
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 13 March 2025
Sec. Heart Valve Disease
Volume 12 - 2025 | https://doi.org/10.3389/fcvm.2025.1537104
This article is part of the Research Topic Frontiers in Cardiovascular Medicine: 10th Year Anniversary View all 3 articles
 Adrien Lupieri1*
Adrien Lupieri1* Prabhash K. Jha1
Prabhash K. Jha1 Victor Nizet2
Victor Nizet2 Walderez O. Dutra3
Walderez O. Dutra3 Maria Carmo P. Nunes4
Maria Carmo P. Nunes4 Robert A. Levine5
Robert A. Levine5 Elena Aikawa1,6*
Elena Aikawa1,6*
Despite being a leading cause of morbidity and mortality among young people, affecting predominantly women, rheumatic heart disease (RHD) remains neglected and understudied. This autoimmune condition arises from a complex continuum that begins with repeated Group A Streptococcal (GAS) pharyngitis, leading to acute rheumatic fever (ARF) that eventually results in damage to the heart, mainly affecting the mitral valve. While RHD has been nearly eradicated in high-income countries, it continues to be a significant and active health issue in low- and middle-income countries. The resolution of this disease faces several challenges, including the difficulty of diagnosis and the lack of access to preventive measures in resource-poor communities. Addressing these issues will require a global health collaboration involving healthcare professionals, policymakers, and advocacy groups. However, over the past two decades, there has been a revival of scientific interest, fostering optimism for the future. Recent research has significantly advanced our understanding of RHD, shedding light on the immune-to-autoimmune transition, neoangiogenesis, and lymphangiogenesis processes. Additionally, perspectives of discoveries in biomarkers and the development of genetic, transcriptomic, and provide a solid foundation for further advancements in the field.
ARF is a complication of untreated nasopharyngeal infection by beta-hemolytic GAS, primarily affecting children and adolescents aged 5–15 years. Although ARF symptoms resolve over weeks to months, ARF can progress to rheumatic heart valve disease (RHVD), which remains a leading cause of acquired heart failure and cardiovascular death, impacting around 40 million people and causes roughly 300,000 annual deaths worldwide (1, 2). RHVD is typically viewed as a condition associated with poverty, overcrowding, inadequate sanitation, and limited access to healthcare. Historically, before the socioeconomic developments of the late 20th century, ARF and RHVD were more prevalent worldwide, affecting young people in both high- and low-income nations. Currently, most patients affected by these conditions live in low- and middle-income countries in Central and South Asia, the South Pacific, and sub-Saharan Africa (3, 4). Although RHVD incidence has declined in high-income countries over the past decades, a recent resurgence has been observed in many of these regions. This increase may partly stem from the influx of migrants and refugees from areas with high RHVD prevalence (5). However, a 2008–2018 study on ARF and RHVD in the USA found that most cases were not linked to foreign exposure but rather to poor living conditions and limited healthcare access in underserved communities (6). RHVD also remains prevalent in some high-income countries, including Australia, New Zealand, and Canada, disproportionately affecting indigenous young people. Higher incidence in these populations has been associated with inadequate housing, restricted access to preventive and primary care and challenges in early diagnosis (7). Ongoing migration crises and the marginalization of vulnerable populations worldwide continue to shape RHVD epidemiology and management, reinforcing its status as a pressing global health issue (8).
Furthermore, while ARF appears to be equally found in males and females, RHVD has a two-fold higher prevalence in women than men (9). The cause behind this sex disparity remains unclear and warrants further investigation of underlying factors. In addition to the higher female prevalence, studies indicate that RHVD in pregnancy is a significant non-obstetric cause of maternal mortality, emphasizing the urgent need for enhanced monitoring and management of affected women (10, 11).
In summary, both the socioeconomic environment and genetic susceptibility play crucial roles in the development of the disease. To effectively control it, we must address key challenges, including ensuring access to existing prevention and treatment options, as well as increasing our understanding of the underlying biological mechanisms.
Most RHVD-associated morbidity and mortality can be effectively managed through existing therapies. However, the high prevalence of this condition in certain regions is primarily due to limited access to healthcare infrastructure and a lack of public education regarding disease prevention. Addressing these barriers is crucial for reducing the impact of RHD and improving health outcomes in impacted communities, which requires enhancing the treatment of GAS infections, ensuring early detection of subclinical RHVD, providing access to surgical interventions in resource-poor regions, developing biomarkers for early disease, and increasing scientific research.
The most effective strategy for preventing RHVD is the timely and adequate treatment of primary GAS-induced pharyngitis and thereby halting the development of ARF. This approach relies on simple and cost-effective antibiotic treatment, such as penicillin, to prevent recurrent episodes of pharyngitis and reduce the risk of ARF. Even in asymptomatic patients with echocardiogram-detected RHVD, secondary penicillin prophylaxis is effective in preventing disease progression (12).
However, ARF is often underdiagnosed for various reasons, including its diverse clinical manifestations, the symptom overlaps with other common diseases like malaria and viral infections, and a lack of health-seeking behavior. Consequently, many patients are diagnosed too late with RHVD at advanced and severe stages resulting in high mortality rates at a young age (13). It is crucial to improve access to diagnosis and guiding antibiotic prophylaxis in remote and underserved communities, particularly during the period between the initial injury and the onset of clinical disease (10, 14).
The overall prevalence of subclinical RHVD, often called “latent” RHVD, is estimated to be seven to eight times higher than clinically manifested RHVD (15), highlighting the critical importance of early detection in the initial stages of RHVD. Notably, echocardiography has been shown to be four times more sensitive in detecting RHVD compared to traditional cardiac auscultation (15). Therefore, improving access to diagnostic tools is essential for effectively identifying early cases of RHVD. This can be achieved through the implementation of echocardiography-based screening in remote community settings, which will facilitate early detection of valvopathies and enhance patient care.
Mitral regurgitation (MR) is the most prevalent valvular dysfunction during the early stages of RHVD, typically associated with structural impairment of the MV apparatus during acute rheumatic inflammation in children (16). Later disease stages are associated with morphological changes of the MV such as thickening and stiffening of leaflets and chordae, ultimately leading to mitral stenosis (MS) (17). These severe valve dysfunctions are strongly associated with functional limitation and patient mortality, so that surgical or catheter-based interventions are required for repair or replacement of the damaged heart valves. However, in resource-poor settings, health facilities and trained personnel capable of performing percutaneous and surgical interventions may be limited. In some cases, patients diagnosed late with advanced-stage RHVD may receive limited benefits from valve intervention due to a poor prognosis with persistent pulmonary hypertension and right-sided heart failure (9, 18).
