
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
SYSTEMATIC REVIEW article
Front. Cardiovasc. Med. , 21 January 2025
Sec. Cardiovascular Metabolism
Volume 11 - 2024 | https://doi.org/10.3389/fcvm.2024.1415547
 Gavin Y. Oudit1,2*
Gavin Y. Oudit1,2* Pronabesh DasMahapatra3
Pronabesh DasMahapatra3 Nicole Lyn3Florence R. Wilson4Adekemi Adeyemi4Chae Sung Lee3
Nicole Lyn3Florence R. Wilson4Adekemi Adeyemi4Chae Sung Lee3 Ana Crespo5
Ana Crespo5 Mehdi Namdar6
Mehdi Namdar6
Background: Agalsidase beta is used to treat Fabry disease (FD); however, data on cardiac and cerebrovascular outcomes with agalsidase beta treatment come from studies with limited numbers of patients.
Methods: A systematic literature review of studies reporting on the efficacy and effectiveness of agalsidase beta in FD was conducted. Studies were identified in searches of MEDLINE, Embase, and the Cochrane Central Register of Controlled Trials from January 2000–June 2022. Outcomes of interest included cardiac structure and mass, cardiac events, and cerebrovascular events.
Results: Fifty-two citations (41 studies) were included. Reductions in interventricular septal thickness (IVST) and/or left ventricular posterior wall thickness (LVPWT) were demonstrated in six studies (follow-up 1–6 years, n = 4 using echocardiography, n = 2 cardiac MRI). IVST ranged from 12.1–14.9 mm at baseline and 10.8–14.1 mm at follow-up (all p < 0.05). LVPWT ranged from 11.7–16.0 mm at baseline and 10.7–13.0 mm at follow-up (all p < 0.05). Significant reductions in cardiac mass were demonstrated after 1 year of treatment in a single-arm study using cardiac MRI [left ventricular mass (LVM) 193–178 g; LVM index 102–94 g/m2; both p < 0.05]. Rates of composite cardiac events (3.8%–24.0%; four studies, follow-up 2–10 years) and cerebrovascular events (0.0%–18.9%; 12 studies, follow-up 1–10 years) were numerically lower than rates for placebo (follow-up 3 years).
Conclusion: Literature over the last 20 years indicates that agalsidase beta treatment may lead to stabilization or regression of cardiac structural thickness and mass, and reduction in cardiac and cerebrovascular events relative to placebo.
Fabry disease (FD) is a rare, X-linked genetic disorder caused by a deficiency in the activity of the lysosomal enzyme alfa-galactosidase A (α-gal A) (1). Over 1,000 pathogenic variants in the α-gal A gene (GLA) have been identified that interfere with the normal function of the α-gal A enzyme, resulting in the progressive lysosomal accumulation of globotriaosylceramide (GL3) and related sphingolipids in various types of cells including endothelial cells, podocytes, smooth muscle cells, cardiomyocytes, and neurons (1, 2). The deposition of GL3 in various organs increases the risk of irreversible damage and renal, cardiac, cardiovascular, and cerebrovascular clinical events, which can lead to death (3, 4).
Phenotypically, FD can be divided into classic or later-onset (non-classic) disease (5, 6). Males with the classic phenotype demonstrate symptoms typically manifesting in childhood, and have markedly reduced α-gal A enzyme activity, resulting in the accumulation of GL3 in the vascular endothelial cells of the heart, kidneys, brain, and skin (6–8). Comparatively, males diagnosed with the later-onset phenotype have relatively higher residual activity of α-gal A and usually present with a milder form of the disease at a later stage (6). In females, the clinical manifestations of FD range from asymptomatic to severe classic disease depending on the type of genetic mutation and the extent of X-chromosome inactivation (Lyonization), with skewed Lyonization significantly impacting phenotype (6, 9, 10).
Cardiac or cardiovascular involvement, which is often evident on electrocardiogram (11–13), typically begins early in patients with FD, involving the accumulation of GL3 in multiple cell types, including cells of the conduction system, cardiomyocytes, endothelial cells, and vascular smooth muscle cells (14). The resulting progressive cardiac damage can manifest as diastolic dysfunction, left ventricular hypertrophy (LVH), reduced myocardial blood flow, microvascular ischemia, fibrosis, arrhythmias, and conduction disturbances (2). These changes serve as indicators of cardiac involvement and measures of cardiac function and outcomes in FD (14). Eventually, the resulting GL3 accumulation and cascade of pathophysiological changes may cause clinical events such as arrhythmias requiring defibrillator and/or pacemaker therapy, syncope, myocardial infarction (MI), heart failure, and sudden cardiac death (2, 4). These serious cardiac manifestations are a leading cause of premature death among patients and one of the main determinants of their prognosis (3, 15). Approximately 50.0% of deaths among males and females with FD are accounted for by cardiovascular manifestations (15). Cerebrovascular complications due to the accumulation of GL3 within the endothelium of intracranial blood vessels are also a major cause of morbidity (16, 17), with stroke and transient ischemic attacks (TIAs) being the most prevalent cerebrovascular events in FD (16–18).
Current therapeutic approaches for the treatment of FD include the reduction of accumulated GL3 through enzyme replacement therapy (ERT) or oral chaperone therapy, along with symptomatic and palliative treatments when needed (5). There are currently three approved ERTs available in a number of countries worldwide: agalsidase beta (Fabrazyme®; Sanofi), pegunigalsidase alfa (Elfabrio®; Chiesi) and agalsidase alfa (Replagal®; Takeda). Agalsidase alfa is currently approved in Europe and is marketed globally including the UK, Australia, Germany, China and Japan, among others (19). However, agalsidase beta (also approved in Europe and Canada, among others) and pegunigalsidase alfa (also approved in Europe, Northern Ireland, and UK, among others) are the only two approved ERTs in the United States, where the former is indicated for patients aged 2 years and older and the latter for adult patients only (19–24). The chaperone therapy migalastat (Galafold®; Amicus Therapeutics) was first conditionally approved in 2018 in the United States and Canada for use in adult patients with amenable variants (25, 26).
Given the rarity of FD, it is challenging to conduct clinical studies with large numbers of patients; consequently, sample sizes are limited in publications on the cardiovascular and cerebrovascular outcomes associated with the treatment of FD. The aim of this systematic literature review (SLR) was to evaluate the clinical efficacy and effectiveness of agalsidase beta on cardiovascular and cerebrovascular outcomes in patients with FD from a collection of all such studies over 20 years.
An SLR was conducted to identify studies on the clinical efficacy and effectiveness, and immunogenicity of agalsidase beta in patients with FD. This paper focuses on cardiac and cerebrovascular outcomes from the search.
Relevant studies were identified by searching MEDLINE, Embase, and the Cochrane Central Register of Controlled Trials (CCTR). Database searches were performed using the Ovid platform. The initial searches were conducted for the period January 2000 to March 2019 with pre-defined search strategies (Supplementary Tables S1–S3). A supplementary search was conducted in May 2019 in which the search strings “exp enzyme replacement/” (Embase), “exp Enzyme Replacement Therapy/” (MEDLINE and CCTR), and “ERT.mp” (all three databases) were added to ensure that all agalsidase beta studies were captured, including those that did not use “Fabrazyme” or “agalsidase beta” as index terms. An update to the SLR was performed in June 2022 for the period March 2019 to June 2022 using the same pre-defined search strategies (Supplementary Tables S4–S6). The database searches were augmented with hand searches of the bibliographies of key studies of interest, including recently published SLRs and meta-analyses.
Study eligibility criteria were defined in terms of the population, interventions, comparators, outcomes, and study design structure outlined in Table 1. These criteria guided the identification and selection of studies relevant for the SLR. The population search terms were adapted from previously published SLRs and included both adult and pediatric study populations with FD (27–29). The intervention of interest was agalsidase beta; however, natural history and placebo could be selected as comparators. Agalsidase alfa or switch data were included if no other data were available. Outcomes of interest presented in this paper included cardiac structure and mass, as well as cardiac and cerebrovascular events (Table 1). Eligible studies included clinical trials and prospective or retrospective observational studies; case reports of a single patient and narrative review articles were excluded. Only full-text publications in English language were included, and therefore conference abstracts and presentations were excluded from the evidence base.
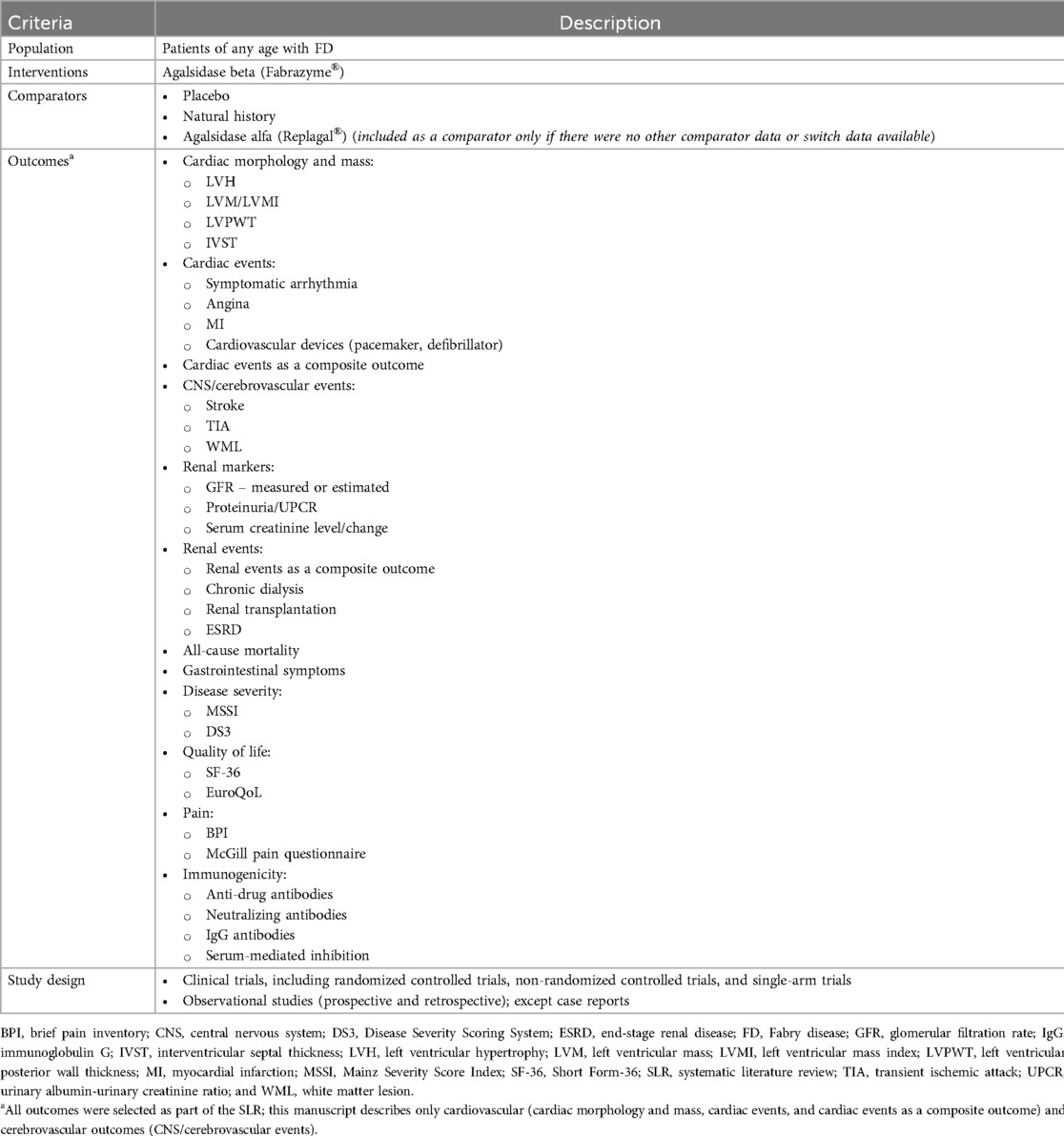
Table 1. Population, interventions, comparators, outcomes, and study design criteria for study selection.
