 Lev ShapiraShaul Lerner
Lev ShapiraShaul Lerner Guila AssayagAlexandra VardiDikla HahamGideon BarVicky Fidelsky KozokaroMaayan Elias RobicsekImmanuel Lerner
Guila AssayagAlexandra VardiDikla HahamGideon BarVicky Fidelsky KozokaroMaayan Elias RobicsekImmanuel Lerner Amit Michaeli*
Amit Michaeli*- Pepticom Ltd., Jerusalem, Israel
Introduction: The COVID-19 pandemic has cast a heavy toll in human lives and global economics. COVID-19 is caused by the SARS-CoV-2 virus, which infects cells via its spike protein binding human ACE2.
Methods: To discover potential inhibitory peptidomimetic macrocycles for the spike/ACE2 complex we deployed Artificial Intelligence guided virtual screening with three distinct strategies: 1) Allosteric spike inhibitors 2) Competitive ACE2 inhibitors and 3) Competitive spike inhibitors. Screening was performed by docking macrocycles to the relevant sites, clustering and synthesizing cluster representatives. Synthesized molecules were screened for inhibition using AlphaLISA and RSV particles.
Results: All three strategies yielded inhibitory peptides, but only the competitive spike inhibitors showed “hit” level activity.
Discussion: These results suggest that direct inhibition of the spike RBD domain is the most attractive strategy for peptidomimetic, “head-to-tail” macrocycle drug development against the ongoing pandemic.
Introduction
The beginning of third decade in the 21st century was marred by the spread of the COVID-19 pandemic. Patients with pneumonia-like symptoms were initially reported in Wuhan, China as early as 8 December 2019 with a rapid, global spread, leading to over 21 million cases and 761,779 deaths reported by the World Health Organization (WHO) in August 2020 (Muralidar et al., 2020), with the WHO reporting global mortality rates of about 6.5 million by October 2022.
COVID-19 is caused by infection of The Severe Acute Respiratory Syndrome-Coronavirus-2 (SARS-CoV-2), which belongs to the family of coronaviruses and is highly homologous to SARS-CoV (Zhou et al., 2020). Infection normally begins with transmission through aerosol droplets followed by a latent period and respiratory tract infection (Salzberger et al., 2021). The majority of infections (∼90%) do not require hospitalization and approximately 40% are asymptomatic (Salzberger et al., 2021). Fatality rates increase with the presence of risk factors, with Case Fatality Rates (CFR) reaching double digit percentages (10%–20%) in patients over 70 years of age (Salzberger et al., 2021). High CFR rates and widespread infection rates have led to control measures such as lockdowns, which proved effective (Salzberger et al., 2021), albeit with a social and economic price tag. The understanding that in addition to the acute disease, tissue damage, autoimmunity and chronic inflammation may result in Long COVID (Yong, 2021), has increased the urgency for drug discovery.
The SARS-CoV-2 virion is made up of four structural proteins: nucleocapsid N), membrane M), envelope E) and spike S). Infection begins with the viral spike protein binding the human angiotensin-converting enzyme 2 (ACE2) receptor (Zhou et al., 2020; Jackson et al., 2022), the same receptor that mediates SARS-CoV infection (Li et al., 2003). The spike protein is a homotrimer in which each monomer is composed of a viral membrane binding S2 domain, and the S1 domain, containing the Receptor Binding Domain (RBD) which binds ACE2 and following furin-mediated cleavage, initiates cell fusion towards infection (Huang et al., 2020). Cryo-electron microscopy has demonstrated that spike can exist with all 3 RBD domains exposed, binding ACE2 receptors (PDB: 7A98), spike with 2 unbound RBDs erect (PDB: 7A93), spike with one RBD exposed, binding one ACE2 receptor (PDB: 7A94) and spike with 2 RBDs binding 2 ACE2 receptors (PDB: 7A97) (Benton et al., 2020). The diversity of the spike/ACE2 complex models raises the questions of biological relevance and implications to inhibitor discovery. In addition to the ACE2 binding conformations, there is also a furin-cleaved closed conformation, with no exposed RBDs (PDB: 6ZGI) (Wrobel et al., 2020). Additionally, it has also been shown that an infection pathway independent of ACE2 exists, in which spike binds activated α5β1 integrins, a process suggested to be more significant following initial ACE2 infection and resulting cytokine mediated integrin activation (Liu et al., 2022). Of the current therapeutic approaches only monoclonal antibodies and Convalescent Plasma Therapy target spike (Raman et al., 2021). These treatments are costly and need to be administered professionally, limiting their application in a future major outbreak, making additional drug discovery essential. The combination of a diverse viral spike protein, capable of infection via two independent pathways makes the discovery of viral fusion inhibitors a challenging task.
Peptides composed of 20 natural amino-acid monomers often suffer poor absorption, low stability and fast clearance that often limits their progress as therapeutic agents, to partially overcome these limitations, “non-natural” amino-acids and backbone modifications have been introduced to form peptidomimetics (Li Petri et al., 2022). The addition of monomers greatly increases the chemical space that needs to be covered by a discovery platform. The polymer chemical space can be described using the equation below:
Where M is the number of monomers available and N is the number of monomers composing the polymer. Non-natural amino-acid monomers are constantly being discovered, increasing M from 20 to over 500. So, for example, a polymer of five amino-acids has gone from a chemical space of approximately 106 natural peptides to over 1013 peptidomimetics! Larger polymers, such as 15-mer macrocycles reach chemical spaces that exceed 1040. Naturally, high-throughput screening systems that deploy robotic physical screening of 105-106 molecules cover too small a fraction of the chemical space to be efficient in peptidomimetic discovery. Even when limited to the natural monomers, the large peptide chemical space was not well explored before phage display libraries (Smith, 1985). Display technologies, have utilized tRNA-monomer attachment to increase chemical space (Kourouklis et al., 2005; Taki et al., 2006; Ohuchi et al., 2007), with some constraint/cyclization capacity (Cary et al., 2017). Nevertheless, physical techniques have a limited capacity to fully explore the chemical space, requiring more flexible screening methods.
Pepticom Ltd. was founded with the vision of allowing in silico peptide, peptidomimetic and macrocycle screening. As the enormous polymer chemical space is too large for explicit screening using standard “docking” software, even in the age of cloud computing, Pepticom took an Artificial Intelligence (AI) approach where reinforcement learning guides cloud based docking algorithms. In this methodology, docking is performed by agents or “bots” that upload to the cloud with a set of docking algorithms, heuristic, grid and sequence preferences. Bots may explore an innovative solution or alternately exploit an existing one. Once bots complete the docking task, they update the system with their findings and get a reward on the quality and originality of their solution. To further enhance this process, the concept of risk/return efficiency was translated from the financial world, making the exploitation of existing docked structures for additional sequences more efficient (Lerner et al., 2017). This screening approach has yielded numerous novel polymers; including GABA-A receptor allosteric modulators (Michaeli et al., 2020), bi-specific MD2/CD14 macrocycle agonists of Toll-Like Receptor 4 (TLR4) (Michaeli et al., 2018) and a rescue peptide for a destabilizing Glycogen Branching Enzyme (GBE1) mutation (Froese et al., 2015). The latter case showed another advantage for in silico screening; as protein rescue assays had to be conducted in vivo, on primary patient cells, barring the option for massive physical screening (Froese et al., 2015).
We used spike and spike/ACE2 protein structures to virtually screen macrocycle peptidomimetics with the potential to inhibit SARS-CoV-2 viral entry.
Materials and methods
AlphaLISA
Reagents
ACE2 (Biotinylated Recombinant Human ACE-2 His-tag Avi-tag Protein (CHO), CF, catalog no AVI10579) and SARS-CoV-2 Spike RBD-Fc (Recombinant SARS-CoV-2 Spike RBD Fc Chimera Protein, CF, catalog no 10499-CV) were acquired from R&D biosystems. IgG1_Fc-Avi-tag (Biotinylated Human IgG1 Fc protein, Avitag™ catalog no. IG1-H8213) was acquired from ACROBiosystems. Alphascreen Streptavidin donor beads (catalog no. 6760002) and AlphaLISA protein A acceptor beads (catalog no. AL101) were acquired from PerkinElmer (Waltham, MA).