Despite its ongoing prevalence in certain regions, research and funding for RHD have lagged significantly, with clear evidence of disproportionate ratio of research funding relative to the disease burden (19).
Historically, the first period from the 1930s and 1950s marked significant advances, including the establishment of the first Jones criteria for ARF, some major advances in bacteriology and the emergence of antibiotics to treat GAS pharyngitis along with secondary antibiotic prophylaxis to prevent ARF recurrence. In the 1960s, targeted advocacy, education, and awareness efforts led by the American Heart Association largely promoted global research effort (Figure 1). However, the subsequent reduction of RHVD incidence and severity in high-income countries resulted in prolonged stagnation of research from the 1970s and 2000s (Figure 1) (20). Recent years have seen the re-emergence of global awareness regarding RHVD (Figure 1), emphasized by several initiatives in Africa, including Drakensberg Declaration (2005) (21), Mosi-o-Tunya Call to Action (2014) (22), Cairo Accord on RHD (2017) (23), Cape Town Declaration on Access to Cardiac Surgery in the Developing World (2018) (24).
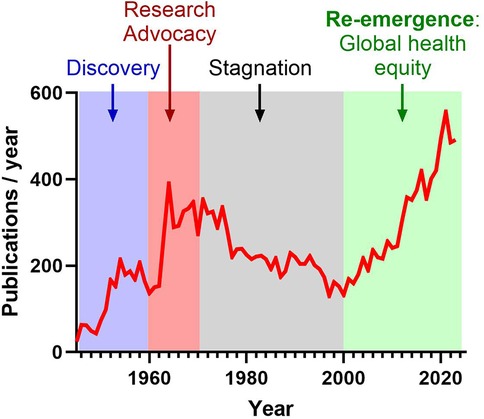
Figure 1. Evolution of publications related to “rheumatic heart disease” referenced in PubMed over the past 80 years. Following an initial phase of significant discoveries, the 1960s witnessed increased advocacy and awareness efforts within the scientific community. However, research stagnated from the 1970s to the 2000s due to the eradication of RHD in high-income countries. Recently, there has been a resurgence of global awareness, underscored by various initiatives and resolutions prioritizing ARF and RHD.
The surge in research interest related to RHD after 2000 can be largely attributed to increased global health prioritization and a focus on health equity (Figure 1). International organizations like the World Health Organization (WHO) and World Heart Federation (WHF) recognized RHD as a major cause of cardiovascular morbidity and mortality, particularly in low- and middle-income countries. This led to heightened awareness, more research, and greater funding for prevention and treatment initiatives. At the same time, attention shifted toward the social determinants of health, with a focus on how poverty, limited healthcare access, and inequities disproportionately affect RHD outcomes, driving further research on its persistence in underserved populations (25, 26). Additionally, In May 2018, the World Health Assembly adopted a resolution (WHA71.14) prioritizing the global response to ARF and RHVD. In 2020, the American Heart Association has issued a call to action aimed at reducing the global impact of RHVD (27). Most recently, in September 2023, the National Institutes of Health, through the National Institute of Allergy and Infectious Diseases and the National Heart, Lung, and Blood Institute, released a notice of special interest (NOT-HL-23-106) aimed at advancing RHVD research. Furthermore, the Leducq Foundation, an international charitable organization, is dedicated to supporting collaborative research on RHVD. This organization provides funding for international networks with three primary goals: identifying biomarkers for ARF, developing a vaccine against GAS, and creating a polymeric heart valve for patients suffering from advanced RHVD.
Collectively, these global initiatives and funding sources are expected to drive significant advancements in research on ARF and RHVD, emphasizing innovative approaches and the development of effective strategies to prevent and cure RHVD.
A single GAS infection triggers a natural immune response. However, repeated or untreated infections can occasionally shift this to an autoimmune reaction, leading to ARF, which affects various organs, including the joints, brain, skin, and heart (Figure 2). This autoimmune response can cause long-term damage to the cardiac valve, known as RHVD. It is established that in this condition triggers an immune response that alters the matrix architecture and cellular components of tissues, particularly the MV leaflets (28, 29). This alteration of MV function is observed in up to 60% of ARF cases, leading to disruption of the normal blood flow through the heart chambers, which can result in heart failure and pulmonary hypertension.
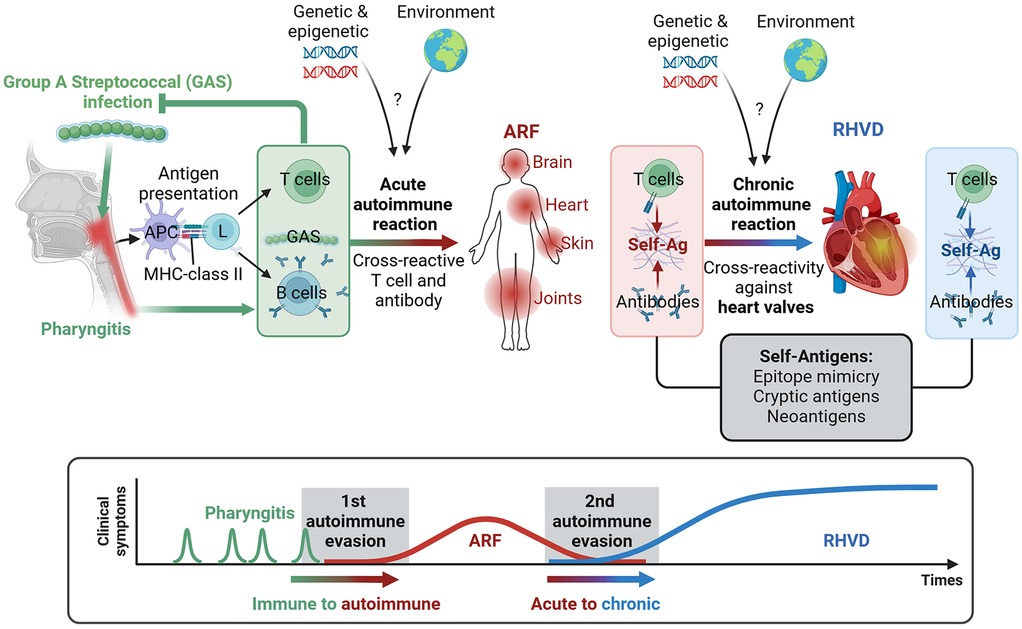
Figure 2. Continuum of disease from group A streptococcus (GAS) infection to autoimmune acute rheumatic fever (ARF) and rheumatic heart valve disease (RHVD). Following GAS pharyngitis, the adaptive immune system processes bacterial antigens, and antigen-presenting cells (APCs) activate lymphocyte responses. T cell clones that recognize GAS epitopes are activated alongside B cells, which produce antibodies targeting GAS. Repeated episodes of GAS pharyngitis, combined with genetic and environmental risk factors, can trigger an autoimmune response affecting the brain, heart, skin, and joints through the cross-reactivity of self-antigens (self-Ag). This autoimmune response may lead to long-term damage, specifically to the cardiac valves, resulting in RHVD. The transition from acute to chronic autoimmune reactions, particularly those targeting the heart valves, may be linked to the evolution of self-antigens, including the release of cryptic antigens and the formation of neoantigens.
