 Wenjing Cao
Wenjing Cao Kui Wang
Kui Wang Jiawei Wang4,†
Jiawei Wang4,† Hanxian Gong
Hanxian Gong Lei Xiao
Lei Xiao Wei Pan
Wei Pan
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 27 August 2024
Sec. Coronary Artery Disease
Volume 11 - 2024 | https://doi.org/10.3389/fcvm.2024.1416112
This article is part of the Research Topic Exploring Immune Cell Roles in Cardiac Repair and Remodeling View all 14 articles
Background: Atherosclerotic plaque rupture is a major cause of heart attack. Previous studies have shown that immune cells are involved in the development of atherosclerosis, but different immune cells play different roles. The aim of this study was to investigate the causal relationship between immunological traits and myocardial infarction (MI).
Methods: To assess the causal association of immunological profiles with myocardial infarction based on publicly available genome-wide studies, we used a two-sample mendelian randomization (MR) approach with inverse variance weighted (IVW) as the main analytical method. Sensitivity analyses were used to assess heterogeneity and horizontal pleiotropy.
Results: A two-sample MR analysis was conducted using IVW as the primary method. At a significance level of 0.001, we identified 47 immunophenotypes that have a significant causal relationship with MI. Seven of these were present in B cells, five in cDC, four in T cells at the maturation stage, six in monocytes, five in myeloid cells, 12 in TBNK cells, and eight in Treg cells. Sensitivity analyses were performed to confirm the robustness of the MR results.
Conclusions: Our results provide strong evidence that multiple immune cells have a causal effect on the risk of myocardial infarction. This discovery provides a new avenue for the development of therapeutic treatments for myocardial infarction and a new target for drug development.
Myocardial infarction (MI) occurs on the basis of atheromatous coronary artery lesions, where there is a dramatic reduction or interruption of coronary blood supply, leading to myocardial ischemia and hypoxia or even necrosis (1, 2). Cardiovascular disease is the leading cause of death in the United States, with a prevalence of myocardial infarction as high as 3.2 per cent among American adults (3). Globally, more than 8 million lives are threatened by AMI each year, resulting in a huge health and economic burden (4–6).
Recent studies have shown that the immune system plays a key role in myocardial infarction development and repair, determining the extent of myocardial damage and prognosis (7–9). Intense aseptic inflammation occurs in the infarcted area, which can be divided into an inflammatory phase, a proliferative phase and a mature phase (10, 11). Neutrophils, macrophages and immune cells such as B and T cells are attracted to the infarcted area by chemokines and produce pro-inflammatory or anti-inflammatory factors to remove and repair damaged tissue, affecting cardiac remodeling and healing (12–14). Macrophages are the most numerous immune cells in myocardial tissue (14, 15). Cardiac macrophages can be divided into two subpopulations depending on their origin (16). Macrophages of monocyte origin are predominantly associated with inflammatory responses and pathogenic properties, while resident macrophages of embryonic yolk sac origin exhibit cardioprotective functions. Early macrophages produce pro-inflammatory cytokines leading to tissue inflammation and over time can polarize into M2 macrophages that release anti-inflammatory cytokines to repair the healing process (14). Studies analyzing the role of different subsets of innate lymphoid cells (ILC) reveal the involvement of ILC1 in the progression of atherosclerosis (AS) and the anti-AS role of ILC2 (17). Rafael Blanco-Domínguez finds that CD69 expression on Tregs increases survival from coronary ligation in mice, and increased myocardial inflammation and deterioration of cardiac function after ischemia in CD69- mice (18). These findings suggest that different subpopulations of immune cells behave differently in the inflammatory response to myocardial infarction (8, 14, 17, 19, 20). It has been found that an overactive inflammatory response leads to excessive fibrosis appearing as heart failure with preserved ejection fraction. The absence of an effective inflammatory response leads to unstable scarring and ventricular rupture (13, 20). However, there are few studies on different immune cell subsets and myocardial infarction. These studies often suffer from the limitations of limited sample size, confounding factors and reverse causality.
Mendelian randomization is a statistical technique. Genetic variants, also known as single nucleotide polymorphisms (SNPs), are used as instrumental variables(IV). MR uses genetic variants as a proxy for exposure to assess whether exposure is associated with an outcome (21). It is well known that genetic variants are randomly assigned at conception. In this way, MR is similar to a randomized controlled trial (RCT), which avoids confounding factors and reverse causality. It follows that MR can provide a causal relationship between exposure and outcome (22–25). The role of different types of immune cells in the development and progression of atherosclerosis has been confirmed in previous observational studies. This study identifies additional immune cells associated with myocardial infarction and elucidates the causal relationship between these immune traits and myocardial infarction. The discovery of the immune cell traits associated with the occurrence of MI will help to develop novel therapeutic targets for therapeutic interventions in myocardial infarction (26).
We analyzed the causal relationship between 731 immune cell traits and myocardial infarction based on two-sample Mendelian randomization (MR). MR studies use genetic variation that is associated with exposure as an instrumental variable (IV) to predict the causal relationship between exposure and outcome (27). The study obtained 47 genetic variants for circulating immune cell traits and their association with the risk of myocardial infarction (MI) from previously published genome-wide association studies (GWAS).The use of MR design reduces the impact of confounding and reverse causation, thereby improving the ability to make causal inferences about associations between exposure and outcome. The use of genetic variants as instrumental variables (IV) to study the effects of altered exposures is advantageous due to their random assignment at the time of conception, making them less susceptible to confounding by environmental factors or reverse causation. This study analyzed datasets from human individuals that are publicly available. The overall design is shown in Figure 1. As it is a secondary analysis of published data, ethical approval is not required.
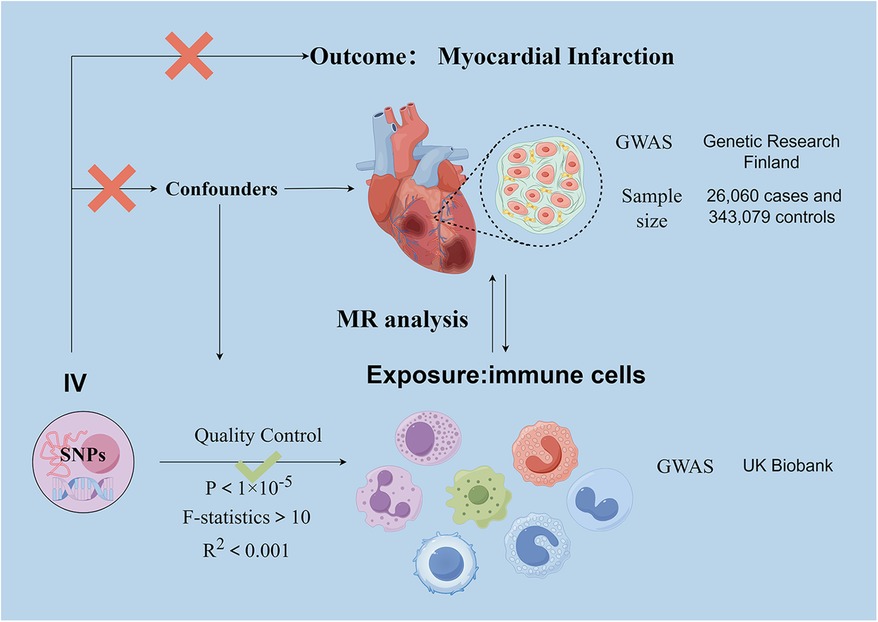
Figure 1. Schematic representation of the research process. Assumption 1, significantly associated with exposure, Assumption 2, not associated with outcome, Assumption 3, not associated with confounders. SNPs, single-nucleotide polymorphisms; MR, Mendelian randomization.
