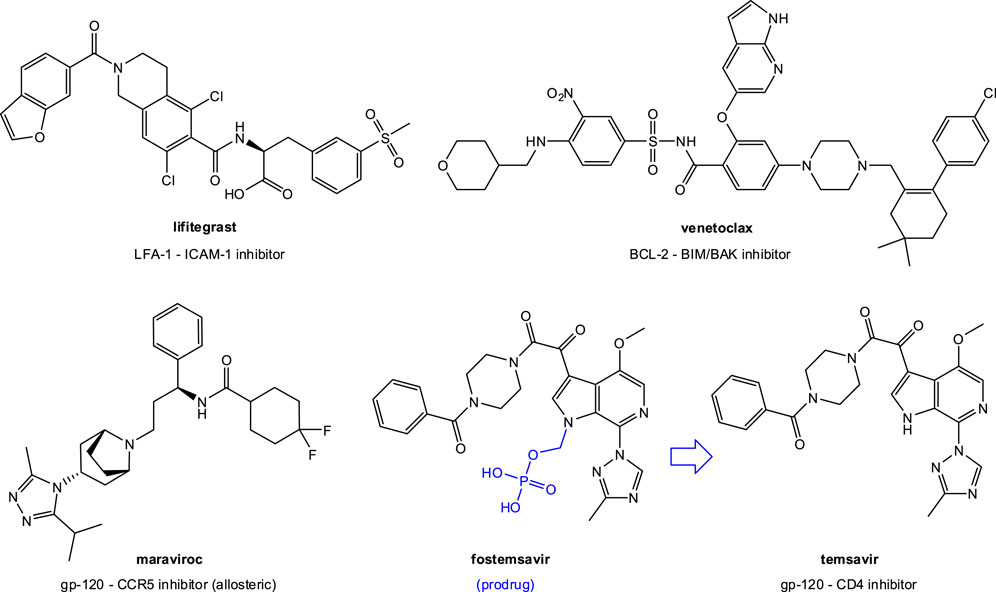
FIGURE 3. SMIs of PPIs approved for clinical use by the FDA. In addition to lifitegrast (an LFA-1–ICAM-1 inhibitor) and venetoclax (a BCL-2–BIM/BAK inhibitor), they include two anti (retro)virals: maraviroc, an allosteric CCR5 inhibitor, and fostemsavir, a prodrug of temsavir, a gp-120–CD4 PPI inhibitor.
While human coronaviruses (CoVs), enveloped positive-stranded RNA viruses mostly responsible for upper respiratory and digestive tract infections, have been circulating for long, SARS-CoV-2 (severe acute respiratory syndrome-coronavirus 2), the most recent one to emerge, became particularly infamous by being the most infectious agent in a century (Tiwari et al., 2020) and the one responsible for the COVID-19 pandemic that caused hundreds of millions of infections and millions of deaths worldwide (Matheson and Lehner, 2020; Shang et al., 2021; V'Kovski et al., 2021). SARS-CoV-2 is one of the seven CoVs known to infect humans, four of which (HCoV 229E, OC43, NL63, and HKU1) are responsible for about a third of the common cold cases and three that are highly pathogenic and caused recent epidemics associated with considerable mortality: SARS-CoV(-1) (2002–2003, ∼10% mortality), MERS-CoV (2012, ∼35% mortality), and now SARS-CoV-2 (2019-), which is less lethal but more transmissible (Guy et al., 2020; Rajgor et al., 2020). While estimates vary, about 3% of the individuals infected with the original SARS-CoV-2 strain needed hospitalization, and the average infection fatality ratio (IFR, percentage of those infected that do not survive) was around 0.5% but in a strongly age-dependent manner increasing exponentially from 0.001 to 0.002% in <20 years old to 10–20% in those >80–90 years old (Salje et al., 2020; O'Driscoll et al., 2021; COVID-19 Forecasting Team, 2022). While difficult to estimate due to changes in vaccination status and treatment options (Bhattacharyya and Hanage, 2022), it has been considerably, several-fold reduced with the more later emerged omicron (B.1.1.529) variant, but likely remained higher than that of influenza (IFR << 0.1%) (Liu et al., 2022; Matsuyama, 2022).
CoVs, which are classified into four genera (α-, ß-, γ-, and δ-CoV), initiate infection with the binding of their spike (S) protein to cell surface receptors followed by membrane fusion and virus entry. For SARS-CoV(-1) and SARS-CoV-2 (as well as HCoV-NL63), the receptor is angiotensin converting enzyme 2 (ACE2) (Lan et al., 2020; Shang et al., 2020; Sivaraman et al., 2021; Zhang et al., 2021). For MERS-CoV, it is dipeptidyl peptidase 4 (DPP4), and for HCoV-229E, human aminopeptidase N (APN; CD13) (V'Kovski et al., 2021). Some ß-coronaviruses (e.g., HCoV-OC43) bind to sialic acid receptors (Tortorici et al., 2019). Thus, blockade of the SARS-CoV-2-S–hACE2 PPI can disrupt infection efficiency, and abrogation of this interaction is a main goal in the development of vaccines and neutralizing antibodies (nAbs) for the COVID-19 pandemic (Jiang et al., 2020; Lv et al., 2020; Tai et al., 2020). In fact, the spike protein is the principal target of nAbs generated following infection by SARS-CoV-2, with the majority of those identified so far recognizing epitopes within the receptor-binding domain (RBD) that binds ACE2 (Sui et al., 2014; Lv et al., 2020; Wang et al., 2020; Wu et al., 2020; Yuan et al., 2020). The spike protein also is the SARS-CoV-2 component of mRNA and adenovirus-based vaccines approved for use (Harvey et al., 2021).
The SARS-CoV-2 spike protein is a homotrimer with monomer units of ∼180 kDa; it is highly glycosylated and is post-translationally cleaved into an S1 and S2 subunit. S1 consists of the amino-terminal domain and the RBD and is responsible for binding to ACE2; S2 includes the trimeric core and is responsible for membrane fusion (Ou et al., 2020; Wang et al., 2020). The RBD located within the S1 domain is known to switch between a standing-up and a lying-down position for receptor binding and immune evasion, respectively (Gil et al., 2020; Shang et al., 2020). Notably, there is a multi-basic furin cleavage site at the S1-S2 boundary, which is unique within b-lineage betacoronaviruses and sarbecoviruses, and is important for the increased infectivity and virulence facilitating the conformational change required for receptor binding (Coutard et al., 2020; Hoffmann et al., 2020; Harvey et al., 2021). It is also an important part of the discussions surrounding the controversies regarding the possible origins of this CoV (Cohen, 2021; Ambati et al., 2022).
There are several possible targets for therapeutic interventions in the CoV lifecycle: viral attachment and entry, uncoating, gRNA replication, translation in ER and Golgi, assembly, and virion release (Guy et al., 2020; V'Kovski et al., 2021; Zhao et al., 2022). Viral attachment and entry are particularly promising among them because they are the first steps in the replication cycle and take place at relatively accessible extracellular sites (Melby and Westby, 2009). They are also well suited for a PPI inhibition focused approach, the subject of the present review. However, targeting viral entry also has its own challenges, as the envelope and fusion glycoproteins are usually the most variable of all virus-encoded proteins. Indeed, the amino acid sequences can vary both within and between individuals, making the spectrum of antiviral activity for any entry inhibitor an important consideration (Melby and Westby, 2009). RNA viruses are known to accumulate mutations over time yielding antibody resistance and requiring the use of antibody cocktails to avoid mutational escape (Baum et al., 2020). Not surprisingly, several SARS-CoV-2 mutants have already emerged some being variants of concern (VOC) with increased transmissibility, higher disease severity, and resistance to neutralizing antibodies, including those elicited by current vaccines (Cai et al., 2021; Gobeil et al., 2021; Harvey et al., 2021; Kupferschmidt, 2021; Wibmer et al., 2021). Currently, as labeled by the WHO, these include alpha (B.1.1.7; first identified in UK, Sep 2020), beta (B.1.351; South Africa, May 2020), gamma (P.1; Brazil, November 2020), delta (B.1.617.2; India, October 2020), and omicron (B.1.1.529; multi/S. Africa, November 2021). Emergence of escape variants is likely to continue as the accumulation of RBD mutations is facilitated by the structural plasticity at the RBD-ACE2 interface, further eroding the activities of therapeutic antibodies and serums of vaccine recipients (Nabel et al., 2022).
Based on the above, it would be particularly important to have broadly cross-reactive agents that can neutralize a wide range of antigenically disparate viruses (Sui et al., 2014). SARS-CoV(-1) and SARS-CoV-2 share close to 80% amino acid identity in their S proteins, raising the possibility of conserved immunogenic surfaces on these antigens, as supported by the identification of some antibodies of possibly broader activity (Lv et al., 2020; Wec et al., 2020; Starr et al., 2021; Martinez et al., 2022; Park et al., 2022) such as the more recently identified RBD-specific antibody DH1047 (Martinez et al., 2022) or the ACE2-mimicking S2K146 (Park et al., 2022). Nevertheless, most SARS-CoV antibodies are not cross-reactive; for example, none of the 206 RBD-specific monoclonal antibodies derived from single B cells of eight SARS-CoV-2 infected individuals in one study cross-reacted with SARS-CoV(-1) or MERS-CoV RBDs (Ju et al., 2020). As already discussed, targeting such PPIs with SMIs is undoubtedly more challenging, but if successful, it could lead to alternative antiviral treatment options with possible benefits including less strain-specific activity.
Despite the undeniable success of the COVID-19 vaccination program, there remains a considerable need to develop new antivirals and especially oral ones, as a significant portion of the population is either unwilling to be vaccinated or unable to do so due to pre-existing medical conditions. Effective oral treatments could have significant impact on this pandemic as they can be taken easily following the first symptoms. Remdesivir, the first small-molecule COVID-19 drug approved by the FDA, must be given intravenously. Considerable effort and financial resources have been invested in the repurposing of approved drugs as possible small-molecule antiviral agents for SARS-CoV-2, but with only minimal success so far. For example, the large WHO Solidarity trial found that repurposed antiviral drugs including hydroxychloroquine, remdesivir, lopinavir, and interferon-β1 had little or no effect on hospitalized COVID-19 patients, as indicated by overall mortality, initiation of ventilation, and duration of hospital stay (WHO Solidarity Trial Consortium et al., 2020). Further, a paper suggested that most drugs identified in many of the screening assays as possibility for being repurposed against SARS-CoV-2 are not working because they inhibit in the in vitro assay by being cationic amphiphilic drugs that cause phospholipidosis, which, however, does not translate into in vivo activity (Tummino et al., 2021). This observation has been questioned and should be treated with caution as many of these molecules have both in vitro and in vivo efficacy with no reported phospholipidosis (Lane and Ekins, 2021).
