 José L. Medina-Franco
José L. Medina-Franco- DIFACQUIM Research Group, Department of Pharmacy, School of Chemistry, National Autonomous University of Mexico, Mexico City, Mexico
Background
Computer-aided drug discovery (CADD) has become an essential part of several projects in different settings and research environments. CADD has largely contributed to identifying and optimizing hit compounds leading them to advanced stages of the drug discovery pipeline or the market (Prieto-Martínez et al., 2019). CADD includes several theoretical disciplines, including chemoinformatics, bioinformatics, molecular modeling, and data mining, among others (López-López et al., 2021). Artificial intelligence (AI) that has been used since the 60 s (Gasteiger, 2020) in drug discovery is regaining momentum, in particular with machine learning (ML) and deep learning (DL) (Bajorath, 2021; Bender and Cortés-Ciriano, 2021). In parallel to the continued contribution of CADD, several methodologies used in CADD have entered the hype cycle with waives of hope, inflated expectations, disappointments, and productive applications. The disillusionments are frequently driven by fashion, exacerbated misuse, and a lack of proper training to interpret the results (Medina-Franco et al., 2021). Examples are quantitative structure-activity relationship studies (QSAR). A few decades ago, there was a hype for QSAR studies; but uneducated use, bad practices, and poor reporting led to inflated expectations and disappointment (Johnson, 2008). As part of the hype, scientific journals containing the word “QSAR” in the title emerged, and years later, some journals were re-named. Molecular docking is another example of a method that is often misused, leading to false expectations and disappointments, not because the technique is not useful but because it is tried to be used for purposes that was not initially designed (e.g., correlation of docking cores with experimental binding affinities). At the time of writing this manuscript, there is a hype for AI, ML, DL; quoting Bajorath, an “AI ecstasy” (Bajorath, 2021).
Despite the contributions of CADD in different stages of the drug discovery pipelines and technological advances, there are challenges that need to be addressed. Table 1 outlines the grand challenges that face drug discovery using in silico methods and AI and are further commented on in this manuscript. The list of topics is not exhaustive; the selected challenges are based on the author’s opinion, and it is intended to be a reference for a continued update. Here, the challenges are organized into six sections. The first two are related to the chemical and biological relevant chemical spaces, respectively; that is, what spaces are being explored? Another section covers methodological challenges: how is being conducted the search for new and better drugs at the intersection of the relevant chemical and biological spaces? The next three sections present hurdles associated with communication and Human interaction in research teams, scientific dissemination, data sharing, and education, respectively. The last section contains the Conclusions.
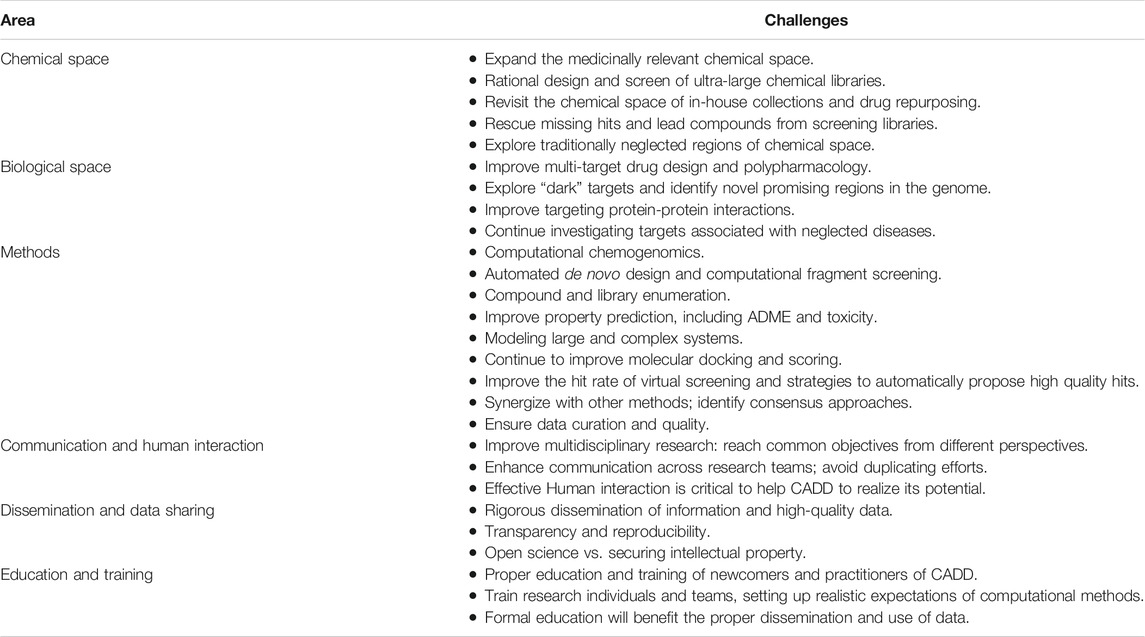
TABLE 1. Grand challenges that face drug discovery and design using in silico methods and AI.
