
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med., 19 February 2025
Sec. Atherosclerosis and Vascular Medicine
Volume 12 - 2025 | https://doi.org/10.3389/fcvm.2025.1538202
 Jonica Campolo1,†
Jonica Campolo1,† Paola Canale2,3,†
Paola Canale2,3,† Gianluca Gazzaniga4,5Marina Parolini1Emanuela Piccaluga6Irene Bossi6Jacopo Oreglia6Andrea Borghini2Irene Marinaro2
Gianluca Gazzaniga4,5Marina Parolini1Emanuela Piccaluga6Irene Bossi6Jacopo Oreglia6Andrea Borghini2Irene Marinaro2 Maria Grazia Andreassi2*
Maria Grazia Andreassi2*
Objective: Mitochondrial dysfunction is associated with increased risk of atherosclerosis by disrupting key cellular processes that contribute to premature vascular ageing. However, the specific role of mitochondrial dysfunction in early-onset coronary artery disease (EOCAD), which is increasing at a particularly alarming rate, remains largely unexplored. This study investigated the association of leukocyte mtDNA-CN and mtDNA4977 deletion with the risk of EOCAD.
Methods: The study included 118 patients (99 men, 51.0 ± 5.6 years) with angiographically EOCAD (≤60 years) and 150 healthy controls (94 men, 49.8 ± 5.8 years). Quantitative RT-PCR was used to quantify mtDNA-CN and mtDNA4977 deletion rate.
Results: The EOCAD group had a higher prevalence of male gender (p < 0.001), smoking (p = 0.001), hypertension (p < 0.001), diabetes mellitus (p = 0.04) and obesity (p < 0.001) than controls. EOCAD patients had lower mtDNA-CN (p < 0.001) and higher mtDNA4977 deletion (p = 0.026). Low mtDNA-CN levels were significantly associated with male gender (p < 0.001), smoking (p = 0.004), hypertension (p = 0.039), hypercholesterolemia (p < 0.001), and obesity (p < 0.001). Increased levels of the mtDNA4977 deletion were significantly higher in males (p = 0.026) and hypercholesterolemic patients (p < 0.001). The ROC curve of mtDNA-CN and mtDNA4977 deletion in predicting EOCAD showed an AUC of 0.902 (95% CI 0.867–0.937, p < 0.001) and 0.762 (95% CI 0.691–0.834, p < 0.001), respectively. Logistic regression analysis adjusted for confounders showed that both mtDNA-CN and mtDNA4977 deletion were independent significant predictors of EOCAD (p < 0.001 and p = 0.001, respectively).
Conclusions: EOCAD is characterized by a high prevalence of modifiable risk factors and mitochondrial damage, underscoring the need for more efforts to reduce the burden of traditional risk factors and highlighting the potential for innovative mitochondrial-targeted therapies.
Coronary artery disease (CAD) remains a leading cause of illness, disability, and mortality worldwide, despite advances in prevention and treatment strategies (1). Alarmingly, recent trends have shown a rise in premature coronary artery disease, also called early-onset CAD (EOCAD), among younger populations (2, 3). Individuals with EOCAD often exhibit a high prevalence of modifiable traditional risk factors such as dyslipidemia, smoking, diabetes mellitus, hypertension, and overweight/obesity (4–8). Non-classical risk factors including inflammation, infections, sleep deprivation, and exposure to air pollution and environmental noise also warrant consideration (1, 9).
Moreover, it is well known that premature CAD is characterized by a rapid progression of the disease and is associated with a poorer long-term prognosis (10, 11). Thus, further research is essential to better understand the risk profile, behaviors, and underlying biological mechanisms contributing to premature CAD in younger individuals (12, 13).
Interestingly, recent research has identified a significant association for the mitochondrial mtDNA 16223T variant and heteroplasmy with an increased presence of cardiovascular risk factors and premature CAD (14).
Indeed, several studies have highlighted the direct role of mtDNA-mediated mitochondrial dysfunction in the initiation and progression of atherosclerosis (15), and the importance of preserving the mitochondrial genome to maintain healthy vessel and to delay vascular aging (16).
Mitochondria, essential cellular organelles, are primarily responsible for energy production through ATP generation via oxidative phosphorylation. They also play key roles in various cellular processes, including modulation of oxidation–reduction status, regulation of senescence and apoptosis. Mitochondria possess their own circular genome, known as mitochondrial DNA (mtDNA), with each mitochondrion containing multiple copies of this genome, referred to as mtDNA copy number (mtDNA-CN). The mtDNA-CN is positively correlated with mitochondrial enzyme activity and ATP production, making it a surrogate biomarker for assessing mitochondrial function (17).
Lower mtDNA-CN has shown associations to several aging-related diseases and all-cause mortality, including cardiovascular disease (18).
Additionally, mtDNA is particularly vulnerable to damage from reactive oxygen species (ROS) due to the absence of histones and a less efficient repair mechanism. Besides rare pathogenic variants, the accumulation of mutations in the mitochondrial genome can further accelerate the development of atherosclerosis (19). Among various mtDNA defects, a large-scale deletion of 4,977 bp, known as the mtDNA4977 common deletion, results in the loss of nearly one-third of the mitochondrial genome. This leads to the accumulation of dysfunctional mitochondrial transcripts and a consequent increase in ROS production (20).
In light of this, we hypothesized that reduced mtDNA integrity and mitochondrial dysfunction may predispose individuals to accelerated atherosclerosis and the premature development of CAD. To test this hypothesis, we investigated the association of leukocyte mtDNA4977 deletion and mtDNA-CN with early-onset coronary artery disease.
