 Steven J. Cassady
Steven J. Cassady Danielle Soldin
Danielle Soldin Gautam V. Ramani
Gautam V. Ramani
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Drug Discov., 15 December 2022
Sec. Cardiovascular and Hematologic Drugs
Volume 2 - 2022 | https://doi.org/10.3389/fddsv.2022.1022971
This article is part of the Research TopicInsights in Cardiovascular and Hematologic Drugs: 2022View all 6 articles
Pulmonary arterial hypertension (PAH), defined as a mean pulmonary artery pressure exceeding 20 mmHg with a pulmonary vascular resistance of 3 or more Wood units, is an incurable and progressive condition. The cornerstone of PAH treatment is pulmonary vasodilators, which act on the pulmonary vasculature to reduce pulmonary pressures and pulmonary vascular resistance and prevent progression to right heart failure. The number of available pulmonary vasodilator therapies has grown markedly in the last 10 years, alongside a rapidly expanding body of literature establishing strategies for their use. Up-front combination therapy, typically with two pulmonary vasodilator medications, has become the standard of care based on landmark trials showing superior outcomes over single therapies alone. Complex risk stratification matrices have begun to see widespread use as tools with which to guide changes in PAH therapies for individual patients. Strategies for using the pulmonary vasodilators in common use continue to be evaluated in trials exploring concepts such as up-front triple combination therapy and substitution of vasodilators for patients not meeting therapeutic goals. Alongside established pulmonary vasodilator therapies for PAH, there is a broad spectrum of experimental therapies that are being studied for the disease. These include both more conventional medications that act on pathways targeted by existing vasodilator therapies as well as non-vasodilator treatments with novel methods of action, that may act both to vasodilate and to address the detrimental changes of pulmonary arterial and right ventricular remodeling. Many of these emerging medications are the focus of active phase 2 and 3 trials. Finally, there has been significant interest in therapeutic pathways that are well established in left heart failure, with the hope of adapting strategies that may be efficacious in PAH and right heart failure as well. These include explorations of pathways treated by goal-directed medical therapy as well as device therapies such as pacing, resynchronization therapy, and cardiac monitoring devices. Many of these options show promise and may represent a complementary approach to treatment of PAH, allowing for multimodal therapy alongside pulmonary vasodilators to improve patient outcomes.
Pulmonary arterial hypertension is a heterogeneous disease that results from pathogenic remodeling of the pulmonary arteries. As of the 6th World Symposium on Pulmonary Hypertension in 2018, the diagnosis of pulmonary hypertension is based on hemodynamic measurements, including mean pulmonary arteriole pressure (mPAP), pulmonary vascular resistance (PVR) and pulmonary artery wedge pressure, to classify patients into pre-capillary, post-capillary, or mixed pre- and post-capillary hypertension (Humbert et al., 2022).
The World Health Organization (WHO) Groups classify pulmonary hypertension into five clinical subgroups: WHO Group I, pulmonary arterial hypertension (PAH); (Table 1) WHO Group II, pulmonary hypertension related to left heart disease; WHO Group III, pulmonary hypertension related to chronic lung disease; WHO Group IV, pulmonary hypertension as a result of pulmonary obstructions (chronic thromboembolic pulmonary hypertension, CTEPH), and WHO Group V, pulmonary hypertension due to multifactorial mechanisms (Humbert et al., 2022).

TABLE 1. Subgroups of WHO group I pulmonary hypertension (Humbert et al., 2022).
PAH is principally a disease of progressive increases in pulmonary vascular resistance, which occurs through several concurrent pathways thought to be triggered by endothelial injury from environmental or genetic insults. These pathways center on remodeling and progressive loss of small pulmonary arteries, with fibrosis of the intima and hypertrophy of media layers (Stacher et al., 2012). The hallmark pathological finding of pulmonary arterial hypertension are plexiform lesions, glomerulus-like vascular structures that result from endothelial and vascular smooth muscle cell proliferation in pre-capillary pulmonary arterioles (Stacher et al., 2012). The process of pulmonary artery remodeling involves numerous cell types, including pulmonary artery smooth muscle cells (PASMCs), endothelial cells, fibroblasts and myofibroblasts, and inflammatory cells including B and T cells, dendritic cells, mast cells, and macrophages (Price et al., 2012). The pathways underlying this remodeling process are highly complex and include by chronic inflammation, oxidative damage, cell proliferation, resistance to apoptosis, insulin resistance, and shifts in metabolic pathways including aerobic respiration. These changes in the pulmonary vasculature in turn induce chronic compensatory and maladaptive changes in the right ventricle, with increasing hypertrophy, myocardial fibrosis, diastolic stiffness, and reduced contractility (van der Bruggen et al., 2017).
The current therapies used to treat pulmonary arterial hypertension act to dilate the pulmonary vasculature, decreasing pulmonary vascular resistance and secondarily improving right ventricular function, thereby improving functional capacity. The medications currently in widespread use for the treatment of pulmonary arterial hypertension target three major pathways: prostacyclin, endothelin, and nitric oxide. Unfortunately, none of the therapies currently in widespread use directly targets the processes of pulmonary vascular or right ventricular remodeling, and the natural course of the disease is progression to right ventricular failure and death (van der Bruggen et al., 2017). This review will briefly discuss the current paradigm of PAH treatment and discuss the multitude of potential and developing therapies to treat pulmonary arterial hypertension, with a focus on those that have progressed to use in human clinical trials.
The general approach to PAH treatment has evolved substantially over the last decade. While pulmonary vasodilators remain the core therapy for the disease, there has been both a wide expansion of treatment options targeting the three primary treatment pathways, an expansion in the use of risk stratification paradigms, and a shift towards combination therapy as the standard of care in PAH. Other adjunct treatments may include use of diuretic therapy, supplemental oxygen, regimented exercise, and vaccination, but are beyond the scope of this review.
Pulmonary vasodilator medications in widespread use at the time of this writing target one of three molecular pathways–nitric oxide (NO), endothelin, and prostacyclin (Figures 1,2). Medications targeting the nitric oxide pathway include phosphodiesterase-5 inhibitors (PDE5i), including sildenafil and tadalafil, and the soluble guanylate cyclase (sGC) stimulator riociguat. PDE5 inhibitors act to prevent breakdown of cyclic GMP (cGMP), while riociguat acts through direct stimulation of sGC, which both produces cGMP and sensitizes the enzyme to endogenous nitric oxide. cGMP activates protein kinase G, resulting in lower intracellular Ca2+ concentrations, and producing smooth muscle relaxation and vasodilatation. Effective use of riociguat was first established by the PATENT-1 and -2 trials, and it additionally carries FDA approval for treatment of inoperable or residual CTEPH (Ghofrani et al., 2013; Rubin et al., 2015). Side effects of concern for both medication types include headache, flushing, back pain, and peripheral hypotension; bleeding risk can also be increased, warranting care when giving these medications alongside antiplatelet or anticoagulant medications.
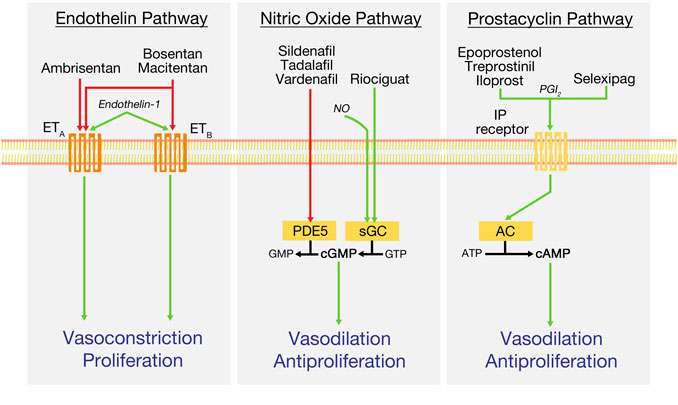
FIGURE 1. Key treatment pathways in the current management of pulmonary arterial hypertension. Green arrows indicate pathway stimulation; red arrows indicate inhibition. AC, adenylyl cyclase; cAMP, cyclic adenosine monophosphate; ATP, adenosine triphosphate; cGMP, cyclic guanosine monophosphate; ETA and ETB, endothelin receptors; GMP, guanosine monophosphate; IP, prostacyclin receptor; NO, nitric oxide; PDE5, phosphodiesterase-5; PGI2, prostacyclin or prostaglandin I2; sGC, soluble guanylyl cyclase.
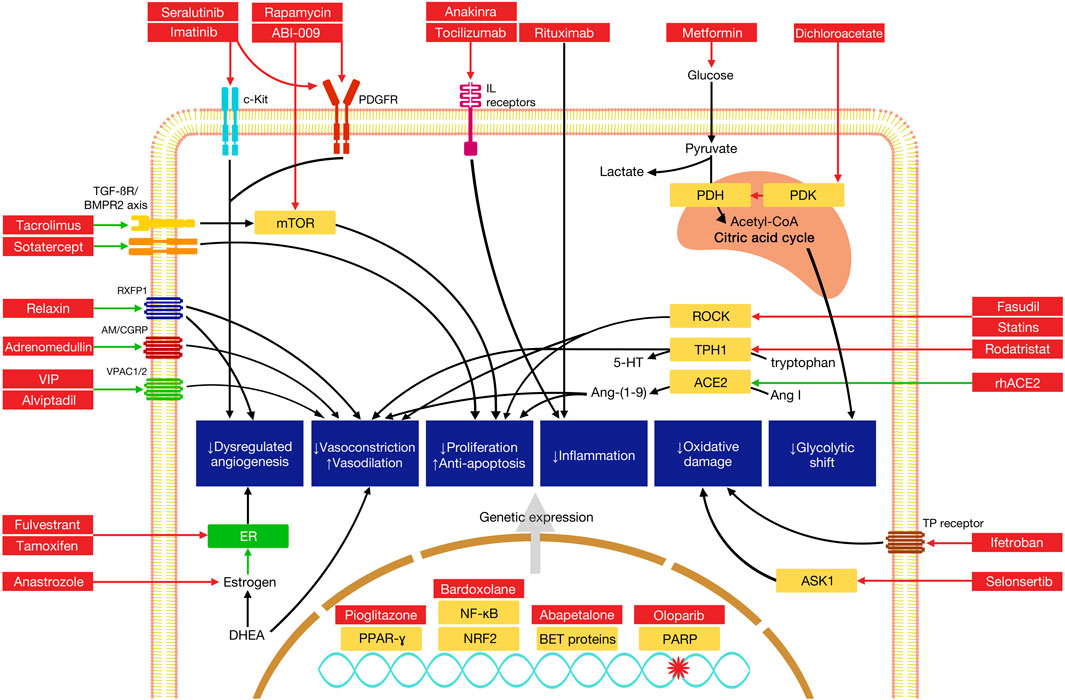
FIGURE 2. Mechanisms of selected novel therapies in the treatment of pulmonary arterial hypertension. Green arrows indicate pathway stimulation; red arrows indicate inhibition. Some pathways have been simplified for the purposes of depiction within the diagram. AM, adrenomedullin receptor; ASK1, apoptosis signal-regulating kinase 1; BET, bromodomain and extaterminal domain; BMPR2, bone morphogenetic protein receptor 2; CGRP, calcitonin gene-related peptide; mTOR, mammalian target of rapamycin; NF-κB, nuclear factor kappa light chain enhancer of activated B cells; Nrf2, nuclear factor erythroid 2-related factor 2; PARP, poly (ADP-ribose) polymerase; PDH, pyruvate dehydrogenase; PDGFR, platelet-derived growth factor receptor; PDK, pyruvate dehydrogenase kinase; PPAR-γ, peroxisome proliferator activated receptor gamma; ROCK, Rho-associated protein kinase; RXFP1, relaxin family peptide receptor 1; TPH1, tryptophan hydroxylase 1; VPAC, vasoactive intestinal polypeptide receptor.
Endothelin receptor antagonists (ERAs), including the dual antagonists bosentan and macitentan and the ETA selective antagonist ambrisentan, block binding of endothelin-1 (ET-1) to its receptors, ETA and ETB. Activation of smooth muscle ETA and ETB by ET-1 produces potent vasoconstriction; notably, activation of endothelial ETB receptors mediates clearance of ET-1 and release of vasodilatory prostacyclin and nitric oxide (Galie et al., 2004). Side effects of concern with ERAs include peripheral edema, anemia, and hepatotoxicity; the latter two effects require serial lab monitoring while on the medication. Additionally, all ERAs are highly teratogenic, necessitating careful birth control practices in women of child-bearing age.
Prostacyclin medications, the oldest group used to treat PAH, act to increase cAMP concentrations to produce pulmonary vasodilation. Epoprostenol, the first drug to be introduced specifically for the treatment of PAH, was approved by the FDA in 1995 and was the first pulmonary vasodilator to show a durable reduction in mortality in a controlled trial of IPAH (Badesch et al., 2000). However, epoprostenol is limited in its versatility by its restriction to intravenous use in the outpatient setting, requiring central venous access; its very short half-life of 3–5 min, and its instability at room temperature, requiring refrigeration.
Since its introduction, epoprostenol has been joined by several different prostacyclin derivatives with a variety of modes of delivery. These include inhaled iloprost and treprostinil, subcutaneous or intravenous treprostinil, oral treprostinil diolamine, and the oral prostacyclin agonist selexipag. In outpatients, intravenous and subcutaneous prostacyclins are delivered by continuous pumps, such as the CADD MS3 or the recently introduced subcutaneous-only Remunity pump. Consensus recommendations continue to support use of these parenteral medications as first-line therapy in WHO Functional Class IV or high-risk patients alongside agents from the other two pathways, as appropriate (Klinger et al., 2019). Side effects of concern include nausea, vomiting, diarrhea, jaw pain, and headaches; in addition, due to the antiplatelet and antithrombotic properties of the prostacyclin family, risk of bleeding complications is increased.
The general overall treatment goals in PAH include improvements in survival, quality of life, exercise capacity, symptom burden, and clinical worsening, with risk stratification tools increasingly used as a vehicle to guide therapy and improve each of these elements. Current recommendations weigh the use of multiple factors, including WHO Functional Class, exercise ability, lab indices, and hemodynamic and echocardiographic variables to establish overall severity of disease and guide intensity of therapy (Humbert et al., 2022). These factors have been codified in risk-stratification matrices such as REVEAL 2.0 and the ERS/ESC criteria, which stratify patients into risk groups based on projected mortality. Initial therapy choices and subsequent changes in therapies are chosen with the goal of achieving a low-risk category.
Primarily beginning with the publication of the landmark AMBITION trial, upfront initiation of combination pulmonary vasodilator therapy has become the state of the art for treatment of PAH (Galie et al., 2015). The AMBITION trial was a multinational, randomized, double-blinded controlled trial evaluating monotherapy with tadalafil or ambrisentan versus up-front combination therapy with both drugs in 500 WHO Functional Class II and III PAH patients, the majority of whom (69%) were WHO Functional Class III. Compared to the monotherapy groups, the combination therapy group showed a significant reduction in time to first clinical failure event, primarily driven by a decrease in hospitalizations. Significant improvements were also noted in nt-pro-BNP levels and proportion of patients that had achieved a satisfactory clinical response by week 24. Importantly, no major safety concerns arose with the use of combination therapy.
Further evidence for the use of combination therapy came with the SERAPHIN and GRIPHON trials. The randomized controlled SERAPHIN trial evaluated macitentan in 742 PAH patients, of whom 63.7% were on non-ERA background therapy, the majority on a PDE5 inhibitor (Pulido et al., 2013). The trial compared placebo to groups on 3mg and 10 mg macitentan and demonstrated statistically significant reductions in a composite end point of first PAH-related event. An average 6-min walk increase of 22 m was demonstrated in the high-dose group. The GRIPHON trial was a phase 2 randomized controlled trial comparing selexipag versus placebo in 1,156 PAH patients, most of whom were on background therapy–PDE5i, ERA, or both. Addition of selexipag showed a significant reduction in the primary composite end point, most of which was due to decreased disease progression and rates of hospitalization (Sitbon et al., 2015).
Contemporary combination therapy in PAH patients frequently mirrors the patterns of medication use in the major trials discussed above. Typical therapy for patients not at high risk and without contraindications consists of a phosphodiesterase-5 inhibitor (PDE5i) and an endothelin-receptor antagonist (ERA) (Galie et al., 2016). For patients with WHO Functional Class IV symptoms or who present in a high-risk category, combination therapy including a parenteral prostacyclin remains the consensus recommendation. General principles of combination therapy mandate avoidance of two overlapping medications that act on the same pathway. Within-class combinations of medications likely provide no additional clinical efficacy and raise the risk of side effects, such as peripheral hypotension. Concurrent medications with different methods of action that act on the same molecular pathways are also avoided, such as riociguat alongside PDE-5 inhibitors, or selexipag alongside prostacyclin therapy.
Strong evidence for the use of dual combination therapy has led to further studies of triple combination therapy. Small uncontrolled trials have demonstrated significant clinical benefits with the use of up-front triple combination therapy (Sitbon et al., 2014). A trial of upfront triple combination therapy with ambrisentan, tadalafil, and subcutaneous treprostinil in 21 PAH patients was found to produce marked hemodynamic improvements, along with echocardiographic findings suggesting reversal of right heart remodeling (D'Alto et al., 2020). Registry data from the French PAH registry published in 2021 demonstrated a significant survival difference in the triple combination group (91% at 5 years) versus the dual and single therapy groups (61% at 5 years, p < 0.001), but notes that the much smaller triple combination group, while higher risk at baseline, was significantly younger overall and had fewer comorbidities (Boucly et al., 2021; Hassoun and Farber, 2021). An association between triple therapy and improved survival was also noted in intermediate-risk patients. Further exploration of these findings with a clinical trial of triple combination therapy with parenteral prostacyclins is necessary. Consideration for the use of triple combination therapy with selexipag in PAH was also supported by an expert consensus survey for Functional Class II patients with higher risk characteristics or CTD-associated PAH, as well as Functional Class III patients overall (McLaughlin et al., 2020).
Controlled trials evaluating up-front triple combination therapy have been more equivocal in their findings. The TRITON trial was a phase 3b randomized controlled trial of 247 PAH patients evaluating combination therapy with macitentan and tadalafil with and without the addition of selexipag (Chin et al., 2021). Both groups were found to have reductions in PVR and nt-pro-BNP as well as increases in 6-min walk distance by week 26, but there were no significant differences found between groups. A non-significant trend towards reduced risk of disease progression was noted in the triple therapy group.
With the increasingly widespread use of combination therapy, and especially with the growing enthusiasm for up-front triple combination therapy, there are concerns about issues such as polypharmacy, substantial associated costs, and medication intolerances, especially in older patients and/or those with significant comorbidities (Hassoun and Farber, 2021). Each medication is associated with substantial costs and significant manpower to obtain insurance approval for what may be three specialty medications started concurrently. Side effect profiles for each of the three pathways are not insubstantial and can lead to issues with compliance, especially in older patients and/or those at risk for other cardiac conditions, such as heart failure with preserved ejection fraction.
With the shift towards combination therapy discussed above, monotherapy with a single pulmonary vasodilator has become less common, with recommended use confined to certain subsets of the PAH population (Gaine and McLaughlin, 2017). These subsets include overall low-risk disease, treatment-naïve low-risk patients who are durable responders to calcium-channel blockers for over 1 year; patients with demonstrated long-term stability on monotherapy, those with concern for pulmonary veno-occlusive disease, and patients with significant tolerability concerns, especially older patients at risk for heart failure with preserved ejection fraction. Additionally, patients with PAH due to HIV and portopulmonary hypertension were largely excluded from major trials of combination therapy and may also be considered for monotherapy as appropriate (Simonneau et al., 2019).
The ongoing role of calcium channel blocker (CCB) use in appropriate patients is less clear as new treatment options continue to emerge. Vasodilator testing, in which patients are challenged with vasodilators such as epoprostenol or nitric oxide during right heart catheterization, continues as a recommendation for certain WHO Group I patients to predict the likelihood of response to CCB therapy. As in the treatment of peripheral hypertension, CCBs act on the pulmonary arteries of acute responders by blocking calcium uptake, resulting in reduction of vascular tone. For patients without contraindications and without evidence of right heart failure who demonstrate acute vasoreactivity, a trial of CCB therapy may be considered, with careful reassessment after 3 months’ time (Klinger et al., 2019). Typical options for CCBs include relatively high doses of nifedipine, amlodipine, or diltiazem (Barst et al., 2009). Rates of acute vasoreactivity are relatively low, with overall response rates of 13–15% and sustained clinical response rates of 6–9% in IPAH and anorexigen-related PAH, the groups with the highest rates of response (Medarov and Judson, 2015). While safe and inexpensive, CCBs are not formally included in contemporary PAH treatment and risk assessment algorithms, and there is a lack of controlled studies of CCB therapy or understanding regarding their synergy with other pulmonary vasodilators. These factors and increasing access to pulmonary vasodilators could make the use of CCBs in PAH obsolete.
