 Michèle Reboud-Ravaux*
Michèle Reboud-Ravaux* Chahrazade El Amri
Chahrazade El Amri- Sorbonne Université, Institut de Biologie Paris Seine (IBPS), CNRS UMR 8256, Inserm ERL U1164, Paris, France
The global spread of severe acute respiratory syndrome corona virus-2 (SARS-CoV-2) variants is alarming. In addition to vaccines, effective antiviral agents are urgently needed to combat corona virus disease 2019 (COVID-19). In this review, we will give insights on several canonical approaches using current medicinal chemistry. They target host (TMPRSS2, cathepsins B/L, furin) and viral (3CLpro and PLPro) proteases involved in virus cell entry and virus production, respectively. Innovative mechanisms of drug action are now explored whereby the drug triggers a cellular event that reduces the level of disease-implicated protein or RNA. The potential therapeutic power of induced degradations of viral proteins by PROTACs and of RNA by RIBOTACs for the treatment of COVID-19 will be discussed. Degraders of host cell RNA-binding proteins (RNA-PROTACs) may also constitute a therapeutical opportunity. First applicated to oncology, these novel technologies may be of a particular interest to obtain therapeutics susceptible to act on mutated viruses.
Introduction
Due to the global outbreak of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), pharmaceutical companies and academic institutions are actively pursuing efforts to develop new vaccines, repurpose existing drugs and discover of small inhibitors. Host and viral proteases have been especially targeted by inhibitors to block their functions in virus entry and cell cycle. High local concentrations of these inhibitors are needed to obtain a large clinical benefit but they could facilitate off-target binding and side-effects (Adjei, 2006). Such occupancy-based strategy is based on inhibitor binding into an active site. Other putative protein targets to combat COVID-19 are devoid of binding site. Their control can be obtained by decreasing their cellular level in so-called event-driven pharmacology (Cromm and Crews, 2017). These novel therapeutic approaches comprise nucleotide-based techniques [small interfering RNA (siRNA), antisens oligonucleotides, genome editing CRISPR-Cas9 strategy], and targeted protein degradation (TPD) techniques. TPDs examplified by PROteolysis TArgeting Chimeras (PROTACs), molecular glues, LYsosome-TArgeting Chimeras (LYTACs) and Antibody-based PROTACs (AbTACs) (Alabi and Crews, 2021; Bond and Crews, 2021) may constitute a new area of drug discovery to combat SARS-CoV-2 and the uncontrolled spread of virus variants. Other new chemical modalities use RNA-PROTACs that target specific RNAs to degrade RNA-binding proteins for their degradation (Ghidini et al., 2021). The degradation of the viral RNA itself can be induced with ribonuclease targeting chimeras (RIBOTACs) (Di Giorgio and Duca, 2020).
Virus Cell Cycle and Therapeutical Approaches
The enveloped SARS-CoV-2 is single-stranded positive-sense RNA virus. Its genome encodes Non-structural proteins Nsps (Nsp1-Nsp16) (replicase complex), nine accessory proteins (ORFs) and four major structural proteins: S (Spike), E (Envelope) M (Membrane), and N (Nucleocapsid) (Arya et al., 2021) (Figure 1A). Nsps are produced by processing by viral proteases, papain-like protease PLpro (Nsp5) and main protease 3CLpro (Nsp3), of the polyproteins pp1a and pp1ab. Virus entry into the host cells occurs via two pathways: endocytosis or membrane fusion after protein S binding to the cellular angiotensin-converting enzyme 2 receptor (ACE2) (Figure 1A). The homo-trimeric spike glycoprotein that protrudes from the viral surface has two major subunits, the S1 subunit implicated in receptor recognition and the membrane-anchored S2 subunit mediating fusion between the viral and the host cell membranes. The host protease cleavage site called S1/S2 is located at the border between S1 and S2 subunits. The concerted action of ACE2 binding and S protein processing by the transmembrane serine protease 2 (TMPSSR2) induces irreversible conformational changes that promote virus-cell fusion (Senapati et al., 2021). Several other host proteases have been suggested to promote cell entry, cathepsins B/L and furin (Figure 1A). The uncoating by nucleocapsid degradation allows the release of the viral RNA into the cytoplasm to be translated. Two overlapping open reading frames (ORFs) encodes for polyproteins pp1a and pp1ab that are processed by the proteases 3CLpro and PLpro leading to Nsp 1–16 which form the replicase/transcriptase complex (RTC). The subgenomic RNAs are translated in the four structural proteins and some accessory proteins. New viral particles are assembled at intracellular membranes. Host and viral proteases as well as RNAs constitute targets to obtain anti-SARS-CoV-2 agents; protein and RNA degraders may also lead to next-generation of drugs (Figure 1A).
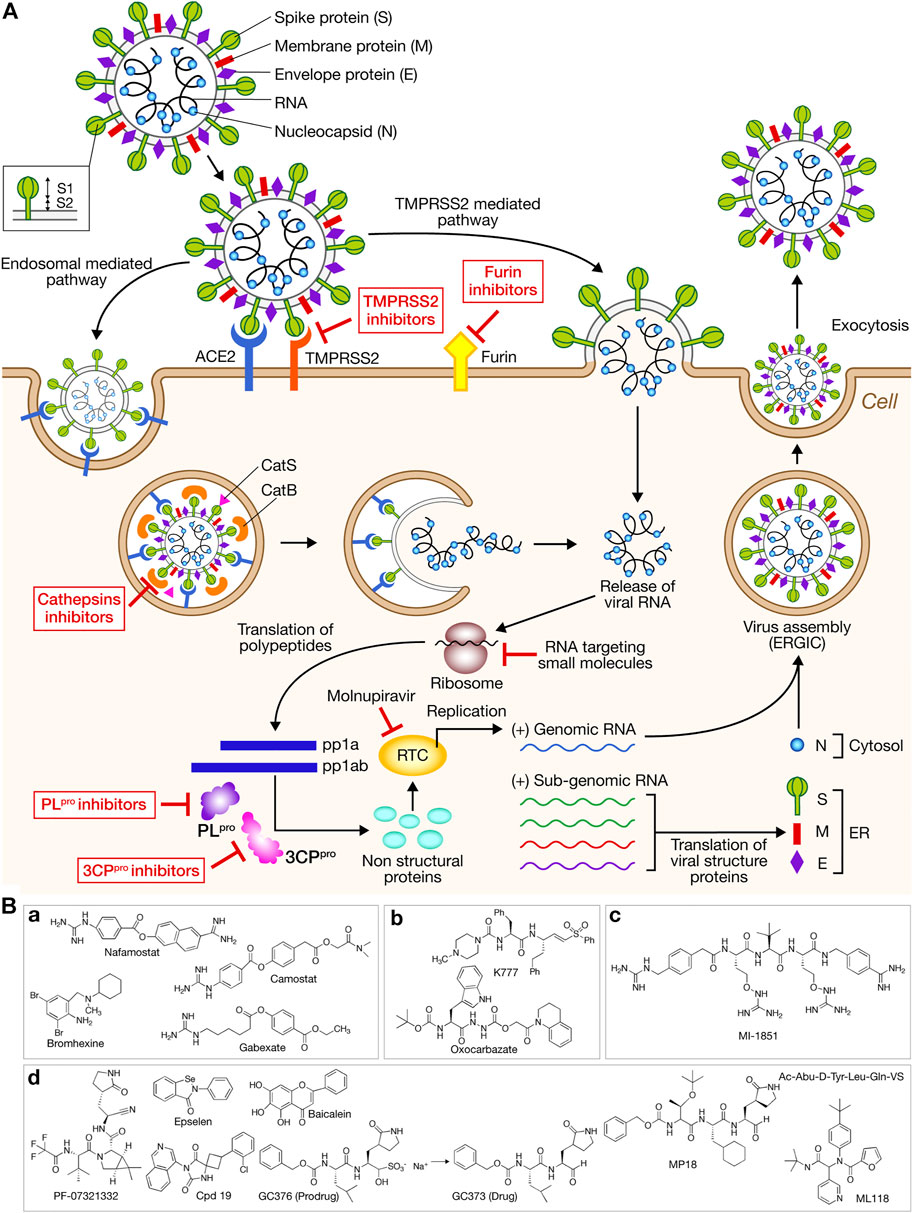
FIGURE 1. (A) Simplified diagram of life cycle of SARS-CoV-2 and host and viral targets for antiviral development. The host’s machinery is used to translate the released single-stranded positive RNA into a large polyprotein. After autocatalytic cleavage of 3CLpro, the polyprotein is cleaved at 14 different sites with 11 of these by 3CLpro. The intracellular virus replication is followed by the release of the newly packaged SARS-CoV-2. Cellular and viral proteases are highlighted as potential targets for antiviral development. PROTACs or molecular glues drugs may potentially target proteases such as 3CPpro, or proteins such as the envelope protein E. RNA targeting molecules such as RIBOTAC and RNA PROTAC may also constitute antivirals. The sites of action of molnupiravir and RNA targeting small molecules are also indicated. ACE2: angiotensin-converting enzyme 2; CatL: cathepsin L; CatS: cathepsin S; pp1a, pp1ab: polyproteins; RTC: replication transcription complex; RER: rough endoplasmic reticulum; ERGIC: endoplasmic-Golgi intermediate compartment. (B) Protease inhibitors. (a) TMPRSS2 inhibitors. (b) Cathepsin L inhibitors. (c) Furin inhibitor MI-1851. (d) 3CLpro inhibitors. PF-0732132 is found in the drug paxlovid. MPI8 inhibits both 3CLpro and CatL.