The genome-wide association study (GWAS) summary data for MI are sourced from the December 2023 released by Genetic Research Finland and can be accessed at https://finngen.gitbook.io/documentation. The FinnGen study has analyzed more than 500,000 biobank samples from Finland. The study correlates genetic variants with health data to understand pathogenesis and identify risk factors. A genome-wide association study (GWAS) was conducted on 118,870 European individuals, with 26,060 cases and 343,079 controls.
Genetic variants associated with immune cells are derived from pooled data from the UK Biobank. This information is available for search on the website https://gwas.mrcieu.ac.uk/datasets/.GWAS statistical summaries for each immunological trait are publicly available from the GWAS catalogue under accession numbers ranging from GCST90001391 to GCST90002121. The GWAS data comprised four cellular parameters: median fluorescence intensity (MFI) representing surface antigen levels (n = 389), absolute cell count (AC) (n = 118), morphological parameters (MP) (n = 32), and relative cell count (RC) (n = 192). Of these, AC, MFI and RC features comprised B cells, CDC, T cell maturation stage, monocytes, myeloid cells, TBNK (T cells, B cells, natural killer cells) and Treg panels, whereas MP features included CDC and TBNK panels.
In causal inference, a valid instrumental variable (IV) must satisfy three key assumptions: (1) the genetic variant is directly related to the exposure (731 circulating immune cell traits); (2) there is no confounding influence between the genetic variant and the outcome; and (3) the genetic variant does not affect the outcome in ways other than through the exposure. Based on recent studies, a significance level of 1 × 10−5 was used for the selection of instrumental variables (IV) associated with each immunological trait. To ensure unbiased results, we applied a chain imbalance threshold of 5,000 kb and an R2 value of less than 0.001 to the chain imbalance distance. The strength of the association between the independent variables (IVs) and exposure was assessed using the F-statistic. IVs with an F-statistic greater than 10 were used to exclude effects due to weak instrumental variables (28).
A variety of MR analyses were conducted, such as MR Egger, weighted median, inverse variance weighted (IVW), simple mode, weighted mode, and MR-PRESSO methods. Each of these methods uses different statistical assumptions to make causal inferences (29). In MR analysis, the primary method for assessing causality is IVW (30). IVW assumes that all SNPs are independent of pleiotropy and depend on the variance of the outcome, and this method has a robust causality detection (31). Even if the results of the weighted median and MR-Egger methods were not significant, we considered them significant if the IVW results were significant (p < 0.05) and consistent with the trend of the other methods (32, 33). The odds ratio (OR) reflects the increase in risk factor levels per standard deviation (SD) (25). We used the Cochrane Q statistic to assess the variability of the instrumental variables, with p-value above 0.05 indicating no heterogeneity. To assess the presence of horizontal pleiotropy, we conducted MR Egger regression analysis. A p-value greater than 0.05 suggests the absence of horizontal pleiotropy (34). A “leave-one-out” analysis evaluate the impact of individual SNPs on the overall MR estimate. Furthermore, scatter plots were utilized to demonstrate that outliers did not impact the outcomes (35).
According to recent studies, the genome-wide significance p-value for each immune cell trait in IVs was set at 5 × 10−6 (36–38). Following F-statistical validation, 985 SNPs were identified as IVs among 731 immune cell SNPs. The retained SNPs were found to be strong instruments, as indicated by F-statistics exceeding 10. These results are presented in Supplementary Table S1.
To investigate the causal relationship between immunophenotype and MI, we conducted a two-sample MR analysis using IVW as the primary method. At a significance level of 0.001, we identified 47 immunophenotypes that have a significant causal relationship with MI. Of these, 7 were found in B cells, 5 in cDC, 4 in the maturation stages of T cells, 6 in monocytes, 5 in myeloid cells, 12 in TBNK cells, and 8 in Treg cells. Detailed findings are presented in Table 1 and Figure 2. We conducted MR-Egger intercept and MR-PRESSO global tests to eliminate SNPs with pleiotropy (MR-PRESSO test P < 0.05, MR-Egger regression P < 0.05). Following Cochran’s Q test, SNPs with heterogeneity (P < 0.05) were excluded, as detailed in Table 1. Furthermore, the leave-one-out analysis indicated that the exclusion of any of the SNPs did not affect the overall results. This suggests a stable causal relationship between immunophenotype and MI (Supplementary Figure S1). In addition, the stability of causality is demonstrated by analysing species using scatterplots and funnel plots. (Supplementary Figure S2).
