
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 17 June 2024
Sec. Atherosclerosis and Vascular Medicine
Volume 11 - 2024 | https://doi.org/10.3389/fcvm.2024.1381520
This article is part of the Research Topic Dysfunction of Vascular Smooth Muscle Cells in Vascular Remodeling Diseases View all 4 articles
 M. T. La Chica Lhoëst1,2A. Martinez1,2L. Claudi1,2E. Garcia1,2A. Benitez-Amaro1,2A. Polishchuk1,2
M. T. La Chica Lhoëst1,2A. Martinez1,2L. Claudi1,2E. Garcia1,2A. Benitez-Amaro1,2A. Polishchuk1,2 J. Piñero3
J. Piñero3 D. Vilades4,5
D. Vilades4,5 J. M. Guerra4,5
J. M. Guerra4,5 F. Sanz3N. Rotllan2,6
F. Sanz3N. Rotllan2,6 J. C. Escolà-Gil2,6
J. C. Escolà-Gil2,6 V. Llorente-Cortés1,2,5*
V. Llorente-Cortés1,2,5*
In recent years, the role of macrophages as the primary cell type contributing to foam cell formation and atheroma plaque development has been widely acknowledged. However, it has been long recognized that diffuse intimal thickening (DIM), which precedes the formation of early fatty streaks in humans, primarily consists of lipid-loaded smooth muscle cells (SMCs) and their secreted proteoglycans. Recent studies have further supported the notion that SMCs constitute the majority of foam cells in advanced atherosclerotic plaques. Given that SMCs are a major component of the vascular wall, they serve as a significant source of microvesicles and exosomes, which have the potential to regulate the physiology of other vascular cells. Notably, more than half of the foam cells present in atherosclerotic lesions are of SMC origin. In this review, we describe several mechanisms underlying the formation of intimal foam-like cells in atherosclerotic plaques. Based on these mechanisms, we discuss novel therapeutic approaches that have been developed to regulate the generation of intimal foam-like cells. These innovative strategies hold promise for improving the management of atherosclerosis in the near future.
Cardiovascular diseases (CVDs) are one of the leading causes of death worldwide, accounting for 32% of all global deaths (1). Among them, atherosclerosis is predominant, leading to tissue ischemia and events such as strokes and heart attacks, depending on the vascular territory affected. Atherosclerosis is characterized by the progressive accumulation of cholesterol and fibromatous material within the tunica intima of arterial walls, resulting in the formation of an atheroma plaque, which can obstruct blood flow (2). Human atherosclerosis is initiated by the retention of low-density lipoproteins (LDLs), positively charged, by negatively charged glucosaminoglycans (GAGs) of proteoglycans (PGs) that structure the extracellular matrix (ECM) in the arterial intima (3). In this context, Camejo et al, pioneers within the field, identified the specific segments of ApoB100 involved in the interaction of LDL with arterial PGs (4), and show that human proteoglycans have high affinity for fused LDL particles (5) and cytokines and growth factors (6).
Previous evidence suggests that diffuse intimal thickening (DIM), preceding early fatty streaks in humans, mainly consists of lipid-loaded SMC and their secreted proteoglycans (7). However, it was widely accepted for several years that macrophages were the major cell type forming foam cells in the arterial intima and the main contributors to atheroma plaque formation (8). Recent studies utilizing lineage-tracing in mice have provided evidence that SMCs are not only the initial foam cells in the lesion but also comprise the majority of foam cells in advanced atherosclerotic plaques (9, 10). In vitro studies have demonstrated that human coronary SMCs readily uptake aggregated LDL (agLDL), leading to foam cell formation (11–13). Upon cholesterol loading, SMCs downregulate contractility markers such as alpha-actin, alpha-tropomyosin, myosin heavy chain, and calponin H1 while upregulating macrophage-related markers CD68, Mac-2, and ABCA1 (14). Cell lineage tracing technology and other cutting-edge methods, such as single-cell RNA sequencing, have enabled the definition of SMC reprogramming towards various phenotypes, including osteochondrogenic, myofibroblastic, and mesenchymal stem cell-like phenotypes, in addition to the macrophage-like phenotype (15, 16). Depending on the activation of specific transcription factors, SMCs switch to dedifferentiated transitional states that can be pathological (such as macrophage-like) (17) or beneficial (such as protective fibromyocytes forming the fibrous cap) (18). Currently, the contribution of SMCs to atherosclerosis is undoubtedly greater than previously thought, as genome-wide association studies (GWAS) have highlighted the pivotal role of SMC-specific gene expression in predicting coronary artery disease risk in humans (19, 20).