Host Proteases
Virus cell entry occurs via two independent pathways, endosomal mediated and TMPRSS2 mediated (Figure 1A). Among type II transmembrane proteases of the human respiratory tract known to cleave surface proteins of respiratory virus, the activity of TMPRSS2 was found the most crucial for SARS-CoV-2 entry and pathogenesis (Hoffmann et al., 2020b). TMPRSS2 aided by TMPRSS4 also facilitates virus entry into human small intestinal enterocytes participating to clinical complications (Zang et al., 2020). By priming the spike protein, TMPRSS2 facilitates the fusion of viral and host membranes whereas endosomal cathepsin B/L facilitates the fusion of viral and endosomal membranes (Figure 1A). These proteases can work independently. Recently, a suboptimal S1/S2 spike cleavage and inability to utilize TMPRSS2 was observed for the Omicron BA.1 variant that bears multiple spike mutations compared to the Delta one, thus favoring the endocytic pathway (Meng et al., 2022).
Camostat mesylate is a clinical TMPSSR2 inhibitor (phase I) that can partially block SARS-CoV-2 entry into cell lung line Calu-3, without cytotoxicity. This TMPSRRS2 inhibitor has the potential to treat COVID-19 in humans (Hoffmann et al., 2020b). Several other TMPRSS2 inhibitors are in clinical or pre-clinical phases: nafanostat approved in Japan (phase II), bromhexine (phase IV) and gabexate (preclinical phase) (Figure 1Ba). The TMPRSS2 main exosite is a novel target for inhibitors (Singh et al., 2020). Whereas TMPRSS2 acts locally at host cell membrane, the cysteine protease cathepsin L (CatL) with its acidic optimum pH is the major protease that cleaves the virus S1 subunit within endosomes (Ou et al., 2020). CatS is the major endosomal protease that mediates antigen presentation and antibody production (Beers et al., 2005). About 10 FDA approved drugs have an inhibitory activity against CatL but no available drug can specifically inhibit CatL (Dana and Pathak, 2020). Among them, are found oxocarbazate and the vinylsulfone K777 (pre-clinical phase) (Figure 1Bb). A combination of TMPRSS2 and cathepsins B/L inhibitors could lead to a complete blockade of viral entry due to a strong synergy (Hoffmann et al., 2020b; Liu T. et al., 2020; Padmanabhan et al., 2020; Hashimoto et al., 2021). Cathepsin L and 3CLpro share common structural and electrochemical similarities. The dual non-covalent inhibitor MPI8 that inhibits the viral 3CLpro and the host cathepsin L selectively versus cathepsins B or K is a potent antiviral in vitro (Cao et al., 2022; Ma et al., 2022) (Figures 1Bb,d).
Emerging evidence suggests that furin plays a critical role in viral entry and propagation. SARS-CoV-2 bears a polybasic sequence PRRAR at the S1/S2 cleavage site that can be cleaved by furin (Hoffmann et al., 2020a; Peacock et al., 2021). Several peptide-based and small-molecule inhibitors of furin have been developed (Dahms et al., 2021; Osman et al., 2022). A combination of the furin inhibitor MI-1851 (Figure 1Bc) with various TMPRSS2 inhibitors enhances the antiviral potency (Bestle et al., 2020). A novel antibody against furin cleavage site constitutes a suitable approach to decrease viral infectivity (Spelios et al., 2022).
Virus Proteases
The viral 3-chymotrypsin-like protease (3CLpro or Mpro) and papain-like protease (PLpro), and the RNA-dependent-RNA polymerase (RdRP) appeared as traditional targets to combat virus. Two oral antiviral treatments have been approved with molnupiravir targeting RdRP developed by Merck, and nirmatrelvir (PF-07321332) in paxlovid targeting 3CLpro developed by Pfizer (Figures 1A,B). The third drug, remdesivir developed by Gilead targeting RNA polymerase is less accessible (expensive intravenous infusions).
3CLpro and PLpro activities represent rate-limiting steps in viral replication (Cannalire et al., 2022). Active 3CLpro is a homodimer containing a noncanonical Cys145-His41 dyad whereas 3CLpro contains the classic Cys112-His273-Asp287 triad for papain-like proteases. 3CLpro cleaves the C-terminal region of the precursor protein at 11 sites whereas PL cleaves the N-terminal region of the viral precursor protein at three sites. 3CLpro shows glutamine-specific cleavage activity not observed in human proteases making it an interesting target (Ullrich and Nitsche 2020). Nearly 200 3D-structures of 3CLpro have been released favoring identification of new inhibitors by structure-based rational design or virtual screening of large collections of molecules (Liu Y. et al., 2020; Zhang et al., 2020; Singh and Villoutreix, 2021; Luttens et al., 2022). A lot of structurally diverse compounds (synthetic or natural) displaying 3CLpro inhibitory activity are summarized in earlier reviews (Akaji and Konno, 2020; Chen et al., 2021) (Figure 1Bd). Many are covalent peptidomimetics such as Ac-Abu-D-Tyr-Leu-Gln-VS (Rut et al., 2021) but non-covalent ones are now reported (Han et al., 2022) (Figure 1Bd). Efforts have been made to avoid peptidyl secondary amides (Yamamoto et al., 2022). Ultra-large virtual screening identifies an inhibitor showing comparable efficacy as PF-07321332 against SARS-CoV-2, and antiviral efficacy against SARS-CoV-1 and MERS (non-covalent Cpd 19) (Luttens et al., 2022). No peptidic compound is reported as orally available.