As noted above, there has been intense interest in the use of up-front triple therapy for appropriately high-risk PAH patients, which has been explored in trials like TRITON. Other trials have explored the role for substituting existing medications for others to further improve treatment responses in PAH. Riociguat has received focus given its potential as substitution for PDE5 inhibitors in patients not meeting treatment targets. The RESPITE study was a 24-week open-label uncontrolled study of 61 WHO Functional Class III PAH patients that substituted riociguat for PDE5 inhibitors (Hoeper et al., 2017). Most patients (82%) were on concurrent endothelin-receptor antagonists. Of the 51 who completed 24 weeks of treatment, there was an increase in 6-min walk distance of 31 +/− 63 m by 24 weeks (p = 0.001), alongside statistically significant reductions in nt-pro-BNP level and pulmonary vascular resistance. This data was followed up on with the REPLACE trial, an open-label, multicenter, randomized controlled trial of 226 PAH patients at intermediate mortality risk (Hoeper et al., 2021). Included patients could be on ERA therapy but were excluded if on any prostacyclin therapy in the 30 days prior to enrollment. Using a composite outcome defined as an absence of clinical worsening and improvement in two of three variables (6-min walk distance, WHO Functional Class, and/or nt-pro-BNP level), there were significantly fewer clinical worsening events in the riociguat group (1% versus 9%, p = 0.0007), with fewer serious adverse events reported. Together these studies appear to demonstrate switching from PDE5 inhibitors to riociguat as a promising option for PAH patients who have failed to progress to low risk status on their current therapies.
Multiple currently active trials are exploring the possibility of new combinations and dosing strategies for established pulmonary vasodilator therapies for PAH. The A-DUE trial, mirroring to some degree the AMBITION trial, is an adaptive phase 3 placebo-controlled trial comparing tadalafil and macitentan monotherapy groups with fixed-dose combination of the two drugs. It seeks to recruit a total of 170 PAH patients. The phase 3 UNISUS trial seeks to evaluate the effect of significantly higher doses of macitentan on reduction in a composite of morbidity and mortality events. The trial is actively recruiting for a planned 900 participants, divided into placebo, currently used dosing of 10 mg, and higher dosage at 37.5 mg and 75 mg, respectively.
The increasing complexity of PAH medication regimens has led to efforts to simplify drug schedules and delivery methods. Given the lifestyle drawbacks of parenteral therapies and challenges of learning how to operate prostacyclin pumps, there may be considerable hesitancy among patients to start these medications. Daily activities such as showering or exercise are much more careful considerations when managing patients on parenteral therapies; streamlining the delivery process may help address the practicality of parenteral prostacyclins considerably (Farber and Gin-Sing, 2016). The subcutaneous-only Remunity pump was introduced in 2021 and represents a sleeker alternative to the MS3 pump currently in common use, with a waterproof casing and handheld remote. It should be noted that the pump’s lower limit of delivery does not allow for very low rates of drug administration in nearly all patients, so that patients will likely need to be transitioned from an MS3 pump to Remunity later in their titration schedule if beginning a prostacyclin at the low basal rates generally used for outpatient initiation. Further efforts to develop medication to simplify pulmonary vasodilator administration for ease of use are described below.
The novel oral prostacyclin receptor agonist ralinepag has shown significant promise as a therapeutic option in PAH. The drug is described as highly selective and potent, with in vitro data suggesting more potent antiproliferative and vasodilatory properties than iloprost or MRE-269, a metabolite of selexipag (Preston et al., 2018). A phase 2 placebo-controlled trial of 61 PAH patients showed a reduction in PVR compared to placebo in patients on background mono or dual therapy (Torres et al., 2019). The drug has a 24-h half-life, making once-daily dosing feasible and providing a potential advantage over the twice-daily dosing of selexipag, currently the only available direct prostacyclin receptor agonist. Two phase 3 trials using once-daily dosing of the medication are ongoing (see Table 2).
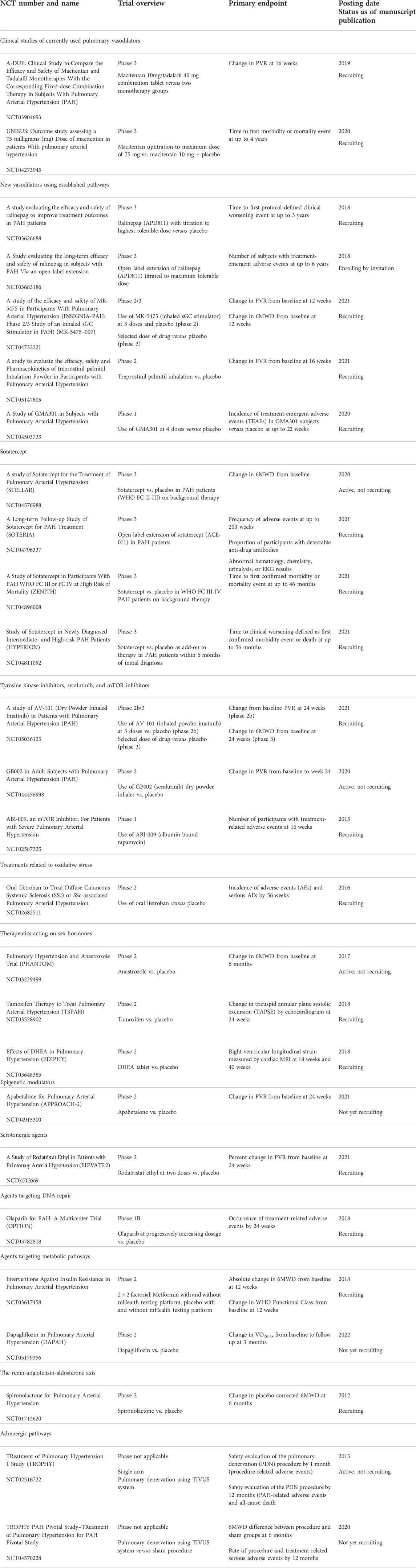
TABLE 2. Selected active therapeutic trials in pulmonary arterial hypertension.
The multicenter Phase 2/3 INSIGNIA-PAH study (NCT04732221), currently active, seeks to evaluate the use of MK-5475 (Merck Sharp & Dohme Corp., Kenilworth, NJ, United States), an inhaled soluble guanylate cyclase inhibitor. The study plans to evaluate efficacy at three different doses of medication versus placebo for a 12-week base period with a 24-month optional extension period, with primary outcomes of improvement in PVR and 6-min walk distance at 12 weeks. Interestingly, while current guidelines do not allow for the use concurrent PDE5 inhibitors and oral sGC stimulators, largely due to the risk of hypotension from additive therapies acting on the same molecular pathway, the INSIGNIA-PAH study does not explicitly exclude patients already on either therapy.
Treprostinil inhalation powder (TreT) is a new method of inhaled treprostinil delivery explored in the open-label BREEZE study, which enrolled 51 patients with PAH (Spikes et al., 2022). The delivery method allows for once daily dosing, a significant decrease from the current four times daily dosing of inhaled treprostinil. Transitions to TreT were well-tolerated, with adverse effects felt to be similar in severity and frequency to that of inhaled treprostinil. A small if statistically significant increase in 6-min walk distance of 11.5 m was seen (p = 0.0217), along with improvement in symptom-based scores and high reports of patient satisfaction by week 3. Additionally, use of doses above the higher range of 9–12 puffs per use was found to be well-tolerated. TreT may offer a more convenient method of prostanoid delivery at least as effective as the inhaled treprostinil currently in widespread use. The Tyvaso dry powder inhaler (DPI) using TreT was approved by the FDA on 24 May 2022.
A second form of treprostinil formulated for dry powder inhalation, treprostinil palmitil (TPIP), is being studied in a placebo-controlled phase 2 trial in PAH patients (NCT05147805). Treprostinil palmitil is a prodrug with efficacy of at least 12 h, with once daily dosing specified in the study (Plaunt et al., 2021). In animal models, TPIP demonstrated a higher cough threshold with administration than inhaled treprostinil, which may improve a common and often significant effect of inhaled treprostinil delivery (Chapman et al., 2021).
In addition to new pulmonary vasodilators for established molecular pathways in PAH, there have been increasing explorations of new molecules for non-PAH applications. The sGC stimulator vericiguat reduced the incidence of death compared to placebo in a cohort of 5,050 patients with heart failure with reduced ejection fraction (less than 45%) in the VICTORIA trial, resulting in its FDA approval for this application (Armstrong et al., 2020). However, this medication has not been formally studied for use in PAH. Several novel endothelin receptor antagonists, namely darusentan and aprocitentan, are currently being studied, though current studies focus on their potential use in essential hypertension and atherosclerosis rather than in PAH (Enevoldsen et al., 2020). ERA derivatives have also been previously explored in treatment of cancer and in diabetic renal disease, the latter with some degree of success (Heerspink et al., 2019).
In addition to the development of new conventional ERAs, both vaccination against ET-A and getagozumab (GMA-301), a humanized monoclonal antibody against endothelin receptor A (ET-A), have been developed with the potential for trials for PAH treatment (Zhang C. et al., 2019; Dai et al., 2019). A phase 1 trial to study dosing, safety, and efficacy in GMA-301 in PAH patients is actively recruiting (NCT04503733).
Relaxin, a family of protein hormones first identified for its role in reproduction, has been shown to have manifold effects on the cardiovascular system, including stimulation of neoangiogenesis through increases in VEGF transcription, vasodilation through stimulation of NO production, and reduction in endothelin-dependent vasoconstriction (Feijoo-Bandin et al., 2017) (Table 3; Figure 2). In a rat model with hypoxia-induced PH, relaxin administration was found to inhibit proliferation based on decreases in collagen and fibronectin formation; high doses of relaxin were subsequently shown to improve pulmonary arterial compliance and suppress VF and asystolic arrest in a Sugen-hypoxia rat model (Tozzi et al., 2005; Martin et al., 2021). Human trials of relaxin have centered on left-sided heart failure; RELAX-AHF, a trial using the recombinant human relaxin 2 serelaxin in acute left-sided heart failure, reported improvements in dyspnea and deaths at 180 days, but the much larger RELAX-AHF2 trial but did not show significant improvements in cardiovascular or all-cause mortality (Teerlink et al., 2013; Metra et al., 2019). Interestingly, higher circulating relaxin levels were separately demonstrated to correlate with higher mPAP and worsened RV dysfunction in a cohort of patients with acute left-sided heart failure due to both HFrEF and HFpEF, suggesting utility as a biomarker (Pintalhao et al., 2017). Relaxin may yet hold promise as a treatment modality in PAH, though no human trials are ongoing at the present time.
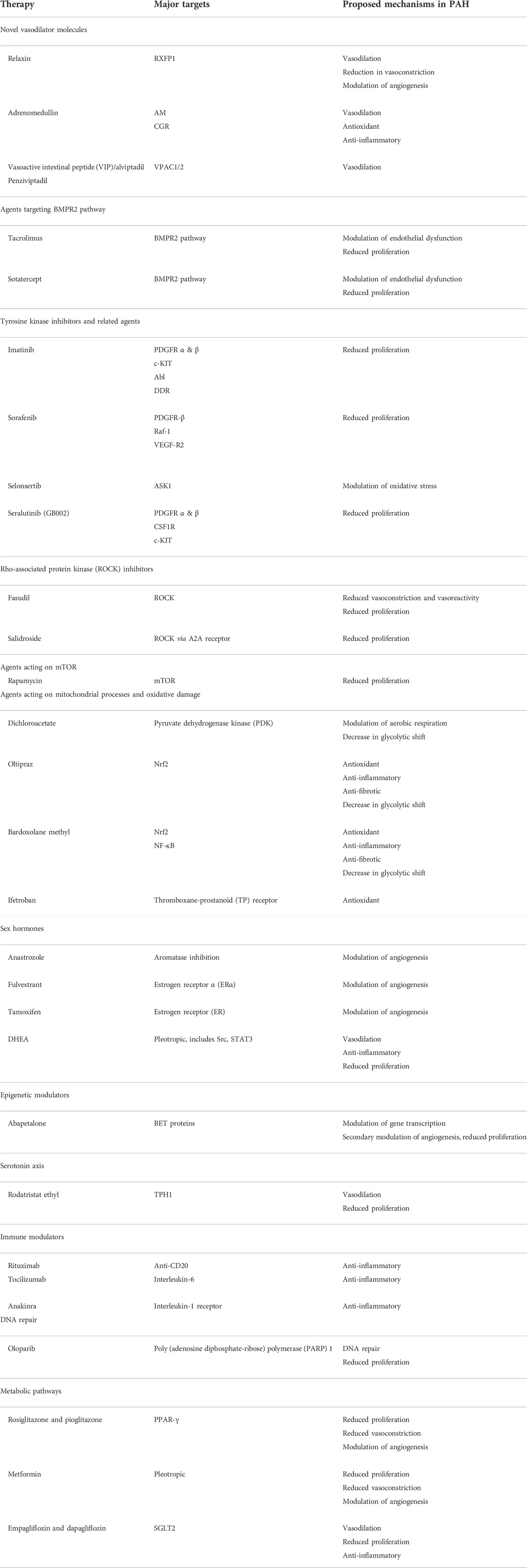
TABLE 3. Selected novel treatment agents for pulmonary arterial hypertension.
Adrenomedullin (ADM) is a peptide hormone with activities that center on regulation of endothelial barrier function and vascular tone, with vasodilatory, anti-inflammatory, and antimicrobial effects, the last of which has raised interest for its use in the treatment of sepsis (Geven et al., 2018). In rat models of PAH, intravenous, inhaled, and cell-based gene transfer delivery of ADM have been shown to improve RV hypertrophy and pulmonary hemodynamics, and a related peptide, intermedin/adrenomedullin-2, has shown potent pulmonary vasodilatory effects through action on calcitonin-like calcitonin receptors in vitro and cellular models (Yoshihara et al., 1998; Nagaya and Kangawa, 2004; Kandilci et al., 2008; Telli et al., 2018; Holmes et al., 2020). Circulating ADM levels are elevated in PAH patients compared to healthy controls, and ADM levels have been shown to correlate with PAH severity based on REVEAL scoring and overall survival (Bouzina and Radegran, 2020). Though the biomarker remains a focus of intensive research in other arenas, there are no human trials presently planned in PAH patients.
Vasoactive intestinal peptide (VIP) induces pulmonary artery smooth muscle relaxation via activation of adenylate cyclase and generation of cAMP. Favorable effects on right ventricular and vascular remodeling have been shown with the administration of VIP to VIP-knockout monocrotaline mice models of PAH (Said et al., 2007). In humans, there appear to be marked reductions in serum VIP levels and decreases in immunoreactive VIP nerve fibers in the media layer of the pulmonary arteries compared to healthy patients; in vitro, VIP was also found to inhibit growth of PASMCs (Petkov et al., 2003). A subsequent study of inhaled VIP in 8 severe primary pulmonary hypertension patients showed significant improvements in mean pulmonary arterial pressure, stroke volume, and cardiac output, as well as an improvement in 6MWD of 113 m at 3 months’ treatment. In a subsequent trial of 20 patients with PH, of which nine had PAH and the remainder either WHO Group III PH due to lung disease or CTEPH, inhalation of a single dose of 100 mcg of aviptadil during right heart catheterization caused a small but significant decrease in mean pulmonary arterial pressure and increases in stroke volume and mixed venous O2 saturation, replicating these hemodynamic findings (Leuchte et al., 2008). However, these results were not replicated in a larger controlled trial; a phase II randomized-controlled trial of inhaled VIP in 56 PAH patients was unable to demonstrate any significant beneficial effects on hemodynamics or exercise capacity (Galie et al., 2010; Said, 2012). A sustained-release analogue of VIP, PB1046, has been evaluated in a group of 3 PAH patients with reductions in mPAP and increases in stroke volume and cardiac output based on implanted CardioMEMS data in 2 of the 3 patients (Benza et al., 2020). A phase 2 trial to study once-weekly subcutaneous PB1046/penziviptadil (NCT03556020) was unfortunately terminated due to COVID-19-related drug resupply problems, and ongoing studies of aviptadil appear to be focused on the treatment of COVID-19. Further studies are needed to evaluate safety, tolerability, and efficacy of VIP analogues in PAH patients.
Mutations in the gene for BMPR2 (bone morphogenic protein receptor type 2) are the most common heritable factor leading to the development of HPAH, with the various described mutations causing dysfunction and/or reduced trafficking of the receptor to the cell surface, leading to markedly reduced levels in the pulmonary vasculature (Morrell, 2006). Reduced expression and downstream signaling of BMPR2 is also observed in rat models of non-heritable idiopathic PAH (Happe et al., 2020). Deficiencies in the BMP pathways have been shown to cause effects that include increased PASMC proliferation, decreased endothelial nitric oxide and eNOS levels, and a shift away from oxidative metabolism towards glycolysis (Dorai et al., 2000; Gangopahyay et al., 2011; Diebold et al., 2015).
Tacrolimus (FK506), a calcineurin inhibitor used commonly for immunosuppression in solid organ transplant patients, is a potent activator of BMPR2, and has shown ability to reverse PAH in various rat models (Spiekerkoetter et al., 2013). Low-dose FK506 was subsequently trialed in 3 patients with end-stage PAH, with significant benefits reported in symptom improvement and reduction of hospitalizations (Spiekerkoetter et al., 2015). TransformPAH, a larger randomized phase IIb trial of FK506 in 23 PAH patients, showed improvement in BMPR2 expression in the treatment group versus placebo (Spiekerkoetter et al., 2017). Some patients with more marked increases in BMPR2 expression with FK506 showed proportionally greater improvements in 6MWD and echocardiographic correlates of RV function, but the overall changes were not significant, noting that the study was not powered for this purpose.
Sotatercept (ActRIIA-Fc) is a novel fusion protein containing the extracellular domain of human activin receptor type IIA and the Fc domain of human IgG1. The protein acts as a ligand “trap” for members of the TGF-β family, which includes BMPR2. Through decreases in circulating TGF-β superfamily members, sotatercept is thought to rebalance the pro-proliferative activin growth differentiation pathway and antiproliferative BMP pathway, decreasing signaling that leads to pulmonary vascular remodeling. Use of a rodent analog of sotatercept in a Sugen-hypoxia rat model of PH showed marked decreases in proinflammatory and pro-proliferative gene profiles assessed by genome-wide expression analysis, and reversed RV dilatation, septal wall flattening, and RV fractional area change (RVFAC), all effects that were not seen with treatment with administration of sildenafil alone (Joshi et al., 2022). A phase 2 randomized, placebo-controlled, multicenter trial, PULSAR, evaluated 163 PAH patients on various baseline therapies, of which 106 were randomized to two doses of the drug or placebo and followed these patients for 24 weeks (Humbert et al., 2021). Subcutaneous injections of the drug were given every 21 days. A significant reduction in the primary outcome of PVR was observed, with secondary improvements in 6MWD (+29.4 m, 95% CI 3.8–55.0) and nt-pro-BNP levels. The phase 3 STELLAR trial, a randomized, double-blind, placebo-controlled trial evaluating change in 6MWD from baseline as its primary outcome, has completed recruitment and is presently active. Three additional trials are recruiting, including an open-label extension of other sotatercept studies (SOTERIA, NCT04796337), a placebo-controlled study of the drug in high-risk WHO Functional Class III and IV PAH patients (ZENITH, NCT04896008), and an additional placebo-controlled trial in PAH patients diagnosed within the prior 6 months (HYPERION, NCT04811092).
Multiple tyrosine kinase inhibitors have been explored for therapeutic use in PAH. These have shown promise in their pleotropic effects on PAH but have significant concerns have been raised regarding their safety and tolerability.