Reagents preparation
SARS-CoV-2 Spike RBD-Fc was reconstituted in sterile PBS pH 7.4 to a final concentration of 9600 nM and was used for standard curve preparation (0nM – 3.231 nM) and preincubation with inhibitors at 0.64 nM IgG1_Fc-Avi-tag was reconstituted in sterile DW to a final concentration of 7017 nM. Assay HEPES based buffer was freshly prepared: HEPES pH 7.5 (Biological Industries Bet HaEmek), 150 mM NaCl, 5 mM CaCl₂, 0.05% (v/v) Tween 20, 2% DMSO, 0.01% BSA.
AlphaLISA Experiment
Inhibitory peptides and controls were dispensed into clear 384-well plate (REF P384120SQC Axygen) in 6 serial dilutions 1:2 in 100% DMSO. Each well content was diluted with assay buffer to achieve DMSO concentration 2% (v/v). Spike standard curve of 12 concentrations was performed in order to quantify IC50 value of each inhibitory compound. Biotinylated ACE2-His-Avi-tag at 15.3 nM was added to each experiment well with exception of wells containing IgG1_Fc-Avi-tag which was used as a maximum energy transfer control. Each sample was tested in triplicate in a white 384-well plate (catalog no. 6007290 PerkinElmer). Donor and acceptor beads at 20 ug/ml were added for 1 h incubation at room temperature (RT). The plate was scanned using VICTOR NivoTM Multimode plate reader (PerkinElmer) using AlphaLISA technology with excitation at 680 nm and emission at 615 nm.
Some compounds can interfere with the detection part of the assay, thereby artificially reducing AlphaLISA signal and leading to false positive hits. In order to exclude such, AlphaLISA TruHits kit (PerkinElmer, AL900D) was used. The kit allows identification of inner filters, light scatterers (insoluble compounds), singlet oxygen quenchers, and biotin mimetics that interfere with the assay detection. Samples were diluted at the same concentrations as in AlphaLISA assay, in triplicates and incubated with AlphaLISA BSA-biotin Acceptor beads for 1 h according to manufacturer’s instructions. Streptavidin Alpha Donor beads were then added (for 30 min) to generate AlphaLISA signal.
Data Interpretation
To determine compound activity in the AlphaLISA assay, the concentration−response data for each sample was plotted and modeled by a four-parameter logistic fit (4 PL) yielding IC50 and efficacy (maximal response) values using Prism (GraphPad, San Diego, CA) software program. Raw plate reads for each concentration point were first normalized relative to positive control (100% activity, full inhibition) and negative control (basal, 0% activity) wells. Data normalization, curve fitting and scheme figures were performed using Microsoft Excel.
Inhibitory peptide neutralizing assay
Cells
The 239T-hsACE2 monoclonal cell line was purchased from Integral Molecular, Inc. (Philadelphia, PA). This cell line was created by transduction of 293 T cells with a second-generation lentiviral vector carrying a transgene for hsACE2 (Accession# NM_021804.3) linked by an IRES to a puromycin resistance gene. Integration and expression were selected for by incubation with Puromycin, and clonal populations developed by limiting dilutions.
Media
Cells were cultured in medium contained of DMEM + 10% FBS + 10 mM HEPES + 1x Penicillin-Streptomycin + 1X L-GLU. Neutralization assays were performed in Low-FBS medium contained of DMEM + 0.5% FBS + 10 mM HEPES + 1x Penicillin-Streptomycin + 1X L-GLU. All tissue culture media and reagents were obtained from BI (Biological industries Beit HaEmek, Israel) except HEPES which was purchased from Sigma Aldrich.
Reporter virus particles (RVPs)
RVPs for this experiment were purchased from Integral Molecular, Inc. (Philadelphia, PA). SARS-CoV-2 RVPs Lot# CL-114B display antigenically correct spike protein on a heterologous virus core and carry a modified genome that expresses a convenient optical reporter gene (luciferase) within 24 h of cellular infection. VSV (vesicular stomatitis virus) RVPs Lot# VL-110B were used as a control to test for non-specific factors that affect virus infectivity. These RVPs display the VSV envelope glycoprotein (VSV-G) pseudo typed on replication-incompetent virus particles that contain a heterologous lentiviral (HIV) core and carry a genome that expresses a luciferase optical reporter gene upon infection.
Inhibitory peptides and spike S1 neutralizing antibody
Inhibitory peptides were designed by Pepticom’s AI platform. Structure-Activity Relationship (SAR) was performed by Pepticom’s chemists. Peptides were synthesized by CPC Scientific, Inc.
Spike S1 neutralizing antibody (catalog no. 220111 BPS Bioscience) was used as positive control in this study. This antibody was derived from COVID-19 patients who have cleared the virus.
Inhibitory peptide neutralizing assay
The assay was performed in cell culture 96 well plates (Greiner Lot# E20123KK). All experimental wells were coated with 50 µL of Poly-l-Lysine 50 μg/ml for better cell adhesion, then incubated in RT for 5 min, aspirated and thoroughly rinsed with 200 µL of sterile tissue culture grade water twice and incubated at room temperature for at least 2.5 h.
Inhibitory peptides were diluted in accordance with manufacturers manual. Water soluble peptides were diluted with Low-FBS cell culture medium. Water insoluble peptides were diluted with 100% DMSO (ARCOS ORGANICS Lot# 2193154) to 6 concentrations (1:1 dilutions). Test concentrations were achieved by dilution of each concentration X50 with Low-FBS cell culture medium to reach 2% DMSO. Neutralizing antibody was tested in 6 1:2 concentrations (25 nM maximal tested concentration) 2% DMSO. The diluent was Low-FBS cell culture medium with 0/2% DMSO (depending on tested inhibitory peptide being water soluble/insoluble). Complexation of RVPs and inhibitory peptide/antibody was performed in a sterile 1.5 Eppendorf vials. The optimal final concentration for RVPs was experimentally determined as 10 µL for a single well of 96-well plate. RVP + inhibitory peptide/antibody complexation mixtures were thoroughly mixed then incubated at room temperature for 1 h.
20,000 239 T-hsACE2 cells, in a 100 µL Low-FBS cell culture medium, were seeded per a single well of a 96-well plate. 100 µL of RVP + inhibitory peptide/antibody complexation mixture were added to each experimental well, bringing the total volume to 200 µL per well. All concentrations were tested in triplicates. All RVP experiments were performed within a BSL2 laboratory environment.
24 h post infection (HPI) Low-FBS cell culture medium was replaced with complete cell culture medium containing 10% FBS. 72 HPI supernatant from all experimental wells was aspirated. 30 µL of Dubecco’s Phosphate Buffered saline (PBS) was added, then additional 30 µL of Luciferase Assay Substrate (Promega) diluted 1:100 in Renilla-Glo™ Luciferase Assay Buffer was added to each well. After 10 min incubation according to manufacturer’s manual, luminescence was measured using Biotek Synergy H1 plate reader.
Data interpretation
Raw plate reads for each concentration point were first normalized relative to positive control (Maximal luciferase activity, no inhibitory compound) and negative control (background luciferase activity) wells. Data normalization, curve fitting and scheme figures were performed using Microsoft Excel. The results were represented as relative luciferase activity as derivative of inhibitory peptide/antibody concentration.
Micro Scale Thermophoresis (MST)
Peptide’s affinity to ACE2-His tag, from BPS Bioscience (catalog number #11003-1) was tested.
Monolith protein labelling kit HIS-tag Red-tris-NTA 2nd generation (MO-L018) from NanoTemper was used to label 10 ug of protein according to manufacturer’s protocol. Labeling was performed after buffer exchange, in the following HEPES-based buffer: 50 mM HEPES, pH 7.4, 50 mM NaCl, 0.2 mM CaCl2, 0.05% Tween20, 2% DMSO. Peptides are dissolved to 20 mM stock in 100% DMSO and then diluted 1:50 to 400 uM, in the same HEPES-based buffer w/o DMSO. Following 15 × 1:1 dilutions the dose response curve is mixed 1:1 with labeled ACE2-His tag at a final concentration of 25 nM. After 15 min incubation, samples were loaded in 16 standard capillaries and MST measurements were performed at high MST and 100% excitation power in Monolith NT.115 device. Data was analyzed using the MO. Control Analysis Software was used to extract Kd.
