
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PROTOCOLS article
Front. Drug Deliv. , 24 May 2022
Sec. Ophthalmic Drug Delivery
Volume 2 - 2022 | https://doi.org/10.3389/fddev.2022.898568
This article is part of the Research Topic Ocular gene therapy: where we are and where to go? View all 4 articles
 Sidsel Alsing1Anna Bøgh Lindholm1Jakob Haldrup1Emilie Grarup Jensen1
Sidsel Alsing1Anna Bøgh Lindholm1Jakob Haldrup1Emilie Grarup Jensen1 Jacob Giehm Mikkelsen1Lars Aagaard1
Jacob Giehm Mikkelsen1Lars Aagaard1 Anne Louise Askou1,2*†
Anne Louise Askou1,2*† Thomas Corydon1,2†
Thomas Corydon1,2†Gene therapy is a promising therapeutic modality for ocular diseases arising in and affecting the retina and choroid. In this context, delivering gene therapy to the multifunctional retinal pigment epithelium (RPE) cells situated between the retina and choroid is desired. Efficacy assessment of any gene therapy strategy, whether it is gene augmentation, inhibition, or editing is initially tested in vitro in cell models, where delivery is simple and efficient. However, efficacy assessment in vivo in animal models is far more complex and several factors can influence the result significantly. Here we report a simple fluorescence activated cell sorting (FACS)-based enrichment method for direct assessment of efficacy and potential off-target effects of gene therapy co-delivered with an eGFP reporter to murine RPE cells using subretinal administration. Isolation of true eGFP+ RPE cells by FACS is notoriously difficult due to their intrinsic autofluorescence resulting in decreased sensitivity and false positives. Combining retinal dissection and harvest of RPE cells with a FACS-gating strategy utilizing the GFP filter and a neighboring filter, to separate the eGFP signal from autofluorescence, allows a significant enrichment of gene therapy-targeted eGFP+ RPE cells. In our hands the method may provide quantitative and qualitative advances in terms of up to 7-fold enrichment of true eGFP+ RPE cells compared to a standard protocol. The isolated cells can subsequently be utilized for reliable assessment of changes in DNA, RNA, or protein. This method allows proof-of-principle analysis of early gene therapy development and investigation of new delivery strategies or therapeutic approaches targeting RPE cells in vivo.
Inherited retinal diseases (IRDs), such as retinitis pigmentosa and macular degenerations, are characterized by progressive vision loss caused by variations in genes encoding proteins responsible for development, function, or survival of the photoreceptor (PR) cells. The PRs are the light-sensitive cells of the retina, initiating visual phototransduction by capturing incoming light and converting the light into electrical signals. Adjacent to the PRs, the RPE cells form the outermost cell layer of the retina. The RPE cells are vital for the survival of PRs, expressing multiple molecules crucial for the health, maintenance, and function of the PR cells, transporting oxygen and nutrients essential from the choroid, and disposing of retinal waste and metabolic end-products from PRs. Patients with biallelic mutations in the RPE65 gene can be treated with Luxturna, which is currently the only FDA-approved gene augmentation therapy for retinal dystrophies, but clinical trials for delivery of other genes to the PRs and the RPE cells are underway (Clinicaltrials.gov identifier: NCT04671433, NCT03496012, NCT02935517). While gene augmentation strategies for treatment of IRDs caused by loss-of-function mutations have led to the early success in the field, efforts are now increasingly focused on development of novel approaches for treatment of the remainder of causative variations, including variations leading to gain-of-function or dominant negative effects. Current gene therapy strategies to modulate expression of detrimental alleles or dysregulated genes include genome editing and RNA interference (RNAi) (Askou et al., 2021). Notably, the recent success in the field have also sparked an interest in gene therapy for the more prevalent acquired retinal disorders characterized by dysfunctional RPE cells, such as age-related macular degeneration (Clinicaltrials.gov identifier: NCT04704921, NCT03585556, NCT04645212, NCT01024998) (Askou, 2014).
Before a new gene therapy is ready for clinical testing, proof-of-principle studies are conducted in small animal models to examine the efficacy. However, since efficacy is strongly correlated with delivery, assessment of efficacy in the rodent eye can be precluded by sub-optimal delivery. Optimal delivery of gene-coding, virus-based vectors to the RPE cells can be achieved by a subretinal injection (Askou et al., 2012; Askou et al., 2015), a micro-precise process with inherent variability between injections (Alsing et al., 2022). In mice, this leads to differences in the size of the transduced area, and thus a variation among mice in the same treatment group. This constitutes a substantial challenge, particularly if the expected in vivo efficacy is low, which is often the case with the novel genome editing tools such as CRISPR-based technology (Benati et al., 2020; Banskota et al., 2022; Jang et al., 2022; Zhi et al., 2022). Therefore, in vivo efficacy is most often determined by assessment of editing in sorted cells (Hung et al., 2016; Latella et al., 2016; Yu et al., 2017; Giannelli et al., 2018; Matsuda and Oinuma, 2019).
Fluorescence-activated cell sorting (FACS) is a well-established and preferred method for cell sorting. Highly pure cell populations can be isolated from tissue composed of several cell types, such as the retina, through antibody-labeled, cell-surface markers. When assessing the efficacy of a novel gene therapy tool, isolation of a specific cell subpopulation is desired; the target cells that received the gene therapy or gene editing tool. By co-delivering genes expressing fluorescent proteins, such as the enhanced green fluorescent protein (eGFP) (Askou and Corydon, 2018), isolation of gene therapy target cells is possible by FACS methods (Holmgaard et al., 2017; Alsing et al., 2022). In this context, isolation of targeted RPE cells can be a hurdle. First of all, to date, no RPE specific cell-surface markers have been identified for flow sorting of primary murine RPE cells. Therefore, prior to target cell sorting, RPE cells must be collected by dissection of the murine eye. Moreover, as opposed to other cells in the retina, RPE cells produce strong autofluorescence which overlaps the excitation/emission spectrum of eGFP. The autofluorescence of a cell is related to the intracellular granule type, and the RPE cells accumulate several different kinds such as lipofuscin and melanolipofuscin, which are highly autofluorescent after blue light excitation (Bermond et al., 2021). Accumulation of lipofuscin increases gradually with age, and represents a marker of retinal stress (Einbock et al., 2005; Ueda et al., 2016; Moreno-Garcia et al., 2018).
Here we present an optimized protocol for autofluorescence-restrictive sorting of eGFP positive (eGFP+) RPE cells following co-delivery of therapeutic molecules with eGFP to murine RPE cells, including delivery with lentiviral vectors (LVs). Our protocol is simple, only requiring standard filter sets for the FACS and allows enrichment of gene therapy targeted cells, where the challenge often lies in contamination of rare, desired events with unwanted cells (Holmgaard et al., 2021). This can advance retinal gene therapy development, particularly for early proof-of-principle assessment of gene therapy targeted to the RPE cells.
Male C57Bl/6J mice (8-weeks-old, weight 24–28 g) were purchased from Janvier Labs (Le Genest-Saint-Isle, France). Animals were handled as previously described (Askou et al., 2019), and kept on a 12 h/12 h light/dark cycle at the Animal Facilities at the Department of Biomedicine, Aarhus University, Denmark. Mice had ad libitum access to water and Altromin maintenance feed (Altromin, Brogaarden, Denmark). Animals were handled in accordance with the “Statement for the Use of Animals in Ophthalmic and Vision Research” from the Association for Research in Vision and Ophthalmology (ARVO). All animal experiments were performed under the approval of The Danish Animal Inspectorate (case# 2020-15-0201-00745 and 2020-15-0201-00556).
0.9% saline solution: Sodium chloride 9 mg/ml (Braun, Melsungen, Germany).
Ketamine: Ketador Vet 100 mg/ml (Richter Pharma AG, Wels, Austria).
Medetomidin hydrochloride: Cepetor Vet 1 mg/ml (ScanVet Animal Health A/S, Fredensborg, Denmark).
1% tropicamide: Mydriacyl® (Alcon Nordic A/S, Copenhagen, Denmark).
Carbomer eye gel: Viscotears® (Alcon Nordic).
Carprofen: Rimadyl Bovis Vet 50 mg/ml (Zoetis Finland Oy, Helsinki, Finland).
Atipamezole: Antisedan 5 mg/ml (Orion Pharma, Copenhagen, Denmark).
ProLong® Gold antifade reagent (Invitrogen, Life Technologies Corporation, Eugene, OR).
4′,6-diamidino-2-phenylindol (DAPI; Sigma, St. Louis, MO, United States).
DNAfree (Thermo Fisher Scientific, Waltham, MA, United States).
RNeasy micro kit (Qiagen, Hilden, Germany).
iScript cDNA synthesis kit (BioRad, Hercules, CA).
RealQ Plus Master Mix Green (Ampliqon, Odense, Denmark).
HBSS: Hanks’ Balanced Salt solution 1X (Gibco; Thermo Fisher Scientific).
Hyaluronidase solution: 1 mg/ml hyaluronidase (Sigma) in HBSS.
Fetal bovine serum (Sigma) is diluted x5 in HBSS to obtain 20% FBS-HBSS.
0.25% Trypsin-EDTA solution (Sigma).
0.25% Trypsin-EDTA solution (Sigma) is diluted x5 in HBSS to obtain 0.05% Trypsin/EDTA.
FACS buffer: 1 w/v% BSA (VWR, Radnor, PA), 2.5 mM EDTA (Sigma), 25 mM HEPES (Alfa Aesar, Haverhill, MA) in HBSS.
TE buffer pH 9.0: 10 mM Tris (Sigma 7-9®; Sigma), 1 mM EDTA (Ethylenediaminetetraacetic acid disodium salt dihydrate; Sigma) in H2O.
PBS: Dulbecco's Phosphate Buffered Saline without magnesium and calcium (BioWest, Nuaillé, France).
Blocking buffer: 3 w/v% BSA (VWR), 1% TritonTM X-100 in PBS.
PFA-PBS: 4% paraformaldehyde (Merck Life Science A/S, Søborg, Denmark) in PBS.
4x Laemmli Sample Buffer (Biorad) with 50 mM DTT (Sigma).
TBS-T: Tris Buffered Saline (Thermo Fisher Scientific) with 0.1% v/v Tween-20 (Sigma).
Blocking buffer: in 5% w/v skim milk powder (VWR) in TBS-T buffer.
Clarity™ Western ECL substrate (BioRad).
Xylene (Supelco®, Radnor, PA).
Ethanol.
Ice.
UF H2O.