Imatinib, the first drug in this class to be FDA-approved, has been studied extensively for PAH treatment. Originally approved for use in treatment of Philadelphia chromosome-positive (Ph+) chronic myelogenous leukemia (CML), it was shown to reverse PAH in monocrotaline and chronic hypoxia rat models (Schermuly et al., 2005). This has been hypothesized to be due to inhibition of both platelet-derived growth factor (PDGF) signaling and of c-Kit, which reduces progenitor cell mobilization and maturation into mast cells, which are contributors to dysregulated angiogenesis in PAH (Farha et al., 2014). After several favorable case reports the drug was used in phase 2 trial of 59 patients with PAH; this showed significant hemodynamic improvements in PVR and cardiac output, but no significant change in 6MWD from baseline at 6 months (Ghofrani et al., 2010). Post-hoc analysis suggested greater hemodynamic improvement in those with higher baseline PVR. This was followed by the stage 3 IMPRES study, which enrolled 202 PAH patients with high baseline PVR (≥800 dyne s cm−5) already on two or more background PAH therapies (Hoeper et al., 2013). This did show a significant improvement in 6MWD from baseline at 24 weeks (32 m, 95% CI 12–52, p = 0.002) but saw a high rate of serious adverse events versus placebo, with high rates of gastrointestinal side effects and 8 patients developing subdural hematoma while on concurrent anticoagulation. The extension for the core study was subsequently discontinued by its parent company, Novartis. A phase 2b/3 study evaluating use of a dry powder form of the drug (AV-101), IMPAHCT (NCT05036135), is currently recruiting, as is a phase 1 study of a nebulized form of the drug (NCT04903730).
Sorafenib, an inhibitor of multiple tyrosine and serine/threonine kinases approved for use in treating multiple cancers, has shown favorable effects on PAH in rat models, including reduction in PA and RV pressures and reduced RV hypertrophy (Moreno-Vinasco and Garcia, 2010). A phase 1b trial of 12 patients, all of whom were on continuous parenteral prostacyclins, did not show improvement in baseline 6MWD at 16 weeks and showed a statistically significant decrease in cardiac output (Gomberg-Maitland et al., 2010). Notably, all subjects experienced at least one drug-related adverse event, with intolerance of the dermatologic side effects seen at usual dosing. A subsequent small human trial of 9 patients, 7 with severe refractory PAH and two with pulmonary veno-occlusive disease (PVOD), showed the drug to improve WHO Functional Class in eight patients and improve mPAP in six (Kimura et al., 2017). Most reported adverse skin reactions, but only 1 patient discontinued the drug for this reason.
Beyond their noted frequency of adverse effects, consideration of TKIs for the treatment of PAH is complicated by findings that some TKIs, particularly dasatinib, have toxic effects on the pulmonary vasculature and in fact appear to induce PAH. Dasatinib, a TKI also in widespread use for the treatment of Ph + CML, is thought to lead to PAH through endothelial toxicity, apoptosis, and by producing an exaggerated response to injurious factors, such as hypoxia (El-Dabh and Acharya, 2019). Thankfully, dasatinib-associated PAH does appear to be at least partially reversible after stopping the drug. Through pharmacovigilance database use, other TKIs show signals of association with PAH, including ruxolitinib and nilotinib (El-Dabh and Acharya, 2019). Nilotinib was explored in a small phase 2 trial for treatment of PAH (NCT01179737) which was terminated early in 2014 for a serious adverse event. Ruxolitinib has been described to exacerbate PAH in case reports, though its effect on modulating Jak2 is thought to hold therapeutic promise in PAH (see below). (Low et al., 2015).
The apoptosis signal-regulating kinase 1 (ASK1) inhibitor selonsertib has similarly shown promise in preclinical rat and mouse models of PAH. The kinase acts principally to modulate oxidative stress and has been shown to slow progression of PH and RV remodeling and to have direct antifibrotic effects on the RV (Budas et al., 2018). The phase 2 ARROW trial employed the drug in 150 patients with PAH treated with background therapy; it did not lead to significant hemodynamic or clinical improvements, though the drug was found to be well-tolerated (Rosenkranz et al., 2022).
Cyclin-dependent kinases (CDKs), which mediate the progression of the cell cycle, have recently emerged as a potential therapeutic target for PAH. CDKs have been shown to have heightened activity in PASMCs derived from IPAH patients, akin to the high activity shown in cancer cells. Inhibition of CDK4 and CDK6 using the breast cancer drug palbociclib was shown to reduce right ventricular systolic pressure and hypertrophy as well as reduce pulmonary vascular remodeling in Mct and Sugen-hypoxia rats, suggesting promise as a treatment strategy in humans (Weiss et al., 2019).
Modification of abnormal growth factor signaling may play a potential role in the treatment of PAH. Seralutinib (formerly known as GB002) is an inhaled, nonselective inhibitor of platelet-derived growth factor receptor (PDGFR) -α and -β, as well as colony stimulating factor 1 receptor (CSF1R) and c-KIT. In Sugen-hypoxia and monocrotaline rat models of PAH, seralutinib increased BMPR2 levels and caused reductions in right ventricular systolic pressure, mean pulmonary artery pressure, and pulmonary arteriolar muscularization (Galkin et al., 2022). The TORREY trial (NCT04456998) is a current phase 2 controlled randomized trial evaluating seralutinib on WHO Functional Class II and III PAH patients, with change in pulmonary vascular resistance at 24 weeks as its primary outcome (Frantz et al., 2021). It has been suggested that seralutinib may be a more easily tolerated therapy due to its inhalational route; its efficacy in humans will hopefully be determined based on the results of the TORREY trial.
Rho-associated protein kinase (ROCK) inhibitors have been explored as an avenue for PAH therapy. ROCK plays a major role in vasoconstriction, vasoreactivity, and proliferation of pulmonary artery smooth muscle cells in animal models of PAH, and the ROCK inhibitor fasudil was demonstrated to reduce pulmonary arterial pressures in rat models of PAH (Fukumoto et al., 2005; Nagaoka et al., 2005; Wei et al., 2019). However, a small subsequent trial of fasudil in PAH patients did not show significant differences versus placebo (Fukumoto et al., 2013). Continued study of various novel ROCK inhibitors for PAH is ongoing and continues to show promising results, though thus far limited to animal models (Cantoni et al., 2019; Yamamura et al., 2021). Activation of the adenosine A2A receptor, which causes downstream inhibition of ROCK pathways, has also shown promise; salidroside, which increases A2A expression, demonstrates inhibition of PAH development and PA remodeling in mouse models (Huang et al., 2015; Alencar et al., 2017).
ROCK has been proposed as a pathway through which statins may elicit a beneficial effect on PAH. Numerous trials of statins in rat models have demonstrated improvement or even reversal of PAH, with such improvements shown to be mediated through ROCK I and II (Girgis et al., 2007). Use of rosuvastatin added to fasudil in a rat model of PAH showed a greater reduction in right ventricular pressures and hypertrophy than fasudil alone (Jasinska-Stroschein et al., 2016). However, two randomized controlled trials in PAH using atorvastatin and simvastatin with aspirin unfortunately did not meet their respective endpoints, and neither showed improvement in 6MWD (Kawut et al., 2011; Zeng et al., 2012). ROCK inhibition was shown to ameliorate dasatinib-induced PAH in a rat model, itself thought to be due to endothelial dysfunction, increased ROS production, apoptosis, and increased levels of adhesion molecules, presenting a potentially beneficial modality specific to this subtype of PAH (Guignabert et al., 2016; Fazakas et al., 2018).
The mammalian target of rapamycin (mTOR), a serine/threonine protein kinase involved in the coordination of growth and metabolism of cells, has been implicated as a potential target for PAH therapeutics. mTOR has a pro-proliferative and anti-apoptotic effect on PASMCs in both animal models and human cells, and mTOR complexes have been found to be upregulated in small remodeled pulmonary arteries and PASMCs isolated from patients with pulmonary hypertension (Goncharov et al., 2014; Babicheva et al., 2021).
Rapamycin, an mTOR inhibitor, has been shown to reduce pulmonary vascular remodeling in animal models of PAH, and to prevent and reverse proliferation of PASMCs derived from monocrotaline rat models of PAH (Paddenberg et al., 2007; Houssaini et al., 2013). Rapamycin also appears to activate PDGFR signaling in PASMCs, the same target pathway targeted by seralutinib (Shi et al., 2021). ABI-009, an albumin-bound form of sirolimus, is currently being used in an active phase 1 trial to determine efficacy and safety in PAH patients. An early abstract report indicated that of six patients, 1 discontinued treatment and 3 of the remaining 5 had a reduction in WHO Functional Class and an increase in 6MWD of over 130 m, suggesting therapeutic promise (Simon et al., 2019).
Oxidative stress appears to have a major role in the development of vasoconstriction and pulmonary vascular remodeling, and it induces the transition of pulmonary endothelial cells to mesenchymal cells. The “Warburg effect” describes the tendency for pulmonary vascular cells and right ventricular myocytes to switch from aerobic respiration to glycolysis, with this modulation of aerobic respiration resulting in increased reactive oxygen species in the process (Chan and Rubin, 2017). This glycolytic shift, analogous to that seen in malignant cells in cancer, results in a state of encouraged cellular proliferation and anti-apoptosis (Archer, 2017). Changes that produce this effect are largely mediated through the master transcription factor hypoxia inducible factor-1 (HIF-1α), which also appears to have a confer resistance to apoptosis when highly expressed (Greijer and van der Wall, 2004).
There have been therapeutic attempts to address the Warburg effect directly through mediation of oxidative metabolism. Dichloracetate (DCA) inhibits pyruvate dehydrogenase kinase, which shunts pyruvate from aerobic respiration to glycolysis and is upregulated in hypoxic settings. DCA significantly reduced PVR, PA remodeling, and right ventricular hypertrophy in chronically hypoxic rats, and has been shown to induce PASMC apoptosis (Michelakis et al., 2002). Use of DCA improved markers of mitochondrial respiration in explanted PAH lungs, and a 4-month open-label phase 1 study showed reductions in mPAP and PVR, albeit with widely varied responses dependent on function of other proteins involved in modulating the shift away from aerobic metabolism (Michelakis et al., 2017). Side effects such as peripheral neuropathy, though typically reversible with drug cessation, are likely to limit the therapeutic potential of DCA (Michelakis et al., 2008). A second strategy to modulate pyruvate management and restore mitochondrial pyruvate oxidation through inhibition of pyruvate carboxylase (PC) is also under research; use of the PC inhibitor phenylacetic acid significantly reduced RVSP in a Sugen/hypoxia rat model through attenuation of the glycolytic shift (Valuparampil Varghese et al., 2020).
Pathways complementary to glycolysis and the tricarboxyclic acid (TCA) cycle may also present targets for therapy. Glutaminolysis, the hydrolysis of glutamine by the mitochondrial enzyme glutaminase, repletes intermediates of the TCA cycle and is well known to provide metabolic fuel for the constitutive growth of cancer cells. Targeting this process has been studied extensively for cancer treatment and holds promise for modulating the unregulated proliferation of pulmonary vascular cells in PAH, in which there is evidence of dysregulated glutamine metabolism possibly mediated by hypoxia and HIF (Wang et al., 2022). Activation of glutaminolysis and subsequent proliferation resulting in pulmonary hypertension has been shown to be provoked by mechanical stiffening, which activates the transcription factors YAP1 and TAZ (Bertero et al., 2016). Simultaneous blockade of YAP1 with verteporfin and glutaminase with CB-839 showed improvements in hemodynamic markers and signs of vascular remodeling and stiffening in a monocrotaline rat model of PAH (Acharya et al., 2021). Glutaminolysis has also been found to be a key metabolic pathway in the formation of myofibroblasts and thus progression of cardiac fibrosis; inhibition using CB-839 or genetic deletion of glutaminase prevented myofibroblast formation in vitro (Gibb et al., 2022). Modulating glutamine metabolism therefore appears to hold promise for addressing both vascular and cardiac sequalae of PAH.
The transcription factor nuclear factor erythroid 2-like 2 (Nrf2) has been shown to be protective against PAH and to have antioxidant, antifibrotic, and anti-inflammatory effects. Nrf2 deficient mice exposed to chronic hypoxia show worsened RVSP and right ventricular hypertrophy, with this affect ameliorated by use of the Nrf2 activator oltipraz (Eba et al., 2013). Activation of Nrf2 with the polyphenol salvianolic acid A reduced reactive oxygen species formation and endothelial-mesenchymal transition in human pulmonary arterial endothelial cells, and reduced inflammation and free radical production in a monocrotaline rat model (Chen et al., 2017).
Bardoxolone methyl, an inducer of Nrf2 and suppressor of NF-κB, has been employed in several clinical trials in PH patients. Because of the widespread array of Nrf2 targets, including endothelial cells and macrophages, the phase 1/2 LARIAT trial was not limited to Group I disease, and included subsets of both PH-ILD (Group III) and sarcoidosis (Group V) patients. An initial report of data from 24 patients in LARIAT suggested the drug was well tolerated and showed a significant increase in 6MWD vs. placebo (21.4 m, p = 0.037), and a subsequent report of data on 22 CTD-PAH patients showed a non-significant placebo-corrected 6MWD increase of 26.1 m (p = 0.06) (Oudiz et al., 2015; Oudiz et al., 2017). The placebo-controlled phase 3 CATALYST trial (NCT02657356) aimed to recruit 202 patients with CTD-PAH but unfortunately was terminated due to COVID-19-related exposure concerns for the study patients. Its long-term study extension, RANGER (NCT03068130) was also stopped for the same reason. Plans to restart these or similar trials are not clear.
The thromboxane-prostanoid (TP) receptor is a signal mediator of 15-F2t isoprostane, a biomarker of oxidative stress that correlates with severity of heart failure and may play a direct role in cardiomyopathy (Nonaka-Sarukawa et al., 2003; West et al., 2016). TP receptor activation in mouse models of hypertension induces cardiac hypertrophy, and receptor density is increased in the human right ventricle in PAH (Katugampola and Davenport, 2001; Francois et al., 2004). Use of the TP receptor antagonist CPI211 in a pulmonary artery banding mouse model of PAH substantially reduced RV fibrosis and hypertrophy; notably, use of a thromboxane synthesis inhibitor or aspirin did not achieve these same effects (West et al., 2016). Ifetroban, a selective thromboxane A2 and prostaglandin H2 receptor antagonist, has been explored for the treatment of numerous diseases, most recently dilated cardiomyopathy in Duchenne muscular dystrophy (Mitchell et al., 2021). Ifetroban is now being used in an active phase 2 placebo-controlled trial of PAH due to systemic sclerosis (NCT02682511), with the primary goal to explore safety and incidence of adverse events.
Ongoing research has explored the “estrogen paradox” of PAH - while estrogen appears to confer a protective effect on outcomes in vitro, PAH has a higher incidence in women (Umar et al., 2012). The overall effects of the estrogen axis on PAH are quite complex. Estradiol (E2) has demonstrated protective effects on the right ventricle, though elevated levels are seen in patients with PAH and are associated with worsened exercise capacity. Other estrogen metabolites demonstrate detrimental pro-proliferative effects, and of the two types of estrogen receptors, ERα and ERβ, the former is pro-proliferative while the latter has protective and anti-proliferative effects on the pulmonary vasculature, as shown by successful use of ERβ-specific phytoestrogens in a rat model of PAH (Homma et al., 2006). In very early experiments in animal models of PAH, E2 administration reversed vascular changes in both male and female animals, and in vitro studies showed the hormone to exert significant antiproliferative and antifibrotic properties, as well as to stimulate nitric oxide production through stimulation of nitric oxide synthase (NOS). (Umar et al., 2011).
However, despite protective effects of estrogen acting on ERβ receptors in animal models, higher circulating levels of estrogen are associated with more severe hemodynamic changes and worsened 6-min walk distance in men and post-menopausal women with PAH (Ventetuolo et al., 2016). This is thought to be due to overexpression of ERα in the pulmonary arteries of women with PAH, through which estrogen mediates neoangiogenesis through higher circulating numbers of hematopoietic progenitor cells (HSCs) (Masuda et al., 2007; Asosingh et al., 2008). Therapeutic trials targeting the estrogen axis in human patients have thus focused on blocking the effects of estrogen. Use of anastrozole, an aromatase inhibitor used in breast cancer that blocks conversion of androgens to estrogen, was explored in a randomized controlled trial of 18 patients with PAH, showing reduced circulating E2 levels and a significant increase in 6MWD of +26 m, compared to -12 m in the control group (Kawut et al., 2017). An ERα-specific inhibitor, fulvestrant, has been evaluated in a small proof-of-concept trial of five women with PAH not otherwise on hormone therapy; a median increase in 6MWD of +31 m was observed, along with a serum decrease in HSCs and pro-proliferative 16-α-hydroxyestradiol (Kawut et al., 2019). Both anastrozole and fulvestrant were well-tolerated in these trials. Further studies investigating use of anastrozole as well as tamoxifen, a selective estrogen receptor modulator (SERM) long established in treatment of breast cancer, are ongoing (see Table 2).
DHEA, a steroid precursor of both estrogen and testosterone, has also been shown to ameliorate PH in animal models of the disease, and lower circulating levels are correlated with worsened RV function and mortality in both men and women (Ventetuolo et al., 2016). DHEA has been shown to directly bind and activate NO synthase, suppress endothelin-1 expression, and stimulate pulmonary artery relaxation; in a trial in healthy adults, it was also shown to reduce IL-6 and TNF-α, inflammatory cytokines involved in PAH pathogenesis (Soon et al., 2010; Walsh et al., 2021). A phase 2 study exploring DHEA’s effects on RV function in PAH, EDIPHY (NCT03648385), is underway and currently recruiting.
The bromodomain and extra-terminal proteins (BET) are a family of epigenetic regulators of gene transcription that modulate numerous cell activities, including cell proliferation, angiogenesis, and apoptosis (Borck et al., 2020). Apabetalone, an orally available selective inhibitor of bromodomain (BRD2-4) and extraterminal proteins, is a first-in-class medication that has been or is being explored for multiple indications, including coronary artery disease (CAD), COVID-19 infection, heart failure after acute coronary syndrome (ACS), and PAH. The molecule targets BET proteins and is thought to inhibit maladaptive gene expression, including those that stimulate atherosclerotic change, and BRD4 in particular has been shown to play a significant role in the development of PAH (Meloche et al., 2015). Early studies of the drug showed improvement in coronary plaque volume and circulating lipoprotein levels, though a large randomized clinical trial of 2425 patients with coronary artery disease and type 2 diabetes mellitus did not demonstrate a benefit (Nicholls et al., 2011; Nicholls et al., 2016; Shishikura et al., 2019; Ray et al., 2020). A prespecified secondary analysis of the study did, however, show a reduction in rates of first and total hospitalizations for heart failure after ACS (Nicholls et al., 2021).
In vitro and in studies of several animal models of PAH, inhibition of BRD4 using apabetalone reversed the PAH phenotype and improved hemodynamics, and there has been benefit suggested by the effects of the drug on coronary disease and lipoprotein levels in earlier studies and the concordantly higher rates of CAD and metabolic syndrome in PAH. An open-label pilot clinical study, APPROACH-p, tested the drug in 7 PAH patients, and demonstrated favorable hemodynamic improvements in PVR, cardiac output, and stroke volume, though no changes to 6MWD or nt-pro-BNP (Provencher et al., 2022). A phase 2 randomized placebo-controlled trial to study the drug in PAH, APPROACH-2 (NCT04915300), is currently recruiting.
Serotonin (5-HT) is an endogenous vasoactive monoamine, and the 5-HT transporter is highly expressed in the lung and in PASMCs. It has been postulated that serotonin may be a viable target for PAH therapy, as several subtypes of serotonin cause pulmonary vasoconstriction and induce PASMC proliferation (Kaumann and Levy, 2006). This association was identified after the emergence of PAH due to extended use of anorexigenic drugs such as fenfluramine, which are indirect serotonergic agonists (Abenhaim et al., 1996). The relationship has been demonstrated in animal models, with inhibition of serotonin receptors inhibiting the development of PAH in mice and rats (Dumitrascu et al., 2011; Delaney et al., 2018). An early study evaluated the 5-HT2A receptor antagonist ketanserin, showing a small vasodilatory response in a group of 20 primary PH patients (McGoon and Vlietstra, 1987). The serotonin receptor 5-HT2A & 5-HT2B inhibitor terguride showed favorable effects in reducing pulmonary vasoconstriction and PASMC proliferation in a monocrotaline rat model of PAH (Dumitrascu et al., 2011). However, in a phase 2a clinical trial, the drug showed no hemodynamic benefit in PAH patients, with high rates of severe adverse events and drug discontinuation (Ghofrani et al., 2012).
Tryptophan hydroxylase 1 (TPH1) is an enzyme involved in the rate-limiting step in serotonin synthesis and has been found to be upregulated in pulmonary arterial endothelial cells in those with PAH. Rodatristat, a peripheral inhibitor of TPH1, has shown efficacy in nonclinical models of PAH (Lazarus et al., 2022). Rodatristat ethyl is a prodrug of rodatristat that is orally bioavailable, reversible, and suppresses serotonin production without CNS effects as it does not cross the blood-brain barrier (Bader, 2020). The Phase 2b, double-blind, multicenter ELEVATE 2 trial (NCT04712669) is currently examining the effects of several doses of rodatristat ethyl in PAH patients (Lazarus et al., 2022).