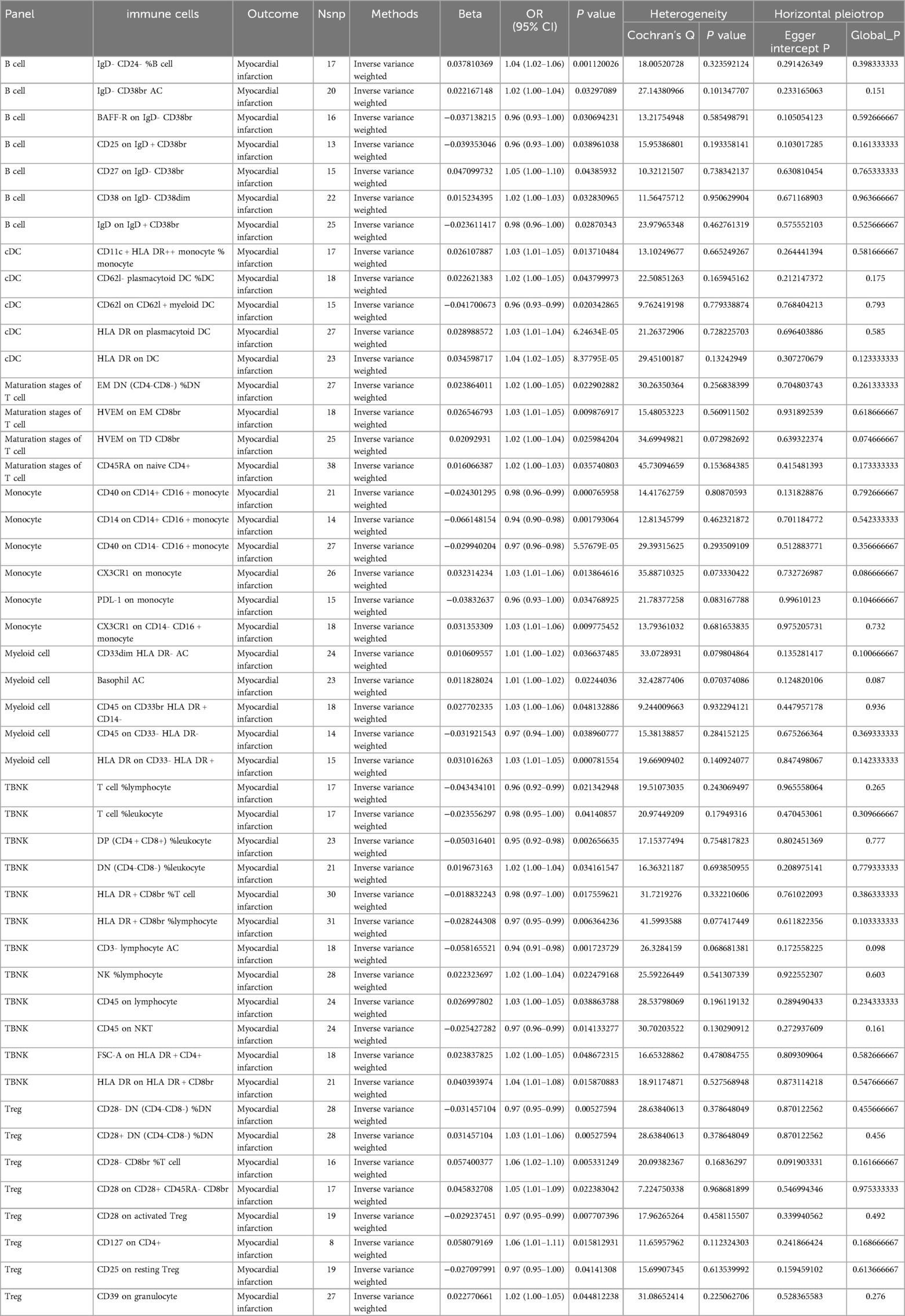
Table 1. Summary of the GWAS included in this two sample Mendelian randomization study.

Figure 2. Forest plot of the MR analysis results.
The study revealed that BAFF-R on IgD- CD38br [OR(95% CI):0.96 (0.93–1.00)], CD25 on IgD + CD38br [OR(95% CI): 0.96 (0.93–1.00)], and IgD on IgD + CD38br [OR(95% CI):0.98 (0.96–1.00)] had a protective effect against MI. The risk factors for MI were IgD- CD24- %B cell [OR (95% CI): 1.04 (1.02–1.06)], IgD- CD38br AC [OR (95% CI): 1.02 (1.00–1.04)], CD27 on IgD- CD38br [OR (95% CI): 1.05 (1.00–1.10)], and CD38 on IgD- CD38dim [OR (95% CI): 1.02 (1.00–1.03)] (Table 1).
The study revealed that four types of DC cells (CD11c + HLA DR++ monocyte % monocyte [OR (95% CI): 1.03 (1.01–1.05)], CD62l- plasmacytoid DC %DC [OR (95% CI): 0.96 (0.93–0.99)], HLA DR on plasmacytoid DC [OR (95% CI): 1.03 (1.01–1.04)], HLA DR on DC [OR (95% CI): 1.03 (1.01–1.04)] were linked to a higher risk of MI. It was also discovered that CD62l on CD62l + myeloid DC [OR (95% CI): 0.96 (0.93–0.99)] was linked to a decreased risk of MI (Table 1).
The study revealed that four types of Maturation stages of T cells (EM DN (CD4-CD8-) %DN [OR (95% CI): 1.02 (1.00–1.05)], HVEM on EM CD8br(OR(95% CI):1.03 (1.01–1.05), HVEM on TD CD8br [OR (95% CI):1.02 (1.00–1.04)], CD45RA on naive CD4+ [OR (95% CI): 1.02 (1.00–1.03)] were linked to a higher risk of MI (Table 1).
The study revealed that CD40 on CD14+ CD16 + monocyte [OR (95% CI):0.98 (0.96–0.99)], CD14 on CD14+ CD16 + monocyte [OR (95% CI):0.94 (0.90–0.98)], CD40 on CD14- CD16 + monocyte [OR (95% CI): 0.97 (0.96–0.98)], PDL-1 on monocyte [OR (95% CI): 0.96 (0.93–1.00)] had a protective effect against MI. The risk factors for MI wereCX3CR1 on monocyte [OR (95% CI): 1.03 (1.01–1.06)], CX3CR1 on CD14- CD16 + monocyte [OR (95% CI): 1.03 (1.01–1.06)] (Table 1).
The study revealed that four types of DC cells CD33dim HLA DR- AC [OR (95% CI): 1.01 (1.00–1.02)], Basophil AC [OR (95% CI): 1.01 (1.00–1.02)], CD45 on CD33br HLA DR + CD14- (OR (95% CI): 1.03 (1.00–1.06)], HLA DR on CD33- HLA DR+ [OR (95% CI): 1.03 (1.01–1.05)] were linked to a higher risk of MI. It was also discovered that CD45 on CD33- HLA DR- [OR(95% CI):0.97 (0.94–1.00)] was linked to a decreased risk of MI (Table 1).
The study revealed that T cell %lymphocyte [OR (95% CI): 0.96 (0.92–0.99)], T cell %leukocyte [OR (95% CI): 0.98 (0.95–1.00)], DP (CD4 + CD8+) %leukocyte [OR (95% CI): 0.95 (0.92–0.98)], HLA DR + CD8br %T cell [OR (95% CI): 0.98 (0.97–1.00)], HLA DR + CD8br %lymphocyte [OR (95% CI): 0.97 (0.95–0.99)], CD3- lymphocyte AC [OR (95% CI): 0.94 (0.91–0.98)], CD45 on NKT [OR (95% CI): 0.97 (0.96–0.99)] had a protective effect against MI. The risk factors for MI were DN (CD4-CD8-) %leukocyte [OR (95% CI): 1.02 (1.00–1.04)], NK %lymphocyte [OR (95% CI): 1.02 (1.00–1.04)], CD45 on lymphocyte [OR (95% CI): 1.03 (1.00–1.05)], FSC-A on HLA DR + CD4+ [OR (95% CI): 1.02 (1.00–1.05)], HLA DR on HLA DR + CD8br [OR (95% CI): 1.04 (1.01–1.08)] (Table 1).