Regardless, there is an ongoing need to not just repurpose existing drugs but develop novel ones that can combat such infections (Zhao et al., 2022). Recently, two new drugs with classic antiviral mechanisms (i.e., inhibition of protease activity or viral reproduction) have shown promise and granted emergency use authorization by the United States Food and Drug Administration (FDA) for the treatment of COVID-19: molnupiravir (Jayk Bernal et al., 2021) and nirmatrelvir (part of the nirmatrelvir/ritonavir combination Paxlovid) (Owen et al., 2021). Molnupiravir is a prodrug of the synthetic nucleoside derivative N4-hydroxycytidine that exerts antiviral action through introduction of copying errors during viral RNA replication. It was developed originally for the treatment of influenza at Emory University and acquired by Ridgeback Biotherapeutics, who later partnered with Merck (Jayk Bernal et al., 2021). Nirmatrelvir (PF-07321332) is an inhibitor of the SARS-CoV-2 main protease (Mpro) developed at Pfizer starting from PF-00835231, an inhibitor of recombinant SARS-CoV(-1) Mpro identified during the response to the 2002 SARS outbreak (Owen et al., 2021). It showed very promising clinical results as Paxlovid (nirmatrelvir/ritonavir). In addition, AT-527, a double prodrug of a guanosine nucleotide analog, derived from Atea Pharmaceuticals’ nucleotide prodrug platform and shown to be efficacious and well tolerated in hepatitis C virus (HCV) infected subjects (Good et al., 2021), was also pursued, but it was not successful in its first clinical trial. Lessons learned from RNA viruses so far proved that the size and quality of existing antiviral libraries needs to be increased and diversified and polymerase and protease drugs need to be complemented with others targeting different viral proteins (Edwards et al., 2022).
Following the outbreak of COVID-19, due to the immense therapeutic need generated by the pandemic it created, tremendous screening and drug discovery efforts were invested into the identification of effective preventive or therapeutic antiviral interventions in both academic and industrial settings (Ghosh et al., 2020; Shyr et al., 2020; Xiu et al., 2020; Su et al., 2021; Zhao et al., 2022). Here, those directed at identifying SMIs of the SARS-CoV-2-S–hACE2 PPI will be highlighted briefly; some of the earliest ones have been summarized in (Chang et al., 2021). Various screening campaigns have been conducted aiming to identify promising hits mainly from repositionable (repurposable) drug and existing chemical libraries. Assays used (often after virtual screening, i.e., in silico preselection typically via molecular docking in AutoDock or Glide) included ELISA (enzyme-linked immunosorbent assay) types (Carino et al., 2020; Bojadzic et al., 2021b; Fu et al., 2021), AlphaLISA (Hanson et al., 2020), Luminex bead-based (Tsegay et al., 2021), surface plasmon resonance (SPR) (Day et al., 2021; Yu et al., 2021; Zhu et al., 2021), affinity selection-mass spectrometry (van Breemen et al., 2022), NanoBiT (Xiong et al., 2021; Yu et al., 2021), CEBIT (condensate-aided enrichment of biomolecular interactions in test tubes) (Pei et al., 2022), and others. Some possible natural product inhibitor have been highlighted in (Ma et al., 2021); however, most are just molecular docking based hypotheses. Considering that several publications relied solely on in silico derived hypotheses or just one in vitro (often cell-free) inhibitory assay, here, only those compounds will be highlighted first that inhibited this PPI in vitro and have concentration-dependent antiviral activity confirmed in a live virus or pseudovirus assay with a sufficiently promising IC50.
In fact, following the emergence of the SARS-CoV(-1) epidemic in the early 2000s, a few groups already performed high-throughput screening (HTS) assays to identify possible antiviral candidates targeting various early steps in its cell invasion. As part of this, some putative SMI candidates of viral entry have been identified, including, for example, SSAA09E2 (from a screening using a SARS/HIV-luc pseudotyped virus infection assay; pseudovirus IC50 9.7 μM) (Adedeji et al., 2013) and VE607 (from a screening using protection from SARS-CoV-induced cytopathic effects, CPE, in Vero cells as a phenotypic indicator; live virus IC50 1.6 μM) (Kao et al., 2004) (see structures in Figure 4). Other inhibitory small-molecule candidates acting by different mechanism have also been identified; they include, for example, SSAA09E1, SSAA09E3 (Adedeji et al., 2013); MP576, HE602 (Kao et al., 2004); ARB 05–018137, ARB 05–090614 (Severson et al., 2007); K22 (Lundin et al., 2014); and others–see reviews in (Du et al., 2009; Gil et al., 2020; Xiu et al., 2020). Most of these had low micromolar activity (Xiu et al., 2020); however, none of them led to approved preventive or curative therapies for human CoV diseases mainly because in addition to their relatively low (i.e., not nanomolar) potency, they were also not particularly suitable for clinical translatability. They could not pass the preclinical development stage and enter clinical trials due to their poor bioavailability, safety, and pharmacokinetics (Xiu et al., 2020).
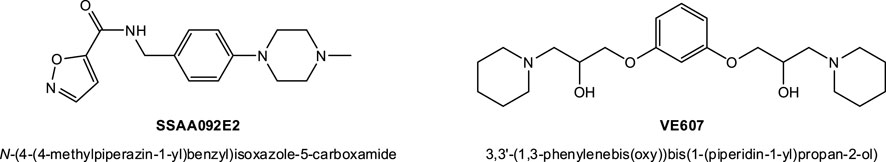
FIGURE 4. Compounds identified before the COVID-19 pandemic as possible SMIs of the SARS-CoV(-1)-S–hACE2 PPI. SSAA09E2 and VE607 have been identified as viral entry inhibitors for SARS-CoV(-1) with low micromolar activity, see text for details.
SMIs of the SARS-CoV-2-S–hACE2 PPI with confirmed antiviral activity in a live virus or pseudovirus assay having IC50 < 30 µM are from the studies listed below in approximate chronological order of their corresponding publications (structures shown in Figure 5). Whenever possible, therapeutic (selectivity) index (TI, SI) estimates are also included as an indicator of the relative safety, as it quantifies the separation between toxic and effective concentrations, TI = TC50/IC50.
• A computational screening interrogating 57,641 compounds followed by SPR screening of a library of 3,141 compounds by Day and co-workers at Griffith University, Australia identified three candidates showing concentration-dependent antiviral activity in vitro: Evans blue, lifitegrast (Figure 3), and lumacaftor (Day et al., 2021) (March 2021). Of these, Evans blue was the most promising candidate and the only one with IC50 < 30 μM; it had a KD of 2 μM for SARS-CoV-2-S and inhibited SARS-CoV-2 infection in Vero E6 cells with an IC50 of 28 μM. According to the authors, it was also non-toxic for up to 1 mM (TC50 > 1,000 μM), suggesting a sufficiently large therapeutic index (TI > 30).
• Our work at the University of Miami, Florida, United States identified several organic dyes (Congo red, direct violet 1, Evans blue) and novel druglike compounds (DRI-C23041, DRI-C91005) that inhibited the interaction of ACE2 with the spike proteins of SARS-CoV-2 as well as SARS-CoV(-1) with low micromolar activity in cell-free ELISA-type assays (IC50’s of 0.2–3.0 μM) (Bojadzic et al., 2021b) (May 2021). Of these, DRI-C23041, Congo red, and direct violet 1 (Figure 5) were also confirmed to inhibit the entry of two different spike-bearing pseudoviruses into HEK293/Vero E6 cells with IC50’s of 5.6/7.4, 20.3/27.4, and 35.8/16.4 μM, respectively. They were also relatively noncytotoxic in the same assay having TC50 > 400 μM for DRI-C23041 (i.e., TI > 70) and >100 μM for Congo red and direct violet 1 (TI > 5). Evans blue, which was the best hit in the work from Day, was identified as an inhibitor, but was not tested here in viral assays as other compounds were more active.
• During this screening, we also identified methylene blue as a SMI and confirmed that it had a quite promising IC50 of 3.5 μM in this viral assay (Bojadzic et al., 2021a) (January 2021). This is of possible interest as a methylene blue is an inexpensive and widely available drug approved by the FDA for the treatment of methemoglobinemia and used for other medical applications. It was also identified by several other groups as having anti-SARS-CoV-2 activity and confirmed to have low micromolar activity in concentration-response studies including with live viruses, possibly due to additional multiple mechanisms of action (Gendrot et al., 2020; Cagno et al., 2021; Gendrot et al., 2021; Murer et al., 2022). Methylene blue seems to be a promiscuous PPI inhibitor with low micromolar activity and a relatively narrow TI, but with multiple evidence suggesting that it clearly inhibits SARS-CoV-2 including VOCs such as delta (B.1.617.2) (Chuang et al., 2022).
• Fu and co-workers at the New York University School of Medicine, United States screened a library of 958 FDA-approved drugs using ELISA-based HTS, and identified five drugs, N-acetylcysteine (NAC), tiopronin (TPR), verteporfin (VP), calcitriol, and racecadotril, to inhibit RBD–ACE2 interaction at both low and high concentrations (Fu et al., 2021) (July 2021). Of these, verteporfin (Visudyne) significantly inhibited pseudovirus entry into hACE2 overexpressing HEK293T cells (IC50 < 0.1 μM) while having a half cytotoxic concentration TC50 ≈ 10 μM (implying TI > 100). Before this work, verteporfin was confirmed by another group to potently inhibit the cytopathic effect produced by SARS-CoV-2 infection with an IC50 < 0.31 μM with indications that the porphyrin ring structure binds the ACE2 receptor (Gu et al., 2021) (December 2020).
• Xiong and co-workers at the Chinese Academy of Sciences, Shanghai and Beijing, China and collaborators virtually screened and filtered compounds from the SPECS database and then purchased 109 selected candidates for follow-up biological testing including NanoBiT and SPR assays to check their ability to block the SARS-CoV-2-S-RBD–ACE2 PPI (Xiong et al., 2021) (Sep 2021). From these, they highlighted two inhibitors as sufficiently promising in pseudovirus assays with some separation between efficacy and cytotoxicity: DC-RA016 (ZINC125276) (IC50 = 22.4 μM) and DC-RA052 (IC50 = 68 μM). The IC50 for DC-RA016 was, however, somewhat overstated due to the way the concentration-response curve was fitted (with a non-zero bottom) as this compound barely caused 50% inhibition at 100 μM (Figure 4B in (Xiong et al., 2021)). Nevertheless, its structure was included here for illustration (Figure 5).
• Finally, van Breeman and co-workers at Oregon State University, Corvallis, OR, United States used affinity selection-mass spectrometry for the discovery of botanical ligands to the SARS-CoV-2 spike protein and found cannabinoid acids from hemp (Cannabis sativa) to be allosteric as well as orthosteric ligands with micromolar affinity for the spike protein (van Breemen et al., 2022) (January 2022). In follow-up virus neutralization assays, cannabigerolic acid and cannabidiolic acid prevented infection of human epithelial cells by a pseudovirus expressing the SARS-CoV-2 spike protein and prevented entry of live SARS-CoV-2 into cells including for variants B.1.1.7 and B.1.351 with IC50’s of 21 and 23 μM (7.7 and 8.4 μg/ml) in the pseudovirus and 67 and 103 μM (24 and 37 μg/ml) in the live virus assay for cannabidiolic and cannabigerolic acid, respectively, where their cytotoxicities were not yet significant (van Breemen et al., 2022). Cannabidiolic acid seems to have a TC50 around 80 μg/ml (Fig. S4 in (van Breemen et al., 2022)) giving TI ≈ 10.