Vectors: Lentiviral transfer vectors encoding either an anti-VEGF RNAi effector or an irrelevant control was engineered, LV/VMD2-miR451-12 and LV/VMD2-miR451-S1, respectively. The shRNA is driven by the RPE specific vitelliform macular dystrophy 2 (VMD2) promoter. Furthermore, the vector contains an expression cassette with eGFP expressed from the ubiquitously active phosphoglycerate kinase 1 (PGK) promoter. The back-to-back orientation of the two expression cassettes as well as the full cloning strategy can be found in (Alsing et al., 2022). VSV-G-pseudotyped LV particles were produced, purified, and the infectious titer determined by RT-qPCR as described in (Alsing et al., 2022). In this paper, LV/shRNAVEGF-eGFP corresponds to LV/VMD2-miR451-12, and LV/shRNAIrr-eGFP corresponds to LV/VMD2-miR451-S1.
Primers (Eurofins Genomics, Ebersberg, Germany): eGFP forward primer: GGTGAACTTCAAGATCCGCC and reverse primer: CTTGTACAGCTCGTCCATGC, product size: 229 bp. Vegfa forward primer: CAGGCTGCTGTAACGATGAA and reverse primer: AATGCTTTCTCCGCTCTGAA, product size: 207 bp. Actb forward primer: CACTGTCGAGTCGCGTCC and reverse primer: TCATCCATGGCGAACTGGTG, product size: 89 bp.
Antibodies: Anti-GFP antibody for IHC (G10362, Invitrogen, 0.2 mg/ml), anti-GFP antibody for WB (ab290, Abcam, 5 mg/ml), anti-RPE65 antibody (ab13826, Abcam, Cambridge, Great Britain, 1 mg/ml), anti-vinculin (V9131, Sigma, 5–10 mg/ml), Alexa 568-conjugated donkey-anti-rabbit (A10042, Invitrogen, 2 mg/ml), HRP-conjugated goat-anti-mouse antibody (P0447, Dako, Agilent, Agilent Technologies, Santa Clara, CA, 1 mg/ml), HRP-conjugated goat-anti-rabbit antibody (P0448, Dako, 0.25 mg/ml).
A fluorescence activated cell sorter, minimum equipped with a 488 nm laser and detectors for GFP/FITC and PE. We used the BD FACSAria III (BD Biosciences, Franklin Lakes, NJ).
LightCycler 480 (Roche, Basel, Switzerland) and software (Roche, release 1.5.1.62).
Incubator 37°C.
Water bath 37°C.
Centrifuge for 50 ml tubes and 1.5 ml tube (up to 1000 g).
Rocking table.
Microwave oven.
Heating block for 1.5 ml tubes (up 100°C).
Jar with a lid for glass slides.
Humidity chamber.
SDS-PAGE gel: Midi-PROTEAN TGX Stain-Free Gels, 4–15%, 18-well comb, 30 µl (Biorad).
Electrophoresis chamber and power supply.
Trans-Blot® Turbo™ Midi 0.2 µm PVDF Transfer Pack (Biorad).
Trans-Blot® Turbo™ Transfer System (Biorad).
ChemiDoc™ MP imaging system (BioRad).
Dissection microscope.
OPMI 1 FR PRO ophthalmic surgical microscope with foldable tube (f170/f260) and KL 1600 LED schott light source (Zeiss, Jena, Germany).
Olympus VS.120 slide scanner (Olympus Corporation, Tokyo, Japan).
Fluorescence microscope for GFP/FITC and TRITC detection. We used a Leica DM IRBE (Leica Microsystems, Wetzlar, Germany) equipped with a mercury lamp and excitation and emission filters were as follows: DAPI: Excitation BP 360/40, dichromatic mirror 400, and suppression filter 470/40; GFP/FITC: excitation BP 480/40, dichromatic mirror 505, and suppression filter 527/30; and TRITC: excitation BP 560/40, dichromatic mirror 595, and suppression filter 645/75.
Hamilton syringe 5 μl, Model 65 RN SYR (P/N 7633-01, Hamilton Company, Bonaduz, Switzerland).
Hamilton RN Needles, 34 gauge/10mm/pst2/tapN (P/N 207434, Hamilton Company).
p10, p100, and p1000 pipettes.
Angled forceps.
Micron IV imaging system (Phoenix Research Laboratories, Pleasanton, CA).
Dumont style 3C Dumoxel® tweezers (Sigma).
Vannas scissors 8 cm, straight, 5 mm blades (Simonsen & Weel A/S, Vallensbæk Strand, Denmark).
SuperFrost®Plus glass slide (Menzel-Glaser, Braunschweig, Germany).
LifterSlip premium printed cover glass (Erie Scientific Company, Portsmouth, NH).
Plastics and consumables 48-well plate, 12-well plate, 100 μm cell strainer (Falcon, Thermo Fisher Scientific), 50 ml tubes, 1.5 ml tubes, 1.5 ml tubes with screw caps, p1000 tips, Petri dishes, plastic coverslips or parafilm, glass coverslips (D 12 mm), Multiply®-μStrip 0.2 ml chain and 8-Lid chain (flat; Sarstedt, Nürnbrecht, Germany), Lightcycler® 480 Multiwell Plate 96 (white; Roche), filterpapers, 1 ml syringes, G27 needles.
This protocol describes delivery of LVs encoding anti-angiogenic RNAi molecules and eGFP to murine RPE cells (Figures 1A,B) as described in (Alsing et al., 2022). Steps involving gene therapy and eGFP co-delivery to murine RPE, in vivo and ex vivo assessment of expression and localization in the retina, and isolation of RPE cells and autofluorescence-restrictive cell sorting (Figure 1C), are outlined in Figure 1C. The details are as follows:
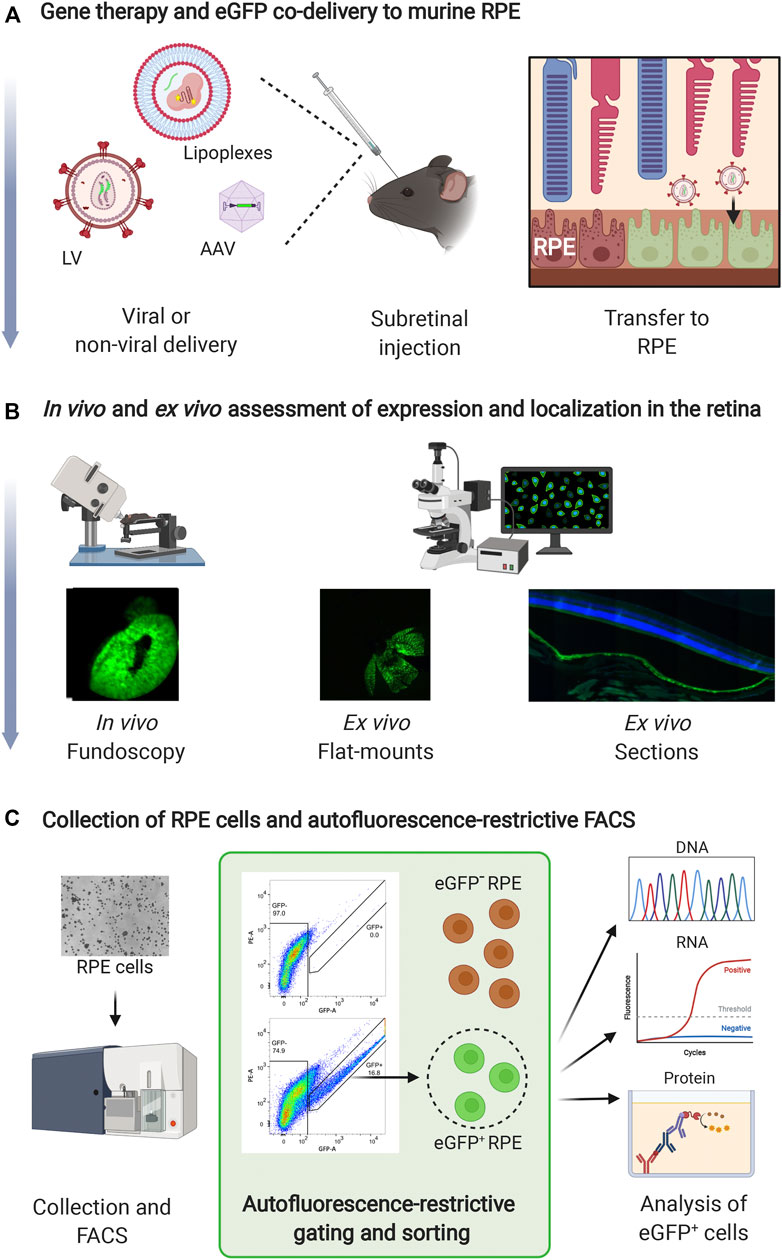
FIGURE 1. Method overview. (A) Gene therapy and eGFP can be co-delivered to murine RPE cells via a subretinal injection in mice. Delivery vehicles can be viral [e.g., LVs or Adeno-Associated Virus (AAV) vectors] or non-viral. Depending on the gene delivery vehicle, eGFP can be delivered as a gene or mRNA. (B) eGFP can be used to easily visualize retinal transduction/transfection in vivo and ex vivo. Fluorescence fundoscopy (in vivo) or RPE/choroid/sclera flat-mounts (ex vivo) can be prepared to examine the size of the transduced area. Paraffin or frozen sections of the eye are used to examine the localization of eGFP expression in the retina. (C) Mice are sacrificed, and RPE cells are collected by dissection and enzymatic digestion. Following collection, eGFP+ RPE cells are isolated by autofluorescence-restrictive gating and sorting, and the eGFP+ RPE cells can be utilized for subsequent downstream DNA, RNA, or protein analysis (indicated by the green square). Furthermore, eGFP− RPE cells can be isolated as control sample. Created with BioRender.com.
1) Mice were anesthetized with an intraperitoneal injection of ketamine (60–100 mg/kg) and medetomidin hydrochloride (0.8–1.2 mg/kg).
2) Pupils were dilated with a drop of 1% tropicamide solution, whereafter carbomer eye gel was used to keep eyes lubricated.
3) 24 mice were subretinally injected with 4.13 ∙ 105 (IUs) LV/shRNAVEGF-eGFP (12 mice, unilateral) or 4.54 ∙ 105 IUs of the LV/shRNAIrr-eGFP (12 mice, unilateral) in a total volume of 2 µl as described previously (Askou et al., 2019). Subretinal injections were performed under an ophthalmic surgical microscope. The contralateral eyes from 6 mice (4 from the LV/shRNAIrr-eGFP group and two from the LV/shRNAVEGF-eGFP group) were used as un-injected controls.
4) For pain relief, mice were treated with a subcutaneous injection of carprofen (5 mg/kg) just before the subretinal injection and during the next 3 days after via their drinking water (3.33 mg/100 ml).
5) Mice were brought out of anesthesia with 0.8–1.2 mg/kg Atipamezole.