The study revealed that three types of Tregs cells (CD28- DN (CD4-CD8-) %DN [OR (95% CI): 0.97 (0.95–0.99)], CD28 on activated Treg [OR (95% CI): 0.97 (0.95–0.99], CD25 on resting Treg [OR (95% CI): 0.97 (0.95–1.00)] were linked to a decreased risk of MI, while five types of Tregs cells(CD28+ DN (CD4-CD8-) %DN [OR (95% CI): 1.03 (1.01–1.06)], CD28- CD8br %T cell [OR (95% CI): 1.06 (1.02–1.10)], CD28 on CD28+ CD45RA- CD8br [OR (95% CI): 1.05 (1.01–1.09)], CD127 on CD4+ [OR (95% CI): 1.06 (1.01–1.11)], CD39 on granulocyte [OR (95% CI): 1.02 (1.00–1.05)] were associated with an increased risk of MI (Table 1).
A large proportion of myocardial infarction (MI) is caused by the rupture or erosion of vulnerable atherosclerotic plaques, leading to occlusion of the coronary arteries (39–41). Recent studies have confirmed that atherogenesis and progression of atherosclerosis are associated with inflammation and autoimmunity (42). The pathology of atherosclerosis is characterized by aseptic inflammation. This is mediated by innate and adaptive immune responses (43, 44). Previous studies have shown that innate immunity, represented by monocytes/macrophages, and adaptive immunity, dominated by T/B cells, can accelerate or inhibit atherosclerosis (45). Using MR analysis, we investigated the causal relationship between MI and 731 immune cell traits, based on a significant amount of publicly available genetic data. As far as we know, this study represents the inaugural MR analysis exploring the causal relationship between multiple immunophenotypes and myocardial infarction. In this study, 47 immune cell traits from seven panels were found to be causally associated with MI.
Macrophages are major players in two major cardiovascular diseases, myocardial infarction and atherosclerosis (46). Previous studies have shown significant heterogeneity in macrophage phenotype and function in infarcted myocardium. In the context of atherosclerotic cardiovascular disease (ASCVD), M1 macrophages produce pro-inflammatory cytokines that initiate and maintain inflammation, whereas M2 macrophages produce growth factors and anti-inflammatory cytokines that suppress the immune response (47, 48). However, this is too simplistic to try to categorise. The function of macrophages in the microenvironment of the infarcted heart is currently unknown. It is now believed that macrophages play different roles at different stages of the cardiac inflammatory response (19). Macrophages are recruited to the damaged myocardium and are regulated by the local microenvironment to differentiate into pro-inflammatory and anti-inflammatory macrophages (49–51). Early pro-inflammatory macrophages produce cytokines (e.g., IL-12, IL-23, IL-27, TNF-α) and chemokines (CXCL9, CXCL10, CXCL11) and other chemokines that exert pro-inflammatory effects (52, 53). In contrast, anti-inflammatory macrophages produce mediators such as IL-10, CCL17, and TGF-β during the value-adding and repair processes to exert anti-inflammatory effects, as well as to promote cell proliferation and angiogenesis, and to remove tissue debris (54). At the same time, macrophages crosstalk with cardiomyocytes, fibroblasts and various immune cells to regulate the process of post-infarction myocardial repair (49, 55). Our study found that CX3CR1 on CD14- CD16 + monocyte, CX3CR1 on monocyte were significantly associated with an increased risk of MI, and 4 cell types of monocytes (CD40 on CD14+ CD16 + monocyte, CD14 on CD14+ CD16 + monocyte, CD40 on CD14- CD16 + monocyte, and PDL-1 on monocyte) were associated with a reduced risk of myocardial infarction. These results illustrate the balance between macrophage populations that influence the development of atherosclerosis. This is consistent with previous studies and provides directional targets for future therapy (56–58).
Stimulated by oxidised LDL, TNF-α and hypoxia, DCs enter the vessel wall to take up, process and deliver antigens (59). Our study revealed that four types of DC cells were linked to a higher risk of MI. DCs produce proinflammatory cytokines that activate T and B cells to initiate, modulate and maintain cardiac immunity (60, 61). Coronary plaque rupture is the leading cause of myocardial infarction.DC cells have a potent antigen-presenting effect, activating CD4 + helper cells and CD8 + cytotoxic T cells, killing plaque-resident cells and increasing the risk of plaque rupture (62–64). DC is also involved in shaping the functional activity of natural killer cells, enhancing their cytotoxic potential against endothelial cells and vascular smooth muscle (65). Secondly, DC induces the production of proteases, such as metalloproteinases, which break down the extracellular matrix (62). We found that CD62l on CD62l + myeloid DC was associated with a reduced risk of MI. A study conducted on mice revealed that a depletion of bone marrow dendritic cells (DCs) resulted in a sustained elevation of pro-inflammatory cytokines and a prolonged duration of the inflammatory response following myocardial infarction (66). These results illustrate that different subpopulations of DCs have a division of labour in the development of atherosclerotic sclerosis. It also implies that direct exposure to antigens or soluble cytokines mediates atherosclerosis. Further studies are required to comprehend the pathogenic antigens and DC-derived cytokines linked to cardiovascular disease.
Our research has found that three types of Tregs cells [CD28- DN (CD4-CD8-) %DN, CD28 on activated Treg, CD25 on resting Treg] were linked to a decreased risk of MI. Tregs are major immunoregulatory cells that secrete suppressor cytokines to maintain immune homeostasis. A study has shown that reduced numbers of Tregs and impaired inhibitory function are associated with the progression of atherosclerosis (67). Treg/Th17 ratio, Treg count and Treg function were significantly decreased in acute coronary syndrome (ACS) patients (68). Tregs have also been shown to have a protective effect against atherosclerosis in some studies (69–73). Consistent with our findings, the mRNA sequencing technique showed that Tregs cells play a role in promoting and resisting atherosclerosis (74). Tregs protect against atherosclerosis in different ways. On the one hand, tregs inhibit the progression of atherosclerosis by suppressing the proliferation of T cells that secrete inhibitory cytokines such as IL-10 and TGF-β (72, 75–77). On the other hand, tregs may promote atherosclerotic plaque resolution by activating macrophage cytotoxicity and upregulating pro-soluble lipids (78). In addition, Tregs are involved in regulating lipid metabolism to attenuate the progression of atherosclerosis (79, 80). However our study found that five types of Tregs cells [CD28+ DN (CD4-CD8-) %DN, CD28- CD8br %T cells, CD28+ CD45RA- CD8br on CD28, CD127 on CD4+, CD39 on granulocytes] were associated with an increased risk of myocardial infarction. These results remind us to reconsider the role of Tregs in atherosclerosis, which needs to be further explored by more research, including clinical trials.