Fewer inhibitors of PLpro are known and potent and specific inhibitors are still needed (Ma and Wang, 2022). Additionally, PLpro contributes to immune escape by cleaving post-translational modified host proteins involved in innate immune response, (ubiquitin and ubiquitin-like protein ISG15 from interferon (IFN) responsive factor) (Freitas et al., 2020).
Targeted Protein Degradations
Protease inhibitors rely on the accessible binding sites. The emerging TPD technologies can target proteins devoid of binding site, categorized as “undruggable” before. They draw inspiration from natural proteasomal protein degradation to specifically eliminate disease-relevant proteins. They are based on the design of small molecules called “degraders” able to induce the proteasomal degradation of the targeted protein. Two major types are known: PROteolysis TArgeting Chimeras (PROTACs) firstly developed in 2001 by Crews and Deshaies (Sakamoto et al., 2001; Sakamoto et al., 2003), and non-chimeric molecules known as molecular glues (Lu et al., 2014). In both cases, the association between the protein of interest (POI) and an E3 ubiquitin ligase is induced allowing the ubiquitin transfer to the POI and its subsequent degradation by proteasome (Figure 2A). The feasibility of PROTAC technology had led to clinical trials using the first oral PROTACs [ARV-110 (phase I) and ARV-471 (phase II)] for prostate and breast cancer treatment (Arvinas, Inc., 2019a; Arvinas, Inc., 2019b), and more recently, KT-474 (Kymera Therapeutics) and NX-2127 (Nurix Therapeutics) for autoimmune disorders and B-cell malignancies treatment, respectively (Qi et al., 2021). Several molecular glues such as the immunomodulatory “ImID” small molecules that bind to E3 ligase cereblon (thalidomide, lenalidomide and pomalidomide) have been approved for liquid cancers and four other ones are in clinical trials (Chamberlain and Hamann, 2019).
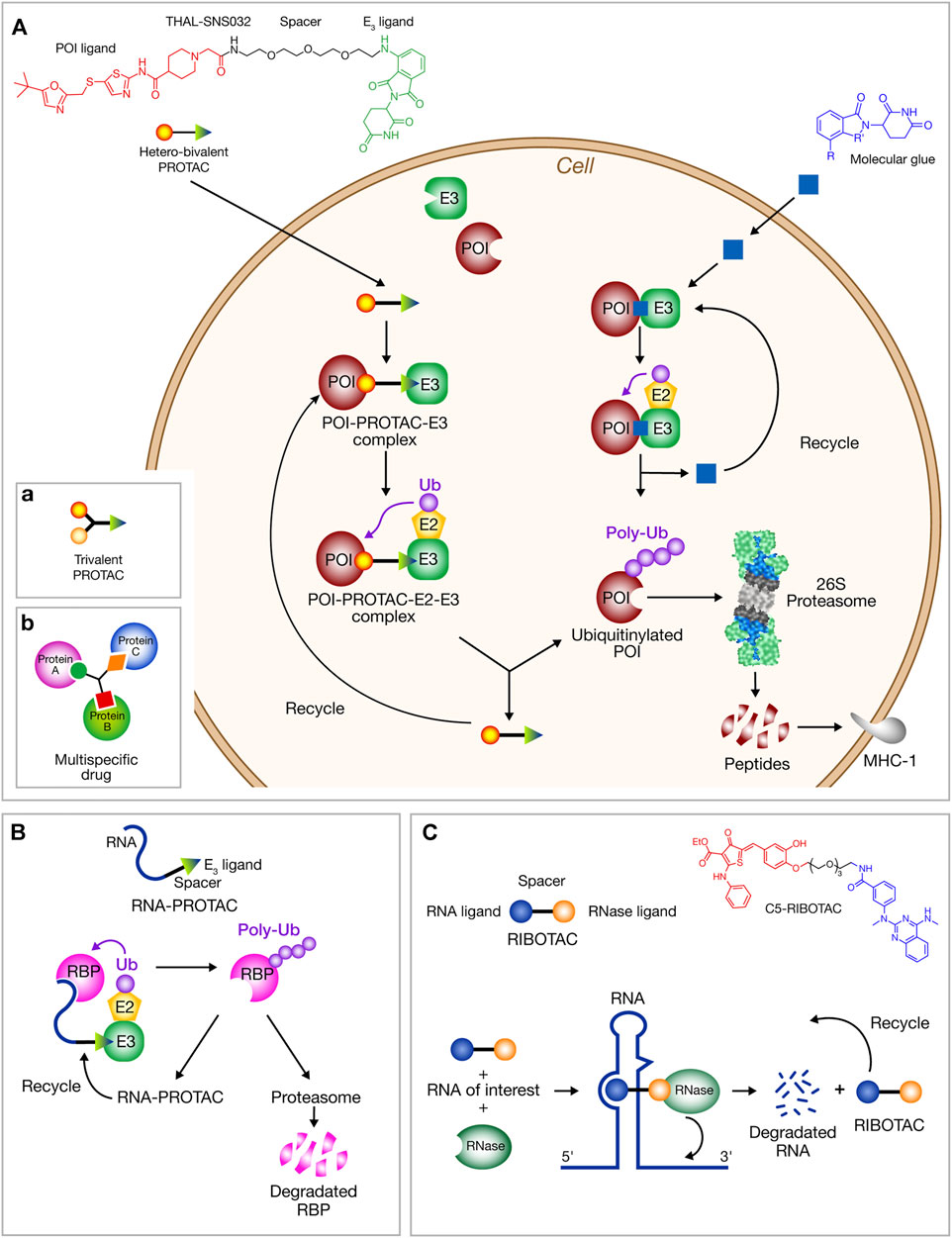
FIGURE 2. Schematic representation of degrader strategies. (A) Targeted protein degradation by chimeric PROTACs and molecular glues. Hetero-bifunctional PROTACs and molecular glues induce the proximity of a protein target (POI) and biological effector (E3) allowing POI ubiquitination and its targeting to proteasome for degradation. PROTAC and molecular glue are released allowing their subsequent use in a new cycle of POI induced degradation. The capability of PROTACs to induce MHC-I peptides is outlined. The chemical structure of THAL-SNS032 (PROTAC) and immunomodulators IMiDs (molecular glues) are shown. Schematic structure of: (a) trivalent PROTACs, (b) multitarget drugs. (B) RNA-PROTACs direct RBPs to proteasomal degradation. A short oligonucleotide binds to the RNA domain of the RBP. Linked by a spacer to a motif susceptible to binds an E3 ligase, it mediates RBP ubiquitination and degradation. (C) RIBOTACs induce the degradation of RNA itself.