Virtual screening
Virtual screening was performed using Pepticom’s software. Screening was performed on input protein structures, focusing around a specific amino-acid centered spherical grid, as specified in the text. Explicit hydrogen atoms were added prior to grid initiation. Several grids were prepared at a 1Å resolution, both with precalculated properties (including a concavity/convexity and “best-case” atom-type docking) and naïve grids used by learning algorithms, as previously described (Lerner et al., 2017).
Docking was performed on a rigid target structure with both ligand backbones and side chains flexible. Docking was performed using virtual agents, or “bots”, on the AWS cloud. Each bot selected a sequence either random (“explore”) or based on a previously docked sequence (“exploit”). Each bot has a predetermined number of steps, uniformly initialized between bots and increased/decreased through relative rewards. Macrocycle conformational flexibility was achieved using the Pepticom’s CYCPEP module database, previously used to discover TLR4 MD2/CD14 agonists (Michaeli et al., 2018). Briefly, the bot selects a macrocycle backbone with dihedral angle compatibility to the selected sequence, according to a predefined complex preference function, so for example, one bot may prefer a backbone rich in internal hydrogen-bonds (a lower desolvation penalty risk), whereas another may prefer a backbone which maximizes expected side-chain flexibility for the selected sequence (a lower structure compatibility risk). Docking is then performed using a variety of docking gids, with each “bot” having a specific grid preference. Grids include a “best case” atom type docking grid, an “average case” atom type docking grid and size specific (see Size Specific Concavity Calculation Section below) concavity docking grids. The combination of grids with “bot” preference and docking heuristic allows fast solutions convergence. Once a docking solution has been reached, the bot can select to thread additional sequences to it (“exploit”) using the risk/return heuristic (Lerner et al., 2017), or seek another docking solution (“explore”). Docking is performed using Ephemeral Super Cluster on Amazon cloud AWS, computing ∼8000 docking solutions in parallel. Ephemeral meaning that the cloud resources are provisioned for computation writing the results to a static data-lake (s3 buckets) and duly released. Upon completion of its tasks, the bot updates the Kubernetes Cluster which updates the existing grids using the Cassandra, distributed Key-value datastore. Successful bots also recombine their preferences to form other potentially successful bots. Docked peptides are evaluated as previously described (Lerner et al., 2017). Since the reward is given to the docking bot, and not the sequence, a sequence may theoretically be selected multiple times, providing alternative docking strategies and solutions. Bot reward functions are composed of the top decile of docked sequences, with a decay penalty for exploited solutions.
Clustering was performed on docked solutions of backbones of the same size, using the K-means algorithm (MacQueen, 1967), performed on the 3D docked backbone coordinates. Cluster representatives were selected as the top scoring sequences in each cluster in the following categories: calculated binding energy, calculated binding energy per mass, calculated electrostatics, calculated electrostatics per mass and backbone hydrogen bond count (both internal and with the target protein). The number of representatives selected was determined by the users and described in the Results section.
Validation docking
To re-asses the docked structures using published scoring functions, docking positions were re-assessed using commercial software. Prior to docking, all peptides and the relevant spike/ACE2 protein structures were prepared using the protein preparation protocol in Schrödinger’s Maestro software (Citation). In the next step with the prepared receptor-peptide complex in the Workspace, Receptor Grid Generation Panel of Schrödinger’s Glide software (Friesner et al., 2004, 2006; Halgren et al., 2004) was used to generate receptor grid for docking. The standard protocol for grid generation was used, and for each grid groups that should be treated as rotatable in the grid generation were chosen. In the final step, all peptides were docked using Ligand Docking Panel of Schrödinger’s Glide software (Friesner et al., 2004, 2006; Halgren et al., 2004). For the docking procedure the SP (standard precision) method was used and post-docking minimization was activated. This option allows to perform minimization following the final docking. Finally, the minimized poses were re-scored using Schrödinger’s Glide proprietary GlideScore function (Friesner et al., 2004, 2006; Halgren et al., 2004). All presented Docking scores were calculated using the methodology described above with the GlideScore serving as the Docking score in the article.
Size specific concavity calculation
To achieve shape complementarity in docking, macrocycle and target protein convexity/concavity were calculated as follows: A central point for docking was selected on the protein and peptide surface. The sum of angles between the other molecule’s central point, the investigated central point and all other surface points of the same structure, within a pre-set radius was calculated. Surfaces with an average angle of about 90° are considered flat, lower than 90° are concave and higher than 90° are convex. This method allows for pre-calculation on a backbone conformation database, and selection of a plausible conformation prior to docking.
Results
Virtual screening on different conformations was performed using Pepticom software. Each screening run focused on a specific area of a solved spike structure. The structures and docking regions were selected according to three strategies:
1) Allosteric inhibition of the spike trimer that keeps spike in the closed conformation, preventing RBD exposure.
2) Direct ACE2 competitive inhibition, targeting the spike binding interface.
3) Direct spike RBD competitive inhibition, targeting the ACE2 interface.
For each virtual screening, a docking area was selected, A.I. screening was performed and the results were clustered into frequently exploited, high scoring conformational clusters. A set of high scoring, diverse, members of each cluster was selected for synthesis and physical screening. The number of molecules per set was determined according to score distribution and diversity.
Allosteric closed conformation inhibitors
To screen for potential allosteric inhibitors, a structure of spike in its closed state was selected (PDB: 6VXX) (Walls et al., 2020). To best interact with the 3 protomer buried RBD domains, a grid encompassing the 15 Å radius from the CA atom of arginine in position 408 of chain A was selected (Figure 1A). The screening process involved N to C cyclic peptide combinations, ranging in size from 5 to 15 amino acid positions and including over 400 “non-natural” amino-acids per position. Screening was performed in 50 cycles interlocking machine learning and virtual screening. The output was then clustered and representative peptides of four clusters were selected for synthesis. Peptides were then screened using Reporter virus particles (RVP) at peptides concentrations of 10 μM and 100 µM (Table 1).
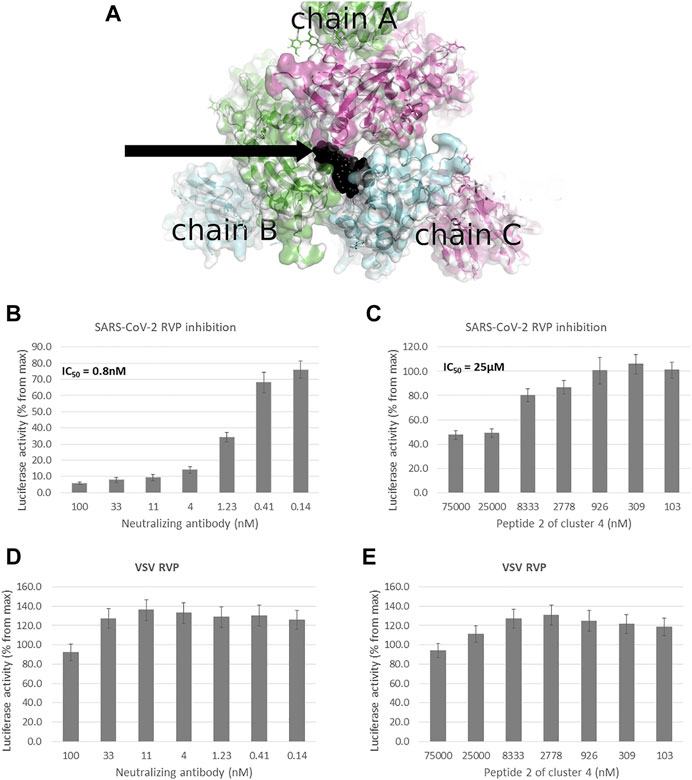
FIGURE 1. Peptidomimetic macrocycles screening for allosteric inhibitors. (A) Allosteric macrocycles were screened on the closed state spike structure (PDB: 6VXX), In the area adjoining the RBD domains of chain A (green), chain B (blue) and chain C (purple). The docked model of cluster 4, peptide 2 is shown in black. To screen molecules for inhibitory activity, SARS-CoV-2 Reporter virus particles (RVP) were used, with VSV RVP used as control. (B) A commercial human monoclonal antibody shows an IC50 of 0.8 nM, with (C) cluster 4, peptide 2 showing an IC50 of 25 µM and incomplete inhibition. Neither the (D) antibody or (E) peptide show inhibitory activity against the VSV RVP (RVP data based on n = 2 biological replicates, 3 technical replicates per experiment, standard mean error (SEM) is represented for each concentration).