This SLR was conducted and reported in accordance with the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) statement (30, 31). Two independent reviewers reviewed all abstracts and proceedings identified by the search according to the selection criteria; however, the outcome criteria were only applied during the screening of full-text publications. All eligible studies identified during abstract screening were then evaluated at full-text stage by two independent reviewers, and a third reviewer was included to reach consensus for any conflicts. The full-text studies identified as eligible were included for data extraction.
Data were extracted from all eligible studies on study characteristics, interventions, patient characteristics, and outcomes. In cases where there were multiple publications from a single study, data were extracted from the publication with the most complete dataset; however, some data were extracted from multiple publications to capture outcomes at different follow-up times or for different subgroups.
Study quality was assessed by two independent reviewers. Following reconciliation between the two reviewers, a third reviewer was included to reach consensus for any remaining discrepancies. The risk of bias in randomized clinical trials was assessed using the Cochrane Collaboration's Risk of Bias tool (Supplementary Table S7) (32). The quality of observational studies and non-randomized clinical trials was assessed using the Newcastle–Ottawa Scale (Supplementary Tables S8 and S9) (33).
Searches were conducted on March 13, 2019 and June 15, 2022 for the period from January 2000 to June 2022. The flow diagram for the study selection process is presented in Figure 1. A total of 83 citations corresponding to 68 studies were eligible for inclusion in the SLR. Of these, 52 citations corresponding to 41 studies reported cardiovascular and/or cerebrovascular outcomes. These comprised nine interventional studies [including four randomized controlled trials (RCTs)] and 32 observational studies (Table 1). Key characteristics of the included studies are summarized in Supplementary Table S10. Most studies were assessed to be of good quality with a low risk of bias (Supplementary Tables S11–S13). There was limited reporting of cardiovascular and cerebrovascular outcome data in five studies (four citations, one article included the results of two studies) (34–37) and one article (38) reported the pooled outcomes of two RCTs; as such, these five citations were not included in the below results. Therefore, cardiovascular and cerebrovascular outcomes were reviewed from 47 citations (36 studies). A summary of baseline patient characteristics for the selected studies is presented in Table 2.
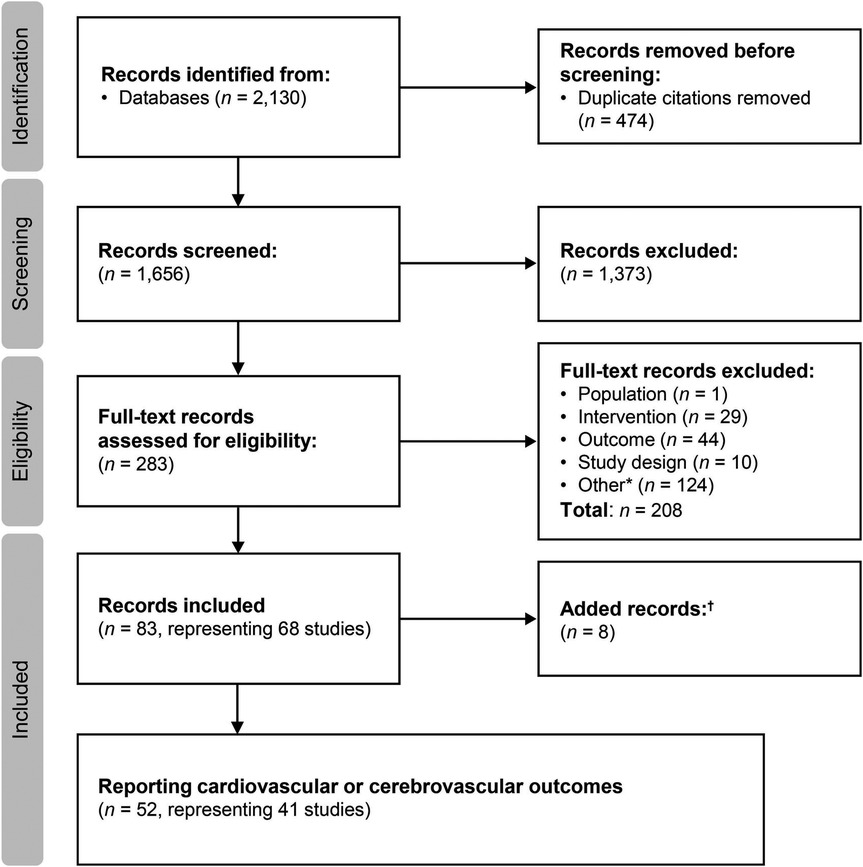
Figure 1. Study selection flow diagram (2000–2022). aIncludes conference abstracts and duplicate records; badditional studies identified through hand searches of key study references, or supplemental searches.
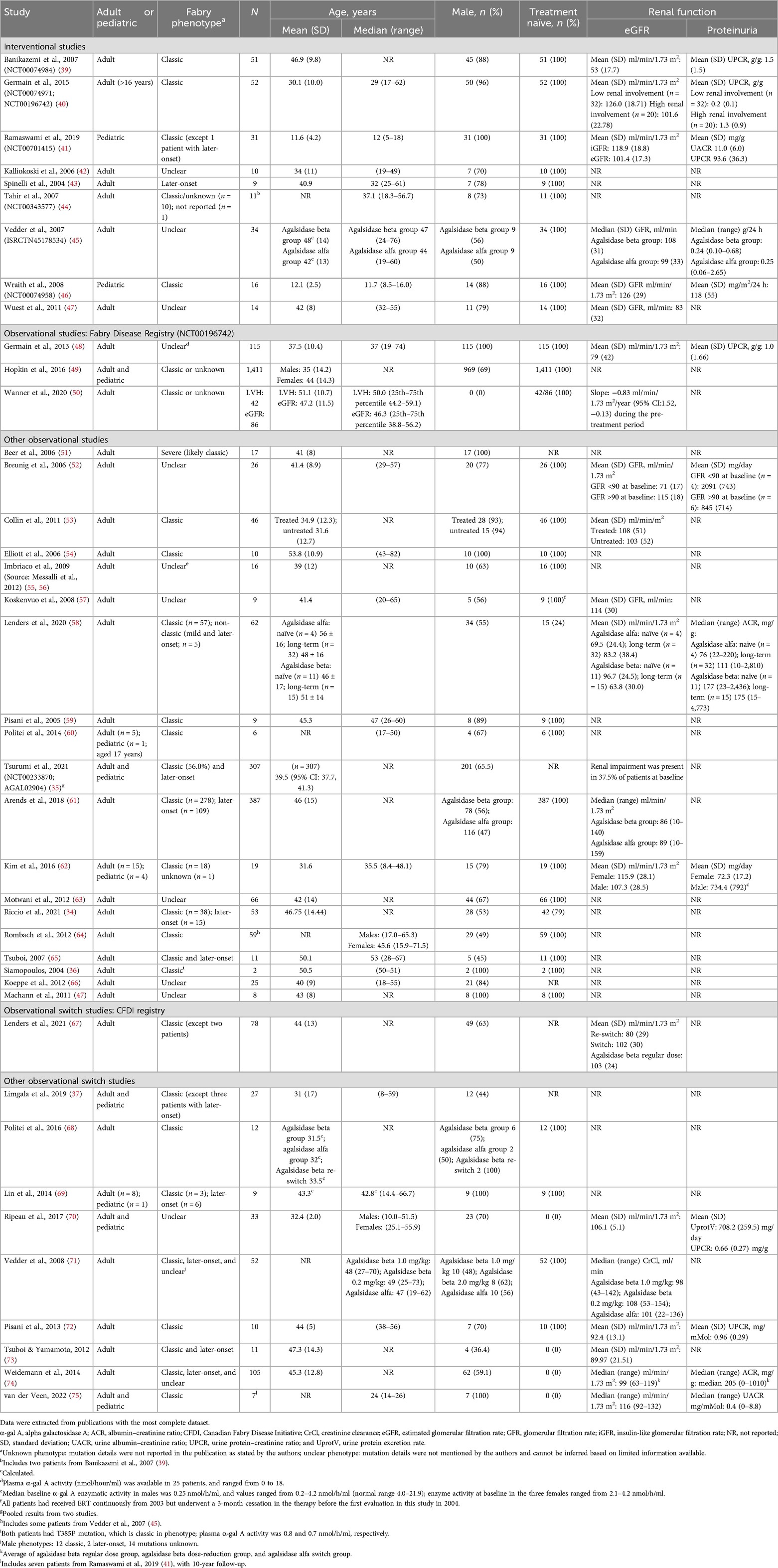
Table 2. Summary of baseline patient characteristics.
A total of 36 publications corresponding to 29 studies reported cardiac structure [interventricular septal (IVST) and left ventricular posterior wall thickness (LVPWT)] and cardiac mass [left ventricular mass (LVM) or LVM index (LVMI)] parameters, including six interventional studies which are summarized in Tables 3, 4. Data from all 29 studies are summarized in Supplementary Tables S14 and S15. Most available data on cardiac structure and mass outcomes were from studies in adults.
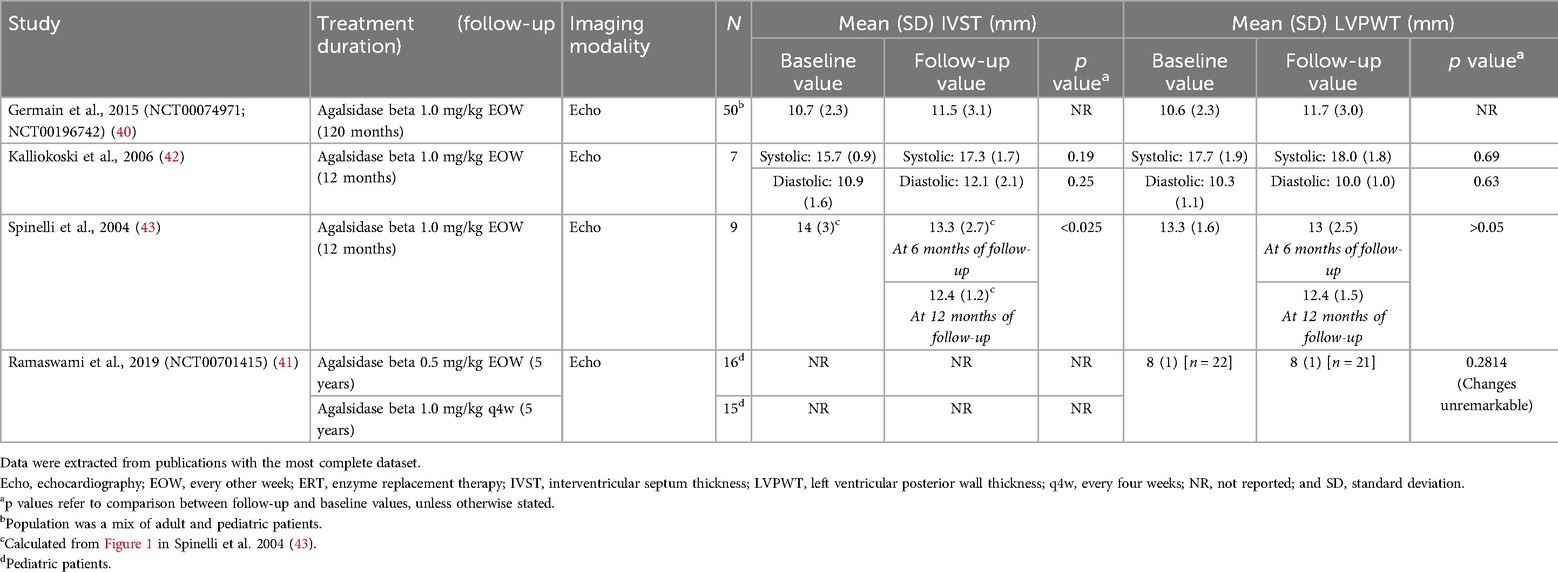
Table 3. IVST and LVPWT in interventional studies of patients with FD treated with agalsidase beta.