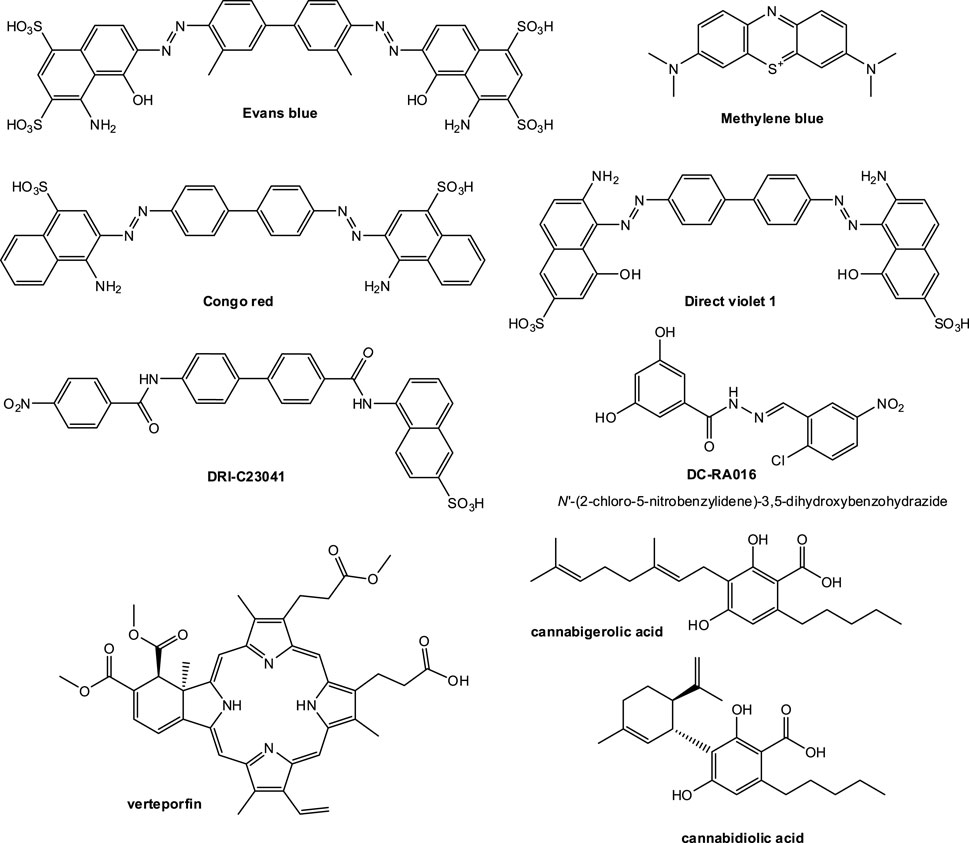
FIGURE 5. Compounds identified so far since the outbreak of COVID-19 as possible SMIs of the SARS-CoV-2-S–hACE2 PPI. Only compounds that have been confirmed to have antiviral activity in a live virus or pseudovirus assay with promising enough activity (IC50 < 30 µM) are shown.
Some of the other works that identified SMI hits but did not include confirmation in viral assay or the inhibitory activity in these assays was not sufficiently potent include (again, in chronological order of their corresponding publications):
• Carino and co-workers at the University of Perugia, Italy used in silico prescreening followed by in vitro confirmation using a commercial SARS-CoV-2 spike inhibitor screening assay kit and found that naturally occurring and clinically available triterpenoids, such as glycyrrhetinic and oleanolic acids, as well as primary and secondary bile acids and their amidated derivatives, such as glyco-ursodeoxycholic acid and semi-synthetic derivatives such as obeticholic acid, reduced the RBD–ACE2 binding (Carino et al., 2020) (October 2020). However, these compounds showed only weak activity and concentration dependence. None of them caused 50% reduction at the highest concentration tested (10 μM). Activities were not confirmed in viral or pseudoviral assays.
• The group of Hanson an co-workers at the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH), Bethesda, MD, United States used an AlphaLISA assay based HTS of 3,384 small-molecule drugs and preclinical compounds suitable for repurposing and identified 25 possible hits (Hanson et al., 2020) (November 2020). However, of these only corilagin was validated in cherry-picking as showing activity against ACE2−RBD with an IC50 of 5.5 μM, and there was no confirmation in viral assays.
• Zhu and co-workers at Peking Union Medical College, Beijing, China used SPR to screen a library of 960 compounds and identified demethylzeylasteral as having promisingly high affinities for S-RBD and ACE2 (KD of 1.0 and 1.7 μM for S-RBD and ACE2, respectively) (Zhu et al., 2021) (Dec. 2020). In a pseudovirus assay, it inhibited entry of SARS-CoV-2 pseudovirus into HEK293T cells to “a certain extent” at nontoxic concentration (7% inhibition at 0.37 μM).
• Yu and co-workers at the Shanghai University of Traditional Chinese Medicine, Shanghai, China used SPR and NanoBit assays to verify the spike protein-binding activity of compounds selected via virtual screening from traditional Chinese medicines and then their inhibitory activities on SARS-CoV-2-S-RBD–ACE2 PPI (Yu et al., 2021) (May 2021). They found glycyrrhizic acid to be the most efficient and nontoxic broad-spectrum anti-CoV SMI with a KD of 0.87 μM toward SARS-CoV-2-S1 as suggested by SPR, but an IC50 of only 22 μM for disrupting the corresponding PPI in the NanoBiT assessment. There was no confirmation in viral assay.
• Tsegay and co-workers at Seattle Children’s Research Institute, Seattle, WA, United States screened 2,701 compounds from an “FDA-approved drug screening library” for their ability to inhibit the binding of recombinant SARS-CoV-2 spike to hACE2 in a Luminex bead-based assay and identified 56 that inhibited in a concentration-dependent manner (June 2021) (Tsegay et al., 2021). Best SMIs were thiostrepton, oxytocin, nilotinib, and hydroxycamptothecin with IC50’s in the 4–9 μM range, but there were no cell-based activity or toxicity assessments.
• Pei and co-workers from Tsinghua University, Beijing, China used CEBIT to screen 2572 FDA approved drugs for their ability to inhibit this PPI and identified six candidate compounds that were confirmed by SPR to bind with KD of 17–780 μM: varenicline, sennoside A, quercetin, quinacrine, methylene blue, and sunitinib (Pei et al., 2022) (March 2022).
In addition to SMIs, peptide-based inhibitors of PPIs are also a possibility–see (Lee et al., 2019; Wang et al., 2021; Trisciuzzi et al., 2022) for recent reviews. Some peptide disruptors have also been reported for SARS-CoV-2–hACE2, but so far none have been very effective (Gil et al., 2020; Xiu et al., 2020; Zhang et al., 2020). A stapled peptide approach carried out at the University of Southern Denmark, Odense, Denmark showed some promise with an IC50 of 3.6 μM for inhibition of the PPI, but no cell-based confirmations were performed (Maas et al., 2021). Relatively high affinity peptide binders of the SARS-CoV-2 spike RBD (KD: 80–970 nM) have been identified by affinity selection-mass spectrometry from a screening of 800 million synthetic peptides at the Massachusetts Institute of Technology (MIT), Cambridge, MA, United States ; however, they turned out to not compete for ACE2 binding (Pomplun et al., 2021). Because of bioavailability, metabolic instability (short half-life), lack of membrane permeability, and other issues, developing peptides into clinically approved drugs is difficult and rarely pursued (Otvos and Wade, 2014; Henninot et al., 2018)—a main reason why we focused here on small-molecule compounds that represent an approach much more likely to ultimately transition into clinical development.
Blocking of PPIs involved in the initiation of cell attachment and entry of CoVs can provide efficient antiviral therapeutics and is the main mechanism of action of biologics such as neutralizing antibodies. SMIs face more challenges to achieve this, as they do for all other PPIs; however, they could lead to new alternative antiviral agents that are suitable for oral administration and act by a different mechanism of action than existing small-molecule antivirals such as protease or viral reproduction inhibitors. Oral bioavailability is highly desirable to achieve widespread usage and compliance (Neklesa et al., 2017), and oral therapeutics are much more suitable for long-term use and/or broadly acceptable preventive use (including for transmission control of viral diseases) than any other routes of administration (Cochrane et al., 1999; Moia et al., 2013). Broadly specific activity is also of considerable interest as it could make possible mutation resistant, multi-strain, or even pan-CoV inhibition. While it is usually difficult if not impossible to achieve with antibodies that tend to be target-specific, it could be more achievable with SMIs. For example, we have shown that while the corresponding antibodies did not cross-react for the human vs. mouse CD40–CD40L PPI, our SMIs did and even maintained similar potencies (Margolles-Clark et al., 2009; Bojadzic and Buchwald, 2019). The impact of SARS-CoV-2 variants on spike and RBD structure and on nAb activity, which could also affect SMIs, has been summarized recently (Nabel et al., 2022). Computational simulations of SARS-CoV-2 spike flexibility and its interactions with other proteins are being carried out and should provide helpful tools for future screening efforts (Pedebos and Khalid, 2022).
SMIs of the SARS-CoV-2-S–hACE2 PPI identified so far and summarized above provide proof-of-principle evidence for the feasibility of such a small-molecule approach, but it remains to be seen if they can ultimately lead to clinically usable therapies as specificity and activity profiles still need improvement. While specific goals vary somewhat depending on the specifics of the project, small-molecule drug candidates are generally expected to have, among others: • potency in at least the hundred-nanomolar range (i.e., IC50 < 100 nM meaning pKi > 7) (the median of existing drugs being ∼20 nM); • adequate selectivity/specificity (>20× versus other targets is a reasonable minimum and >100× is desirable); • good safety profile (TI > 30 and optimally >100 in early studies plus passing of all toxicity studies); • adequate solubility and partition properties (needed to achieve acceptable formulation and desired delivery to the intended target); and • acceptable oral bioavailability and duration of action (somewhat flexible, but oral bioavailability F% > 30% and elimination half-life t1/2 > 4 h are reasonable goals) (Williams, 2005; Smith and O’Donnell, 2007; Bodor and Buchwald, 2012). Some of these are undoubtedly more difficult to achieve with small molecules targeting PPIs than with those targeting classic drug targets such as GPCRs, ion channels, and enzymes that have pre-formed domains (pockets) to bind their natural ligands with good affinity and specificity. Problems related to lack of good binding pockets and thus a relatively low ligand efficiency (LE) have been reviewed briefly earlier (Small-Molecule Inhibitors of Protein-Protein Interactions; see also illustration in Figure 2). Because of this, SMIs of PPIs tend to be larger structures than classic drugs (Neugebauer et al., 2007), and it is now well-recognized that the chemical space of existing drugs and corresponding screening libraries does not correspond well with that of promising SMIs of PPIs (Pagliaro et al., 2004; Neugebauer et al., 2007; Reynès et al., 2010; Sperandio et al., 2010; Morelli et al., 2011). Fortunately, computational prescreening including exploration of relevant physicochemical properties can provide valuable information (Villoutreix et al., 2012; Trisciuzzi et al., 2019) and there are now databases, such as TIMBAL (Higueruelo et al., 2009), 2P2I (Bourgeas et al., 2010), or iPPI-DB (Labbe et al., 2016; Torchet et al., 2021), that contain an increasing number of 3D structures for protein-protein and protein-inhibitor complexes. These can make computationally enriched library selection much more successful, which has been shown to accelerate hit discovery (Milhas et al., 2016). A chemical library of >10,000 compounds dedicated to PPI inhibition has been developed (Fr-PPIChem) and is freely available upon request for experimental screening against PPIs (Bosc et al., 2020).
