 Marcus A. C. Williams
Marcus A. C. Williams Bairavi Shankar
Bairavi Shankar Joban Vaishnav
Joban Vaishnav Mark J. Ranek
Mark J. Ranek- 1Department of Medicine, Division of Cardiology, Johns Hopkins University, Baltimore, MD, United States
- 2Department of Pharmacology and Molecular Sciences, Johns Hopkins University, Baltimore, MD, United States
Cardiac amyloidosis is a progressive disorder caused by the deposition of amyloid, abnormal proteins that aggregate to form insoluble plaques in the myocardium resulting in restrictive cardiomyopathy. The two most common subtypes of cardiac amyloidosis are immunoglobulin light chain (AL) and transthyretin (TTR) amyloid cardiomyopathy (ATTR-CM). ATTR-CM can further be subdivided into two main categories, wild-type or hereditary TTR. TTR is a homotetrameric protein complex that is synthesized in the liver and is secreted into the circulation for retinol and vitamin A transfer. Genetic mutations in the TTR gene can disrupt the thermodynamic stability of the homotetrameric complex causing dissociation into monomers that, when taken up by the myocardium, will aggregate to form insoluble fibers. Though the mechanism of wild-type TTR is not fully elucidated, it is thought to be an age-related process. Myocardial uptake and aggregation of TTR monomeric subunits result in cytotoxicity, impaired cardiac function, and eventually heart failure. Historically, ATTR-CM had a poor prognosis, with no therapeutics available to specifically target ATTR-CM and treatment focused on managing symptoms and disease-related complications. In 2019, the FDA approved the first-in-class TTR stabilizer for ATTR-CM, which has led to improved outcomes. In recent years, several promising novel therapies have emerged which aim to target various points of the ATTR-CM amyloidogenic cascade. In this review, we discuss the mechanistic underpinnings of ATTR-CM, review current FDA-approved strategies for treatment, and highlight ongoing research efforts as potential therapeutic options in the future.
Introduction
Amyloidosis can occur from the deposition of multiple proteins within a variety of organs including the heart, kidneys, gastrointestinal tract, and peripheral nervous system among others (Hazenberg, 2013; Wechalekar et al., 2016; Dogan, 2017; Picken, 2020). In the context of cardiac amyloidosis (CA), myocardial infiltration of amyloid proteins leads to restrictive cardiomyopathy (Bhogal et al., 2018; Martinez-naharro et al., 2018; Ternacle et al., 2019). CA is subdivided based on the types of proteins being deposited within the myocardium. Though over 30 proteins have been identified to cause amyloidosis, the two most common subtypes are immunoglobulin light chain (AL) (Falk et al., 2016; Joshi et al., 2020; Palladini et al., 2020; Nasrullah et al., 2021) and transthyretin (TTR) (Lane et al., 2019; Hanna et al., 2020; Kittleson et al., 2020). Here, we focus on the latter, detailing the underlying pathogenesis of the disease, current therapeutic regimens, novel research efforts, and potential new strategies for the management of amyloid transthyretin cardiomyopathy (ATTR-CM).
TTR is synthesized by the liver and secreted into circulation to function as a homotetrameric protein complex to transport small molecules, such as retinol and tetraiodothyroxine (T4), vital for proper bodily function (Monaco, 2000; Zanotti and Berni, 2004; Alemi et al., 2021). The dissociation of the tetrameric TTR complex into individual monomers underlies the development and progression of ATTR-CM. ATTR-CM is further subdivided based on the presence or absence of a hereditary mutation, known as heredity ATTR-CM (hATTR-CM) or wild-type ATTR-CM (wtATTR-CM), respectively (Rader et al., 2019; Chacko et al., 2020; He et al., 2020; Law et al., 2020; Law et al., 2022). The mechanisms causing wtATTR-CM remain incompletely understood (Bulawa et al., 2012; Sekijima, 2014; Sekijima, 2015; Bezerra et al., 2020). Studies have attributed this disruption to increased temperature, less than optimal pH, or oxidative stress (Khurana et al., 2001; Eisenberg and Jucker, 2012). Genetic mutations in TTR reduce the thermodynamic stability of the TTR tetracomplex further influencing dissociation in addition to increased temperature, pH changes, or oxidative stress (Sekijima et al., 2005). For both forms of ATTR-CM, dissociated TTR in circulation can be taken up by various organs, including the heart. Following organ infiltration, the now-exposed hydrophobic residues may polymerize mainly via the hydrophobic effect in tandem with other factors such as external force, local extracellular matrix (ECM), or metals that may modulate fibril formation (Hipp et al., 2019; Van Gils et al., 2020; Griffin et al., 2021). Continual dissociation, deposition, and polymerization of TTR creates a proteinopathy—an accumulation of a specific protein or proteins in excess with altered conformations that facilitate aggregation and pathogenesis (Figure 1). This proteotoxic stress increases cellular damage and eventually cytotoxicity that in conjunction with TTR deposition and fibrosis, results in reduced cardiac function and ultimately cardiac failure.
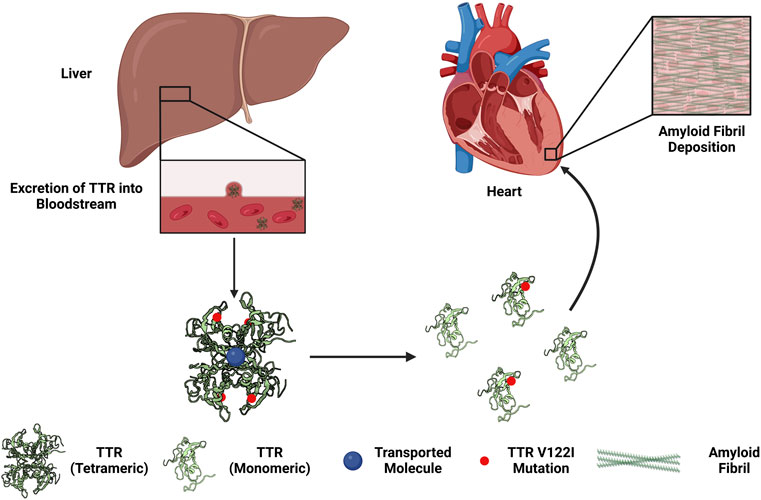
FIGURE 1. Overview of the development of hereditary transthyretin cardiac amyloidosis. The liver synthesizes and excretes transthyretin (TTR) into the circulation to function as a transport complex. Mutations in TTR (e.g., Val122Iso, V122I) increases the propensity of the TTR tetrameric complex to dissociate into monomers. TTR monomers can by taken up by the myocardium, aggregated, and form amyloid fibrils. The deposition of amyloid fibrils in the heart may lead to restrictive cardiomyopathy.
In recent years, the field has experienced an exciting increase in our understanding of ATTR-CM pathogenesis, novel treatment options, and promising targets of interest for future therapies. Here, we review the clinical presentation and management of ATTR-CM, our current mechanistic insight into the disease, and new therapies on the horizon.