To detect eGFP expression in vivo, fluorescence fundoscopy was carried out with the Micron IV imaging system, Figure 1B and Figures 2A,B. Mice were anesthetized and pupils dilated as described in step (1)−(2). We could not detect eGFP by fluorescence fundoscopy 3 days post injection (dpi), but 13 dpi eGFP was detectable (Figure 2B), and at 126 dpi sustained eGFP expression was evident (data not shown).
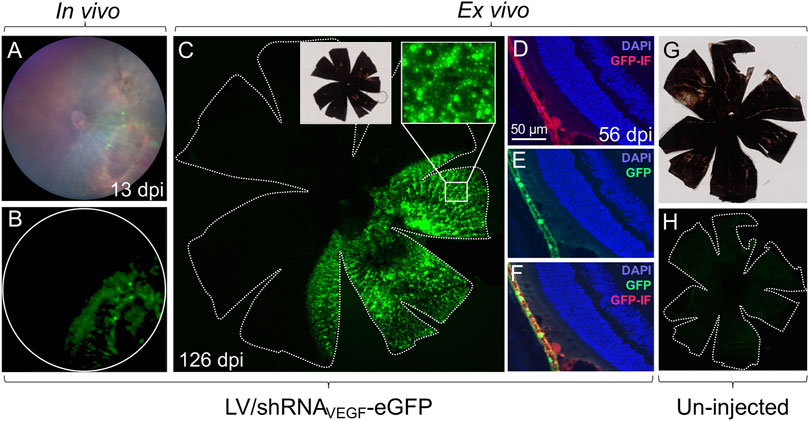
FIGURE 2. Localization of eGFP expression in the mouse retina. Different methods can be used to visualize eGFP expression in the mouse retina following co-delivery of eGFP and gene therapy. (A,B) In vivo, fundoscopic examination can be performed with the Micron IV imaging system. Images show a representative fundus of a mouse, 13 dpi of 4.13 × 105 IU of LV/shRNAVEGF-eGFP. (C) RPE/choroid/sclera flat-mount of the eye shown in (A,B). The mouse was sacrificed 126 dpi. Images were captured using an Olympus VS.120 slide scanner (×20 and ×40 objectives). The contour of the flat-mount is marked by a dotted line. (D–F) Images of eGFP (green) and eGFP immunofluorescence (red) on paraffin sections of a mouse 56 dpi of 4.13 × 105 IU of LV/shRNAVEGF-eGFP. (G,H) RPE/choroid/sclera flat-mount of un-injected control eye captured using an Olympus VS.120 slide scanner (×20 objective) and with the same settings as in (C). The contour of the flat-mount is marked by a dotted line.
6) Mice were sacrificed at 126 dpi. Using angled forceps, the eyes were enucleated and immediately transferred to 4% PFA-PBS for fixation. Eyes were fixed at 4°C for 24 h. Note, all work with 4% formalin solution should be performed in a ventilated cabinet or fume hood.
7) Eyes were transferred and rinsed in PBS. Following fixation, eyes can be kept in PBS at 4°C for several days. Note, if the dissection described in the following is performed immediately after fixation, it should be performed in a ventilated cabinet or fume hood to avoid fumes from the fixative. Excess fixative can be removed by placing the eyes in PBS for 24 h at 4°C.
8) For dissection, the eyes were placed on a filter paper in a Petri dish soaked in ice-cold PBS (Figure 3A) and dissected using a microscope. For the dissection, we used Vannas scissors. Periocular tissue was carefully removed without making any cuts in the sclera, the overlying choroid, or RPE layer. Note, to be able to mount the RPE/choroid/sclera flat, it is important, that all connective tissue, muscles etc. is completely cleaned away (Figure 3B).
9) The cornea, lens, and vitreous were removed by making a circumferential incision just behind the sclera-cornea divide (Figures 3C,D).
10) The eyecup was grabbed by the optic nerve and transferred to a new Petri dish with ice-cold PBS. The protruding parts of the optic nerve were cut off. Note, in subsequent steps, hold on to the eyecup by the edge and avoid touching the tissue, to avoid damaging the RPE cells.
11) The eyecup was placed with the neuroretina facing downward. The RPE/choroid/sclera layers were gently peeled off (Figure 3E) and pulled away from the neuroretina using Dumont tweezers and Vannas scissors. The internal attachment to the optic nerve was cut with Vannas scissors (Figure 3F).
12) Four incisions were placed in the eye cup from the periphery to the optic nerve (Figure 3G1). Additionally, four incisions can be placed from the periphery halfway to the optic nerve between the initial four cuts.
13) The RPE/choroid/sclera flat-mounts were gently transferred to a SuperFrost®Plus glass slide using the Dumont tweezers. The flat-mounts were gently unfolded with the RPE-cell layer facing upwards using the Dumont tweezers. Remaining PBS was drained from the flat-mount by approaching the side of the tissue with filter paper.
14) Cover glass was mounted using ProLong® Gold antifade reagent (Figure 3H1). Flat-mounts from injected and un-injected eyes were analyzed for eGFP expression by fluorescence microscopy using an Olympus VS.120 slide scanner (×20 and ×40 objectives) and associated VS-ASW imaging software. Representative brightfield and eGFP images of LV/shRNAVEGF-eGFP and un-injected eyes are presented in Figures 2C,G,H.
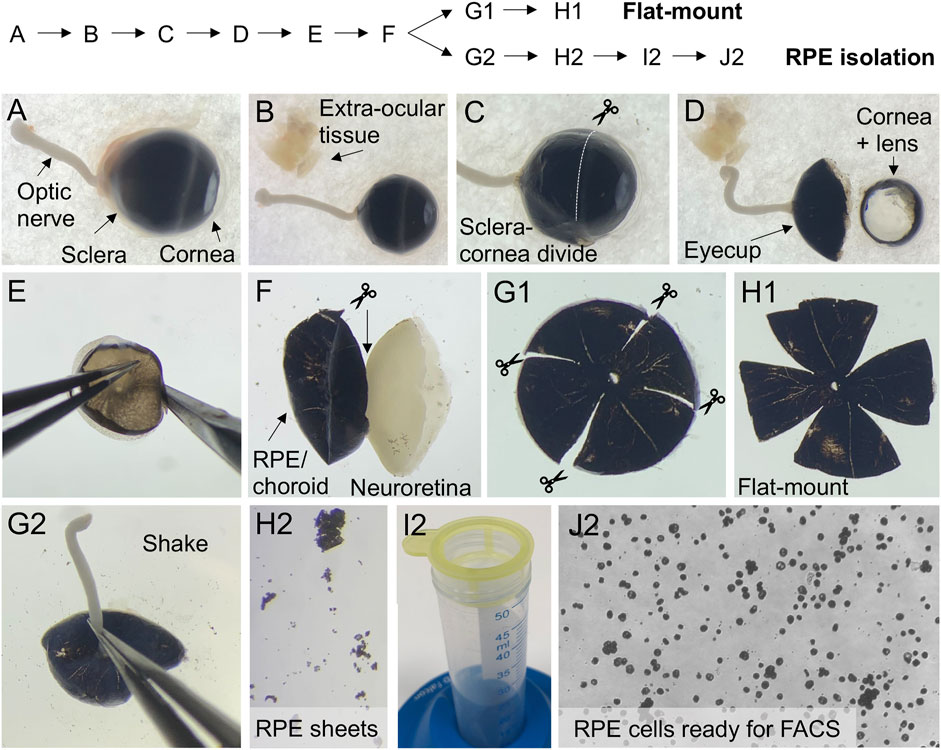
FIGURE 3. Dissection of murine eyecups for flat-mounts and collection of RPE cells for FACS. The flow chart indicates the order of steps involved in dissection of murine eyecups (A–F) to obtain flat-mounts (G1,H1) or to isolate RPE cells for FACS (G2–J2). (A) Enucleated eye with arrows pointing to the optic nerve, the posterior (sclera) and anterior (cornea) part of the eye. (B) Extra-ocular tissue is removed. (C) Image showing where to cut, to remove the anterior part of the eye. (D) Dissected eyecup, and the removed cornea and lens. (E) Image showing how to position the eyecup, while gently peeling the RPE/choroid/sclera of the neuroretina. (F) When the RPE/choroid/sclera is peeled of, cut the internal attachment to the optic nerve. (G1) For ex vivo examination of eGFP expression, make four radial incisions in the RPE/choroid/sclera eyecup. (H1) RPE/choroid/sclera flat-mount. (G2) For FACS of eGFP+ RPE cells, the eyecup is placed face down in 20% FBS after trypsin digestion. Hold it by the optic nerve and gently shake it vertically to detach the RPE. (H2) Detached RPE sheets. (I2) Sieve the RPE cell suspension using a cell strainer to prepare for FACS. (J2) Collected single-cell RPE suspension ready for FACS.
15) Mice were sacrificed at 57 dpi, and using angled forceps, the eyes were enucleated and immediately transferred to 4% PFA-PBS for fixation. Eyes were fixed for 24–48 h at 4°C. Note, all work with 4% formalin solution should be performed in a ventilated cabinet or fume hood.
16) After fixation, eyes were embedded in paraffin and sectioned.
17) 5 µm thick paraffin sections from LV/shRNAVEGF-eGFP and LV/shRNAIrr-eGFP transduced mouse retinas were thawed, and paraffin was removed by placing the sections in xylene overnight at room temperature (RT).
18) Sections were rehydrated through graded dilutions of ethanol in water.
19) Target antigen retrieval was performed by placing the sections in TE buffer pH 9.4 and heated in the microwave oven for 3 min at low effect. Subsequently, the jar with slides were placed on a rocking table for at least 30 min to cool down.
20) Sections were washed in PBS.
21) Slides were placed in a humidity chamber, and 200 µl blocking buffer was added to each slide. Slides were covered with plastic coverslips or parafilm, to make sure the buffer was evenly distributed. Slides were incubated with blocking buffer for 30 min at RT.
22) The coverslips were removed, and 150 µl of 1:100 diluted anti-GFP antibody in PBS containing 1 w/v% BSA and 1 v/v% Triton was added to each slide. Slides were covered with plastic coverslips and incubated in a humidity chamber at 4 overnight.
23) Sections were placed at RT for 30–60 min, and washed 3x in PBS, before incubation with 1:400 diluted Alexa 568 conjugated donkey anti-rabbit in PBS containing 1 w/v % BSA. 150 µl antibody solution was added to each slide placed in a humidity chamber for 60 min at RT.
24) Slides were washed 3x in PBS and stained with DAPI.
25) Slides were washed 2x in UF H20, dipped in 70% ethanol and dried for 30 min at RT, before mounting a coverslip with two drops of ProlongTM Gold antifade reagent.