Larger structures, often with multiple aromatic rings, are usually better suited for effective PPI inhibition (Che et al., 2006; Fletcher and Hamilton, 2006; Hershberger et al., 2007); however, these tend to violate the widely used “rule-of-five” (Ro5) criteria, which includes MW < 500 (Lipinski et al., 1997; Lipinski, 2004) and has been widely used to guide candidate selection and ensure adequate oral bioavailability and ADME (absorption, distribution, metabolism, and excretion) profile. Nevertheless, an increasing number of new drugs have been launched lately (including venetoclax and fostemsavir discussed earlier) that significantly violate these empirical rules proving that oral bioavailability can be achieved even in the “beyond the rule-of-five” chemical space (DeGoey et al., 2017; Doak and Kihlberg, 2017). Along these lines, it is instructive to highlight that the first promising lead during the development of venetoclax (ABT-199) was ABT-737, which was so far from being suitable for formulation as a drug that one of its developers jokingly described it as having “the biophysical properties of brick dust” (Mullard, 2016b). Similarly, the incredibly tedious process of medicinal chemistry optimization that was required to make the original lead of the series that ultimately led to fostemsavir (BMS-663068) as a clinical product is nicely described in detail in (Meanwell et al., 2018).
Hits obtained so far for this PPI (Figure 5) reemphasize that our approach relying on the chemical space of organic dyes as a starting point when screening for SMIs of PPIs makes sense. For example, Evans blue which was the best hit identified from a HTS of >3,000 compounds selected after in silico prescreening of ∼60,000 structures (Day et al., 2021), came up as a hit from our screening of a much smaller library of <100 dyes (Bojadzic et al., 2021b). For obvious reasons, organic dyes have good affinity for proteins (Hunger, 2003), and they contain privileged structures for protein binding (Che et al., 2006; Fletcher and Hamilton, 2006; Hershberger et al., 2007). Thus, contrary to commonly available drug-like libraries, they are a good starting point to identify SMIs of PPIs. We have even found organic dyes that are promiscuous PPI inhibitors (Ganesan et al., 2011; Ganesan and Buchwald, 2013). Of course, organic dyes are not particularly suitable for therapeutic development because of their strong color (plus, for azo dyes, their quick metabolic degradation (Levine, 1991; Feng et al., 2012)). Nevertheless, we have shown in at least one case (the CD40–CD40L PPI, a member of the TNF superfamily) that new drug-like SMIs can be developed by first using this chemical space to identify the molecular scaffold required for activity and then removing the color-causing chromophore(s) while retaining PPI inhibitory activity (Chen et al., 2017; Bojadzic et al., 2018). Specificity can be an issue with dyes, and indeed most dyes found here as promising SMIs of the SARS-CoV-2-S–hACE PPI seem to be quite non-specific as the specificity plot shown in Figure 6 illustrates (data from (Bojadzic et al., 2021b)). Also, many azo-containing dyes are likely PAINS (pan-assay interference compounds) that can show up as false positives in screening assays (Baell and Walters, 2014; Aldrich et al., 2017); thus, they need to be treated carefully to ensure, for example, that the PPI inhibitory activity seen is not due to aggregation/polymolecular conglomeration. Nevertheless, medicinal chemistry optimization for specificity should still be feasible, and it is worth remembering that modern medicinal chemistry emerged in the early 20th century from the synthetic dye industry of the late 19th century (mostly in Germany at that time) (Paterson, 1984). Following the discovery of the first synthetic dye in 1856, Paul Ehrlich (1854–1915) (Drews, 2004; Bosch and Rosich, 2008), who acquired his medical doctor’s degree with a thesis on “the theory and practice of histological staining”, laid the foundations of chemotherapy. The analogy between the azo –N=N– bond common in many of these dyes and the arsenic bond –As=As– led to his search of arsenicals and ultimately to the discovery of arsphenamine (Salvarsan, no. 606), the first modern antimicrobial, in 1909. A few years later, the testing of thousands of azo dye related compounds and the contributions of Gerhard Domagk (1895–1964) led to the discovery of Prontosil (1932), the first effective sulfonamide antibacterial.
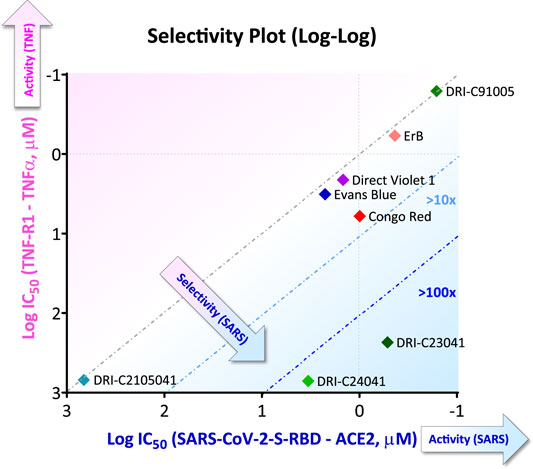
FIGURE 6. Illustrative selectivity plot comparing inhibitory activity of SMIs against two PPIs. Selectivity plot showing the inhibitory activity of some of the compounds found to inhibit the SARS-CoV-2-S-RBD–hACE2 as the targeted PPI (quantified as log IC50) versus that against another non-targeted PPI (here, TNF-R1–TNF-α PPI). More active and selective compounds are clustered toward the lower right corner. Figure prepared with data from (Bojadzic et al., 2021b).
None of the SMIs of SARS-CoV-2-S–hACE2 identified so far and discussed here (Figure 5) have reached clinical development (Zhao et al., 2022); in fact, none seem to have even been evaluated in existing preclinical animal models for SARS-CoV-2 (Takayama, 2020; Saravanan et al., 2022). Methylene blue, a phenothiazine dye we have identified as such an SMI (Bojadzic et al., 2021a), is, in fact, included in the WHO List of Essential Medicines and is orally bioactive; thus it might have some potential for repositioning for COVID-19 prevention and treatment especially as its low micromolar anti-CoV activity, possibly due to multiple mechanisms of action, has been confirmed by several other groups (Chuang et al., 2022). Overall, results summarized here provide proof-of-principle evidence for the feasibility of such SMI approaches toward antivirals that inhibit CoV attachment and entry, and they serve as a first guide of the chemical space needed to achieve this.
PB is the sole author; he conceived and wrote the manuscript.
The author declares the following competing financial interest(s): The University of Miami has filed a patent on some of the DRI-C compounds discussed here and their use for potential antiviral applications with PB as inventor.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The author is grateful to people in his lab (Oscar Alcazar, Damir Bojadzic, and Sung-Ting Chuang) for their dedicated work on this and other projects despite the challenges imposed by the COVID-19 pandemic and the related restrictions and lockdowns.
Adedeji, A. O., Severson, W., Jonsson, C., Singh, K., Weiss, S. R., and Sarafianos, S. G. (2013). Novel Inhibitors of Severe Acute Respiratory Syndrome Coronavirus Entry that Act by Three Distinct Mechanisms. J. Virol. 87 (14), 8017–8028. doi:10.1128/jvi.00998-13
Aldrich, C., Bertozzi, C., Georg, G. I., Kiessling, L., Lindsley, C., Liotta, D., et al. (2017). The Ecstasy and Agony of Assay Interference Compounds. J. Med. Chem. 60 (6), 2165–2168. doi:10.1021/acs.jmedchem.7b00229
Ambati, B. K., Varshney, A., Palú, G., Uhal, B. D., Uversky, V. N., et al. (2022). MSH3 Homology and Potential Recombination Link to SARS-CoV-2 Furin Cleavage Site. Front.Virol. 2, 834808. doi:10.3389/fviro.2022.834808
Andrei, S. A., Sijbesma, E., Hann, M., Davis, J., O’Mahony, G., Perry, M. W. D., et al. (2017). Stabilization of Protein-Protein Interactions in Drug Discovery. Expert Opin. Drug Discov. 12 (9), 925–940. doi:10.1080/17460441.2017.1346608
Arkin, M. R., Tang, Y., and Wells, J. A. (2014). Small-molecule Inhibitors of Protein-Protein Interactions: Progressing toward the Reality. Chem. Biol. 21 (9), 1102–1114. doi:10.1016/j.chembiol.2014.09.001
Arkin, M. R., and Wells, J. A. (2004). Small-molecule Inhibitors of Protein-Protein Interactions: Progressing towards the Dream. Nat. Rev. Drug Discov. 3 (4), 301–317. doi:10.1038/nrd1343
Baell, J., and Walters, M. A. (2014). Chemistry: Chemical con artists foil drug discovery. Nature 513 (7519), 481–483. doi:10.1038/513481a
Baum, A., Fulton, B. O., Wloga, E., Copin, R., Pascal, K. E., Russo, V., et al. (2020). Antibody Cocktail to SARS-CoV-2 Spike Protein Prevents Rapid Mutational Escape Seen with Individual Antibodies. Science 369 (6506), 1014–1018. doi:10.1126/science.abd0831
Bhattacharyya, R. P., and Hanage, W. P. (2022). Challenges in Inferring Intrinsic Severity of the SARS-CoV-2 Omicron Variant. N. Engl. J. Med. 386 (7), e14. doi:10.1056/nejmp2119682
Birkinshaw, R. W., Gong, J.-n., Luo, C. S., Lio, D., White, C. A., Anderson, M. A., et al. (2019). Structures of BCL-2 in Complex with Venetoclax Reveal the Molecular Basis of Resistance Mutations. Nat. Commun. 10 (1), 2385. doi:10.1038/s41467-019-10363-1