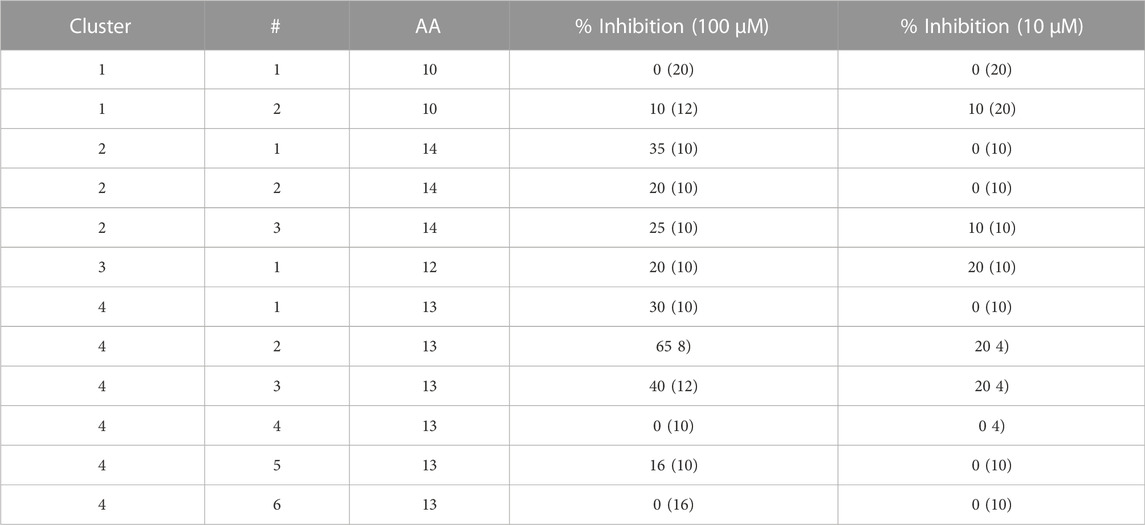
TABLE 1. Peptidomimetics with allosteric spike binding model. Macrocycles representative of four clusters, were synthesized and screened at 10 μM and 100 µM against a SARS-CoV-2 RVP. The percent inhibition is shown with numbers in parenthesis showing the standard deviation of the mean (n = 3).
Of the 12 peptides screened in the RVP assay, only two, peptides 2 and 3 of cluster 4, showed consistent inhibition at both 10 μM and 100 µM (Table 1). The most potent peptide, peptide 2 of cluster 4, was selected for dose response analysis, compared to a SARS-CoV-2 neutralizing antibody serving as a positive control and a VSV RVP serving as a negative control. Both peptide and antibody showed no significant inhibitory activity on the control VSV RVP, with less than 10% inhibition at 100 µM and mildly increasing infection at lower concentrations (Figures 1D, E). On the SARS-CoV-2 RVP, the macrocycle showed inhibition with a partial dose response, maximizing at 50% inhibition at 25 μM, without significant increase at higher concentrations (Figure 1C). In contrast, the antibody showed a complete dose response, with over 94% inhibition reached at 100 µM (Figure 1B).
ACE2 competitive inhibitors
Screening for both spike RBD domain and ACE2 binding peptides was performed using the spike/ACE2 complex structure (PDB: 6VW1) (Shang et al., 2020). To screen for potential ACE2 binders, a 15 Å radius grid was centered around the CA atom of leucine 79 of chain A, covering the ACE2 area that binds the spike RBD loop between residues 470–492 in the complex structure (chain E) (Figure 2A). Peptidomimetics were screened using the same molecular parameters and run protocol as above. The output yielded only one cluster, of 15-mer peptidomimetics with Glide scores indicating inhibitory potential (−4.6 to −7.3 Kcal/mol).
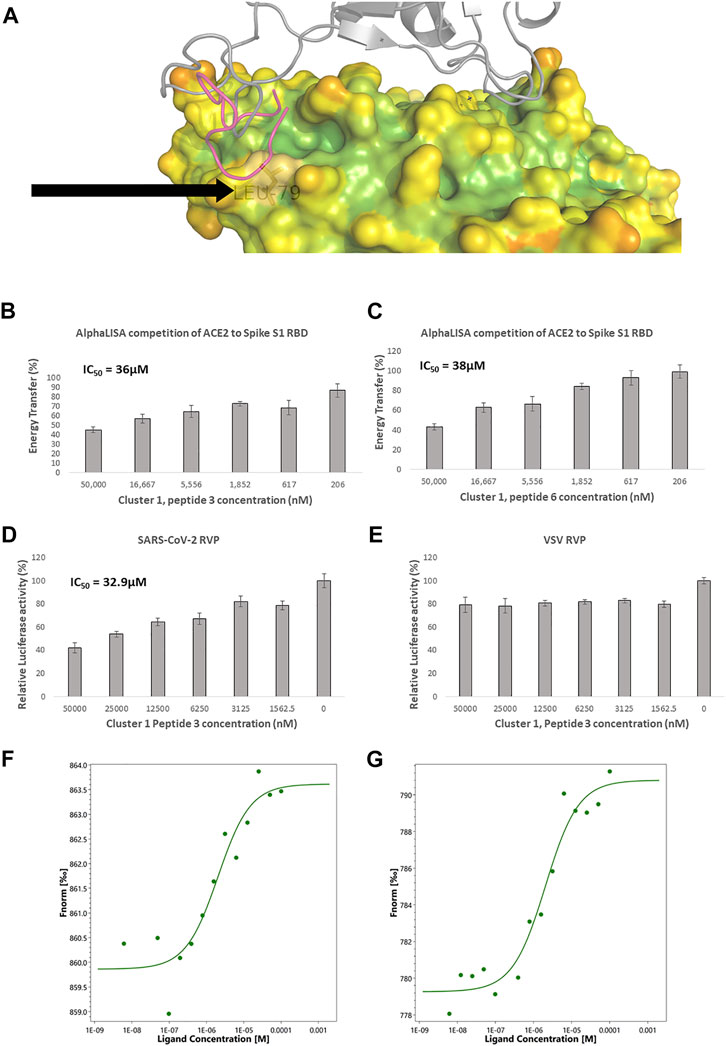
FIGURE 2. Competitive ACE2 targeted screening. (A) A heat diagram analysis of RBD surface concavity (PDB: 6VW1, chain A), (blue = concave, red = convex), spike RBD is shown as a grey cartoon. Leucine 79, chosen as the center of the screening grid, is shown with the black arrow. Cluster 1, peptide 3 docking model is shown as purple cartoon. AlphaLISA analysis was used to detect the interaction between the spike protein and the hACE2 receptor. Characterization of inhibitory effect on the interaction between spike and hACE2 proteins. IC50 was calculated for (B) Cluster 1, peptide 3 and (C) cluster 1 peptide 6 (n = 2 biological replicates, 3 technical replicates per experiment, standard mean error (SEM) is represented for each concentration) SARS-CoV-2 and control lentivirus were incubated with Cluster 1, peptide 3 and cells stably expressing human ACE2 in low serum media. After 24 h the media was replaced with fully supplemented media for additional 48 h (D) Inhibitory effect on the infection of SARS-CoV-2 lentivirus in 6 concentrations are presented. n = 1 (3 technical replicates) (E) Inhibitory effect on the infection of control lentivirus in 6 concentrations are presented. n = 1 (3 technical replicates) for VSV. IC50 is calculated. The standard mean error (SEM) is represented for each concentration. Micro Scale Thermophoresis (MST) was used to analyze ACE2 binding (F) Cluster 1 peptide 3, shows a dissociation constant (Kd) of 2.02 µM (Amplitude 3.8, Signal to Noise Ratio 8) and (G) Cluster 1, peptide 6 a Kd of 2.03 µM. (Amplitude 11.5, Signal to Noise Ratio 11.6).