Selective serotonin reuptake inhibitors (SSRIs) are theoretically promising agents that intervene along this pathway, but they have so far not shown meaningful results. One SSRI, escitalopram, was studied in PAH patients in 2008, but no results were published. A small open-label pilot study of fluoxetine did not find changes in hemodynamics, 6MWD, or clinical worsening events with fluoxetine at 12 or 24 weeks, with side effects limiting uptitration in 5 of 13 patients (Sodimu et al., 2020).
Autoantibodies are a major contributor to the development of pulmonary arterial hypertension, particularly in connective tissue disease. Agents felt to have clinical utility in addressing the underlying autoinflammation seen in diseases like systemic sclerosis have been explored to determine if they have efficacy in treating CTD-PAH as well. Rituximab, a monoclonal anti-CD20 antibody introduced for the treatment of lymphoma but now employed in a wide variety of autoimmune diseases, acts to deplete B cells, which express CD20 on their outer surfaces. Use of anti-CD20 antibodies in a novel ovalbumin inhalation rat model of PAH produced decreases in several pro-proliferative and pro-inflammatory markers, including IL-6, VEGF, and HIF-1α, and ultimately led to decreases in mPAP and RVH (Mizuno et al., 2012). A subsequent randomized controlled trial of rituximab in 57 CTD-PAH patients from 2010 to 2018 showed a trend towards 6MWD increase with the drug but did not reach significance at the planned primary outcome endpoint of 24 weeks, though differences were statistically significant by 48 weeks (Zamanian et al., 2021). Rituximab was well tolerated, with slightly more frequent serious adverse events in the rituximab group though not beyond what was expected.
Interleukin-6 (IL-6), a major pro-inflammatory cytokine produced during inflammation in acute infection, appears to play an important role in PAH, where higher serum levels are observed (Soon et al., 2010). Overexpression of IL-6 causes acceleration of pulmonary vascular disease in a rat hypoxia model of PAH, and overexpression of the receptor for IL-6 in human PASMCs has a pro-proliferative effect (Steiner et al., 2009; Tamura et al., 2018). Registry data has shown a correlation between higher IL-6 levels and worsened RV function and RV-PA coupling (Prins et al., 2018). A biobank study of over 2000 PAH patients showed much higher IL-6 expression in PASMCs than endothelial cells in PAH patients with no difference in controls, and an overall negative association between IL-6 and survival (Simpson et al., 2020). Notably, IL-6 levels were highest in portopulmonary hypertension and CTD-PAH, both higher risk PAH phenotypes. Given the increasingly clear role of IL-6 in the pathogenesis of PAH, directly targeting this cytokine using tocilizumab, an anti-IL-6 monoclonal antibody, may hold promise.
An open-label phase 2 trial, TRANSFORM-UK, administered 8 mg/kg of intravenous tocilizumab (up to an 800 mg maximum dose) monthly for up to 6 months to 29 PAH patients, with primary outcomes of safety based on adverse events and change in PVR by 6 months (Hernandez-Sanchez et al., 2018). While decreases in C-reactive protein and IL-6 were seen, there was no significant decrease in PVR and no change in other secondary outcomes; however, prespecified subgroup analysis suggested a decrease in PVR in the CTD-PAH patients (Toshner and Rothman, 2020; Toshner et al., 2022). In a separate Mendelian randomization study of nearly 12,000 subjects, a genetic variant of the IL-6 receptor associated with higher circulating IL-6R levels was unexpectedly found not to affect risk of PAH (Toshner et al., 2022). Based on this data, further exploration of IL-6 as a therapeutic target in CTD-PAH may have more promise than overall WHO Group I disease.
Interleukin-1 (IL-1) has also been shown to be involved in the aberrant inflammatory signaling of PAH, and to correlate with severity of RV failure in PAH (Soon et al., 2010). Favorable protective effects of IL-1 blockade using anakinra, a recombinant IL-1 receptor antagonist, led to an open-label phase IB/II pilot study of anakinra as add-on therapy in which 6 patients with stable PAH were ultimately enrolled and completed the trial protocol (Voelkel et al., 1994; Trankle et al., 2019). Anakinra use was safe and produced decreased CRP levels and a trend towards decreased IL-6 levels but was likely underpowered to show differences in the cardiopulmonary exercise testing and right ventricular echocardiographic parameters it measured.
Higher levels of Janus kinase activity 2 (JAK2) has been found in human PASMCs in PAH, and the use of the JAK2 inhibitor ruxolitinib reduced proliferation of PASMCs and improved both hemodynamics and RV function and remodeling in two rat models (Yerabolu et al., 2021). No current human studies of JAK2 inhibition in PAH appear to be in progress, but this preclinical data may warrant further exploration of ruxolitinib and other JAK2 inhibitors such as baricitinib.
Poly (adenosine diphosphate-ribose) polymerase (PARP) is an enzyme involved in DNA repair that allows for ongoing cell survival despite DNA damage (Meloche et al., 2014). The persistent inflammatory state and oxidative damage that occurs in PAH leads to DNA damage; despite this, PASMCs in PAH exist in a pro-proliferative and anti-apoptotic state, leading to further progression of disease. PARP-1 has also been shown to upregulate proinflammatory cytokines like IL-6, which further contributes to apoptosis resistance through HIF-1α. PARP inhibition would therefore allow dysfunctional but proliferative PASMCs to proceed to cell death.
In vitro studies of human pulmonary artery explants showed significantly increased levels of DNA damage in PAH versus controls (Malenfant et al., 2013). PARP inhibition using ABT-888 decreased PASMC proliferation and increased levels of apoptosis, and showed reversal of downregulation of miR-204, a microRNA involved in reducing levels of pro-proliferative transcription factors NFAT and HIF-1α. The same agent showed reversal of PAH when administered to monocrotaline and Sugen/hypoxia rat PAH models.
Based on these findings, PARP inhibition appears to hold promise in the treatment of PAH. Oloparib is an orally available poly (adenosine diphosphate-ribose) polymerase (PARP) 1 inhibitor with efficacy as monotherapy or combination therapy for BRCA-positive advanced ovarian cancer, metastatic pancreatic adenocarcinoma, and HER2-negative high-risk early breast cancer (Moore et al., 2018; Golan et al., 2019; Tutt et al., 2021). A multicenter phase 1B trial in 20 PAH patients to evaluate safety and efficacy, OPTION (NCT03782818), is currently recruiting, with plans for a phase 2 trial to follow.
Cellular senescence, a phenomenon of arrested cell growth and division where cells do not progress through the cellular cycle and has been studied extensively as a process underpinning aging and age-associated diseases. Senescence is triggered by genotoxic stressors that induce a persistent activation of the cellular DNA damage response, resulting in a phenotype that both exits the cell cycle and does not progress to apoptosis while producing a specific set of inflammatory mediators that suppress regenerative growth (Culley and Chan, 2022). Senescence of endothelial cells has been implicated in PH and senescent profiles have been observed in both in vitro and animal models of PAH and appears to be associated with deficiency of frataxin. Frataxin deficiency results in a deficiency of endothelial iron-sulfur clusters and a heightened DNA damage response resulting in endothelial senescence, which leads to vessel remodeling and PAH (Culley et al., 2021). The senolytic ABT263 (navitoclax), which induces apoptosis specifically in senescent cells, successfully reversed the hemodynamic changes of PAH in a monocrotaline rate model of PAH with aortocaval shunting (van der Feen et al., 2020). Navitoclax is being employed in multiple solid and hematologic cancer treatment trials and may hold promise for human trials treating PAH.
Insulin resistance appears to be more common in PAH patients than in the general population and represents a potential risk factor for more severe disease. The presence of insulin resistance has been associated with worsened WHO Functional Class and left ventricular diastolic function in PAH (Zamanian et al., 2009; Brunner et al., 2014). Metabolic dysfunction and insulin resistance, replicated in animal models through use of a high-fat diet, have also been shown to adversely affect the pulmonary vasculature by inducing oxidative stress, vascular remodeling, inflammation, and alterations in lipid metabolism (Trammell et al., 2017; Brittain et al., 2019). The numerous agents used to address diabetes mellitus thus may hold promise for the treatment of PAH.
A strong correlation has been shown between circulating insulin levels and PAH severity in a BMPR2 mutant model of PAH. Insulin sensitivity in ApoE-deficient mice who develop PAH after exposure to a high fat diet improves with administration of the peroxisome proliferator-activated receptor-gamma (PPAR-γ) binding agent rosiglitazone (Hansmann et al., 2007). Rosiglitazone also has been shown to reduce PASMC proliferation and to reduce endothelin-1 and VEGF levels in a chronic hypoxia rat model of PH (Hansmann et al., 2007; Kim et al., 2010). Promising findings of normalization of PA pressures and improved RV function with pioglitazone in rat models have also been shown (Legchenko et al., 2018). Though promising, the connection between the thiazolidenediones and heart failure and fluid retention may dampen promise as PAH treatment options (Hernandez et al., 2011). However, pioglitazone has an overall better side effect profile and has prevented development of HFpEF in a rat model and may be a stronger choice for future studies (Kamimura et al., 2016; Hansmann et al., 2020). Promisingly, pioglitazone use in the IRIS trial showed decreased rates of MI and stroke vs. placebo without increased rates of heart failure (Kernan et al., 2016).
The biguanide drug metformin, in widespread use for treatment of type 2 diabetes mellitus, has been studied as a treatment for an extremely wide spectrum of other diseases. The drug has broadly pleiotropic effects; in addition to its effects on hyperglycemia through increases in peripheral insulin sensitivity and decreases in intestinal glucose absorption and hepatic gluconeogenesis, it has been shown to have both antiproliferative activity and favorable effects on the endothelium (Agard et al., 2009). Further study has shown it exerts anti-remodeling and anti-apoptotic effects through action on AMPK, through which it may decrease endothelin-1 levels (Yoshida et al., 2020). Interestingly, it has also been shown to reduce PAH through reduction in circulating estrogen via inhibition of aromatase synthesis (Dean et al., 2016).
In hypoxia and monocrotaline rat models of PAH, metformin has been shown to prevent the onset of PAH; however, results have been more equivocal in Sugen hypoxia rat models of PAH, in which trials have shown both reversal of PAH or no significant effect on amelioration or prevention of PAH (Agard et al., 2009; Goncharov et al., 2018). More promise has perhaps been suggested in PH-LHD, which is closely tied with insulin resistance (Mulkareddy and Simon, 2020). A small phase 2 trial of metformin in 20 patients with IPAH showed it to be well-tolerated and safe and suggested a small improvement in right ventricular fractional area change but was not powered to demonstrate changes in clinical outcomes (Brittain et al., 2020). An earlier attempt at a phase 3 trial studying metformin in PAH, MetHAP, was unsuccessful; a larger placebo-controlled trial comparing metformin with or without a mobile health intervention is currently recruiting.
Sodium glucose cotransporter 2 (SGLT2) inhibitors have been the target of intense interest in the treatment of heart failure, with great successes demonstrated in trials studying empagliflozin in both HFrEF (EMPEROR-Reduced) and HFpEF (EMPEROR-Preserved), both of which showed significant reductions in cardiovascular death and hospitalization (Packer et al., 2020; Anker et al., 2021). Trials in left sided heart failure have also demonstrated improvements in pulmonary hemodynamics that may translate to efficacy in PAH. The EMBRACE-HF trial, a placebo-controlled trial used CardioMEMS monitoring in patients with stable left-sided heart failure, showed a significant reduction in pulmonary artery diastolic pressure and a trend towards reduced mean pulmonary arterial pressures independent of loop diuretic use (Nassif et al., 2021). An open-label trial of dapagliflozin in 78 patients with stable NYHA Class I-II heart failure showed reductions in development of exercise-induced pulmonary hypertension (Kayano et al., 2020).
In PAH models, the medication class appears to have favorable vasodilatory, antiproliferative and anti-inflammatory effects, and they may act to directly prevent cardiac remodeling (Lee et al., 2019; King and Brittain, 2022). Empagliflozin has been shown to prevent progression of PAH in a monocrotaline rat model (Chowdhury et al., 2020). A phase 2 trial of dapagliflozin in PAH (DAPAH, NCT05179356) is preparing to begin recruitment, with change in VO2max from baseline to 3 months as its primary outcome.
Glucagon-like peptide-1 (GLP1) agonists, in widespread use in the treatment of diabetes mellitus and in increasing use as potent agents for weight loss, may hold promise for treatment of PAH as well. GLP1 agonists have multiple metabolic effects, including insulin regulation at the pancreatic level, induction of brown fat thermogenesis, and signaling of satiety at the hypothalamus (Andersen et al., 2018). GLP1 agonist therapy increased nitric oxide production and reduced right ventricular systolic pressures and pulmonary arteriolar thickness in a mouse model of PAH; studies in rat models show GLP1 agonism reduces PASMC proliferation and inflammatory signaling. These promising findings may be used to justify studies in humans in the future (Honda et al., 2018).
Numerous strategies successfully employed in management of left heart failure have also been studied as therapeutic targets for PAH and right ventricular failure. These include blockade at various points along the renin-angiotensin-aldosterone system (RAAS), including use of angiotensin converting enzyme (ACE) inhibitors, angiotensin receptor blockers (ARBs), aldosterone blockade, beta blockade, and cardiac resynchronization therapy. Additionally, some promise has been shown in modulating sympathetic and parasympathetic autonomic nervous activity. These pathways are logical targets for therapy given the contribution of RAAS system activation and elevated sympathetic tone to PAH, though the amount of promise shown as treatment targets has been variable (Ciarka et al., 2010; de Man et al., 2012).
Addressing RAAS dysregulation with ACE inhibitors, ARBs, and aldosterone blocking agents has long been an established element of goal-directed medical therapy in left heart failure. Studies of IPAH patients have also shown dysregulation of the RAAS and elevated levels of renin, angiotensin I, and angiotensin II, with higher levels correlated to disease progression and mortality, and higher expression of AT1 receptors is observed in pulmonary vascular smooth muscle cells in IPAH (Orte et al., 2000; de Man et al., 2012). Animal studies have shown that use of ACE inhibitors reduces pulmonary vascular medial thickening in chronically hypoxic rats with pulmonary hypertension, and several studies using ARBs in animal models of PAH have shown benefits including decreased right ventricular afterload, improved right ventricular-arterial coupling and diastolic function, and prevention of pulmonary vascular remodeling (Abraham et al., 1995; Rondelet et al., 2005; Kato et al., 2008; de Man et al., 2012; Li et al., 2014). However, in an older study in 7 women with PAH, captopril showed no improvement in hemodynamics or exercise tolerance, and a study of ARB use in COPD-PH showed no clear clinical benefits (Leier et al., 1983; Morrell et al., 2005). The role of ACE inhibitors or ARBs in PAH patients remains unclear aside from their use in treatment of concomitant essential hypertension.
More recently, there has been increasing interest in ACE2 as a therapeutic target, with recent discoveries of ACE2 deficiency rendering mice more prone to develop hypoxia-induced PH and an imbalance between ACE and ACE2 levels in a cohort of PAH patients (Hemnes et al., 2018; Zhang et al., 2018). These findings were channeled into studies of ACE2 gene transfer and ACE2 agonists in animal models, then into use of recombinant human ACE2 infusion (GSK2586881) in a Phase I study of five PAH patients who showed improvements in cardiac output and PVR and decreases in markers of oxidant stress (Yamazato et al., 2009; Breitling et al., 2015; Hemnes et al., 2018). A subsequent Phase II study (NCT03177603) has completed, but it is unclear if there are further development plans for the drug. Separately, successful efforts to stabilize the ACE2 and Ang-(1–7) proteins for delivery using plant cells also hold promise for its use as a potential oral therapy (Shenoy et al., 2014; Daniell et al., 2020).
Aldosterone blockade has also been a target of interest with mixed results. The ARIES trials used a combination of ambrisentan and spironolactone in a small group of patients with PAH and demonstrated improvements in nt-proBNP and 6MWD compared to ambrisentan alone, but the trends were not statistically significant, and no functional measures of the right heart were included (Maron et al., 2013). A single-center study of PAH patients taking aldosterone receptor antagonists had a significantly greater rate of hospitalizations than those who did not; however, a subsequent cross-over study of spironolactone use in 42 PAH patients showed the drug was well-tolerated but demonstrated no differences in the primary outcome of collagen biomarkers or secondary outcomes, including 6MWD and hemodynamic parameters (Corkish et al., 2019; Safdar et al., 2020). Additional trials to study aldosterone blockade in PAH and stable right heart failure are actively recruiting. Finally, with the successful adoption of sacubitril-valsartan in HFrEF management, there is interest in use of neprilysin inhibition in PAH. Sacubitril-valsartan has been shown to reduce RVSP and RV wall thickness and to increase cGMP levels in a rat model of PAH, and a proof-of-concept trial using racecadotril in 21 PAH patients showing that the drug was well-tolerated and produced significant decreases in PVR (Clements et al., 2019; Hobbs et al., 2019).
Beta-adrenergic blockers, while a cornerstone treatment in left heart failure, have shown mixed results in studies of pre-capillary pulmonary hypertension and right heart failure. A 2010 study of beta-blockade in a rat model demonstrated beneficial effects on the right ventricle in the form of decreased fibrosis, apoptosis, and capillary loss in right ventricles with pressure overload, though such a benefit was not seen in a later study of bisoprolol and losartan in RV hypertrophy (Bogaard et al., 2010; Andersen et al., 2014). Clinical studies have also been equivocal; detrimental effects of beta-blockade were seen in a trial of portopulmonary hypertension patients, while a 2017 study of 30 patients, most of whom had WHO Group I PH, found improved heart rate recovery after exercise and no decrease in cardiac output with the use of carvedilol (Provencher et al., 2006; Farha et al., 2017). Beta-blockers remain indicated in patients with PAH only for treatment of appropriate comorbidities such as coronary artery disease or arrhythmias (Perros et al., 2017).
More direct interventions to reduce heightened sympathetic activity in PH have also been explored. Sympathetic ganglion block was explored in a monocrotaline rat model of PAH and demonstrated decreased right ventricular hypertrophy and pressures along with a measurable decrease in oxidative stress (Na et al., 2014). Pulmonary artery denervation, a minimally invasive endovascular procedure, is a subject of ongoing research and has been tested both in animal models and in patients. A phase II trial of 66 patients revealed improvement in 6MWD and cardiac output along with decreases in mPAP, PVR, and RVSP that were maintained at 6 and 12 months from the procedure, though the study did not include a comparator group (Chen et al., 2015). Favorable improvements in 6MWD and PVR have been reported in a small trial of CpC-PH, but its applicability to PAH patients is unclear, and trials exploring pulmonary artery denervation in PAH are ongoing (Zhang H. et al., 2019; Rothman et al., 2020). A related strategy, renal denervation of sympathetic fibers, has previously been explored as a treatment for refractory systemic hypertension and shown beneficial effects on the RV and pulmonary vascular remodeling in monocrotaline rat models, though decreases in systemic blood pressure would be a significant concern in PAH patients (Liu et al., 2017; Vaillancourt et al., 2017).
With the establishment of elevated sympathetic nervous system activity as an element in the pathogenesis of PH, the parasympathetic nervous system, which has shown concurrent decreases in activity in PAH, has also emerged as a target for treatment (da Silva Goncalves Bos et al., 2018; Hemnes and Brittain, 2018). Use of the acetylcholinesterase inhibitor pyridostigmine in a rat model of PAH demonstrated decreased right ventricular hypertrophy, fibrosis, and stiffness in a pressure-volume loop analysis (da Silva Goncalves Bos et al., 2018). Direct modulation of the parasympathetic nervous system through vagal nerve stimulation has also been studied in animal models, with a 2018 study finding decreases in pulmonary artery pressure, vascular remodeling, and RVEDP and increases in cardiac index (Yoshida et al., 2018).
There has been ongoing interest in strategies that directly affect the timing of the contractility of the right atrium and ventricle in pulmonary arterial hypertension, namely right atrial pacing and cardiac resynchronization therapy.
Right atrial pacing has been employed in cases of right ventricular infarction and after cardiac surgery to improve cardiac output, and in the latter has been shown to increase RV contractility (Lancon et al., 1994; Vrouchos et al., 1997). In a 2021 study, patients with PAH referred for right heart catheterization underwent evaluation with a pressure-volume catheter before and after introduction of a pacing wire into the right atrium, with measurements of pressure-volume loops, right ventricular end-diastolic volume (RVEDV), cardiac output, and stroke volume (Khural et al., 2021). 32 subjects were enrolled, of which 16 were ultimately included in the study. Increasing heart rates through RA pacing produced significant improvements in measures of RV contractile force, decreases in RV end-diastolic pressure and volume, and increased cardiac output, though a significant decline in stroke volume was seen. Right atrial pacing was thought to improve interaction between the right and left ventricles to allow optimization of LV filling, thereby increasing cardiac output. Though the study was small and limited to a brief period on the day of right heart catheterization, it may represent a promising strategy to treat a failing right ventricle in the setting of PAH. The implications of chronic pacing will need to be explored in further studies; the authors assert that continuous hemodynamic monitoring would likely be necessary, and the effect of higher basal heart rates resulting in higher baseline myocardial oxygen demand over the long term is a major consideration.