B cells are considered a key immune cell type in the regulation of atherosclerosis (81–85). They control the cellular immune response through cell-to-cell contact, antigen presentation, and cytokine production (84). B cell effects mediated by antibodies and cytokines are subpopulation specific. Previous studies have shed light on B cell subpopulations and their functions, demonstrating that distinct subpopulations of B cells have specific effects on atherosclerosis (86, 87). Our study found that 4 B cell types (IgD- CD24- %B cell, IgD- CD38br AC, CD27 on IgD- CD38br, CD38 on IgD- CD38dim) were associated with an increased risk of MI, and 3 B cell types (BAFF-R on IgD- CD38br, CD25 on IgD + CD38br, IgD on IgD + CD38br) reduced the risk of MI.B cells were classified as B1 or B2 cells based on the presence or absence of CD5 expression. Previous studies have shown that B1 cells can improve atherosclerosis by secreting IgM, while B2 cells have proatherogenic effects (84, 88–90). However, B2 cells constitute the majority of B cells and differentiate into follicular B cells (FOB) and marginal zone B cells (MZB). Nus et al. discovered that MZB cells regulate helper T cell responses in hypercholesterolaemic mice and have a protective effect against atherosclerosis (91). B2 cells differentiate into either antibody-producing plasma cells or memory B cells, which secrete antibodies to mediate the humoral immune response. However, studies investigating the correlation between IgG levels and myocardial infarction have produced conflicting results (83). This may be indicative of a specific subtype of IgG. Furthermore, patients with myocardial infarction exhibited elevated plasma IgE levels, indicating a potential proatherosclerotic function for IgE-mediated immune responses (92). Due to the complexity of humoral immunity mediated by B cells that release antigen-specific antibodies, this study provides a new direction for B cell-targeted interventions.
The study is statistically efficient as it is based on the results of a large published GWAS cohort. Two-sample MR analyses and causal inference using multiple MR analyses were used to exclude potential confounders and reverse causation.
However, it is important to note that our study has limitations. However, it must be noted that our study has its limitations. Firstly, Mendelian randomization studies use genetic variation as a natural instrumental variable to infer causality. The presence of pleiotropy means that genetic variation can affect multiple biological pathways or phenotypes, not just the exposure of interest in the study. This can make it difficult to determine whether the effect of a genetic variant is direct or indirect, potentially leading to a misjudgment of the causal relationship between exposure and outcome. To exclude SNPs with pleiotropic effects, we employed the MR-Egger intercept and the MR-PRESSO global test. However, these methods still did not fully control for unknown or unmeasured confounding factors.
Secondly, the relationship between immune responses and cardiovascular health is intricate. In some instances, there may be a linear correlation between immune responses and cardiovascular health; for example, levels of certain inflammatory markers might increase in direct proportion to the rise in cardiovascular risk. The immune system interacts with the cardiovascular system through a variety of cell types and molecular pathways, and these interactions could exhibit different effect patterns at various levels of immune activation. The impact of different types of immune cells on cardiovascular diseases may be independent or interwoven. Moreover, the influence of immune cells on cardiovascular diseases may vary with changes in thresholds, individuals, stages of disease, and other factors. Further research is still needed to address the nonlinear interactions between immunity and cardiovascular diseases.
Additionally, the GWAS data utilized in our study were derived from individuals of European ancestry, hence further investigation is warranted to determine the applicability of our findings to other ethnic groups. The absence of detailed individual information precluded us from conducting refined stratified analyses of the data. Since our study focused solely on specific genetic instruments, it may not fully capture the genetic influence on immune cell activity and the risk of myocardial infarction (MI). With the increasing abundance of genomics data, multi-gene Mendelian randomization studies are demonstrating substantial potential in disease risk prediction, personalized medicine, and the formulation of public health strategies.
In conclusion, this study provides strong evidence that multiple immune cells have a causal effect on the risk of myocardial infarction. Furthermore, this study highlights the intricate nature of immune cells involved in the development of atherosclerosis and myocardial infarction. This discovery provides a new avenue for the development of therapeutic treatments for myocardial infarction and a new target for drug development.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethical approval was not required for the studies involving humans because All the data utilized in this investigation are publicly accessible and fall within the public domain. All participants granted informed consent, and the study protocols received approval from their respective local ethical committees. The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from The genome-wide association study (GWAS) summary data for myocardial infarction are sourced from the December 2023 release by Genetic Research Finland and can be accessed at https://finngen.gitbook.io/documentation. Genetic variants associated with immune cells are derived from pooled data from the UK Biobank. This information is available for search on the website https://gwas.mrcieu.ac.uk/datasets/. Written informed consent to participate in this study was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and the institutional requirements.
WC: Conceptualization, Data curation, Formal Analysis, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. KW: Data curation, Formal Analysis, Investigation, Software, Visualization, Writing – original draft, Writing – review & editing. JW: Software, Validation, Writing – original draft, Writing – review & editing. YC: Writing – review & editing. HG: Investigation, Supervision, Writing – review & editing. LX: Investigation, Supervision, Writing – review & editing. WP: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Funding was supported by Foshan 14th Five-Year Plan Medical Cultivated Specialized Project grants.
We are very grateful to FinnGen database and public catalog (GCST90001391 to GCST90002121) GWAS Catalog database. We would like to thank Figdraw (www.figdraw.com) for helping us to draw the figure.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2024.1416112/full#supplementary-material
Supplementary Figure S1 | Leave-one-out analysis for the impact of individual SNPs on the association between immune cells and MI risk.
Supplementary Figure S2 | The scatter plots and Scatterplots of associations between immune cells and MI risk.