Eight representative molecules were selected and synthesized. Screening was performed using AlphaLISA, to determine biochemical potency. Additionally, molecules were tested using the TruHits kits, as a control for false positive signal. None of the active molecules showed a TruHits false positive signal. Three out of the eight molecules showed spike RBD/ACE2 inhibitory activities, with IC50 values ∼40 µM (Table 2). Cluster 1 peptide 3 and cluster 1 peptide 5 showed a shallow inhibition slope (Figures 2B, C), and cluster 1 peptide 3 showed a similar inhibition profile in the SARS-CoV-2 RVP (Figure 2D). Cluster 1 peptide 3 showed no significant inhibition in the control VSV RVP (Figure 2E). Peptide 5 of the same cluster showed no inhibitory activity on either SAS-CoV-2 or VSV RVP. As the selected docking area corresponds to a relatively flexible RBD loop, we tested the binding of inhibitory peptide 1 and inactive peptide 2 using Micro Scale Thermophoresis (MST). Both peptides showed Kd values of approximately 2 µM (Figures 2F, G). To attempt to overcome this obstacle, another grid was selected for screening, with a similar radius around the ACE2 methionine 383 (chain A), which corresponds to the more rigid area of spike. However, screened molecules drifted towards an area that is not predicted to inhibit the spike complex formation.
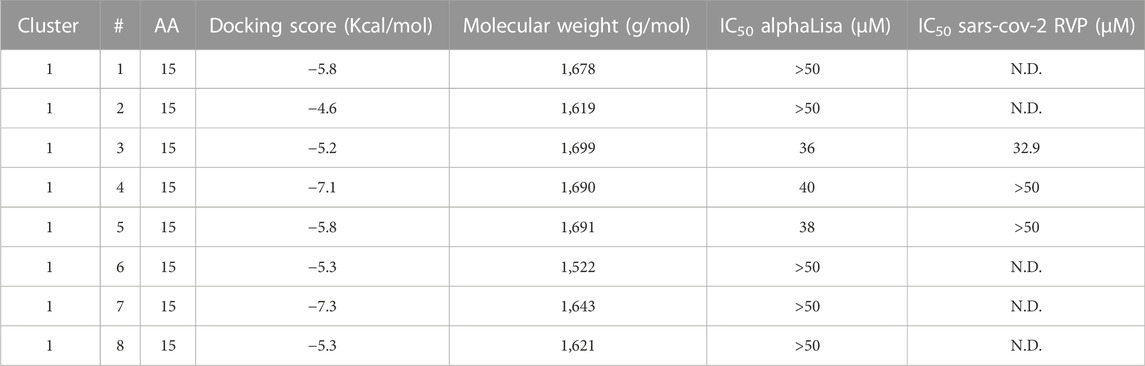
TABLE 2. Peptidomimetics designed to target ACE2 at the spike interface. Macrocycles representative of one cluster were synthesized and screened for spike RBD/ACE2 complex formation inhibition using AlphaLISA. Measured AlphaLISA IC50 values are shown. Macrocycles showing inhibitory activity were selected for SARS-CoV-2 RVP inhibition assays. N.D. = Not Done. Values with a greater than (>) symbol indicate that IC50 was not reached at maximal tested dosage.
Spike competitive inhibitors
Screening for direct spike RBD inhibitors was performed using a 15 Å radius grid was centered around the CA atom of tyrosine 453 of chain E (Figure 3A), with similar run parameters. High scoring output molecules were clustered into 5 clusters, with cluster representatives synthesized and screened using the AlphaLISA assay, with TruHits control assay (Table 3). None of the active molecules showed a TruHits false positive signal.
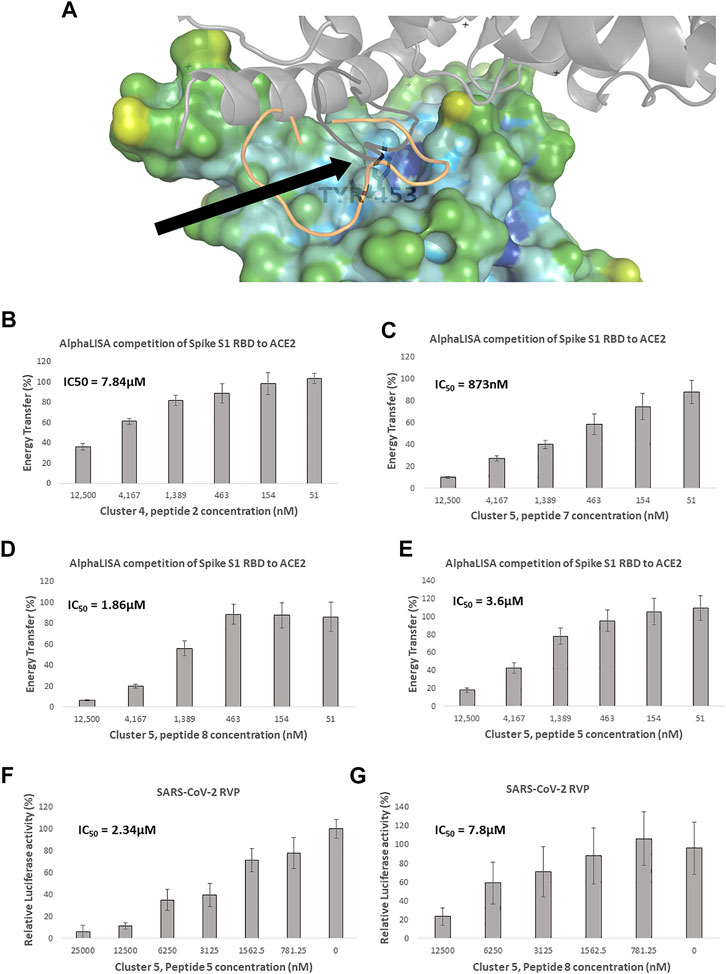
FIGURE 3. Competitive spike RBD targeted screening. (A) A heat diagram analysis of RBD surface concavity (PDB: 6VW1, chain E), (blue = concave, red = convex), the ACE2 receptor is shown as a grey cartoon. Tyrosine 453, chosen as the center of the screening grid, is shown with the black arrow. Cluster 4, peptide 7 docking model is shown as orange cartoon and Cluster 4, peptide 7 in black cartoon. AlphaLISA analysis was used to detect the interaction between the spike protein and the hACE2 receptor. Characterization of inhibitory effect on the interaction between spike and hACE2 proteins. IC50 was calculated for (B) Cluster 4, peptide 2 and (C) Cluster 5, peptide 7 (D) Cluster 5, peptide 8 and (E) Cluster 5, peptide 5 (n = 2 biological replicates, 3 technical replicates per experiment, standard mean error (SEM) is represented for each concentration) Of these macrocycles, only (F) Cluster 5, peptide 5 n = 3 (9 technical replicates) and (G) Cluster 5, peptide 8 n = 3 (9 technical replicates), showed specific SARS-CoV-2 RVP inhibition. SARS-CoV-2 and control lentivirus were incubated with the tested peptide and cells stably expressing human ACE2 in low serum media. After 24 h the media was replaced with fully supplemented media for additional 48 h, IC50 was calculated. The standard mean error (SEM) is represented for each concentration.