Similar lines of thought have driven consideration of cardiac resynchronization therapy (CRT) as a therapy in right ventricular failure in PAH. CRT therapy in LHD primarily functions to resynchronize ventricular contraction but also appears to reverse long-term maladaptive cardiac remodeling (Jones et al., 2017). In the face of chronically elevated pulmonary vascular resistance, maladaptive remodeling of the right ventricular myocardium appears to lead to delayed intraventricular conduction and interventricular dysynchrony, like that observed in the left ventricle. This conduction delay in turn affects LV performance through multiple mechanisms, including increasing septal bowing into the left ventricle, compromising diastolic filling, and increasing left chamber pressures (Rasmussen et al., 2014). By ameliorating this dysynchrony, CRT has been proposed as a mechanism to improve biventricular performance and thus reverse RV remodeling.
Use of CRT was first explored successfully in PAH due to congenital heart disease by Dubin et al. (2003), though these 7 patients demonstrated dysynchrony due to overt conduction delay from right bundle branch block, felt to be more akin to left heart failure than PAH. This was explored first in animal models and then as proof of concept by Hardziyenka et al. (2011) in a cohort of 14 CTEPH patients, all of whom had long diastolic interventricular delays. These patients underwent temporary RV pacing using gradually shortened atrioventricular delay. The group showed significant decreases in diastolic interventricular delay, enhanced RV contractility, and increases in stroke volume. Despite evaluating HFrEF patients, a study of 31 patients by Martens et al. (2018) demonstrated improvements in pulmonary pressures and a long-term improvement in RV-PA coupling. Larger, long-term trials of CRT therapy in PAH are warranted, both to further explore the durable effects of this modality on the right ventricle and to determine the characteristics of PAH patients likely to respond best to the therapy.
Remote monitoring devices have also been explored as an adjunctive tool in the management of PAH. Remotely assessing 6-min walk distance, a cornerstone metric in evaluating PAH severity and response to therapy, has been a common goal. Trials using monitoring of physical activity via commercially available smartwatch and smartphone platforms have demonstrated feasibility in PAH patients and showed moderate to good correlation between traditional and digital 6-min walk distances (Glinskii et al., 2021; Stollfuss et al., 2021). Invasive monitoring systems such as the CardioMEMS sensor can assess pulmonary arterial pressures to allow real-time hemodynamics to guide heart failure therapy and have been explored primarily in left-sided heart failure. Safe long-term use of the CardioMEMS device has been demonstrated in a group of 26 PAH patients, with promise for larger scale testing, and the device has begun to be introduced for real-time measurement in pharmaceutical trials (Benza et al., 2019).
The broad landscape of new therapies being studied for pulmonary arterial hypertension promises both a bright future and growing complexity in how to approach its treatment. In the last several years, there have been further refinements to the risk stratification tools that guide current pulmonary vasodilator therapy in PAH. With the expected introduction of therapies like sotatercept, which utilize pathways more complex than the approved pulmonary vasodilators, these tools will have to adapt to fit a more intricate approach, and further phenotyping and personalization may be required to optimize treatment strategies. Additionally, further tools to better phenotype complex precapillary pulmonary hypertension could open treatment pathways to certain patients with multifactorial PH from several WHO Groups.
In this review, several promising treatments are discussed that showed significant promise at the preclinical level but went on to show no net benefits in phase 2 clinical trials. Further improvements in murine and rat models of PAH that are more reliably predictive of the treatment response in humans may allow for more effective focus on truly promising treatments. Exploration of meaningful treatment targets for human clinical trials, adapted to account for the relatively small numbers of subjects in PAH trials and potentially employing new biomarkers and genetic assay techniques, will also be critical to establish true benefit of emerging therapies (Sitbon et al., 2019).
Finally, there are great opportunities for PAH treatment within the field of new drug discovery and smart drug design. Numerous therapies discussed here were first established as treatments for cancer. With better understanding of the complex molecular pathways underlying PAH and as the potential of targeting these pathways becomes increasingly clear, computational techniques and artificial intelligence can be used to find existing drugs with compatible targets for treating PAH (Negi et al., 2021).
Therapeutics in pulmonary arterial hypertension has been an area of explosive research over roughly the last 20 years and new developments continue to emerge. Pulmonary vasodilator medications have become more diverse, easier to deliver, and guided by a much more sophisticated risk-stratified paradigm to guide their use. Beyond pulmonary vasodilators, the field is beginning to see the first medications targeting alternative pathways for treating PAH, promising both greater abilities to treat the disease and increasing complexity in the decisions surrounding starting and adjusting treatments. The wealth of treatment options currently being explored at the phase 1 and 2 levels promises continuing breakthroughs in PAH in the coming years.
SC and DS contributed to the design of the manuscript. DS provided the initial outline and structure of the manuscript. SC wrote the draft manuscript and subsequent revisions. SC created the images. SC, DS, and GR wrote sections of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Partial funding for open access was provided by the University of Maryland Health Sciences and Human Services Library’s Open Access Fund.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor declared a shared affiliation, though no other collaboration, with several of the authors SC, DS, GR at the time of the review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abenhaim, L., Moride, Y., Brenot, F., Rich, S., Benichou, J., Kurz, X., et al. (1996). Appetite-suppressant drugs and the risk of primary pulmonary hypertension. International primary pulmonary hypertension study group. N. Engl. J. Med. 335 (9), 609–616. doi:10.1056/NEJM199608293350901
Abraham, W. T., Raynolds, M. V., Gottschall, B., Badesch, D. B., Wynne, K. M., Groves, B. M., et al. (1995). Importance of angiotensin-converting enzyme in pulmonary hypertension. Cardiology 86 (1), 9–15. doi:10.1159/000176939
Acharya, A. P., Tang, Y., Bertero, T., Tai, Y. Y., Harvey, L. D., Woodcock, C. C., et al. (2021). Simultaneous pharmacologic inhibition of yes-associated protein 1 and glutaminase 1 via inhaled poly(lactic-co-glycolic) acid-encapsulated microparticles improves pulmonary hypertension. J. Am. Heart Assoc. 10 (12), e019091. doi:10.1161/JAHA.120.019091
Agard, C., Rolli-Derkinderen, M., Dumas-de-La-Roque, E., Rio, M., Sagan, C., Savineau, J. P., et al. (2009). Protective role of the antidiabetic drug metformin against chronic experimental pulmonary hypertension. Br. J. Pharmacol. 158 (5), 1285–1294. doi:10.1111/j.1476-5381.2009.00445.x
Alencar, A. K. N., Montes, G. C., Barreiro, E. J., Sudo, R. T., and Zapata-Sudo, G. (2017). Adenosine receptors as drug targets for treatment of pulmonary arterial hypertension. Front. Pharmacol. 8, 858. doi:10.3389/fphar.2017.00858
Andersen, A., Lund, A., Knop, F. K., and Vilsboll, T. (2018). Glucagon-like peptide 1 in health and disease. Nat. Rev. Endocrinol. 14 (7), 390–403. doi:10.1038/s41574-018-0016-2
Andersen, S., Schultz, J. G., Andersen, A., Ringgaard, S., Nielsen, J. M., Holmboe, S., et al. (2014). Effects of bisoprolol and losartan treatment in the hypertrophic and failing right heart. J. Card. Fail. 20 (11), 864–873. doi:10.1016/j.cardfail.2014.08.003
Anker, S. D., Butler, J., Filippatos, G., Ferreira, J. P., Bocchi, E., Bohm, M., et al. (2021). Empagliflozin in heart failure with a preserved ejection fraction. N. Engl. J. Med. 385 (16), 1451–1461. doi:10.1056/NEJMoa2107038
Archer, S. L. (2017). Pyruvate kinase and Warburg metabolism in pulmonary arterial hypertension: Uncoupled glycolysis and the cancer-like phenotype of pulmonary arterial hypertension. Circulation 136 (25), 2486–2490. doi:10.1161/CIRCULATIONAHA.117.031655
Armstrong, P. W., Pieske, B., Anstrom, K. J., Ezekowitz, J., Hernandez, A. F., Butler, J., et al. (2020). Vericiguat in patients with heart failure and reduced ejection fraction. N. Engl. J. Med. 382 (20), 1883–1893. doi:10.1056/NEJMoa1915928
Asosingh, K., Aldred, M. A., Vasanji, A., Drazba, J., Sharp, J., Farver, C., et al. (2008). Circulating angiogenic precursors in idiopathic pulmonary arterial hypertension. Am. J. Pathol. 172 (3), 615–627. doi:10.2353/ajpath.2008.070705
Babicheva, A., Makino, A., and Yuan, J. X. (2021). mTOR signaling in pulmonary vascular disease: Pathogenic role and therapeutic target. Int. J. Mol. Sci. 22 (4), 2144. doi:10.3390/ijms22042144
Bader, M. (2020). Inhibition of serotonin synthesis: A novel therapeutic paradigm. Pharmacol. Ther. 205, 107423. doi:10.1016/j.pharmthera.2019.107423
Badesch, D. B., Tapson, V. F., McGoon, M. D., Brundage, B. H., Rubin, L. J., Wigley, F. M., et al. (2000). Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann. Intern. Med. 132 (6), 425–434. doi:10.7326/0003-4819-132-6-200003210-00002
Barst, R. J., Gibbs, J. S. R., Ghofrani, H. A., Hoeper, M. M., McLaughlin, V. V., Rubin, L. J., et al. (2009). Updated evidence-based treatment algorithm in pulmonary arterial hypertension. J. Am. Coll. Cardiol. 54 (1), S78–S84. doi:10.1016/j.jacc.2009.04.017
Benza, R. L., Doyle, M., Lasorda, D., Parikh, K. S., Correa-Jaque, P., Badie, N., et al. (2019). Monitoring pulmonary arterial hypertension using an implantable hemodynamic sensor. Chest 156 (6), 1176–1186. doi:10.1016/j.chest.2019.06.010
Benza, R. L. S., Chakinala, M., Correa, P., Lee, J., and White, J. (2020). “Case study of long-term safety, tolerability, and hemodynamic response of Pb1046, A sustained-release analogue for vasoactive intestinal peptide (VIP), in an adult subject with pulmonary arterial hypertension (PAH),” in Pulmonary vascular research institute (PVRI) World congress (Lima, Peru.
Bertero, T., Oldham, W. M., Cottrill, K. A., Pisano, S., Vanderpool, R. R., Yu, Q., et al. (2016). Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Invest.. 126 (9), 3313–3335. doi:10.1172/JCI86387
Bogaard, H. J., Natarajan, R., Mizuno, S., Abbate, A., Chang, P. J., Chau, V. Q., et al. (2010). Adrenergic receptor blockade reverses right heart remodeling and dysfunction in pulmonary hypertensive rats. Am. J. Respir. Crit. Care Med. 182 (5), 652–660. doi:10.1164/rccm.201003-0335OC
Borck, P. C., Guo, L. W., and Plutzky, J. (2020). BET epigenetic reader proteins in cardiovascular transcriptional programs. Circ. Res. 126 (9), 1190–1208. doi:10.1161/CIRCRESAHA.120.315929
Boucly, A., Savale, L., Jais, X., Bauer, F., Bergot, E., Bertoletti, L., et al. (2021). Association between initial treatment strategy and long-term survival in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 204 (7), 842–854. doi:10.1164/rccm.202009-3698OC
Bouzina, H., and Radegran, G. (2020). Plasma adrenomedullin peptides and precursor levels in pulmonary arterial hypertension disease severity and risk stratification. Pulm. Circ. 10 (3), 2045894020931317. doi:10.1177/2045894020931317
Breitling, S., Krauszman, A., Parihar, R., Walther, T., Friedberg, M. K., and Kuebler, W. M. (2015). Dose-dependent, therapeutic potential of angiotensin-(1-7) for the treatment of pulmonary arterial hypertension. Pulm. Circ. 5 (4), 649–657. doi:10.1086/683696
Brittain, E. L., Niswender, K., Agrawal, V., Chen, X., Fan, R., Pugh, M. E., et al. (2020). Mechanistic phase II clinical trial of metformin in pulmonary arterial hypertension. J. Am. Heart Assoc. 9 (22), e018349. doi:10.1161/JAHA.120.018349
Brittain, E. L., Talati, M., Fortune, N., Agrawal, V., Meoli, D. F., West, J., et al. (2019). Adverse physiologic effects of western diet on right ventricular structure and function: Role of lipid accumulation and metabolic therapy. Pulm. Circ. 9 (1), 2045894018817741. doi:10.1177/2045894018817741
Brunner, N. W., Skhiri, M., Fortenko, O., Hsi, A., Haddad, F., Khazeni, N., et al. (2014). Impact of insulin resistance on ventricular function in pulmonary arterial hypertension. J. Heart Lung Transpl. 33 (7), 721–726. doi:10.1016/j.healun.2014.02.016
Budas, G. R., Boehm, M., Kojonazarov, B., Viswanathan, G., Tian, X., Veeroju, S., et al. (2018). ASK1 inhibition halts disease progression in preclinical models of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 197 (3), 373–385. doi:10.1164/rccm.201703-0502OC
Cantoni, S., Cavalli, S., Pastore, F., Accetta, A., Pala, D., Vaccaro, F., et al. (2019). Pharmacological characterization of a highly selective Rho kinase (ROCK) inhibitor and its therapeutic effects in experimental pulmonary hypertension. Eur. J. Pharmacol. 850, 126–134. doi:10.1016/j.ejphar.2019.02.009
Chan, S. Y., and Rubin, L. J. (2017). Metabolic dysfunction in pulmonary hypertension: From basic science to clinical practice. Eur. Respir. Rev. 26 (146), 170094. doi:10.1183/16000617.0094-2017
Chapman, R. W., Corboz, M. R., Fernandez, C., Sullivan, E., Stautberg, A., Plaunt, A. J., et al. (2021). Characterisation of cough evoked by inhaled treprostinil and treprostinil palmitil. ERJ Open Res. 7 (1), 00592–02020. doi:10.1183/23120541.00592-2020
Chen, S. L., Zhang, H., Xie, D. J., Zhang, J., Zhou, L., Rothman, A. M., et al. (2015). Hemodynamic, functional, and clinical responses to pulmonary artery denervation in patients with pulmonary arterial hypertension of different causes: Phase II results from the pulmonary artery denervation-1 study. Circ. Cardiovasc. Interv. 8 (11), e002837. doi:10.1161/CIRCINTERVENTIONS.115.002837
Chen, Y., Yuan, T., Zhang, H., Yan, Y., Wang, D., Fang, L., et al. (2017). Activation of Nrf2 attenuates pulmonary vascular remodeling via inhibiting endothelial-to-mesenchymal transition: An insight from a plant polyphenol. Int. J. Biol. Sci. 13 (8), 1067–1081. doi:10.7150/ijbs.20316
Chin, K. M., Sitbon, O., Doelberg, M., Feldman, J., Gibbs, J. S. R., Grunig, E., et al. (2021). Three- versus two-drug therapy for patients with newly diagnosed pulmonary arterial hypertension. J. Am. Coll. Cardiol. 78 (14), 1393–1403. doi:10.1016/j.jacc.2021.07.057
Chowdhury, B., Luu, A. Z., Luu, V. Z., Kabir, M. G., Pan, Y., Teoh, H., et al. (2020). The SGLT2 inhibitor empagliflozin reduces mortality and prevents progression in experimental pulmonary hypertension. Biochem. Biophys. Res. Commun. 524 (1), 50–56. doi:10.1016/j.bbrc.2020.01.015
Ciarka, A., Doan, V., Velez-Roa, S., Naeije, R., and van de Borne, P. (2010). Prognostic significance of sympathetic nervous system activation in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 181 (11), 1269–1275. doi:10.1164/rccm.200912-1856OC
Clements, R. T., Vang, A., Fernandez-Nicolas, A., Kue, N. R., Mancini, T. J., Morrison, A. R., et al. (2019). Treatment of pulmonary hypertension with angiotensin II receptor blocker and neprilysin inhibitor sacubitril/valsartan. Circ. Heart Fail. 12 (11), e005819. doi:10.1161/CIRCHEARTFAILURE.119.005819
Corkish, M., Devine, L., Clarke, M., Murray, B., and Rose-Jones, L. (2019). Express: Rates of hospitalization associated with the use of aldosterone receptor antagonists in patients with pulmonary arterial hypertension. Pulm. Circ. 9, 2045894019868422. doi:10.1177/2045894019868422
Culley, M. K., and Chan, S. Y. (2022). Endothelial senescence: A new age in pulmonary hypertension. Circ. Res. 130 (6), 928–941. doi:10.1161/CIRCRESAHA.121.319815
Culley, M. K., Zhao, J., Tai, Y. Y., Tang, Y., Perk, D., Negi, V., et al. (2021). Frataxin deficiency promotes endothelial senescence in pulmonary hypertension. J. Clin. Invest.. 131 (11), e136459. doi:10.1172/JCI136459
D'Alto, M., Badagliacca, R., Argiento, P., Romeo, E., Farro, A., Papa, S., et al. (2020). Risk reduction and right heart reverse remodeling by upfront triple combination therapy in pulmonary arterial hypertension. Chest 157 (2), 157376–157383. doi:10.1016/j.chest.2019.09.009
da Silva Goncalves Bos, D., Van Der Bruggen, C. E. E., Kurakula, K., Sun, X. Q., Casali, K. R., Casali, A. G., et al. (2018). Contribution of impaired parasympathetic activity to right ventricular dysfunction and pulmonary vascular remodeling in pulmonary arterial hypertension. Circulation 137 (9), 910–924. doi:10.1161/CIRCULATIONAHA.117.027451
Dai, Y., Chen, X., Song, X., Chen, X., Ma, W., Lin, J., et al. (2019). Immunotherapy of endothelin-1 receptor type A for pulmonary arterial hypertension. J. Am. Coll. Cardiol. 73 (20), 2567–2580. doi:10.1016/j.jacc.2019.02.067
Daniell, H., Mangu, V., Yakubov, B., Park, J., Habibi, P., Shi, Y., et al. (2020). Investigational new drug enabling angiotensin oral-delivery studies to attenuate pulmonary hypertension. Biomaterials 233, 119750. doi:10.1016/j.biomaterials.2019.119750
de Man, F. S., Tu, L., Handoko, M. L., Rain, S., Ruiter, G., Francois, C., et al. (2012). Dysregulated renin-angiotensin-aldosterone system contributes to pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 186 (8), 780–789. doi:10.1164/rccm.201203-0411OC
Dean, A., Nilsen, M., Loughlin, L., Salt, I. P., and MacLean, M. R. (2016). Metformin reverses development of pulmonary hypertension via aromatase inhibition. Hypertension 68 (2), 446–454. doi:10.1161/HYPERTENSIONAHA.116.07353
Delaney, C., Sherlock, L., Fisher, S., Maltzahn, J., Wright, C., and Nozik-Grayck, E. (2018). Serotonin 2A receptor inhibition protects against the development of pulmonary hypertension and pulmonary vascular remodeling in neonatal mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 314 (5), L871–L881. doi:10.1152/ajplung.00215.2017
Diebold, I., Hennigs, J. K., Miyagawa, K., Li, C. G., Nickel, N. P., Kaschwich, M., et al. (2015). BMPR2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab. 21 (4), 596–608. doi:10.1016/j.cmet.2015.03.010
Dorai, H., Vukicevic, S., and Sampath, T. K. (2000). Bone morphogenetic protein-7 (osteogenic protein-1) inhibits smooth muscle cell proliferation and stimulates the expression of markers that are characteristic of SMC phenotype in vitro. J. Cell. Physiol. 184 (1), 37–45. doi:10.1002/(SICI)1097-4652(200007)184:1<37::AID-JCP4>3.0.CO;2-M
Dubin, A. M., Feinstein, J. A., Reddy, V. M., Hanley, F. L., Van Hare, G. F., and Rosenthal, D. N. (2003). Electrical resynchronization: A novel therapy for the failing right ventricle. Circulation 107 (18), 2287–2289. doi:10.1161/01.CIR.0000070930.33499.9F
Dumitrascu, R., Kulcke, C., Konigshoff, M., Kouri, F., Yang, X., Morrell, N., et al. (2011). Terguride ameliorates monocrotaline-induced pulmonary hypertension in rats. Eur. Respir. J. 37 (5), 1104–1118. doi:10.1183/09031936.00126010
Eba, S., Hoshikawa, Y., Moriguchi, T., Mitsuishi, Y., Satoh, H., Ishida, K., et al. (2013). The nuclear factor erythroid 2-related factor 2 activator oltipraz attenuates chronic hypoxia-induced cardiopulmonary alterations in mice. Am. J. Respir. Cell Mol. Biol. 49 (2), 324–333. doi:10.1165/rcmb.2011-0396OC
El-Dabh, A., and Acharya, D. (2019). Express: Pulmonary hypertension with dasatinib and other tyrosine kinase inhibitors. Pulm. Circ. 9, 2045894019865704. doi:10.1177/2045894019865704
Enevoldsen, F. C., Sahana, J., Wehland, M., Grimm, D., Infanger, M., and Kruger, M. (2020). Endothelin receptor antagonists: Status quo and future perspectives for targeted therapy. J. Clin. Med. 9 (3), 824. doi:10.3390/jcm9030824
Farber, H. W., and Gin-Sing, W. (2016). Practical considerations for therapies targeting the prostacyclin pathway. Eur. Respir. Rev. 25 (142), 418–430. doi:10.1183/16000617.0083-2016
Farha, S., Dweik, R., Rahaghi, F., Benza, R., Hassoun, P., Frantz, R., et al. (2014). Imatinib in pulmonary arterial hypertension: c-Kit inhibition. Pulm. Circ. 4 (3), 452–455. doi:10.1086/677359
Farha, S., Saygin, D., Park, M. M., Cheong, H. I., Asosingh, K., Comhair, S. A., et al. (2017). Pulmonary arterial hypertension treatment with carvedilol for heart failure: A randomized controlled trial. JCI Insight 2 (16), e95240. doi:10.1172/jci.insight.95240
Fazakas, C., Nagaraj, C., Zabini, D., Vegh, A. G., Marsh, L. M., Wilhelm, I., et al. (2018). Rho-kinase inhibition ameliorates dasatinib-induced endothelial dysfunction and pulmonary hypertension. Front. Physiol. 9, 537. doi:10.3389/fphys.2018.00537
Feijoo-Bandin, S., Aragon-Herrera, A., Rodriguez-Penas, D., Portoles, M., Rosello-Lleti, E., Rivera, M., et al. (2017). Relaxin-2 in cardiometabolic diseases: Mechanisms of action and future perspectives. Front. Physiol. 8, 599. doi:10.3389/fphys.2017.00599
Francois, H., Athirakul, K., Mao, L., Rockman, H., and Coffman, T. M. (2004). Role for thromboxane receptors in angiotensin-II-induced hypertension. Hypertension 43 (2), 364–369. doi:10.1161/01.HYP.0000112225.27560.24
Frantz, R. P., Benza, R. L., Channick, R. N., Chin, K., Howard, L. S., McLaughlin, V. V., et al. (2021). TORREY, a Phase 2 study to evaluate the efficacy and safety of inhaled seralutinib for the treatment of pulmonary arterial hypertension. Pulm. Circ. 11 (4), 20458940211057071. doi:10.1177/20458940211057071
Fukumoto, Y., Matoba, T., Ito, A., Tanaka, H., Kishi, T., Hayashidani, S., et al. (2005). Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart 91 (3), 391–392. doi:10.1136/hrt.2003.029470
Fukumoto, Y., Yamada, N., Matsubara, H., Mizoguchi, M., Uchino, K., Yao, A., et al. (2013). Double-blind, placebo-controlled clinical trial with a rho-kinase inhibitor in pulmonary arterial hypertension. Circ. J. 77 (10), 2619–2625. doi:10.1253/circj.cj-13-0443
Gaine, S., and McLaughlin, V. (2017). Pulmonary arterial hypertension: Tailoring treatment to risk in the current era. Eur. Respir. Rev. 26 (146), 170095. doi:10.1183/16000617.0095-2017
Galie, N., Barbera, J. A., Frost, A. E., Ghofrani, H. A., Hoeper, M. M., McLaughlin, V. V., et al. (2015). Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N. Engl. J. Med. 373 (9), 834–844. doi:10.1056/NEJMoa1413687
Galie, N. B., Ewert, R., Gomez-Sanchez, M. A., Barbera, J. A., Torbicki, A., Bremer, H., et al. (2010). “Effects of inhaled avaptadil (vasoactive intestinal peptide) in patients with pulmonary arterial hypertension (PAH),” in American thoracic society (New Orleans, LA, USA: Am J Respir Crit Care Med).