The study employed a case-control design, involving 118 patients (99 males, mean age 51.0 ± 5.6 years) with EOCAD (≤60 years) recruited from the GENOCOR (Genetic Mapping for Assessment of Cardiovascular Risk) and VICTORIA (Vascular Senescence and Atherosclerotic Plaque Vulnerability) databases (21, 22). Cases included patients with angiographically confirmed CAD, defined by significant coronary stenosis in at least one vessel (>50% lumen reduction). The severity of CAD was classified based on the number of vessels involved (one-, two-, or three-vessel disease). All subjects had a diagnosis of stable angina pectoris or acute coronary syndrome (myocardial infarction or unstable angina). Exclusion criteria comprised patients with known malignancy, end-stage renal disease, or other severe illnesses that could confound study outcomes.
A control group comprised 150 healthy individuals (94 males, mean age 49.8 ± 5.8 years), who were members of the medical and technical staff at our institution and exhibited no clinical evidence of cardiovascular disease. Classic cardiovascular risk factors were recorded for all participants, including age, gender, diabetes (fasting plasma glucose >120 mg/dl or on glucose-lowering medication), hypercholesterolemia (plasma cholesterol >220 mg/dl or on lipid-lowering therapy), obesity (body mass index >30 kg/m2), and arterial hypertension (systolic blood pressure >140 mmHg and/or diastolic pressure >90 mmHg, or on antihypertensive therapy). Smoking status was categorized as current smokers (at least 3 cigarettes per day), past smokers (abstinent for at least 6 months), and non-smokers (never smoked). For analysis, smoking patients included both past and current smokers.
Blood samples were taken from participants on admission, prior to any diagnostic or therapeutic procedure, and DNA was extracted using established protocols (23, 24).
The mtDNA-CN and mtDNA4977 deletion levels were quantified using quantitative real-time PCR (CFX384 Touch Real-time PCR detection system, Bio-Rad, Hercules, CA, USA).
To determine the levels of the mtDNA4977 deletion (spanning nucleotides 8,470–13,447 bp), we amplified the NDI1 gene located in an undeleted region of mtDNA (mtNDI1) along with the remaining fragment after the mtDNA4977 deletion. The difference in the average threshold cycle (Ct) values was used to measure the relative content. The percentage of the mtDNA4977 deletion was calculated as follows: 2–ΔCt × 100%, where ΔCT = Ct × mtDNA4977−Ct × mtNDI1. Additionally, ΔCt values were computed as the difference between the Ct of the ß-globin gene and the Ct of the NDI1 gene, which was used to measure mtDNA-CN relative to genomic DNA (gDNA). mtDNA-CN was then calculated by using (2-ΔCt) method where ΔCt = Ct mtNDI1—Ct gDNA. Each sample was run in triplicate to ensure intra-assay precision. Because there were insufficient amounts of DNA for some participants, we had measurements mtDNA-CN on 149 controls and 117 cases, and measurements on mtDNA deletion on 119 controls and 93 cases.
Values are presented as mean ± standard deviation (SD), median with interquartile range (IQR) or percent, according to the nature of the data.
Categorical data, expressed as frequencies and percentages, were compared using the Chi-square or Fisher's exact test when appropriate, while continuous variables using the Student's t-test and Mann–Whitney U test for parametric and non-parametric data, respectively.
The association between mtDNA4977 deletion, mtDNA-CN and other continuous parameters was assessed using Spearman rank correlation.
The receiver operating characteristic (ROC) curve was used as a measure of diagnostic accuracy of mtDNA4977 deletion and mtDNA-CN and the area under the curve (AUC) along with 95% confidence intervals (95% CI), was calculated to evaluate the specificity, sensitivity and accuracy for predicting EOCAD, and optimal cutoff values were determined by the Youden index.
Univariate and multivariate logistic regression analyses confined to subjects with complete dataset were also performed to determine odds ratios (ORs) and 95% CIs. mtDNA4977 deletion and mtDNA-CN were analyzed as continuous variables, and as categorical variables dichotomized according to the optimal cut-off values identified by ROC curves.
Age, gender, smoking, hypertension, hypercholesterolemia, diabetes, obesity were considered as potential confounders, and were therefore tested in univariate and multivariate models.
Power analysis showed that our sample size of 118 EOCAD and 150 healthy controls allowed to detect a medium effect size (d = 0.5) at 0.05 significance level and 80% power using G*Power software. A p-value of <0.05 was considered statistically significant in this study. Statistical analyses were performed using SPSS ver. 24.0 software package (IBM SPSS, New York, USA).
The majority of patients presented with stable angina (82%), and most had single-vessel obstructive CAD (61%). A detailed description of the study population is presented in Table 1. The average age of patients with EOCAD and controls was not significantly different. As expected, the prevalence of male gender (p < 0.001), smoking (p = 0.001), hypertension (p < 0.001), diabetes mellitus (p = 0.04) and obesity (p < 0.001) was significantly higher in the EOCAD group compared to healthy controls. As shown in Figure 1, mtDNA-CN levels were significantly lower in patients with EOCAD compared to controls (p < 0.001). Similarly, the mtDNA4977 deletion was higher in the EOCAD group than in the control group (p < 0.001).
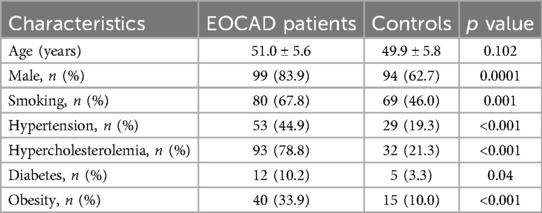
Table 1. Demographic and clinical characteristics of the study population.