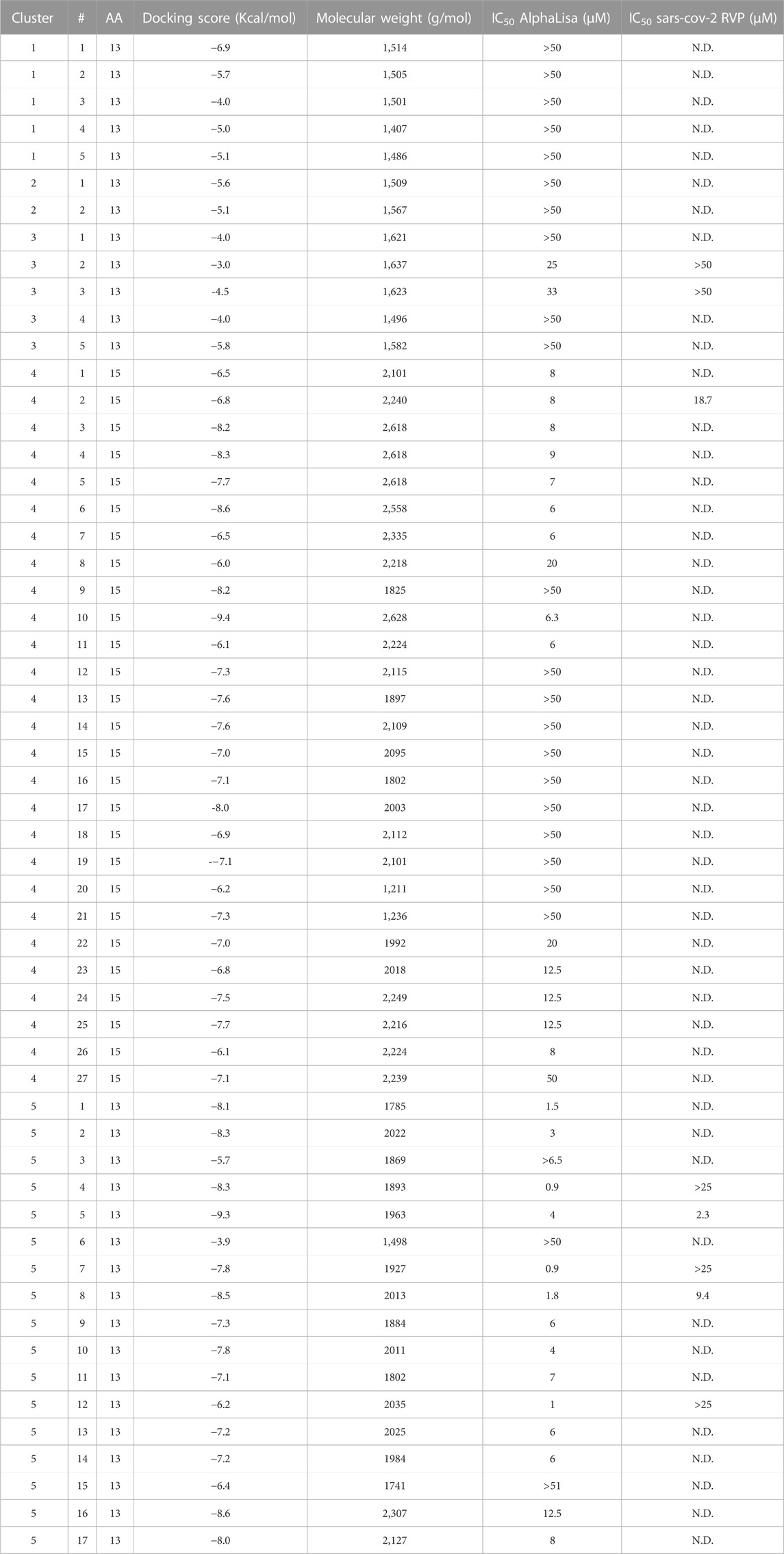
TABLE 3. Peptidomimetics designed to target spike at the ACE2 interface. Macrocycles representative of five clusters were synthesized and screened for spike RBD/ACE2 complex formation inhibition using AlphaLISA. AlphaLISA IC50 values are shown. Representative macrocycles showing inhibitory activity were selected for SARS-CoV-2 RVP inhibition assays. N.D. = Not Done. Values with a greater than (>) symbol indicate that IC50 was not reached at maximal tested dosage.
Clusters 1, 2 and 3 had the poorest Glide Scores (−3 to −6.9, with an average of −4.9 Kcal/mol), with clusters 1 and 2 showing no inhibitory activity, while cluster 3 molecules included weak inhibitors, numbers 2 and 3, with weak Glide scores (−3 and −4.5 Kcal/mol, respectively) showing AlphaLISA inhibition (IC50 values of 25 and 33 μM, respectively) and no inhibitory activity at the cell RVP level.
Clusters 4 and 5 showed better Glide scores, ranging from −3.9 to −9.4, with an average of −7.3 Kcal/mol. These clusters included several inhibitors with AlphaLISA IC50 values lower than 10 µM (Table 3). Molecule 2 of cluster 4 showed an IC50 of 7.84 µM in AlphaLISA, with a shallow dose response (Figure 3B), and showed an IC50 of 18.7 µM when tested against SARS-CoV-2 with some inhibition of the control VSV RVPs. The Glide score of Molecule 2 of cluster 4 was −6.8 Kcal/mol, with a binding model relatively poor in hydrogen bonds (8 for a 15 AA macrocycle), that together with the observed shallow does response and VSV activity suggested some non-specific activity.
The most potent AlphaLISA inhibitor of cluster 5, peptide 7 showed an IC50 of 873 nM in AlphaLISA (Figure 3C), but no SARS-CoV-2 RVP inhibition. Peptides 8 and 5 showed AlphaLISA IC50 values of 1.86 µM and 3.6 µM, respectively (Figures 3D, E). When screened against SARS-CoV-2 RVP, peptide 5 showed an IC50 of 2.34 µM and peptide 8 an IC50 of 9.4 µM (Figures 3F, G), both peptides showed no significant inhibition of the control VSV RVP. Macrocycles 5, 7 and 8 had calculated Glide scores of −9.3, −7.8 and −8.5 Kcal/mol, respectively. Unlike the incomplete AlphaLISA inhibition exhibited by ACE2 targeted macrocycles (Figures 2B, C), some spike targeting molecules showed a steeper dose response, reaching 80%–90% inhibition at 12.5 µM (Figures 3C–E). A similar pattern was observed in the RVP assays, with a shallow dose response for the ACE2 binding inhibitor (Figure 2D) and a steeper dose response in the spike RBD binding inhibitors (Figures 3F, G). The amino-acid composition of cluster 5 was heavily leaning towards non-natural amino-acids, with peptide 5, 7 and 8 having 10, 9 and 10 “non-natural” amino-acids out of a 13 AA macrocycle. Cluster structures used 2-amino-2-indancarboxylic-acid (Aic) and l-Pipecolic acid (Pip) for structure rigidification, potentially reducing the loss of entropy upon binding and lowering enzyme susceptibility.
Discussion
In this study we performed virtual screening of small (5–15 amino-acid), N to C cyclic, peptidomimetics for three different inhibition strategies for the spike/ACE2 complex: allosteric inhibition of the spike trimer that keep spike in the closed conformation; direct ACE2 inhibition; and direct spike RBD inhibition. Output peptidomimetics for each strategy were clustered and representatives of each cluster were selected for synthesis and screening. Biochemical screening was performed using a spike RBD/ACE2 AlphaLISA assay and functional screening was performed using the SARS-CoV-2 RVP.
Of the three strategies, allosteric inhibition had the most attractive stoichiometry. Whereas, in the RBD competitive binding strategy, complete inhibition requires all spike RBDs to be bound, allosteric inhibitors can theoretically lock all the spike RBDs with one molecule. The ACE2 strategy is challenged by the broad tissue expression of ACE2. The critical morbidity factor in SARS-CoV-2 infection arises from the lungs, ACE2 is however, broadly expressed, in tissues including the small intestine, colon, gallbladder and others (Hikmet et al., 2020). Systematic administration of molecules that bind ACE2, will also bind ACE2 at sites remote to viral infection, requiring a high dosage. The allosteric strategy can theoretically deploy one peptidomimetic to constrict all 3 RBDs to the closed state, making it attractive. Another theoretical advantage of this approach, is the non-competitive mode of action, allowing a relatively small molecule to inhibit a high affinity protein-protein complex.