Galie, N., Humbert, M., Vachiery, J. L., Gibbs, S., Lang, I., Torbicki, A., et al. (2016). 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: The joint task force for the diagnosis and treatment of pulmonary hypertension of the European society of cardiology (ESC) and the European respiratory society (ERS): Endorsed by: Association for European paediatric and congenital cardiology (AEPC), international society for heart and lung transplantation (ISHLT). Eur. Heart J. 37 (1), 67–119. doi:10.1093/eurheartj/ehv317
Galie, N., Manes, A., and Branzi, A. (2004). The endothelin system in pulmonary arterial hypertension. Cardiovasc. Res. 61 (2), 227–237. doi:10.1016/j.cardiores.2003.11.026
Galkin, A., Sitapara, R., Clemons, B., Garcia, E., Kennedy, M., Guimond, D., et al. (2022). Inhaled seralutinib exhibits potent efficacy in models of pulmonary arterial hypertension. Eur. Respir. J., 2102356. doi:10.1183/13993003.02356-2021
Gangopahyay, A., Oran, M., Bauer, E. M., Wertz, J. W., Comhair, S. A., Erzurum, S. C., et al. (2011). Bone morphogenetic protein receptor II is a novel mediator of endothelial nitric-oxide synthase activation. J. Biol. Chem. 286 (38), 33134–33140. doi:10.1074/jbc.M111.274100
Geven, C., Kox, M., and Pickkers, P. (2018). Adrenomedullin and adrenomedullin-targeted therapy as treatment strategies relevant for sepsis. Front. Immunol. 9, 292. doi:10.3389/fimmu.2018.00292
Ghofrani, H. A. A.-H., Vonk-Noordegraaf, A., Behr, J., Neurohr, C., Jansa, P., Wilkens, H., et al. (2012). “Proof-of-concept study to investigate the efficacy, hemodynamics and tolerability of terguride vs. Placebo in subjects with pulmonary arterial hypertension: Results of A double blind, randomised, prospective phase IIa study,” in American thoracic society (San Francisco, CA, USA).
Ghofrani, H. A., Galie, N., Grimminger, F., Grunig, E., Humbert, M., Jing, Z. C., et al. (2013). Riociguat for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 369 (4), 330–340. doi:10.1056/NEJMoa1209655
Ghofrani, H. A., Morrell, N. W., Hoeper, M. M., Olschewski, H., Peacock, A. J., Barst, R. J., et al. (2010). Imatinib in pulmonary arterial hypertension patients with inadequate response to established therapy. Am. J. Respir. Crit. Care Med. 182 (9), 1171–1177. doi:10.1164/rccm.201001-0123OC
Gibb, A. A., Huynh, A. T., Gaspar, R. B., Ploesch, T. L., Lombardi, A. A., Lorkiewicz, P. K., et al. (2022). Glutamine uptake and catabolism is required for myofibroblast formation and persistence. J. Mol. Cell. Cardiol. 172, 78–89. doi:10.1016/j.yjmcc.2022.08.002
Girgis, R. E., Mozammel, S., Champion, H. C., Li, D., Peng, X., Shimoda, L., et al. (2007). Regression of chronic hypoxic pulmonary hypertension by simvastatin. Am. J. Physiol. Lung Cell. Mol. Physiol. 292 (5), L1105–L1110. doi:10.1152/ajplung.00411.2006
Glinskii, V. A., Lopez, A., Lin, A., Del Rosario, P., Hershman, S., Hess, J., et al. (2021). Feasibility and validation testing of consumer wearables-based platform for remote monitoring of the six minute walk test in pulmonary arterial hypertension. American Thoracic Society.
Golan, T., Hammel, P., Reni, M., Van Cutsem, E., Macarulla, T., Hall, M. J., et al. (2019). Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N. Engl. J. Med. 381 (4), 317–327. doi:10.1056/NEJMoa1903387
Gomberg-Maitland, M., Maitland, M. L., Barst, R. J., Sugeng, L., Coslet, S., Perrino, T. J., et al. (2010). A dosing/cross-development study of the multikinase inhibitor sorafenib in patients with pulmonary arterial hypertension. Clin. Pharmacol. Ther. 87 (3), 303–310. doi:10.1038/clpt.2009.217
Goncharov, D. A., Goncharova, E. A., Tofovic, S. P., Hu, J., Baust, J. J., Pena, A. Z., et al. (2018). Metformin therapy for pulmonary hypertension associated with heart failure with preserved ejection fraction versus pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 198 (5), 681–684. doi:10.1164/rccm.201801-0022LE
Goncharov, D. A., Kudryashova, T. V., Ziai, H., Ihida-Stansbury, K., DeLisser, H., Krymskaya, V. P., et al. (2014). Mammalian target of rapamycin complex 2 (mTORC2) coordinates pulmonary artery smooth muscle cell metabolism, proliferation, and survival in pulmonary arterial hypertension. Circulation 129 (8), 864–874. doi:10.1161/CIRCULATIONAHA.113.004581
Greijer, A. E., and van der Wall, E. (2004). The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 57 (10), 1009–1014. doi:10.1136/jcp.2003.015032
Guignabert, C., Phan, C., Seferian, A., Huertas, A., Tu, L., Thuillet, R., et al. (2016). Dasatinib induces lung vascular toxicity and predisposes to pulmonary hypertension. J. Clin. Invest.. 126 (9), 3207–3218. doi:10.1172/JCI86249
Hansmann, G., Calvier, L., Risbano, M. G., and Chan, S. Y. (2020). Activation of the metabolic master regulator PPARγ: A potential PIOneering therapy for pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 62 (2), 143–156. doi:10.1165/rcmb.2019-0226PS
Hansmann, G., Wagner, R. A., Schellong, S., Perez, V. A., Urashima, T., Wang, L., et al. (2007). Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation 115 (10), 1275–1284. doi:10.1161/CIRCULATIONAHA.106.663120
Happe, C., Kurakula, K., Sun, X. Q., da Silva Goncalves Bos, D., Rol, N., Guignabert, C., et al. (2020). The BMP receptor 2 in pulmonary arterial hypertension: When and where the animal model matches the patient. Cells 9 (6), 1422. doi:10.3390/cells9061422
Hardziyenka, M., Surie, S., de Groot, J. R., de Bruin-Bon, H. A., Knops, R. E., Remmelink, M., et al. (2011). Right ventricular pacing improves haemodynamics in right ventricular failure from pressure overload: An open observational proof-of-principle study in patients with chronic thromboembolic pulmonary hypertension. Europace 13 (12), 1753–1759. doi:10.1093/europace/eur189
Hassoun, P. M., and Farber, H. W. (2021). Hit early and hit hard in pulmonary arterial hypertension? Not so fast. Am. J. Respir. Crit. Care Med. 204 (7), 755–756. doi:10.1164/rccm.202107-1570ED
Heerspink, H. J. L., Parving, H. H., Andress, D. L., Bakris, G., Correa-Rotter, R., Hou, F. F., et al. (2019). Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): A double-blind, randomised, placebo-controlled trial. Lancet 393 (10184), 1937–1947. doi:10.1016/S0140-6736(19)30772-X
Hemnes, A. R., and Brittain, E. L. (2018). Autonomic nervous system in pulmonary arterial hypertension: Time to rest and digest. Circulation 137 (9), 925–927. doi:10.1161/CIRCULATIONAHA.117.032355
Hemnes, A. R., Rathinasabapathy, A., Austin, E. A., Brittain, E. L., Carrier, E. J., Chen, X., et al. (2018). A potential therapeutic role for angiotensin-converting enzyme 2 in human pulmonary arterial hypertension. Eur. Respir. J. 51 (6), 1702638. doi:10.1183/13993003.02638-2017
Hernandez, A. V., Usmani, A., Rajamanickam, A., and Moheet, A. (2011). Thiazolidinediones and risk of heart failure in patients with or at high risk of type 2 diabetes mellitus: A meta-analysis and meta-regression analysis of placebo-controlled randomized clinical trials. Am. J. Cardiovasc. Drugs 11 (2), 115–128. doi:10.2165/11587580-000000000-00000
Hernandez-Sanchez, J., Harlow, L., Church, C., Gaine, S., Knightbridge, E., Bunclark, K., et al. (2018). Clinical trial protocol for transform-UK: A therapeutic open-label study of tocilizumab in the treatment of pulmonary arterial hypertension. Pulm. Circ. 8 (1), 2045893217735820. doi:10.1177/2045893217735820
Hobbs, A. J., Moyes, A. J., Baliga, R. S., Ghedia, D., Ochiel, R., Sylvestre, Y., et al. (2019). Neprilysin inhibition for pulmonary arterial hypertension: A randomized, double-blind, placebo-controlled, proof-of-concept trial. Br. J. Pharmacol. 176 (9), 1251–1267. doi:10.1111/bph.14621
Hoeper, M. M., Al-Hiti, H., Benza, R. L., Chang, S. A., Corris, P. A., Gibbs, J. S. R., et al. (2021). Switching to riociguat versus maintenance therapy with phosphodiesterase-5 inhibitors in patients with pulmonary arterial hypertension (REPLACE): A multicentre, open-label, randomised controlled trial. Lancet. Respir. Med. 9 (6), 573–584. doi:10.1016/S2213-2600(20)30532-4
Hoeper, M. M., Barst, R. J., Bourge, R. C., Feldman, J., Frost, A. E., Galie, N., et al. (2013). Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: Results of the randomized IMPRES study. Circulation 127 (10), 1128–1138. doi:10.1161/CIRCULATIONAHA.112.000765
Hoeper, M. M., Simonneau, G., Corris, P. A., Ghofrani, H. A., Klinger, J. R., Langleben, D., et al. (2017). Respite: Switching to riociguat in pulmonary arterial hypertension patients with inadequate response to phosphodiesterase-5 inhibitors. Eur. Respir. J. 50 (3), 1602425. doi:10.1183/13993003.02425-2016
Holmes, D., Corr, M., Thomas, G., Harbinson, M., Campbell, M., Spiers, P., et al. (2020). Protective effects of intermedin/adrenomedullin-2 in a cellular model of human pulmonary arterial hypertension. Peptides 126, 170267. doi:10.1016/j.peptides.2020.170267
Homma, N., Morio, Y., Takahashi, H., Yamamoto, A., Suzuki, T., Sato, K., et al. (2006). Genistein, a phytoestrogen, attenuates monocrotaline-induced pulmonary hypertension. Respiration. 73 (1), 105–112. doi:10.1159/000088946
Honda, J., Kimura, T., Sakai, S., Maruyama, H., Tajiri, K., Murakoshi, N., et al. (2018). The glucagon-like peptide-1 receptor agonist liraglutide improves hypoxia-induced pulmonary hypertension in mice partly via normalization of reduced ET(B) receptor expression. Physiol. Res. 67 (1), S175–S184. doi:10.33549/physiolres.933822
Houssaini, A., Abid, S., Mouraret, N., Wan, F., Rideau, D., Saker, M., et al. (2013). Rapamycin reverses pulmonary artery smooth muscle cell proliferation in pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 48 (5), 568–577. doi:10.1165/rcmb.2012-0429OC
Huang, X., Zou, L., Yu, X., Chen, M., Guo, R., Cai, H., et al. (2015). Salidroside attenuates chronic hypoxia-induced pulmonary hypertension via adenosine A2a receptor related mitochondria-dependent apoptosis pathway. J. Mol. Cell. Cardiol. 82, 153–166. doi:10.1016/j.yjmcc.2015.03.005
Humbert, M., Kovacs, G., Hoeper, M. M., Badagliacca, R., Berger, R. M. F., Brida, M., et al. (2022). 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur. Respir. J., 2200879. doi:10.1183/13993003.00879-2022
Humbert, M., McLaughlin, V., Gibbs, J. S. R., Gomberg-Maitland, M., Hoeper, M. M., Preston, I. R., et al. (2021). Sotatercept for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 384 (13), 1204–1215. doi:10.1056/NEJMoa2024277
Jasinska-Stroschein, M., Owczarek, J., Soltysiak, U., and Orszulak-Michalak, D. (2016). Rosuvastatin intensifies the beneficial effects of rho-kinase inhibitor in reversal of monocrotaline-induced pulmonary hypertension. Arch. Med. Sci. 12 (4), 898–905. doi:10.5114/aoms.2015.49740
Jones, S., Lumens, J., Sohaib, S. M. A., Finegold, J. A., Kanagaratnam, P., Tanner, M., et al. (2017). Cardiac resynchronization therapy: Mechanisms of action and scope for further improvement in cardiac function. Europace 19 (7), 1178–1186. doi:10.1093/europace/euw136
Joshi, S. R., Liu, J., Bloom, T., Karaca Atabay, E., Kuo, T. H., Lee, M., et al. (2022). Sotatercept analog suppresses inflammation to reverse experimental pulmonary arterial hypertension. Sci. Rep. 12 (1), 7803. doi:10.1038/s41598-022-11435-x
Kamimura, D., Uchino, K., Ishigami, T., Hall, M. E., and Umemura, S. (2016). Activation of peroxisome proliferator-activated receptor gamma prevents development of heart failure with preserved ejection fraction; inhibition of wnt-beta-catenin signaling as a possible mechanism. J. Cardiovasc. Pharmacol. 68 (2), 155–161. doi:10.1097/FJC.0000000000000397
Kandilci, H. B., Gumusel, B., and Lippton, H. (2008). Intermedin/adrenomedullin-2 (IMD/AM2) relaxes rat main pulmonary arterial rings via cGMP-dependent pathway: Role of nitric oxide and large conductance calcium-activated potassium channels (BK(Ca)). Peptides 29 (8), 1321–1328. doi:10.1016/j.peptides.2008.04.008
Kato, T., Nasu, T., Sonoda, H., Ito, K. M., Ikeda, M., and Ito, K. (2008). Evaluation of olmesartan medoxomil in the rat monocrotaline model of pulmonary hypertension. J. Cardiovasc. Pharmacol. 51 (1), 18–23. doi:10.1097/FJC.0b013e318159b01c
Katugampola, S. D., and Davenport, A. P. (2001). Thromboxane receptor density is increased in human cardiovascular disease with evidence for inhibition at therapeutic concentrations by the AT(1) receptor antagonist losartan. Br. J. Pharmacol. 134 (7), 1385–1392. doi:10.1038/sj.bjp.0704416
Kaumann, A. J., and Levy, F. O. (2006). 5-hydroxytryptamine receptors in the human cardiovascular system. Pharmacol. Ther. 111 (3), 674–706. doi:10.1016/j.pharmthera.2005.12.004
Kawut, S. M., Archer-Chicko, C. L., DeMichele, A., Fritz, J. S., Klinger, J. R., Ky, B., et al. (2017). Anastrozole in pulmonary arterial hypertension. A randomized, double-blind, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 195 (3), 360–368. doi:10.1164/rccm.201605-1024OC
Kawut, S. M., Bagiella, E., Lederer, D. J., Shimbo, D., Horn, E. M., Roberts, K. E., et al. (2011). Randomized clinical trial of aspirin and simvastatin for pulmonary arterial hypertension: ASA-STAT. Circulation 123 (25), 2985–2993. doi:10.1161/CIRCULATIONAHA.110.015693
Kawut, S. M., Pinder, D., Al-Naamani, N., McCormick, A., Palevsky, H. I., Fritz, J., et al. (2019). Fulvestrant for the treatment of pulmonary arterial hypertension. Ann. Am. Thorac. Soc. 16 (11), 1456–1459. doi:10.1513/AnnalsATS.201904-328RL
Kayano, H., Koba, S., Hirano, T., Matsui, T., Fukuoka, H., Tsuijita, H., et al. (2020). Dapagliflozin influences ventricular hemodynamics and exercise-induced pulmonary hypertension in type 2 diabetes patients- A randomized controlled trial. Circ. J. 84 (10), 1807–1817. doi:10.1253/circj.CJ-20-0341
Kernan, W. N., Viscoli, C. M., Furie, K. L., Young, L. H., Inzucchi, S. E., Gorman, M., et al. (2016). Pioglitazone after ischemic stroke or transient ischemic attack. N. Engl. J. Med. 374 (14), 1321–1331. doi:10.1056/NEJMoa1506930
Khural, J. S., Houston, B. A., Leary, P. J., Mathai, S. C., Kolb, T. M., Damico, R., et al. (2021). Right atrial pacing to improve acute hemodynamics in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 203 (4), 508–511. doi:10.1164/rccm.202006-2278LE
Kim, E. K., Lee, J. H., Oh, Y. M., Lee, Y. S., and Lee, S. D. (2010). Rosiglitazone attenuates hypoxia-induced pulmonary arterial hypertension in rats. Respirology 15 (4), 659–668. doi:10.1111/j.1440-1843.2010.01756.x
Kimura, G., Kataoka, M., Inami, T., Fukuda, K., Yoshino, H., and Satoh, T. (2017). Sorafenib as a potential strategy for refractory pulmonary arterial hypertension. Pulm. Pharmacol. Ther. 44, 46–49. doi:10.1016/j.pupt.2017.03.009
King, N. E., and Brittain, E. (2022). Emerging therapies: The potential roles SGLT2 inhibitors, GLP1 agonists, and ARNI therapy for ARNI pulmonary hypertension. Pulm. Circ. 12 (1), e12028. doi:10.1002/pul2.12028
Klinger, J. R., Elliott, C. G., Levine, D. J., Bossone, E., Duvall, L., Fagan, K., et al. (2019). Therapy for pulmonary arterial hypertension in adults: Update of the CHEST guideline and expert panel report. Chest 155 (3), 565–586. doi:10.1016/j.chest.2018.11.030
Lancon, J. P., Pillet, M., Gabrielle, F., Fayolle, J. L., and Tatou, E. (1994). Effects of atrial pacing on right ventricular contractility after coronary artery surgery. J. Cardiothorac. Vasc. Anesth. 8 (5), 536–540.