Chemical Space
The chemical space itself, including its visualization, has been fundamental in decision making and analyzing the advances in the drug discovery process (Reymond, 2015). The searchable chemical space offers a vast number of possibilities to find hit compounds, in particular with the advent of approaches to increase its size rapidly. In this context, the grand challenge question could be framed as follow: which regions of the chemical scape should be investigated to identify potential drug candidates? Searching on the traditional, drug-like medicinally relevant chemical space has not led to the identification of many new chemical entities (Mullard, 2021). As such exploring novel regions might help to identify novel starting points. Challenges in this direction include but are not limited to expand the searchable drug-like chemical space (e.g., take advantage of the ultra-large chemical libraries); revisit and repurpose existing chemical libraries, including revisiting large corporate collections and the SAR of in-house data: rescue missing hits; search traditional chemical spaces with novel techniques; revisit “neglected chemical spaces,” e.g., type of molecules poorly explored: peptides, peptidomimetics, macrocycles, biologics, metallodrugs (Anthony et al., 2020; Pandya and Patravale, 2021). These types of molecules could lead to promising starting points to develop drug candidates for the novel or difficult targets. To this end, novel computational approaches will have to be developed.
Biological Space
A crucial quest in drug discovery is identifying and validating molecular targets with clinical applications. The grand challenge is in what regions of the biologically relevant chemical space look for? Identification of the therapeutically relevant targets has been the subject of recent comprehensive analysis (Oprea et al., 2018). Further, it is also of major importance to study polypharmacology and try to predict the interactions of a drug candidate with all its putative target partners (Méndez-Lucio et al., 2017). Challenges of CADD methods in this area include but are not limited to improving multi-target drug design and multi-target property prediction (Zhang et al., 2017); identifying better ways to address protein-protein interactions (Lu et al., 2020); and continue exploring molecular targets associated with neglected diseases. Computational methods can also help to identify druggable binding pockets and allosteric sites, and transient binding sites that can be exploited for drug discovery.
Methods
In parallel to defining the chemical and biological spaces to be addressed for drug discovery comes the grand challenge: which computational methods could be used to help designing and optimizing drug candidates? General avenues are continually developing and improving computational chemogenomics (Jacoby and Brown, 2018). On this regard, it is also crucial to keep refining the selection of descriptors to delineate the chemical and biological spaces.
There are ongoing challenges in CADD, such as improving the hit rate of virtual screening; improve molecular docking, including docking of flexible compounds and macrocycles; covalent and protein-protein docking; molecular modeling of large and complex systems; prediction of druggable pockets, binding sites, including allosteric binding sites; and accurate prediction of ADMETox-related properties, among others. Another major and continued grand challenge is data curation; ensure data quality, including that stored in public databases. Such data is a key to develop reliable models and information.
There are also newer or emerging challenges, a number of which are driven by AI (Schneider et al., 2020), such as (fully) automated de novo design (Schneider and Clark, 2019); improve the performance and demonstrate experimentally the practical applications of predictive models including models generated with ML and DL. A challenge is to continue enforcing proper model validation: one of the key aspects that have been discussed since the emergence of traditional QSAR modeling (Muratov et al., 2020). Another current challenge is expanding the searchable chemical space through ML-based compound and library enumeration, the rational design, and mining of large and ultra-large chemical libraries (Irwin et al., 2020; Coley, 2021).
In the author’s opinion, one should avoid pursuing the single “best” or “universal CADD approach” that performs well in all cases. Likely, such a method does not exist. Instead, consider combining different and complementary approaches so as to try to solve the scientific questions more fully and efficiently (Schneider, 2018). All in all, a grand challenge involves also the selection and rational use of the best AI, ML, DL, and related trendy methods (Medina-Franco et al., 2021).
Communication and Human Interaction
Lack of communication between research individuals and teams hampers drug discovery progress and might become one of the most significant drivers of project failures. Poor communication between principal investigators, postgraduate and undergraduate students, etc., has been a constant challenge that warrants improvement. The efforts of computational teams or individuals might be futile if there is no open dialog and credibility among research teams. It is desirable to work synergistically with experts across different research disciplines, each contributing to the projects goals from different perspectives and complementary areas of expertise. Close collaboration and effective communication will help to reduce the chances of duplicating efforts and waste valuable resources.