Of the 12 peptides representing 4 clusters in this effort, we reached one inhibitor that showed specific SARS-CoV-2 RVP inhibition. We believe that the partial inhibition was caused by partial RBD escape from inhibition that is most likely caused by lack of shape complementarity between the three-pointed star shape of the pocket and the relatively flat small macrocycle conformation (Figure 1A). Other options, such as branching new chains out of macrocycle side-chains are currently being explored to address this issue and divide the modeled binding energy somewhat more equally between the monomers. Another possible cause for potential RBD escape is the lack of symmetry between the calculated binding free-energy between the RBD petals. So, for example, in peptide 2 of cluster 4, the aspartate at position 405 of the closed structure (PDB: 6VXX) is met by a macrocycle salt bridge on chain C, but not chains A or B, while sterically preventing the access of another identical peptide to these positions. An alternative solution to this issue could be screening for molecules that bind the corners in which 2 petaLs meet. Such potential solutions could provide a better free energy distribution, albeit, requiring 3 peptides to inhibit spike/ACE2 complex formation.
The human receptor ACE2, spike binding region is rather convex in relation to a typical 5–15 AA macrocycle (Figure 2A), when compared to the corresponding spike region (Figure 3A). As small macrocycles are rather rigid, they are less likely to bind a convex region. The ACE2 convex geometry is further complexed by more concave areas that are near the spike binding interface, these cause the virtual screening agents to “runoff” away from areas where they can have complete efficacy. Our efforts yielded one cluster of weak inhibitors, with similar IC50 values in AlphaLISA and cellular RVP assay, suggesting that our cell assay lacked integrin mediated penetration. Interestingly, the small, linear, fibronectin derived, non-competitive α5β1 integrin binding peptide, ATN-161 can inhibit complex formation and decrease infection at the cellular level (Beddingfield et al., 2021). In an in silico screening effort followed by lab validation, Merck’s antifungal macrocycle, Caspofungin, was also predicted to bind the ACE2 spike binding site at a low-level micromolar activity, but showed no lab screening activity, whereas the non-peptide bases macrocycle Zotarolimus, showed a low micromolar Kd and inhibitory activity (Day et al., 2021). Small molecules of the same screening effort such as the amino acid Levodopa showed an ACE2 Kd in the double digit nanomolar range, with over 36% competition with spike-RBD at 1 μM (Day et al., 2021). These published results concur with our findings that macrocycles are less than optimal for ACE2 targeted SARS-CoV-2 viral entry inhibitors.
Targeting the spike RBD in its ACE2 binding region yielded more clusters at the computational stage than in the corresponding ACE2 region, with RBD clusters 4 and 5 showing several molecules with better docking scores than the top scoring molecules o the ACE2 cluster (Tables 2, 3), suggesting it is a more plausible macrocycle target. Of the five selected clusters, two clusters, cluster 4 and 5, showed molecules with single digit micromolar inhibition constants in both AlphaLISA and SARS-CoV-2 RVP. These molecules initiated our ongoing effort to reach lead stage molecules.
Computationally derived, 56 amino acid RBD binding miniproteins, based on ACE2 helices, yielded inhibition constants reaching the picomolar range with proven live virus efficacy (Cao et al., 2020). Nevertheless, in our opinion, miniproteins do not offer distinct pharmaceutical differentiation from antibodies, some of which are in clinical use (Raman et al., 2021), or their derived nanobodies which show similar potency (Xiang et al., 2020). Hence, achieving smaller, non-natural molecules, could produce more novel drug candidates. A discovery effort for small peptide inhibitor, based on the ACE2 interface helix yielded a 13 amino acid helical peptide with ∼40% inhibition at 100 µM (Rajpoot et al., 2021). Like macrocycles, helical peptides “suffer” from geometric constraints that limit their flexibility in interacting with the target protein, albeit with lower loss of entropy, as was shown using molecular dynamics (Rajpoot et al., 2021). Small molecules screened against the spike/ACE2 complex also showed a proof of concept that drug candidates without a tertiary structure are possible (Bojadzic et al., 2021; Day et al., 2021). The physical screening of molecules in the organic dye chemical space yielded several low micromolar inhibitors, shown to bind the spike RBD domain and provide similar simulated pseudo viral entry blockage as cluster 4 and 5 RBD targeting macrocycles (Bojadzic et al., 2021). Interestingly, Lifitegrast was shown to bind spike RBD at ∼ 2 nM, and inhibit complex formation, with a millimolar viral infection IC50 (Day et al., 2021). The Evans Blue dye was also shown to bind both ACE2 (Kd ∼2 nM) and spike RBD (Kd ∼2 μM), with a viral inhibition IC50 of ∼28 μM. With the exception of Evans Blue these small molecules all share a relatively poor abundance of hydrogen bond donors and acceptors and that can possibly lead to multiple non-specific interactions when assayed in biological assays. We feel that the data presented here, along with published efforts suggest that spike RBD can be useful drug target, in which a compromise between size, target specific binding and efficacy can produce innovative drug candidates.
Data availability statement
The datasets presented in this article are not readily available because the dataset is composed of internal molecular data that will be available to the public upon patent protection. Requests to access the datasets should be directed to YW1pdC5taWNoYWVsaUBwZXB0aWNvbS5jb20=.
Author contributions
AM is the head of research and technology at Pepticom. LS performed the cell RVP assays and concentrated the data for this paper. SL is the head of biology at Pepticom and performed the Micro Scale Thermophoresis experiments. GA and DH performed the AlphaLISA biochemical screening assay. AV and VK supervised software run parameters, output clustering and molecular synthesis. GB managed cloud computing operations and clustering scripts. MR assisted in background research, target selection and strategy proposition. IL is Pepticom’s CEO, he initiated the discovery effort and secured the funds to perform it.
Acknowledgments
We thank Dr. Waleed Danho and Dr. Tomi Sawyer for fruitful discussion and advice. We also thank Dr. Lior Nissim from the Hebrew university for the useful advice, participation and scientific supervision in our inhibitory peptide neutralizing assay.
Conflict of interest
LS, SL, GA, AV, DH, GB, VK, MR, IL, and AM were employed by the company Pepticom Ltd. The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Beddingfield, B. J., Iwanaga, N., Chapagain, P. P., Zheng, W., Roy, C. J., Hu, T. Y., et al. (2021). The integrin binding peptide, ATN-161, as a novel Therapy for SARS-CoV-2 infection. JACC. Basic Transl. Sci. 6 (1), 1–8. doi:10.1016/j.jacbts.2020.10.003
Benton, D. J., Wrobel, A. G., Xu, P., Roustan, C., Martin, S. R., Rosenthal, P. B., et al. (2020). Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 588, 327–330. doi:10.1038/s41586-020-2772-0
Bojadzic, D., Alcazar, O., Chen, J., Chuang, S. T., Condor Capcha, J. M., Shehadeh, L. A., et al. (2021). Small-molecule inhibitors of the coronavirus spike: ACE2 protein-protein interaction as blockers of viral attachment and entry for SARS-CoV-2. ACS Infect. Dis. 7 (6), 1519–1534. doi:10.1021/acsinfecdis.1c00070
Cao, L., Goreshnik, I., Coventry, B., Case, J. B., Miller, L., Kozodoy, L., et al. (2020). De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Sci. (New York, N.Y.) 370 (6515), 426–431. doi:10.1126/science.abd9909
Cary, D. R., Ohuchi, M., Reid, P. C., and Masuya, K. (2017). Constrained peptides in drug discovery and development. J. Synth. Org. Chem. Jpn. 75 (11), 1171–1178. doi:10.5059/yukigoseikyokaishi.75.1171
Day, C. J., Bailly, B., Guillon, P., Dirr, L., Jen, F. E. C., Spillings, B. L., et al. (2021). Multidisciplinary approaches identify compounds that bind to human ACE2 or SARS-CoV-2 spike protein as candidates to block SARS-CoV-2-ACE2 receptor interactions. Mbio. U. S. 12 (2), 036811. doi:10.1128/mBio.03681-20
Friesner, R. A., Banks, J. L., Murphy, R. B., Halgren, T. A., Klicic, J. J., Mainz, D. T., et al. (2004). Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47 (7), 1739–1749. doi:10.1021/jm0306430
Friesner, R. A., Murphy, R. B., Repasky, M. P., Frye, L. L., Greenwood, J. R., Halgren, T. A., et al. (2006). Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 49 (21), 6177–6196. doi:10.1021/jm051256o
Froese, D. S., Michaeli, A., McCorvie, T. J., Krojer, T., Sasi, M., Melaev, E., et al. (2015). Structural basis of glycogen branching enzyme deficiency and pharmacologic rescue by rational peptide design. Hum. Mol. Genet. 24 (20), 5667–5676. doi:10.1093/hmg/ddv280
Halgren, T. A., Murphy, R. B., Friesner, R. A., Beard, H. S., Frye, L. L., Pollard, W. T., et al. (2004). Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 47 (7), 1750–1759. doi:10.1021/jm030644s
Hikmet, F., Mear, L., Edvinsson, A., Micke, P., Uhlen, M., and Lindskog, C. (2020). The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 16 (7), e9610. doi:10.15252/msb.20209610
Huang, Y., Yang, C., Xu, X. F., Xu, W., and Liu, S. W. (2020). Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 41 (9), 1141–1149. doi:10.1038/s41401-020-0485-4
Jackson, C. B., Farzan, M., Chen, B., and Choe, H. (2022). Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 23 (1), 3–20. doi:10.1038/s41580-021-00418-x
Kourouklis, D., Murakami, H., and Suga, H. (2005). Programmable ribozymes for mischarging tRNA with nonnatural amino acids and their applications to translation. Methods (San Diego, Calif.) 36 (3), 239–244. doi:10.1016/j.ymeth.2005.04.001
Lerner, I., Goldblum, A., Rayan, A., and Vardi, A. (2017). From finance to molecular modeling algorithms: The risk and return heuristic. Curr. Top. Peptide Protein Res. 18, 117–131.