26) Sections were analyzed by fluorescence microscopy (Leica DM IRBE). Images were captured with a Leica DFC 360 FX camera and associated software (Leica Application Suite v3), Figures 2D–F.
The 12 mice injected with LV/shRNAIrr-eGFP were divided into 4 groups (G1−4) of three mice, corresponding to mouse 1–12, and the 12 mice injected with LV/shRNAVEGF-eGFP were divided into 4 groups (G5−8) of three mice, corresponding to mouse 13–24 (Supplementary Figure S1). Each group produced a sample consisting of RPE cells pooled from three injected eyes. Furthermore, three groups of cells pooled from two un-injected eyes each were produced (N1−3).
27) At 14 dpi, mice were sacrificed by cervical dislocation. Using angled forceps, the eyes were enucleated, and placed in HBSS on ice. The best results will be obtained, if the mouse is sacrificed just prior to enucleation. Note, extended incubation in HBSS on ice will cause the RPE cells to die.
28) The eyes were placed on a filter paper in a Petri dish soaked in ice-cold HBSS (Figure 3A) and dissected using a microscope. For the dissection, we used Vannas scissors. Periocular tissue was removed (Figure 3B), and subsequently, the cornea, lens and vitreous were removed by a circumferential incision just behind the sclera-cornea divide (Figures 3C,D).
29) The eyecups were placed in one well of a 48-well plate containing 0.5 ml hyaluronidase solution and incubated at 37°C for 45 min.
30) The eyecups were transferred to a new 48-well plate containing HBSS and incubated on ice for 30 min, to stop the hyaluronidase activity.
31) The eyecups were transferred to a Petri dish containing fresh HBSS buffer, where the neural retina was gently removed (Figures 3E,F).
32) The remaining eyecup was transferred to a 48-well plate containing 0.5 ml of 0.25% trypsin/EDTA solution and incubated at 37°C for 45 min.
33) To stop the trypsin activity, the eyecups were transferred to a 48-well plate containing 0.5 ml FCS.
34) To detach the RPE cells, each eyecup was placed with the RPE side downwards. The eyecup was held by the external attachment of the optic nerve, and gently shaken vertically into a 12-well plate containing 0.5 ml of 20% FCS in HBSS until all the RPE cells were detached, Figure 3G2. Detached RPE sheets from pigmented mice can be easily visualized, by placing a white filter paper underneath the dish, Figure 3H2.
35) The solution with the RPE cells, the trypsin/EDTA solution, and the FCS were transferred to a 15 ml Falcon tube with a p1000 pipette. RPE cells from three injected eyes or two uninjected eyes were pooled in the same tube.
36) The RPE cell pools were centrifuged at 1000 g for 2 min at RT and the supernatant was discarded.
37) The pellet was carefully resuspended in a 0.05% trypsin/EDTA solution and incubated in a water bath at 37°C for 1 min.
38) Four ml of FACS buffer was added to the RPE cell solution, which was transferred to a 100 μm cell strainer (Figure 3I2). The strainer was washed 3 times with 1 ml FACS buffer
39) The RPE cell solution was centrifuged for 2 min at 1000 g.
40) The RPE cell pellet was resuspended in 300 µl FACS buffer, and the solution was kept at 4°C until analysis (Figure 3J2).
The collected RPE cells were sorted by FACS using a BD FACSAria™ III cell sorter equipped with 3-lasers (375, 488 and 637 nm). In this protocol, the 488 nm laser was used for excitation and 530/30 (eGFP detector) and 585/42 (PE detector) bandpass filters were used for detection. Dissociated cells were maintained in the sample chamber at 4°C and collection tubes were also kept at 4°C. PBS was used as sheath fluid. The FACSAria was set up with a 100 µm nozzle and 20 psi. Instrument quality control is performed daily after fluidics startup by the core facility staff, per manufacturer instructions. Subsequently, the sorter was additionally cleaned because the sorted material was used for RNA purification. The sort chamber and holder were cleaned in RNAse Zapp, as well as the table and pipettes for sample handling. In addition, 5 min FACS Clean at flow rate 11 was acquired through the sample line, followed by 1 min sample line backflush and 5 min autoclaved miliQ water at flow rate 11. Accudrop was run using a vial of sterile accudrop and side streams were adjusted for a two-way sorting into 1.5 ml tubes.
41) The cell population was identified based on forward scatter (FSC) and side scatter (SSC), Figure 4A. Debris was removed and a rough percentage of cells is indicated in the plot.
42) FSC and SSC doublets were excluded by gating on area vs. height plots for FSC and SSC, respectively, Figures 4B,C. The percentage of FSC singlets and SSC singlets are indicated on the plots. Figures 4A–C are representative plots, and gating of all four samples analyzed in Figures 4D–K is shown in Supplementary Figure S2.
43) The standard method for isolating eGFP+ cells is to sort according to the detected eGFP levels either on a histogram or vs. SSC-A or FSC-A, Figures 4D,F,H,J. The percentage of RPE cells in the eGFP+ gate is indicated on the plots. The eGFP− cell population is also indicated on the plots.
44) To enrich for true eGFP+ cells and avoid autofluorescent cells, we identified the eGFP+ cell population based on fluorescence measured in the eGFP detector and in the neighboring PE detector. The gating strategy is shown in Figures 4E,G,I,K. The percentage of eGFP+ RPE cells are indicated on the plots. The eGFP− cell population is also indicated on the plots.
45) eGFP+ and eGFP− cells were collected in separate 1.5 ml tubes with screw caps in 50 µl HBSS. Note, collect cells in a buffer compatible with the downstream analysis you wish to perform.
46) The collected RPE cells were centrifuged for 3 × 5 min at 300, 500, and 1000 g, respectively. Then the supernatant was removed, and the cell pellet was frozen at −80°C.
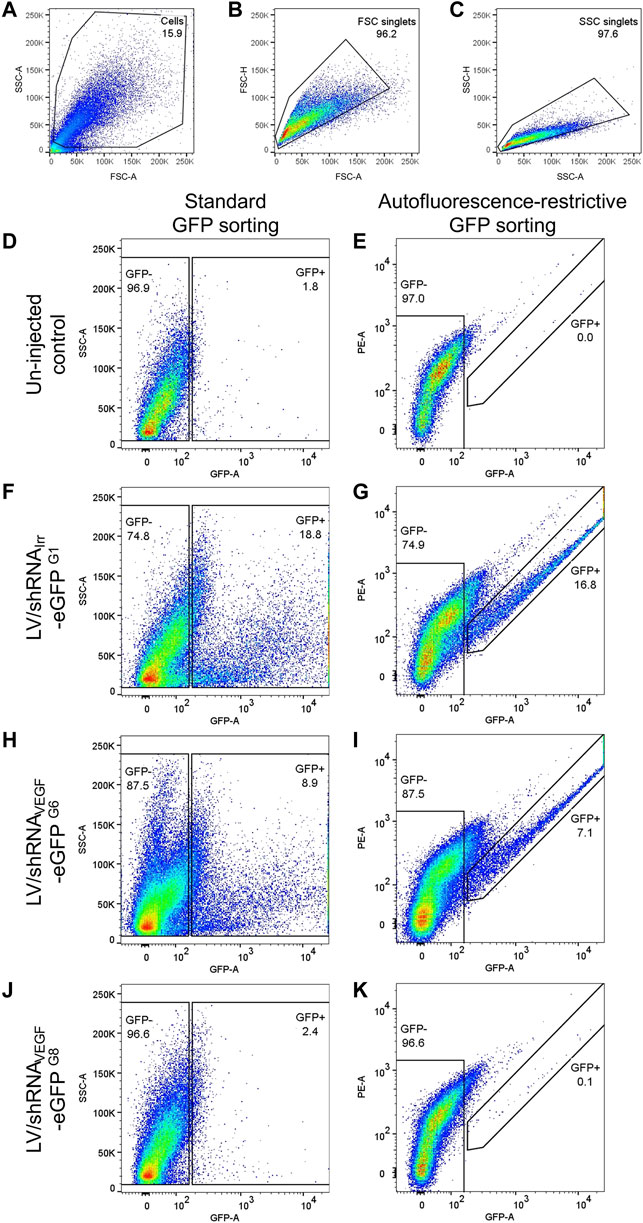
FIGURE 4. Autofluorescence-restrictive gating strategy. Representative examples of the gating strategy for isolation of eGFP+ RPE cells from mice injected with LV particles and un-injected controls. (A) The RPE cell population was identified based on forward scatter (FSC) and side scatter (SSC). (B) FSC doublets, and (C) SSC doublets were excluded. (D,F,H,J) Standard isolation according to eGFP levels measured in the eGFP detector (530/30 nm). (E,G,I,K) To exclude autofluorescent RPE cells, the eGFP+ cells were identified based on fluorescence measured in the eGFP detector (530/30 nm) and in the neighboring PE detector (585/42 nm). Cells in the eGFP− gate was isolated as a control. (D,E) Gating of eGFP+ RPE cells from two un-injected eyes. (F,G) gating of eGFP+ RPE cells from three eyes injected with the LV/shRNAIrr-eGFP (Supplementary Figures S1, G1, m1−m3). (H,I) gating of eGFP+ RPE cells from three eyes injected with the LV/shRNAVEGF-eGFP (Supplementary Figures S1, G6, m16−m18), and (J,K) gating of eGFP+ RPE cells from three eyes injected with the LV/shRNAVEGF-eGFP, where the eGFP signal was undetectable by fluorescence fundoscopy (Supplementary Figures S1, G8, m22−m24). Gating of all four samples analyzed in (D–K) is shown in Supplementary Figure S2. The percentage of eGFP+ and eGFP− RPE cells for each cell pool is indicated. FSC, SSC, and applied detector are indicated on the plots.
Following FACS, G2 and G8 did not yield sufficient eGFP+ cells for purification of RNA, therefore RNA was purified from the eGFP+ cell pools from G1 and G3−7 and eGFP− cell pools from G1−8 (Supplementary Figure S1), and from the eGFP− cell pools from N1−3.
47) RNA was purified using the RNeasy micro kit according to the manufacturer’s protocol. In brief, addition of lysis buffer followed by vortexing was used to disrupt and homogenize the samples. On-column DNase digestion was omitted. If recovery of protein is desired, the flow-through that is obtained following binding of RNA to the column, is kept on ice and the steps (57)−(59) is followed to precipitate protein. The RNA was eluted with 14 µl of RNase-free water.
48) The eluate was treated with DNAfree according to the manufacturer’s protocol, resulting in a final volume of approximately 11 µl.
49) cDNA was synthesized with 5 µl of the DNase-treated RNA in a 10 µl reaction volume using the iScript cDNA synthesis kit. This allows RT-qPCR analysis of 4–5 genes with the method described in step (50)−(56). For all samples, a DNA contamination control without reverse transcriptase (-RT) was included, using 2.5 µl of the DNase-treated RNA in a total volume of 5 µl.