Table 4. LVM and LVMI in interventional studies of patients with FD treated with agalsidase beta.
In adult patients, IVST and LVPWT was reported in 22 publications, and LVM or LVMI were reported in 32 publications (Supplementary Tables S14 and S15). Overall, most patients treated with agalsidase beta showed stabilization or improvement in cardiac structure or cardiac mass parameters, demonstrated by non-significant changes or reductions from study baseline in IVST, LVPWT, and LVM or LVMI.
Four interventional studies investigated IVST or LVPWT in patients treated with agalsidase beta (Table 3) (40–43). In a single-arm study of nine patients in Italy with later-onset disease, IVST was significantly reduced from 14.0 mm at baseline to 12.4 mm after 12 months of follow-up (p < 0.025) (43). A numerical reduction in LVPWT from 13.3 mm to 12.4 mm was also demonstrated; however, this change was not statistically significant (43). Three further interventional studies reported stabilization of cardiac wall thickness after treatment with agalsidase beta, with no significant changes observed between baseline and follow-up (range 1–10 years) (40–42). These included a 10-year follow-up of 50 patients (classic phenotype) from the Fabry Registry who had participated in the phase 3 pivotal trial for agalsidase beta and its open-label extension; non-significant changes from baseline to 10 years in IVST (10.7–11.5 mm) and LVPWT (10.6–11.7 mm) were demonstrated, with both measures remaining within the normal range (40). Patients with cardiomyopathy in these studies presented with a range of hypertrophic phenotypes and baseline characteristics, with individual variation in the response to ERT but an overall improvement in LV mass and LV stiffness (40–42).
A total of 13 observational studies investigated the effects of agalsidase beta on IVST and LVPWT, and these generally showed consistent results with the interventional studies (Supplementary Table S14) (50, 52, 55, 56, 58–60, 66, 76, 77). Of note, significant reductions in both IVST (13.5–11.9 mm; p < 0.0001) and LVPWT (13.2–11.4 mm; p < 0.0001) were reported at 72-month follow-up in a prospective cohort study in Germany (77). Whereas data from the Fabry Registry demonstrated that LVPWT and IVST stabilized during 3.6 years of treatment in female patients treated with agalsidase beta, regardless of renal involvement or antiproteinuric agent use (50). Several other observational studies also reported significant improvements or non-significant increases in IVST and/or LVPWT in patients receiving agalsidase beta (Supplementary Table S14) (55, 56, 58–60, 66), regardless of sex (58). For example, an open-label study assessing the long-term effects of agalsidase beta also reported significantly reduced LVPWT after 48 months of treatment (16.0 mm vs. 13.0 mm; p < 0.001) (55).
Five interventional studies investigated cardiac mass in patients treated with agalsidase beta (Table 4) (41–43, 45, 47). In one study, significant reductions from baseline in cardiac mass were demonstrated in a cohort of nine patients with FD (43). At 12 months of follow-up, LVMI (calculated from echocardiographs using the Penn convention) was significantly reduced from 183 g/m2 to 169.5 g/m2 (p < 0.01) (43). A single-arm study of 14 patients in Germany also demonstrated significant reductions in LVM and LVMI [determined based on cardiac magnetic resonance imaging (MRI)] at 13 months (LVM: 193.0–178.0 g; LVMI: 102–94 g/m2; both p < 0.05) (47). A further two interventional studies reported no significant changes in cardiac mass at 12 or 24 months of follow-up (42, 45). These included an RCT of 34 patients treated with either agalsidase beta or agalsidase alfa in which there was no significant reduction in LVM (determined by echocardiography) from baseline to 24 months, and no significant difference between treatments (agalsidase beta: median 313–308 g; agalsidase alfa: 279–294 g) (45).
Seven observational studies demonstrated significant improvements in cardiac mass from baseline with agalsidase beta treatment (Supplementary Table S15) (47, 51, 53, 55, 56, 63, 66, 76, 77), regardless of sex (63). In a retrospective cohort study of 387 patients with classic or later-onset disease, there was no significant difference in LVMI (calculated from echocardiographs using the Devereux formula) between those treated with agalsidase beta compared with agalsidase alfa over the first year of treatment (p = 0.15) (61). However, a higher proportion of patients treated with agalsidase beta had a reduction in LVMI (79.0%) at 1 year compared with patients treated with agalsidase alfa [62.0%; odds ratio (OR): 2.27, 95% confidence interval (CI): 1.11, 4.86; p = 0.03] (61). The proportion of males in the agalsidase alfa (47%) and agalsidase beta (56%) groups may confound these results and should be interpreted with caution due to the observational nature of the study, since female patients generally have less LVH, may have different clinical presentations and response patterns to treatment with ERT (61). A prospective cohort study of nine patients also showed that after 24 months of therapy with agalsidase beta, progression of LVMI decreased in all but two patients (59). Prior to initiation of agalsidase beta, the average percentage increase per year in LVMI was 6.0%, which decreased to 3.0% during the 3 years of treatment (59). Another study assessed the natural progression of LVH in untreated male patients with FD and the left ventricular changes after 2 years of treatment of agalsidase beta (48). The results showed that LVM progressively increased in untreated males; however, males who began treatment with agalsidase beta at an earlier age experienced significant decline in LVM compared with untreated males (mean LVM slope for patients who initiated treatment before the age of 30 was −3.6 g/year compared with +9.5 g/year in untreated males within the same age bracket) (48).
Several studies demonstrated differences in cardiac outcomes with agalsidase beta depending on disease severity at baseline (Supplementary Tables S14 and S15) (51, 52, 63, 66, 76). For example, an observational study reported reduction in LVPWT at 24-month follow-up, but significance was only reached in the subgroup of patients with normal renal function (estimated glomerular filtration rate >90 ml/min/1.73 m2; p = 0.017) (52). Three studies were identified in which patients were classified based on the presence of myocardial fibrosis visualized using MRI with late enhancement (LE) (51, 66, 76). Two studies showed significant improvements in IVST and LVPWT or cardiac mass in patients without cardiac fibrosis at baseline after 12–36 months of agalsidase beta treatment (51, 76). In these studies, improvements in IVST and LVPWT or cardiac mass were also observed in patients with mild or severe fibrosis, or LE-positive MRI; however, these changes were not statistically significant (51, 76). A further prospective case-control study demonstrated that LVM was significantly higher in patients who were LE positive compared with LE negative at baseline (p < 0.05) (66). After 12 months of agalsidase beta treatment, mean LVM decreased in LE positive and LE negative patients; however, the difference between baseline and follow-up values was not statistically significant (66). A retrospective study of registry data from England reported that in 42 patients with LVH at baseline, LVMI was significantly reduced by agalsidase beta; however, there was no change for those patients without baseline LVH (63).
Three studies were identified that reported follow-up data for cardiac structural outcomes in pediatric patients treated with agalsidase beta (Supplementary Tables S14 and S15) (41, 62, 75). An open-label study of 31 males aged 5–18 years treated with 0.5 mg/kg EOW or 1.0 mg/kg every 4 weeks (q4w) of agalsidase beta demonstrated unremarkable changes in wall thickness and LVMI after 5 years (41). Cardiac mass (LVMI determined by echocardiogram) was reported to remain within normal range after 5–10 years of treatment for four male South Korean patients (aged 8–17 years) in one retrospective cohort study (62). In addition, significantly lower LVM was observed in a cohort of seven males with classic disease who had been treated with agalsidase beta for approximately 10 years since childhood, compared with untreated controls (median 80 g/m2 vs. 94 g/m2 by echocardiography; median 53 g/m2 vs. 68 g/m2 by cardiac MRI; both p = 0.02) (75).
A total of 17 publications from 14 studies reported cardiac events (Tables 4, 5; Supplementary Table S16).
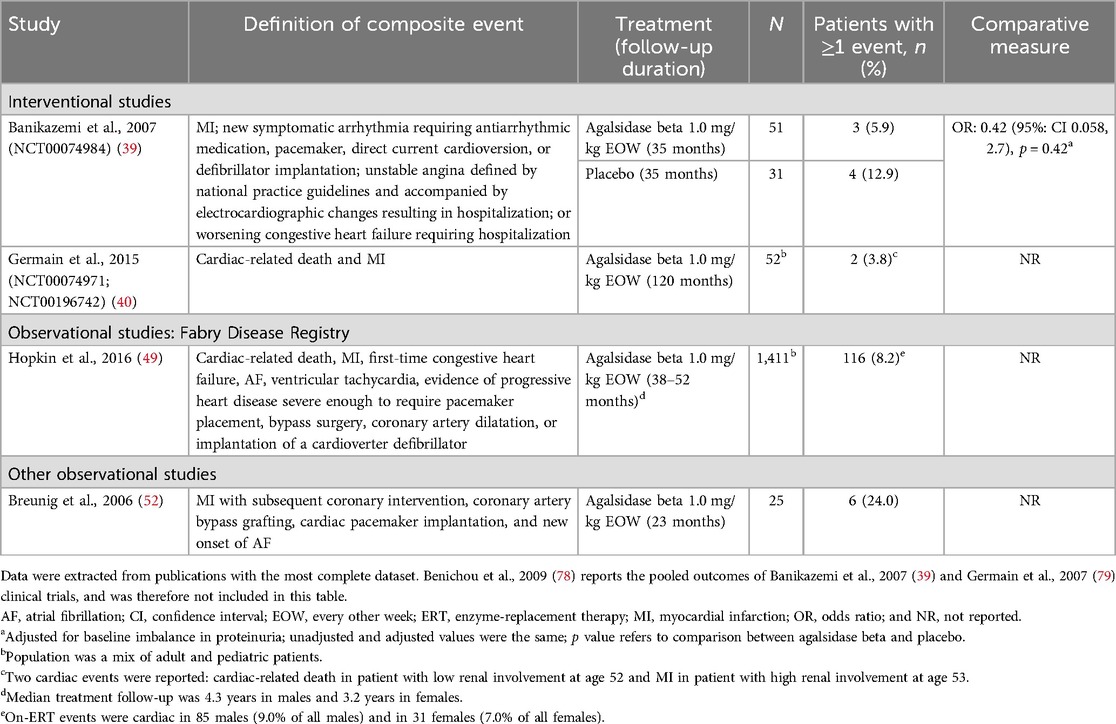
Table 5. Composite cardiac events reported between baseline and follow-up in patients with FD treated with agalsidase beta.
Two interventional studies and two observational studies reported composite cardiac events in adult patients treated with agalsidase beta, using varying definitions of composite events (Table 5) (39, 40, 49, 52). Across studies, the incidence of composite cardiac events in agalsidase beta-treated patients ranged from 3.8%–24.0%. Data from the phase 3 pivotal trial for agalsidase beta showed that patients treated with agalsidase beta experienced fewer composite cardiac events over 35 months of follow-up than patients treated with placebo [5.9% vs. 12.9%; OR (95% CI): 0.42 (0.058, 2.7)], although the comparison was not statistically significant (p = 0.42) (39). The 10-year follow-up results from the phase 3 study of agalsidase beta showed that the majority (50/52; 96.2%) of patients with classic FD did not experience any cardiac events; only one patient experienced cardiac-related death and one patient experienced an MI (40). In a large prospective cohort study of the Fabry Registry, over 90.0% of patients did not experience a cardiac event while on agalsidase beta treatment over 3 to 4 years of follow-up; however, compared with other events, cardiac events in this study were more common than non-cardiac events (49). The proportion of male patients who experienced cardiac events (9.0%) was slightly greater than in female patients (7.0%) (49). Of note, patients who experienced cardiac events while on agalsidase beta treatment were more likely to have experienced a cardiac event pre-treatment than those without cardiac events (49).