PROTACs are hetero-bifunctional small molecules comprising two ligands connected by an organic linker, a ligand targets the POI and the other an E3 ligase (Bondeson et al., 2018; Burslem and Crews, 2020; Reboud-Ravaux, 2021). Compared to nucleic-acid strategies, PROTACs have the advantage to lead to an acute and reversible reduction of the targeted protein cell level and a reversible chemical extinction of all properties of POI (Verma et al., 2020). Molecular glues are monovalent small molecules (MW < 200 Da) that do not require a binding pocket on the POI and are good therapeutic candidates for undruggable proteins. They reshape the surface of an E3 ligase inducing the binding of a protein leading to assembly of a possibly cooperative ternary complex. Novel mechanisms for their action have been described (Alabi, 2021) such as polymerization enhancing interaction with a E3 ligase or, for bulky and aggregated proteins, an induced degradation via autophagy (Alabi, 2021). New degrader technologies such as LYTACs and AbTACs are developed to broaden the spectrum of protein targets to extracellular and membrane proteins (Alabi, 2021; Lin et al., 2021).
The TPD technologies can now be considered as newly emerging antiviral strategies that may counteract pathogen viruses by inducing the degradation of either viral or host protein targets (Alabi and Crews, 2021; Verma, 2021; Grohmann et al., 2021; Desantis and Giracci, 2022). For example, a NS3/4A protease degrader of the hepatitis C virus (HCV) has been reported (de Wispelaere et al., 2019). Anti-influenza activity was reported for oseltamivir-based PROTAC derivatives (Zhou et al., 2021). Protein degraders for SARS-CoV-2 3CLpro were hypothesized (Liu Y. et al., 2020). Design of PROTAC structures were obtained by computer modeling of the interaction between 3CLpro and cereblon E3 ligase (Shaheer et al., 2021). The countereffect of potential deubiquitinase action of PLpro is not experimentally evidenced. In parallel, a novel and potential capability of PROTAC compounds as anti-SARS-CoV-2 has been reported with an antiviral PROTAC targeting the envelope protein E that acts as viroporin. (Martinez-Ortiz and Zhou, 2020). This non-glycosylated envelope protein is a feasible target since inhibition of SARS coronavirus envelope protein ion channel affects several virus functions such as virulence, membrane permeabilizing activity and the viral assembly (Pervushin et al., 2009). Moreover, viral epitopes derived from the proteasomal degradation of protein E can be presented to MHC-I and promote the generation of antibodies against the viral protein (Jensen et al., 2018) (Figure 2A). This may result in the development of host T-cell activity against the viral protein. In view to combat resistance to viral mutations, host-directed antivirals are also promising. Four first-in-class indomethacin (INM)-based-PROTACs inhibit SARS-CoV-2 replication and exhibit broad-spectrum anti-viral activity in the Coronaviridae family (Desantis et al., 2021). THAL-SNS032 is a commercial cyclin-dependent-kinase 9 (CDK9)-directed PROTAC that has anti-human cytomegalovirus (HCV) activity. It inhibits SARS-CoV-2 replication (Hahn et al., 2021). Targeting androgen regulation of TMPRSS2 and ACE2 is a possible strategy to combat COVID-19 (Qiao et al., 2020; Deng et al., 2021). Androgen receptor-inhibitory therapies might reduce susceptibility to COVID-19 symptoms and mortality (Stopsack et al., 2020). Besides inhibitors (e.g., darolutamide, enzalutamide, flutamide and apalutamide), PROTACs targeting the androgen receptor could be protective against COVID since increased mortality and morbidity is observed in men (Wadman, 2020).
RNA-PROTACs and RIBOTACs
Targeting conserved viral RNA structures and sequences is a novel approach to inhibit viral infection and progression. Interactions between RNA and small molecules are poorly understood rendering RNA difficult to target (Costales et al., 2020; Hegde et al., 2021). Nevertheless, RNA-PROTACs have been designed and synthesized producing degraders of RNA-binding proteins (RBPs) whose defects are observed in many diseases (Figure 2B) (Ghidini et al., 2021). The RNA binding site of RBPs can be used to produce RNA-PROTACs. These chimeric structures are composed of a small-RNA mimics that docks the RNA binding site of the RBP and of a peptide able to recruit the E3 ligase. This delivery of peptide to its target site by the oligonucleotide successfully provokes RBP degradation in cancer cells (Ghidini et al., 2021). Several RBPs in the host cells are predicted to bind to the SARS-CoV-2 RNA genome (Sun et al., 2021).
Ribonuclease targeting chimeras (RIBOTACs) are chimeric molecules inducing the degradation of RNA itself (Figure 2C), able to destroy cancer associated RNA (Kargbo, 2020) or SARS-CoV-2 RNA structures sequences (Di Giorgio and Duca, 2020; Haniff et al., 2020; Hegde et al., 2021). The PROTAC concept to the RNA field, firstly developed by the Disney group has now been extended (Costales et al., 2020; Liu Y. et al., 2020). The small molecule RIBOTAC has been shown to reduce SARS-CoV-2 RNA levels in a cellular model (Haniff et al., 2020). Using a 15-nucleotide complementary antisense oligonucleotide (ASO) linked to an RNase L recruiter the viral titer was reduced in virus-infected Vero E6 cells (Su et al., 2021).
Discussion
Intensive efforts of the scientific community are needed to face potential future pandemics in order to discover broad-spectrum antiviral agents with new scaffolds and better resistance profiles. Viral 3CLPro and RdRP show the highest degree of conservation across different CoVs and are among the most characterized SARS-CoV-2 targets. Two small molecules nirmatrelvir (PF-07321332) in paxlovid targeting 3CLpro and molnupiravir targeting RdRP were recently introduced in clinics. Having no known homolog in host cell, 3CLpro remains an ideal target to identify new efficient inhibitors with broad-spectrum activity against coronavirus. Being catalytically inactive in the monomeric form, the development of dimerization inhibitors may also potentially lead to a new class of compounds as previously observed with the homodimeric HIV-1 protease (Bannwarth et al., 2009) and recently for SARS-CoV main protease (Goyal and Goyal, 2020). The underexplored PLpro is less conserved across CoVs family and could appear as less attractive in view of future CoV outbreaks. The antiviral efficacy of protease inhibitors could benefit of the use of drug combination therapy (host TMPRRS2 and cathepsins B/L) (Padmanabhan et al., 2020) or of multitarget drugs binding simultaneously to viral 3CLpro and host cathepsin L (Ma et al., 2022).