The primer sequences for eGFP, Vegfa, and Actb can be found in the Materials and Equipment section. Further details on primer design and choice of reference gene can be found in (Alsing et al., 2022). Data analysis was performed using the LightCycler 480 software.
50) RealQ Plus Master Mix Green was prepared according to the manufacturer’s protocol in a total reaction volume of 10 µl. A primer concentration of 0.5 µM was used.
51) 7 µl of the Master Mix was dispensed in white 96 well plates. 3 μl of diluted (1:6) cDNA synthesis reaction was used as template and transferred to the wells. All reactions, except negative controls (-RT, UF H2O) were analyzed in technical triplicates.
52) A standard curve was made using a mix of cDNA samples harvested from RPE cells from whole eyecups in 5-fold serial dilutions and included to determine the PCR efficiency. The efficiency was 92% for eGFP, 98% for Vegfa and 98% for Actb.
53) The qPCR was performed using the LightCycler 480 instrument and software. Cycling conditions were set to 10 s denaturation, 20 s annealing, and 30 s elongation for 50 cycles. Annealing temperatures for Vegfa and Atcb primer pairs were set to 58°C, and to 60°C for eGFP primers.
54) Cq values were determined with the 2nd derivative max method, and Cq values above 35 were not considered. If the difference in the technical triplicates exceeded 0.5 Cq values, the most extreme value was excluded. The range of Cq values was between 22 and 34.5 for Atcb and Vegfa, and between 23 and 29 for eGFP in the eGFP+ samples.
55) For each of the genes, concentrations were calculated from the Cq values using the efficiency calculated from the standard curve. For Vegfa and Actb the relative quantity (RQ) was calculated by relating the concentrations for each sample to the concentration of the control samples, and for eGFP the concentration was related to the eGFP+ sample with the lowest expression of eGFP. The RQ for Vegfa and eGFP were then normalized using the RQ for Actb. Results are presented on Figures 5A,B. The -RT DNA contamination controls were analyzed in parallel, providing Cq values close to or above 35, or >5 Cq values larger than the corresponding positive sample, indicating no DNA contamination.
56) The specificity of the primer pairs was confirmed using Tm analysis and/or gel electrophoresis.
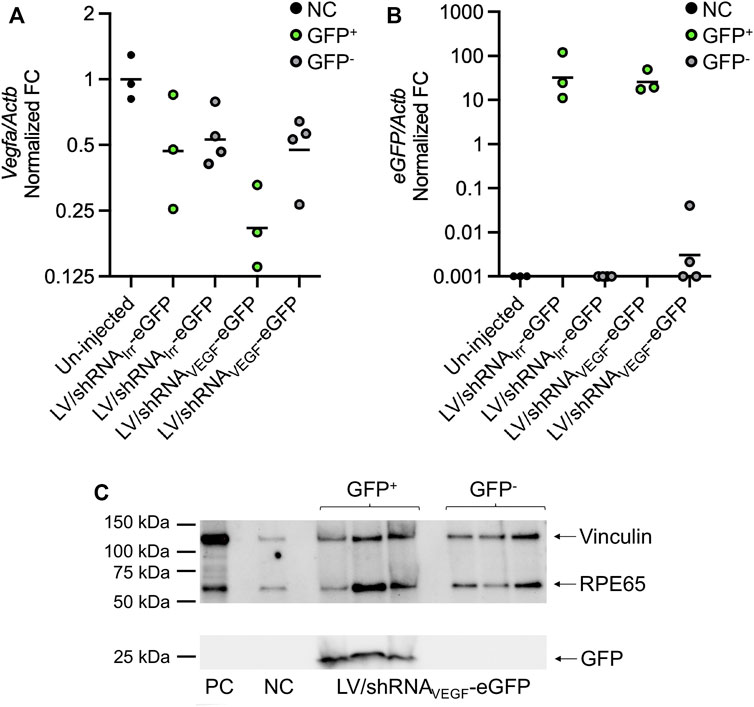
FIGURE 5. Analysis of eGFP and Vegfa expression in eGFP+ and eGFP− RPE cell pools by RT-qPCR and detection of GFP and RPE65 by Western blotting. RPE cells from eyes injected with LV/shRNAIrr-eGFP or LV/shRNAVEGF-eGFP, or from un-injected control eyes were harvested at 14 dpi. RNA was purified from the eGFP+ RPE pools from G1 and G3−7 (green dots; Supplementary Figure S1) and eGFP− cell pools from G1−8 (grey dots; Supplementary Figure S1), and Vegfa and eGFP mRNA was quantified using RT-qPCR. (A) Actb-normalized Vegfa fold change (FC) relative to un-injected control eyes (black dots), and (B) Actb-normalized eGFP FC relative to the eGFP+ sample with the lowest expression of eGFP with the geometric mean indicated. Samples with Cq values >35 were assigned the FC value of 0.001. Each data point represents a pool of FACS isolated RPE cells. (C) Western blot-detection of RPE65, GFP, and vinculin (loading control) in eGFP− and eGFP+ RPE pools isolated from LV/shRNAVEGF-eGFP injected mice and un-injected control (NC). PC, positive control, cell lysate from primary, murine RPE cells (no cell sorting). (A) is a reprint from (Alsing et al., 2022) with permission from the American Society of Gene & Cell Therapy.
Protein from flow-through collected in step (47) was precipitated by acetone as described in the following steps. eGFP+ and eGFP− RPE cell pools from LV/shRNAVEGF-eGFP injected eyes as well as from one of the negative control samples (un-inj.) were analyzed by Western blotting. As a positive control (PC), we used lysates from RPE cells collected from mice (no cell sorting).
57) 4 volumes of ice-cold acetone were added to the flow-through, and then incubated for 30 min on ice.
58) The precipitates were centrifuged for 10 min at full speed and the supernatant was discarded.
59) The precipitate was stored at -80°C until analysis. The pellet was washed with 100 µl of ice-cold ethanol and air-dried.
60) Each pellet was resuspended in 19.5 µl PBS. For each of the three eGFP+ RPE cell pools, 19.5 µl resuspended protein was transferred to a new 1.5 ml tube with 6.5 µl 4x Laemmli Sample buffer. For the three eGFP− RPE cell pools, 1.95 µl resuspended protein was transferred to a new tube, with 17.55 PBS and 6.5 µl 4x Laemmli Sample buffer. For the eGFP− RPE cell pool isolated from un-injected eyes, 2.93 µl resuspended protein was transferred to a new tube, with 16.57 PBS and 6.5 µl 4x Laemmli Sample buffer.
61) Samples were boiled for 7 min, and 25 µl of each sample were immediately loaded on an SDS-PAGE gel and electrophoresed. Note, pellets of precipitated protein can contain traces of guanidine thiocyanate from the buffers in the RNeasy micro kit. Addition of sample buffer containing sodium dodecyl sulfate (SDS) can cause guanidine salts to precipitate. This was particularly evident in the undiluted samples from the three eGFP+ RPE cell pools.
62) After electrophoresis, the gel was activated for 45 s using the ChemiDoc MP imaging system before transfer to a PVDF membrane. Total protein was visualized using the ChemiDoc MP imaging system, but was undetectable.
63) The membrane was transferred to blocking buffer and placed on a rocking table for 1 hour at RT.
64) The membrane was incubated overnight at 4°C with an anti-RPE65 antibody at a concentration of 1:1000 with constant rocking.
65) The membrane was washed in TBS-T 3 times for 5 min and incubated with an HRP-conjugated goat-anti-mouse antibody in a concentration of 1:10,000 for 1 hour at RT placed on a rocking table.
66) Detection was done using Clarity Western ECL substrate and imaging was done using the ChemiDoc MP imaging system.
67) If immunolabeling of other proteins is desired, wash the membrane in TBS-T 3 times for 5 min and repeat step (64)−(66). We re-probed the membrane with anti-vinculin (primary antibody concentration 1:10,000), followed by anti-GFP (primary antibody concentration 1:10,000). The result is presented in Figure 5C.
Co-delivery of eGFP allows easy visualization of the transduced area in the retina as well as the localization of the gene therapy. There are different methods, which can be used to assess eGFP expression (Figure 1B). In this protocol we have performed in vivo fluorescence fundoscopy and ex vivo examination of RPE/choroid/sclera flat-mounts as well as paraffin retinal sections (Figure 2, Figure 6 and Supplementary Figure S1). Retinal sections confirm RPE specific eGFP expression following subretinal delivery of VSV-G pseudo-typed LVs (Figure 2D-F). At 13 dpi, we were able to detect eGFP expression in vivo. However, the timepoint for detection will depend on severable variables, e.g., delivery vehicle, amount, and, in the case of eGFP gene delivery, the promoter. In our experience, following delivery of the eGFP gene by high amounts of VSV-G-LV or single-stranded AAV2.5, eGFP fluorescence can be detected by in vivo fluorescence fundoscopy as early as 6 (Askou et al., 2017) and seven dpi (unpublished data), respectively, whereas delivery of eGFP mRNA by lipoplexes (Holmgaard et al., 2021) and the eGFP gene by lentiviral particles [LPs; integrase-deficient LVs adapted as carriers of non-viral proteins (Cai et al., 2014; Mikkelsen, 2018)] was below detection limits when examined in vivo at 4 and 5 dpi, respectively. However, ex vivo examination of these retinas revealed eGFP expression in RPE cells at 4 and 5 dpi (Figures 6A–C,G–I).