Data on individual cardiac events (angina, MI, and pacemaker/defibrillator implantation events) were reported in eight studies in adults (Supplementary Table S16). Two interventional studies with follow-up of ≥12 months to 35 months reported angina events in adult patients; however, neither study described how angina was defined or measured (39, 42). In both studies, no events of angina were reported in patients treated with regular-dose agalsidase beta (n = 51 and n = 10) (39, 42); however, one angina event was reported in a placebo arm (n = 31) (39). Low rates of MI (1.9%–4.0%) were reported in three studies, with only one agalsidase beta-treated patient in each study experiencing an MI event (39, 40, 52). In the 10-year follow-up of the phase 3 study of agalsidase beta, one of 52 patients with classic FD (1.9%) treated with agalsidase beta experienced an MI (40). The incidence of pacemaker and/or defibrillator implantation events in adults treated with agalsidase beta was reported in six studies; across all study designs and follow-up durations (ranging from 12–88 months), the incidence of events was 0.0%–27.3% (37, 44, 52, 65, 67, 77, 80). Two studies reporting cardiac events in pediatric patients reported normal cardiac function during treatment with agalsidase beta (46, 62).
The incidence of arrhythmias, atrial fibrillation (AF), and other electrocardiogram (ECG) abnormalities in adult patients treated with agalsidase beta was reported in seven studies (follow-up ≥12 months to 8.1 years) and ranged from 3.9%–66.7% (Table 6) (39, 44, 45, 52, 62, 67, 75, 81). Of these, four studies evaluated the new onset of AF in adult patients (incidence of 5.6%–12.5% over 23–36 months follow-up) (Table 6) (44, 45, 52, 73). In a randomized open-label study, new-onset AF in patients receiving ERT for more than 24 months was reported in two patients treated with agalsidase beta (n = 2/16; after 42 and 36 months) and one patient treated with agalsidase alfa (n = 1/18; after 30 months) (45). While in a placebo-controlled phase 3 pivotal trial for agalsidase beta with over 35 months of follow-up, a higher proportion of patients treated with placebo experienced events of new-onset symptomatic arrhythmia requiring medication compared with patients treated with agalsidase beta (9.7% vs. 3.9%) (39). Data from a long-term prospective cohort study with median follow-up of 6 years, showed new-onset ventricular tachycardia in 30.0% of patients treated with agalsidase beta, but the event rate was not reported for the natural history control group (77). In this study, almost all patients with documented ventricular tachycardia had LVH with myocardial fibrosis, and all patients in the agalsidase beta group who died because of cardiac arrest had ventricular tachycardia (77). Other cardiac arrhythmias were detected in 66.7% of adult patients prior to initiating agalsidase beta and persisted throughout treatment in a small retrospective observational study in South Korea (n = 15 adults; mean follow-up 8 years; Table 6).
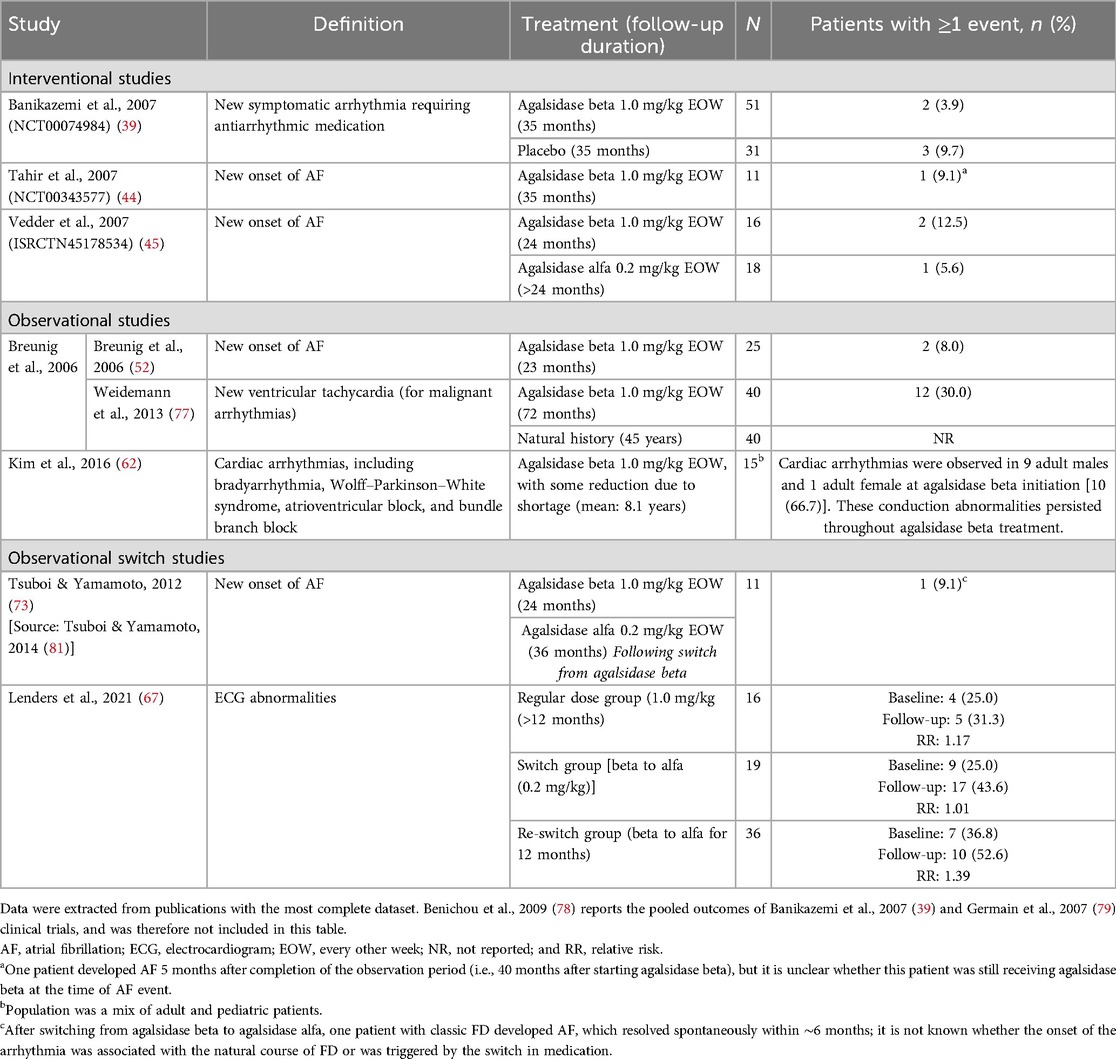
Table 6. Arrhythmia and AF events reported between baseline and follow-up in patients with FD treated with agalsidase beta.
Limited data were available on arrhythmias and AF events in pediatric patients receiving ERT with agalsidase beta. An observational study of seven male pediatric patients with classic disease after 10 years of treatment with agalsidase beta at a lower than standard dose (0.5 mg/kg EOW or 1.0 mg/kg q4w) did not demonstrate incomplete right bundle branch block in five patients with available data and sinus bradycardia in 14.3%; corresponding rates for the untreated comparison group were 31.3% and 52.4%, respectively (75). Low rates of sinus arrhythmia or conduction abnormalities were observed in an interventional study in 16 pediatric patients (0.0% at 48 weeks of follow-up) (46); similarly all pediatric patients (n = 4) showed normal cardiac function during treatment with agalsidase beta in a retrospective cohort study (5–10 year follow-up) (62).
A total of 18 publications based on 12 studies reported cerebrovascular ischemia events in patients treated with agalsidase beta. Across these studies, the rate of cerebrovascular ischemia events (defined as hemorrhagic or ischemic stroke, or TIA) in adult patients ranged from 0%–18.9% over follow-up periods from ≥12 months to 10 years (Table 7). In comparative studies, the rate of cerebrovascular ischemia events with agalsidase beta was similar or lower than the control group (Table 7) (39, 77, 82). The 35 months of follow-up results from the phase 3 pivotal trial for agalsidase beta showed that 6.5% of the patients in the placebo group experienced a cerebrovascular event compared with 0.0% of the patients in the agalsidase beta group [OR (95% CI): 0.00 (0.00, 3.20); p = 0.14] (39). In addition, in an observational study of 40 patients with FD in Germany, a numerically lower rate of stroke events in patients treated with agalsidase beta (10.0%; 72 months of follow-up duration) was reported compared with the natural history of disease (25.0%; 45 years of follow-up duration) (77). In one study of 16 pediatric patients, no stroke or TIA events were reported after 12 months of agalsidase beta treatment (46).
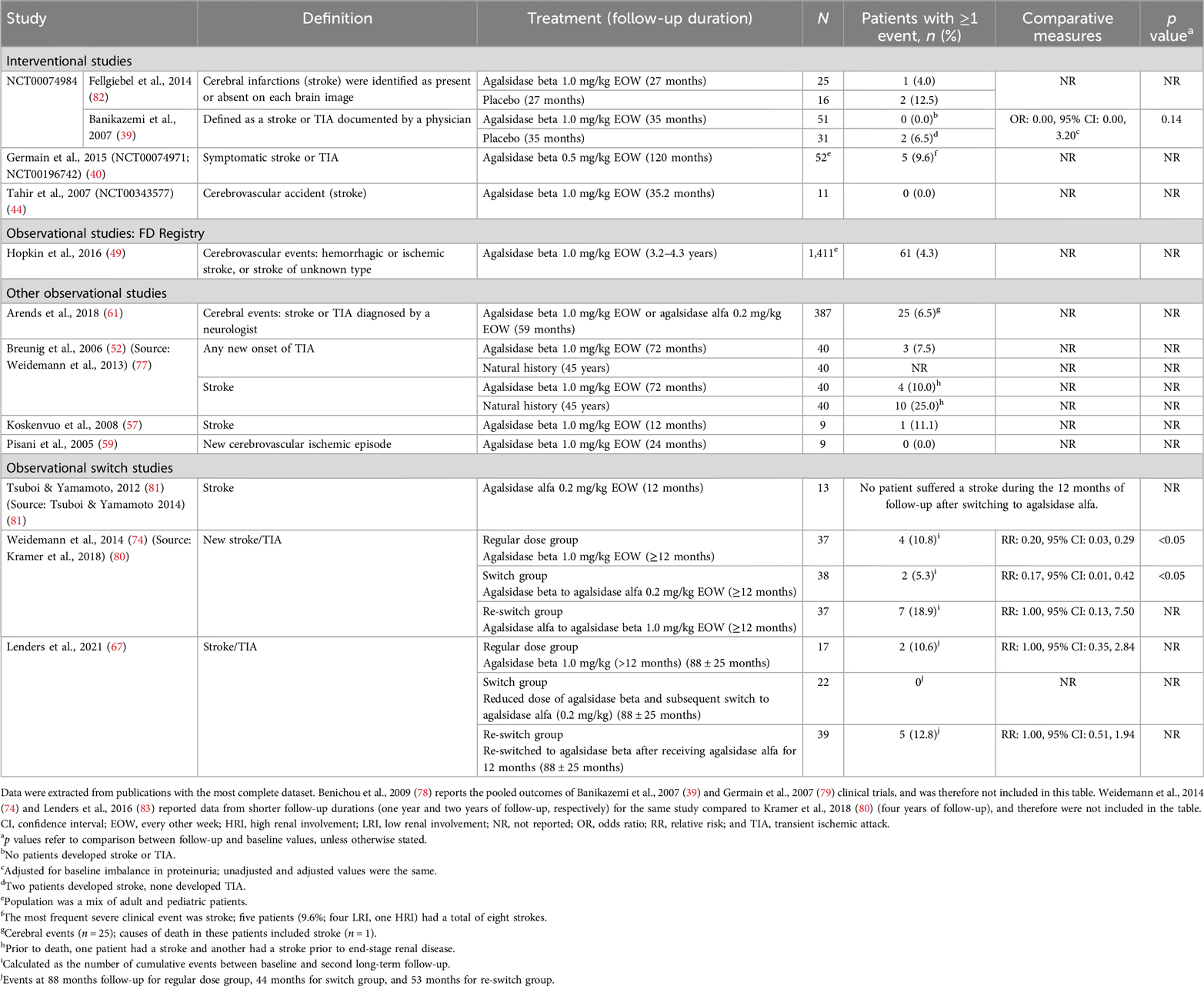
Table 7. Cerebrovascular ischemia events reported between baseline and follow in patients with FD treated with agalsidase beta.