The increasing understanding of proteasomal protein degradation and RNA biology provides powerful and practical opportunities for the development of novel anti-SARS-CoV-2 agents. TPD using PROTACs and molecular glues may target not only viral or host enzymes but also a large variety of “undruggable” proteins such as structural proteins as previously suggested (Martinez-Ortiz and Zhou, 2020) or potentially many other ones. PROTACs have been applied to degrade a large variety of proteins, cytoplasmic, nuclear, membrane-bound and multipass transmembranes ones as well as “hard to drug” proteins (e.g., KRAS and Myc families) (Bond and Crews, 2021). In the case of 3CLpro, developing PROTACs using existing enzyme inhibitors could combine occupancy-driven and event-driven technologies avoiding enzyme accumulation in infected cells and lowering side-effects since PROTACs are recycled. They have many advantages over traditional protein inhibitors (Burslem and Crews, 2020). Acting in a catalytic manner allowing for substoichiometric usage, their effect persists until the POI reaccumulates. Off-target side effects and toxicity may be reduced. Nevertheless, the PROTAC large molecular weight (“beyond rule of 5”) may result in low bioavalaibility. Modifications of the linkers can efficiently enhance cell permeability and activity (Klein et al., 2021). Click-chemistry can also be used for intracellular PROTAC synthesis (Lebraud et al., 2016; Schiedel et al., 2018). Recent developments of PROTAC technology (kinetic and pharmacokinetic studies, covalence, resistance, new ligands) favor the increase of PROTAC repertoire (Hu and Crews, 2022). Pro-drug PROTACs could be beneficial by improving clinical delivery and metabolic stability (Wei et al., 2021). Recently trivalent PROTACs with their branched trifunctional scaffold (Figure 2Aa) were designed and proved to enhance protein degradation via combined avidity and cooperativity (Imaide et al., 2021; Zheng et al., 2021). By augmenting the binding valency, various multispecific agents are now prospectively developed opening up to various applications beyond PROTACs (Figure 2Ab) (Deshaies, 2020). Such newly created drugs acting on different proteins may possibly perturb the viral cycle for example at the protein assembly level. They could be directed towards distinct domains of the same protein, or even two distinct proteins belonging to a multiprotein complex. The low-molecular-weight molecular glues may have also utility by targeting undruggable proteins and complexes implicated in virus cell cycle. They can be utilized according different mechanisms to induce degradation of neosubstrates (Alabi, 2021). A better understanding of protein-protein interfaces will greatly facilitate rational design of molecular glues (Kozicka and Thomä, 2021).
Structure-function studies of viral non-structural, structural and accessory proteins as well as those implicated in interaction with cell proteins are essential to select potential targets for drug development against COVID-19 (Gorgulla et al., 2021; Singh and Villoutreix, 2021). The discovery of small ligands is needed, even very poor binding ones susceptible to be introduced in PROTACs, molecular glues or other multispecific agents (Mayor-Ruiz et al., 2020). The avidity and cooperativity in ternary complexes (POI-PROTAC-E3) compensate for low binary binding affinities or poor cellular permability allowing for the use of weak, non-functional ligands (Imaide et al., 2021). In silico screening platforms comprising ultra-large-scale ones, artificial intelligence and machine learning techniques are essential to discover novel protein ligands. (Gupta and Mohanty, 2021; Villoutreix, 2021; Luttens et al., 2022). As well, the innovative RNA-targeting strategies could lead to promising developments that will benefit of RNA-targeting drug discovery platforms (Warner et al., 2018).
Author Contributions
MR-R conceived, planned and wrote the manuscript. CE edited and contributed to the manuscript organization. The two authors critically revised the paper and approved the submitted version.
Funding
This work was supported by the Institut National pour Recherche Médicale (INSERM), Sorbonne-Université and the Centre National de la Recherche Scientifique (CNRS).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We wish to thank Sophie Gournet (Plate-forme Illustration & Graphisme; IBPS, Sorbonne Université) for performing schematics and drawings for this mini-review.
References
Adjei, A. A. (2006). What Is the Right Dose? the Elusive Optimal Biologic Dose in Phase I Clinical Trials. J. Clin. Oncol. 24, 4054–4055. doi:10.1200/JCO.2006.07.4658
Akaji, K., and Konno, H. (2020). Design and Evaluation of Anti-SARS-coronavirus Agents Based on Molecular Interactions with the Viral Protease. Molecules 25, 3920. doi:10.3390/molecules25173920
Alabi, S. B., and Crews, C. M. (2021). Major Advances in Targeted Protein Degradation: PROTACs, LYTACs, and MADTACs. J. Biol. Chem. 296, 100647. doi:10.1016/j.jbc.2021.100647
Alabi, S. (2021). Novel Mechanisms of Molecular Glue-Induced Protein Degradation. Biochemistry 60, 2371–2373. doi:10.1021/acs.biochem.1c00353
Arvinas, Inc. (2019a) Clinical Trial of ARV-471 in Patients wither/HER2-Locally Advanced or Metastatic Breast Cancer (mBC). In Patients with Metastatic Castration Resistant Prostate Cancer (mCRPC). Available at: https//clinical trials.gov/ct2/show/NCT94072952. (October 23).
Arvinas, Inc. (2019b) Trial of ARV-110 in Patients with Metastatic Castration Resistant Prostate Cancer (mCRPC). Available at: https//clinical trials.gov/ct2/show/NCT03888612. (March, 2019).
Arya, R., Kumari, S., Pandey, B., Mistry, H., Bihani, S. C., Das, A., et al. (2021). Structural Insights into SARS-CoV-2 Proteins. J. Mol. Biol. 433, 166725. doi:10.1016/j.jmb.2020.11.024
Bannwarth, L., Rose, T., Dufau, L., Vanderesse, R., Dumond, J., Jamart-Grégoire, B., et al. (2009). Dimer Disruption and Monomer Sequestration by Alkyl Tripeptides Are Successful Strategies for Inhibiting Wild-type and Multidrug-Resistant Mutated HIV-1 Proteases. Biochemistry 48, 379–387. doi:10.1021/bi801422u