ARF is an acute autoimmune response triggered by the infection with group A Streptococcus (GAS). It is estimated that over 600 million cases of symptomatic GAS pharyngitis occur annually (25). However, only 3% to 6% of patients infected by GAS develop ARF (4), indicating that factors such as genetic susceptibility, socioeconomic conditions, and the virulence of GAS may play a role in its development. Patients with ARF can experience a variety of clinical manifestations that may affect the skin, subcutaneous tissues, joints, heart, and brain, though these effects can vary significantly among individuals.
Primary GAS pharyngeal infection induces innate immune reaction and phagocytosis of GAS by antigen-presenting cells, such as macrophages and dendritic cells, for further antigens presentation, and subsequent clone selection and activation of both T and B cells (17). Consequently, the initiation of the adaptive immunity through the activation of specific repertoire of activated T cells and B cells will fight the primary infection targeting GAS-specific antigens (Figures 2, 3). However, the mechanism turning the immune reaction targeting GAS into an autoimmune reaction affecting host tissues is not entirely understood.
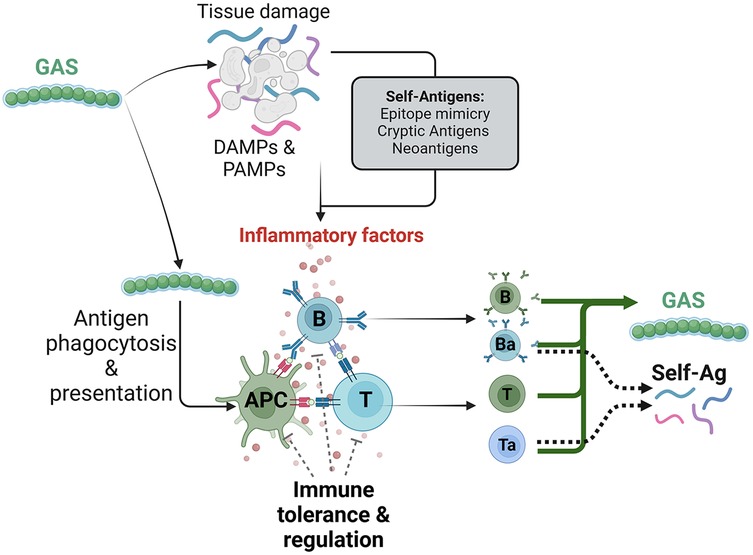
Figure 3. Generation of autoimmune immune response by group A streptococcus (GAS) infection. GAS infection triggers an immune response that leads to tissue damage and the release of damage- and pathogen-associated molecular patterns (DAMPs and PAMPs). Concomitantly, GAS antigens are processed and presented by antigen presenting cells (APCs), activating reactive T and B cell clones. However, the combination of repeated infectious episode and a deficiency in immune tolerance and T cell regulation processes, along with the generation of cross-reactive self-antigens, may contribute to the maturation and activation of autoreactive clones of T cells (Ta) and B cells (Ba), which react to both GAS and self-antigens.
Among the key mechanisms driving autoimmunity, molecular mimicry is the most frequently reported (8, 30–32). The current paradigm of ARF suggests that the similarity of epitopes allows T cells and B cells to recognize both GAS-specific antigens and tissue specific self-proteins through receptors (T-Cell Receptors, TCR; B-Cell Receptors, BCR). This cross-reactivity leads to the production of antibodies that target not only GAS-specific antigens, but also self-proteins found in the brain, heart, skin, and joints.
Several examples of molecular mimicry have been identified in the various organs affected during ARF. For instance, neurological manifestation (chorea syndrome) is associated with cross-reacting antibodies for group A carbohydrate epitope N-acetyl-β-d-glucosamine (GlcNAc) and antigens found in the brain such as the D1 and D2 dopamine receptors, lysoganglioside and tubulin (17). Rheumatic carditis has been associated with cross-reactive antibodies targeting group A carbohydrate or streptococcal M protein and cardiac myosin and valvular laminin (17). In addition, collagen-reactive antibodies and T cells have also been observed in patients with ARF, potentially due to epitope similarities with M protein (33) or streptococcal collagen-like proteins (34). Other studies suggested that these anti-collagen antibodies are induced by the exposure of cryptic collagen epitopes resulting from collagen degradation or the aggregation of collagen by certain streptococcal serotypes (17).
Nevertheless, epitopes mimicry is commonly found in nature, but cross-reactivity requires self-reactive lymphocytes to escape the robust mechanism of thymic and peripheral tolerance (35). This process of immune tolerance prevents lymphocytes with a high affinity for self-antigens from functional differentiation through several mechanisms. These include inhibitory molecules that downregulate immune responses (such as cytotoxic T-lymphocyte associated protein 4, CTLA-4; Programmed cell Death protein 1, PD-1); anergy, where T cells become unresponsive without co-stimulatory signals; ignorance, which keeps certain self-antigens hidden from the immune system; and active suppression by regulatory T cells (Tregs) that inhibit autoreactive lymphocytes (Figure 3). Together, these mechanisms help maintain self-tolerance and protect against autoimmune diseases (36). However, lymphocytes with low affinity for self-antigens that react weakly, can escape thymic selection and persist in the periphery (37), potentially becoming activated through repeated stimulation and loss of tolerance. In this model, in which a clone of lymphocyte reacting to GAS infection but also to self-antigen(s) with low affinity are activated and able to mature into memory lymphocyte, recurrent GAS pharyngitis episodes might increase its reactivity and lead to ARF through the recognition of self-antigens (Figure 3). Mathematical modeling of autoimmune disease initiated by pathogen infection has suggested that cross-reactivity of antigen should be minimal to escape the processes of negative selection and T-cell activation, but needs to be strong enough for eliminating pathogenic antigens (38).
Moreover, some hypotheses suggest that specific strains of GAS may play a role in the induction of ARF. The virulence of these GAS strains has been linked to the high variability of the M protein present on their surface (39). This variability may induce different potential cross-reactivity with self-antigens. However, a definitive identification of rheumatogenic GAS strains has not been established. Additionally, other theories proposing that neo-antigens are formed from the combination of the PARF motif on the M protein bound to the CB3 region of type IV collagen (40). The immunogenicity of this chimeric complex leads to the production of autoantibodies targeting type IV collagen, possibly cross-reacting with other collagen types, including type I collagen present in the valve.