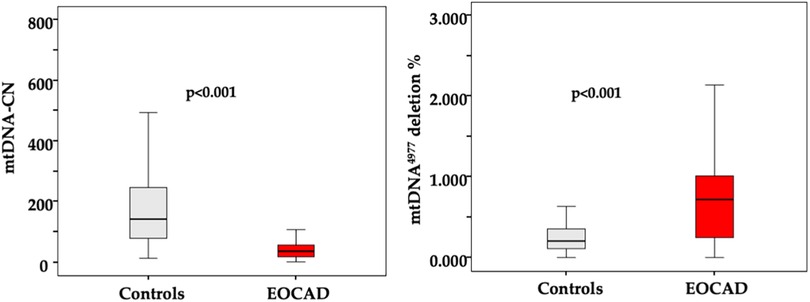
Figure 1. Box and whisker plot showing the levels of mtDNA CN and mtDNA4977 deletion in the leukocytes of the control group and the EOCAD group. Interquartile range, median, maximum and minimum values are shown in the box-and-whisker plots.
Across the entire sample, there was a significant inverse correlation between mtDNA4977 deletion and mtDNA-CN levels (Spearman's rho = −0.372, p < 0.001).
In the overall study population, mtDNA-CN and the mtDNA4977 deletion did not show a significant correlation with chronological age. However, in patients with EOCAD, a significant positive correlation between mtDNA-CN and age was observed (Spearman's rho = 0.321, p < 0.001).
In terms of cardiovascular risk factors, lower mtDNA-CN levels (Figure 2) were significantly associated with male gender (p < 0.001), smoking (p = 0.004), hypertension (p = 0.039), hypercholesterolemia (p < 0.001), and obesity (p < 0.001), while higher mtDNA4977 deletion levels (Figure 3) were significantly higher in male gender (p = 0.026) and hypercholesterolemic patients (p < 0.001).
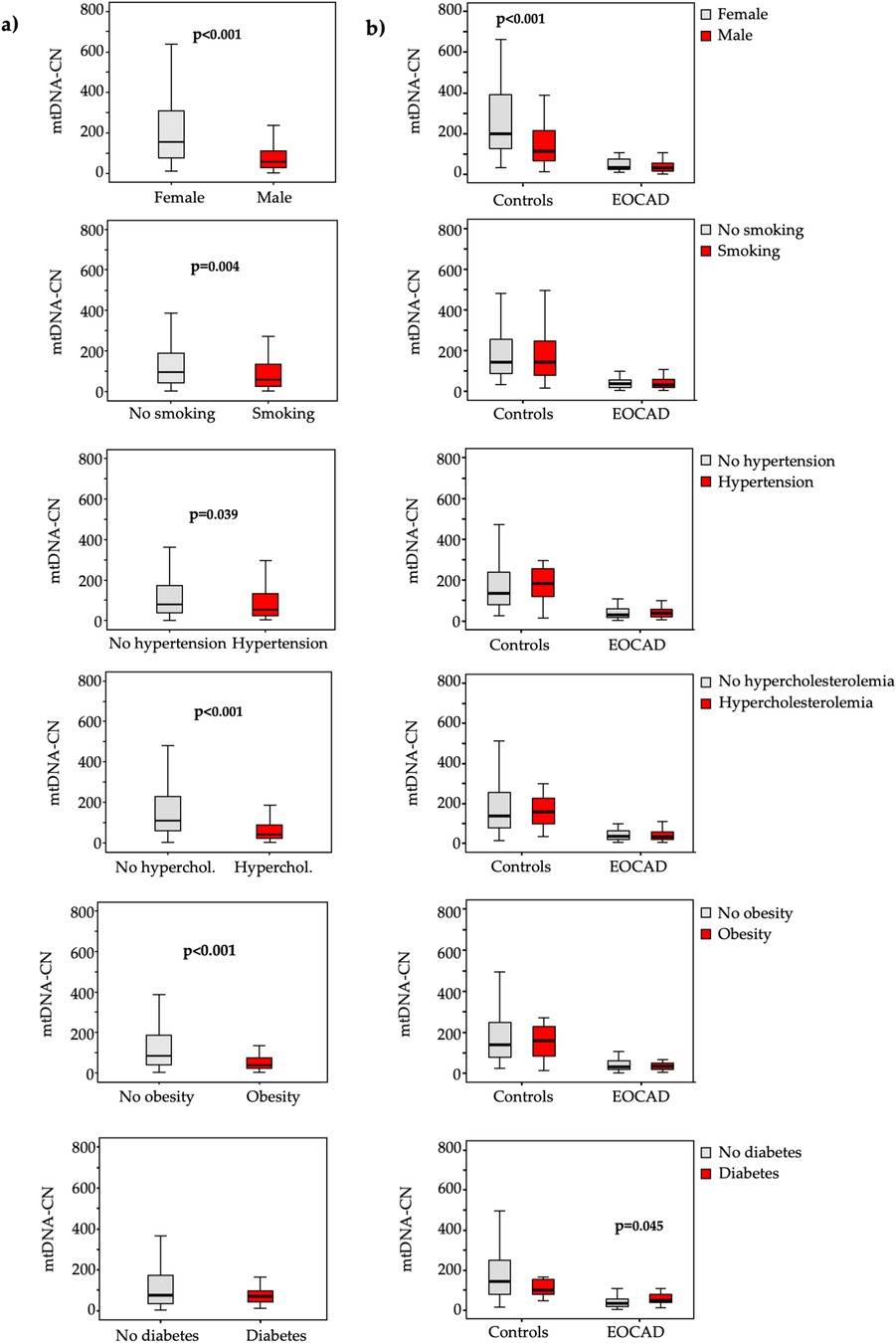
Figure 2. Box and whiskers plot of the mtDNA-CN in the total study population (a) and in each group (b) according to the absence (grey bars) or presence (red bars) of various traditional vascular risk factors.