Beers, C., Burich, A., Kleijmeer, M. J., Griffith, J. M., Wong, P., and Rudensky, A. Y. (2005). Cathepsin S Controls MHC Class II-Mediated Antigen Presentation by Epithelial Cells In Vivo. J. Immunol. 174 (3), 1205–1212. doi:10.4049/jimmunol.174.3.1205
Bestle, D., Heindl, M. R., Limburg, H., Van Lam van, T., Pilgram, O., Moulton, H., et al. (2020). TMPRSS2 and Furin Are Both Essential for Proteolytic Activation of SARS-CoV-2 in Human Airway Cells. Life Sci. Alliance 3, e202000786. doi:10.26508/lsa.202000786
Bond, M. J., and Crews, C. M. (2021). Proteolysis Targeting Chimeras (PROTACs) Come of Age: Entering the Third Decade of Targeted Protein Degradation. RSC Chem. Biol. 2, 725–742. doi:10.1039/d1cb00011j
Bondeson, D. P., Smith, B. E., Burslem, G. M., Buhimschi, A. D., Hines, J., Jaime-Figueroa, S., et al. (2018). Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cel Chem. Biol. 25, 78–87. doi:10.1016/j.chembiol.2017.09.010
Burslem, G. M., and Crews, C. M. (2020). Proteolysis-targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 181, 102–114. doi:10.1016/j.cell.2019.11.031
Cannalire, R., Cerchia, C., Beccari, A. R., Di Leva, F. S., and Summa, V. (2022). Targeting SARS-CoV-2 Proteases and Polymerase for COVID-19 Treatment: State of the Art and Future Opportunities. J. Med. Chem. 65, 2716–2746. doi:10.1021/acs.jmedchem.0c01140
Cao, W., Cho, C.-C. D., Geng, Z. Z., Shaabani, N., Ma, X. R., Vatansever, E. C., et al. (2022). Evaluation of SARS-CoV-2 Main Protease Inhibitors Using a Novel Cell-Based Assay. ACS Cent. Sci. 8, 192–204. doi:10.1021/acscentsci.1c00910
Chamberlain, P. P., and Hamann, L. G. (2019). Development of Targeted Protein Degradation Therapeutics. Nat. Chem. Biol. 15, 937–944. doi:10.1038/s41589-019-0362-y
Chen, W., Wang, Z., Wang, Y., and Li, Y. (2021). Natural Bioactive Molecules as Potential Agents against SARS-CoV-2. Front. Pharmacol. 12, 702472. doi:10.3389/fphar.2021.702472
Costales, M. G., Aikawa, H., Li, Y., Childs-Disney, J. L., Abegg, D., Hoch, D. G., et al. (2020). Small-molecule Targeted Recruitment of a Nuclease to Cleave an Oncogenic RNA in a Mouse Model of Metastatic Cancer. Proc. Natl. Acad. Sci. U.S.A. 117, 2406–2411. doi:10.1073/pnas.1914286117
Cromm, P. M., and Crews, C. M. (2017). Targeted Protein Degradation: from Chemical Biology to Drug Discovery. Cel Chem. Biol. 24, 1181–1190. doi:10.1016/j.chembiol.2017.05.024
Dahms, S. O., Haider, T., Klebe, G., Steinmetzer, T., and Brandstetter, H. (2021). OFF-State-Specific Inhibition of the Proprotein Convertase Furin. ACS Chem. Biol. 16, 1692–1700. doi:10.1021/acschembio.1c00411
Dana, D., and Pathak, S. K. (2020). A Review of Small Molecule Inhibitors and Functional Probes of Human Cathepsin L. Molecules 25, 698. doi:10.3390/molecules25030698
de Wispelaere, M., Du, G., Donovan, K. A., Zhang, T., Eleuteri, N. A., Yuan, J. C., et al. (2019). Small Molecule Degraders of the Hepatitis C Virus Protease Reduce Susceptibility to Resistance Mutations. Nat. Commun. 10, 3468. doi:10.1038/s41467-019-11429-w
Deng, Q., Rasool, R. U., Russell, R. M., Natesan, R., and Asangani, I. A. (2021). Targeting Androgen Regulation of TMPRSS2 and ACE2 as a Therapeutic Strategy to Combat COVID-19. iScience 24, 102254. doi:10.1016/j.isci.2021.102254
Desantis, J., and Goracci, L. (2022). Proteolysis Targeting Chimeras in Antiviral Research. Future Med. Chem. 14 (7), 459–462. doi:10.4155/fmc-2022-0005
Desantis, J., Mercorelli, B., Celegato, M., Croci, F., Bazzacco, A., Baroni, M., et al. (2021). Indomethacin-based PROTACs as Pan-Coronavirus Antiviral Agents. Eur. J. Med. Chem. 226, 113814. doi:10.1016/j.ejmech.2021.113814
Deshaies, R. J. (2020). Multispecific Drugs Herald a New Era of Biopharmaceutical Innovation. Nature 580, 329–338. doi:10.1038/s41586-020-2168-1
Di Giorgio, A., and Duca, M. (2020). New Chemical Modalities Enabling Specific RNA Targeting and Degradation: Application to SARS-CoV-2 RNA. ACS Cent. Sci. 6, 1647–1650. doi:10.1021/acscentsci.0c01187
Freitas, B. T., Durie, I. A., Murray, J., Longo, J. E., Miller, H. C., Crich, D., et al. (2020). Characterization and Noncovalent Inhibition of the Deubiquitinase and DeISGylase Activity of SARS-CoV-2 Papain-like Protease. ACS Infect. Dis. 6, 2099–2109. doi:10.1021/acsinfecdis.0c00168
Ghidini, A., Cléry, A., Halloy, F., Allain, F. H. T., and Hall, J. (2021). RNA-PROTACs: Degraders of RNA-Binding Proteins. Angew. Chem. Int. Ed. 60, 3163–3169. doi:10.1002/anie.202012330
Gorgulla, C., Padmanabha Das, K. M., Leigh, K. E., Cespugli, M., Fischer, P. D., Wang, Z.-F., et al. (2021). A Multi-Pronged Approach Targeting SARS-CoV-2 Proteins Using Ultra-large Virtual Screening. iScience 24 (2), 102021. doi:10.1016/j.isci.2020.102021
Goyal, B., and Goyal, D. (2020). Targeting the Dimerization of the Main Protease of Coronaviruses: A Potential Broad-Spectrum Therapeutic Strategy. ACS Comb. Sci. 22 (6), 297–305. doi:10.1021/acscombsci.0c00058
Grohmann, C., Marapana, D. S., and Ebert, G. (2021). Targeted Protein Degradation at the Host-Pathogen Interface. Mol. Microbiol. 117, 670–681. doi:10.1111/mmi.14849
Gupta, P., and Mohanty, D. (2021). SMMPPI: a Machine Learning-Based Approach for Prediction of Modulators of Protein-Protein Interactions and its Application for Identification of Novel Inhibitors for RBD:hACE2 Interactions in SARS-CoV-2. Brief Bioinform 22, bbab111. doi:10.1093/bib/bbab111
Hahn, F., Hamilton, S. T., Wangen, C., Wild, M., Kicuntod, J., Brückner, N., et al. (2021). Development of a PROTAC-Based Targeting Strategy Provides a Mechanistically Unique Mode of Anti-cytomegalovirus Activity. Int. J. Mol. Sci. 22, 12858. doi:10.3390/ijms222312858
Han, S. H., Goins, C. M., Arya, T., Shin, W.-J., Maw, J., Hooper, A., et al. (2022). Structure-Based Optimization of ML300-Derived, Noncovalent Inhibitors Targeting the Severe Acute Respiratory Syndrome Coronavirus 3CL Protease (SARS-CoV-2 3CLpro). J. Med. Chem. 65, 2880–2904. doi:10.1021/acs.jmedchem.1c00598
Haniff, H. S., Tong, Y., Liu, X., Chen, J. L., Suresh, B. M., Andrews, R. J., et al. (2020). Targeting the SARS-CoV-2 RNA Genome with Small Molecule Binders and Ribonuclease Targeting Chimera (RIBOTAC) Degraders. ACS Cent. Sci. 6, 1713–1721. doi:10.1021/acscentsci.0c00984