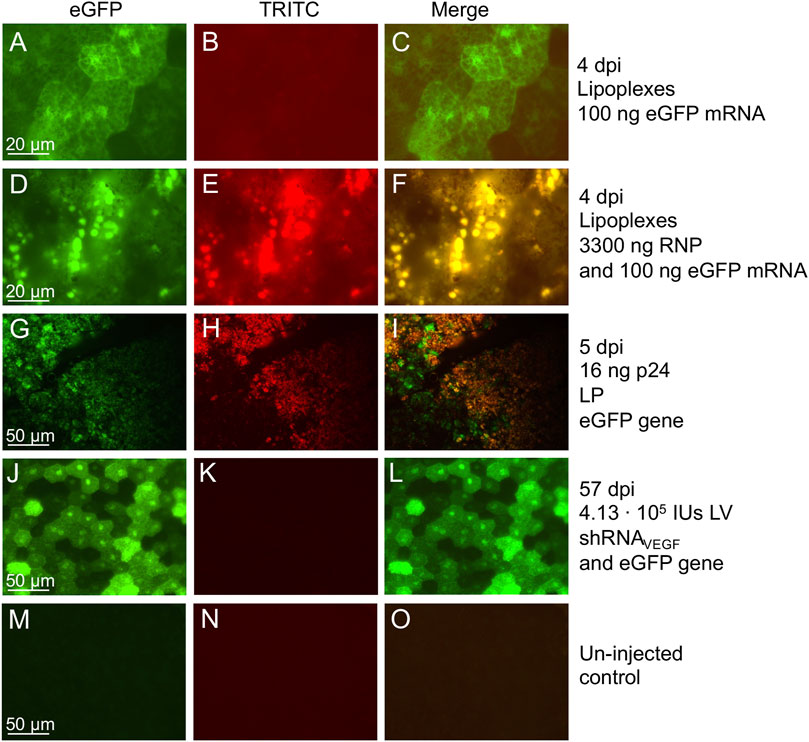
FIGURE 6. RPE autofluorescence following subretinal delivery of lipoplexes, LPs, and LVs. Fluorescence microscopy examination of RPE/choroid/sclera flat-mounts from mice injected with viral or non-viral vectors reveals autofluorescence in the RPE cells illustrated by red in the TRICT images and yellow in the merged images. True eGFP+ signal can be visualized as green in the merged images. (A–C) Lipoplexes containing 100 ng eGFP mRNA, and examined at 4 dpi. Exposure time: 2 s eGFP and TRITC (Holmgaard et al., 2021). (D–F) Lipoplexes containing 100 ng eGFP mRNA and 3300 ng RNP, and examined at 4 dpi. Exposure time: 2 s eGFP and TRITC (Holmgaard et al., 2021). (G–I) 16 ng p24 of LP encoding eGFP, and examined at 5 dpi. Exposure time: 1.5 s eGFP and TRITC (ongoing study). (J–L) 4.13 ⋅ 105 IU of LV/shRNAVEGF-eGFP encoding eGFP, and examined at 57 dpi. Exposure time: 0.7 s eGFP and 0.2 s TRITC (Alsing et al., 2022). (M–O) Un-injected control eye, age-matched to mice shown in (A–F). Exposure time: 2 s eGFP and TRITC (Holmgaard et al., 2021).
The simple gating strategy that allows enrichment of true eGFP+ RPE cells by excluding autofluorescent cells is shown in Figure 4. Autofluorescent RPE cells will emit light with equal intensity as measured in the GFP and PE detector when excited by the 488 nm laser, thus placing the cells in the diagonal of the plot, whereas emission from eGFP+ cells will be red-shifted in the PE detector. Representative data following FACS of RPE cells collected from two groups of mice are shown: three eyes injected with the LV/shRNAIrr-eGFP (Figures 4F,G and Supplementary Figures S1, G1, m1−m3) and from three eyes injected with the LV/shRNAVEGF-eGFP (Figures 4H,I and Supplementary Figures S1, G6, m16−m18). For comparison, and to investigate background autofluorescence in the retina of mice injected with LV particles, we included sorting of two un-injected eyes (Figures 4D,E) and three eyes injected with the LV/shRNAVEGF-eGFP, where the eGFP signal was undetectable by in vivo fluorescence fundoscopy (Figures 4J,K and Supplementary Figures S1, G8, m22−m24). Application of this protocol enabled isolation in the range of 1000–5000 eGFP+ RPE cells/group of three retinas from the mice shown in Supplementary Figures S1, G1 and G3−G7.
In our recently published paper, the autofluorescence-restrictive sorting protocol (Figure 1C) was applied to FACS isolate eGFP+ RPE cells (Alsing et al., 2022). In the study, we did not detect a significant reduction of Vegfa mRNA levels following isolation of the total RPE cell population (no cell sorting) from murine eyecups injected with LV particles encoding anti-VEGF RNAi-molecules and subsequent RNA purification and RT-qPCR analysis. However, when we collected RPE cells, and applied the FACS method described here to isolate and enrich for eGFP+ RPE cells, we found a 55% reduction in the Vegfa mRNA levels in the eGFP+ RPE cells from mice injected with the LV/shRNAVEGF-eGFP compared to Vegfa mRNA levels in the eGFP+ RPE cells from mice injected with the LV/shRNAIrr-eGFP (Figure 5A) (Alsing et al., 2022). We used RT-qPCR to confirm that the eGFP+ RPE cell populations express eGFP mRNA (Figure 5B). Detection of small amounts of eGFP mRNA in the eGFP− cell pool from mice injected with LV/shRNAVEGF-eGFP (Figure 5B) is believed to be due to imperfect sorting. The sorting efficiency was not determined since this would reduce the number of isolated cells. Further to this, we confirmed by Western blotting of extract from eGFP+ RPE cells that the isolated cells express eGFP as well as RPE65, which primarily is expressed in an RPE-specific manner (Hamel et al., 1993; Nicoletti et al., 1995) (Figure 5C). Contamination with other cell types is not expected when collecting RPE cells as described in the protocol, but it can be verified by evaluating the expression of markers for other cell types by RT-qPCR.
Previously, we have experienced increased autofluorescence in the RPE cells following subretinal injection of both viral and non-viral vectors in mice (Figure 6). The most extensive increase in autofluorescence was found, when treated mice were examined shortly (4–5 dpi) after a subretinal injection of high amounts of Cas9-ribonucleoprotein (RNP) delivered by lipoplexes (Figures 6D–I). The increase in autofluorescence was not due to the injection, the delivery of eGFP mRNA or the transfection reagent applied (Lipofectamine 3000; compare Figures 6D–F). The increase in autofluorescence hampered the FACS procedure, when we isolated RPE cells according to eGFP intensity. This is illustrated in Figure 7, where we have marked autofluorescent, contaminating cells in the GFP+ gates. In all four cases (un-injected control, LV/shRNAIrr-eGFP G1, LV/shRNAVEGF-eGFP G6, and LV/shRNAVEGF-eGFP G8) a significant number of autofluorescent cells is observed, which will dilute the outcome measure e.g., the editing or knock-down efficiency provided by the transferred gene therapy tool. We further validated the gating strategy by backgating the cell sort data from FACS isolation of eGFP+ RPE cells collected from LP-injected mice (ongoing study) 5 dpi on a GFP vs. PE plot (Figure 8). Comparing the percentage of eGFP+ cells that would have been obtained by the new gating strategy (0.4%) to the percentage of eGFP+ cells that were in fact isolated in the study based on eGFP intensity alone (2.8%), indicates that our method could have enriched the sample by a factor 7.
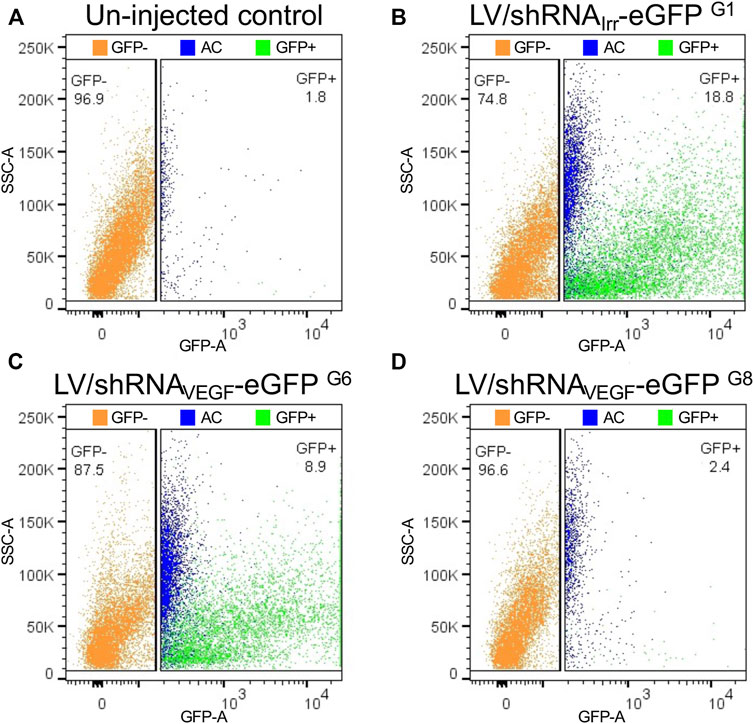
FIGURE 7. Backgating and pseudo-coloring on GFP vs. SSC-A plots to illustrate autofluorescent cells, when RPE cells are sorted according to eGFP intensity. Cells in the eGFP− and eGFP+ gates illustrated in Figures 4E,G,I,K (un-injected control group, G1, G6, and G8 (Supplementary Figure S1) have been plotted on GFP vs. SSC-A plots corresponding to (A–D), respectively. Blue dots in the annotated eGFP+ cell pool represent autofluorescent cells (AC) from the eGFP+ gate shown in Figures 4D,F,H,J. The percentage of eGFP+ (green) and eGFP− (orange) RPE cells for each cell pool is indicated. SSC, side scatter.
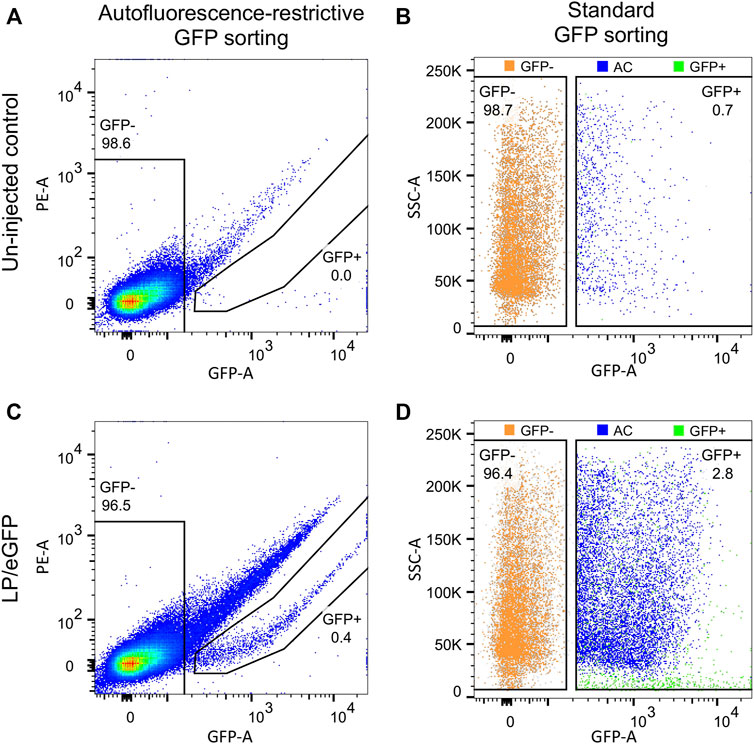
FIGURE 8. Application of GFP vs. PE gating strategy on FACS data set from eGFP+ RPE cells isolated from LP-injected mice. RPE cells from mice injected with LPs encoding eGFP were collected as described in step (27)−(40) at 5 dpi (ongoing study). The RPE cell population from (A) un-injected and (C) LP-injected mice were backgated on GFP vs. PE plots for autofluorescence-restrictive GFP sorting. (B) and (D) illustrate autofluorescent cells (AC; blue dots), when RPE cells are isolated according to standard GFP sorting. The eGFP− (orange) and eGFP+ (green) cell pools as indicated by gates in (A) and (C). The percentage of eGFP+ and eGFP− RPE cells for each cell pool is indicated. SSC, side scatter.