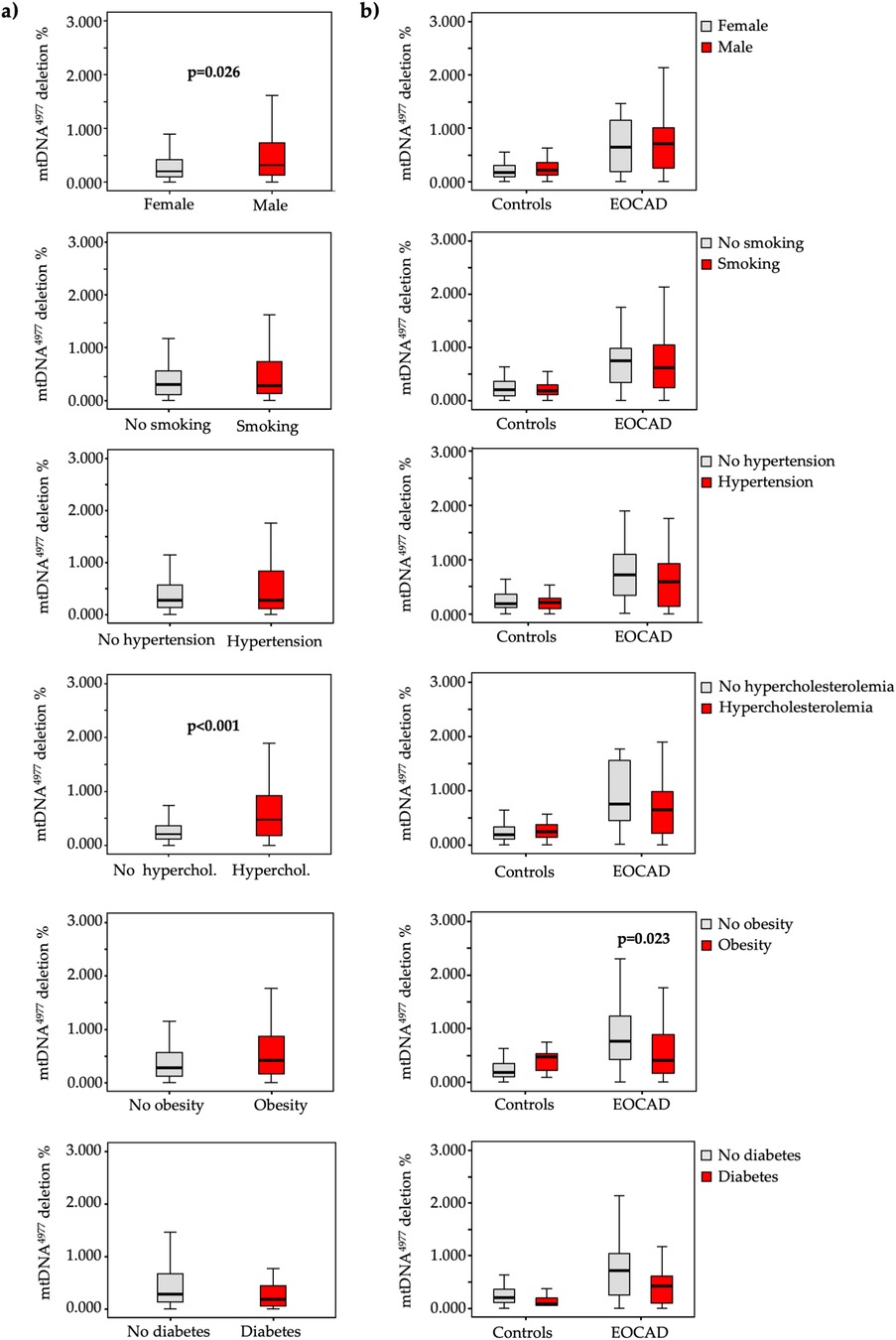
Figure 3. Box and whiskers plot of the mtDNA4977 deletion in the total study population (a) and in each group (b) according to the absence (grey bars) or presence (red bars) of various traditional vascular risk factors.
A similar pattern emerged when these analyses were conducted separately for cases and controls (Figures 2, 3).
No significant differences in mtDNA-CN or mtDNA4977 deletion were found among patients with two- or three-vessel disease compared to those with single-vessel disease.
ROC curve analyses indicated that each marker significantly predicted EOCAD (Figure 4).
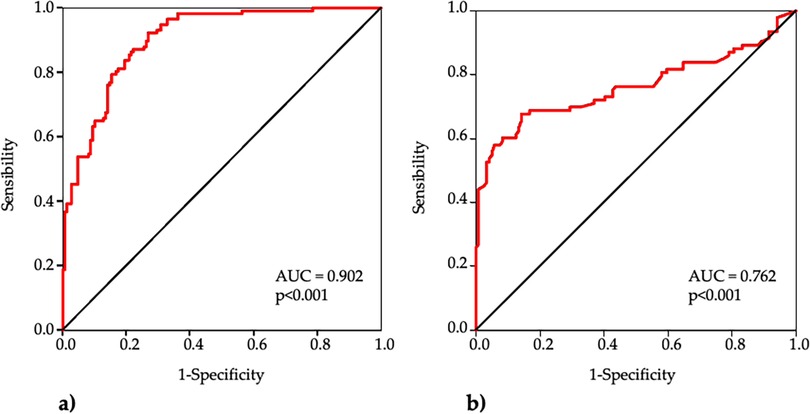
Figure 4. Receiver operating characteristic (ROC) curve analysis of mtDNA CN (a) and mtDNA4977 deletion (b) for the diagnosis of EOCAD.