Five publications corresponding to four studies investigated white matter lesions [WMLs] burden over follow-up periods of ≥12 months to 12 years (Table 8) (41, 64, 68, 75, 82). There was no clear evidence to suggest a treatment benefit of either agalsidase beta or agalsidase alfa on WML burden. One placebo-controlled study showed a slight worsening in WML burden over 27 months in both agalsidase beta and placebo groups (mean increases of 0.7 and 1.0, respectively; difference not significant) (82). In another observational switch study a worsening of WML burden was reported in four patients, and one patient developed a new lesion during the 12 years of follow-up on treatment with either agalsidase beta or agalsidase alfa (68). Two studies of agalsidase beta were identified that reported on WML in pediatric patients (41, 75). In a RCT of 31 male pediatric patients, WMLs of unknown etiology were reported in one patient and remained stable over the 5-year follow-up; no new WML development was observed in any patient (41). In the subsequent retrospective cross-sectional study with 10-year follow-up, new WML events were reported in one of seven patients (14.3%) treated with agalsidase beta (75).
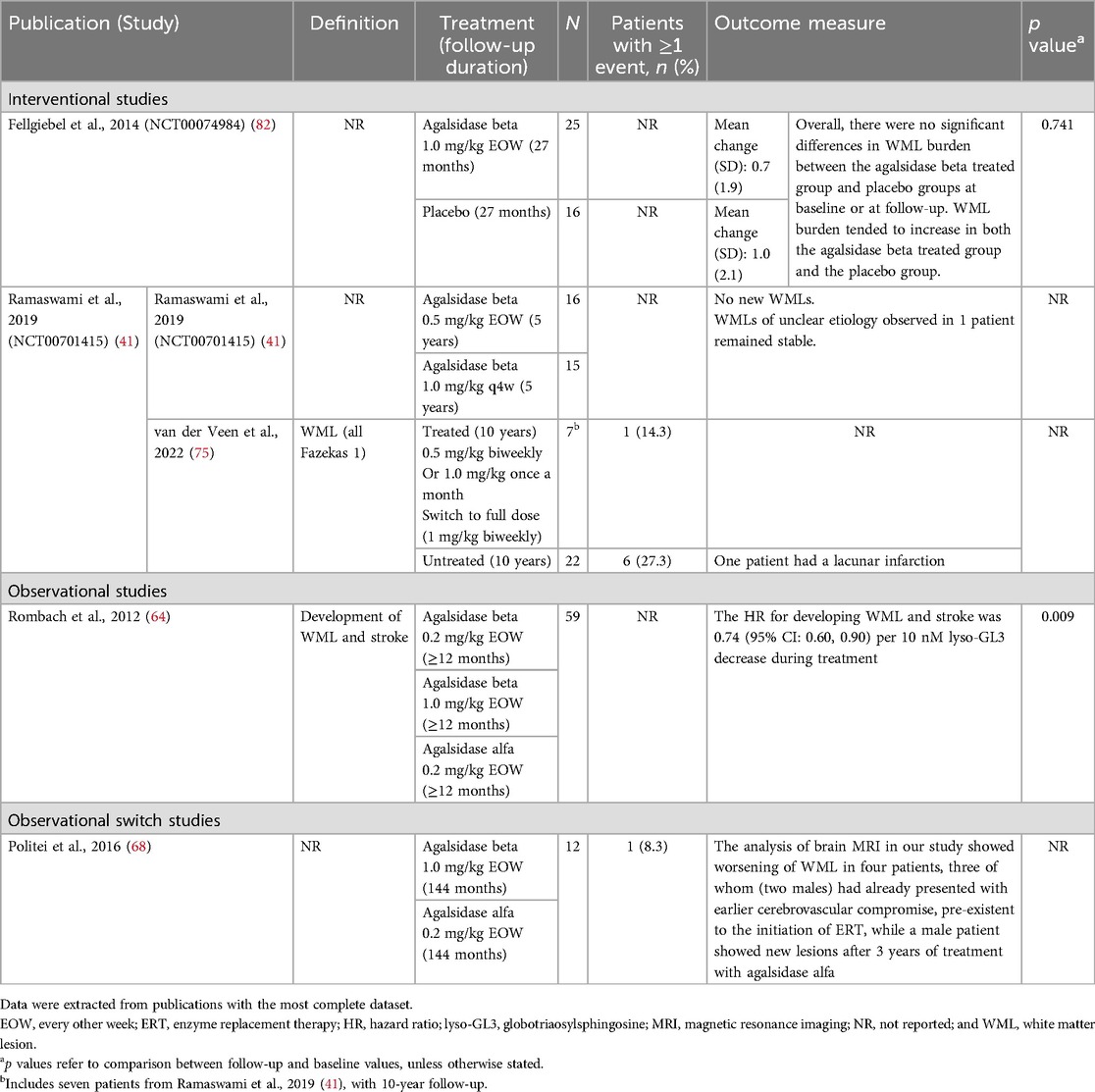
Table 8. WMLs reported between baseline and follow up in patients with FD treated with agalsidase beta.
The natural history of target organ injury in FD progresses slowly in patients with the classic phenotype and even more slowly in those with later-onset FD. Chronologically, the first natural history events in males with classic disease are typically kidney related (84). Structural tissue changes in the heart often occur as an early manifestation of FD due to accumulation of GL3 and related sphingolipids in cardiac cells and tissues, which are associated with progression to cardiac events (2). Subsequent cardiac events usually occur later in classic FD (median age 45 years in males and 54 years in females) and even later in later-onset disease (85). In assessing the natural disease progression, it takes approximately four decades for patients with kidney disease to reach end-stage kidney disease and even longer for cardiac events to occur (84, 85). Moreover, the early detection and management of cardiovascular risk factors is associated with low adverse outcomes in patients with FD (86). Agalsidase beta was first approved in the United States in 2003 and in Europe in 2001 based on the results from a phase 3, multi-center, randomized, placebo-controlled trial and its extension study in which 58 participants with classical FD received 1 mg/kg agalsidase beta EOW (21, 87–89). The primary outcome was the percentage of patients whose tissues were completely cleared of GL3 deposits at 20 weeks follow-up (89). The study reported complete or almost complete clearance of renal endothelial GL3 deposits in 69% of patients in the agalsidase beta group, compared with 0.0% of patients in the placebo group (89). Furthermore, evaluation of heart capillary endothelial cells showed that most agalsidase beta patients with biopsies at baseline and Month 6 had nearly complete clearance of GL3 from the capillary endothelium (89). With sample size limitations due to the low prevalence of FD, in addition to the timeline of progression to clinical events, a comprehensive evaluation of clinical studies is necessary to evaluate the effect of agalsidase beta on clinical outcomes. Since the approval and marketing authorization of agalsidase beta, dating back more than 20 years, observational studies have generated a body of evidence, complementary to that from clinical trials, that provides insights into the effectiveness on different target organ outcomes (kidney, heart, and brain). A meta-analysis published using an expansive evidence base has conclusively shown the clinical benefit of agalsidase beta on renal function (90). This SLR attempts to systematically present published data over the last two decades on the clinical efficacy and effectiveness of agalsidase beta on cardiovascular and cerebrovascular outcomes in patients with FD.
Overall, the SLR comprised 41 studies published since 2000, including four RCTs, five interventional studies, and 32 observational studies. The majority of studies investigated the regular dose of agalsidase beta, i.e., 1.0 mg/kg EOW, and included adult patients. Other study characteristics, such as numbers of patients, follow-up time, FD phenotype, and disease severity were more heterogeneous, providing a diverse evidence base. Published data identified in this SLR showed that agalsidase beta therapy may lead to stabilization or improvement in cardiac wall hypertrophy and mass, with data from 13 studies showing statistically significant reductions in IVST and/or LVPWT, or cardiac mass. While most studies which assessed cardiac mass utilized transthoracic echocardiography, a significant reduction in cardiac mass was also reported where gold-standard cardiac MRI was used (47). Data from several studies, including one with 10-year follow-up (40), also showed stabilization of cardiac wall thickness and mass with agalsidase beta treatment. These findings are consistent with a recent meta-analysis of MRI studies which found ERT was associated with stabilization of LVM and maximum left ventricular wall thickness (91).
The available evidence suggests a benefit of early initiation of ERT with agalsidase beta before the occurrence of clinical events. For example, an observational study of patients in the Fabry Registry demonstrated significant reduction in cardiac mass (LVM) in patients treated with agalsidase beta compared with untreated patients (48). However, this effect was only evident in the subgroup that initiated treatment between 18 and 29 years of age, while groups that initiated agalsidase beta when they were older showed no significant difference vs. controls (48). In the limited studies of agalsidase beta conducted exclusively in pediatric populations, improvement or stabilization of cardiac structure or mass was also evident up to 10 years after treatment initiation in childhood (41, 75). Data also showed that patients with less advanced disease may also derive more benefit from agalsidase beta. Three studies stratified cardiac structure and mass outcomes according to whether there was evidence of myocardial fibrosis on MRI (51, 66, 76). Those patients without fibrosis were more likely to have significant reductions in cardiac wall thickness or mass than those with detectable fibrosis, even of mild severity (51, 66, 76). A greater treatment benefit with agalsidase beta was also reported in patients without renal impairment at study baseline (52). This shows that patients with impaired renal function may display more severe cardiac hypertrophy, as assessed by LVPWT, and may develop cardiovascular progression despite being treated with agalsidase beta (52). Likewise, cardiac disease may also affect renal function, in a reciprocal relationship termed cardio-renal syndrome (92). These findings highlight the importance of early and routine monitoring of cardiac outcomes in patients with FD, especially when renal function is impaired. This is also consistent with a recent study that was published after the cut-off date for this SLR, which demonstrated a reduction in the incidence of cardiac complications and cardiac mortality with early ERT intervention in patients with FD (86).
Analysis of cardiac events, including MI, angina, and implantable cardioverter defibrillator/pacemaker implantation, indicated a generally low rate of these events (0.0%–27.3%) in patients treated with agalsidase beta. This suggests that agalsidase beta treatment may slow the progression to cardiac events in patients with FD. Incidence of new-onset arrhythmias or AF in interventional studies with up to 3 years of follow-up was generally low (3.9%–12.5%) and comparable to that reported for placebo (9.7%) (39, 44, 45). However, monitoring of arrhythmic events was likely based on electrocardiograms and Holter monitoring at scheduled study visits, rather than long-term monitoring using event monitors and implantable loop recorders, and therefore, these may be an underestimation of actual rates.