Rheumatic carditis affects both the myocardium and the heart valves. In the early stages of the disease, pan-carditis is observed. However, as the disease progresses, it predominantly impacts the left heart valves, particularly the MV, leading to RHVD. However, the underlying pathological mechanisms of this preferential MV involvement during the chronic phase remain unclear.
Rheumatic MV damage is initiated through the binding of GAS-specific antibodies that cross-react with valvular endothelium molecules, including laminin and other glycoproteins. This interaction activates valvular endothelial cells, leading to the upregulation of vascular cell adhesion molecule 1 (VCAM-1) on the cell surface. The subsequent rearrangements in the endothelium facilitate immune cell infiltration (Figure 4) (17, 41). The resulting intra-valvular inflammation, supported by chronic activation by self-antigens, produces cytokines, cytotoxins and proteases that contribute to structural alterations in the valve tissue, ultimately causing valve dysfunction.

Figure 4. Schematic representation of the pathogenesis of rheumatic heart valve disease (RHVD). Antibodies targeting Group A Streptococcus (GAS) epitope can cross-react with autoantigens of the valve endothelium by molecular mimicry, leading to endothelial activation and subsequent infiltration of immune cells, including macrophages, B cells, and T cells. The local inflammation, coupled with the presence of autoantibodies cross-reacting with valve components, promotes the release of cytokines and matrix metalloproteinases (MMPs), contributing to the generation of new self-antigens (both cryptic and neoantigens). Additionally, inflammatory factors such as TGFβ, promote valvular interstitial cell (VIC)-mediated fibrosis and calcification, leading to valve dysfunction. The formation of neovessels within the inflamed leaflets may play a crucial role in sustaining chronic inflammation in the mitral valve (MV) by supporting the trafficking of immune cells, antigen-presenting cells (APCs) and self-antigens through lymphatic neovessels to the lymph nodes. This process allows autoreactive cells to recirculation into the bloodstream and subsequently remigration to the leaflets via neocapillaries. Furthermore, local factors, such as prothymosin alpha (PTMA), may enhance autoimmune responses by increasing T cell activity and their ability to bind collagen.
Similarly to the triggering of the acute autoimmune reaction, the chronic phase has been associated with immune cross-reactivity. While only cardiac myosin and M protein mimicry have been associated with cardiac valvulitis in animal studies (42, 43), other forms of molecular mimicry likely play a significant role in triggering rheumatic carditis. This include the exposure of cryptic collagen epitopes (17), and the molecular mimicry between type I collagen-derived peptides and GAS epitopes (33). However, another study reported the presence of autoantibodies targeting collagen I in patients with ARF, although these antibodies do not cross-react with GAS-derived antigens (44). This lack of cross-reactivity suggests an alternative mechanism for autoimmunity that operates independently of molecular mimicry (Figure 4).
Recent studies have revealed that protein prothymosin-alpha (PTMA) levels are elevated in the mitral valves of RHVD patients. This protein has been shown to promote both CD8+ T-cell cytotoxicity and recognition of human type I collagen via Very Late Antigen α2β1 (VLA-2), suggesting a potential role in promoting autoimmune responses (33).
Cardiac valve leaflets are largely avascular structures under normal physiological conditions as thin leaflets get their nutrients through the process of diffusion. Neovascularization has been described as a key feature of valvular heart disease, including RHVD (41, 45–47). This phenomenon could provide valuable mechanistic insights into the progression of RHVD. The primary functions of neovessels are to supply oxygen, metabolites, and support immune cell trafficking. This process is regulated by several growth factors including vascular endothelial growth factors (VEGFs), and by extracellular matrix remodeling, which aids endothelial cell migration.
However, dysfunctional angiogenesis can also compromise valve tissue structure and mechanical properties and could lead to blood leakage into the tissue. Of note, immature vessels were observed in human RHVD MVs (41), suggesting that aberrant vessel formation may contribute to disease pathogenesis, either by disrupting the normal valve architecture or supporting infiltration of immune cells and soluble inflammatory factors. Furthermore, studies have demonstrated that preventing angiogenesis may serve as a potential therapeutic strategy to mitigate valve disease (46), emphasizing the crucial role of valvular neoangiogenesis in the progression of RHVD (Figure 4).
The role of lymphatic vessels in the progression of valvular disease is not yet well established. The lymphatic system consists of blind-ended vessels that form a one-way network essential for draining interstitial fluid and supporting adaptive immune responses (48). Its primary immune function is to transport antigens and activated antigen-presenting cells (APCs) to the lymph nodes, thereby supporting the activation of adaptive immune effector cells and humoral responses (Figure 4). Remarkably, de-novo lymphangiogenesis has been implicated in chronic inflammation, as seen in conditions such as psoriasis, rheumatoid arthritis, and allograft transplantation (49). These conditions often feature significant lymphangiogenesis and immune cell trafficking mediated by vascular endothelial growth factor receptor 3 (VEGFR3)-dependent CCL21 production (50). In such cases, inhibiting VEGFR-3 has been shown to reduce lymphangiogenesis and decrease CCL21-mediated adaptive immune responses (49, 50).
Moreover, while the development of lymphatic vessels in mitral valves of patients with RHVD has been previously observed (41, 51, 52), a recent study has elegantly characterized the development of lymphatic vessels (LYVE1+) in the mitral valves during autoimmune valvular carditis in K/B.g7 mice (53). These mice naturally produce autoantibodies targeting the widely expressed glycolytic enzyme glucose-6-phosphate isomerase (GPI), which leads to severe autoimmune arthritis and valvular carditis similarly to human ARF and RHVD (54).
Although ARF appears to affect males and females similarly (17), RHVD is more prevalent in women (relative risk of 1.6–2) (9). Epidemiological data on RHVD align with patterns observed in other autoimmune diseases, which have an approximately two-fold higher prevalence in women (36). This sexual difference has been linked to sex hormones and the X chromosome. Estrogen, the predominant female sex hormone, is known to promote antibody and autoantibody responses in females through B cell activation, hypermutation, and class switch recombination (55). Importantly, estrogen has been found to regulate T cell development and enhance the immune response by increasing the expression of pro-inflammatory factors (55). Additionally, it decreases the expression of the autoimmune regulator (AIRE) gene, thereby increasing the susceptibility to developing autoreactive lymphocyte (55). This dysregulation can contribute to the development of autoimmune disorders (36).