Bodor, N., and Buchwald, P. (2012). Retrometabolic Drug Design and Targeting. Hoboken, NJ: Wiley. doi:10.1002/9781118407738Retrometabolic Drug Design and Targeting
Bojadzic, D., Alcazar, O., and Buchwald, P. (2021a). Methylene Blue Inhibits the SARS-CoV-2 Spike-ACE2 Protein-Protein Interaction-A Mechanism that Can Contribute to its Antiviral Activity against COVID-19. Front. Pharmacol. 11 (1), 600372. doi:10.3389/fphar.2020.600372
Bojadzic, D., Alcazar, O., Chen, J., Chuang, S.-T., Condor Capcha, J. M., Shehadeh, L. A., et al. (2021b). Small-Molecule Inhibitors of the Coronavirus Spike: ACE2 Protein-Protein Interaction as Blockers of Viral Attachment and Entry for SARS-CoV-2. ACS Infect. Dis. 7 (6), 1519–1534. doi:10.1021/acsinfecdis.1c00070
Bojadzic, D., and Buchwald, P. (2019). CD40-targeting KGYY15 Peptides Do Not Efficiently Block the CD40-CD40L Interaction. Diabetologia 62 (11), 2158–2160. doi:10.1007/s00125-019-04996-6
Bojadzic, D., Chen, J., Alcazar, O., and Buchwald, P. (2018). Design, Synthesis, and Evaluation of Novel Immunomodulatory Small Molecules Targeting the CD40-CD154 Costimulatory Protein-Protein Interaction. Molecules 23 (5), 1153. doi:10.3390/molecules23051153
Bosc, N., Muller, C., Hoffer, L., Lagorce, D., Bourg, S., Derviaux, C., et al. (2020). Fr-PPIChem: An Academic Compound Library Dedicated to Protein-Protein Interactions. ACS Chem. Biol. 15 (6), 1566–1574. doi:10.1021/acschembio.0c00179
Bosch, F. e. l., and Rosich, L. (2008). The Contributions of Paul Ehrlich to Pharmacology: a Tribute on the Occasion of the Centenary of His Nobel Prize. Pharmacology 82 (3), 171–179. doi:10.1159/000149583
Bourgeas, R., Basse, M.-J., Morelli, X., and Roche, P. (2010). Atomic Analysis of Protein-Protein Interfaces with Known Inhibitors: the 2P2I Database. PLoS One 5 (3), e9598. doi:10.1371/journal.pone.0009598
Buchwald, P. (2019). A Receptor Model with Binding Affinity, Activation Efficacy, and Signal Amplification Parameters for Complex Fractional Response versus Occupancy Data. Front. Pharmacol. 10, 605. doi:10.3389/fphar.2019.00605
Buchwald, P. (2008). Glucocorticoid Receptor Binding: a Biphasic Dependence on Molecular Size as Revealed by the Bilinear LinBiExp Model. Steroids 73 (2), 193–208. doi:10.1016/j.steroids.2007.10.001
Buchwald, P. (2010). Small-molecule Protein-Protein Interaction Inhibitors: Therapeutic Potential in Light of Molecular Size, Chemical Space, and Ligand Binding Efficiency Considerations. IUBMB Life 62 (10), 724–731. doi:10.1002/iub.383
Cagno, V., Medaglia, C., Cerny, A., Cerny, T., Zwygart, A. C.-A., Cerny, E., et al. (2021). Methylene Blue Has a Potent Antiviral Activity against SARS-CoV-2 and H1N1 Influenza Virus in the Absence of UV-Activation In Vitro. Sci. Rep. 11 (1), 14295. doi:10.1038/s41598-021-92481-9
Cai, Y., Zhang, J., Xiao, T., Lavine, C. L., Rawson, S., Peng, H., et al. (2021). Structural Basis for Enhanced Infectivity and Immune Evasion of SARS-CoV-2 Variants. Science 373 (6555), 642–648. doi:10.1126/science.abi9745
Carino, A., Moraca, F., Fiorillo, B., Marchianò, S., Sepe, V., Biagioli, M., et al. (2020). Hijacking SARS-CoV-2/ace2 Receptor Interaction by Natural and Semi-synthetic Steroidal Agents Acting on Functional Pockets on the Receptor Binding Domain. Front. Chem. 8, 572885. doi:10.3389/fchem.2020.572885
C. G. Smith, and J. T. O’Donnell (Editors) (2007). The Process of New Drug Discovery and Development (New York: Informa Healthcare). doi:10.1201/9781420004236
Chang, C.-k., Lin, S.-M., Satange, R., Lin, S.-C., Sun, S.-C., Wu, H.-Y., et al. (2021). Targeting Protein-Protein Interaction Interfaces in COVID-19 Drug Discovery. Comput. Struct. Biotechnol. J. 19, 2246–2255. doi:10.1016/j.csbj.2021.04.003
Che, Y., Brooks, B. R., and Marshall, G. R. (2006). Development of Small Molecules Designed to Modulate Protein-Protein Interactions. J. Comput. Aided Mol. Des. 20 (2), 109–130. doi:10.1007/s10822-006-9040-8
Chen, J., Song, Y., Bojadzic, D., Tamayo-Garcia, A., Landin, A. M., Blomberg, B. B., et al. (2017). Small-molecule Inhibitors of the CD40-Cd40l Costimulatory Protein-Protein Interaction. J. Med. Chem. 60 (21), 8906–8922. doi:10.1021/acs.jmedchem.7b01154
Cheng, F., Desai, R. J., Handy, D. E., Wang, R., Schneeweiss, S., Barabási, A.-L., et al. (2018). Network-based Approach to Prediction and Population-Based Validation of In Silico Drug Repurposing. Nat. Commun. 9 (1), 2691. doi:10.1038/s41467-018-05116-5
Chuang, S.-T., Papp, H., Kuczmog, A., Eells, R., Condor Capcha, J. M., Shehadeh, L. A., et al. (2022). Methylene Blue Is a Nonspecific Protein-Protein Interaction Inhibitor with Potential for Repurposing as an Antiviral for COVID-19. bioRxiv, 2022.2003. doi: doi:10.1101/2022.03.22.485299
Cochrane, G. M., Horne, R., and Chanez, P. (1999). Compliance in Asthma. Respir. Med. 93 (11), 763–769. doi:10.1016/s0954-6111(99)90260-3
Coutard, B., Valle, C., de Lamballerie, X., Canard, B., Seidah, N. G., and Decroly, E. (2020). The Spike Glycoprotein of the New Coronavirus 2019-nCoV Contains a Furin-like Cleavage Site Absent in CoV of the Same Clade. Antiviral Res. 176, 104742. doi:10.1016/j.antiviral.2020.104742
Couzin-Frankel, J. (2013). Cancer Immunotherapy. Science 342 (6165), 1432–1433. doi:10.1126/science.342.6165.1432
COVID-19 Forecasting Team (2022). Variation in the COVID-19 Infection-Fatality Ratio by Age, Time, and Geography during the Pre-vaccine Era: a Systematic Analysis. Lancet 399 epub. doi:10.1016/s0140-6736(21)02867-1
Day, C. J., Bailly, B., Guillon, P., Dirr, L., Jen, F. E.-C., Spillings, B. L., et al. (2021). Multidisciplinary Approaches Identify Compounds that Bind to Human ACE2 or SARS-CoV-2 Spike Protein as Candidates to Block SARS-CoV-2-ACE2 Receptor Interactions. mBio 12 (2), e03681–03620. doi:10.1128/mBio.03681-20
DeGoey, D. A., Chen, H.-J., Cox, P. B., and Wendt, M. D. (2017). Beyond the Rule of 5: Lessons Learned from AbbVie's Drugs and Compound Collection. J. Med. Chem. 61 (7), 2636–2651. doi:10.1021/acs.jmedchem.7b00717
Doak, B. C., and Kihlberg, J. (2017). Drug Discovery beyond the Rule of 5 - Opportunities and Challenges. Expert Opin. Drug Discov. 12 (2), 115–119. doi:10.1080/17460441.2017.1264385
Downing, N. S., Shah, N. D., Aminawung, J. A., Pease, A. M., Zeitoun, J.-D., Krumholz, H. M., et al. (2017). Postmarket Safety Events Among Novel Therapeutics Approved by the US Food and Drug Administration between 2001 and 2010. JAMA 317 (18), 1854–1863. doi:10.1001/jama.2017.5150
Drews, J. (2004). Paul Ehrlich: Magister Mundi. Nat. Rev. Drug Discov. 3 (9), 797–801. doi:10.1038/nrd1498
Du, L., He, Y., Zhou, Y., Liu, S., Zheng, B.-J., and Jiang, S. (2009). The Spike Protein of SARS-CoV - a Target for Vaccine and Therapeutic Development. Nat. Rev. Microbiol. 7 (3), 226–236. doi:10.1038/nrmicro2090
Edwards, A. M., Baric, R. S., Saphire, E. O., and Ulmer, J. B. (2022). Stopping Pandemics before They Start: Lessons Learned from SARS-CoV-2. Science 375 (6585), 1133–1139. doi:10.1126/science.abn1900
Feng, J., Cerniglia, C. E., and Chen, H. (2012). Toxicological Significance of Azo Dye Metabolism by Human Intestinal Microbiota. Front. Biosci. E4, 568–586. doi:10.2741/400
Fletcher, S., and Hamilton, A. D. (2006). Targeting Protein-Protein Interactions by Rational Design: Mimicry of Protein Surfaces. J. R. Soc. Interf. 3 (7), 215–233. doi:10.1098/rsif.2006.0115
Fu, W., Chen, Y., Wang, K., Hettinghouse, A., Hu, W., Wang, J.-Q., et al. (2021). Repurposing FDA-Approved Drugs for SARS-CoV-2 through an ELISA-Based Screening for the Inhibition of RBD/ACE2 Interaction. Protein Cell 12 (7), 586–591. doi:10.1007/s13238-020-00803-w
Fuller, J. C., Burgoyne, N. J., and Jackson, R. M. (2009). Predicting Druggable Binding Sites at the Protein-Protein Interface. Drug Discov. Today 14 (3-4), 155–161. doi:10.1016/j.drudis.2008.10.009
Gadek, T. R., Burdick, D. J., McDowell, R. S., Stanley, M. S., Marsters, J. C., Paris, K. J., et al. (2002). Generation of an LFA-1 Antagonist by the Transfer of the ICAM-1 Immunoregulatory Epitope to a Small Molecule. Science 295 (5557), 1086–1089. doi:10.1126/science.295.5557.1086
Ganesan, L., and Buchwald, P. (2013). The Promiscuous Protein Binding Ability of Erythrosine B Studied by Metachromasy (Metachromasia). J. Mol. Recognit. 26 (4), 181–189. doi:10.1002/jmr.2263
Ganesan, L., Margolles-Clark, E., Song, Y., and Buchwald, P. (2011). The Food Colorant Erythrosine Is a Promiscuous Protein-Protein Interaction Inhibitor. Biochem. Pharmacol. 81 (6), 810–818. doi:10.1016/j.bcp.2010.12.020
Gendrot, M., Andreani, J., Duflot, I., Boxberger, M., Le Bideau, M., Mosnier, J., et al. (2020). Methylene Blue Inhibits Replication of SARS-CoV-2 In Vitro. Int. J. Antimicrob. Agents 56, 106202. doi:10.1016/j.ijantimicag.2020.106202