Li Petri, G., Di Martino, S., and De Rosa, M. (2022). Peptidomimetics: An overview of recent medicinal Chemistry efforts toward the discovery of novel small molecule inhibitors. J. Med. Chem. 65 (11), 7438–7475. doi:10.1021/acs.jmedchem.2c00123
Li, W., Moore, M. J., Vasilieva, N., Sui, J., Wong, S. K., Berne, M. A., et al. (2003). Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426 (6965), 450–454. doi:10.1038/nature02145
Liu, J., Lu, F., Chen, Y., Plow, E., and Qin, J. (2022). Integrin mediates cell entry of the SARS-CoV-2 virus independent of cellular receptor ACE2. J. Biol. Chem. 298 (3), 101710. doi:10.1016/j.jbc.2022.101710
MacQueen, J. B. (1967). “Some methods for classification and analysis of MultiVariate observations,” in Proc. Of the fifth berkeley symposium on mathematical statistics and probability. Editors L. M. Le Cam, and J. Neyman (University of California Press), 281–297.
Michaeli, A., Lerner, I., Zatsepin, M., Mezan, S., and Kilshtain, A. V. (2020). Discovery of novel GABAAR allosteric modulators through reinforcement learning.’, Current pharmaceutical design. Curr. Pharm. Des. 26 (44), 5713–5719. doi:10.2174/1381612826666201113104150
Michaeli, A., Mezan, S., Kuhbacher, A., Finkelmeier, D., Elias, M., Zatsepin, M., et al. (2018). Computationally designed bispecific MD2/CD14 binding peptides show TLR4 agonist activity. J. Immunol. 201, 3383–3391. doi:10.4049/jimmunol.1800380
Muralidar, S., Ambi, S. V., Sekaran, S., and Krishnan, U. M. (2020). The emergence of COVID-19 as a global pandemic: Understanding the epidemiology, immune response and potential therapeutic targets of SARS-CoV-2. Biochimie 179, 85–100. doi:10.1016/j.biochi.2020.09.018
Ohuchi, M., Murakami, H., and Suga, H. (2007). In situ generation of aminoacyl-tRNAs assisted by ribozymes in translation apparatus. Nucleic Acids Symp. Ser. (Oxf). 51 (51), 115–116. doi:10.1093/nass/nrm058
Rajpoot, S., Ohishi, T., Kumar, A., Pan, Q., Banerjee, S., Zhang, K. Y. J., et al. (2021). A novel therapeutic peptide blocks SARS-CoV-2 spike protein binding with host cell ACE2 receptor. Drugs R. D. 21 (3), 273–283. doi:10.1007/s40268-021-00357-0
Raman, R., Patel, K. J., and Ranjan, K. (2021). COVID-19: Unmasking emerging SARS-CoV-2 variants, vaccines and therapeutic strategies. Biomolecules 11 (7), 993. doi:10.3390/biom11070993
Salzberger, B., Buder, F., Lampl, B., Ehrenstein, B., Hitzenbichler, F., Holzmann, T., et al. (2021). Epidemiology of SARS-CoV-2. Infection 49 (2), 233–239. doi:10.1007/s15010-020-01531-3
Shang, J., Ye, G., Shi, K., Wan, Y., Luo, C., Aihara, H., et al. (2020). Structural basis of receptor recognition by SARS-CoV-2. Nature 581, 221–224. doi:10.1038/s41586-020-2179-y
Smith, G. P. (1985). Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 228 (4705), 1315–1317. doi:10.1126/science.4001944
Taki, M., Kuno, A., Matoba, S., Kobayashi, Y., Futami, J., Murakami, H., et al. (2006). Leucyl/Phenylalanyl-tRNA-protein transferase-mediated chemoenzymatic coupling of N-terminal Arg/Lys units in post-translationally processed proteins with non-natural amino acids. Chembiochem a Eur. J. Chem. Biol. 7 (11), 1676–1679. doi:10.1002/cbic.200600181
Walls, A. C., Park, Y. J., Tortorici, M. A., Wall, A., McGuire, A. T., and Veesler, D. (2020). Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 181 (2), 281–292. e6. doi:10.1016/j.cell.2020.02.058
Wrobel, A. G., Benton, D. J., Xu, P., Roustan, C., Martin, S. R., Rosenthal, P. B., et al. (2020). SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat. Struct. Mol. Biol. 27 (8), 763–767. doi:10.1038/s41594-020-0468-7
Xiang, Y., Nambulli, S., Xiao, Z., Liu, H., Sang, Z., Duprex, W. P., et al. (2020). Versatile and multivalent nanobodies efficiently neutralize SARS-CoV-2. Sci. (New York, N.Y.) 370 (6523), 1479–1484. doi:10.1126/science.abe4747
Yong, S. J. (2021). Long COVID or post-COVID-19 syndrome: Putative pathophysiology, risk factors, and treatments. Infect. Dis. 53 (10), 737–754. doi:10.1080/23744235.2021.1924397
Keywords: macrocycle, peptidomimetic, SARS-CoV-2, inhibitor, screening, spike, ACE2
Citation: Shapira L, Lerner S, Assayag G, Vardi A, Haham D, Bar G, Kozokaro VF, Robicsek ME, Lerner I and Michaeli A (2022) Discovery of novel spike/ACE2 inhibitory macrocycles using in silico reinforcement learning. Front. Drug. Discov. 2:1085701. doi: 10.3389/fddsv.2022.1085701
Received: 31 October 2022; Accepted: 01 December 2022;
Published: 23 December 2022.
Edited by:
Giuseppe Felice Mangiatordi, National Research Council (CNR), ItalyReviewed by:
Francois Berenger, The University of Tokyo, JapanMaria Cristina De Rosa, Institute of Chemical Sciences and Technologies “Giulio Natta” (SCITEC) - CNR, Italy
Conrad Veranso Simoben, University of Buea, Cameroon
Copyright © 2022 Shapira, Lerner, Assayag, Vardi, Haham, Bar, Kozokaro, Robicsek, Lerner and Michaeli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amit Michaeli, YW1pdC5taWNoYWVsaUBwZXB0aWNvbS5jb20=