WMLs are frequently detected in brain scans of patients with FD (93); however, there was no evidence of improvement or worsening in WML burden in patients receiving agalsidase beta. It should be noted that the clinical consequence of WML in patients with FD is poorly understood and the interpretation of these data is therefore challenging. In addition, ERT does not cross the blood-brain barrier. Other central nervous system events assessed in this SLR included cerebrovascular ischemic events of stroke and TIA, which may have been influenced by supportive therapy. A low risk of these events was also demonstrated (0.0%–18.9% over follow-up periods of ≥12 months to 10 years), with similar or lower rates than comparators in interventional studies.
Strengths of this SLR include comprehensive searches of peer-reviewed literature and the involvement of two independent researchers in the study selection and data extraction, in accordance with PRISMA guidelines (30, 31). In addition, most studies included in the SLR were considered to be of good quality with a low risk of bias based on established criteria (32, 33). Previous SLRs of the efficacy of ERT with agalsidase beta in FD have focused on efficacy in specific study populations (e.g., males, females, pediatrics) (94, 95) and, to our knowledge, this is the first to focus on cardiac and cerebrovascular outcomes. A limitation, however, is that the majority of the studies identified were observational studies and many of these had retrospective designs, which may have influenced quality. Differences in the methodologies used in different studies to assess cardiac parameters (e.g., MRI vs. echocardiography), must also be considered when interpreting these results (96). Some of the studies did not clearly describe whether the study population had classic or later-onset FD, which may confound the measured outcome. In terms of study outcomes, specific cardiac outcomes, such as heart failure and syncope, were not extracted unless part of composite cardiac outcomes. Overall, the heterogeneity between studies in terms of populations, outcomes, follow-up, and imaging modalities limit the strength of the results. In addition, limited data were available from pediatric patients with FD; however, the lack of large controlled prospective trials in adults or pediatrics was not unexpected given the rarity of FD. Finally, the study selection criteria did not include conference materials, meaning that some recently completed studies may have been excluded, and the search was completed in 2022, excluding more recent publications.
Data from other existing and emerging therapies may be of interest in a future review to assess the relative impact of treatment options on patients with FD. However, comparative assessments prove challenging as a result of phenotypic heterogeneity and varying methods of measurement (e.g., echocardiography or cardiac MRI) (96). Future studies with systematic data collection in a controlled setting are required to corroborate and strengthen the evidence shown here. In addition, the search strategy utilized for this SLR was not designed to capture studies on the effects of agalsidase beta on GL3 and related sphingolipid deposits in cardiac tissue, or its effects on endothelial dysfunction and inflammation, although these data are limited. Data from an RCT of agalsidase beta in patients with classic FD showed significantly greater clearance of microvascular endothelial deposits of GL3 in cardiac tissue vs. placebo (89). However, preliminary data from a small cohort study of older patients with more severe cardiac disease at baseline did not find evidence of any improvement in coronary microvasculature dysfunction (based on myocardial blood flow and coronary flow reserve) with agalsidase beta treatment (54). Further studies on this in broader patient populations are warranted. Future studies on the effect of agalsidase beta on specific cardiac biomarkers such as high-sensitivity cardiac troponin I, an independent marker of major adverse cardiovascular events, would also be of value in patients with FD (97–99). Available data from a small cohort study of ERT (agalsidase alfa or agalsidase beta) have demonstrated that established ERT use does not normalize high-sensitivity troponin T levels in patients with advanced, but stable, disease (100). While a prospective observational clinical study of 36 ERT-naïve patients reported that in patients with high-sensitivity troponin I levels within the normal range at baseline (n = 20), levels remained within this range after agalsidase alfa treatment; whereas in patients with abnormally high levels at baseline (n = 16), levels generally remained stable (101). However, patients with FD who are eligible to receive ERT in clinical practice are likely to have more advanced disease.
Nonetheless, our study attempts to provide a comprehensive assessment of the available published evidence to evaluate the impact of agalsidase beta on clinical benefit in the cardiovascular and cerebrovascular systems of patients with FD. In conclusion, this SLR supports that agalsidase beta treatment may be associated with stabilization or regression of cardiac structural parameters and mass, and a reduction in cardiac and cerebrovascular events relative to controls. For patients who have already experienced cardiac disease progression, stabilization of cardiac structure and mass on agalsidase beta treatment may be a sufficient demonstration of treatment effect. Overall, agalsidase beta may be most effective if initiated when the progression is reversible prior to the onset of advanced cardiac fibrosis. How the impact of emerging therapies compares with that of agalsidase beta on cardiac and cerebrovascular aspects of FD remains a topic of interest in this field.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
GO: Conceptualization, Funding acquisition, Investigation, Methodology, Supervision, Validation, Writing – original draft, Writing – review & editing. PD: Conceptualization, Funding acquisition, Investigation, Methodology, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. NL: Investigation, Methodology, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. FW: Conceptualization, Data curation, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. AA: Conceptualization, Data curation, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. CL: Conceptualization, Investigation, Methodology, Writing – original draft, Writing – review & editing. AC: Conceptualization, Methodology, Visualization, Writing – original draft, Writing – review & editing. MN: Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by Sanofi. MN has received honoraria and research funds from Sanofi and Takeda. GO has received speaker fees and granting support from Sanofi and Takeda.
Medical writing support for the development of this manuscript, under the direction of the authors, was provided by Amy Watkins, PhD, of Ashfield MedComms, an Inizio company, and funded by Sanofi in accordance with Good Publication Practice guidelines.
AC, PD, CL, and NL are employees and may hold stock and/or stock options in Sanofi. FW is an employee of PRECISIONheor which received funding from Sanofi. AA was an employee of PRECISIONheor, which received funding from Sanofi at the time the research took place and is now an employee of Nassau University Medical Center. Sanofi funded the study, the funder was involved in the following: medical writing support, study design, data collection and analysis.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2024.1415547/full#supplementary-material
2. Pieroni M, Moon JC, Arbustini E, Barriales-Villa R, Camporeale A, Vujkovac AC, et al. Cardiac involvement in Fabry disease: JACC review topic of the week. J Am Coll Cardiol. (2021) 77(7):922–36. doi: 10.1016/j.jacc.2020.12.024
3. Mehta A, Clarke JT, Giugliani R, Elliott P, Linhart A, Beck M, et al. Natural course of Fabry disease: changing pattern of causes of death in FOS - Fabry outcome survey. J Med Genet. (2009) 46(8):548–52. doi: 10.1136/jmg.2008.065904
4. Schiffmann R, Warnock DG, Banikazemi M, Bultas J, Linthorst GE, Packman S, et al. Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol Dial Transplant. (2009) 24(7):2102–11. doi: 10.1093/ndt/gfp031
5. Ortiz A, Germain DP, Desnick RJ, Politei J, Mauer M, Burlina A, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. (2018) 123(4):416–27. doi: 10.1016/j.ymgme.2018.02.014
6. Arends M, Wanner C, Hughes D, Mehta A, Oder D, Watkinson OT, et al. Characterization of classical and nonclassical Fabry disease: a multicenter study. J Am Soc Nephrol. (2017) 28(5):1631–41. doi: 10.1681/ASN.2016090964
7. Eng CM, Banikazemi M, Gordon RE, Goldman M, Phelps R, Kim L, et al. A phase 1/2 clinical trial of enzyme replacement in Fabry disease: pharmacokinetic, substrate clearance, and safety studies. Am J Hum Genet. (2001) 68(3):711–22. doi: 10.1086/318809
8. Tøndel C, Thurberg BL, DasMahapatra P, Lyn N, Maski M, Batista JL, et al. Clinical relevance of globotriaosylceramide accumulation in Fabry disease and the effect of agalsidase beta in affected tissues. Mol Genet Metab. (2022) 137(4):328–41. doi: 10.1016/j.ymgme.2022.10.005
9. Hopkin RJ, Germain DP, Bichet DG, Gruskin DJ, Lemay RM, Oliveira JP, et al. A survivor analysis for major clinical events in heterozygous female patients with Fabry disease using group consensus phenotype classifications from hemizygous male patients. Mol Genet Metab. (2018) 123(2):S65–S6. doi: 10.1016/j.ymgme.2017.12.160
10. Echevarria L, Benistan K, Toussaint A, Dubourg O, Hagege AA, Eladari D, et al. X-chromosome inactivation in female patients with Fabry disease. Clin Genet. (2016) 89(1):44–54. doi: 10.1111/cge.12613
11. Namdar M, Richardot P, Johner N, Shah D, Nordbeck P, Olivotto I, et al. Recognition of pre-hypertrophic cardiac involvement in Fabry disease based on automated electrocardiographic measures. Int J Cardiol. (2021) 338:121–6. doi: 10.1016/j.ijcard.2021.06.032
12. Figliozzi S, Camporeale A, Boveri S, Pieruzzi F, Pieroni M, Lusardi P, et al. ECG-based score estimates the probability to detect Fabry disease cardiac involvement. Int J Cardiol. (2021) 339:110–7. doi: 10.1016/j.ijcard.2021.07.022
13. Augusto JB, Johner N, Shah D, Nordin S, Knott KD, Rosmini S, et al. The myocardial phenotype of Fabry disease pre-hypertrophy and pre-detectable storage. Eur Heart J Cardiovasc Imaging. (2021) 22(7):790–9. doi: 10.1093/ehjci/jeaa101
14. Linhart A, Elliott PM. The heart in Anderson-Fabry disease and other lysosomal storage disorders. Heart. (2007) 93(4):528–35. doi: 10.1136/hrt.2005.063818
15. Waldek S, Patel MR, Banikazemi M, Lemay R, Lee P. Life expectancy and cause of death in males and females with Fabry disease: findings from the Fabry registry. Genet Med. (2009) 11(11):790–6. doi: 10.1097/GIM.0b013e3181bb05bb
16. Burlina A, Politei J. The central nervous system involvement in Fabry disease:a review. J Inborn Errors Metab Screen. (2016) 4:e160039. doi: 10.1177/2326409816661361
17. Sims K, Politei J, Banikazemi M, Lee P. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry registry. Stroke. (2009) 40(3):788–94. doi: 10.1161/STROKEAHA.108.526293
18. Kolodny E, Fellgiebel A, Hilz MJ, Sims K, Caruso P, Phan TG, et al. Cerebrovascular involvement in Fabry disease: current status of knowledge. Stroke. (2015) 46(1):302–13. doi: 10.1161/STROKEAHA.114.006283
19. Takeda pharmaceuticals. Takeda pharmaceuticals, Agalsidase alfa (2021). Available online at: https://www.globaldata.com/data-insights/healthcare/number-of-ongoing-clinical-trials-for-drugs-involving-fabry-disease-by-phase-503350/ (Accessed December 9, 2024).
20. European Medicines Agency. EPAR Agalsidase Beta (2024). Available online at: https://www.ema.europa.eu/en/documents/product-information/fabrazyme-epar-product-information_en.pdf (Accessed December 9, 2024).
21. FDA. Highlights of prescribing information (Fabrazyme) (2024). Available online at: https://products.sanofi.us/fabrazyme/fabrazyme.pdf (Accessed December 9, 2024).
22. British National Formulary. Pegunigalsidase alfa for treating Fabry disease (2023). Available online at: https://www.nice.org.uk/guidance/ta915 (Accessed December 9, 2024).
23. Pharmaceuticals and Medicines Agency (PMDA) Japan. Annual Report FY 2018 (2018). Available online at: https://www.pmda.go.jp/files/000232603.pdf (Accessed December 9, 2024).
24. Health Canada Product Monograph. Product Monograph - Patient Medical Information (2022). Available online at: https://pdf.hres.ca/dpd_pm/00068925.PDF (Accessed December 9, 2024).
25. McCafferty EH, Scott LJ. Migalastat: a review in Fabry disease. Drugs. (2019) 79(5):543–54. doi: 10.1007/s40265-019-01090-4
26. FDA. Highlights of prescribing information (Galafold) (2024). Available online at: https://amicusrx.com/pi/galafold.pdf (Accessed December 9, 2024).