The AUC of mtDNA-CN was 0.902 (95% CI 0.867–0.937, p < 0.001) and the best cut-off was <85 with sensitivity 92%, specificity 73% and accuracy 82%. The AUC of mtDNA4977 deletion was 0.762 (95% CI 0.691–0.834, p < 0.001), and the best cut-off was >0.40 with sensitivity 68%, specificity 86% and accuracy 78%.
Logistic regression analysis adjusted for confounders revealed that increased mtDNA-CN levels were associated to a decreased risk of developing EOCAD with an OR of 0.96 (95% CI: 0.95–0.98; p < 0.001). Similarly, we found an association between high levels of mtDNA4977 deletion and increased risk of EOCAD (OR = 26.1, 95% CI: 3.81–178.51; p = 0.001). We also performed a multiple logistic regression analysis using each mitochondrial biomarker dichotomized on the optimal cutoff value determined by the ROC analysis, and the results support the previous observations obtained as continuous variables (Figure 5).
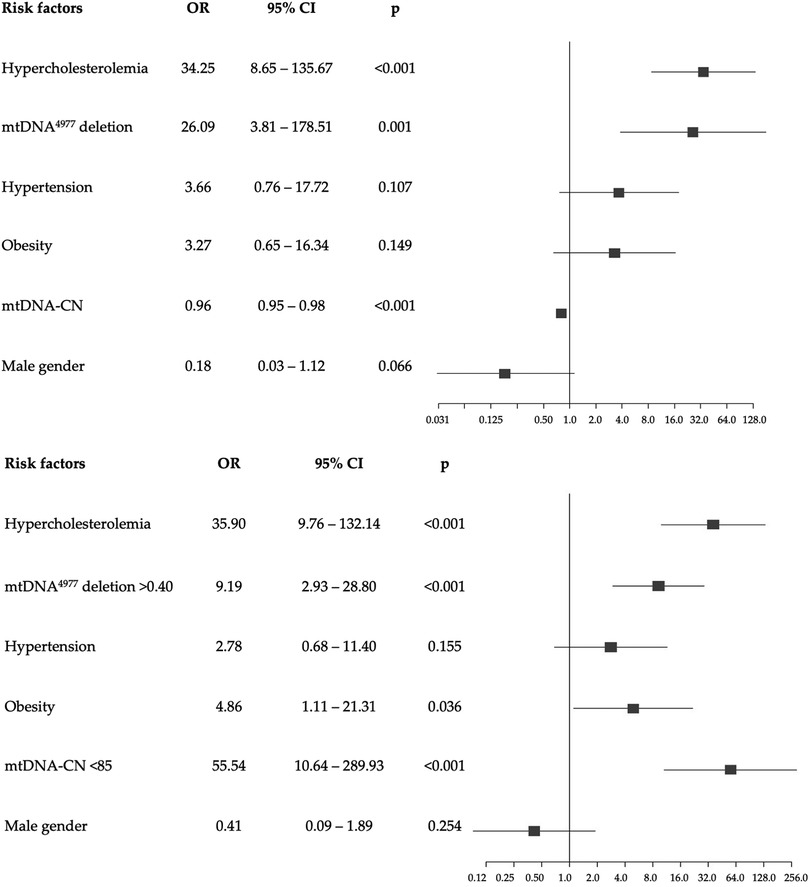
Figure 5. Forest plots of multivariable logistic regression analysis with odds ratio (95% confidence interval) of the association of each potential risk factor compared to none (reference) with the development of EOCAD. The top figure shows the model with the mtDNA4977 deletion and the mtDNA-CN as continuous variables, while the bottom figure refers to the model with categorical variables, dichotomized according to the optimal thresholds identified by the ROC curves.
To the best of our knowledge, this is the first attempt to explore the interaction between mitochondrial damage and dysfunction in blood, and early-onset of coronary artery disease. We identified an independent, significant association between both mtDNA4977 deletion and mtDNA-CN and incident EOCAD after accounting for traditional risk factors. In addition, our results showed significant associations between traditional cardiovascular risk factors and mitochondrial dysfunction, suggesting that the mtDNA damage and dysfunction induced by atherosclerosis-related risk factors may play a crucial role in the early initiation and progression of atherosclerosis (25). In addition, our results agree with previous findings suggesting that mitochondrial dysfunction may have a gender-specific component influenced by hormonal and metabolic factors. In fact, women's mitochondria are better able to deal with stressful situations and conditions, and are more resistant to DNA damage and mutations (17, 18).
Previous studies have found that lower mtDNA-CN in blood is consistently associated with higher risk of CAD, reflecting mitochondrial dysfunction and oxidative stress, both of which are critical in CAD pathogenesis (26).
In particular, three large prospective studies suggested that mtDNA-CN in circulating leukocytes may have clinical utility in improving CVD risk classification (27).
Interestingly, a case-control study also found a significant joint effect between low levels of mtDNA-CN and risk factors like plasma LDL-C or smoking with increasing the incidence of CAD (28).
Moreover, cardiometabolic risk factors such as high blood pressure, obesity, and dyslipidemia have been shown to impact mtDNA-CN (29–31).
Regarding mtDNA damage, previous studies showed that mtDNA4977 deletion accumulates with age in normal hearts (19, 20) and that the levels of mtDNA4977 deletion are increased in human atherosclerotic lesions (32).
Our previous study also reported significant associations between lower mtDNA4977 deletion levels and poorer CVD outcomes as well as increased all-cause mortality in patients with stable CAD, confirming the critical role of mitochondrial dysfunction in atherosclerosis (24).
Remarkably, experimental studies in animal models further support the role of mtDNA damage in atherosclerosis. For instance, mtDNA damage has been observed during the early stages of atherogenesis in ApoE-/- mice and correlates with the severity of the disease (33). In double knock-out ATM+/-/ApoE-/- mice, an increased frequency of the mouse mitochondrial “common” deletion was associated with impaired respiratory complex activity (34). These mitochondrial alterations accelerated the development of atherosclerosis and were linked to key metabolic syndrome features such as hypertension, hypercholesterolemia, and obesity (35).