Background of transthyretin cardiac amyloidosis
TTR was first identified in 1942 from human plasma (Seibert and Nelson, 1942) and cerebral spinal fluid (Kabat et al., 1942) during electrophoresis where TTR was observed to precede albumin, thus earning the name prealbumin. Further characterization of this novel protein determined the role of TTR as a transport protein for thyroid hormone and retinol and was aptly renamed transthyretin (PRE-ALBUMIN: A THYROXINE-BINDING PROTEIN OF HUMAN PLASMA, 1958; Raz and Goodman, 1969; Nomenclature committee of the International Union of Biochemistry (NC-IUB), 1981). TTR found in human plasma was produced by the liver and excreted into the circulation where the TTR complex functions to carry approximately 15% of the secreted thyroid hormone T4 or also bind with retinol-binding proteins for retinol transport (Goodman, 1984; Bartalena, 1990; Noy and Xu, 1990). In human cerebral spinal fluid, TTR is produced by the choroid plexus and accounts for 80% of T4 transport (Chanoine and Braverman, 1992).
There are over 120 identified single-nucleotide mutations in TTR worldwide, ∼80 of which lead to pathogenesis (Benson and Kincaid, 2007; Vieira and Saraiva, 2014). The first report of ATTR-CM came from Soyka in 1876 and was deemed to be resultant of increasing age (Ruberg and Berk, 2012). Over the next century, similar case reports followed this initial finding and were eventually linked to the deposition of TTR within the myocardium (Pomerance, 1965; Cornwell et al., 1983; Tanskanen et al., 2008). The most common variant that affects the myocardium is the valine to isoleucine 122 substitution, a mutation that is found in ∼4% of African Americans (Gorevic et al., 1989; Jacobson et al., 1997; Connors et al., 20032003). In contrast, the valine to methionine substitution at position 30 most commonly affects the nervous system and leads to familial amyloidotic polyneuropathy (ANDRADE, 1952; Dyck and Lambert, 1969; Sousa and Saraiva, 2003; Coelho et al., 2012a). Despite these two commonly referred to and distinct mutations, many give rise to multiple comorbidities and varying cardiomyopathy and peripheral neuropathy phenotypes (Kalkunte et al., 2013; Sekijima, 2021).
Transthyretin cardiac amyloidosis clinical presentation, prevalence, and management
Patients with ATTR-CM often present with symptoms characteristic of heart failure, including dyspnea, fatigue, and lower extremity edema. Thus, the backbone of management for patients with ATTR-CM, and for all patients with cardiac amyloidosis, is loop diuretics to relieve volume overload (Maurer et al., 2019; Stern and Kittleson, 2021). Notably, given their restrictive cardiac physiology, these patients often sit in a tenuous space between hyper- and hypovolemia. Close monitoring of blood pressure, symptoms of congestion, and renal function are crucial. Patients with ATTR-CM who also have heart failure with a reduced ejection fraction (EF ≤ 40%) do not benefit from guideline-directed medical therapy (Longhi et al., 2016; Maurer et al., 2019). Beta-blockers and vasodilators could potentially exacerbate hypotension by reducing cardiac output in light of a fixed stroke volume, thereby worsening the existing condition.
ATTR-CM is associated with a significant burden of both tachy- and brady-arrhythmias. This phenomenon is due to structural cardiac disease, such as bi-atrial enlargement and left ventricular hypertrophy, as well as infiltrative conduction disease. Atrial fibrillation is common in these patients. However, there is scant evidence regarding control strategies (rate vs. rhythm control)—there are no data to suggest one strategy is superior to the other (Maurer et al., 2019). Amiodarone is often employed for rate and rhythm control; digoxin is also used, albeit sparingly due to the increased risk of digoxin toxicity in this population. Ablation and cardioversion are considered on a case-by-case basis. Ventricular arrhythmias are less common, but given their significant mortality risk, implantable cardiac defibrillators (ICDs) can be used for primary prevention of sudden cardiac death. Importantly, the actual mortality benefit of ICD implantation is unclear (Maurer et al., 2019). Patients with ATTR-CM should still have ICDs implanted for secondary prevention if anticipated survival is more than 1 year.
Valvular disease is common as both a sign and sequela of ATTR-CM. It has been previously suggested that some complications seen in aortic valve replacement could be attributed to concomitant cardiac amyloidosis (Castano et al., 2017). Small prospective studies and case series have diagnosed ATTR-CM in 12%–33% of patients with aortic stenosis (AS) undergoing valve replacement (Castano et al., 2017). These patients tend to be more symptomatic and have severe AS. Additionally, ATTR-CM was associated specifically with low-flow, low-gradient AS (Nietlispach et al., 2012).
There are a variety of imaging features to aid in the diagnosis and monitoring of ATTR-CM. Low QRS complex voltages on electrocardiogram (ECG) are classically associated with ATTR-CM, but this finding has low sensitivity (Quarta et al., 2014; Towbin et al., 2019; Cimiotti et al., 2021). Other potential findings include a pseudo-infarct pattern and conduction disease (Cyrille et al., 2014). Echocardiography typically shows increased LV wall thickness, and LV hypertrophy in a patient over 65 years with symptoms of heart failure should prompt consideration of cardiac amyloidosis. Discordance between ECG and echocardiogram (that is, low voltages on ECG with evidence of hypertrophy) should also increase suspicion of ATTR-CM (Gillmore et al., 2016; Longhi et al., 2016).
There are several important non-cardiac manifestations of the disease. Peripheral polyneuropathy is common in patients with hATTR-CM—patients with a family history of polyneuropathy, particularly with heart failure, should receive genetic screening (Castano et al., 2017). Carpal tunnel syndrome is present in 60% of patients with wtATTR-CM, and a history of bilateral carpal tunnel release may be a key clue in diagnosis (Connors et al., 2016; Sperry et al., 2018; aus dem Siepen et al., 2019). Similarly, 33% of patients with wtATTR-CM experience biceps tendon rupture, and 23% report prior hip and/or knee arthroplasty (Westermark et al., 2014; Geller et al., 2017; Rubin et al., 2017). Orthostatic hypotension, erectile dysfunction, gastrointestinal motility issues, and urinary retention are other conditions associated with hATTR-CM (Kim et al., 2020).
Diagnosis of ATTR-CM requires detailed interpretation of several laboratories and imaging modalities. When cardiac amyloidosis is suspected, the first step in diagnosis is to rule out plasma cell dyscrasia that may be consistent with AL amyloidosis by laboratory screening. If screening blood work is normal, then cardiac scintigraphy (i.e., 99mTc-PYP scan) is the preferred diagnostic test for ATTR-CM. Cardiac MRI may be used if there is concern for infiltrative cardiomyopathy and will show late gadolinium enhancement in patients with ATTR-CM. However, this is not specific to amyloid infiltration, and thus 99mTc-PYP scan is the preferred imaging test 13. If the 99mTc-PYP scan is abnormal, TTR gene sequencing should be performed to further determine whether the patient suffers hATTR-CM or wtATTR-CM (Ruberg et al., 2012; Pinney et al., 2013; Longhi et al., 2015; Donnellan et al., 2019). If a 99mTc-PYP scan is not available, or testing is inconclusive, endomyocardial biopsy with immunostaining and mass spectrometry is the final step in this diagnostic algorithm for ATTR-CM. A false-negative 99mTc-PYP scan is possible in early ATTR-CM with relatively low cardiac infiltration, or with specific genetic mutations. Thus, endomyocardial biopsy remains the gold standard for the diagnosis of cardiac amyloidosis.