Hashimoto, R., Sakamoto, A., Deguchi, S., Yi, R., Sano, E., Hotta, A., et al. (2021). Dual Inhibition of TMPRSS2 and Cathepsin B Prevents SARS-CoV-2 Infection in iPS Cells. Mol. Ther. - Nucleic Acids 26, 1107–1114. doi:10.1016/j.omtn.2021.10.016
Hegde, S., Tang, Z., Zhao, J., and Wang, J. (2021). Inhibition of SARS-CoV-2 by Targeting Conserved Viral RNA Structures and Sequences. Front. Chem. 9, 802766. doi:10.3389/fchem.2021.802766
Hoffmann, M., Kleine-Weber, H., and Pöhlmann, S. (2020a). A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cel 78, 779–784. e5. doi:10.1016/j.molcel.2020.04.022
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Krüger, N., Herrler, T., Erichsen, S., et al. (2020b). SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 181, 271–280. e8. doi:10.1016/j.cell.2020.02.052
Hu, Z., and Crews, C. M. (2022). Recent Developments in PROTAC-Mediated Protein Degradation: From Bench to Clinic. Chembiochem 23, e202100270. doi:10.1002/cbic.202100270
Imaide, S., Riching, K. M., Makukhin, N., Vetma, V., Whitworth, C., Hughes, S. J., et al. (2021). Trivalent PROTACs Enhance Protein Degradation via Combined Avidity and Cooperativity. Nat. Chem. Biol. 17, 1157–1167. doi:10.1038/s41589-021-00878-4
Jensen, S. M., Potts, G. K., Ready, D. B., and Patterson, M. J. (2018). Specific MHC-I Peptides Are Induced Using PROTACs. Front. Immunol. 9, 2697. doi:10.3389/fimmu.2018.02697
Kargbo, R. B. (2020). SMARCA2/4 PROTAC for Targeted Protein Degradation and Cancer Therapy. ACS Med. Chem. Lett. 11, 1797–1798. doi:10.1021/acsmedchemlett.0c00347
Klein, V. G., Bond, A. G., Craigon, C., Lokey, R. S., and Ciulli, A. (2021). Amide-to-Ester Substitution as a Strategy for Optimizing PROTAC Permeability and Cellular Activity. J. Med. Chem. 64, 18082–18101. doi:10.1021/acs.jmedchem.1c01496
Kozicka, Z., and Thomä, N. H. (2021). Haven't Got a Glue: Protein Surface Variation for the Design of Molecular Glue Degraders. Cel Chem. Biol. 28, 1032–1047. doi:10.1016/j.chembiol.2021.04.009
Lebraud, H., Wright, D. J., Johnson, C. N., and Heightman, T. D. (2016). Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2 (12), 927–934. doi:10.1021/acscentsci.6b00280
Lin, J., Jin, J., Shen, Y., Zhang, L., Gong, G., Bian, H., et al. (2021). Emerging Protein Degradation Strategies: Expanding the Scope to Extracellular and Membrane Proteins. Theranostics 11, 8337–8349. doi:10.7150/thno.62686
Liu, T., Luo, S., Libby, P., and Shi, G.-P. (2020). Cathepsin L-Selective Inhibitors: A Potentially Promising Treatment for COVID-19 Patients. Pharmacol. Ther. 213, 107587. doi:10.1016/j.pharmthera.2020.107587
Liu, Y., Liang, C., Xin, L., Ren, X., Tian, L., Ju, X., et al. (2020). The Development of Coronavirus 3C-like Protease (3CLpro) Inhibitors from 2010 to 2020. Eur. J. Med. Chem. 206, 112711. doi:10.1016/j.ejmech.2020.112711
Lu, G., Middleton, R. E., Sun, H., Naniong, M., Ott, C. J., Mitsiades, C. S., et al. (2014). The Myeloma Drug Lenalidomide Promotes the Cereblon-dependent Destruction of Ikaros Proteins. Science 343, 305–309. doi:10.1126/science10.1126/science.1244917
Luttens, A., Gullberg, H., Abdurakhmanov, E., Vo, D. D., Akaberi, D., Talibov, V. O., et al. (2022). Ultralarge Virtual Screening Identifies SARS-CoV-2 Main Protease Inhibitors with Broad-Spectrum Activity against Coronaviruses. J. Am. Chem. Soc. 144, 2905–2920. doi:10.1021/jacs.1c08402
Ma, C., and Wang, J. (2022). Validation and Invalidation of SARS-CoV-2 Papain-like Protease Inhibitors. ACS Pharmacol. Transl. Sci. 5, 102–109. doi:10.1021/acsptsci.1c00240
Ma, X. R., Alugubelli, Y. R., Ma, Y., Vatansever, E. C., Scott, D. A., Qiao, Y., et al. (2022). MPI8 Is Potent against SARS-CoV-2 by Inhibiting Dually and Selectively the SARS-CoV-2 Main Protease and the Host Cathepsin L**. ChemMedChem 17, e202100456. doi:10.1002/cmdc.202100456
Martinez-Ortiz, W., and Zhou, M.-M. (2020). Could PROTACs Protect Us from COVID-19? Drug Discov. Today 25, 1894–1896. doi:10.1016/j.drudis.2020.08.007
Mayor-Ruiz, C., Bauer, S., Brand, M., Kozicka, Z., Siklos, M., Imrichova, H., et al. (2020). Rational Discovery of Molecular Glue Degraders via Scalable Chemical Profiling. Nat. Chem. Biol. 16, 1199–1207. doi:10.1038/s41589-020-0594-x
Meng, B., Abdullahi, A., Ferreira, I. A. T. M., Goonawardane, N., Saito, A., Kimura, I., et al. (2022). Altered TMPRSS2 Usage by SARS-CoV-2 Omicron Impacts Tropism and Fusogenicity. Nature 603 (7902), 706–714. doi:10.1038/s41586-022-04474-x
Osman, E. E. A., Rehemtulla, A., and Neamati, N. (2022). Why All the Fury over Furin? J. Med. Chem. 65, 2747–2784. doi:10.1021/acs.jmedchem.1c00518
Ou, X., Liu, Y., Lei, X., Li, P., Mi, D., Ren, L., et al. (2020). Characterization of Spike Glycoprotein of SARS-CoV-2 on Virus Entry and its Immune Cross-Reactivity with SARS-CoV. Nat. Commun. 11, 1620. doi:10.1038/s41467-020-15562-9
Padmanabhan, P., Desikan, R., and Dixit, N. M. (2020). Targeting TMPRSS2 and Cathepsin B/L Together May Be Synergistic against SARS-CoV-2 Infection. Plos Comput. Biol. 16, e1008461. doi:10.1371/journal.pcbi.1008461
Peacock, T. P., Goldhill, D. H., Zhou, J., Baillon, L., Frise, R., Swann, O. C., et al. (2021). The Furin Cleavage Site in the SARS-CoV-2 Spike Protein Is Required for Transmission in Ferrets. Nat. Microbiol. 6, 899–909. doi:10.1038/s41564-021-00908-w
Pervushin, K., Tan, E., Parthasarathy, K., Lin, X., Jiang, F. L., Yu, D., et al. (2009). Structure and Inhibition of the SARS Coronavirus Envelope Protein Ion Channel. Plos Pathog. 5, e1000511. doi:10.1371/journal.ppat.1000511
Qi, S.-M., Dong, J., Xu, Z.-Y., Cheng, X.-D., Zhang, W.-D., Qin, J.-J., et al. (2021). PROTAC: An Effective Targeted Protein Degradation Strategy for Cancer Therapy. Front. Pharmacol. 12, 692574. doi:10.3389/fphar.2021.692574
Qiao, Y., Wang, X.-M., Mannan, R., Pitchiaya, S., Zhang, Y., Wotring, J. W., et al. (2020). Targeting Transcriptional Regulation of SARS-CoV-2 Entry Factors ACE2 and TMPRSS2. Proc. Natl. Acad. Sci. U.S.A. 118, e2021450118. doi:10.1073/pnas.2021450118