In response to reactive oxygen species, human vascular endothelial and smooth muscle cells accumulate more mtDNA damage than nuclear DNA, impairing key cellular processes like growth, signaling, and apoptosis. This cell dysfunction can, in turn, contribute to the initiation and progression of atherosclerotic lesions (35).
In addition, point mtDNA mutations in blood cells, especially rare variants and heteroplasmy levels (a mixture of both wild-type and mutant species of mtDNA), are being actively investigated in relation to atherosclerosis, coronary heart disease and various atherosclerosis-related diseases such as arterial hypertension and diabetes mellitus, and the list of identified mutations continues to grow (14, 36, 37). These mutations cause structural and functional changes in the mtDNA OXPHOS-related and mt-tRNA genes, which in turn can affect mitochondrial protein synthesis and lead to mitochondrial dysfunction.
Altogether, our findings, along with the evidence summarized above, strongly suggest that mtDNA plays a major role in the pathogenesis of atherosclerosis directly regulating the onset and progression of early vascular aging (16).
In patients with early-onset CAD, we hypothesize that the cumulative burden of traditional clinical risk factors may lead to more aggressive premature atherosclerosis through mechanisms such as oxidative stress and sustained inflammation, which are central to the progression of the mtDNA damage and dysfunction.
These observations raise significant clinical questions regarding broader preventive strategies to reduce the risk of premature coronary artery disease. First, our study confirms a high rate of concomitant modifiable cardiovascular risk factors in individuals with EOCAD (4–8), emphasizing the importance of increased efforts to reduce the burden of these risk factors and premature clinical events (11).
Second, ROC curve analysis suggested that both mitochondrial damage and dysfunction might be considered a biomarker reflecting the early diagnosis and prediction of EOCAD The evaluation of mtDNA could also serve as an easy and accessible biomarker that could be exploited for cardiovascular prognosis or for assessing responses to therapies (24). The incorporation of mitochondrial biomarkers into traditional risk assessments may have the potential to improve the accuracy of prediction. For example, it may help to reclassify intermediate risk patients into higher or lower risk categories. However, their integration into clinical practice will depend on standardization of measurement techniques and confirmation of their predictive value by robust evidence.
Third, targeting of mitochondrial dysfunction may be a feasible strategy to provide a more personalized approach to vascular risk managemen (38). Potential therapies include mitochondrial antioxidative therapies and lifestyle interventions (e.g., exercise, diet) aimed at mitigating mitochondrial dysfunction and ROS production. In addition, mitochondrial biogenesis enhancers may offer promising avenues for treating vascular damage caused by mitochondrial dysfunction by repairing mtDNA, such as increasing the activity of mitochondrial-specific enzymes (e.g., OGG1), or enhancing mitochondrial biogenesis. In addition, new gene-editing techniques such as CRISPR hold promise to correct mtDNA mutations (38).
Our study has some potential limitations. First, the sample size of patients is relatively small, which limits the precision of the associations. As result, the estimated ORs with the wide confidence intervals for risk factors should be interpreted cautiously. Second, the case-control study design comes with inherent sources of confounding and unmeasured biases. For instance, we did not measure family history or potential social and psychological biases. Third, the cohort comprised patients admitted to the hospital for coronary angiography, and some already had existing CAD or previous myocardial infarction and were receiving pharmacological treatment. Finally, mtDNA4977 deletion and mtDNA-CN were measured only at enrollment, without capturing potential variability over time. Indeed, there is potential uncertainty about the temporal variability of biomarkers when relying on measurements from a single point in time. Mitochondrial biomarkers may fluctuate with circadian rhythms, transient physiological states or in response to environmental exposures, which could affect their predictive utility. Future studies should, therefore, be focused on assessing mitochondrial biomarkers longitudinally in order to evaluate their temporal dynamics and improve the accuracy of risk prediction.