To our knowledge, we are the first to describe a method for FACS isolation of eGFP+ primary murine RPE cells. Several protocols exist for FACS isolation of cultured RPE cells, iPSC-differentiated RPE cells, and FACS isolation of other cell types in the retina, e.g., retinal ganglion cells or PRs (Westenskow et al., 2012; Chintalapudi et al., 2016; Giannelli et al., 2018; Plaza Reyes et al., 2020). Furthermore, cell specific surface markers have not been identified for isolation of primary murine RPE cells. Thus, we present a simple and robust method, without the need for additional expensive filter sets for the cell sorter, transgenic mice expressing fluorescent proteins in the RPE, or optimization of antibody-based staining protocols prior to FACS. Most published FACS protocols facilitate isolation of cells, where it is possible to increase the intensity of the fluorescent signal or to decrease the autofluorescence. However, when we rely on the signal from eGFP co-delivered with gene therapy tools, we have intrinsic low intensity of the fluorescent signal. This is because we aim to deliver a low copy number per cell to avoid toxicity, when we provide the gene or mRNA encoding eGFP to living cells in the mouse. Standard FACS protocols rely on antibody-labelling of surface markers following cell collection from the animal. Autofluorescence is cell type specific and can for instance be induced in the murine RPE cells shortly after high amounts of RNPs are delivered in the subretinal space (Holmgaard et al., 2021). The number of autofluorescent RPE cells can be reduced by prolonging the time from subretinal injection to harvest and by ensuring ultra-clean vector preparations for injection. However, in the case of eGFP mRNA delivery, the possibility of extending the timepoint for harvest is limited by the in vivo half-life of eGFP mRNA and protein.
Co-delivery of eGFP, either as mRNA or the gene, not only allows easy visualization of gene transfer, but also allows isolation of targeted RPE cells for downstream analysis of DNA, RNA, or protein (Figure 1). We demonstrated the success of the isolation protocol using LV injected eyes from 10-weeks-old C57BL/6J mice to analyze gene expression (Alsing et al., 2022). However, the method is widely applicable to other mouse strains, for delivery using other vectors such as AAV, and to analyze DNA following RPE isolation (Holmgaard et al., 2021). Different RPE collection methods can be applied prior to FACS isolation as long as the RPE cells remain intact. We have adapted a protocol for collection of RPE cells utilizing hyaluronidase to detach the neural retina from the RPE layer followed by enzymatic digestion using a reduced concentration of trypsin combined with shaking of the eyecup to gently detach the RPE cells from the Bruch’s membrane (Fernandez-Godino et al., 2016). The collection of RPE cells takes approximately 3 h and this protocol was designed to collect RPE cells from mice at any age from 10 days to 12 months (Fernandez-Godino et al., 2016).
Expression of fluorescent reporter genes in murine retinal target cells and the subsequent isolation of these, have previously been applied to show proof-of-principle for in vivo gene knock-out using CRISPR/Cas9 (Hung et al., 2016; Latella et al., 2016; Yu et al., 2017; Giannelli et al., 2018; Koo et al., 2018), genome editing by HDR (Matsuda and Oinuma, 2019; Vagni et al., 2019), and base editing (Levy et al., 2020). Recently, novel, prime editor-based tools have been delivered to the mouse retina by co-injection of AAV8-mediated split-intein prime editors. At 6 weeks post injection, genomic DNA was isolated from the mouse retina without sorting. Next generation sequencing revealed an average editing efficiency of 1.71 ± 1.35% in the Dnmt1 locus (Zhi et al., 2022). In another study, two trans-splicing prime editor-expressing AAVs were delivered together with an AAV expressing the Atp7b-targeting pegRNA and sgRNA into the retina via intravitreal injection (Jang et al., 2022). The retinal cells were collected for deep sequencing 6 weeks post injection, which revealed an average editing efficiency of 1.87% in retinal cells. Again, no cell sorting was applied.
The protocol provides a useful method for isolation of true eGFP+ cells, which can be used to evaluate gene transfer and gene editing in murine RPE cells, without the need of expensive deep sequencing or the use of transgenic animals. It is particularly useful for proof-of-principle studies for new gene therapy tools or delivery methods, where the expected outcome measure is relatively small, such as the low rate of in vivo editing. Methods to deliver gene editing tools as RNPs offer advantages such as increased safety and packaging capacity. Several promising vehicles for protein delivery have emerged including LPs (Cai et al., 2014), nanoblades (Mangeot et al., 2019) and most recently, virus-like particles (VLPs) (Banskota et al., 2022). However, protein delivery of editing tools to the target tissue has been shown to be a limitation that significantly impacts the in vivo editing efficiency of these techniques. Taking these findings in consideration, we believe that our autofluorescence-restrictive FACS-based protocol (Figure 1C), providing up to 7-fold enrichment of the sample consisting of true eGFP+ RPE cells, which express RPE65, could greatly improve the future development of novel delivery vehicles for retinal gene therapy.
One disadvantage of this method is that it requires co-delivery of eGFP. Even though eGFP is the most widely used gene expression marker and cellular tag, evidence suggest immunogenicity and cytotoxicity of eGFP (Ansari et al., 2016). Therefore, all cells originally marked with eGFP will not survive, which will lead to a deterioration of the eGFP expression over time. Application of the method described in this protocol will only enable isolation of RPE cells with eGFP expression at the time of harvest. Moreover, since we did not isolate the autofluorescent cell (AC) population lying on the diagonal of the GFP vs. PE plot, we cannot rule out that we have excluded some RPE cells which were both autofluorecent and eGFP+. Furthermore, the inclusion of a marker gene in a vector with a strict limitation of packaging capacity such as the popular AAV vector, may require optimization of the vector design.
This protocol was only tested with eGFP, since its use is the most widespread among fluorescent proteins due to its superior stability and sensitivity. Switching to another fluorescent protein with an excitation/emission spectrum that does not overlap with the RPE autofluorescence, could potentially solve the problem. Additionally, a limitation of the protocol is the high number of animals that must be used, if a high number of isolated eGFP+ RPE cells is required for the downstream analysis. However, the number of isolated eGFP+ RPE cells will depend on the size of the transduced area, and thus can be increased by optimizing the delivery. Notably, we have shown in this protocol that RNA purified from as little as 745 true eGFP+ RPE cells are applicable in conventional RT-qPCR analysis of up to 5 genes and for protein detection by Western blot analysis.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The animal study was reviewed and approved by The Danish Animal Inspectorate (case# 2020-15-0201-00745 and 2020-15-0201-00556).
SA, EGJ, JH, and ALA performed the experiments and produced the data, and ABL assisted with data processing. SA, LAA, ALA, and TC interpreted experimental results. ALA drafted the manuscript with assistance of SA and TC. ALA revised the manuscript with the assistance of SA, ABL, EGJ, JH, JGM, LAA, and TC. All authors approved the final version of the manuscript.
This work was supported by the Faculty of Health Sciences at Aarhus University (PhD scholarship to SA), the Danish Eye Research Foundation (TC), Aase and Ejnar Danielsen’s Foundation (TC), Bagenkop-Nielsens Myopifond, Svend Helge Schrøder og hustru Ketty Lydia Larsen Schrøders fond (TC), Maskinfabrikant Jochum Jensen og hustru Mette Marie Jensen F. Poulsens Mindelegat (SA), and The Velux Foundation (Grant number 00038189, TC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors would like to thank Tina Hindkjaer, Kamilla Zahll Hornbek and Pernille Svarrer Sørensen for their excellent technical support. Cell sorting was performed at the FACS Core Facility, Aarhus University, Denmark, and the authors would like to thank the staff at the core facility for their support. Furthermore, the authors acknowledge AU Health Bioimaging Core Facility for the use of equipment and support of the imaging facility, and the Department of Pathology, Aarhus University Hospital for paraffin embedding and sectioning.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fddev.2022.898568/full#supplementary-material
Alsing, S., Doktor, T. K., Askou, A. L., Jensen, E. G., Ahmadov, U., Kristensen, L. S., et al. (2022). VEGFA-Targeting miR-agshRNAs Combine Efficacy with Specificity and Safety for Retinal Gene Therapy. Mol. Ther. - Nucleic Acids 28, 58–76. doi:10.1016/j.omtn.2022.02.019
Ansari, A. M., Ahmed, A. K., Matsangos, A. E., Lay, F., Born, L. J., Marti, G., et al. (2016). Cellular GFP Toxicity and Immunogenicity: Potential Confounders in In Vivo Cell Tracking Experiments. Stem Cell Rev. Rep. 12 (5), 553–559. doi:10.1007/s12015-016-9670-8