Lazarus, H. M., Denning, J., Wring, S., Palacios, M., Hoffman, S., Crizer, K., et al. (2022). A trial design to maximize knowledge of the effects of rodatristat ethyl in the treatment of pulmonary arterial hypertension (ELEVATE 2). Pulm. Circ. 12 (2), e12088. doi:10.1002/pul2.12088
Lee, H. C., Shiou, Y. L., Jhuo, S. J., Chang, C. Y., Liu, P. L., Jhuang, W. J., et al. (2019). The sodium-glucose co-transporter 2 inhibitor empagliflozin attenuates cardiac fibrosis and improves ventricular hemodynamics in hypertensive heart failure rats. Cardiovasc. Diabetol. 18 (1), 45. doi:10.1186/s12933-019-0849-6
Legchenko, E., Chouvarine, P., Borchert, P., Fernandez-Gonzalez, A., Snay, E., Meier, M., et al. (2018). PPARγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci. Transl. Med. 10 (438), eaao0303. doi:10.1126/scitranslmed.aao0303
Leier, C. V., Bambach, D., Nelson, S., Hermiller, J. B., Huss, P., Magorien, R. D., et al. (1983). Captopril in primary pulmonary hypertension. Circulation 67 (1), 155–161. doi:10.1161/01.cir.67.1.155
Leuchte, H. H., Baezner, C., Baumgartner, R. A., Bevec, D., Bacher, G., Neurohr, C., et al. (2008). Inhalation of vasoactive intestinal peptide in pulmonary hypertension. Eur. Respir. J. 32 (5), 1289–1294. doi:10.1183/09031936.00050008
Li, H., Lu, W., Cai, W. W., Wang, P. J., Zhang, N., Yu, C. P., et al. (2014). Telmisartan attenuates monocrotaline-induced pulmonary artery endothelial dysfunction through a PPAR gamma-dependent PI3K/Akt/eNOS pathway. Pulm. Pharmacol. Ther. 28 (1), 17–24. doi:10.1016/j.pupt.2013.11.003
Liu, Q., Song, J., Lu, D., Geng, J., Jiang, Z., Wang, K., et al. (2017). Effects of renal denervation on monocrotaline induced pulmonary remodeling. Oncotarget 8 (29), 46846–46855. doi:10.18632/oncotarget.15154
Low, A. T., Howard, L., Harrison, C., and Tulloh, R. M. (2015). Pulmonary arterial hypertension exacerbated by ruxolitinib. Haematologica 100 (6), e244–e245. doi:10.3324/haematol.2014.120816
Malenfant, S., Neyron, A. S., Paulin, R., Potus, F., Meloche, J., Provencher, S., et al. (2013). Signal transduction in the development of pulmonary arterial hypertension. Pulm. Circ. 3 (2), 278–293. doi:10.4103/2045-8932.114752
Maron, B. A., Waxman, A. B., Opotowsky, A. R., Gillies, H., Blair, C., Aghamohammadzadeh, R., et al. (2013). Effectiveness of spironolactone plus ambrisentan for treatment of pulmonary arterial hypertension (from the [ARIES] study 1 and 2 trials). Am. J. Cardiol. 112 (5), 720–725. doi:10.1016/j.amjcard.2013.04.051
Martens, P., Verbrugge, F. H., Bertrand, P. B., Verhaert, D., Vandervoort, P., Dupont, M., et al. (2018). Effect of cardiac resynchronization therapy on exercise-induced pulmonary hypertension and right ventricular-arterial coupling. Circ. Cardiovasc. Imaging 11 (9), e007813. doi:10.1161/CIRCIMAGING.118.007813
Martin, B., Vanderpool, R. R., Henry, B. L., Palma, J. B., Gabris, B., Lai, Y. C., et al. (2021). Relaxin inhibits ventricular arrhythmia and asystole in rats with pulmonary arterial hypertension. Front. Cardiovasc. Med. 8, 668222. doi:10.3389/fcvm.2021.668222
Masuda, H., Kalka, C., Takahashi, T., Yoshida, M., Wada, M., Kobori, M., et al. (2007). Estrogen-mediated endothelial progenitor cell biology and kinetics for physiological postnatal vasculogenesis. Circ. Res. 101 (6), 598–606. doi:10.1161/CIRCRESAHA.106.144006
McGoon, M. D., and Vlietstra, R. E. (1987). Acute hemodynamic response to the S2-serotonergic receptor antagonist, ketanserin, in patients with primary pulmonary hypertension. Int. J. Cardiol. 14 (3), 303–309. doi:10.1016/0167-5273(87)90200-2
McLaughlin, V. V., Channick, R., De Marco, T., Farber, H. W., Gaine, S., Galie, N., et al. (2020). Results of an expert consensus survey on the treatment of pulmonary arterial hypertension with oral prostacyclin pathway agents. Chest 157 (4), 955–965. doi:10.1016/j.chest.2019.10.043
Medarov, B. I., and Judson, M. A. (2015). The role of calcium channel blockers for the treatment of pulmonary arterial hypertension: How much do we actually know and how could they be positioned today? Respir. Med. 109 (5), 557–564. doi:10.1016/j.rmed.2015.01.004
Meloche, J., Pflieger, A., Vaillancourt, M., Paulin, R., Potus, F., Zervopoulos, S., et al. (2014). Role for DNA damage signaling in pulmonary arterial hypertension. Circulation 129 (7), 786–797. doi:10.1161/CIRCULATIONAHA.113.006167
Meloche, J., Potus, F., Vaillancourt, M., Bourgeois, A., Johnson, I., Deschamps, L., et al. (2015). Bromodomain-containing protein 4: The epigenetic origin of pulmonary arterial hypertension. Circ. Res. 117 (6), 525–535. doi:10.1161/CIRCRESAHA.115.307004
Metra, M., Teerlink, J. R., Cotter, G., Davison, B. A., Felker, G. M., Filippatos, G., et al. (2019). Effects of serelaxin in patients with acute heart failure. N. Engl. J. Med. 381 (8), 716–726. doi:10.1056/NEJMoa1801291
Michelakis, E. D., Gurtu, V., Webster, L., Barnes, G., Watson, G., Howard, L., et al. (2017). Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci. Transl. Med. 9 (413), eaao4583. doi:10.1126/scitranslmed.aao4583
Michelakis, E. D., McMurtry, M. S., Wu, X. C., Dyck, J. R., Moudgil, R., Hopkins, T. A., et al. (2002). Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: Role of increased expression and activity of voltage-gated potassium channels. Circulation 105 (2), 244–250. doi:10.1161/hc0202.101974
Michelakis, E. D., Webster, L., and Mackey, J. R. (2008). Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br. J. Cancer 99 (7), 989–994. doi:10.1038/sj.bjc.6604554
Mitchell, R., Frederick, N. E., Holzman, E. R., Agobe, F., Allaway, H. C. M., and Bagher, P. (2021). Ifetroban reduces coronary artery dysfunction in a mouse model of Duchenne muscular dystrophy. Am. J. Physiol. Heart Circ. Physiol. 321 (1), H52–H58. doi:10.1152/ajpheart.00180.2021
Mizuno, S., Farkas, L., Al Husseini, A., Farkas, D., Gomez-Arroyo, J., Kraskauskas, D., et al. (2012). Severe pulmonary arterial hypertension induced by SU5416 and ovalbumin immunization. Am. J. Respir. Cell Mol. Biol. 47 (5), 679–687. doi:10.1165/rcmb.2012-0077OC
Moore, K., Colombo, N., Scambia, G., Kim, B. G., Oaknin, A., Friedlander, M., et al. (2018). Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N. Engl. J. Med. 379 (26), 2495–2505. doi:10.1056/NEJMoa1810858
Moreno-Vinasco, L., and Garcia, J. G. (2010). Receptor tyrosine kinase inhibitors in rodent pulmonary hypertension. Adv. Exp. Med. Biol. 661, 419–434. doi:10.1007/978-1-60761-500-2_27
Morrell, N. W., Higham, M. A., Phillips, P. G., Shakur, B. H., Robinson, P. J., and Beddoes, R. J. (2005). Pilot study of losartan for pulmonary hypertension in chronic obstructive pulmonary disease. Respir. Res. 6, 88. doi:10.1186/1465-9921-6-88
Morrell, N. W. (2006). Pulmonary hypertension due to BMPR2 mutation: A new paradigm for tissue remodeling? Proc. Am. Thorac. Soc. 3 (8), 680–686. doi:10.1513/pats.200605-118SF
Mulkareddy, V., and Simon, M. A. (2020). Metformin in pulmonary hypertension in left heart disease. Front. Med. 7, 425. doi:10.3389/fmed.2020.00425
Na, S., Kim, O. S., Ryoo, S., Kweon, T. D., Choi, Y. S., Shim, H. S., et al. (2014). Cervical ganglion block attenuates the progression of pulmonary hypertension via nitric oxide and arginase pathways. Hypertension 63 (2), 309–315. doi:10.1161/HYPERTENSIONAHA.113.01979
Nagaoka, T., Fagan, K. A., Gebb, S. A., Morris, K. G., Suzuki, T., Shimokawa, H., et al. (2005). Inhaled Rho kinase inhibitors are potent and selective vasodilators in rat pulmonary hypertension. Am. J. Respir. Crit. Care Med. 171 (5), 494–499. doi:10.1164/rccm.200405-637OC
Nagaya, N., and Kangawa, K. (2004). Adrenomedullin in the treatment of pulmonary hypertension. Peptides 25 (11), 2013–2018. doi:10.1016/j.peptides.2004.07.007
Nassif, M. E., Qintar, M., Windsor, S. L., Jermyn, R., Shavelle, D. M., Tang, F., et al. (2021). Empagliflozin effects on pulmonary artery pressure in patients with heart failure: Results from the EMBRACE-HF trial. Circulation 143 (17), 1673–1686. doi:10.1161/CIRCULATIONAHA.120.052503
Negi, V., Yang, J., Speyer, G., Pulgarin, A., Handen, A., Zhao, J., et al. (2021). Computational repurposing of therapeutic small molecules from cancer to pulmonary hypertension. Sci. Adv. 7 (43), eabh3794. doi:10.1126/sciadv.abh3794
Nicholls, S. J., Gordon, A., Johansson, J., Wolski, K., Ballantyne, C. M., Kastelein, J. J., et al. (2011). Efficacy and safety of a novel oral inducer of apolipoprotein a-I synthesis in statin-treated patients with stable coronary artery disease a randomized controlled trial. J. Am. Coll. Cardiol. 57 (9), 1111–1119. doi:10.1016/j.jacc.2010.11.015
Nicholls, S. J., Puri, R., Wolski, K., Ballantyne, C. M., Barter, P. J., Brewer, H. B., et al. (2016). Effect of the BET protein inhibitor, RVX-208, on progression of coronary atherosclerosis: Results of the phase 2b, randomized, double-blind, multicenter, ASSURE trial. Am. J. Cardiovasc. Drugs 16 (1), 55–65. doi:10.1007/s40256-015-0146-z
Nicholls, S. J., Schwartz, G. G., Buhr, K. A., Ginsberg, H. N., Johansson, J. O., Kalantar-Zadeh, K., et al. (2021). Apabetalone and hospitalization for heart failure in patients following an acute coronary syndrome: A prespecified analysis of the BETonMACE study. Cardiovasc. Diabetol. 20 (1), 13. doi:10.1186/s12933-020-01199-x
Nonaka-Sarukawa, M., Yamamoto, K., Aoki, H., Takano, H., Katsuki, T., Ikeda, U., et al. (2003). Increased urinary 15-F2t-isoprostane concentrations in patients with non-ischaemic congestive heart failure: A marker of oxidative stress. Heart 89 (8), 871–874. doi:10.1136/heart.89.8.871
Orte, C., Polak, J. M., Haworth, S. G., Yacoub, M. H., and Morrell, N. W. (2000). Expression of pulmonary vascular angiotensin-converting enzyme in primary and secondary plexiform pulmonary hypertension. J. Pathol. 192 (3), 379–384. doi:10.1002/1096-9896(2000)9999:9999<::AID-PATH715>3.0.CO;2-Q
Oudiz, R. J. M., Chin, M., Feldman, J., Goldsberry, A., McConnell, J. W., McCullough, P., et al. (2017). Results of interim analysis of the efficacy and safety of bardoxolone. Washington, DC): American Thoracic Society.Methyl in patients with pulmonary arterial hypertension associated with connective tissue disease (CTD) (the LARIAT study)
Oudiz, R. M., Chin, M., Feldman, J., Goldsberry, A., McConnell, J., McCullough, P., et al. (2015). Initial data report from "LARIAT": A phase 2 study of bardoxolone methyl in PAH patients on stable background therapy". Montreal, QC: Chest.
Packer, M., Anker, S. D., Butler, J., Filippatos, G., Pocock, S. J., Carson, P., et al. (2020). Cardiovascular and renal outcomes with empagliflozin in heart failure. N. Engl. J. Med. 383 (15), 1413–1424. doi:10.1056/NEJMoa2022190
Paddenberg, R., Stieger, P., von Lilien, A. L., Faulhammer, P., Goldenberg, A., Tillmanns, H. H., et al. (2007). Rapamycin attenuates hypoxia-induced pulmonary vascular remodeling and right ventricular hypertrophy in mice. Respir. Res. 8, 15. doi:10.1186/1465-9921-8-15
Perros, F., de Man, F. S., Bogaard, H. J., Antigny, F., Simonneau, G., Bonnet, S., et al. (2017). Use of beta-blockers in pulmonary hypertension. Circ. Heart Fail. 10 (4), e003703. doi:10.1161/CIRCHEARTFAILURE.116.003703
Petkov, V., Mosgoeller, W., Ziesche, R., Raderer, M., Stiebellehner, L., Vonbank, K., et al. (2003). Vasoactive intestinal peptide as a new drug for treatment of primary pulmonary hypertension. J. Clin. Invest.. 111 (9), 1339–1346. doi:10.1172/JCI17500
Pintalhao, M., Castro-Chaves, P., Vasques-Novoa, F., Goncalves, F., Mendonca, L., Fontes-Carvalho, R., et al. (2017). Relaxin serum levels in acute heart failure are associated with pulmonary hypertension and right heart overload. Eur. J. Heart Fail. 19 (2), 218–225. doi:10.1002/ejhf.611
Plaunt, A. J., Islam, S., Macaluso, T., Gauani, H., Baker, T., Chun, D., et al. (2021). Development and characterization of treprostinil palmitil inhalation aerosol for the investigational treatment of pulmonary arterial hypertension. Int. J. Mol. Sci. 22 (2), 548. doi:10.3390/ijms22020548
Preston, I. S., Patel, J., Sood, N., Torres, F., Cadaret, L., Adams, J., et al. (2018). Ralinepag reduces pulmonary vascular resistance (PVR) in a phase 2 study confirming preclinical findings on prostacyclin (IP) receptors in human tissues. Chest 154, 990A–991A. doi:10.1016/j.chest.2018.08.900
Price, L. C., Wort, S. J., Perros, F., Dorfmuller, P., Huertas, A., Montani, D., et al. (2012). Inflammation in pulmonary arterial hypertension. Chest 141 (1), 210–221. doi:10.1378/chest.11-0793
Prins, K. W., Archer, S. L., Pritzker, M., Rose, L., Weir, E. K., Sharma, A., et al. (2018). Interleukin-6 is independently associated with right ventricular function in pulmonary arterial hypertension. J. Heart Lung Transpl. 37 (3), 376–384. doi:10.1016/j.healun.2017.08.011
Provencher, S., Herve, P., Jais, X., Lebrec, D., Humbert, M., Simonneau, G., et al. (2006). Deleterious effects of beta-blockers on exercise capacity and hemodynamics in patients with portopulmonary hypertension. Gastroenterology 130 (1), 120–126. doi:10.1053/j.gastro.2005.10.013
Provencher, S., Potus, F., Blais-Lecours, P., Bernard, S., Martineau, S., Breuils-Bonnet, S., et al. (2022). BET protein inhibition for pulmonary arterial hypertension: A pilot clinical trial. Am. J. Respir. Crit. Care Med. 205 (11), 1357–1360. doi:10.1164/rccm.202109-2182LE
Pulido, T., Adzerikho, I., Channick, R. N., Delcroix, M., Galie, N., Ghofrani, H. A., et al. (2013). Macitentan and morbidity and mortality in pulmonary arterial hypertension. N. Engl. J. Med. 369 (9), 809–818. doi:10.1056/NEJMoa1213917
Rasmussen, J. T., Thenappan, T., Benditt, D. G., Weir, E. K., and Pritzker, M. R. (2014). Is cardiac resynchronization therapy for right ventricular failure in pulmonary arterial hypertension of benefit? Pulm. Circ. 4 (4), 552–559. doi:10.1086/678470
Ray, K. K., Nicholls, S. J., Buhr, K. A., Ginsberg, H. N., Johansson, J. O., Kalantar-Zadeh, K., et al. (2020). Effect of apabetalone added to standard therapy on major adverse cardiovascular events in patients with recent acute coronary syndrome and type 2 diabetes: A randomized clinical trial. JAMA 323 (16), 1565–1573. doi:10.1001/jama.2020.3308
Rondelet, B., Kerbaul, F., Van Beneden, R., Hubloue, I., Huez, S., Fesler, P., et al. (2005). Prevention of pulmonary vascular remodeling and of decreased BMPR-2 expression by losartan therapy in shunt-induced pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 289 (6), H2319–H2324. doi:10.1152/ajpheart.00518.2005
Rosenkranz, S., Feldman, J., McLaughlin, V. V., Rischard, F., Lange, T. J., White, R. J., et al. (2022). Selonsertib in adults with pulmonary arterial hypertension (ARROW): A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. Respir. Med. 10 (1), 35–46. doi:10.1016/S2213-2600(21)00032-1
Rothman, A. M. K., Vachiery, J. L., Howard, L. S., Mikhail, G. W., Lang, I. M., Jonas, M., et al. (2020). Intravascular ultrasound pulmonary artery denervation to treat pulmonary arterial hypertension (TROPHY1): Multicenter, early feasibility study. JACC. Cardiovasc. Interv. 13 (8), 989–999. doi:10.1016/j.jcin.2019.12.027
Rubin, L. J., Galie, N., Grimminger, F., Grunig, E., Humbert, M., Jing, Z. C., et al. (2015). Riociguat for the treatment of pulmonary arterial hypertension: A long-term extension study (PATENT-2). Eur. Respir. J. 45 (5), 1303–1313. doi:10.1183/09031936.00090614
Safdar, Z., Frost, A., Basant, A., Deswal, A., O'Brian Smith, E., and Entman, M. (2020). Spironolactone in pulmonary arterial hypertension: Results of a cross-over study. Pulm. Circ. 10 (2), 2045894019898030. doi:10.1177/2045894019898030
Said, S. I., Hamidi, S. A., Dickman, K. G., Szema, A. M., Lyubsky, S., Lin, R. Z., et al. (2007). Moderate pulmonary arterial hypertension in male mice lacking the vasoactive intestinal peptide gene. Circulation 115 (10), 1260–1268. doi:10.1161/CIRCULATIONAHA.106.681718
Said, S. I. (2012). Vasoactive intestinal peptide in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 185 (7), 786. author reply 786. doi:10.1164/ajrccm.185.7.786
Schermuly, R. T., Dony, E., Ghofrani, H. A., Pullamsetti, S., Savai, R., Roth, M., et al. (2005). Reversal of experimental pulmonary hypertension by PDGF inhibition. J. Clin. Invest.. 115 (10), 2811–2821. doi:10.1172/JCI24838
Shenoy, V., Kwon, K. C., Rathinasabapathy, A., Lin, S., Jin, G., Song, C., et al. (2014). Oral delivery of Angiotensin-converting enzyme 2 and Angiotensin-(1-7) bioencapsulated in plant cells attenuates pulmonary hypertension. Hypertension 64 (6), 1248–1259. doi:10.1161/HYPERTENSIONAHA.114.03871