In conclusion, this study revealed that premature EOCAD is characterized by a high prevalence of modifiable cardiovascular risk factors and elevated mitochondrial damage and dysfunction. This underscores the need for more comprehensive efforts to reduce the burden of traditional risk factors and highlights the potential for innovative mitochondrial-targeted therapies to address vascular damage resulting from mitochondrial dysfunction.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving humans were approved by Comitato Etico Milano Area 3 ASST Grande Ospedale Metropolitano di Niguarda. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
JC: Data curation, Formal Analysis, Writing – original draft. PC: Data curation, Formal Analysis, Writing – original draft. GG: Data curation, Formal Analysis, Writing – review & editing. MP: Data curation, Formal Analysis, Writing – review & editing. EP: Data curation, Writing – review & editing. IB: Data curation, Writing – review & editing. JO: Data curation, Writing – review & editing. AB: Data curation, Writing – review & editing. IM: Writing – review & editing, Project administration. MA: Writing – review & editing, Conceptualization, Resources, Supervision.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Project Age-It (Aging Well in an Aging Society) GAE 492 PNRR PE_8 SPOKE 2.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Mensah GA, Fuster V, Murray CJL, Roth GA, Global Burden of Cardiovascular Diseases and Risks Collaborators. Global burden of cardiovascular diseases and risks, 1990–2022. J Am Coll Cardiol. (2023) 82(25):2350–473. doi: 10.1016/j.jacc.2023.11.007
2. Mousavi I, Suffredini J, Virani SS, Ballantyne C, Michos ED, Misra A, et al. Early onset atherosclerotic cardiovascular disease. Eur J Prev Cardiol. (2025) 32:100. doi: 10.1093/eurjpc/zwae240
3. Vasan RS, Song RJ, van den Heuvel ER. Temporal trends in incidence of premature cardiovascular disease over the past 7 decades: the framingham heart study. J Am Heart Assoc. (2022) 11(19):e026497. doi: 10.1161/JAHA.122.026497
4. Arora S, Stouffer GA, Kucharska-Newton AM, Qamar A, Vaduganathan M, Pandey A, et al. Twenty years trends and sex differences in young adults hospitalized with acute myocardial infarction. Circulation. (2019) 139(8):1047–56. doi: 10.1161/CIRCULATIONAHA.118.037137
5. Gupta A, Wang Y, Spertus JA, Geda M, Lorenze N, Nkonde-Price C, et al. Trends in acute myocardial infarction in young patients and differences by sex and race, 2001 to 2010. J Am Coll Cardiol. (2014) 64(4):337–45. doi: 10.1016/j.jacc.2014.04.054
6. Sagris M, Antonopoulos AS, Theofilis P, Oikonomou E, Siasos G, Tsalamandris S, et al. Risk factors profile of young and older patients with myocardial infarction. Cardiovasc Res. (2022) 118(10):2281–92. doi: 10.1093/cvr/cvab264
7. Bugiardini R, Cenko E, Yoon J, Bergami M, Vasiljevic Z, Mendieta G, et al. Traditional risk factors and premature acute coronary syndromes in South Eastern Europe: a multinational cohort study. Lancet Reg Health Eur. (2024) 38:100824. doi: 10.1016/j.lanepe.2023.100824
8. Dugani SB, Hydoub YM, Ayala AP, Reka R, Nayfeh T, Ding JF, et al. Risk factors for premature myocardial infarction: a systematic review and meta-analysis of 77 studies. Mayo Clin Proc Innov Qual Outcomes. (2021) 5(4):783–94. doi: 10.1016/j.mayocpiqo.2021.03.009
9. Banach M, Surma S. Monitoring of traditional atherosclerosis cardiovascular disease risk factors—is it enough to prevent premature acute coronary syndrome? Lancet Reg Health Eur. (2024) 38:100866. doi: 10.1016/j.lanepe.2024.100866
10. Cole JH, Miller JI 3rd, Sperling LS, Weintraub WS. Long-term follow-up of coronary artery disease presenting in young adults. J Am Coll Cardiol. (2003) 41(4):521–8. doi: 10.1016/s0735-1097(02)02862-0
11. Zeitouni M, Clare RM, Chiswell K, Abdulrahim J, Shah N, Pagidipati NP, et al. Risk factor burden and long-term prognosis of patients with premature coronary artery disease. J Am Heart Assoc. (2020) 9(24):e017712. doi: 10.1161/JAHA.120.017712
12. Menezes Fernandes R, Mota T, Costa H, Bispo J, Azevedo P, Bento D, et al. Portuguese registry of acute coronary syndromes (ProACS) investigators. Premature acute coronary syndrome: understanding the early onset. Coron Artery Dis. (2022) 33(6):456–64. doi: 10.1097/MCA.0000000000001141
13. Le A, Peng H, Golinsky D, Di Scipio M, Lali R, Paré G. What causes premature coronary artery disease? Curr Atheroscler Rep. (2024) 26(6):189–203. doi: 10.1007/s11883-024-01200-y
14. Lorca R, Aparicio A, Gómez J, Álvarez-Velasco R, Pascual I, Avanzas P, et al. Mitochondrial heteroplasmy as a marker for premature coronary artery disease: analysis of the poly-C tract of the control region sequence. J Clin Med. (2023) 12(6):2133. doi: 10.3390/jcm12062133
15. Madamanchi NR, Runge MS. Mitochondrial dysfunction in atherosclerosis. Circ Res. (2007) 100(4):460–73. doi: 10.1161/01.RES.0000258450.44413.96
16. Foote K, Reinhold J, Yu EPK, Figg NL, Finigan A, Murphy MP, et al. Restoring mitochondrial DNA copy number preserves mitochondrial function and delays vascular aging in mice. Aging Cell. (2018) 17(4):e12773. doi: 10.1111/acel.12773
17. Jeng JY, Yeh TS, Lee JW, Lin SH, Fong TH, Hsieh RH. Maintenance of mitochondrial DNA copy number and expression are essential for preservation of mitochondrial function and cell growth. J Cell Biochem. (2008) 103(2):347–57. doi: 10.1002/jcb.21625
18. Castellani CA, Longchamps RJ, Sun J, Guallar E, Arking DE. Thinking outside the nucleus: mitochondrial DNA copy number in health and disease. Mitochondrion. (2020) 53:214–23. doi: 10.1016/j.mito.2020.06.004