Therapeutic options
Transthyretin silencers
Reducing or eliminating TTR expression with so-called TTR silencers is one approach to slow down the progression of ATTR-CM. While effectively reducing TTR levels, this approach also removes the ability of TTR to serve as a transport protein. Due to TTR’s vital role, mainly in retinol transport, vitamin A supplementation is required when utilizing silencers (Maestro et al., 2021). There are two main classes of therapies: antisense oligonucleotides (ASOs) and small interfering RNA (siRNA). Both strategies bind TTR mRNA and induce subsequent degradation, albeit by distinct intracellular mechanisms (Aimo et al., 2022).
ASOs are single-stranded DNA molecules that can enter cells (e.g., hepatocytes) via clathrin or caveolin-mediated uptake and are transported to the nucleus where the ASOs can bind to processed mRNA and induce degradation through the RNase H2 endonuclease degradation pathway (Crooke et al., 2017; Lai et al., 2020). ASOs can bind surface proteins and subsequently be taken up and distributed throughout the body. A first-generation ASO for TTR amyloidosis, inotersen, was designed as an ASO to target processed TTR mRNA. Inotersen also contains a 2′-O-methoxyethyl modification providing resistance to endogenous degradation making it longer lasting. The NEURO-TTR trial enrolled 172 patients with familial amyloid polyneuropathy ± ATTR-CM to evaluate inotersen. Patients receiving inotersen had a mean reduction of 74% of TTR serum levels which reached a steady state by week 13; however, adverse side effects included glomerulonephritis and thrombocytopenia halted its development in phase II clinical trials (Benson et al., 2018). A second-generation ASO, AKCEA-TTR-LRx, shares the same base sequence as inotersen, conserving its mechanism of action, but is conjugated to N-acetyl galactosamine to improve its specificity to hepatocytes (Prakash et al., 2014). In the ION-682884-CS1 study, AKCEA-TTR-LRx was reported to have a 51-fold greater potency than inotersen with a greater than 85% reduction in TTR serum levels in ATTR-CM patients without the adverse side effects reported in the NEURO-TTR trial (Viney et al., 2021). Due to the success of AKCEA-TTR-LRx, a phase III clinical trial will take place and plan to enroll approximately 700 patients with ATTR-CM (Falk et al., 2019).
Another approach to reducing TTR expression is with siRNAs; double-stranded RNA containing both the sense and antisense strand of the target mRNA. Once the double-stranded siRNA enters a cell, it becomes incorporated into an RNA-induced silencing complex (RISC). This complex then interacts with Ago2, an RNA endonuclease, in which the sense strand is degraded, thus exposing the antisense strand-Ago2 complex so that it may bind the target mRNA and facilitate degradation. However, siRNAs have characteristic low cellular uptake and organ specificity, so they often require carriers for target therapy (Crooke et al., 2018; Deshayes et al., 2020; Alshaer et al., 2021; Zhang et al., 2021). A first-generation siRNA, patisiran, utilizes lipid nanoparticle delivery to overcome poor cellular uptake and target hepatocyte uptake. In a phase-II clinical trial in 2015, it was reported that following the second dose of patisiran, mean TTR serum levels were reduced by approximately 80% when doses were given at 3-week intervals (Suhr et al., 2015). In the phase III study APOLLO, patients given patisiran had reductions in LV wall thickness (∼1 mm) and NT-proBNP levels (∼55%), showing promise with this approach for cardiovascular disease (Richards and Troughton, 2004; Perry and Selvanayagam, 2019; Gertz et al., 2020). A second-generation siRNA therapeutic for ATTR-CM, vutrisiran, aimed to further improve its specificity and potency by conjugating N-acetyl galactosamine to the lipid nanoparticle carrier. The high specificity and potency of the drug allow patients to receive a markedly smaller dose (∼6-fold less) and be administered once every 3 months. In a phase I study, it was reported that a single dose of vutrisiran reduced serum TTR levels by 83% at 6 weeks and was sustained for an additional 90 days before climbing back to baseline (Adams and Verena, 2019).
A new silencing approach that has shown early promise for ATTR-CM utilizes CRISPR/Cas9 gene editing technology to reduce TTR levels (Gupta et al., 2019). Lipid nanoparticle carriers containing guide RNA and Cas9 mRNA (LNP-INT01) are used to deliver and initiate the reaction. In mice, it was demonstrated that a single dose of LNP-INT01 was able to reduce serum levels of TTR by greater than 97%, which was sustained for 12 months post-administration (Finn et al., 2018). In a clinical trial, a CRISPR/Cas9 lipid nanoparticle (NTLA-2001) was designed for use in ATTR-CM. Two different doses were tested in patients, namely, 0.1 and 0.3 mg/kg, yielding a reduction of serum TTR on day 28 by 52% and 87%, respectively. Such a marked reduction in serum TTR levels gives promise that this may be an efficacious and safe method for managing ATTR-CM (Gillmore et al., 2021).
Transthyretin stabilizers
Dissociation of the TTR homotetrameric complex and subsequent uptake by an organ is the driving force of amyloidogenesis. To this end, there have been efforts to develop strategies to stabilize the TTR homotetrameric complex (termed TTR stabilizers), thereby attenuating disease progression by blocking the rate-limiting step of the amyloidogenesis cascade.
Tolcapone is a catechol-O-methyltransferase inhibitor that was developed as a treatment for Parkinson’s disease acting against inhibition of dopamine degradation; however, it was shown that tolcapone may work as a TTR stabilizer. Catechol-O-methyltransferase has markedly high activity in the liver which may partially explain tolcapone’s serendipitous association with TTR and ATTR-CM (Müller, 2015). Tolcapone has a particularly high affinity for the thyroxine (T4)-binding sites on TTR, which stabilizes the complex (Sant’Anna et al., 2016). The long-term use of tolcapone may not be efficacious in the treatment for ATTR-CM due to its short half-life and association with acute and potentially fatal liver failure.
Diflunisal is a nonsteroidal, anti-inflammatory drug (NSAID), a drug generally used for chronic inflammatory diseases, and was found to bind to the T4-binding sites on TTR and aid in stabilizing the homotetrameric complex preventing fibril polymerization (Sekijima et al., 2006; Adam et al., 2012). In a small single-arm trial, it was shown that a dose of 250 mg twice daily over a 12-month course slows disease progression as evidenced by better maintained cardiac function and structure (Berk et al., 2013). Diflunisal is associated with renal dysfunction and fluid retention which may worsen heart failure in patients with ATTR-CM, thus limiting its use.