Shi, Y., Gu, C., Zhao, T., Jia, Y., Bao, C., Luo, A., et al. (2021). Combination therapy with rapamycin and low dose imatinib in pulmonary hypertension. Front. Pharmacol. 12, 758763. doi:10.3389/fphar.2021.758763
Shishikura, D., Kataoka, Y., Honda, S., Takata, K., Kim, S. W., Andrews, J., et al. (2019). The effect of bromodomain and extra-terminal inhibitor apabetalone on attenuated coronary atherosclerotic plaque: Insights from the ASSURE trial. Am. J. Cardiovasc. Drugs 19 (1), 49–57. doi:10.1007/s40256-018-0298-8
Simon, M. G.-M., Oudiz, R. J., Machado, R., Rischard, F., Elinoff, J. M., Grigorian, B., et al. (2019). Severe pulmonary arterial hypertension treated with ABI-009, nab-sirolimus, an mTOR inhibitor. J. Heart Lung Transpl. 38 (4), S487. doi:10.1016/j.healun.2019.01.1238
Simonneau, G., Montani, D., Celermajer, D. S., Denton, C. P., Gatzoulis, M. A., Krowka, M., et al. (2019). Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 53 (1), 1801913. doi:10.1183/13993003.01913-2018
Simpson, C. E., Chen, J. Y., Damico, R. L., Hassoun, P. M., Martin, L. J., Yang, J., et al. (2020). Cellular sources of interleukin-6 and associations with clinical phenotypes and outcomes in pulmonary arterial hypertension. Eur. Respir. J. 55 (4), 1901761. doi:10.1183/13993003.01761-2019
Sitbon, O., Channick, R., Chin, K. M., Frey, A., Gaine, S., Galie, N., et al. (2015). Selexipag for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 373 (26), 2522–2533. doi:10.1056/NEJMoa1503184
Sitbon, O., Gomberg-Maitland, M., Granton, J., Lewis, M. I., Mathai, S. C., Rainisio, M., et al. (2019). Clinical trial design and new therapies for pulmonary arterial hypertension. Eur. Respir. J. 53 (1), 1801908. doi:10.1183/13993003.01908-2018
Sitbon, O., Jais, X., Savale, L., Cottin, V., Bergot, E., Macari, E. A., et al. (2014). Upfront triple combination therapy in pulmonary arterial hypertension: A pilot study. Eur. Respir. J. 43 (6), 1691–1697. doi:10.1183/09031936.00116313
Sodimu, A., Bartolome, S., Igenoza, O. P., and Chin, K. M. (2020). Hemodynamic effects of fluoxetine in pulmonary arterial hypertension: An open label pilot study. Pulm. Circ. 10 (4), 2045894020971954. doi:10.1177/2045894020971954
Soon, E., Holmes, A. M., Treacy, C. M., Doughty, N. J., Southgate, L., Machado, R. D., et al. (2010). Elevated levels of inflammatory cytokines predict survival in idiopathic and familial pulmonary arterial hypertension. Circulation 122 (9), 920–927. doi:10.1161/CIRCULATIONAHA.109.933762
Spiekerkoetter, E., Sung, Y. K., Sudheendra, D., Bill, M., Aldred, M. A., van de Veerdonk, M. C., et al. (2015). Low-dose FK506 (tacrolimus) in end-stage pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 192 (2), 254–257. doi:10.1164/rccm.201411-2061LE
Spiekerkoetter, E., Sung, Y. K., Sudheendra, D., Scott, V., Del Rosario, P., Bill, M., et al. (2017). Randomised placebo-controlled safety and tolerability trial of FK506 (tacrolimus) for pulmonary arterial hypertension. Eur. Respir. J. 50 (3), 1602449. doi:10.1183/13993003.02449-2016
Spiekerkoetter, E., Tian, X., Cai, J., Hopper, R. K., Sudheendra, D., Li, C. G., et al. (2013). FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J. Clin. Invest.. 123 (8), 3600–3613. doi:10.1172/JCI65592
Spikes, L. A., Bajwa, A. A., Burger, C. D., Desai, S. V., Eggert, M. S., El-Kersh, K. A., et al. (2022). Breeze: Open-label clinical study to evaluate the safety and tolerability of treprostinil inhalation powder as Tyvaso DPI in patients with pulmonary arterial hypertension. Pulm. Circ. 12 (2), e12063. doi:10.1002/pul2.12063
Stacher, E., Graham, B. B., Hunt, J. M., Gandjeva, A., Groshong, S. D., McLaughlin, V. V., et al. (2012). Modern age pathology of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 186 (3), 261–272. doi:10.1164/rccm.201201-0164OC
Steiner, M. K., Syrkina, O. L., Kolliputi, N., Mark, E. J., Hales, C. A., and Waxman, A. B. (2009). Interleukin-6 overexpression induces pulmonary hypertension. Circ. Res. 104 (2), 236–244. 228p following 244. doi:10.1161/CIRCRESAHA.108.182014
Stollfuss, B., Richter, M., Dromann, D., Klose, H., Schwaiblmair, M., Gruenig, E., et al. (2021). Digital tracking of physical activity, heart rate, and inhalation behavior in patients with pulmonary arterial hypertension treated with inhaled iloprost: Observational study (VENTASTEP). J. Med. Internet Res. 23 (10), e25163. doi:10.2196/25163
Tamura, Y., Phan, C., Tu, L., Le Hiress, M., Thuillet, R., Jutant, E. M., et al. (2018). Ectopic upregulation of membrane-bound IL6R drives vascular remodeling in pulmonary arterial hypertension. J. Clin. Invest.. 128 (5), 1956–1970. doi:10.1172/JCI96462
Teerlink, J. R., Cotter, G., Davison, B. A., Felker, G. M., Filippatos, G., Greenberg, B. H., et al. (2013). Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): A randomised, placebo-controlled trial. Lancet 381 (9860), 29–39. doi:10.1016/S0140-6736(12)61855-8
Telli, G., Erac, Y., Tel, B. C., and Gumusel, B. (2018). Mechanism of adrenomedullin 2/intermedin mediated vasorelaxation in rat main pulmonary artery. Peptides 103, 65–71. doi:10.1016/j.peptides.2018.03.015
Torres, F., Farber, H., Ristic, A., McLaughlin, V., Adams, J., Zhang, J., et al. (2019). Efficacy and safety of ralinepag, a novel oral IP agonist, in PAH patients on mono or dual background therapy: Results from a phase 2 randomised, parallel group, placebo-controlled trial. Eur. Respir. J. 54 (4), 1901030. doi:10.1183/13993003.01030-2019
Toshner, M., Church, C., Harbaum, L., Rhodes, C., Villar Moreschi, S. S., Liley, J., et al. (2022). Mendelian randomisation and experimental medicine approaches to interleukin-6 as a drug target in pulmonary arterial hypertension. Eur. Respir. J. 59 (3), 2002463. doi:10.1183/13993003.02463-2020
Toshner, M., and Rothman, A. (2020). IL-6 in pulmonary hypertension: Why novel is not always best. Eur. Respir. J. 55 (4), 2000314. doi:10.1183/13993003.00314-2020
Tozzi, C. A., Poiani, G. J., McHugh, N. A., Shakarjian, M. P., Grove, B. H., Samuel, C. S., et al. (2005). Recombinant human relaxin reduces hypoxic pulmonary hypertension in the rat. Pulm. Pharmacol. Ther. 18 (5), 346–353. doi:10.1016/j.pupt.2005.01.003
Trammell, A. W., Talati, M., Blackwell, T. R., Fortune, N. L., Niswender, K. D., Fessel, J. P., et al. (2017). Pulmonary vascular effect of insulin in a rodent model of pulmonary arterial hypertension. Pulm. Circ. 7 (3), 624–634. doi:10.1086/689908
Trankle, C. R., Canada, J. M., Kadariya, D., Markley, R., De Chazal, H. M., Pinson, J., et al. (2019). IL-1 blockade reduces inflammation in pulmonary arterial hypertension and right ventricular failure: A single-arm, open-label, phase IB/II pilot study. Am. J. Respir. Crit. Care Med. 199 (3), 381–384. doi:10.1164/rccm.201809-1631LE
Tutt, A. N. J., Garber, J. E., Kaufman, B., Viale, G., Fumagalli, D., Rastogi, P., et al. (2021). Adjuvant olaparib for patients with BRCA1- or BRCA2-mutated breast cancer. N. Engl. J. Med. 384 (25), 2394–2405. doi:10.1056/NEJMoa2105215
Umar, S., Iorga, A., Matori, H., Nadadur, R. D., Li, J., Maltese, F., et al. (2011). Estrogen rescues preexisting severe pulmonary hypertension in rats. Am. J. Respir. Crit. Care Med. 184 (6), 715–723. doi:10.1164/rccm.201101-0078OC
Umar, S., Rabinovitch, M., and Eghbali, M. (2012). Estrogen paradox in pulmonary hypertension: Current controversies and future perspectives. Am. J. Respir. Crit. Care Med. 186 (2), 125–131. doi:10.1164/rccm.201201-0058PP
Vaillancourt, M., Chia, P., Sarji, S., Nguyen, J., Hoftman, N., Ruffenach, G., et al. (2017). Autonomic nervous system involvement in pulmonary arterial hypertension. Respir. Res. 18 (1), 201. doi:10.1186/s12931-017-0679-6
Valuparampil Varghese, M., James, J., Eccles, C. A., Niihori, M., Rafikova, O., and Rafikov, R. (2020). Inhibition of anaplerosis attenuated vascular proliferation in pulmonary arterial hypertension. J. Clin. Med. 9 (2), 443. doi:10.3390/jcm9020443
van der Bruggen, C. E. E., Tedford, R. J., Handoko, M. L., van der Velden, J., and de Man, F. S. (2017). RV pressure overload: From hypertrophy to failure. Cardiovasc. Res. 113 (12), 1423–1432. doi:10.1093/cvr/cvx145
van der Feen, D. E., Bossers, G. P. L., Hagdorn, Q. A. J., Moonen, J. R., Kurakula, K., Szulcek, R., et al. (2020). Cellular senescence impairs the reversibility of pulmonary arterial hypertension. Sci. Transl. Med. 12 (554), eaaw4974. doi:10.1126/scitranslmed.aaw4974
Ventetuolo, C. E., Baird, G. L., Barr, R. G., Bluemke, D. A., Fritz, J. S., Hill, N. S., et al. (2016). Higher estradiol and lower dehydroepiandrosterone-sulfate levels are associated with pulmonary arterial hypertension in men. Am. J. Respir. Crit. Care Med. 193 (10), 1168–1175. doi:10.1164/rccm.201509-1785OC
Voelkel, N. F., Tuder, R. M., Bridges, J., and Arend, W. P. (1994). Interleukin-1 receptor antagonist treatment reduces pulmonary hypertension generated in rats by monocrotaline. Am. J. Respir. Cell Mol. Biol. 11 (6), 664–675. doi:10.1165/ajrcmb.11.6.7946395
Vrouchos, G. T., Kiulpalis, A., Trullakis, G. A., Stasinos, P. G., Ellinkakis, S. G., Koumatzias, N. C., et al. (1997). High-rate cardiac pacing increases blood pressure and decreases right atrial pressure in patients with hemodynamic significant acute right ventricular myocardial infarction and bradyarrhythmia. Clin. Cardiol. 20 (1), 41–46. doi:10.1002/clc.4960200110
Walsh, T. P., Baird, G. L., Atalay, M. K., Agarwal, S., Arcuri, D., Klinger, J. R., et al. (2021). Experimental design of the effects of dehydroepiandrosterone in pulmonary hypertension (EDIPHY) trial. Pulm. Circ. 11 (2), 2045894021989554. doi:10.1177/2045894021989554
Wang, S., Yan, Y., Xu, W. J., Gong, S. G., Zhong, X. J., An, Q. Y., et al. (2022). The role of glutamine and glutaminase in pulmonary hypertension. Front. Cardiovasc. Med. 9, 838657. doi:10.3389/fcvm.2022.838657
Wei, H., Zhang, D., Liu, L., Xia, W., and Li, F. (2019). Rho signaling pathway enhances proliferation of PASMCs by suppressing nuclear translocation of Smad1 in PAH. Exp. Ther. Med. 17 (1), 71–78. doi:10.3892/etm.2018.6942
Weiss, A., Neubauer, M. C., Yerabolu, D., Kojonazarov, B., Schlueter, B. C., Neubert, L., et al. (2019). Targeting cyclin-dependent kinases for the treatment of pulmonary arterial hypertension. Nat. Commun. 10 (1), 2204. doi:10.1038/s41467-019-10135-x
West, J. D., Voss, B. M., Pavliv, L., de Caestecker, M., Hemnes, A. R., and Carrier, E. J. (2016). Antagonism of the thromboxane-prostanoid receptor is cardioprotective against right ventricular pressure overload. Pulm. Circ. 6 (2), 211–223. doi:10.1086/686140
Yamamura, A., Nayeem, M. J., and Sato, M. (2021). The Rho kinase 2 (ROCK2)-specific inhibitor KD025 ameliorates the development of pulmonary arterial hypertension. Biochem. Biophys. Res. Commun. 534, 795–801. doi:10.1016/j.bbrc.2020.10.106
Yamazato, Y., Ferreira, A. J., Hong, K. H., Sriramula, S., Francis, J., Yamazato, M., et al. (2009). Prevention of pulmonary hypertension by Angiotensin-converting enzyme 2 gene transfer. Hypertension 54 (2), 365–371. doi:10.1161/HYPERTENSIONAHA.108.125468
Yerabolu, D., Weiss, A., Kojonazarov, B., Boehm, M., Schlueter, B. C., Ruppert, C., et al. (2021). Targeting jak-stat signaling in experimental pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 64 (1), 100–114. doi:10.1165/rcmb.2019-0431OC
Yoshida, K., Saku, K., Kamada, K., Abe, K., Tanaka-Ishikawa, M., Tohyama, T., et al. (2018). Electrical vagal nerve stimulation ameliorates pulmonary vascular remodeling and improves survival in rats with severe pulmonary arterial hypertension. JACC. Basic Transl. Sci. 3 (5), 657–671. doi:10.1016/j.jacbts.2018.07.007
Yoshida, T., Matsuura, K., Goya, S., Ma, D., Shimada, K., Kitpipatkun, P., et al. (2020). Metformin prevents the development of monocrotaline-induced pulmonary hypertension by decreasing serum levels of big endothelin-1. Exp. Ther. Med. 20 (6), 149. doi:10.3892/etm.2020.9278
Yoshihara, F., Nishikimi, T., Horio, T., Yutani, C., Takishita, S., Matsuo, H., et al. (1998). Chronic infusion of adrenomedullin reduces pulmonary hypertension and lessens right ventricular hypertrophy in rats administered monocrotaline. Eur. J. Pharmacol. 355 (1), 33–39. doi:10.1016/s0014-2999(98)00475-0
Zamanian, R. T., Badesch, D., Chung, L., Domsic, R. T., Medsger, T., Pinckney, A., et al. (2021). Safety and efficacy of B-cell depletion with rituximab for the treatment of systemic sclerosis-associated pulmonary arterial hypertension: A multicenter, double-blind, randomized, placebo-controlled trial. Am. J. Respir. Crit. Care Med. 204 (2), 209–221. doi:10.1164/rccm.202009-3481OC
Zamanian, R. T., Hansmann, G., Snook, S., Lilienfeld, D., Rappaport, K. M., Reaven, G. M., et al. (2009). Insulin resistance in pulmonary arterial hypertension. Eur. Respir. J. 33 (2), 318–324. doi:10.1183/09031936.00000508
Zeng, W. J., Xiong, C. M., Zhao, L., Shan, G. L., Liu, Z. H., Xue, F., et al. (2012). Atorvastatin in pulmonary arterial hypertension (APATH) study. Eur. Respir. J. 40 (1), 67–74. doi:10.1183/09031936.00149011
Zhang, C., Wang, X., Zhang, H., Yao, C., Pan, H., Guo, Y., et al. (2019a). Therapeutic monoclonal antibody antagonizing endothelin receptor A for pulmonary arterial hypertension. J. Pharmacol. Exp. Ther. 370 (1), 54–61. doi:10.1124/jpet.118.252700
Zhang, H., Zhang, J., Chen, M., Xie, D. J., Kan, J., Yu, W., et al. (2019b). Pulmonary artery denervation significantly increases 6-min walk distance for patients with combined pre- and post-capillary pulmonary hypertension associated with left heart failure: The PADN-5 study. JACC. Cardiovasc. Interv. 12 (3), 274–284. doi:10.1016/j.jcin.2018.09.021
Zhang, J., Dong, J., Martin, M., He, M., Gongol, B., Marin, T. L., et al. (2018). AMP-Activated protein kinase phosphorylation of angiotensin-converting enzyme 2 in endothelium mitigates pulmonary hypertension. Am. J. Respir. Crit. Care Med. 198 (4), 509–520. doi:10.1164/rccm.201712-2570OC
5-HT 5-hydroxytryptamine (serotonin)
6MWD 6-min walk distance
AC adenylate cyclase
ACE angiotensin converting enzyme
AM adrenomedullin receptor
ASK1 apoptosis signal-regulating kinase 1
BET bromodomain and extraterminal domain
BMPR2 bone morphogenetic protein receptor 2
BRD4 bromodomain-containing protein 4
cAMP cyclic adenosine monophosphate
cGMP cyclic guanosine monophosphate
CGRP calcitonin gene-related peptide
DHEA dehydroepiandrosterone
ERAs endothelin receptor antagonists
ETA & ETB endothelin receptors A & B
HIF hypoxia-inducible factor
IL interleukin
IP prostacyclin receptor
mPAP mean pulmonary arterial pressure
mTOR mammalian target of rapamycin
NF-κB nuclear factor kappa light chain enhancer of activated B cells
NO nitric oxide
Nrf2 nuclear factor erythroid 2-related factor 2
PA pulmonary artery
PAH pulmonary arterial hypertension
PARP poly (ADP-ribose) polymerase
PASMCs pulmonary arterial smooth muscle cells
PAWP pulmonary arterial wedge pressure
PDE5 phosphodiesterase 5
PDH pyruvate dehydrogenase
PDGFR platelet-derived growth factor receptor
PDK pyruvate dehydrogenase kinase
PGI2 prostaglandin I2
PH pulmonary hypertension
PPAR-γ peroxisome proliferator activated receptor gamma
PVR pulmonary vascular resistance
ROCK Rho-associated protein kinase
RV right ventricle
RXFP1 relaxin family peptide receptor 1
sGC soluble guanylate cyclase
TAZ transcriptional coactivator with PDZ-binding motif
TGF-βR TGF-β receptor
TP thromboxane receptor
TPH1 tryptophan hydroxylase 1
VPAC vasoactive intestinal polypeptide receptor
VIP vasoactive intestinal peptide
YAP1 Yes-associated protein 1
Keywords: pulmonary hypertension, pulmonary arterial hypertension, preclinical studies, novel therapies, emerging therapies
Citation: Cassady SJ, Soldin D and Ramani GV (2022) Novel and emerging therapies in pulmonary arterial hypertension. Front. Drug. Discov. 2:1022971. doi: 10.3389/fddsv.2022.1022971
Received: 19 August 2022; Accepted: 23 November 2022;
Published: 15 December 2022.
Edited by:
Charles C. Hong, University of Maryland, United StatesReviewed by:
Stephen Y. Chan, University of Pittsburgh Medical Center, United StatesCopyright © 2022 Cassady, Soldin and Ramani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven J. Cassady, c3RldmVuLmNhc3NhZHlAc29tLnVtYXJ5bGFuZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.