Askou, A. L., Aagaard, L., Kostic, C., Arsenijevic, Y., Hollensen, A. K., Bek, T., et al. (2015). Multigenic Lentiviral Vectors for Combined and Tissue-specific Expression of miRNA- and Protein-Based Antiangiogenic Factors. Mol. Ther. - Methods Clin. Dev. 2, 14064. doi:10.1038/mtm.2014.64
Askou, A. L., Alsing, S., Benckendorff, J. N. E., Holmgaard, A., Mikkelsen, J. G., Aagaard, L., et al. (2019). Suppression of Choroidal Neovascularization by AAV-Based Dual-Acting Antiangiogenic Gene Therapy. Mol. Ther. - Nucleic Acids 16, 38–50. doi:10.1016/j.omtn.2019.01.012
Askou, A. L., Benckendorff, J. N. E., Holmgaard, A., Storm, T., Aagaard, L., Bek, T., et al. (2017). Suppression of Choroidal Neovascularization in Mice by Subretinal Delivery of Multigenic Lentiviral Vectors Encoding Anti-Angiogenic MicroRNAs. Hum. Gene Ther. Methods 28 (4), 222–233. doi:10.1089/hgtb.2017.079
Askou, A. L., and Corydon, T. J. (2018). Development of Multigenic Lentiviral Vectors for Cell-Specific Expression of Antiangiogenic miRNAs and Protein Factors. Methods Mol. Biol. 1715, 47–60. doi:10.1007/978-1-4939-7522-8_4
Askou, A. L. (2014). Development of Gene Therapy for Treatment of Age-Related Macular Degeneration. Acta Ophthalmol. 92, 1–38. doi:10.1111/aos.12452
Askou, A. L., Jakobsen, T. S., and Corydon, T. J. (2021). Retinal Gene Therapy: an Eye-Opener of the 21st Century. Gene Ther. 28 (5), 209–216. doi:10.1038/s41434-020-0168-2
Askou, A. L., Pournaras, J.-A. C., Pihlmann, M., Svalgaard, J. D., Arsenijevic, Y., Kostic, C., et al. (2012). Reduction of Choroidal Neovascularization in Mice by Adeno-Associated Virus-Delivered Anti-vascular Endothelial Growth Factor Short Hairpin RNA. J. Gene Med. 14 (11), 632–641. doi:10.1002/jgm.2678
Banskota, S., Raguram, A., Suh, S., Du, S. W., Davis, J. R., Choi, E. H., et al. (2022). Engineered Virus-like Particles for Efficient In Vivo Delivery of Therapeutic Proteins. Cell 185 (2), 250–265.e16. doi:10.1016/j.cell.2021.12.021
Benati, D., Patrizi, C., and Recchia, A. (2020). Gene Editing Prospects for Treating Inherited Retinal Diseases. J. Med. Genet. 57 (7), 437–444. doi:10.1136/jmedgenet-2019-106473
Bermond, K., Berlin, A., Tarau, I.-S., Wobbe, C., Heintzmann, R., Curcio, C. A., et al. (2021). Characteristics of Normal Human Retinal Pigment Epithelium Cells with Extremes of Autofluorescence or Intracellular Granule Count. Ann. Eye Sci. 6, 3. doi:10.21037/aes-2021-01
Cai, Y., Bak, R. O., Krogh, L. B., Staunstrup, N. H., Moldt, B., Corydon, T. J., et al. (2014). DNA Transposition by Protein Transduction of the piggyBac Transposase from Lentiviral Gag Precursors. Nucleic Acids Res. 42 (4), e28. doi:10.1093/nar/gkt1163
Chintalapudi, S. R., Djenderedjian, L., Stiemke, A. B., Steinle, J. J., Jablonski, M. M., and Morales-Tirado, V. M. (2016). Isolation and Molecular Profiling of Primary Mouse Retinal Ganglion Cells: Comparison of Phenotypes from Healthy and Glaucomatous Retinas. Front. Aging Neurosci. 8, 93. doi:10.3389/fnagi.2016.00093
Einbock, W., Moessner, A., Schnurrbusch, U. E. K., Holz, F. G., Wolf, S., and Group, F. A. M. S. (2005). Changes in Fundus Autofluorescence in Patients with Age-Related Maculopathy. Correlation to Visual Function: a Prospective Study. Graefe's Arch. Clin. Exp. Ophthalmol. 243 (4), 300–305. doi:10.1007/s00417-004-1027-3
Fernandez-Godino, R., Garland, D. L., and Pierce, E. A. (2016). Isolation, Culture and Characterization of Primary Mouse RPE Cells. Nat. Protoc. 11 (7), 1206–1218. doi:10.1038/nprot.2016.065
Giannelli, S. G., Luoni, M., Castoldi, V., Massimino, L., Cabassi, T., Angeloni, D., et al. (2018). Cas9/sgRNA Selective Targeting of the P23H Rhodopsin Mutant Allele for Treating Retinitis Pigmentosa by Intravitreal AAV9.PHP.B-Based Delivery. Hum. Mol. Genet. 27 (5), 761–779. doi:10.1093/hmg/ddx438
Hamel, C. P., Tsilou, E., Pfeffer, B. A., Hooks, J. J., Detrick, B., and Redmond, T. M. (1993). Molecular Cloning and Expression of RPE65, a Novel Retinal Pigment Epithelium-Specific Microsomal Protein that Is Post-transcriptionally Regulated In Vitro. J. Biol. Chem. 268 (21), 15751–15757. doi:10.1016/s0021-9258(18)82319-5
Holmgaard, A., Askou, A. L., Benckendorff, J. N. E., Thomsen, E. A., Cai, Y., Bek, T., et al. (2017). In Vivo Knockout of the Vegfa Gene by Lentiviral Delivery of CRISPR/Cas9 in Mouse Retinal Pigment Epithelium Cells. Mol. Ther. - Nucleic Acids 9, 89–99. doi:10.1016/j.omtn.2017.08.016
Holmgaard, A. B., Askou, A. L., Jensen, E. G., Alsing, S., Bak, R. O., Mikkelsen, J. G., et al. (2021). Targeted Knockout of the Vegfa Gene in the Retina by Subretinal Injection of RNP Complexes Containing Cas9 Protein and Modified sgRNAs. Mol. Ther. 29 (1), 191–207. doi:10.1016/j.ymthe.2020.09.032
Hung, S. S. C., Chrysostomou, V., Li, F., Lim, J. K. H., Wang, J.-H., Powell, J. E., et al. (2016). AAV-Mediated CRISPR/Cas Gene Editing of Retinal Cells In Vivo. Invest. Ophthalmol. Vis. Sci. 57 (7), 3470–3476. doi:10.1167/iovs.16-19316
Jang, H., Jo, D. H., Cho, C. S., Shin, J. H., Seo, J. H., Yu, G., et al. (2022). Application of Prime Editing to the Correction of Mutations and Phenotypes in Adult Mice with Liver and Eye Diseases. Nat. Biomed. Eng. 6 (2), 181–194. doi:10.1038/s41551-021-00788-9
Koo, T., Park, S. W., Jo, D. H., Kim, D., Kim, J. H., Cho, H.-Y., et al. (2018). CRISPR-LbCpf1 Prevents Choroidal Neovascularization in a Mouse Model of Age-Related Macular Degeneration. Nat. Commun. 9 (1), 1855. doi:10.1038/s41467-018-04175-y
Latella, M. C., Di Salvo, M. T., Cocchiarella, F., Benati, D., Grisendi, G., Comitato, A., et al. (2016). In Vivo Editing of the Human Mutant Rhodopsin Gene by Electroporation of Plasmid-Based CRISPR/Cas9 in the Mouse Retina. Mol. Ther. - Nucleic Acids 5 (11), e389. doi:10.1038/mtna.2016.92
Levy, J. M., Yeh, W.-H., Pendse, N., Davis, J. R., Hennessey, E., Butcher, R., et al. (2020). Cytosine and Adenine Base Editing of the Brain, Liver, Retina, Heart and Skeletal Muscle of Mice via Adeno-Associated Viruses. Nat. Biomed. Eng. 4 (1), 97–110. doi:10.1038/s41551-019-0501-5
Mangeot, P. E., Risson, V., Fusil, F., Marnef, A., Laurent, E., Blin, J., et al. (2019). Genome Editing in Primary Cells and In Vivo Using Viral-Derived Nanoblades Loaded with Cas9-sgRNA Ribonucleoproteins. Nat. Commun. 10 (1), 45. doi:10.1038/s41467-018-07845-z
Matsuda, T., and Oinuma, I. (2019). Optimized CRISPR/Cas9-mediated In Vivo Genome Engineering Applicable to Monitoring Dynamics of Endogenous Proteins in the Mouse Neural Tissues. Sci. Rep. 9 (1), 11309. doi:10.1038/s41598-019-47721-4
Mikkelsen, J. G. (2018). Viral Delivery of Genome-Modifying Proteins for Cellular Reprogramming. Curr. Opin. Genet. Dev. 52, 92–99. doi:10.1016/j.gde.2018.06.005
Moreno-García, A., Kun, A., Calero, O., Medina, M., and Calero, M. (2018). An Overview of the Role of Lipofuscin in Age-Related Neurodegeneration. Front. Neurosci. 12, 464. doi:10.3389/fnins.2018.00464
Nicoletti, A., Wong, D. J., Kawase, K., Gibson, L. H., Yang-Feng, T. L., Richards, J. E., et al. (1995). Molecular Characterization of the Human Gene Encoding an Abundant 61 kDa Protein Specific to the Retinal Pigment Epithelium. Hum. Mol. Genet. 4 (4), 641–649. doi:10.1093/hmg/4.4.641
Plaza Reyes, A., Petrus-Reurer, S., Padrell Sánchez, S., Kumar, P., Douagi, I., Bartuma, H., et al. (2020). Identification of Cell Surface Markers and Establishment of Monolayer Differentiation to Retinal Pigment Epithelial Cells. Nat. Commun. 11 (1), 1609. doi:10.1038/s41467-020-15326-5
Ueda, K., Zhao, J., Kim, H. J., and Sparrow, J. R. (2016). Photodegradation of Retinal Bisretinoids in Mouse Models and Implications for Macular Degeneration. Proc. Natl. Acad. Sci. U.S.A. 113 (25), 6904–6909. doi:10.1073/pnas.1524774113
Vagni, P., Perlini, L. E., Chenais, N. A. L., Marchetti, T., Parrini, M., Contestabile, A., et al. (2019). Gene Editing Preserves Visual Functions in a Mouse Model of Retinal Degeneration. Front. Neurosci. 13, 945. doi:10.3389/fnins.2019.00945
Westenskow, P. D., Moreno, S. K., Krohne, T. U., Kurihara, T., Zhu, S., Zhang, Z.-n., et al. (2012). Using Flow Cytometry to Compare the Dynamics of Photoreceptor Outer Segment Phagocytosis in iPS-Derived RPE Cells. Invest. Ophthalmol. Vis. Sci. 53 (10), 6282–6290. doi:10.1167/iovs.12-9721
Yu, W., Mookherjee, S., Chaitankar, V., Hiriyanna, S., Kim, J.-W., Brooks, M., et al. (2017). Nrl Knockdown by AAV-Delivered CRISPR/Cas9 Prevents Retinal Degeneration in Mice. Nat. Commun. 8, 14716. doi:10.1038/ncomms14716
Keywords: RPE, retinal gene therapy, gating strategy, FACS, eGFP, autofluorescence
Citation: Alsing S, Lindholm AB, Haldrup J, Jensen EG, Mikkelsen JG, Aagaard L, Askou AL and Corydon T (2022) Simple Autofluorescence-Restrictive Sorting of eGFP+ RPE Cells Allows Reliable Assessment of Targeted Retinal Gene Therapy. Front. Drug. Deliv. 2:898568. doi: 10.3389/fddev.2022.898568
Received: 17 March 2022; Accepted: 20 April 2022;
Published: 24 May 2022.
Edited by:
Heather Sheardown, McMaster University, CanadaReviewed by:
Torben Moos, Aalborg University, DenmarkCopyright © 2022 Alsing, Lindholm, Haldrup, Jensen, Mikkelsen, Aagaard, Askou and Corydon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anne Louise Askou, YWxhQGNsaW4uYXUuZGs=
†These authors have contributed equally to this work and share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.