Tafamidis (Vyndamax) is a small-molecule analog of diflunisal that does not carry the same NSAID properties, making it safer for patients with ATTR-CM. Similar to diflunisal, tafamidis binds to the T4-binding site of TTR, stabilizing the protein and preventing the dissociation of both wt-TTR and h-TTR, albeit with a higher binding affinity than diflunisal (Spoladore et al., 2021). These attributes make tafamidis more efficacious for a diverse population with multiple genetic variants. In a landmark phase II clinical trial, it was observed that tafamidis significantly delays amyloidogenesis and improves quality of life in patients with associated polyneuropathy (Coelho et al., 2012a). In a seminal clinical trial, ATTR-ACT, 441 patients were enrolled and randomized to receive either 80 mg, 20 mg, or a placebo once per day over the course of 30 months. It was reported that patients receiving tafamidis had an associated 13.9% reduction in all-cause mortality when compared to control and a 32% reduction in cardiovascular-related hospitalization (Maurer et al., 2018). However, it should be noted that patients with NYHA class III symptoms had higher rates of cardiovascular hospitalization and did not have any benefit from therapeutic intervention with tafamidis (Maurer et al., 2018). In 2019, tafamidis became the first FDA-approved therapeutic for the treatment of ATTR-CM.
AG10 is a novel, potent, and selective TTR stabilizer that has been shown to outperform tafamidis in regard to TTR stabilization (Penchala et al., 2013). In a phase II study, AG10-201 was reported to achieve ∼90% stabilization of TTR in patient serum. Following 28 days of 800 mg twice daily administration of AG10, it was observed that stable tetrameric TTR levels had risen to 51% (Judge et al., 2019). A phase-III trial enlisting approximately 510 patients who will be dosed with 800 mg AG10 or placebo twice daily is currently underway (NCT03860935).
Transthyretin degraders
Slowing the progression or completely halting the progression of ATTR-CM has proven to be a significant challenge in itself as both TTR silencers and stabilizers 1) do not address the damage that has already been created to the myocardium nor 2) enhance the removal of newly dissociating TTR which still occurs with both treatments albeit at a slower pace. Therapeutic options that may degrade amyloid plaques already deposited in the myocardium or newly dissociated TTR infiltrating the myocardium are an intriguing approach.
The combination of doxycycline and tauroursodeoxycholic acid (TUDCA) has been explored as a TTR degrader approach. Doxycycline has been associated with disruption of preformed fibrils prior to deposition, and tauroursodeoxycholic acid has been shown to disrupt prefibrillar deposits (Macedo et al., 2008). In a familial amyloidotic polyneuropathy mouse model, the combination therapy yielded promising results with increased clearance of TTR. However, in a small open-label phase II human study, it was reported that over the course of 12 months the combination therapy was only able to slow the progression or halt ATTR-CM, but there was no observed clearance of TTR (Obici et al., 2012).
Epigallocatechin-3-gallate (EGCG) is a naturally occurring polyphenol extract of green tea that has been hypothesized to disrupt fibril formation and increase the clearance of misfolded or polymerized fibrils. EGCG has previously been shown to disrupt amyloid fibril formation in vitro (Ferreira et al., 2012) and in vivo (mice) (aus dem Siepen et al., 2015a). Small open-label studies have reported both marginal decreases and increases in myocardial mass in specific patient populations. In addition, NT-proBNP levels and LV measurements have variable changes from patient to patient (Kristen et al., 2012; aus dem Siepen et al., 2015a; 2015b).
Conclusion
The awareness and subsequent testing for ATTR-CM have increased in the last decade, which has highlighted various issues: the prevalence of the disease is much greater than previously thought, early-stage diagnosis is critical, and improved treatment options are sorely needed. This review focused on the current therapeutic approaches and ongoing efforts in research and development for ATTR-CM. Many strategies for ATTR-CM are being tested that interrogate distinct stages of amyloidogenesis, with varying degrees of efficacy and side effects. To date, tafamidis is the only FDA-approved therapeutic for ATTR-CM; however, more approaches are on the horizon. Seminal basic science studies and clinical trials have provided the field with exciting and promising data going forward. The field is rapidly evolving, and it is possible that ATTR-CM, a disease that once was associated with a poor prognosis and mortality rate, will continue to be more manageable and potentially even curable.
Author contributions
MR and JV conceived the original idea. MW and BS wrote the manuscript. MW took the lead in writing the manuscript. MR and JV edited the manuscript. MR supervised the project. All authors provided critical feedback and helped shape the manuscript.
Funding
This work was supported by funds from the NIH PREP Program (MACW), NIH Johns Hopkins PREP: R25GM109441 (MACW), the Amyloidosis Foundation, and the American Heart Association (Grant Nos. #18CDA34110140 and #20TPA35500008, MR).
Acknowledgments
The illustration in Figure 1 was created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adam, C., Helmke, S., Alvarez, J., Delisle, S., and Maurer, M. S. (2012). Diflunisal for ATTR cardiac amyloidosis. Congest. Heart Fail. 18 315–319. doi:10.1111/j.1751-7133.2012.00303.x
Adams, D., and Verena, K. (2019). Phase 1 study of ALN-TTRsc02, a subcutaneously administered investigational RNAi therapeutic for the treatment of transthyretinmediated amyloidosis. Rev. Neurol. Paris. 175, S129. doi:10.1016/j.neurol.2019.01.339
Aimo, A., Castiglione, V., Rapezzi, C., Franzini, M., Panichella, G., Vergaro, G., et al. (2022). RNA-targeting and gene editing therapies for transthyretin amyloidosis. Nat Rev Cardiol. 19 (10), 655–667. doi:10.1038/s41569-022-00683-z
Alemi, M., Oliveira, A., Tavares, S. C., Vieira, J. R., Alves, M. G., Oliveira, P. F., et al. (2021). Exploring the physiological role of transthyretin in glucose metabolism in the liver. Int. J. Mol. Sci. 22, 6073. doi:10.3390/ijms22116073
Alshaer, W., Zureigat, H., Al Karaki, A., Al-Kadash, A., Gharaibeh, L., Hatmal, M. M., et al. (2021). siRNA: Mechanism of action, challenges, and therapeutic approaches. Eur. J. Pharmacol. 905, 174178. doi:10.1016/j.ejphar.2021.174178
Andrade, C. A. (1952). Peculiar form of peripheral neuropathy: Familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain. 75, 408–427. doi:10.1093/brain/75.3.408