Gendrot, M., Jardot, P., Delandre, O., Boxberger, M., Andreani, J., Duflot, I., et al. (2021). In Vitro evaluation of the Antiviral Activity of Methylene Blue Alone or in Combination against SARS-CoV-2. J. Clin. Med. 10, 3007. doi:10.3390/jcm10143007
Ghosh, A. K., Brindisi, M., Shahabi, D., Chapman, M. E., and Mesecar, A. D. (2020). Drug Development and Medicinal Chemistry Efforts toward SARS‐Coronavirus and Covid‐19 Therapeutics. ChemMedChem 15 (11), 907–932. doi:10.1002/cmdc.202000223
Gil, C., Ginex, T., Maestro, I., Nozal, V., Barrado-Gil, L., Cuesta-Geijo, M. Á., et al. (2020). COVID-19: Drug Targets and Potential Treatments. J. Med. Chem. 63 (21), 12359–12386. doi:10.1021/acs.jmedchem.0c00606
Gobeil, S. M.-C., Janowska, K., McDowell, S., Mansouri, K., Parks, R., Stalls, V., et al. (2021). Effect of Natural Mutations of SARS-CoV-2 on Spike Structure, Conformation, and Antigenicity. Science 373 (6555). doi:10.1126/science.abi6226
Good, S. S., Westover, J., Jung, K. H., Zhou, X.-J., Moussa, A., La Colla, P., et al. (2021). AT-527, a Double Prodrug of a Guanosine Nucleotide Analog, Is a Potent Inhibitor of SARS-CoV-2 In Vitro and a Promising Oral Antiviral for Treatment of COVID-19. Antimicrob. Agents Chemother. 65 (4), e02479–02420. doi:10.1128/aac.02479-20
Gu, C., Wu, Y., Guo, H., Zhu, Y., Xu, W., Wang, Y., et al. (2021). Protoporphyrin IX and Verteporfin Potently Inhibit SARS-CoV-2 Infection In Vitro and in a Mouse Model Expressing Human ACE2. Sci. Bull. 66 (9), 925–936. doi:10.1016/j.scib.2020.12.005
Guy, R. K., DiPaola, R. S., Romanelli, F., and Dutch, R. E. (2020). Rapid Repurposing of Drugs for COVID-19. Science 368 (6493), 829–830. doi:10.1126/science.abb9332
Hajduk, P. J. (2006). Fragment-based Drug Design: How Big Is Too Big? J. Med. Chem. 49 (24), 6972–6976. doi:10.1021/jm060511h
Hanson, Q. M., Wilson, K. M., Shen, M., Itkin, Z., Eastman, R. T., Shinn, P., et al. (2020). Targeting ACE2-RBD Interaction as a Platform for COVID-19 Therapeutics: Development and Drug-Repurposing Screen of an AlphaLISA Proximity Assay. ACS Pharmacol. Transl. Sci. 3, 1352–1360. doi:10.1021/acsptsci.0c00161
Harvey, W. T., Carabelli, A. M., Jackson, B., Gupta, R. K., Thomson, E. C., Harrison, E. M., et al. (2021). SARS-CoV-2 Variants, Spike Mutations and Immune Escape. Nat. Rev. Microbiol. 19 (7), 409–424. doi:10.1038/s41579-021-00573-0
Henninot, A., Collins, J. C., and Nuss, J. M. (2018). The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 61 (4), 1382–1414. doi:10.1021/acs.jmedchem.7b00318
Hershberger, S., Lee, S.-G., and Chmielewski, J. (2007). Scaffolds for Blocking Protein-Protein Interactions. Curr. Top. Med. Chem. 7 (10), 928–942. doi:10.2174/156802607780906726
Higueruelo, A. P., Schreyer, A., Bickerton, G. R. J., Pitt, W. R., Groom, C. R., and Blundell, T. L. (2009). Atomic Interactions and Profile of Small Molecules Disrupting Protein-Protein Interfaces: the TIMBAL Database. Chem. Biol. Drug Des. 74 (5), 457–467. doi:10.1111/j.1747-0285.2009.00889.x
Hoffmann, M., Kleine-Weber, H., and Pöhlmann, S. (2020). A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cel 78 (4), 779–784. doi:10.1016/j.molcel.2020.04.022
Hopkins, A. L., Groom, C. R., and Alex, A. (2004). Ligand Efficiency: a Useful Metric for lead Selection. Drug Discov. Today 9 (10), 430–431. doi:10.1016/S1359-6446(04)03069-7
Hopkins, A. L., and Groom, C. R. (2002). The Druggable Genome. Nat. Rev. Drug Discov. 1 (9), 727–730. doi:10.1038/nrd892
Huck, B. R., Kötzner, L., and Urbahns, K. (2018). Small Molecules Drive Big Improvements in Immuno-Oncology Therapies. Angew. Chem. Int. Ed. 57 (16), 4412–4428. doi:10.1002/anie.201707816
Hughes, J., Rees, S., Kalindjian, S., and Philpott, K. (2011). Principles of Early Drug Discovery. Br. J. Pharmacol. 162 (6), 1239–1249. doi:10.1111/j.1476-5381.2010.01127.x
Jayk Bernal, A., Gomes da Silva, M. M., Musungaie, D. B., Kovalchuk, E., Gonzalez, A., Delos Reyes, V., et al. (2022). Molnupiravir for Oral Treatment of COVID-19 in Nonhospitalized Patients. N. Engl. J. Med. 386, 509–520. doi:10.1056/nejmoa2116044
Jiang, S., Zhang, X., Yang, Y., Hotez, P. J., and Du, L. (2020). Neutralizing Antibodies for the Treatment of COVID-19. Nat. Biomed. Eng. 4 (12), 1134–1139. doi:10.1038/s41551-020-00660-2
Ju, B., Zhang, Q., Ge, J., Wang, R., Sun, J., Ge, X., et al. (2020). Human Neutralizing Antibodies Elicited by SARS-CoV-2 Infection. Nature 584, 115–119. doi:10.1038/s41586-020-2380-z
J. Wilson, A. (2009). Inhibition of Protein-Protein Interactions Using Designed Molecules. Chem. Soc. Rev. 38 (12), 3289–3300. doi:10.1039/b807197g
Kao, R. Y., Tsui, W. H. W., Lee, T. S. W., Tanner, J. A., Watt, R. M., Huang, J.-D., et al. (2004). Identification of Novel Small-Molecule Inhibitors of Severe Acute Respiratory Syndrome-Associated Coronavirus by Chemical Genetics. Chem. Biol. 11 (9), 1293–1299. doi:10.1016/j.chembiol.2004.07.013
K. Hunger (Editor) (2003). Industrial Dyes. Chemistry, Properties, Applications (Weinheim: Wiley VCH). doi:10.1002/3527602011
Kupferschmidt, K. (2021). Evolving Threat. Science 373 (6557), 844–849. doi:10.1126/science.373.6557.844
Labbé, C. M., Kuenemann, M. A., Zarzycka, B., Vriend, G., Nicolaes, G. A. F., Lagorce, D., et al. (2016). iPPI-DB: an Online Database of Modulators of Protein-Protein Interactions. Nucleic Acids Res. 44 (D1), D542–D547. doi:10.1093/nar/gkv982
Lan, J., Ge, J., Yu, J., Shan, S., Zhou, H., Fan, S., et al. (2020). Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 581 (7807), 215–220. doi:10.1038/s41586-020-2180-5
Lane, T. R., and Ekins, S. (2021). Defending Antiviral Cationic Amphiphilic Drugs that May Cause Drug-Induced Phospholipidosis. J. Chem. Inf. Model. 61 (9), 4125–4130. doi:10.1021/acs.jcim.1c00903
Leader, B., Baca, Q. J., and Golan, D. E. (2008). Protein Therapeutics: a Summary and Pharmacological Classification. Nat. Rev. Drug Discov. 7 (1), 21–39. doi:10.1038/nrd2399
Lee, A. C.-L., Harris, J. L., Khanna, K. K., and Hong, J.-H. (2019). A Comprehensive Review on Current Advances in Peptide Drug Development and Design. Int. J. Mol. Sci. 20 (10), 2383. doi:10.3390/ijms20102383
Levine, W. G. (1991). Metabolism of Azo Dyes: Implication for Detoxication and Activation. Drug Metab. Rev. 23 (3-4), 253–309. doi:10.3109/03602539109029761
Lipinski, C. A. (2004). Lead- and Drug-like Compounds: the Rule-Of-Five Revolution. Drug Discov. Today Tech. 1 (4), 337–341. doi:10.1016/j.ddtec.2004.11.007
Lipinski, C. A., Lombardo, F., Dominy, B. W., and Feeney, P. J. (1997). Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 23, 3–25. doi:10.1016/s0169-409x(00)00129-010.1016/s0169-409x(96)00423-1
Liu, Y., Yu, Y., Zhao, Y., and He, D. (2022). Reduction in the Infection Fatality Rate of Omicron (B.1.1.529) Variant Compared to Previous Variants in South Africa. SSRN. doi:10.2139/ssrn.4010080
Lundin, A., Dijkman, R., Bergström, T., Kann, N., Adamiak, B., Hannoun, C., et al. (2014). Targeting Membrane-Bound Viral RNA Synthesis Reveals Potent Inhibition of Diverse Coronaviruses Including the Middle East Respiratory Syndrome Virus. Plos Pathog. 10 (5), e1004166. doi:10.1371/journal.ppat.1004166
Lv, Z., Deng, Y.-Q., Ye, Q., Cao, L., Sun, C.-Y., Fan, C., et al. (2020). Structural Basis for Neutralization of SARS-CoV-2 and SARS-CoV by a Potent Therapeutic Antibody. Science 369 (6510), 1505–1509. doi:10.1126/science.abc5881
Ma, L.-l., Liu, H.-m., Liu, X.-m., Yuan, X.-y., Xu, C., Wang, F., et al. (2021). Screening S Protein - ACE2 Blockers from Natural Products: Strategies and Advances in the Discovery of Potential Inhibitors of COVID-19. Eur. J. Med. Chem. 226, 113857. doi:10.1016/j.ejmech.2021.113857
Maas, M. N., Hintzen, J. C. J., Löffler, P. M. G., and Mecinović, J. (2021). Targeting SARS-CoV-2 Spike Protein by Stapled hACE2 Peptides. Chem. Commun. 57 (26), 3283–3286. doi:10.1039/d0cc08387a
Margolles-Clark, E., Umland, O., Kenyon, N. S., Ricordi, C., and Buchwald, P. (2009). Small-molecule Costimulatory Blockade: Organic Dye Inhibitors of the CD40-Cd154 Interaction. J. Mol. Med. 87 (11), 1133–1143. doi:10.1007/s00109-009-0519-3
Martinez, D. R., Schäfer, A., Gobeil, S., Li, D., De la Cruz, G., Parks, R., et al. (2022). A Broadly Cross-Reactive Antibody Neutralizes and Protects against Sarbecovirus challenge in Mice. Sci. Transl. Med. 14 (629), eabj7125. doi:10.1126/scitranslmed.abj7125
Matheson, N. J., and Lehner, P. J. (2020). How Does SARS-CoV-2 Cause COVID-19? Science 369 (6503), 510–511. doi:10.1126/science.abc6156
Matsuyama, K. (2022). Omicron May Be Less Lethal Than Other COVID-19 Variants but It’s Still 40% Deadlier Than the Flu, Scientists Say. Fortune. Available at: https://fortune.com/2022/03/03/omicron-less-lethal-covid-19-variants-40-deadlier-than-flu/March 3.