27. Alegra T, Vairo F, de Souza MV, Krug BC, Schwartz IV. Enzyme replacement therapy for Fabry disease: a systematic review and meta-analysis. Genet Mol Biol. (2012) 35(4 (suppl)):947–54. doi: 10.1590/S1415-47572012000600009
28. Dib R E, Gomaa H, Carvalho RP, Camargo SE, Bazan R, Barretti P, et al. Enzyme replacement therapy for Anderson-Fabry disease. Cochrane Database Syst Rev. (2016) 7(7):Cd006663. doi: 10.1002/14651858.CD006663.pub4
29. El Dib R, Gomaa H, Ortiz A, Politei J, Kapoor A, Barreto F. Enzyme replacement therapy for Anderson-Fabry disease: a complementary overview of a Cochrane publication through a linear regression and a pooled analysis of proportions from cohort studies. PLoS One. (2017) 12(3):e0173358. doi: 10.1371/journal.pone.0173358
30. Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. (2009) 6(7):e1000097. doi: 10.1371/journal.pmed.1000097
31. Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ (Clin Res Ed). (2021) 372:n71. doi: 10.1136/bmj.n71
32. Higgins JP, Altman DG, Gøtzsche PC, Jüni P, Moher D, Oxman AD, et al. The Cochrane collaboration’s tool for assessing risk of bias in randomised trials. BMJ (Clin Res Ed). (2011) 343:d5928. doi: 10.1136/bmj.d5928
33. Wells GS, O'Connell D, Peterson J, Welch V, Losos M. The Newcastle-Ottawa Scale (NOS) for assessing the quality of nonrandomised studies in meta-analyses [November 04, 2022] (2013). https://www.ohri.ca/programs/clinical_epidemiology/oxford.asp (Accessed December 9, 2024).
34. Riccio E, Zanfardino M, Franzese M, Capuano I, Buonanno P, Ferreri L, et al. Stepwise shortening of agalsidase beta infusion duration in Fabry disease: clinical experience with infusion rate escalation protocol. Mol Genet Genomic Med. (2021) 9(5):e1659. doi: 10.1002/mgg3.1659
35. Tsurumi M, Suzuki S, Hokugo J, Ueda K. Long-term safety and efficacy of agalsidase beta in Japanese patients with Fabry disease: aggregate data from two post-authorization safety studies. Expert Opin Drug Saf. (2021) 20(5):589–601. doi: 10.1080/14740338.2021.1891221
36. Siamopoulos KC. Fabry disease: kidney involvement and enzyme replacement therapy. Kidney Int. (2004) 65(2):744–53. doi: 10.1111/j.1523-1755.2004.00440.x
37. Limgala RP, Jennelle T, Plassmeyer M, Boutin M, Lavoie P, Abaoui M, et al. Altered immune phenotypes in subjects with Fabry disease and responses to switching from agalsidase alfa to agalsidase beta. Am J Transl Res. (2019) 11(3):1683–96.30972193
38. Bénichou B, Goyal S, Sung C, Norfleet AM, O'Brien F. A retrospective analysis of the potential impact of IgG antibodies to agalsidase beta on efficacy during enzyme replacement therapy for Fabry disease. Mol Genet Metab. (2009) 96(1):4–12. doi: 10.1016/j.ymgme.2008.10.004
39. Banikazemi M, Bultas J, Waldek S, Wilcox WR, Whitley CB, McDonald M, et al. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med. (2007) 146(2):77–86. doi: 10.7326/0003-4819-146-2-200701160-00148
40. Germain DP, Charrow J, Desnick RJ, Guffon N, Kempf J, Lachmann RH, et al. Ten-year outcome of enzyme replacement therapy with agalsidase beta in patients with Fabry disease. J Med Genet. (2015) 52(5):353–8. doi: 10.1136/jmedgenet-2014-102797
41. Ramaswami U, Bichet DG, Clarke LA, Dostalova G, Fainboim A, Fellgiebel A, et al. Low-dose agalsidase beta treatment in male pediatric patients with Fabry disease: a 5-year randomized controlled trial. Mol Genet Metab. (2019) 127(1):86–94. doi: 10.1016/j.ymgme.2019.03.010
42. Kalliokoski RJ, Kantola I, Kalliokoski KK, Engblom E, Sundell J, Hannukainen JC, et al. The effect of 12-month enzyme replacement therapy on myocardial perfusion in patients with Fabry disease. J Inherit Metab Dis. (2006) 29(1):112–8. doi: 10.1007/s10545-006-0221-3
43. Spinelli L, Pisani A, Sabbatini M, Petretta M, Andreucci MV, Procaccini D, et al. Enzyme replacement therapy with agalsidase beta improves cardiac involvement in Fabry’s disease. Clin Genet. (2004) 66(2):158–65. doi: 10.1111/j.1399-0004.2004.00284.x
44. Tahir H, Jackson LL, Warnock DG. Antiproteinuric therapy and Fabry nephropathy: sustained reduction of proteinuria in patients receiving enzyme replacement therapy with agalsidase-beta. J Am Soc Nephrol. (2007) 18(9):2609–17. doi: 10.1681/ASN.2006121400
45. Vedder AC, Linthorst GE, Houge G, Groener JE, Ormel EE, Bouma BJ, et al. Treatment of Fabry disease: outcome of a comparative trial with agalsidase alfa or beta at a dose of 0.2 mg/kg. PLoS One. (2007) 2(7):e598. doi: 10.1371/journal.pone.0000598
46. Wraith JE, Tylki-Szymanska A, Guffon N, Lien YH, Tsimaratos M, Vellodi A, et al. Safety and efficacy of enzyme replacement therapy with agalsidase beta: an international, open-label study in pediatric patients with Fabry disease. J Pediatr. (2008) 152(4):563–70.e1. doi: 10.1016/j.jpeds.2007.09.007
47. Wuest W, Machann W, Breunig F, Weidemann F, Koestler H, Hahn D, et al. Right ventricular involvement in patients with Fabry’s disease and the effect of enzyme replacement therapy. ROFO Fortschr Geb Rontgenstr Nuklearmed. (2011) 183(11):1037–42. doi: 10.1055/s-0031-1281744
48. Germain DP, Weidemann F, Abiose A, Patel MR, Cizmarik M, Cole JA, et al. Analysis of left ventricular mass in untreated men and in men treated with agalsidase-beta: data from the Fabry registry. Genet Med. (2013) 15(12):958–65. doi: 10.1038/gim.2013.53
49. Hopkin RJ, Cabrera G, Charrow J, Lemay R, Martins AM, Mauer M, et al. Risk factors for severe clinical events in male and female patients with Fabry disease treated with agalsidase beta enzyme replacement therapy: data from the Fabry registry. Mol Genet Metab. (2016) 119(1-2):151–9. doi: 10.1016/j.ymgme.2016.06.007
50. Wanner C, Feldt-Rasmussen U, Jovanovic A, Linhart A, Yang M, Ponce E, et al. Cardiomyopathy and kidney function in agalsidase beta-treated female fabry patients: a pre-treatment vs. Post-treatment analysis. ESC heart Failure. (2020) 7(3):825–34. doi: 10.1002/ehf2.12647
51. Beer M, Weidemann F, Breunig F, Knoll A, Koeppe S, Machann W, et al. Impact of enzyme replacement therapy on cardiac morphology and function and late enhancement in Fabry’s cardiomyopathy. Am J Cardiol. (2006) 97(10):1515–8. doi: 10.1016/j.amjcard.2005.11.087
52. Breunig FW, Strotmann J, Knoll A, Wanner C. Clinical benefit of enzyme replacement therapy in Fabry disease. Kidney Int. (2006) 69(7):1216–21. doi: 10.1038/sj.ki.5000208
53. Collin C, Briet M, Tran TC, Beaussier H, Benistan K, Bensalah M, et al. Long-term changes in arterial structure and function and left ventricular geometry after enzyme replacement therapy in patients affected with Fabry disease. Eur J Prev Cardiol. (2011) 19(1):43–54. doi: 10.1177/1741826710391118
54. Elliott PM, Kindler H, Shah JS, Sachdev B, Rimoldi OE, Thaman R, et al. Coronary microvascular dysfunction in male patients with Anderson-Fabry disease and the effect of treatment with alpha galactosidase A. Heart. (2006) 92(3):357–60. doi: 10.1136/hrt.2004.054015
55. Messalli G, Imbriaco M, Avitabile G, Russo R, Iodice D, Spinelli L, et al. Role of cardiac MRI in evaluating patients with Anderson-Fabry disease: assessing cardiac effects of long-term enzyme replacement therapy. Radiol Med. (2012) 117(1):19–28. doi: 10.1007/s11547-011-0710-9
56. Imbriaco M, Pisani A, Spinelli L, Cuocolo A, Messalli G, Capuano E, et al. Effects of enzyme-replacement therapy in patients with Anderson-Fabry disease: a prospective long-term cardiac magnetic resonance imaging study. Heart. (2009) 95(13):1103–7. doi: 10.1136/hrt.2008.162800
57. Koskenvuo JW, Hartiala JJ, Nuutila P, Kalliokoski R, Viikari JS, Engblom E, et al. Twenty-four-month alpha-galactosidase A replacement therapy in Fabry disease has only minimal effects on symptoms and cardiovascular parameters. J Inherit Metab Dis. (2008) 31(3):432–41. doi: 10.1007/s10545-008-0848-3
58. Lenders M, Brand E. FAbry STabilization indEX (FASTEX): clinical evaluation of disease progression in Fabry patients. Mol Genet Metab. (2020) 129(2):142–9. doi: 10.1016/j.ymgme.2019.12.010
59. Pisani A, Spinelli L, Sabbatini M, Andreucci MV, Procaccini D, Abbaterusso C, et al. Enzyme replacement therapy in Fabry disease patients undergoing dialysis: effects on quality of life and organ involvement. Am J Kidney Dis. (2005) 46(1):120–7. doi: 10.1053/j.ajkd.2005.03.016
60. Politei J, Hernan A, Beatriz SA, Gustavo C, Antonio M, Eduardo T, et al. Fabry disease: multidisciplinary evaluation after 10 years of treatment with agalsidase Beta. JIMD Rep. (2014) 16:7–14. doi: 10.1007/8904_2014_310
61. Arends M, Biegstraaten M, Wanner C, Sirrs S, Mehta A, Elliott PM, et al. Agalsidase alfa versus agalsidase beta for the treatment of fabry disease: an international cohort study. J Med Genet. (2018) 55(5):351–8. doi: 10.1136/jmedgenet-2017-104863
62. Kim JH, Lee BH, Hyang Cho J, Kang E, Choi JH, Kim GH, et al. Long-term enzyme replacement therapy for Fabry disease: efficacy and unmet needs in cardiac and renal outcomes. J Hum Genet. (2016) 61(11):923–9. doi: 10.1038/jhg.2016.78
63. Motwani M, Banypersad S, Woolfson P, Waldek S. Enzyme replacement therapy improves cardiac features and severity of Fabry disease. Mol Genet Metab. (2012) 107(1-2):197–202. doi: 10.1016/j.ymgme.2012.05.011
64. Rombach SM, Aerts JM, Poorthuis BJ, Groener JE, Donker-Koopman W, Hendriks E, et al. Long-term effect of antibodies against infused alpha-galactosidase A in Fabry disease on plasma and urinary (lyso)Gb3 reduction and treatment outcome. PLoS One. (2012) 7(10):e47805. doi: 10.1371/journal.pone.0047805
65. Tsuboi K. Enzyme replacement therapy in patients with Fabry’s disease. J Int Med Res. (2007) 35(4):574–81. doi: 10.1177/147323000703500418
66. Koeppe S, Neubauer H, Breunig F, Weidemann F, Wanner C, Sandstede J, et al. MR-based analysis of regional cardiac function in relation to cellular integrity in Fabry disease. Int J Cardiol. (2012) 160(1):53–8. doi: 10.1016/j.ijcard.2011.03.023