aus dem Siepen, F., Bauer, R., Aurich, M., Buss, S. J., Steen, H., Altland, K., et al. (2015). Green tea extract as a treatment for patients with wildtype transthyretin amyloidosis: An observational study. Drug Des. devel. Ther. 9, 6319–6325. doi:10.2147/DDDT.S96893
aus dem Siepen, F., Buss, S. J., Andre, F., Seitz, S., Giannitsis, E., Steen, H., et al. (2015). Extracellular remodeling in patients with wild-type amyloidosis consuming epigallocatechin-3-gallate: Preliminary results of T1 mapping by cardiac magnetic resonance imaging in a small single center study. Clin. Res. Cardiol. 104, 640–647. doi:10.1007/s00392-015-0826-3
aus dem Siepen, F., Hein, S., Prestel, S., Baumgartner, C., Schonland, S., Hegenbart, U., et al. (2019). Carpal tunnel syndrome and spinal canal stenosis: Harbingers of transthyretin amyloid cardiomyopathy? Clin. Res. Cardiol. 108, 1324–1330. doi:10.1007/s00392-019-01467-1
Bartalena, L. (1990). Recent achievements in studies on thyroid hormone-binding proteins. Endocr. Rev. 11, 47–64. doi:10.1210/edrv-11-1-47
Benson, M. D., and Kincaid, J. C. (2007). The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve 36, 411–423. doi:10.1002/mus.20821
Benson, M. D., Waddington-Cruz, M., Berk, J. L., Polydefkis, M., Dyck, P. J., Wang, A. K., et al. (2018). Inotersen treatment for patients with hereditary transthyretin amyloidosis. N. Engl. J. Med. 379, 22–31. doi:10.1056/NEJMoa1716793
Berk, J. L., Suhr, O. B., Obici, L., Sekijima, Y., Zeldenrust, S. R., Yamashita, T., et al. (2013). Repurposing diflunisal for familial amyloid polyneuropathy: A randomized clinical trial. JAMA - J. Am. Med. Assoc. 310, 2658–2667. doi:10.1001/jama.2013.283815
Bezerra, F., Saraiva, M. J., and Almeida, M. R. (2020). Modulation of the mechanisms driving transthyretin amyloidosis. Front. Mol. Neurosci. 13, 592644. doi:10.3389/fnmol.2020.592644
Bhogal, S., Ladia, V., Sitwala, P., Cook, E., Bajaj, K., Ramu, V., et al. (2018). Cardiac amyloidosis: An updated review with emphasis on diagnosis and future directions. Curr. Probl. Cardiol. 43, 10–34. doi:10.1016/j.cpcardiol.2017.04.003
Bulawa, C. E., Connelly, S., Devit, M., Wang, L., Weigel, C., Fleming, J. A., et al. (2012). Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc. Natl. Acad. Sci. U. S. A. 109, 9629–9634. doi:10.1073/pnas.1121005109
Castano, A., Narotsky, D. L., Hamid, N., Khalique, O. K., Morgenstern, R., DeLuca, A., et al. (2017). Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur. Heart J. 38 (38), 2879–2887. doi:10.1093/eurheartj/ehx350
Chacko, L., Martone, R., Bandera, F., Lane, T., Martinez-Naharro, A., Boldrini, M., et al. (2020). Echocardiographic phenotype and prognosis in transthyretin cardiac amyloidosis. Eur. Heart J. 41, 1439–1447a. doi:10.1093/eurheartj/ehz905
Chanoine, J. P., and Braverman, L. E. (1992). The role of transthyretin in the transport of thyroid hormone to cerebrospinal fluid and brain. Acta Med. Austriaca 19, 25–28.
Cimiotti, D., Budde, H., Hassoun, R., and Jaquet, K. (2021). Genetic restrictive cardiomyopathy: Causes and consequences an integrative approach. Int. J. Mol. Sci. 22, 558. doi:10.3390/ijms22020558
Coelho, T., Maia, L. F., Martins da Silva, A., Waddington Cruz, M., Plante-Bordeneuve, V., Lozeron, P., et al. (2012). Tafamidis for transthyretin familial amyloid polyneuropathy: A randomized, controlled trial. Neurology 79, 785. doi:10.1212/WNL.0b013e3182661eb1
Coelho, T., Maurer, M. S., and Suhr, O. B. (2012). Thaos – the transthyretin AmyloidosisOutcomes survey: Initial report on clinical manifestations in patients with hereditary and wild-type transthyretin amyloidosis. Curr. Med. Res. Opin. 29, 63–76. doi:10.1185/03007995.2012.754348
Connors, L. H., Lim, A., Prokaeva, T., Roskens, V. A., and Costello, C. E. (20032003). Tabulation of human transthyretin (TTR) variants, 2003. Amyloid 10, 160–184. doi:10.3109/13506120308998998
Connors, L. H., Sam, F., Skinner, M., Salinaro, F., Sun, F., Ruberg, F. L., et al. (2016). Heart failure resulting from age-related cardiac amyloid disease associated with wild type transthyretin: A prospective, observational cohort study. Circulation 133, 282–290. doi:10.1161/CIRCULATIONAHA.115.018852
Cornwell, G. G., Murdoch, W. L., Kyle, R. A., Westermark, P., and Pitkänen, P. (1983). Frequency and distribution of senile cardiovascular amyloid: A clinicopathologiccorrelation. Am. J. Med. 75, 618–623. doi:10.1016/0002-9343(83)90443-6
Crooke, S. T., Wang, S., Vickers, T. A., Shen, W., and Liang, X. H. (2017). Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 35, 230–237. doi:10.1038/nbt.3779
Crooke, S. T., Witztum, J. L., Bennett, C. F., and Baker, B. F. (2018) Perspective RNATargeted therapeutics, Cell Metab., 27, 714–739. doi:10.1016/j.cmet.2018.03.004
Cyrille, N. B., Goldsmith, J., Alvarez, J., and Maurer, M. S. (2014). Prevalence and prognostic significance of low QRS voltage among the three main types of cardiac amyloidosis. Am. J. Cardiol. 114, 1089–1093. doi:10.1016/j.amjcard.2014.07.026
Deshayes, S., Konate, K., Dussot, M., Chavey, B., Vaissiere, A., Van, T. N. N., et al. (2020). Deciphering the internalization mechanism of WRAP:siRNA nanoparticles. Biochim. Biophys. Acta. Biomembr. 1862, 183252. doi:10.1016/j.bbamem.2020.183252
Dogan, A. Amyloidosis (2017). Amyloidosis: Insights from proteomics. Annu. Rev. Pathol. 12, 277–304. doi:10.1146/annurev-pathol-052016-100200
Donnellan, E., Wazni, O. M., Saliba, W. I., Baranowski, B., Hanna, M., Martyn, M., et al. (2019). Cardiac devices in patients with transthyretin amyloidosis: Impact on functional class, left ventricular function, mitral regurgitation, and mortality. J. Cardiovasc. Electrophysiol. 30, 2427–2432. doi:10.1111/jce.14180
Dyck, P. J., and Lambert, E. H. (1969). Dissociated sensation in amylidosis. Compound action potential, quantitative histologic and teased-fiber, and electron microscopic studies of sural nerve biopsies. Arch. Neurol. 20, 490–507. doi:10.1001/archneur.1969.00480110054005
Eisenberg, D., and Jucker, M. (2012). The amyloid state of proteins in human diseases. Cell 148, 1188–1203. doi:10.1016/j.cell.2012.02.022
Falk, R. H., Alexander, K. M., Liao, R., and Dorbala, S. A. L. (2016). AL (Light-Chain) cardiac amyloidosis: A review of diagnosis and therapy. J. Am. Coll. Cardiol. 68, 1323–1341. doi:10.1016/j.jacc.2016.06.053
Falk, R. H., Gertz, M. A., Benson, M. D., Buchele, G., Brambatti, M., Tsimikas, S., et al. (2019). Rationale and design of a phase 3 study to evaluate the efficacy and safety of ION-682884 in patients with transthyretin-mediated amyloid cardiomyopathy (ATTR-CM). Blood 134, 5764–5764. doi:10.1182/blood-2019-129269