The presence or absence of a second X chromosome is strongly associated with susceptibility to autoimmune disease, as the inconsistency of the X chromosome inactivation induces an imbalance of gene expression, affecting approximately 15% of X-linked genes (56). Several of these escaped X-linked genes are involved in various processes of immune activation and regulation (e.g., CD40l, CXCR3, OGT, FOXP3, TLR7, TLR8, IL2RG, BTK, and IL9R) (57). Similarly, other regulatory mechanisms, such as non-coding transcripts, could play important roles. For instance, XIST lncRNA, a key regulator of the X-chromosome inactivation, has been shown to promote the splenic expression of TLR7, TLR8 and CXCR3, and increase levels of TNF, IL-1β, and IL-6 (58). Furthermore, it is estimated that the X-chromosome carries approximately 10% of non-coding miRNA, and several estrogen-regulated miRNA has been identified, which may be an important source of sex discrepancies and warrants further investigation (55).
The importance of these sex-dependent mechanisms in RHVD remains unexplored. However, new insights into the sex-specific pathobiology of RHVD are gradually emerging. Recent findings highlight that the stimulation CD8+ T-cell cytotoxicity by PTMA was linked with estrogen receptor-alpha activity, thus suggesting a plausible role of this and other factors in the sex predisposition in RHVD (33).
In addition to socioeconomic and environmental risk factors, current knowledge indicates a strong genetic susceptibility to ARF. A study on twin pairs estimated the heritability component of susceptibility to ARF at 60% (59). Epidemiologic studies reported that among the more than 600 million cases of symptomatic GAS infection, only 3%–6% of individuals will develop ARF (60). This suggests that the autoimmune disorder only develops in genetically predisposed individuals. Furthermore, it is estimated that 60% of patients with ARF will develop RHVD. Although there is a well-established association between GAS infection and RHVD, RHVD seems to be induced autonomously even after the initial stimulus is removed, thus suggesting that host-factors, such as genetic and epigenetic influences, play a crucial role (25). However, risk factors promoting the transition from acute to chronic autoimmune condition remain unknown. Therefore, genetic studies to identify individuals at high-risk genes could help in developing effective prevention and control strategies, enhancing the management of RHVD.
Genetic susceptibility for RHVD development has been studied in genome-wide association studies (GWASs). Significant association have been demonstrated in class II human leukocyte antigen (HLA) loci (HLA-DQA1, HLA-DQB1, HLA-DRB1) (60), which are expressed on the surface of APCs and support T cell receptor-mediated immune responses. Additionally, class I HLA (HLA-B) has been associated with ARF (60). Similarly, immunoglobin heavy locus (IGHV4-61) was linked to an increased risk of RHVD in Oceanian countries (60).
Furthermore, several polymorphisms in genes coding for immune-related proteins have been associated with ARF and RHD susceptibility. These proteins include toll-like receptor 2 (TLR-2), interleukin 1 beta (IL-1β), interleukin 6 (IL-6), interleukin-10 (IL-10), tumor necrosis factor alpha (TNF-α), transforming growth factor beta 1 (TGF-β1), angiotensin I converting enzyme (ACE), mannose-binding lectin 2 (MBL2), and mannose-binding protein-associated serine protease 2 (MASP2) (61, 62). Although other gene variants have been associated with ARF or RHVD, most have been observed only in single studies, making further meta-analysis impossible (61).
Published GWASs in ARF/RHVD are relatively small and lack the statistical power to characterize reliable and reproducible risk factors, underscoring the need for large-scale, multicenter studies across diverse populations. Additionally, current research has disproportionately focused on European and Asian cohorts, with limited assessment of sub-Saharan populations, who are dramatically affected by these diseases. Among the promising upcoming studies, the RHDGen network GWAS study stands out. It combines echocardiography screening for enrolling RHVD cases and ethnically matched controls, a genetic trio (both parents and offspring) replication, and plasma collection. This presents a unique opportunity to identify relevant genetic factors and biomarkers associated with differential RHD risk in African populations (63, 64). The emergence of robust new knowledge on the genetic susceptibility to RHVD will pave the way for more precise, effective, and individualized prevention and treatment strategies.
The major strategies the scientific community can implement to combat the ARF and RHD can be divided into five key areas: (I) Development of diagnostic tools and strategies, including accessible echocardiography to identify prognostic tissue properties (65), point-of-care rapid tests and portable methods for detecting GAS-antigens, and biomarkers for identifying ARF and latent RHVD. The emerging artificial intelligence-based classification of echocardiography could also provide robust support for detecting early ARF and latent RHVD (66), (II) Large scale epidemiologic and genetic studies of ARF and RHVD with aims to characterize demographic features, outcomes, and genetic markers. (III) Multi-omics approaches, integrating proteomics and transcriptomics at both tissue and single-cell levels (e.g., plasma, peripheral blood mononuclear cells, and valve tissue) are essential to provide functional understanding of the disease. The emergence of open-source omics datasets will provide opportunities for secondary and meta-analysis, even in resource-poor settings. Moreover, single cell omics will help appreciate these changes in the cellular landscape of the disease. Multimodal analysis will offer crucial insights into potential biomarker candidates and pathophysiology, paving the way for new therapeutic strategies, including CRISPR-based gene editing therapies targeting immune cells (67). (IV) Development of preventive treatment, including long-acting penicillin to improve secondary prophylaxis and the development of a vaccine against GAS to transform the primary prevention. (V) Development of education and awareness campaigns to improve affected population understanding of the danger of GAS infection, ARF and RHVD. These initiatives should be implemented through a variety of multimedia campaigns (printed materials, social media, television, radio), school-based educational programs, and training for community workers with hand-held ultrasound screening (68, 69).
These efforts collectively aim to enhance the early detection, prevention, and management of ARF and RHD. Addressing these challenges requires sustained commitment from healthcare professionals, policymakers, and advocacy groups, including non-governmental organizations, to drive long-term change and improve outcomes worldwide.
AL: Writing – original draft. PJ: Writing – review & editing. VN: Writing – review & editing. WD: Writing – review & editing. MN: Writing – review & editing. RL: Writing – review & editing. EA: Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. EA lab is partially supported by NIH R01 HL174066, Leducq Foundation network grant 22ARF02 and a research grant from Pfizer.
The authors acknowledge the Leducq Foundation for the support and commitment to fostering international collaboration in rheumatic heart disease research with the authors in the Leducq PRIMA network. The figures were generated using BioRender.