In this review, we will provide an overview of the main factors modulating the formation of foam-like cells, particularly foam-like SMCs, and the mechanisms involved in their contribution to the progression of atherosclerosis. Additionally, we will review innovative proposals to reduce the onset and/or progression of atherosclerosis by modulating foam-like cell generation. These new therapeutic approaches could be essential for improving coronary artery disease management and preventing coronary acute events in the near future.
In response to microenvironmental factors, SMCs undergo transcriptional reprogramming, often referred to as phenotypic modulation, and migrate from the media to the inner membrane. This process involves the loss of classic contractile function and an increase in proliferative and synthetic abilities (9, 21). The role of phenotypically modulated SMCs in atherosclerosis can be both beneficial and detrimental. On one hand, SMCs contribute to the thickness of the fibrous cap through ECM synthesis, promoting plaque stability (18). On the other hand, a plethora of new SMC phenotypes including foam-cell like phenotype coexist in the arterial intima and contribute to the formation of the necrotic core and the progression of atherosclerosis (17, 22). Statins, the most prescribed hypolipemiants in the world, reduce the number of SMC in the arterial intima of hyperlipidemic rabbits (23) and reduce calcification via restoration of the Gas6-Axl pathway in vascular SMC (24). However, treatments targeting exclusively SMC phenotypic modulation and/or transdifferentiation are currently lacking. Interventions aimed at regulating the SMC phenotype have the potential to alter the progression of atherosclerotic plaques, as demonstrated in murine experimental models lacking specific transcription factors (17, 25–28).
A crucial transcription factor for lipid-induced transdifferentiation of SMCs into macrophage-like cells is the zinc finger transcription factor Krüppel-like factor 4 (KLF4). Lineage-tracing studies in ApoE−/− mice have shown that SMC-specific conditional deficiency in KLF4 (SMC YFP+/+ Klf4Δ/Δ Apoe−/−) as well as loss of one Klf4 allele globally in all cell types (ERT-Cre + Klf4Δ/WT Apoe−/−) decreased lesion size, apoptosis, and proliferation activities in the lesion. These models also exhibited increased plaque stability, including an increase in the thickness of the fibrous cap, indicating that KLF4 deficiency has mitigating effects on plaque development and progression. This suggests KLF4 in SMC as a potential therapeutic target for inducing smaller and more stable lesions (25). Furthermore, the KLF4 reduction promoted by angiopoietin-like 4 (ANGPTL4)-transforming necrosis factor alpha (TNFα)-NADPH oxidase 1 (NOX1) axis plays a role in the protective effect of the anti-inflammatory ANGPTL4 protein in atherosclerosis. Among other anti-atherogenic effects such as reduction of cardiac and vascular inflammation, ANGPTL4 reduces the phenotypic transition of SMCs into macrophage-like cells in the ApoE−/− mouse model of atherosclerosis (29).
Although there is no doubt that KLF4 plays a critical role in regulating phenotypic transitions of arterial smooth muscle cells (SMCs) and overall atherosclerotic lesion pathogenesis, additional studies in the SMC YFP+/+ Klf4Δ/Δ Apoe−/Δ murine model have evidenced a cardioprotective role of KLF4. This role was observed in reducing peripheral vascular resistance, dilated heart, and increased vascular leakage, all of which are changes related to the dysregulation of platelet-derived growth factor (PDGF) and fibroblast growth factor (FGF) signaling in association with KLF deficiency in SMCs. These results have implications regarding the essential role of KLF4 in the maintenance of microvascular SMC structure and function, as well as in the engraftment of supporting arteries (30). In line with the potential protective role of KLFs in the microvasculature, extracellular vesicles enriched in miR-143/145 derived from KLF2-expressing endothelial cells have been shown to reduce atherosclerotic lesion formation in the aorta of ApoE(−/−) mice (31). Additionally, in vitro studies in endothelial-SMC co-cultures have demonstrated that miR-143/145-enriched extracellular vesicles from endothelial cells target crucial atherosclerotic genes in SMCs such as ELK1 (32), KLF4 (33), CAMK2d (32), and SSH2 (33). The authors proposed that the modulation of atheroprotective miRNA- and extracellular-vesicle-mediated mechanisms could be a promising strategy to manage atherosclerosis. Controversially, the inhibition of miR-143/145 in western diet-fed LDLR KO mice (Ldlr−/−-miR-143/145−/−) has been shown to reduce atherosclerosis progression through switching from a contractile/non-proliferative state to a migratory/proliferative SMC state and hepatic ABCA1 expression (34). Consistent with these results, previous studies have evidenced increased levels of miR-145 in human carotid atherosclerotic plaques from symptomatic patients (35). These controversial results about the beneficial or pathological effect of miR-145 require the reevaluation of miR-145 effects in the context of dyslipidemia and cardiovascular-related diseases before assessing its potential clinical utility.