Ferreira, N., Saraiva, M. J., and Almeida, M. R. (2012). Epigallocatechin-3-gallate as a potential therapeutic drug for TTR-related amyloidosis: ‘in vivo’ evidence from FAP mice models. PLoS One 7, e29933–14. doi:10.1371/journal.pone.0029933
Finn, J. D., Smith, A. R., Patel, M. C., Shaw, L., Youniss, M. R., van Heteren, J., et al. (2018). A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Rep. 22, 2227–2235. doi:10.1016/j.celrep.2018.02.014
Geller, H. I., Singh, A., Alexander, K. M., Mirto, T. M., and Falk, R. H. (2017). Association between ruptured distal biceps tendon and wild-type transthyretin cardiac amyloidosis. JAMA - J. Am. Med. Assoc. 318, 962–963. doi:10.1001/jama.2017.9236
Gertz, M., Adams, D., Ando, Y., Beirão, J. M., Bokhari, S., Coelho, T., et al. (2020). Avoiding misdiagnosis: expert consensus recommendations for the suspicion and diagnosis of transthyretin amyloidosis for the general practitioner. BMC Fam. Pract. 21, 198. doi:10.1186/s12875-020-01252-4
Gillmore, J. D., Gane, E., Taubel, J., Kao, J., Fontana, M., Maitland, M. L., et al. (2021). CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 385, 493–502. doi:10.1056/NEJMoa2107454
Gillmore, J. D., Maurer, M. S., Falk, R. H., Merlini, G., Damy, T., Dispenzieri, A., et al. (2016). Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation 133, 2404–2412. doi:10.1161/CIRCULATIONAHA.116.021612
Goodman, D. S. (1984). Vitamin A and retinoids in health and disease. N. Engl. J. Med. 310, 1023–1031. doi:10.1056/NEJM198404193101605
Gorevic, P. D., Prelli, F. C., Wright, J., Pras, M., and Frangione, B. (1989). Systemic senileamyloidosis. Identification of a new prealbumin (Transthyretin) variant in cardiactissue: Immunologic and biochemical similarity to one form of familial amyloidoticpolyneuropathy. J. Clin. Invest. 83, 836–843. doi:10.1172/JCI113966
Griffin, J. M., Rosenblum, H., and Maurer, M. S. (2021). Pathophysiology and therapeutic approaches to cardiac amyloidosis. Circ. Res. 128, 1554–1575. doi:10.1161/CIRCRESAHA.121.318187
Gupta, D., Bhattacharjee, O., Mandal, D., Sen, M. K., Dey, D., Dasgupta, A., et al. (2019). CRISPR-Cas9 system: A new-fangled dawn in gene editing. Life Sci. 232, 116636. doi:10.1016/j.lfs.2019.116636
Hanna, M., Ruberg, F. L., Maurer, M. S., Dispenzieri, A., Dorbala, S., Falk, R. H., et al. (2020). Cardiac scintigraphy with technetium-99m-labeled bone- seeking tracers for suspected amyloidosis: JACC review topic of the week. J. Am. Coll. Cardiol. 75, 2851–2862. doi:10.1016/j.jacc.2020.04.022
HazenbergAmyloidosis, B. P. C. (2013). Amyloidosis: A clinical overview. Rheum. Dis. Clin. North Am. 39, 323–345. doi:10.1016/j.rdc.2013.02.012
He, S., Jin, Y., Tian, Z., Hua, T., Xing, H., Zhuang, S., et al. (2020). Establishment of an induced pluripotent stem cell line PUMCHi004-A from a hereditary transthyretin amyloid cardiomyopathy patient with transthyretin (TTR) p.Asp38Asn mutation. Stem Cell Res. 49, 102022–102052. doi:10.1016/j.scr.2020.102022
Hipp, M. S., Kasturi, P., and Hartl, F. U. (2019). The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 20, 421–435. doi:10.1038/s41580-019-0101-y
Jacobson, D. R., Pastore, R. D., Yaghoubian, R., Kane, I., Gallo, G., Buck, F. S., et al. (1997). Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N. Engl. J. Med. 336, 466–473. doi:10.1056/NEJM199702133360703
Joshi, S., Evangelisti, A., Liao, R., and Alexander, K. M. (2020). Harnessing cardiac regeneration as a potential therapeutic strategy for AL cardiac amyloidosis. Curr. Cardiol. Rep. 22, 1. doi:10.1007/s11886-020-1252-3
Judge, D. P., Heitner, S. B., Falk, R. H., Maurer, M. S., Shah, S. J., Witteles, R. M., et al. (2019). Transthyretin stabilization by AG10 in symptomatic transthyretin amyloid cardiomyopathy. J. Am. Coll. Cardiol. 74, 285–295. doi:10.1016/j.jacc.2019.03.012
Kabat, E. A., Moore, D. H., and Landow, H. (1942). An electrophoretic study of the protein components in cerebrospinal fluid and their relationship to the serum proteins. J. Clin. Invest. 21, 571–577. doi:10.1172/JCI101335
Kalkunte, S. S., Neubeck, S., Norris, W. E., Cheng, S. B., Kostadinov, S., Vu Hoang, D., et al. (2013). Transthyretin is dysregulated in preeclampsia, and its native form prevents the onset of disease in a preclinical mouse model. Am. J. Pathol. 183, 1425\–1436. doi:10.1016/j.ajpath.2013.07.022
Khurana, R., Gillespie, J. R., TAlApAtrA, A., Minert, L. J., IonesCu-Zanetti, C., MIllett, I., et al. (2001). Partially folded intermediates as critical precursors of light chain amyloid fibrils and amorphous aggregates. Biochemistry 40, 3525–3535. doi:10.1021/bi001782b
Kim, E. J., Holmes, B. B., Huang, S., Lugo, R., Al Aboud, A., Goodman, S., et al. (2020). Outcomes in patients with cardiac amyloidosis and implantablecardioverter-defibrillator. Europace 22, 1216–1223. doi:10.1093/europace/euaa094
Kittleson, M. M., Maurer, M. S., Ambardekar, A. V., Bullock-Palmer, R. P., Chang, P. P., Eisen, H. J., et al. (2020). Cardiac amyloidosis: Evolving diagnosis and management: A scientific statement from the American heart association. Circulation 142, e7–e22. doi:10.1161/CIR.0000000000000792
Kristen, A. V., Lehrke, S., Buss, S., Mereles, D., Steen, H., Ehlermann, P., et al. (2012). Green tea halts progression of cardiac transthyretin amyloidosis: An observational report. Clin. Res. Cardiol. 101, 805–813. doi:10.1007/s00392-012-0463-z
Lai, F., Damle, S. S., Ling, K. K., and Rigo, F. (2020). Directed RNase H cleavage of nascent transcripts causes transcription termination. Mol. Cell 77, 1032–1043. (e4) doi:10.1016/j.molcel.2019.12.029
Lane, T., Fontana, M., Martinez-Naharro, A., Quarta, C. C., Whelan, C. J., Petrie, A., et al. (2019). Natural history, quality of life, and outcome in cardiac transthyretin amyloidosis. Circulation 140, 16–26. doi:10.1161/CIRCULATIONAHA.118.038169
Law, S., Petrie, A., Chacko, L., Cohen, O. C., Ravichandran, S., Gilbertson, J. A., et al. (2022). Change in N-Terminal pro-B-Type natriuretic peptide at 1 year predicts mortality in wild-Type transthyretin amyloid cardiomyopathy. Heart 108, 474–478. doi:10.1136/heartjnl-2021-319063
Law, S., Petrie, A., Chacko, L., Cohen, O. C., Ravichandran, S., Gilbertson, J. A., et al. (2020). Disease progression in cardiac transthyretin amyloidosis is indicated by serial calculation of National Amyloidosis Centre transthyretin amyloidosis stage. Esc. Heart Fail. 7, 3942–3949. doi:10.1002/ehf2.12989
Longhi, S., Lorenzini, M., Gagliardi, C., Milandri, A., Marzocchi, A., Marrozzini, C., et al. (2016). Coexistence of degenerative aortic stenosis and wild-type transthyretin-related cardiac amyloidosis. JACC. Cardiovasc. Imaging 9 (3), 325–327. doi:10.1016/j.jcmg.2015.04.012