EA is a member of the scientific board of Elastrin Therapeutics, Inc. and a holder of a research grant from Pfizer.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor MA declared a past co-authorship with the author EA.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Disease GBD, Injury I, Prevalence C. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the global burden of disease study 2017. Lancet. (2018) 392:1789–858. doi: 10.1016/S0140-6736(18)32279-7
2. Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: update from the gbd 2019 study. J Am Coll Cardiol. (2020) 76:2982–3021. doi: 10.1016/j.jacc.2020.11.010
3. Watkins DA, Beaton AZ, Carapetis JR, Karthikeyan G, Mayosi BM, Wyber R, et al. Rheumatic heart disease worldwide: Jacc scientific expert panel. J Am Coll Cardiol. (2018) 72:1397–416. doi: 10.1016/j.jacc.2018.06.063
4. Dougherty S, Okello E, Mwangi J, Kumar RK. Rheumatic heart disease: Jacc focus seminar 2/4. J Am Coll Cardiol. (2023) 81:81–94. doi: 10.1016/j.jacc.2022.09.050
5. Ojha U, Marshall DC, Salciccioli JD, Al-Khayatt BM, Hammond-Haley M, Goodall R, et al. Temporal trend analysis of rheumatic heart disease burden in high-income countries between 1990 and 2019. Eur Heart J Qual Care Clin Outcomes. (2024) 10:108–20. doi: 10.1093/ehjqcco/qcac083
6. de Loizaga SR, Arthur L, Arya B, Beckman B, Belay W, Brokamp C, et al. Rheumatic heart disease in the United States: forgotten but not gone: results of a 10 year multicenter review. J Am Heart Assoc. (2021) 10:e020992. doi: 10.1161/JAHA.120.020992
7. Wyber R, Wade V, Anderson A, Schreiber Y, Saginur R, Brown A, et al. Rheumatic heart disease in indigenous young peoples. Lancet Child Adolesc Health. (2021) 5:437–46. doi: 10.1016/S2352-4642(20)30308-4
8. Mutagaywa RK, Wind AM, Kamuhabwa A, Cramer MJ, Chillo P, Chamuleau S. Rheumatic heart disease anno 2020: impacts of gender and migration on epidemiology and management. Eur J Clin Invest. (2020) 50:e13374. doi: 10.1111/eci.13374
9. Zuhlke L, Engel ME, Karthikeyan G, Rangarajan S, Mackie P, Cupido B, et al. Characteristics, complications, and gaps in evidence-based interventions in rheumatic heart disease: the global rheumatic heart disease registry (the remedy study). Eur Heart J. (2015) 36:1115–22. doi: 10.1093/eurheartj/ehu449
10. Passos LSA, Nunes MCP, Zilla P, Yacoub MH, Aikawa E. Raising awareness for rheumatic mitral valve disease. Glob Cardiol Sci Pract. (2020) 2020:e202026. doi: 10.21542/gcsp.2020.26
11. Diao M, Kane A, Ndiaye MB, Mbaye A, Bodian M, Dia MM, et al. Pregnancy in women with heart disease in sub-saharan Africa. Arch Cardiovasc Dis. (2011) 104:370–4. doi: 10.1016/j.acvd.2011.04.001
12. Beaton A, Okello E, Rwebembera J, Grobler A, Engelman D, Alepere J, et al. Secondary antibiotic prophylaxis for latent rheumatic heart disease. N Engl J Med. (2022) 386:230–40. doi: 10.1056/NEJMoa2102074
13. Zuhlke L, Karthikeyan G, Engel ME, Rangarajan S, Mackie P, Cupido-Katya Mauff B, et al. Clinical outcomes in 3343 children and adults with rheumatic heart disease from 14 low- and middle-income countries: two-year follow-up of the global rheumatic heart disease registry (the remedy study). Circulation. (2016) 134:1456–66. doi: 10.1161/CIRCULATIONAHA.116.024769
14. Passos LSA, Nizet V, Levine RA, Aikawa E. Can we diagnose acute rheumatic fever early to maximize the success of secondary prophylaxis in rheumatic heart valve disease? Cardiovasc Res. (2022) 118:e62–5. doi: 10.1093/cvr/cvac071
15. Rothenbuhler M, O’Sullivan CJ, Stortecky S, Stefanini GG, Spitzer E, Estill J, et al. Active surveillance for rheumatic heart disease in endemic regions: a systematic review and meta-analysis of prevalence among children and adolescents. Lancet Glob Health. (2014) 2:e717–726. doi: 10.1016/S2214-109X(14)70310-9
16. Gomes NFA, Silva VR, Levine RA, Esteves WAM, de Castro ML, Passos LSA, et al. Progression of mitral regurgitation in rheumatic valve disease: role of left atrial remodeling. Front Cardiovasc Med. (2022) 9:862382. doi: 10.3389/fcvm.2022.862382
17. Carapetis JR, Beaton A, Cunningham MW, Guilherme L, Karthikeyan G, Mayosi BM, et al. Acute rheumatic fever and rheumatic heart disease. Nat Rev Dis Primers. (2016) 2:15084. doi: 10.1038/nrdp.2015.84
18. Sagie A, Schwammenthal E, Palacios IF, King ME, Leavitt M, Freitas N, et al. Significant tricuspid regurgitation does not resolve after percutaneous balloon mitral valvotomy. J Thorac Cardiovasc Surg. (1994) 108:727–35. doi: 10.1016/S0022-5223(94)70300-0
19. Macleod CK, Bright P, Steer AC, Kim J, Mabey D, Parks T. Neglecting the neglected: the objective evidence of underfunding in rheumatic heart disease. Trans R Soc Trop Med Hyg. (2019) 113:287–90. doi: 10.1093/trstmh/trz014
20. Rwebembera J, Nascimento BR, Minja NW, de Loizaga S, Aliku T, Dos Santos LPA, et al. Recent advances in the rheumatic fever and rheumatic heart disease continuum. Pathogens. (2022) 11:179. doi: 10.3390/pathogens11020179
21. Mayosi B, Robertson K, Volmink J, Adebo W, Akinyore K, Amoah A, et al. The Drakensberg declaration on the control of rheumatic fever and rheumatic heart disease in Africa. S Afr Med J. (2006) 96:246.16610104
22. Mayosi BM, Gamra H, Dangou JM, Kasonde J. Rheumatic heart disease in Africa: the mosi-o-tunya call to action. Lancet Glob Health. (2014) 2:e438–439. doi: 10.1016/S2214-109X(14)70234-7
23. Kotit S, Phillips DIW, Afifi A, Yacoub M. The “Cairo accord”- towards the eradication of rhd: an update. Front Cardiovasc Med. (2021) 8:690227. doi: 10.3389/fcvm.2021.690227