Another transcription factor crucially involved in the phenotypic transition of SMCs into CD68 + macrophage-like cells is BCLAF1 (BCL2-associated transcription factor 1). Integrating data from in vitro and in vivo experimental models and from humans, it has been demonstrated that BCLAF1 is implicated in the lipid-induced transdifferentiation of SMCs into macrophage-like cells, as it was shown to control lipid uptake in SMC via regulation of CD36 and CD68 scavenger receptors (36). Immunopositivity of BCLAF1 was found to colocalize with BCL2-positive areas but not with apoptotic areas of the necrotic core in the atherosclerotic plaques of SMC lineage-tracing atherosclerotic mice. Furthermore, in humans, downregulation of BCLAF1 was associated with plaque vulnerability parameters and higher cardiovascular risk in patients with carotid atherosclerosis (36). The transcriptional modulation of SMC phenotype is influenced by various environmental cues such as lipids, inflammatory cytokines, and altered cell-cell interactions. Indeed, the heterogeneity of SMCs in the arterial intima may reflect, at least in part, the dynamic nature of SMC responses to the atherosclerotic microenvironment. In the following sections, we will review the reported effects of some of these environmental cues on SMC phenotype.
Retinoic acid (RA) is a metabolite of vitamin A and a morphogen that governs cell differentiation and embryonic patterning in early developmental stages. Specifically, all-trans retinoic acid (tRA) inhibits proliferation while increasing migration and tissue-type plasminogen activator activity in two phenotypically distinct rat SMC populations, cultured respectively from the normal aortic media and from the intimal thickening (IT) after endothelial injury (37). These authors also demonstrated that retinoids reduce IT induced by balloon endothelium injury in a rat experimental model. Further studies explored the effects of tRA, either alone or in combination with basic fibroblast growth factor (bFGF), on the expression of plasminogen activator inhibitor-1 (PAI-1) in cultured vascular SMCs. PAI-1 expression in SMCs regulates the extracellular matrix composition, which, in turn, influences SMC migration, among other relevant aspects for atherosclerosis. tRA alone or in combination with bFGF upregulates PAI-1 synthesis by rabbit aortic SMCs, consequently enhancing SMC migration (38).
Recent studies have explored how the stiffness of the extracellular environment influences cellular processes and have pointed to discoidin receptor 1 (DDR1) as a crucial perceptive receptor whose phosphorylation, aggregation, and oligomerization depend on ECM stiffness. DDR1 activation was observed in collagen (Col)-coated gels and in calcified atherosclerotic plaques, resulting in changes in SMC genes modulated through extracellular signal-regulated kinases (ERKs) and p53 pathways, and related to inflammation and ECM homeostasis (39). These results have implications for the development of innovative ECM-based therapies useful for improving the management of atherosclerosis, as well as other prevalent pathologies such as cancer and fibrosis.
Besides interactions with neighboring cells and the ECM, inflammatory mediators released in response to injury or disease are key drivers of SMC phenotypic modulation (40). Basic fibroblast growth factor (bFGF) and heparin-binding epidermal growth factor-like growth factor (HB-EGF) induce morphological changes, disrupt SMC monolayers, and promote proliferation and dedifferentiation of SMCs (41). Interestingly, activating TGF-β signaling by inhibiting bFGF signaling prevents the switch from a contractile to a proliferative phenotype in SMCs and significantly reduces atherosclerotic plaque size in male mice fed a Western diet (42). These beneficial effects were linked to features of plaque stability sch as increased fibrous cap and reduced necrotic core in this experimental model. Together, they suggest a potential therapeutic effect of TGF-β/bFGF axis for managing coronary artery disease. Additionally, TGF-beta and Notch signaling pathways cooperatively promote a SMC contractile phenotype by co-regulating Smad activity at SMC promoters (43). Interferon-γ (IFN-γ), primarily secreted by macrophages and T cells, plays a crucial role in immune function against pathogens. IFN-γ stimulates SMC migration to the arterial intima, increases the number of intimal SMC-derived foam cells, promotes oxidative stress, and contributes to plaque formation and rupture (44). Conversely, IFN-γ has also been reported to reduce LDL uptake by macrophages, suggesting an anti-atherogenic role during foam cell formation (45). IL-1β and TNF-α enhance macrophage foam cell formation, accelerate the enlargement of unstable lipid-rich atherosclerotic plaques, and decrease lipid efflux, thereby promoting macrophage foam cell formation (46).