Dissemination and Data Sharing
Proper documentation and peer-review have been critical components in science to disseminate information and knowledge. A current challenge that is being addressed is proper data and method sharing in favor of transparency and reproducibility. Peer-reviewed journals are enforcing these practices, including the FAIR principles (Wilkinson et al., 2016). A sensitive point is when the information, data, and methods can be openly shared partially only when intellectual property is at stake. Good editorial practices, proper peer-review, and the author's ethics and responsibility should work synergistically to disseminate high-quality data essential for several CADD approaches, those that are data-driven.
Education and Training
Proper education and formal training are necessary for students, young and established investigators from different disciplines moving to CADD or collaborating with computational teams. This is key to help to improve multidisciplinary work and maximize the CADD’s benefits. There are efforts towards this endeavor (Guha and Willighagen, 2020; López-López et al., 2021). A challenge is continuing the formal training of theoretical disciplines. In particular, because there are now several easy-to-use and freely available applications, their misapplication and flawed interpretation of the results can lead to false expectations and perceived CADD’s disappointments (Medina-Franco et al., 2021).
Conclusion
CADD has been successful but must improve by addressing old challenges and facing new ones. Technological advances are promising to revisit and expand the medicinally relevant chemical space, help to guide the repurposing of approved drugs and existent chemical libraries, and rescuing missing hits hidden as “treasures to be discovered” in the existing SAR of corporate or public data. CADD methods are promising to explore novel chemical and biological spaces relevant to drug discovery. Data-driven approaches such as ML and DL require high-quality data, which generation and dissemination is another major challenge. Researchers working directly with CADD methods or collaborators with computational teams should recognize and pursue realistic expectations to keep the rationale over fashion and hype (Medina-Franco et al., 2021). Another challenge is to continue improving traditional approaches in drug discovery; synergize and mutually improve and benefit from other traditional and successful techniques in drug discovery (for example, natural products, combinatorial chemistry, and traditional medicinal chemistry). Enhancing communication in research teams is critical to achieve and maximize the potential of the computational approaches. Proper scientific dissemination, data sharing, and education are other challenges that need to be addressed to avoid false expectations but maximize the productivity of CADD.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
JM-F research is currently supported in part by CONACyT (Mexico) project 282785, UNAM, DGAPA projects IN201321, IV200121; and Miztly project LANCAD-UNAM-DGTIC-335.
Conflict of Interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The valuable feedback and suggestions of Fernanda I. Saldívar-González are greatly acknowledged.
References
Anthony, E. J., Bolitho, E. M., Bridgewater, H. E., Carter, O. W. L., Donnelly, J. M., Imberti, C., et al. (2020). Metallodrugs Are Unique: Opportunities and Challenges of Discovery and Development. Chem. Sci. 11 (48), 12888–12917. doi:10.1039/D0SC04082G
Bajorath, J. (2021). State-of-the-art of Artificial Intelligence in Medicinal Chemistry. Future Sci. OA. 7, FSO702. doi:10.2144/fsoa-2021-0030
Bender, A., and Cortés-Ciriano, I. (2021). Artificial Intelligence in Drug Discovery: What is Realistic, What Are Illusions? Part 1: Ways to Make an Impact, and Why We are Not There yet. Drug Discov. Today. 26 (2), 511–524. doi:10.1016/j.drudis.2020.12.009
Coley, C. W. (2021). Defining and Exploring Chemical Spaces. Trends Chem. 3 (2), 133–145. doi:10.1016/j.trechm.2020.11.004