1. Thygesen K, Alpert JS, White HD. Universal definition of myocardial infarction. J Am Coll Cardiol. (2007) 50:2173–95. doi: 10.1016/j.jacc.2007.09.011
2. Levine GN, Bates ER, Bittl JA, Brindis RG, Fihn SD, Fleisher LA, et al. 2016 ACC/AHA guideline focused update on duration of dual antiplatelet therapy in patients with coronary artery disease: a report of the American college of cardiology/American heart association task force on clinical practice guidelines: an update of the 2011 ACCF/AHA/SCAI guideline for percutaneous coronary intervention, 2011 ACCF/AHA guideline for coronary artery bypass graft surgery, 2012 ACC/AHA/ACP/AATS/PCNA/SCAI/STS guideline for the diagnosis and management of patients with stable ischemic heart disease, 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction, 2014 AHA/ACC guideline for the management of patients with non-ST-elevation acute coronary syndromes, and 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery. Circulation. (2016) 134:e123–155. doi: 10.1161/cir.0000000000000404
3. Martin SS, Aday AW, Almarzooq ZI, Anderson CAM, Arora P, Avery CL, et al. 2024 heart disease and stroke statistics: a report of US and global data from the American heart association. Circulation. (2024) 149(8):e347–913. doi: 10.1161/cir.0000000000001209
4. Liu M, Patel VR, Salas RN, Rice MB, Kazi DS, Zheng ZN, et al. Neighborhood environmental burden and cardiovascular health in the US. JAMA Cardiol. (2023) 9: 153–163. doi: 10.1001/jamacardio.2023.4680
5. Zhang T, Guan YZ, Liu H. Association of acidemia with short-term mortality of acute myocardial infarction: a retrospective study base on MIMIC-III database. Clin Appl Thromb Hemost. (2020) 26:1076029620950837. doi: 10.1177/1076029620950837
6. Report on cardiovascular health and diseases in China 2021. J Cardiovasc Pulm Dis. (2023) 42:1191–8. doi: 10.3967/bes2022.079
7. Zhang RYK, Cochran BJ, Thomas SR, Rye KA. Impact of reperfusion on temporal immune cell dynamics after myocardial infarction. J Am Heart Assoc. (2023) 12:e027600. doi: 10.1161/jaha.122.027600
8. Kologrivova I, Shtatolkina M, Suslova T, Ryabov V. Cells of the immune system in cardiac remodeling: main players in resolution of inflammation and repair after myocardial infarction. Front Immunol. (2021) 12:664457. doi: 10.3389/fimmu.2021.664457
9. Zhuang L, Wang Y, Chen Z, Li Z, Wang Z, Jia K, et al. Global characteristics and dynamics of single immune cells after myocardial infarction. J Am Heart Assoc. (2022) 11:e027228. doi: 10.1161/jaha.122.027228
10. Kubota A, Frangogiannis NG. Macrophages in myocardial infarction. Am J Physiol Cell Physiol. (2022) 323:C1304–c1324. doi: 10.1152/ajpcell.00230.2022
11. Sun K, Li YY, Jin J. A double-edged sword of immuno-microenvironment in cardiac homeostasis and injury repair. Signal Transduct Target Ther. (2021) 6:79. doi: 10.1038/s41392-020-00455-6
12. Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res. (2016) 119:91–112. doi: 10.1161/circresaha.116.303577
13. Anzai A, Ko S, Fukuda K. Immune and inflammatory networks in myocardial infarction: current research and its potential implications for the clinic. Int J Mol Sci. (2022) 23:5214. doi: 10.3390/ijms23095214
14. Sansonetti M, Al Soodi B, Thum T, Jung M. Macrophage-based therapeutic approaches for cardiovascular diseases. Basic Res Cardiol. (2024) 119:1–33. doi: 10.1007/s00395-023-01027-9
15. Jung SH, Hwang BH, Shin S, Park EH, Park SH, Kim CW, et al. Spatiotemporal dynamics of macrophage heterogeneity and a potential function of Trem2(hi) macrophages in infarcted hearts. Nat Commun. (2022) 13:4580. doi: 10.1038/s41467-022-32284-2
16. Zaman R, Epelman S. Resident cardiac macrophages: heterogeneity and function in health and disease. Immunity. (2022) 55:1549–63. doi: 10.1016/j.immuni.2022.08.009
17. Yang W, Lin J, Zhou J, Zheng Y, Jiang S, He S, et al. Innate lymphoid cells and myocardial infarction. Front Immunol. (2021) 12:758272. doi: 10.3389/fimmu.2021.758272
18. Blanco-Domínguez R, De la Fuente H, Rodríguez C, Martín-Aguado L, Sánchez-Díaz R, Jiménez-Alejandre R, et al. CD69 expression on regulatory T cells protects from immune damage after myocardial infarction. J Clin Invest. (2022) 132(21):e152418. doi: 10.1172/jci152418
19. Jin K, Gao S, Yang PH, Guo R, Li D, Zhang Y, et al. Single-Cell RNA sequencing reveals the temporal diversity and dynamics of cardiac immunity after myocardial infarction. Small Methods. (2022) 6:e2100752. doi: 10.1002/smtd.202100752
20. Feng Q, Li QR, Zhou H, Sun L, Lin C, Jin Y, Wang DX, et al. The role of major immune cells in myocardial infarction. Front Immunol. (2022) 13:1084460. doi: 10.3389/fimmu.2022.1084460
21. Smith GD, Ebrahim S. ‘Mendelian randomization': can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. (2003) 32:1–22. doi: 10.1093/ije/dyg070
22. Davey Smith G, Hemani G. Mendelian Randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. (2014) 23:R89–98. doi: 10.1093/hmg/ddu328
23. Wang S, Wang K, Chen X, Lin S. The relationship between autoimmune thyroid disease, thyroid nodules and sleep traits: a Mendelian randomization study. Front Endocrinol (Lausanne). (2023) 14:1325538. doi: 10.3389/fendo.2023.1325538
24. Wang K, Wang S, Qin X, Chen Y, Chen Y, Wang J, et al. The causal relationship between gut microbiota and biliary tract cancer: comprehensive bidirectional Mendelian randomization analysis. Front Cell Infect Microbiol. (2024) 14:1308742. doi: 10.3389/fcimb.2024.1308742
25. Wang K, Wang J, Chen Y, Long H, Pan W, Liu Y, et al. Causal relationship between gut microbiota and risk of esophageal cancer: evidence from Mendelian randomization study. Aging (Albany NY). (2024) 16:3596–611. doi: 10.18632/aging.205547
26. Nian W, Huang Z, Fu C. Immune cells drive new immunomodulatory therapies for myocardial infarction: from basic to clinical translation. Front Immunol. (2023) 14:1097295. doi: 10.3389/fimmu.2023.1097295
27. Verduijn M, Siegerink B, Jager KJ, Zoccali C, Dekker FW. Mendelian randomization: use of genetics to enable causal inference in observational studies. Nephrol Dial Transplant. (2010) 25:1394–8. doi: 10.1093/ndt/gfq098
28. Chen Y, Yang L, Wang K, An Y, Wang Y, Zheng Y, et al. Relationship between fatty acid intake and aging: a Mendelian randomization study. Aging (Albany NY). (2024) 16:5711–39. doi: 10.18632/aging.205674