24. Zilla P, Bolman RM, Yacoub MH, Beyersdorf F, Sliwa K, Zuhlke L, et al. The Cape Town declaration on access to cardiac surgery in the developing world. S Afr Med J. (2018) 108:702–4. doi: 10.7196/SAMJ.2018.v108i9.13102
25. Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group a streptococcal diseases. Lancet Infect Dis. (2005) 5:685–94. doi: 10.1016/S1473-3099(05)70267-X
26. Hu Y, Tong Z, Huang X, Qin JJ, Lin L, Lei F, et al. The projections of global and regional rheumatic heart disease burden from 2020 to 2030. Front Cardiovasc Med. (2022) 9:941917. doi: 10.3389/fcvm.2022.941917
27. Beaton A, Kamalembo FB, Dale J, Kado JH, Karthikeyan G, Kazi DS, et al. The American Heart Association’s call to action for reducing the global burden of rheumatic heart disease: a policy statement from the American Heart Association. Circulation. (2020) 142:e358–68. doi: 10.1161/CIR.0000000000000922
28. Cunningham MW. Pathogenesis of group a streptococcal infections. Clin Microbiol Rev. (2000) 13:470–511. doi: 10.1128/CMR.13.3.470
29. Guilherme L, Cury P, Demarchi LM, Coelho V, Abel L, Lopez AP, et al. Rheumatic heart disease: proinflammatory cytokines play a role in the progression and maintenance of valvular lesions. Am J Pathol. (2004) 165:1583–91. doi: 10.1016/S0002-9440(10)63415-3
30. Guilherme L, Cunha-Neto E, Coelho V, Snitcowsky R, Pomerantzeff PMA, Assis RV, et al. Human heart–infiltrating t-cell clones from rheumatic heart disease patients recognize both streptococcal and cardiac proteins. Circulation. (1995) 92:415–20. doi: 10.1161/01.CIR.92.3.415
31. Gray L-A, D’Antoine HA, Tong SYC, McKinnon M, Bessarab D, Brown N, et al. Genome-wide analysis of genetic risk factors for rheumatic heart disease in aboriginal Australians provides support for pathogenic molecular mimicry. J Infect Dis. (2017) 216:1460–70. doi: 10.1093/infdis/jix497
32. Guilherme L, Kalil J, Cunningham M. Molecular mimicry in the autoimmune pathogenesis of rheumatic heart disease. Autoimmunity. (2006) 39:31–9. doi: 10.1080/08916930500484674
33. Passos LSA, Jha PK, Becker-Greene D, Blaser MC, Romero D, Lupieri A, et al. Prothymosin alpha: a novel contributor to estradiol receptor alpha-mediated cd8(+) t-cell pathogenic responses and recognition of type 1 collagen in rheumatic heart valve disease. Circulation. (2022) 145:531–48. doi: 10.1161/CIRCULATIONAHA.121.057301
34. Pilapitiya DH, Harris PWR, Hanson-Manful P, McGregor R, Kowalczyk R, Raynes JM, et al. Antibody responses to collagen peptides and streptococcal collagen-like 1 proteins in acute rheumatic fever patients. Pathog Dis. (2021) 79:ftab033. doi: 10.1093/femspd/ftab033
35. Rose NR. Negative selection, epitope mimicry and autoimmunity. Curr Opin Immunol. (2017) 49:51–5. doi: 10.1016/j.coi.2017.08.014
36. Theofilopoulos AN, Kono DH, Baccala R. The multiple pathways to autoimmunity. Nat Immunol. (2017) 18:716–24. doi: 10.1038/ni.3731
37. Enouz S, Carrie L, Merkler D, Bevan MJ, Zehn D. Autoreactive t cells bypass negative selection and respond to self-antigen stimulation during infection. J Exp Med. (2012) 209:1769–79. doi: 10.1084/jem.20120905
38. Hara A, Iwasa Y. Autoimmune diseases initiated by pathogen infection: mathematical modeling. J Theor Biol. (2020) 498:110296. doi: 10.1016/j.jtbi.2020.110296
39. Metzgar D, Zampolli A. The m protein of group a streptococcus is a key virulence factor and a clinically relevant strain identification marker. Virulence. (2011) 2:402–12. doi: 10.4161/viru.2.5.16342
40. Dinkla K, Talay SR, Morgelin M, Graham RM, Rohde M, Nitsche-Schmitz DP, et al. Crucial role of the cb3-region of collagen iv in parf-induced acute rheumatic fever. PLoS One. (2009) 4:e4666. doi: 10.1371/journal.pone.0004666
41. Passos LSA, Nunes MCP, Aikawa E. Rheumatic heart valve disease pathophysiology and underlying mechanisms. Front Cardiovasc Med. (2020) 7:612716. doi: 10.3389/fcvm.2020.612716
42. Galvin JE, Hemric ME, Kosanke SD, Factor SM, Quinn A, Cunningham MW. Induction of myocarditis and valvulitis in lewis rats by different epitopes of cardiac myosin and its implications in rheumatic carditis. Am J Pathol. (2002) 160:297–306. doi: 10.1016/S0002-9440(10)64373-8
43. Quinn A, Kosanke S, Fischetti VA, Factor SM, Cunningham MW. Induction of autoimmune valvular heart disease by recombinant streptococcal m protein. Infect Immun. (2001) 69:4072–8. doi: 10.1128/IAI.69.6.4072-4078.2001
44. Martins TB, Hoffman JL, Augustine NH, Phansalkar AR, Fischetti VA, Zabriskie JB, et al. Comprehensive analysis of antibody responses to streptococcal and tissue antigens in patients with acute rheumatic fever. Int Immunol. (2008) 20:445–52. doi: 10.1093/intimm/dxn004
45. Sekulic SP, Sekulic M. Neovascularization of native cardiac valves, and correlation with histopathologic, clinical, and radiologic features. Cardiovasc Pathol. (2024) 69:107605. doi: 10.1016/j.carpath.2024.107605
46. Yoshioka M, Yuasa S, Matsumura K, Kimura K, Shiomi T, Kimura N, et al. Chondromodulin-i maintains cardiac valvular function by preventing angiogenesis. Nat Med. (2006) 12:1151–9. doi: 10.1038/nm1476
47. Rajamannan NM, Nealis TB, Subramaniam M, Pandya S, Stock SR, Ignatiev CI, et al. Calcified rheumatic valve neoangiogenesis is associated with vascular endothelial growth factor expression and osteoblast-like bone formation. Circulation. (2005) 111:3296–301. doi: 10.1161/CIRCULATIONAHA.104.473165
48. Oliver G, Kipnis J, Randolph GJ, Harvey NL. The lymphatic vasculature in the 21(st) century: novel functional roles in homeostasis and disease. Cell. (2020) 182:270–96. doi: 10.1016/j.cell.2020.06.039