Gasteiger, J. (2020). Chemistry in Times of Artificial Intelligence. ChemPhysChem 21 (20), 2233–2242. doi:10.1002/cphc.202000518
Guha, R., and Willighagen, E. (2020). Learning Cheminformatics. J. Cheminform. 12 (1), 4. doi:10.1186/s13321-019-0406-z
Irwin, J. J., Tang, K. G., Young, J., Dandarchuluun, C., Wong, B. R., Khurelbaatar, M., et al. (2020). ZINC20-A Free Ultralarge-Scale Chemical Database for Ligand Discovery. J. Chem. Inf. Model. 60 (12), 6065–6073. doi:10.1021/acs.jcim.0c00675
Jacoby, E., and Brown, J. B. (2018). The Future of Computational Chemogenomics. Methods Mol. Biol. 1825, 425–450. doi:10.1007/978-1-4939-8639-2_15
Johnson, S. R. (2008). The Trouble With QSAR (Or How I Learned to Stop Worrying and Embrace Fallacy). J. Chem. Inf. Model. 48 (1), 25–26. doi:10.1021/ci700332k
López-López, E., Bajorath, J., and Medina-Franco, J. L. (2021). Informatics for Chemistry, Biology, and Biomedical Sciences. J. Chem. Inf. Model. 61 (1), 26–35. doi:10.1021/acs.jcim.0c01301
Lu, H., Zhou, Q., He, J., Jiang, Z., Peng, C., Tong, R., et al. (2020). Recent Advances in the Development of Protein-Protein Interactions Modulators: Mechanisms and Clinical Trials. Sig Transduct Target. Ther. 5 (1), 213. doi:10.1038/s41392-020-00315-3
Medina-Franco, J. L., Martinez-Mayorga, K., Fernández-de Gortari, E., Kirchmair, J., and Bajorath, J. (2021). Rationality Over Fashion and Hype in Drug Design [Version 1; Peer Review: 1 Approved]. F1000Res. 10 (397), 397. doi:10.12688/f1000research.52676.1
Méndez-Lucio, O., Naveja, J. J., Vite-Caritino, H., Prieto-Martínez, F. D., and Medina-Franco, J. L. (2017). Polypharmacology in Drug Discovery. Drug Selectivity., 1–29. doi:10.1002/9783527674381.ch1
Mullard, A. (2021). 2020 FDA Drug Approvals. Nat. Rev. Drug Discov. 20 (2), 85–90. doi:10.1038/d41573-021-00002-0
Muratov, E. N., Bajorath, J., Sheridan, R. P., Tetko, I. V., Filimonov, D., Poroikov, V., et al. (2020). QSAR without Borders. Chem. Soc. Rev. 49, 3525–3564. doi:10.1039/D0CS00098A
Oprea, T. I., Bologa, C. G., Brunak, S., Campbell, A., Gan, G. N., Gaulton, A., et al. (2018). Unexplored Therapeutic Opportunities in the Human Genome. Nat. Rev. Drug Discov. 17 (5), 317–332. doi:10.1038/nrd.2018.14
Pandya, A. K., and Patravale, V. B. (2021). Computational Avenues in Oral Protein and Peptide Therapeutics. Drug Discov. Today. 26 (6), 1510–1520. doi:10.1016/j.drudis.2021.03.003
Prieto-Martínez, F. D., López-López, E., Eurídice Juárez-Mercado, K., and Medina-Franco, J. L. (2019). “Computational Drug Design Methods-Current and Future Perspectives,” in In Silico Drug Design. Editor K. Roy (Academic Press), 19–44. doi:10.1016/b978-0-12-816125-8.00002-x
Reymond, J.-L. (2015). The Chemical Space Project. Acc. Chem. Res. 48 (3), 722–730. doi:10.1021/ar500432k
Schneider, G. (2018). Automating Drug Discovery. Nat. Rev. Drug Discov. 17 (2), 97–113. doi:10.1038/nrd.2017.232
Schneider, G., and Clark, D. E. (2019). Automated De Novo Drug Design: Are We Nearly There yet?. Angew. Chem. Int. Ed. 58 (32), 10792–10803. doi:10.1002/anie.201814681
Schneider, P., Walters, W. P., Plowright, A. T., Sieroka, N., Listgarten, J., Goodnow, R. A., et al. (2020). Rethinking Drug Design in the Artificial Intelligence Era. Nat. Rev. Drug Discov. 19 (5), 353–364. doi:10.1038/s41573-019-0050-3
Wilkinson, M. D., Dumontier, M., Aalbersberg, I. J., Appleton, G., Axton, M., Baak, A., et al. (2016). The FAIR Guiding Principles for Scientific Data Management and Stewardship. Sci. Data. 3 (1), 160018. doi:10.1038/sdata.2016.18
Keywords: artificial intelligence, biological space, chemical space, chemoinformatics, computer-aided drug discovery, molecular modeling, virtual screening
Citation: Medina-Franco JL (2021) Grand Challenges of Computer-Aided Drug Design: The Road Ahead. Front. Drug. Discov. 1:728551. doi: 10.3389/fddsv.2021.728551
Received: 21 June 2021; Accepted: 13 July 2021;
Published: 28 July 2021.
Edited and reviewed by:
Bruno Villoutreix, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCopyright © 2021 Medina-Franco. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: José L. Medina-Franco, bWVkaW5hamxAdW5hbS5teA==