67. Lenders M, Nordbeck P, Canaan-Kühl S, Kreul L, Duning T, Lorenz L, et al. Treatment switch in Fabry disease- a matter of dose? J Med Genet. (2021) 58(5):342–50. doi: 10.1136/jmedgenet-2020-106874
68. Politei J, Schenone AB, Cabrera G, Heguilen R, Szlago M. Fabry disease and enzyme replacement therapy in classic patients with same mutation: different formulations–different outcome? Clin Genet. (2016) 89(1):88–92. doi: 10.1111/cge.12590
69. Lin H-Y, Huang Y-H, Liao HC, Liu HC, Hsu TR, Shen CI, et al. Clinical observations on enzyme replacement therapy in patients with Fabry disease and the switch from agalsidase beta to agalsidase alfa. J Chin Med Assoc. (2014) 77(4):190–7. doi: 10.1016/j.jcma.2013.11.006
70. Ripeau D, Amartino H, Cedrolla M, Urtiaga L, Urdaneta B, Cano M, et al. Switch from agalsidase beta to agalsidase alfa in the enzyme replacement therapy of patients with Fabry disease in Latin America. Medicina (B Aires). (2017) 77(3):173–9.28643672
71. Vedder AC, Breunig F, Donker-Koopman WE, Mills K, Young E, Winchester B, et al. Treatment of Fabry disease with different dosing regimens of agalsidase: effects on antibody formation and GL-3. Mol Genet Metab. (2008) 94(3):319–25. doi: 10.1016/j.ymgme.2008.03.003
72. Pisani A, Spinelli L, Visciano B, Capuano I, Sabbatini M, Riccio E, et al. Effects of switching from agalsidase Beta to agalsidase alfa in 10 patients with anderson-Fabry disease. JIMD Rep. (2013) 9:41–8. doi: 10.1007/8904_2012_177
73. Tsuboi K, Yamamoto H. Clinical observation of patients with Fabry disease after switching from agalsidase beta (fabrazyme) to agalsidase alfa (replagal). Genet Med. (2012) 14(9):779–86. doi: 10.1038/gim.2012.39
74. Weidemann F, Krämer J, Duning T, Lenders M, Canaan-Kuhl S, Krebs A, et al. Patients with Fabry disease after enzyme replacement therapy dose reduction versus treatment switch. J Am Soc Nephrol. (2014) 25(4):837–49. doi: 10.1681/ASN.2013060585
75. van der Veen SJ, Körver S, Hirsch A, Hollak CEM, Wijburg FA, Brands MM, et al. Early start of enzyme replacement therapy in pediatric male patients with classical Fabry disease is associated with attenuated disease progression. Mol Genet Metab. (2022) 135(2):163–9. doi: 10.1016/j.ymgme.2021.12.004
76. Weidemann F, Niemann M, Breunig F, Herrmann S, Beer M, Stork S, et al. Long-term effects of enzyme replacement therapy on Fabry cardiomyopathy: evidence for a better outcome with early treatment. Circulation. (2009) 119(4):524–9. doi: 10.1161/CIRCULATIONAHA.108.794529
77. Weidemann F, Niemann M, Stork S, Breunig F, Beer M, Sommer C, et al. Long-term outcome of enzyme-replacement therapy in advanced Fabry disease: evidence for disease progression towards serious complications. J Intern Med. (2013) 274(4):331–41. doi: 10.1111/joim.12077
78. Lubanda JC, Anijalg E, Bzduch V, Thurberg BL, Benichou B, Tylki-Szymanska A. Evaluation of a low dose, after a standard therapeutic dose, of agalsidase beta during enzyme replacement therapy in patients with Fabry disease. Genet Med. (2009) 11(4):256–64. doi: 10.1097/GIM.0b013e3181981d82
79. Germain DP, Waldek S, Banikazemi M, Bushinsky DA, Charrow J, Desnick RJ, et al. Sustained, long-term renal stabilization after 54 months of agalsidase beta therapy in patients with Fabry disease. J Am Soc Nephrol. (2007) 18(5):1547–57. doi: 10.1681/ASN.2006080816
80. Kramer J, Lenders M, Canaan-Kuhl S, Nordbeck P, Uceyler N, Blaschke D, et al. Fabry disease under enzyme replacement therapy-new insights in efficacy of different dosages. Nephrol Dial Transplant. (2018) 33(8):1362–72. doi: 10.1093/ndt/gfx319
81. Tsuboi K, Yamamoto H. Clinical course of patients with Fabry disease who were switched from agalsidase-beta to agalsidase-alpha. Genet Med. (2014) 16(10):766–72. doi: 10.1038/gim.2014.28
82. Fellgiebel A, Gartenschlager M, Wildberger K, Scheurich A, Desnick RJ, Sims K. Enzyme replacement therapy stabilized white matter lesion progression in Fabry disease. Cerebrovasc Dis. (2014) 38(6):448–56. doi: 10.1159/000369293
83. Lenders M, Canaan-Kühl S, Kramer J, Duning T, Reiermann S, Sommer C, et al. Patients with Fabry disease after enzyme replacement therapy dose reduction and switch-2-year follow-up. J Am Soc Nephrol. (2016) 27(3):952–62. doi: 10.1681/ASN.2015030337
84. Ortiz A, Cianciaruso B, Cizmarik M, Germain DP, Mignani R, Oliveira JP, et al. End-stage renal disease in patients with Fabry disease: natural history data from the Fabry registry. Nephrol Dial Transplant. (2010) 25(3):769–75. doi: 10.1093/ndt/gfp554
85. Patel MR, Cecchi F, Cizmarik M, Kantola I, Linhart A, Nicholls K, et al. Cardiovascular events in patients with Fabry disease natural history data from the Fabry registry. J Am Coll Cardiol. (2011) 57(9):1093–9. doi: 10.1016/j.jacc.2010.11.018
86. Perera K, Kashyap N, Wang K, Omar F, Prosia E, Thompson RB, et al. Integrating cardiac MRI imaging and multidisciplinary clinical care is associated with improved outcomes in patients with Fabry disease. Curr Probl Cardiol. (2023) 48(2):101476. doi: 10.1016/j.cpcardiol.2022.101476
87. European Medicines Agency. Summary of Product Characteristics (Fabrazyme) (2006). Available online at: https://www.ema.europa.eu/en/documents/product-information/fabrazyme-epar-product-information_en.pdf (Accessed December 9, 2024).
88. Desnick RJ. Enzyme replacement therapy for Fabry disease: lessons from two alpha-galactosidase A orphan products and one FDA approval. Expert Opin Biol Ther. (2004) 4(7):1167–76. doi: 10.1517/14712598.4.7.1167
89. Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, et al. Safety and efficacy of recombinant human alpha-galactosidase A–replacement therapy in Fabry’s disease. N Engl J Med. (2001) 345(1):9–16. doi: 10.1056/NEJM200107053450102
90. Ortiz A, Kanters S, Hamed A, DasMahapatra P, Poggio E, Maski M, et al. Agalsidase beta treatment slows estimated glomerular filtration rate loss in classic fabry disease patients: results from an individual patient data meta-analysis. Clin Kidney J. (2021) 14(4):1136–46. doi: 10.1093/ckj/sfaa065
91. Figliozzi S, Kollia E, Simistiras A, Camporeale A, Stankowski K, Masci PG, et al. Effects of enzyme replacement therapy on cardiac MRI findings in Fabry disease: a systematic review and meta-analysis. Radiol Cardiothorac Imaging. (2024) 6(3):e230154. doi: 10.1148/ryct.230154
92. Siegenthaler M, Huynh-Do U, Krayenbuehl P, Pollock E, Widmer U, Debaix H, et al. Impact of cardio-renal syndrome on adverse outcomes in patients with Fabry disease in a long-term follow-up. Int J Cardiol. (2017) 249:261–7. doi: 10.1016/j.ijcard.2017.09.027
93. Körver S, Vergouwe M, Hollak CEM, van Schaik IN, Langeveld M. Development and clinical consequences of white matter lesions in Fabry disease: a systematic review. Mol Genet Metab. (2018) 125(3):205–16. doi: 10.1016/j.ymgme.2018.08.014
94. Germain DP, Elliott PM, Falissard B, Fomin VV, Hilz MJ, Jovanovic A, et al. The effect of enzyme replacement therapy on clinical outcomes in male patients with Fabry disease: a systematic literature review by a European panel of experts. Mol Genet Metab Rep. (2019) 19:100454. doi: 10.1016/j.ymgmr.2019.100454
95. Germain DP, Arad M, Burlina A, Elliott PM, Falissard B, Feldt-Rasmussen U, et al. The effect of enzyme replacement therapy on clinical outcomes in female patients with fabry disease - a systematic literature review by a European panel of experts. Mol Genet Metab. (2019) 126(3):224–35. doi: 10.1016/j.ymgme.2018.09.007
96. Hazari H, Belenkie I, Kryski A, White JA, Oudit GY, Thompson R, et al. Comparison of cardiac magnetic resonance imaging and echocardiography in assessment of left ventricular hypertrophy in Fabry disease. Can J Cardiol. (2018) 34(8):1041–7. doi: 10.1016/j.cjca.2018.03.011
97. Januzzi JL J, Canty JM, Das S, DeFilippi CR, Gintant GA, Gutstein DE, et al. Gaining efficiency in clinical trials with cardiac biomarkers: JACC review topic of the week. J Am Coll Cardiol. (2021) 77(15):1922–33. doi: 10.1016/j.jacc.2021.02.040
98. Bay B, Goßling A, Blaum CM, Kroeger F, Koppe L, Lorenz T, et al. Association of high-sensitivity troponin T and I blood concentrations with all-cause mortality and cardiovascular outcome in stable patients-results from the INTERCATH cohort. J Am Heart Assoc. (2022) 11(17):e024516. doi: 10.1161/JAHA.121.024516
99. Yogasundaram H, Nikhanj A, Putko BN, Boutin M, Jain-Ghai S, Khan A, et al. Elevated inflammatory plasma biomarkers in patients with Fabry disease: a critical link to heart failure with preserved ejection fraction. J Am Heart Assoc. (2018) 7(21):e009098. doi: 10.1161/JAHA.118.009098
100. Nordin S, Kozor R, Vijapurapu R, Augusto JB, Knott KD, Captur G, et al. Myocardial storage, inflammation, and cardiac phenotype in Fabry disease after one year of enzyme replacement therapy. Circulation Cardiovascular Imaging. (2019) 12(12):e009430. doi: 10.1161/CIRCIMAGING.119.009430
Keywords: Fabry disease, agalsidase beta, genetic disorders, cardiac, cardiovascular, enzyme replacement therapy
Citation: Oudit GY, DasMahapatra P, Lyn N, Wilson FR, Adeyemi A, Lee CS, Crespo A and Namdar M (2025) A systematic literature review to evaluate the cardiac and cerebrovascular outcomes of patients with Fabry disease treated with agalsidase Beta. Front. Cardiovasc. Med. 11:1415547. doi: 10.3389/fcvm.2024.1415547
Received: 10 April 2024; Accepted: 28 November 2024;
Published: 21 January 2025.
Edited by:
Umberto Campia, Brigham and Women's Hospital and Harvard Medical School, United StatesReviewed by:
Stefano Figliozzi, St Thomas' Hospital, United KingdomCopyright: © 2025 Oudit, DasMahapatra, Lyn, Wilson, Adeyemi, Lee, Crespo and Namdar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gavin Y. Oudit, Z2F2aW4ub3VkaXRAdWFsYmVydGEuY2E=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.