Longhi, S., Lorenzini, M., Gagliardi, C., Milandri, A., Marzocchi, A., Marrozzini, C., et al. (2016). Coexistence of degenerative aortic stenosis and wild-type transthyretin-related cardiac amyloidosis. JACC. Cardiovasc. Imaging 9, 325–327. doi:10.1016/j.jcmg.2015.04.012
Longhi, S., Quarta, C. C., Milandri, A., Lorenzini, M., Gagliardi, C., Manuzzi, L., et al. (2015). Atrial fibrillation in amyloidotic cardiomyopathy: Prevalence, incidence, risk factors and prognostic role. Amyloid 22, 147–155. doi:10.3109/13506129.2015.1028616
Macedo, B., Batista, A. R., Ferreira, N., Almeida, M. R., and Saraiva, M. J. (2008). Antiapoptotic treatment reduces transthyretin deposition in a transgenic mouse model of Familial Amyloidotic Polyneuropathy. Biochim. Biophys. Acta 1782, 517–522. doi:10.1016/j.bbadis.2008.05.005
Maestro, S., Weber, N. D., Zabaleta, N., Aldabe, R., and Gonzalez-Aseguinolaza, G. (2021). Novel vectors and approaches for gene therapy in liver diseases. JHEP Rep. 3 (4), 100300. doi:10.1016/j.jhepr.2021.100300
Martinez-naharro, A., Hawkins, P. N., and Fontana, M. (2018). Cardiac amyloidosis. Clin. Med. 18, 30–35. doi:10.7861/clinmedicine.18-2-s30
Maurer, M. S., Bokhari, S., Damy, T., Dorbala, S., Drachman, B. M., Fontana, M., et al. (2019). Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ. Heart Fail. 12 (9), e006075. doi:10.1161/CIRCHEARTFAILURE.119.006075
Maurer, M. S., Schwartz, J. H., Gundapaneni, B., Elliott, P. M., Merlini, G., Waddington-Cruz, M., et al. (2018). Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N. Engl. J. Med. 379, 1007–1016. doi:10.1056/NEJMoa1805689
Monaco, H. L. (2000). The transthyretin-retinol-binding protein complex. Biochim. Biophys. Acta 1482, 65–72. doi:10.1016/s0167-4838(00)00140-0
Müller, T. (2015). Catechol-O-methyltransferase inhibitors in Parkinson’s disease. Drugs 75, 157–174. doi:10.1007/s40265-014-0343-0
Nasrullah, A., Javed, A., Jayakrishnan, T. T., Brumbaugh, A., Sandhu, A., and Hardman, B. (2021). AL type cardiac amyloidosis: A devastating fatal disease. J. Community Hosp. Intern. Med. Perspect. 11, 407–412. doi:10.1080/20009666.2021.1915547
Nietlispach, F., Webb, J. G., Ye, J., Cheung, A., Lichtenstein, S. V., Carere, R. G., et al. (2012). Pathology of transcatheter valve therapy. JACC. Cardiovasc. Interv. 5 (5), 582–590. doi:10.1016/j.jcin.2012.03.012
Nomenclature committee of the International Union of Biochemistry (NC-IUB) (1981). Enzyme nomenclature. Recommendations 1978. Supplement 2: Corrections and additions. Eur. J. Biochem. 116, 423–435.
Noy, N., and Xu, Z. J. (1990). Interactions of retinol with binding proteins: Implications for the mechanism of uptake by cells. Biochemistry 29, 3878–3883. doi:10.1021/bi00468a012
Obici, L., Cortese, A., Lozza, A., Lucchetti, J., Gobbi, M., Palladini, G., et al. (2012). Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: A phase II study. Amyloid 19, 19 34–36. doi:10.3109/13506129.2012.678508
Palladini, G., Milani, P., and Merlini, G. (2020). Management of AL amyloidosis in 2020. Blood 136, 2620–2627. doi:10.1182/blood.2020006913
Penchala, S. C., Connelly, S., Wang, Y., Park, M. S., Zhao, L., Baranczak, A., et al. (2013). AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc. Natl. Acad. Sci. U. S. A. 110, 9992–9997. doi:10.1073/pnas.1300761110
Perry, R., and Selvanayagam, J. B. (2019). Echocardiography in infiltrative cardiomyopathy. Heart Lung Circ. 28, 1365–1375. doi:10.1016/j.hlc.2019.04.017
Pharmaceuticals, A. (2015). Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 379, 11–21. doi:10.1056/NEJMoa1716153
Picken, M. M. (2020). The pathology of amyloidosis in classification: A review. Acta Haematol. 143, 322–334. doi:10.1159/000506696
Pinney, J. H., Whelan, C. J., Petrie, A., Dungu, J., Banypersad, S. M., Sattianayagam, P., et al. (2013). Senile systemic amyloidosis: Clinical features at presentation and outcome. J. Am. Heart Assoc. 2, e000098–11. doi:10.1161/JAHA.113.000098
Prakash, T. P., Graham, M. J., Yu, J., Carty, R., Low, A., Chappell, A., et al. (2014). Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 42, 8796–8807. doi:10.1093/nar/gku531
Quarta, C. C., Solomon, S. D., Uraizee, I., Kruger, J., Longhi, S., Ferlito, M., et al. (2014). Left ventricular structure and function in transthyretin-related versus light-chain cardiac amyloidosis. Circulation 129, 1840–1849. doi:10.1161/CIRCULATIONAHA.113.006242
Rader, D. J., Chaudhary, K., Cho, J. H., Liang, L. W., Argulian, E., Chan, L., et al. (2019). Association of the V122I hereditary transthyretin amyloidosis genetic variant with heart failure among individuals of african or hispanic/latino ancestry. JAMA - J. Am. Med. Assoc. 322, 2191–2202. doi:10.1001/jama.2019.17935
Raz, A., and Goodman, D. S. (1969). The interaction of thyroxine with human plasma prealbumin and with the prealbumin-retinol-binding protein complex. J. Biol. Chem. 244, 3230–3237. doi:10.1016/s0021-9258(18)93118-2
Richards, M., and Troughton, R. W. (2004). NT-proBNP in heart failure: Therapy decisions and monitoring. Eur. J. Heart Fail. 6, 351–354. doi:10.1016/j.ejheart.2004.01.003
Ruberg, F. L., and BerkTransthyretin, J. L. T. T. R. (2012). Transthyretin (TTR) cardiac amyloidosis. Circulation 126, 1286–1300. doi:10.1161/CIRCULATIONAHA.111.078915
Ruberg, F. L., Maurer, M. S., Judge, D. P., Zeldenrust, S., Skinner, M., Kim, A. Y., et al. (2012). Prospective evaluation of the morbidity and mortality of wildtype and V122I mutant transthyretin amyloid cardiomyopathy: The Transthyretin Amyloidosis Cardiac Study (TRACS). Am. Heart J. 164, 222–228. doi:10.1016/j.ahj.2012.04.015
Rubin, J., Alvarez, J., Teruya, S., Castano, A., Lehman, R. A., Weidenbaum, M., et al. (2017). Hip and knee arthroplasty are common among patients with transthyretin cardiac amyloidosis, occurring years before cardiac amyloid diagnosis: Can we identify affected patients earlier? Amyloid 24, 226–230. doi:10.1080/13506129.2017.1375908
Sant’Anna, R., Gallego, P., Robinson, L. Z., Pereira-Henriques, A., Ferreira, N., Pinheiro, F., et al. (2016). Repositioning tolcapone as a potent inhibitor of transthyretin amyloidogenesis and associated cellular toxicity. Nat. Commun. 7, 10787. doi:10.1038/ncomms10787
Seibert, F. B., and Nelson, J. W. (1942). Electrophoretic study of the blood protein response in tuberculosis. J. Biol. Chem. 143, 29–38. doi:10.1016/s0021-9258(18)72655-0
Sekijima, Y., Dendle, M. A., and Kelly, J. W. (2006). Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid 13, 236–249. doi:10.1080/13506120600960882
Sekijima, Y. (2021). “Hereditary transthyretin amyloidosis,” in GeneReviews. Editors Adam, M. P., Everman, D. B., Mirzaa, G. M., et al. (Seattle, WA: University of Washington), 1993–2022. Available at: https://www.ncbi.nlm.nih.gov/books/NBK1194/.