Meanwell, N. A., Krystal, M. R., Nowicka-Sans, B., Langley, D. R., Conlon, D. A., Eastgate, M. D., et al. (2018). Inhibitors of HIV-1 Attachment: the Discovery and Development of Temsavir and its Prodrug Fostemsavir. J. Med. Chem. 61 (1), 62–80. doi:10.1021/acs.jmedchem.7b01337
Melby, T., and Westby, M. (2009). Inhibitors of Viral Entry. Handb. Exp. Pharmacol. 189, 177–202. doi:10.1007/978-3-540-79086-0_7
Milhas, S., Raux, B., Betzi, S., Derviaux, C., Roche, P., Restouin, A., et al. (2016). Protein-protein Interaction Inhibition (2P2I)-Oriented Chemical Library Accelerates Hit Discovery. ACS Chem. Biol. 11 (8), 2140–2148. doi:10.1021/acschembio.6b00286
Milroy, L.-G., Grossmann, T. N., Hennig, S., Brunsveld, L., and Ottmann, C. (2014). Modulators of Protein-Protein Interactions. Chem. Rev. 114 (9), 4695–4748. doi:10.1021/cr400698c
Moia, M., Mantovani, L. G., Carpenedo, M., Scalone, L., Monzini, M. S., Cesana, G., et al. (2013). Patient Preferences and Willingness to Pay for Different Options of Anticoagulant Therapy. Intern. Emerg. Med. 8 (3), 237–243. doi:10.1007/s11739-012-0844-3
Morelli, X., Bourgeas, R., and Roche, P. (2011). Chemical and Structural Lessons from Recent Successes in Protein-Protein Interaction Inhibition (2P2I). Curr. Opin. Chem. Biol. 15 (4), 475–481. doi:10.1016/j.cbpa.2011.05.024
Mullard, A. (2016a). 2015 FDA Drug Approvals. Nat. Rev. Drug Discov. 15 (2), 73–76. doi:10.1038/nrd.2016.15
Mullard, A. (2020). 2019 FDA Drug Approvals. Nat. Rev. Drug Discov. 19 (2), 79–84. doi:10.1038/d41573-020-00001-7
Mullard, A. (2016b). Pioneering Apoptosis-Targeted Cancer Drug Poised for FDA Approval. Nat. Rev. Drug Discov. 15 (3), 147–149. doi:10.1038/nrd.2016.23
Munos, B. (2009). Lessons from 60 Years of Pharmaceutical Innovation. Nat. Rev. Drug Discov. 8 (12), 959–968. doi:10.1038/nrd2961
Murer, L., Volle, R., Andriasyan, V., Petkidis, A., Gomez-Gonzalez, A., Yang, L., et al. (2022). Identification of Broad Anti-coronavirus Chemical Agents for Repurposing against SARS-CoV-2 and Variants of Concern. Curr. Res. Virol. Sci. 3, 100019. doi:10.1016/j.crviro.2022.100019
Nabel, K. G., Clark, S. A., Shankar, S., Pan, J., Clark, L. E., Yang, P., et al. (2022). Structural Basis for Continued Antibody Evasion by the SARS-CoV-2 Receptor Binding Domain. Science 375 (6578), eabl6251. doi:10.1126/science.abl6251
Neklesa, T. K., Winkler, J. D., and Crews, C. M. (2017). Targeted Protein Degradation by PROTACs. Pharmacol. Ther. 174, 138–144. doi:10.1016/j.pharmthera.2017.02.027
Neugebauer, A., Hartmann, R. W., and Klein, C. D. (2007). Prediction of Protein−Protein Interaction Inhibitors by Chemoinformatics and Machine Learning Methods. J. Med. Chem. 50 (19), 4665–4668. doi:10.1021/jm070533j
O’Driscoll, M., Ribeiro Dos Santos, G., Wang, L., Cummings, D. A. T., Azman, A. S., Paireau, J., et al. (2021). Age-specific Mortality and Immunity Patterns of SARS-CoV-2. Nature 590 (7844), 140–145. doi:10.1038/s41586-020-2918-0
Otvos, L., and Wade, J. D. (2014). Current Challenges in Peptide-Based Drug Discovery. Front. Chem. 2, 62. doi:10.3389/fchem.2014.00062
Ou, X., Liu, Y., Lei, X., Li, P., Mi, D., Ren, L., et al. (2020). Characterization of Spike Glycoprotein of SARS-CoV-2 on Virus Entry and its Immune Cross-Reactivity with SARS-CoV. Nat. Commun. 11 (1), 1620. doi:10.1038/s41467-020-15562-9
Overington, J. P., Al-Lazikani, B., and Hopkins, A. L. (2006). How many Drug Targets Are There? Nat. Rev. Drug Discov. 5 (12), 993–996. doi:10.1038/nrd2199
O. Villoutreix, B., M. Labbe, C., Lagorce, D., Laconde, G., and Sperandio, O. (2012). A Leap into the Chemical Space of Protein-Protein Interaction Inhibitors. Cpd 18 (30), 4648–4667. doi:10.2174/138161212802651571
Owen, D. R., Allerton, C. M. N., Anderson, A. S., Aschenbrenner, L., Avery, M., Berritt, S., et al. (2021). An Oral SARS-CoV-2 M Pro Inhibitor Clinical Candidate for the Treatment of COVID-19. Science 374 (6575), 1586–1593. doi:10.1126/science.abl4784
Pagliaro, L., Felding, J., Audouze, K., Nielsen, S. J., Terry, R. B., Krog-Jensen, C., et al. (2004). Emerging Classes of Protein-Protein Interaction Inhibitors and New Tools for Their Development. Curr. Opin. Chem. Biol. 8 (4), 442–449. doi:10.1016/j.cbpa.2004.06.006
WHO Solidarity Trial Consortium Pan, H., Peto, R., Henao-Restrepo, A. M., Preziosi, M. P., Sathiyamoorthy, V., et al. (2021). Repurposed Antiviral Drugs for Covid-19 - Interim WHO Solidarity Trial Results. N. Engl. J. Med. 384, 497–511. doi:10.1056/nejmoa2023184
M. E. Wolff (Editor) (1995). Burger's Medicinal Chemistry and Drug Discovery (New York: Wiley-Interscience).
Park, Y.-J., De Marco, A., Starr, T. N., Liu, Z., Pinto, D., Walls, A. C., et al. (2022). Antibody-mediated Broad Sarbecovirus Neutralization through ACE2 Molecular Mimicry. Science 375 (6579), 449–454. doi:10.1126/science.abm8143
Paterson, G. R. (1984). Relationships between Synthetic Dyes and Drug Entities. Can. Bull. Med. Hist. 1 (2), 1–23. doi:10.3138/cbmh.1.2.1
Paul, S. M., Mytelka, D. S., Dunwiddie, C. T., Persinger, C. C., Munos, B. H., Lindborg, S. R., et al. (2010). How to Improve R&D Productivity: the Pharmaceutical Industry's Grand challenge. Nat. Rev. Drug Discov. 9 (3), 203–214. doi:10.1038/nrd3078
Pedebos, C., and Khalid, S. (2022). Simulations of the Spike: Molecular Dynamics and SARS-CoV-2. Nat. Rev. Microbiol. 20 (4), 192. doi:10.1038/s41579-022-00699-9
Pei, G., Xu, W., Lan, J., Wang, X., and Li, P. (2022). CEBIT Screening for Inhibitors of the Interaction between SARS-CoV-2 Spike and ACE2. Fundam. Res. ePub. doi:10.1016/j.fmre.2022.01.034
Pomplun, S., Jbara, M., Quartararo, A. J., Zhang, G., Brown, J. S., Lee, Y.-C., et al. (2021). De Novo discovery of High-Affinity Peptide Binders for the SARS-CoV-2 Spike Protein. ACS Cent. Sci. 7 (1), 156–163. doi:10.1021/acscentsci.0c01309
Proudfoot, J. R. (2002). Drugs, Leads, and Drug-Likeness: an Analysis of Some Recently Launched Drugs. Bioorg. Med. Chem. Lett. 12, 1647–1650. doi:10.1016/s0960-894x(02)00244-5
Rajgor, D. D., Lee, M. H., Archuleta, S., Bagdasarian, N., and Quek, S. C. (2020). The many Estimates of the COVID-19 Case Fatality Rate. Lancet Infect. Dis. 20 (7), 776–777. doi:10.1016/S1473-3099(20)30244-9
Reuben, B. G. (1996). “The Consumption and Production of Pharmaceuticals,” in The Practice of Medicinal Chemistry. Editor C. G. Wermuth (London: Academic Press), 903–938.
Reynès, C., Host, H., Camproux, A.-C., Laconde, G., Leroux, F., Mazars, A., et al. (2010). Designing Focused Chemical Libraries Enriched in Protein-Protein Interaction Inhibitors Using Machine-Learning Methods. Plos Comput. Biol. 6 (3), e1000695. doi:10.1371/journal.pcbi.1000695
Reynolds, C. H., Bembenek, S. D., and Tounge, B. A. (2007). The Role of Molecular Size in Ligand Efficiency. Bioorg. Med. Chem. Lett. 17 (15), 4258–4261. doi:10.1016/j.bmcl.2007.05.038
Russ, A. P., and Lampel, S. (2005). The Druggable Genome: an Update. Drug Discov. Today 10 (23-24), 1607–1610. doi:10.1016/s1359-6446(05)03666-4
Salje, H., Tran Kiem, C., Lefrancq, N., Courtejoie, N., Bosetti, P., Paireau, J., et al. (2020). Estimating the burden of SARS-CoV-2 in France. Science 369 (6500), 208–211. doi:10.1126/science.abc3517
Santos, R., Ursu, O., Gaulton, A., Bento, A. P., Donadi, R. S., Bologa, C. G., et al. (2017). A Comprehensive Map of Molecular Drug Targets. Nat. Rev. Drug Discov. 16 (1), 19–34. doi:10.1038/nrd.2016.230
Saravanan, U. B., Namachivayam, M., Jeewon, R., Huang, J.-D., and Durairajan, S. S. K. (2022). Animal Models for SARS-CoV-2 and SARS-CoV-1 Pathogenesis, Transmission and Therapeutic Evaluation. Wjv 11 (1), 40–56. doi:10.5501/wjv.v11.i1.40
Scannell, J. W., Blanckley, A., Boldon, H., and Warrington, B. (2012). Diagnosing the Decline in Pharmaceutical R&D Efficiency. Nat. Rev. Drug Discov. 11 (3), 191–200. doi:10.1038/nrd3681
Scott, D. E., Bayly, A. R., Abell, C., and Skidmore, J. (2016). Small Molecules, Big Targets: Drug Discovery Faces the Protein-Protein Interaction challenge. Nat. Rev. Drug Discov. 15 (8), 533–550. doi:10.1038/nrd.2016.29
Severson, W. E., Shindo, N., Sosa, M., Fletcher, T., White, E. L., Ananthan, S., et al. (2007). Development and Validation of a High-Throughput Screen for Inhibitors of SARS CoV and its Application in Screening of a 100,000-compound Library. J. Biomol. Screen. 12 (1), 33–40. doi:10.1177/1087057106296688
Shang, J., Wan, Y., Luo, C., Ye, G., Geng, Q., Auerbach, A., et al. (2020). Cell Entry Mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. U.S.A. 117 (21), 11727–11734. doi:10.1073/pnas.2003138117
Shang, Z., Chan, S. Y., Liu, W. J., Li, P., and Huang, W. (2021). Recent Insights into Emerging Coronavirus: SARS-CoV-2. ACS Infect. Dis. 7 (6), 1369–1388. doi:10.1021/acsinfecdis.0c00646
Shyr, Z. A., Gorshkov, K., Chen, C. Z., and Zheng, W. (2020). Drug Discovery Strategies for SARS-CoV-2. J. Pharmacol. Exp. Ther. 375 (1), 127–138. doi:10.1124/jpet.120.000123
Sivaraman, H., Er, S. Y., Choong, Y. K., Gavor, E., and Sivaraman, J. (2021). Structural Basis of SARS-CoV-2- and SARS-CoV-Receptor Binding and Small-Molecule Blockers as Potential Therapeutics. Annu. Rev. Pharmacol. Toxicol. 61, 465–493. doi:10.1146/annurev-pharmtox-061220-093932
Song, Y., and Buchwald, P. (2015). TNF Superfamily Protein-Protein Interactions: Feasibility of Small-Molecule Modulation. Curr. Drug. Targets 16, 393–408. doi:10.2174/1389450116666150223115628
Souers, A. J., Leverson, J. D., Boghaert, E. R., Ackler, S. L., Catron, N. D., Chen, J., et al. (2013). ABT-199, a Potent and Selective BCL-2 Inhibitor, Achieves Antitumor Activity while Sparing Platelets. Nat. Med. 19 (2), 202–208. doi:10.1038/nm.3048