Reboud-Ravaux, M. (2021). Dégradation induite des protéines par des molécules PROTAC et stratégies apparentées : développements à visée thérapeutique. Biologie Aujourd'hui 215, 25–43. doi:10.1051/jbio/2021007
Rut, W., Groborz, K., Zhang, L., Sun, X., Zmudzinski, M., Pawlik, B., et al. (2021). SARS-CoV-2 Mpro Inhibitors and Activity-Based Probes for Patient-Sample Imaging. Nat. Chem. Biol. 17 (2), 222–228. doi:10.1038/s41589-020-00689-z
Sakamoto, K. M., Kim, K. B., Kumagai, A., Mercurio, F., Crews, C. M., and Deshaies, R. J. (2001). Protacs: Chimeric Molecules that Target Proteins to the Skp1-Cullin-F Box Complex for Ubiquitination and Degradation. Proc. Natl. Acad. Sci. U.S.A. 98, 8554–8559. doi:10.1073/pnas.141230798
Sakamoto, K. M., Kim, K. B., Verma, R., Ransick, A., Stein, B., Crews, C. M., et al. (2003). Development of Protacs to Target Cancer-Promoting Proteins for Ubiquitination and Degradation. Mol. Cell Proteomics 2, 1350–1358. doi:10.1074/mcp.t300009-mcp200
Schiedel, M., Herp, D., Hammelmann, S., Swyter, S., Lehotzky, A., Robaa, D., et al. (2018). Chemically Induced Degradation of Sirtuin 2 (Sirt2) by a Proteolysis Targeting Chimera (PROTAC) Based on Sirtuin Rearranging Ligands (SirReals). J. Med. Chem. 61, 482–491. doi:10.1021/acs.jmedchem.6b01872
Senapati, S., Banerjee, P., Bhagavatula, S., Kushwaha, P. P., and Kumar, S. (2021). Contributions of Human ACE2 and TMPRSS2 in Determining Host-Pathogen Interaction of COVID-19. J. Genet. 100, 12. doi:10.1007/s12041-021-01262-w
Shaheer, M., Singh, R., and Sobhia, M. E. (2021). Protein Degradation: a Novel Computational Approach to Design Protein Degrader Probes for Main Protease of SARS-CoV-2. J. Biomol. Struct. Dyn. 30, 1–13. doi:10.1080/07391102.2021.1953601
Singh, N., Decroly, E., Khatib, A.-M., and Villoutreix, B. O. (2020). Structure-based Drug Repositioning over the Human TMPRSS2 Protease Domain: Search for Chemical Probes Able to Repress SARS-CoV-2 Spike Protein Cleavages. Eur. J. Pharm. Sci. 153, 105495. doi:10.1016/j.ejps.2020.105495
Singh, N., and Villoutreix, B. O. (2021). Resources and Computational Strategies to advance Small Molecule SARS-CoV-2 Discovery: Lessons from the Pandemic and Preparing for Future Health Crises. Comput. Struct. Biotechnol. J. 19, 2537–2548. doi:10.1016/j.csbj.2021.04.059
Spelios, M. G., Capanelli, J. M., and Li, A. W. (2022). A Novel Antibody against the Furin Cleavage Site of SARS-CoV-2 Spike Protein: Effects on Proteolytic Cleavage and ACE2 Binding. Immunol. Lett. 242, 1–7. doi:10.1016/j.imlet.2022.01.002
Stopsack, K. H., Mucci, L. A., Antonarakis, E. S., Nelson, P. S., and Kantoff, P. W. (2020). TMPRSS2 and COVID-19: Serendipity or Opportunity for Intervention? Cancer Discov. 10, 779–782. doi:10.1158/2159-8290.CD-20-0451
Su, X., Ma, W., Feng, D., Cheng, B., Wang, Q., Guo, Z., et al. (2021). Efficient Inhibition of SARS-CoV-2 Using Chimeric Antisense Oligonucleotides through RNase L Activation**. Angew. Chem. Int. Ed. 60, 21662–21667. doi:10.1002/anie.202105942
Sun, L., Li, P., Ju, X., Rao, J., Huang, W., Ren, L., et al. (2021). In Vivo Structural Characterization of the SARS-CoV-2 RNA Genome Identifies Host Proteins Vulnerable to Repurposed Drugs. Cell 184, 1865–1883. e20. doi:10.1016/j.cell.2021.02.008
Ullrich, S., and Nitsche, C. (2020). The SARS-CoV-2 Main Protease as Drug Target. Bioorg. Med. Chem. Lett. 30, 127377. doi:10.1016/j.bmcl.2020.127377
Verma, R. (2021). Exploiting Ubiquitin Ligases for Induced Target Degradation as an Antiviral Strategy. Adv. Exp. Med. Biol. 1322, 339–357. doi:10.1007/978-981-16-0267-2_13
Verma, R., Mohl, D., and Deshaies, R. J. (2020). Harnessing the Power of Proteolysis for Targeted Protein Inactivation. Mol. Cel 77, 446–460. doi:10.1016/j.molcel.2020.01.010
Villoutreix, B. O. (2021). Post-pandemic Drug Discovery and Development: Facing Present and Futures Challenges. Front. Drug Discov., 1. article 728469. doi:10.3389/fddsv.2021.728469
Wadman, M. (2020). Sex Hormones Signal Why Virus Hits Men Harder. Science 368, 1038–1039. doi:10.1126/science.368.6489.35610.1126/science.368.6495.1038
Warner, K. D., Hajdin, C. E., and Weeks, K. M. (2018). Principles for Targeting RNA with Drug-like Small Molecules. Nat. Rev. Drug Discov. 17, 547–558. doi:10.1038/nrd.2018.93
Wei, M., Zhao, R., Cao, Y., Wei, Y., Li, M., Dong, Z., et al. (2021). First Orally Bioavailable Prodrug of Proteolysis Targeting Chimera (PROTAC) Degrades Cyclin-dependent Kinases 2/4/6 In Vivo. Eur. J. Med. Chem. 209, 112903. doi:10.1016/j.ejmech.2020.112903
Yamamoto, K. Z., Yasuo, N., and Sekijima, M. (2022). Screening for Inhibitors of Main Protease in SARS-CoV-2: In Silico and In Vitro Approach Avoiding Peptidyl Secondary Amides. J. Chem. Inf. Model. 62, 350–358. doi:10.1021/acs.jcim.1c01087
Zang, R., Castro, M. F. G., McCune, B. T., Zeng, Q., Rothlauf, P. W., Sonnek, N. M., et al. (2020). TMPRSS2 and TMPRSS4 Promote SARS-CoV-2 Infection of Human Small Intestinal Enterocytes. Sci. Immunol. 5, eabc3582. doi:10.1126/sciimmunol.abc3582
Zhang, L., Lin, D., Sun, X., Curth, U., Drosten, C., Sauerhering, L., et al. (2020). Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-ketoamide Inhibitors. Science 368, 409–412. doi:10.1126/science.abb3405
Zheng, M., Huo, J., Gu, X., Wang, Y., Wu, C., Zhang, Q., et al. (2021). Rational Design and Synthesis of Novel Dual PROTACs for Simultaneous Degradation of EGFR and PARP. J. Med. Chem. 64, 7839–7852. doi:10.1021/acs.jmedchem.1c00649
Keywords: SARS-CoV-2, host proteases, virus proteases, inhibitors, degraders, PROTACs, RIBOTACs
Citation: Reboud-Ravaux M and El Amri C (2022) COVID-19 Therapies: Protease Inhibitions and Novel Degrader Strategies. Front. Drug. Discov. 2:892057. doi: 10.3389/fddsv.2022.892057
Received: 08 March 2022; Accepted: 24 March 2022;
Published: 13 April 2022.
Edited by:
Bruno Villoutreix, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceReviewed by:
Caroline Demeret, Institut Pasteur, FranceAbdel-Majid Khatib, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Copyright © 2022 Reboud-Ravaux and El Amri. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michèle Reboud-Ravaux, bWljaGVsZS5yZWJvdWRAc29yYm9ubmUtdW5pdmVyc2l0ZS5mcg==