Sekijima, Y. (2014). Recent progress in the understanding and treatment of transthyretin amyloidosis. J. Clin. Pharm. Ther. 39, 225–233. doi:10.1111/jcpt.12145
Sekijima, Y. (2015). Transthyretin (ATTR) amyloidosis: Clinical spectrum, molecular pathogenesis and disease-modifying treatments. J. Neurol. Neurosurg. Psychiatry 86, 1036–1043. doi:10.1136/jnnp-2014-308724
Sekijima, Y., Wiseman, R. L., Matteson, J., Hammarstrom, P., Miller, S. R., Sawkar, A. R., et al. (2005). The biological and chemical basis for tissue-selective amyloid disease. Cell 121, 73–85. doi:10.1016/j.cell.2005.01.018
Sousa, M. M., and Saraiva, M. J. (2003). Neurodegeneration in familial amyloid polyneuropathy: From pathology to molecular signaling. Prog. Neurobiol. 71, 385–400. doi:10.1016/j.pneurobio.2003.11.002
Sperry, B. W., Reyes, B. A., Ikram, A., Donnelly, J. P., Phelan, D., Jaber, W. A., et al. (2018). Tenosynovial and cardiac amyloidosis in patients undergoing carpal tunnel release. J. Am. Coll. Cardiol. 72, 2040–2050. doi:10.1016/j.jacc.2018.07.092
Spoladore, R., Falasconi, G., Marcatti, M., Di Maio, S., Fiore, G., Slavich, M., et al. (2021). Advances in pharmacotherapy for cardiac amyloidosis. Expert Opin. Pharmacother. 22, 469–481. doi:10.1080/14656566.2020.1836159
Stern, L. K., and Kittleson, M. M. (2021). Updates in cardiac amyloidosis diagnosis and treatment. Curr. Oncol. Rep. 23 (4), 47–12. doi:10.1007/s11912-021-01028-8
Suhr, O. B., Coelho, T., Buades, J., Pouget, J., Conceicao, I., Berk, J., et al. (2015). Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: A phase II multi-dose study. Orphanet J. Rare Dis. 10, 109–9. doi:10.1186/s13023-015-0326-6
Tanskanen, M., Peuralinna, T., Polvikoski, T., Notkola, I. L., Sulkava, R., Hardy, J., et al. (2008). Senile systemic amyloidosis affects 25% of the very aged and associates with genetic variation in alpha2-macroglobulin and tau: A population-based autopsy study. Ann. Med. 40, 232–239. doi:10.1080/07853890701842988
Ternacle, J., Krapf, L., Mohty, D., Magne, J., Nguyen, A., Galat, A., et al. (2019). Aortic stenosis and cardiac amyloidosis: JACC review topic of the week. J. Am. Coll. Cardiol. 74, 2638–2651. doi:10.1016/j.jacc.2019.09.056
Towbin, J. A., McKenna, W. J., Abrams, D. J., Ackerman, M. J., Calkins, H., Darrieux, F. C. C., et al. (20192019). 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy: Executive summary. Heart rhythm. 16, e373–e407. doi:10.1016/j.hrthm.2019.09.019
Van Gils, J. H. M., van Dijk, E., Peduzzo, A., Hofmann, A., Vettore, N., Schutzmann, M. P., et al. (2020). The hydrophobic effect characterises the thermodynamic signature of amyloid fibril growth. PLoS Comput. Biol. 16, e1007767–25. doi:10.1371/journal.pcbi.1007767
Vieira, M., and Saraiva, M. J. Transthyretin (2014). Transthyretin: A multifaceted protein. Biomol. Concepts 5, 45–54. doi:10.1515/bmc-2013-0038
Viney, N. J., Guo, S., Tai, L. J., Baker, B. F., Aghajan, M., Jung, S. W., et al. (2021). Ligand conjugated antisense oligonucleotide for the treatment of transthyretin amyloidosis: Preclinical and phase 1 data. Esc. Heart Fail. 8, 652–661. doi:10.1002/ehf2.13154
Wechalekar, A. D., Gillmore, J. D., and Hawkins, P. N. (2016). Systemic amyloidosis. Lancet 387, 2641–2654. doi:10.1016/S0140-6736(15)01274-X
Westermark, P., Westermark, G. T., Suhr, O. B., and Berg, S. (2014). Transthyretin-derived amyloidosis: Probably a common cause of lumbar spinal stenosis. Ups. J. Med. Sci. 119, 223–228. doi:10.3109/03009734.2014.895786
Zanotti, G., and Berni, R. (2004). Plasma retinol-binding protein: Structure and interactions with retinol, retinoids, and transthyretin. Vitam. Horm. 69, 271–295. doi:10.1016/S0083-6729(04)69010-8
Keywords: heart, proteinopathies, amyloid, therapeutic targets, heart disease, Transthyretin
Citation: Williams MAC, Shankar B, Vaishnav J and Ranek MJ (2022) Current and potential therapeutic strategies for transthyretin cardiac amyloidosis. Front. Drug. Discov. 2:1015545. doi: 10.3389/fddsv.2022.1015545
Received: 09 August 2022; Accepted: 25 August 2022;
Published: 12 October 2022.
Edited by:
Charles C. Hong, University of Maryland, United StatesReviewed by:
Charles Williams, University of Maryland, Baltimore, United StatesCopyright © 2022 Williams, Shankar, Vaishnav and Ranek. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark J. Ranek, bXJhbmVrMUBqaG1pLmVkdQ==, bXJhbmVrMUBqaC5lZHU=