29. Pang L, Ding Z, Chai H, Shuang W. The causal relationship between immune cells and different kidney diseases: a Mendelian randomization study. Open Med (Wars). (2023) 18:20230877. doi: 10.1515/med-2023-0877
30. Wang K, Wang S, Chen Y, Lu X, Wang D, Zhang Y, et al. Causal relationship between gut microbiota and risk of gastroesophageal reflux disease: a genetic correlation and bidirectional Mendelian randomization study. Front Immunol. (2024) 15:1327503. doi: 10.3389/fimmu.2024.1327503
31. Tian Z, Jiang Z, Hu S, Shi L. Immune factors have a complex causal regulation on pulmonary fibrosis: insights from a two-sample Mendelian randomization analysis. Medicine (Baltimore). (2023) 102:e36781. doi: 10.1097/md.0000000000036781
32. Burgess S, Thompson SG. Interpreting findings from Mendelian randomization using the MR-egger method. Eur J Epidemiol. (2017) 32:377–89. doi: 10.1007/s10654-017-0255-x
33. Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. (2017) 46:1985–98. doi: 10.1093/ije/dyx102
34. Wang S, Wang K, Chen X, Chen D, Lin S. Autoimmune thyroid disease and myasthenia gravis: a study bidirectional Mendelian randomization. Front Endocrinol (Lausanne). (2024) 15:1310083. doi: 10.3389/fendo.2024.1310083
35. Wang K, Qin X, Ran T, Pan Y, Hong Y, Wang J, et al. Causal link between gut microbiota and four types of pancreatitis: a genetic association and bidirectional Mendelian randomization study. Front Microbiol. (2023) 14:1290202. doi: 10.3389/fmicb.2023.1290202
36. Xue H, Chen J, Zeng L, Fan W. Causal relationship between circulating immune cells and the risk of Alzheimer’s disease: a Mendelian randomization study. Exp Gerontol. (2024) 187:112371. doi: 10.1016/j.exger.2024.112371
37. Fei Y, Yu H, Wu Y, Gong S. The causal relationship between immune cells and ankylosing spondylitis: a bidirectional Mendelian randomization study. Arthritis Res Ther. (2024) 26:24. doi: 10.1186/s13075-024-03266-0
38. Shi X, Wang T, Teng D, Hou S, Lin N. A Mendelian randomization study investigates the causal relationship between immune cell phenotypes and cerebral aneurysm. Front Genet. (2024) 15:1333855. doi: 10.3389/fgene.2024.1333855
39. Frangogiannis NG. Pathophysiology of myocardial infarction. Compr Physiol. (2015) 5:1841–75. doi: 10.1002/cphy.c150006
40. White SJ, Newby AC, Johnson TW. Endothelial erosion of plaques as a substrate for coronary thrombosis. Thromb Haemost. (2016) 115:509–19. doi: 10.1160/th15-09-0765
41. Finn AV, Nakano M, Narula J, Kolodgie FD, Virmani R. Concept of vulnerable/unstable plaque. Arterioscler Thromb Vasc Biol. (2010) 30:1282–92. doi: 10.1161/atvbaha.108.179739
42. He X, Liang B, Gu N. Th17/treg imbalance and atherosclerosis. Dis Markers. (2020) 2020:8821029. doi: 10.1155/2020/8821029
43. Kong P, Cui ZY, Huang XF, Zhang DD, Guo RJ, Han M, et al. Inflammation and atherosclerosis: signaling pathways and therapeutic intervention. Signal Transduct Target Ther. (2022) 7:131. doi: 10.1038/s41392-022-00955-7
44. Wolf D, Ley K. Immunity and inflammation in atherosclerosis. Circ Res. (2019) 124:315–27. doi: 10.1161/circresaha.118.313591
45. Hansson GK, Robertson AK, Söderberg-Nauclér C. Inflammation and atherosclerosis. Annu Rev Pathol. (2006) 1:297–329. doi: 10.1146/annurev.pathol.1.110304.100100
46. Schelemei P, Wagner E, Picard FSR, Winkels H. Macrophage mediators and mechanisms in cardiovascular disease. Faseb J. (2024) 38:e23424. doi: 10.1096/fj.202302001R
47. Barrett TJ. Macrophages in atherosclerosis regression. Arterioscler Thromb Vasc Biol. (2020) 40:20–33. doi: 10.1161/atvbaha.119.312802
48. Xu H, Jiang J, Chen W, Li W, Chen Z. Vascular macrophages in atherosclerosis. J Immunol Res. (2019) 2019:4354786. doi: 10.1155/2019/4354786
49. Jian Y, Zhou X, Shan W, Chen C, Ge W, Cui J, et al. Crosstalk between macrophages and cardiac cells after myocardial infarction. Cell Commun Signal. (2023) 21:109. doi: 10.1186/s12964-023-01105-4
50. Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. (2007) 204:3037–47. doi: 10.1084/jem.20070885
51. Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. (2011) 11:750–61. doi: 10.1038/nri3088
52. Dassanayaka S, Jones SP. Recent developments in heart failure. Circ Res. (2015) 117:e58–63. doi: 10.1161/circresaha.115.305765
53. Peet C, Ivetic A, Bromage DI, Shah AM. Cardiac monocytes and macrophages after myocardial infarction. Cardiovasc Res. (2020) 116:1101–12. doi: 10.1093/cvr/cvz336
54. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. (2003) 19:71–82. doi: 10.1016/s1074-7613(03)00174-2
55. Yap J, Irei J, Lozano-Gerona J, Vanapruks S, Bishop T, Boisvert WA. Macrophages in cardiac remodelling after myocardial infarction. Nat Rev Cardiol. (2023) 20:373–85. doi: 10.1038/s41569-022-00823-5
56. Bobryshev YV, Nikiforov NG, Elizova NV, Orekhov AN. Macrophages and their contribution to the development of atherosclerosis. Results Probl Cell Differ. (2017) 62:273–98. doi: 10.1007/978-3-319-54090-0_11
57. Tong Y, Cai L, Yang S, Liu S, Wang Z, Cheng J. The research progress of vascular macrophages and atherosclerosis. Oxid Med Cell Longev. (2020) 2020:7308736. doi: 10.1155/2020/7308736
58. Wculek SK, Dunphy G, Heras-Murillo I, Mastrangelo A, Sancho D. Metabolism of tissue macrophages in homeostasis and pathology. Cell Mol Immunol. (2022) 19:384–408. doi: 10.1038/s41423-021-00791-9
59. Subramanian M, Tabas I. Dendritic cells in atherosclerosis. Semin Immunopathol. (2014) 36:93–102. doi: 10.1007/s00281-013-0400-x
60. Dieterlen MT, John K, Reichenspurner H, Mohr FW, Barten MJ. Dendritic cells and their role in cardiovascular diseases: a view on human studies. J Immunol Res. (2016) 2016:5946807. doi: 10.1155/2016/5946807
61. Zhao Y, Zhang J, Zhang W, Xu Y. A myriad of roles of dendritic cells in atherosclerosis. Clin Exp Immunol. (2021) 206:12–27. doi: 10.1111/cei.13634
62. Niessner A, Weyand CM. Dendritic cells in atherosclerotic disease. Clin Immunol. (2010) 134:25–32. doi: 10.1016/j.clim.2009.05.006
63. Forte E, Perkins B, Sintou A, Kalkat HS, Papanikolaou A, Jenkins C, et al. Cross-priming dendritic cells exacerbate immunopathology after ischemic tissue damage in the heart. Circulation. (2021) 143:821–36. doi: 10.1161/circulationaha.120.044581