49. Alitalo K. The lymphatic vasculature in disease. Nat Med. (2011) 17:1371–80. doi: 10.1038/nm.2545
50. Nykanen AI, Sandelin H, Krebs R, Keranen MA, Tuuminen R, Karpanen T, et al. Targeting lymphatic vessel activation and ccl21 production by vascular endothelial growth factor receptor-3 inhibition has novel immunomodulatory and antiarteriosclerotic effects in cardiac allografts. Circulation. (2010) 121:1413–22. doi: 10.1161/CIRCULATIONAHA.109.910703
51. Kholova I, Dragneva G, Cermakova P, Laidinen S, Kaskenpaa N, Hazes T, et al. Lymphatic vasculature is increased in heart valves, ischaemic and inflamed hearts and in cholesterol-rich and calcified atherosclerotic lesions. Eur J Clin Invest. (2011) 41:487–97. doi: 10.1111/j.1365-2362.2010.02431.x
52. Lupieri A LSAP, Levine RA, Nizet V, Aikawa E. Lymphangiogenesis in rheumatic heart valve disease: a new factor in the pathogenic conundrum. Arterioscler Thromb Vasc Biol. (2024) 44:822–5. doi: 10.1161/ATVBAHA.124.320708
53. Osinski V, Yellamilli A, Firulyova MM, Zhang MJ, Peck AL, Auger JL, et al. Pro-fibrotic vegfr3-dependent lymphatic vessel growth in autoimmune valvular carditis. Arterioscler, Thromb, Vasc Biol. (2024) 44(4):807–21. doi: 10.1161/ATVBAHA.123.320326
54. Binstadt BA, Hebert JL, Ortiz-Lopez A, Bronson R, Benoist C, Mathis D. The same systemic autoimmune disease provokes arthritis and endocarditis via distinct mechanisms. Proc Natl Acad Sci U S A. (2009) 106:16758–63. doi: 10.1073/pnas.0909132106
55. Fairweather D, Beetler DJ, McCabe EJ, Lieberman SM. Mechanisms underlying sex differences in autoimmunity. J Clin Invest. (2024) 134:e180076. doi: 10.1172/JCI180076
56. Carrel L, Willard HF. X-inactivation profile reveals extensive variability in x-linked gene expression in females. Nature. (2005) 434:400–4. doi: 10.1038/nature03479
57. Rubtsova K, Marrack P, Rubtsov AV. Sexual dimorphism in autoimmunity. J Clin Invest. (2015) 125:2187–93. doi: 10.1172/JCI78082
58. Huret C, Ferraye L, David A, Mohamed M, Valentin N, Charlotte F, et al. Altered x-chromosome inactivation predisposes to autoimmunity. Sci Adv. (2024) 10:eadn6537. doi: 10.1126/sciadv.adn6537
59. Engel ME, Stander R, Vogel J, Adeyemo AA, Mayosi BM. Genetic susceptibility to acute rheumatic fever: a systematic review and meta-analysis of twin studies. PLoS One. (2011) 6:e25326. doi: 10.1371/journal.pone.0025326
60. Abdallah AM, Abu-Madi M. The genetic control of the rheumatic heart: closing the genotype-phenotype gap. Front Med (Lausanne). (2021) 8:611036. doi: 10.3389/fmed.2021.611036
61. Muhamed B, Shaboodien G, Engel ME. Genetic variants in rheumatic fever and rheumatic heart disease. Am J Med Genet C Semin Med Genet. (2020) 184:159–77. doi: 10.1002/ajmg.c.31773
62. Tormin J, Nascimento BR, Sable CA, da Silva JLP, Brandao-de-Resende C, Rocha LPC, et al. Cytokine gene functional polymorphisms and phenotypic expression as predictors of evolution from latent to clinical rheumatic heart disease. Cytokine. (2021) 138:155370. doi: 10.1016/j.cyto.2020.155370
63. Salie MT, Yang J, Ramirez Medina CR, Zuhlke LJ, Chishala C, Ntsekhe M, et al. Data-independent acquisition mass spectrometry in severe rheumatic heart disease (rhd) identifies a proteomic signature showing ongoing inflammation and effectively classifying rhd cases. Clin Proteomics. (2022) 19:7. doi: 10.1186/s12014-022-09345-1
64. Machipisa T, Chishala C, Shaboodien G, Zuhlke LJ, Muhamed B, Pandie S, et al. Rationale, design, and the baseline characteristics of the rhdgen (the genetics of rheumatic heart disease) network studydagger. Circ Genom Precis Med. (2023) 16:e003641. doi: 10.1161/CIRCGEN.121.003641
65. Nunes MCP, Tan TC, Elmariah S, Lodi-Junqueira L, Nascimento BR, do Lago R, et al. Net atrioventricular compliance is an independent predictor of cardiovascular death in mitral stenosis. Heart. (2017) 103:1891–8. doi: 10.1136/heartjnl-2016-310955
66. Naser JA, Lee E, Pislaru SV, Tsaban G, Malins JG, Jackson JI, et al. Artificial intelligence-based classification of echocardiographic views. Eur Heart J Digit Health. (2024) 5:260–9. doi: 10.1093/ehjdh/ztae015
67. Lee MH, Shin JI, Yang JW, Lee KH, Cha DH, Hong JB, et al. Genome editing using crispr-cas9 and autoimmune diseases: a comprehensive review. Int J Mol Sci. (2022) 23(3):1337. doi: 10.3390/ijms23031337
68. Nascimento BR, Nunes MCP, Lima EM, Sanyahumbi AE, Wilson N, Tilton E, et al. Outcomes of echocardiography-detected rheumatic heart disease: validating a simplified score in cohorts from different countries. J Am Heart Assoc. (2021) 10:e021622. doi: 10.1161/JAHA.121.021622
Keywords: acute rheumatic fever, rheumatic heart disease, autoimmune disease, neoangiogenesis, lymphangiogenesis
Citation: Lupieri A, Jha PK, Nizet V, Dutra WO, Nunes MCP, Levine RA and Aikawa E (2025) Rheumatic heart valve disease: navigating the challenges of an overlooked autoimmune disorder. Front. Cardiovasc. Med. 12:1537104. doi: 10.3389/fcvm.2025.1537104
Received: 29 November 2024; Accepted: 3 March 2025;
Published: 13 March 2025.
Edited by:
Mabruka Alfaidi, University of Nebraska Medical Center, United StatesReviewed by:
Susy Kotit, Magdi Yacoub Heart Foundation-Aswan Heart Centre, EgyptCopyright: © 2025 Lupieri, Jha, Nizet, Dutra, Nunes, Levine and Aikawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adrien Lupieri, YWRyaWVuLmx1cGllcmlAZ21haWwuY29t; Elena Aikawa, ZWFpa2F3YUBid2guaGFydmFyZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.