Sperandio, O., Reynès, C. H., Camproux, A.-C., and Villoutreix, B. O. (2010). Rationalizing the Chemical Space of Protein-Protein Interaction Inhibitors. Drug Discov. Today 15 (5-6), 220–229. doi:10.1016/j.drudis.2009.11.007
Starr, T. N., Czudnochowski, N., Liu, Z., Zatta, F., Park, Y.-J., Addetia, A., et al. (2021). SARS-CoV-2 RBD Antibodies that Maximize Breadth and Resistance to Escape. Nature 597, 97–102. doi:10.1038/s41586-021-03807-6
Stumpf, M. P. H., Thorne, T., de Silva, E., Stewart, R., An, H. J., Lappe, M., et al. (2008). Estimating the Size of the Human Interactome. Proc. Natl. Acad. Sci. U.S.A. 105 (19), 6959–6964. doi:10.1073/pnas.0708078105
Su, H., Zhou, F., Huang, Z., Ma, X., Natarajan, K., Zhang, M., et al. (2021). Molecular Insights into Small‐Molecule Drug Discovery for SARS‐CoV‐2. Angew. Chem. Int. Ed. 60 (18), 9789–9802. doi:10.1002/anie.202008835
Sui, J., Deming, M., Rockx, B., Liddington, R. C., Zhu, Q. K., Baric, R. S., et al. (2014). Effects of Human Anti-spike Protein Receptor Binding Domain Antibodies on Severe Acute Respiratory Syndrome Coronavirus Neutralization Escape and Fitness. J. Virol. 88 (23), 13769–13780. doi:10.1128/jvi.02232-14
Suntharalingam, G., Perry, M. R., Ward, S., Brett, S. J., Castello-Cortes, A., Brunner, M. D., et al. (2006). Cytokine Storm in a Phase 1 Trial of the Anti-CD28 Monoclonal Antibody TGN1412. N. Engl. J. Med. 355 (10), 1018–1028. doi:10.1056/nejmoa063842
Tai, W., He, L., Zhang, X., Pu, J., Voronin, D., Jiang, S., et al. (2020). Characterization of the Receptor-Binding Domain (RBD) of 2019 Novel Coronavirus: Implication for Development of RBD Protein as a Viral Attachment Inhibitor and Vaccine. Cell. Mol. Immunol. 17 (6), 613–620. doi:10.1038/s41423-020-0400-4
Takayama, K. (2020). In Vitro and Animal Models for SARS-CoV-2 Research. Trends Pharmacol. Sci. 41 (8), 513–517. doi:10.1016/j.tips.2020.05.005
Tan, Q., Zhu, Y., Li, J., Chen, Z., Han, G. W., Kufareva, I., et al. (2013). Structure of the CCR5 Chemokine Receptor-HIV Entry Inhibitor Maraviroc Complex. Science 341 (6152), 1387–1390. doi:10.1126/science.1241475
Thiel, P., Kaiser, M., and Ottmann, C. (2012). Small-molecule Stabilization of Protein-Protein Interactions: an Underestimated Concept in Drug Discovery? Angew. Chem. Int. Ed. 51 (9), 2012–2018. doi:10.1002/anie.201107616
Tiwari, V., Beer, J. C., Sankaranarayanan, N. V., Swanson-Mungerson, M., and Desai, U. R. (2020). Discovering Small-Molecule Therapeutics against SARS-CoV-2. Drug Discov. Today 25 (8), 1535–1544. doi:10.1016/j.drudis.2020.06.017
Torchet, R., Druart, K., Ruano, L. C., Moine-Franel, A., Borges, H., Doppelt-Azeroual, O., et al. (2021). The iPPI-DB Initiative: a Community-Centered Database of Protein-Protein Interaction Modulators. Bioinformatics 37, 89–96. doi:10.1093/bioinformatics/btaa1091
Tortorici, M. A., Walls, A. C., Lang, Y., Wang, C., Li, Z., Koerhuis, D., et al. (2019). Structural Basis for Human Coronavirus Attachment to Sialic Acid Receptors. Nat. Struct. Mol. Biol. 26 (6), 481–489. doi:10.1038/s41594-019-0233-y
Trisciuzzi, D., Nicolotti, O., Miteva, M. A., and Villoutreix, B. O. (2019). Analysis of Solvent-Exposed and Buried Co-crystallized Ligands: a Case Study to Support the Design of Novel Protein-Protein Interaction Inhibitors. Drug Discov. Today 24 (2), 551–559. doi:10.1016/j.drudis.2018.11.013
Trisciuzzi, D., Siragusa, L., Baroni, M., Autiero, I., Nicolotti, O., and Cruciani, G. (2022). Getting Insights into Structural and Energetic Properties of Reciprocal Peptide-Protein Interactions. J. Chem. Inf. Model. 62 (4), 1113–1125. doi:10.1021/acs.jcim.1c01343
Tsegay, K. B., Adeyemi, C. M., Gniffke, E. P., Sather, D. N., Walker, J. K., and Smith, S. E. P. (2021). A Repurposed Drug Screen Identifies Compounds that Inhibit the Binding of the COVID-19 Spike Protein to ACE2. Front. Pharmacol. 12, 685308. doi:10.3389/fphar.2021.685308
Tummino, T. A., Rezelj, V. V., Fischer, B., Fischer, A., O’Meara, M. J., Monel, B., et al. (2021). Drug-induced Phospholipidosis Confounds Drug Repurposing for SARS-CoV-2. Science 373 (6554), 541–547. doi:10.1126/science.abi4708
van Breemen, R. B., Muchiri, R. N., Bates, T. A., Weinstein, J. B., Leier, H. C., Farley, S., et al. (2022). Cannabinoids Block Cellular Entry of SARS-CoV-2 and the Emerging Variants. J. Nat. Prod. 85 (1), 176–184. doi:10.1021/acs.jnatprod.1c00946
Verdine, G. L., and Walensky, L. D. (2007). The challenge of Drugging Undruggable Targets in Cancer: Lessons Learned from Targeting BCL-2 Family Members. Clin. Cancer Res. 13 (24), 7264–7270. doi:10.1158/1078-0432.ccr-07-2184
V’kovski, P., Kratzel, A., Steiner, S., Stalder, H., and Thiel, V. (2021). Coronavirus Biology and Replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 19 (3), 155–170. doi:10.1038/s41579-020-00468-6
Wadman, M. (2006). London's Disastrous Drug Trial Has Serious Side Effects for Research. Nature 440 (7083), 388–389. doi:10.1038/440388a
Walters, W. P., Green, J., Weiss, J. R., and Murcko, M. A. (2011). What Do Medicinal Chemists Actually Make? A 50-year Retrospective. J. Med. Chem. 54 (19), 6405–6416. doi:10.1021/jm200504p
Wang, C., Li, W., Drabek, D., Okba, N. M. A., van Haperen, R., Osterhaus, A. D. M. E., et al. (2020). A Human Monoclonal Antibody Blocking SARS-CoV-2 Infection. Nat. Commun. 11 (1), 2251. doi:10.1038/s41467-020-16256-y
Wang, X., Ni, D., Liu, Y., and Lu, S. (2021). Rational Design of Peptide-Based Inhibitors Disrupting Protein-Protein Interactions. Front. Chem. 9, 682675. doi:10.3389/fchem.2021.682675
Wec, A. Z., Wrapp, D., Herbert, A. S., Maurer, D. P., Haslwanter, D., Sakharkar, M., et al. (2020). Broad Neutralization of SARS-Related Viruses by Human Monoclonal Antibodies. Science 369 (6504), 731–736. doi:10.1126/science.abc7424
Wells, J. A., and McClendon, C. L. (2007). Reaching for High-Hanging Fruit in Drug Discovery at Protein-Protein Interfaces. Nature 450 (7172), 1001–1009. doi:10.1038/nature06526
Wibmer, C. K., Ayres, F., Hermanus, T., Madzivhandila, M., Kgagudi, P., Oosthuysen, B., et al. (2021). SARS-CoV-2 501Y.V2 Escapes Neutralization by South African COVID-19 Donor Plasma. Nat. Med. 27, 622–625. doi:10.1038/s41591-021-01285-x
Williams, M. (2005). Systems and Integrative Biology as Alternative Guises for Pharmacology: Prime Time for an iPharm Concept? Biochem. Pharmacol. 70 (12), 1707–1716. doi:10.1016/j.bcp.2005.08.019
Wu, Y., Wang, F., Shen, C., Peng, W., Li, D., Zhao, C., et al. (2020). A Noncompeting Pair of Human Neutralizing Antibodies Block COVID-19 Virus Binding to its Receptor ACE2. Science 368 (6496), 1274–1278. doi:10.1126/science.abc2241
Xiong, J., Xiang, Y., Huang, Z., Liu, X., Wang, M., Ge, G., et al. (2021). Structure-based Virtual Screening and Identification of Potential Inhibitors of SARS-CoV-2 S-RBD and ACE2 Interaction. Front. Chem. 9, 740702. doi:10.3389/fchem.2021.740702
Xiu, S., Dick, A., Ju, H., Mirzaie, S., Abdi, F., Cocklin, S., et al. (2020). Inhibitors of SARS-CoV-2 Entry: Current and Future Opportunities. J. Med. Chem. 63 (21), 12256–12274. doi:10.1021/acs.jmedchem.0c00502
Yu, S., Zhu, Y., Xu, J., Yao, G., Zhang, P., Wang, M., et al. (2021). Glycyrrhizic Acid Exerts Inhibitory Activity against the Spike Protein of SARS-CoV-2. Phytomedicine 85, 153364. doi:10.1016/j.phymed.2020.153364
Yuan, M., Liu, H., Wu, N. C., Lee, C.-C. D., Zhu, X., Zhao, F., et al. (2020). Structural Basis of a Shared Antibody Response to SARS-CoV-2. Science 369 (6507), 1119–1123. doi:10.1126/science.abd2321
Zhang, G., Pomplun, S., Loftis, A. R., Tan, X., Loas, A., and Pentelute, B. L. (2020). Investigation of ACE2 N-Terminal Fragments Binding to SARSCoV-2 Spike RBD. bioRxiv, 999318. doi:10.1101/2020.03.19.999318
Zhang, J., Zhang, K., Gao, Z.-G., Paoletta, S., Zhang, D., Han, G. W., et al. (2014). Agonist-bound Structure of the Human P2Y12 Receptor. Nature 509 (7498), 119–122. doi:10.1038/nature13288
Zhang, Q. C., Petrey, D., Deng, L., Qiang, L., Shi, Y., Thu, C. A., et al. (2012). Structure-based Prediction of Protein-Protein Interactions on a Genome-wide Scale. Nature 490 (7421), 556–560. doi:10.1038/nature11503
Zhang, Q., Xiang, R., Huo, S., Zhou, Y., Jiang, S., Wang, Q., et al. (2021). Molecular Mechanism of Interaction between SARS-CoV-2 and Host Cells and Interventional Therapy. Sig. Transduct. Target. Ther. 6 (1), 233. doi:10.1038/s41392-021-00653-w
Zhao, L., Li, S., and Zhong, W. (2022). Mechanism of Action of Small-Molecule Agents in Ongoing Clinical Trials for SARS-CoV-2: a Review. Front. Pharmacol. 13, 840639. doi:10.3389/fphar.2022.840639
Zhong, M., Gadek, T. R., Bui, M., Shen, W., Burnier, J., Barr, K. J., et al. (2012). Discovery and Development of Potent LFA-1/ICAM-1 Antagonist SAR 1118 as an Ophthalmic Solution for Treating Dry Eye. ACS Med. Chem. Lett. 3 (3), 203–206. doi:10.1021/ml2002482
Keywords: antiviral, coronavirus, fostemsavir, methylene blue, protein-protein interaction, SARS-cov-2, spike protein, variants of concern
Citation: Buchwald P (2022) Developing Small-Molecule Inhibitors of Protein-Protein Interactions Involved in Viral Entry as Potential Antivirals for COVID-19. Front. Drug. Discov. 2:898035. doi: 10.3389/fddsv.2022.898035
Received: 16 March 2022; Accepted: 31 March 2022;
Published: 25 April 2022.
Edited by:
Bruno Villoutreix, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Daniela Trisciuzzi, University of Bari Aldo Moro, ItalyCopyright © 2022 Buchwald. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peter Buchwald, cGJ1Y2h3YWxkQG1lZC5taWFtaS5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.