64. Gewaltig J, Kummer M, Koella C, Cathomas G, Biedermann BC. Requirements for CD8T-cell migration into the human arterial wall. Hum Pathol. (2008) 39:1756–62. doi: 10.1016/j.humpath.2008.04.018
65. Bobryshev YV, Lord RS. Co-accumulation of dendritic cells and natural killer T cells within rupture-prone regions in human atherosclerotic plaques. J Histochem Cytochem. (2005) 53:781–5. doi: 10.1369/jhc.4B6570.2005
66. Anzai A, Anzai T, Nagai S, Maekawa Y, Naito K, Kaneko H, et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation. (2012) 125:1234–45. doi: 10.1161/circulationaha.111.052126
67. Baardman J, Lutgens E. Regulatory T cell metabolism in atherosclerosis. Metabolites. (2020) 10:279. doi: 10.3390/metabo10070279
68. Li Q, Wang Y, Wang Y, Chen K, Zhou Q, Wei W, et al. Treg/Th17 ratio acts as a novel indicator for acute coronary syndrome. Cell Biochem Biophys. (2014) 70:1489–98. doi: 10.1007/s12013-014-9993-5
69. Albany CJ, Trevelin SC, Giganti G, Lombardi G, Scottà C. Getting to the heart of the matter: the role of regulatory T-cells (tregs) in cardiovascular disease (CVD) and atherosclerosis. Front Immunol. (2019) 10:2795. doi: 10.3389/fimmu.2019.02795
70. Grönberg C, Nilsson J, Wigren M. Recent advances on CD4(+) T cells in atherosclerosis and its implications for therapy. Eur J Pharmacol. (2017) 816:58–66. doi: 10.1016/j.ejphar.2017.04.029
71. Ley K. Fortified Tregs to fight atherosclerosis. Cardiovasc Res. (2021) 117:1987–8. doi: 10.1093/cvr/cvab103
72. Spitz C, Winkels H, Bürger C, Weber C, Lutgens E, Hansson GK, et al. Regulatory T cells in atherosclerosis: critical immune regulatory function and therapeutic potential. Cell Mol Life Sci. (2016) 73:901–22. doi: 10.1007/s00018-015-2080-2
73. Tanaka T, Sasaki N, Rikitake Y. Recent advances on the role and therapeutic potential of regulatory T cells in atherosclerosis. J Clin Med. (2021) 10:5907. doi: 10.3390/jcm10245907
74. Saigusa R, Winkels H, Ley K. T cell subsets and functions in atherosclerosis. Nat Rev Cardiol. (2020) 17:387–401. doi: 10.1038/s41569-020-0352-5
75. Kuan R, Agrawal DK, Thankam FG. Treg cells in atherosclerosis. Mol Biol Rep. (2021) 48:4897–910. doi: 10.1007/s11033-021-06483-x
76. Meng X, Yang J, Dong M, Zhang K, Tu E, Gao Q, et al. Regulatory T cells in cardiovascular diseases. Nat Rev Cardiol. (2016) 13:167–79. doi: 10.1038/nrcardio.2015.169
77. Ou HX, Guo BB, Liu Q, Li YK, Yang Z, Feng WJ, et al. Regulatory T cells as a new therapeutic target for atherosclerosis. Acta Pharmacol Sin. (2018) 39:1249–58. doi: 10.1038/aps.2017.140
78. Sharma M, Schlegel MP, Afonso MS, Brown EJ, Rahman K, Weinstock A, et al. Regulatory T cells license macrophage pro-resolving functions during atherosclerosis regression. Circ Res. (2020) 127:335–53. doi: 10.1161/circresaha.119.316461
79. Von Der Thüsen JH, Kuiper J, Fekkes ML, De Vos P, Van Berkel TJ, Biessen EA. Attenuation of atherogenesis by systemic and local adenovirus-mediated gene transfer of interleukin-10 in LDLr-/- mice. Faseb J. (2001) 15:2730–2. doi: 10.1096/fj.01-0483fje
80. Klingenberg R, Gerdes N, Badeau RM, Gisterå A, Strodthoff D, Ketelhuth DFJ, et al. Depletion of FOXP3 + regulatory T cells promotes hypercholesterolemia and atherosclerosis. J Clin Invest. (2013) 123:1323–34. doi: 10.1172/jci63891
81. Ammirati E, Moroni F, Magnoni M, Camici PG. The role of T and B cells in human atherosclerosis and atherothrombosis. Clin Exp Immunol. (2015) 179:173–87. doi: 10.1111/cei.12477
82. Ma SD, Mussbacher M, Galkina EV. Functional role of B cells in atherosclerosis. Cells. (2021) 10:270. doi: 10.3390/cells10020270
83. Porsch F, Mallat Z, Binder CJ. Humoral immunity in atherosclerosis and myocardial infarction: from B cells to antibodies. Cardiovasc Res. (2021) 117:2544–62. doi: 10.1093/cvr/cvab285
84. Sage AP, Tsiantoulas D, Binder CJ, Mallat Z. The role of B cells in atherosclerosis. Nat Rev Cardiol. (2019) 16:180–96. doi: 10.1038/s41569-018-0106-9
85. Srikakulapu P, McNamara CA. B cells and atherosclerosis. Am J Physiol Heart Circ Physiol. (2017) 312:H1060–H1067. doi: 10.1152/ajpheart.00859.2016
86. Mangge H, Prüller F, Schnedl W, Renner W, Almer G. Beyond macrophages and T cells: b cells and immunoglobulins determine the fate of the atherosclerotic plaque. Int J Mol Sci. (2020) 21:4082. doi: 10.3390/ijms21114082
87. Pattarabanjird T, Li C, McNamara C. B cells in atherosclerosis: mechanisms and potential clinical applications. JACC Basic Transl Sci. (2021) 6:546–63. doi: 10.1016/j.jacbts.2021.01.006
88. Adamo L, Rocha-Resende C, Mann DL. The emerging role of B lymphocytes in cardiovascular disease. Annu Rev Immunol. (2020) 38:99–121. doi: 10.1146/annurev-immunol-042617-053104
89. Sage AP, Tsiantoulas D, Baker L, Harrison J, Masters L, Murphy D, et al. BAFF Receptor deficiency reduces the development of atherosclerosis in mice–brief report. Arterioscler Thromb Vasc Biol. (2012) 32:1573–6. doi: 10.1161/atvbaha.111.244731
90. Kyaw T, Cui P, Tay C, Kanellakis P, Hosseini H, Liu E, et al. BAFF Receptor mAb treatment ameliorates development and progression of atherosclerosis in hyperlipidemic ApoE(-/-) mice. PLoS One. (2013) 8:e60430. doi: 10.1371/journal.pone.0060430
91. Nus M, Sage AP, Lu Y, Masters L, Lam BYH, Newland S, et al. Marginal zone B cells control the response of follicular helper T cells to a high-cholesterol diet. Nat Med. (2017) 23:601–10. doi: 10.1038/nm.4315
Keywords: causal inference, myocardial infarction, genome-wide association study, MR analysis, immunity
Citation: Cao W, Wang K, Wang J, Chen Y, Gong H, Xiao L and Pan W (2024) Causal relationship between immune cells and risk of myocardial infarction: evidence from a Mendelian randomization study. Front. Cardiovasc. Med. 11:1416112. doi: 10.3389/fcvm.2024.1416112
Received: 11 April 2024; Accepted: 1 August 2024;
Published: 27 August 2024.
Edited by:
Vinay Kumar, The Pennsylvania State University, United StatesReviewed by:
Satish Raut, The Ohio State University, United StatesCopyright: © 2024 Cao, Wang, Wang, Chen, Gong, Xiao and Pan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Pan, d2VpcGFuMTk3N2ZzQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.