19. Hu H, Lin Y, Xu X, Lin S, Chen X, Wang S. The alterations of mitochondrial DNA in coronary heart disease. Exp Mol Pathol. (2020) 114:104412. doi: 10.1016/j.yexmp.2020.104412
20. Vecoli C, Borghini A, Andreassi MG. The molecular biomarkers of vascular aging and atherosclerosis: telomere length and mitochondrial DNA4977 common deletion. Mutat Res Rev Mutat Res. (2020) 784:108309. doi: 10.1016/j.mrrev.2020.108309
21. Andreassi MG, Adlerstein D, Carpeggiani C, Shehi E, Fantinato S, Ghezzi E, et al. Individual and summed effects of high-risk genetic polymorphisms on recurrent cardiovascular events following ischemic heart disease. Atherosclerosis. (2012) 223(2):409–15. doi: 10.1016/j.atherosclerosis.2012.05.029
22. Campolo J, Canale P, Piccaluga E, Bossi I, Gazzaniga G, Parolini M, et al. Vascular senescence and atherosclerotic plaque vulnerability: investigating the telomere-mitochondria crosstalk—rationale and design of the VICTORIA study. Explor Cardiol. (2024) 2:168–77. doi: 10.37349/ec.2024.00030
23. Vecoli C, Borghini A, Pulignani S, Mercuri A, Turchi S, Picano E, et al. Independent and combined effects of telomere shortening and mtDNA4977 deletion on long-term outcomes of patients with coronary artery disease. Int J Mol Sci. (2019) 20(21):5508. doi: 10.3390/ijms20215508
24. Vecoli C, Borghini A, Pulignani S, Mercuri A, Turchi S, Carpeggiani C, et al. Prognostic value of mitochondrial DNA4977 deletion and mitochondrial DNA copy number in patients with stable coronary artery disease. Atherosclerosis. (2018) 276:91–7. doi: 10.1016/j.atherosclerosis.2018.07.015
25. Qu K, Yan F, Qin X, Zhang K, He W, Dong M, et al. Mitochondrial dysfunction in vascular endothelial cells and its role in atherosclerosis. Front Physiol. (2022) 13:1084604. doi: 10.3389/fphys.2022.1084604
26. Li X, Liu X, Chen X, Wang Y, Wu S, Li F, et al. Leukocyte mitochondrial DNA copy number and cardiovascular disease: a systematic review and meta-analysis of cohort studies. iScience. (2024) 27(9):110522. doi: 10.1016/j.isci.2024.110522
27. Ashar FN, Zhang Y, Longchamps RJ, Lane J, Moes A, Grove ML, et al. Association of mitochondrial DNA copy number with cardiovascular disease. JAMA Cardiol. (2017) 2(11):1247–55. doi: 10.1001/jamacardio.2017.3683
28. Chen S, Xie X, Wang Y, Gao Y, Xie X, Yang J, et al. Association between leukocyte mitochondrial DNA content and risk of coronary heart disease: a case-control study. Atherosclerosis. (2014) 237(1):220–6. doi: 10.1016/j.atherosclerosis.2014.08.051
29. Révész D, Verhoeven JE, Picard M, Lin J, Sidney S, Epel ES, et al. Associations between cellular aging markers and metabolic syndrome: findings from the CARDIA study. J Clin Endocrinol Metab. (2018) 103(1):148–57. doi: 10.1210/jc.2017-01625
30. Huang CH, Su SL, Hsieh MC, Cheng WL, Chang CC, Wu HL, et al. Depleted leukocyte mitochondrial DNA copy number in metabolic syndrome. J Atheroscler Thromb. (2011) 18(10):867–73. doi: 10.5551/jat.8698
31. Hang D, Nan H, Kværner AS, De Vivo I, Chan AT, Hu Z, et al. Longitudinal associations of lifetime adiposity with leukocyte telomere length and mitochondrial DNA copy number. Eur J Epidemiol. (2018) 33(5):485–95. doi: 10.1007/s10654-018-0382-z
32. Bogliolo M, Izzotti A, De Flora S, Carli C, Abbondandolo A, Degan P. Detection of the “4977 bp” mitochondrial DNA deletion in human atherosclerotic lesions. Mutagenesis. (1999) 14(1):77–82. doi: 10.1093/mutage/14.1.77
33. Ballinger SW, Patterson C, Knight-Lozano CA, Burow DL, Conklin CA, Hu Z, et al. Mitochondrial integrity and function in atherogenesis. Circulation. (2002) 106(5):544–9. doi: 10.1161/01.cir.0000023921.93743.89
34. Mercer JR, Cheng KK, Figg N, Gorenne I, Mahmoudi M, Griffin J, et al. DNA damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome. Circ Res. (2010) 107(8):1021–31. doi: 10.1161/CIRCRESAHA.110.218966
35. Ballinger SW, Patterson C, Yan CN, Doan R, Burow DL, Young CG, et al. Hydrogen peroxide and peroxynitrite induced mitochondrial DNA damage and dysfunction in vascular endothelial and smooth muscle cells. Circ Res. (2000) 86(9):960–6. doi: 10.1161/01.res.86.9.960
36. Ding Y, Gao BB, Huang JY. The role of mitochondrial DNA mutations in coronary heart disease. Eur Rev Med Pharmacol Sci. (2020) 24(16):8502–9. doi: 10.26355/eurrev_202008_22647
37. Dabravolski SA, Khotina VA, Sukhorukov VN, Kalmykov VA, Mikhaleva LM, Orekhov AN. The role of mitochondrial DNA mutations in cardiovascular diseases. Int J Mol Sci. (2022) 23(2):952 doi: 10.3390/ijms23020952
Keywords: mitochondrial dysfunction, atherosclerosis, premature vascular ageing, early coronary artery disease, mtDNA-CN, mtDNA4977
Citation: Campolo J, Canale P, Gazzaniga G, Parolini M, Piccaluga E, Bossi I, Oreglia J, Borghini A, Marinaro I and Andreassi MG (2025) The mitochondrial dysfunction, alongside the modifiable burden of traditional risk factors, drives the development of early-onset coronary artery disease. Front. Cardiovasc. Med. 12:1538202. doi: 10.3389/fcvm.2025.1538202
Received: 2 December 2024; Accepted: 10 February 2025;
Published: 19 February 2025.
Edited by:
Carlo Palombo, University of Pisa, ItalyReviewed by:
Baiba Vilne, Riga Stradiņš University, LatviaCopyright: © 2025 Campolo, Canale, Gazzaniga, Parolini, Piccaluga, Bossi, Oreglia, Borghini, Marinaro and Andreassi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Grazia Andreassi, bWFyaWFncmF6aWEuYW5kcmVhc3NpQGNuci5pdA==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.