Reactive oxygen species (ROS) production is increased in the vessel walls in pathological conditions such as hypertension, diabetes, smoking, and dyslipidemia (47). ROS induces proliferation (48, 49), and phenotypic switching (50) of SMC, through different mechanisms. ROS upregulates the production of growth factors and hypertrophic hormones including phenylephrine, thrombin, vascular epidermal growth factor (VEGF), basic fibroblast growth factor (bFGF), PDGF, insulin-like growth factor-I (IGF-I), and angiotensin II by vascular cells (48, 51–53). In addition, ROS induces SMC migration through the activation of matrix metalloproteinase (MMP) (54), responsible for matrix degradation and reorganization, facilitating SMC migration and proliferation (55, 56).
NADPH oxidase (NOX) enzymes present in the vascular wall are major contributors to ROS production and signaling in the vasculature (57). Multiple studies have evidenced an association between hyperactivation of NADPH oxidases and vascular pathologies, including atherosclerosis and restenosis (58). NOX1 is the NADPH oxidase isoform mainly expressed by SMC and one of the primary enzymes regulating SMC activation (59). NOX1-induced ROS production upregulates vascular inflammation in early atherosclerotic lesions of ApoE−/− mice (60). In line, NOX1/NOX4 pharmacological inhibition (61) and Nox1 deficiency (62) significantly attenuate vascular ROS levels and atherosclerosis burden in ApoE−/− mice.
NOX activator 1 (NOXA1), a critical functional homolog of p67phox for NADPH oxidase activation in mouse VSMC (63), is upregulated by TNF-α and angiotensin 2 in SMC, causing SMC activation (64). NOXA1-dependent NOX1 activation promotes SMC proliferation and migration and KLF4-mediated transition to macrophage-like cells, plaque inflammation, and expansion (65). These results support a critical role of NOX1 in the pathogenesis and progression of atherosclerosis. NADPH oxidase 4 (Nox4) is a constitutively active ROS-producing enzyme that is highly expressed in the vascular endothelium. The NOX4 isoform constitutively generates hydrogen peroxide (H2O2) in normal non-disease conditions. H2O2 production is tightly controlled in terms of levels and subcellular locations to mediate various physiological processes in vascular cells including the maintenance of the contractile SMC phenotype (66). Under hyperglycemic/diabetic conditions, NOX4 overexpression in endothelium is associated with atherosclerosis reduction in ApoE−/− mice (67). Di Marco et al. showed that NOX4 counteracts hyperglycemia-induced SMC proliferation and fibrosis and maintains a differentiated SMC phenotype in ApoE−/− mice (68). In these diabetic ApoE−/− mice, genetic deletion of NOX4 increases PDGF, osteopontin (OPN), and ECM protein fibronectin in aortic SMC concomitantly with elevated NOX1-associated ROS levels. Additional studies have consistently shown that genetic deletion of NOX4 increases atherosclerosis in ApoE−/− mice (69).
Previous studies have demonstrated that aggregated LDL is a potent stimulus for foam cell formation from human coronary SMCs (11–13). Early research indicated that when LDL aggregates were removed from LDL preparations, LDL did not induce intracellular cholesteryl ester accumulation in SMC (70–72). Currently, individual susceptibility to LDL aggregation has been directly linked to future cardiovascular events (73), underscoring the crucial relevance of the interaction between aggregated LDL and SMC for atherosclerosis progression.
The susceptibility of LDL particles to aggregate is strongly associated with the particle's lipid composition, as evidenced by samples from a nested case–control study designed from the Finnish Corogene study (73). In this study, human LDL particles prone to aggregation were those enriched in sphingomyelins (SMs) and ceramides (Cer), with fewer phosphatidylcholine (PC) and triglyceride (TG) species compared to aggregation-resistant LDL particles. Additionally, dietary factors and/or medication can modify susceptibility to LDL aggregation. Increasing dietary intake of vegetable oils and plant stanol-enriched spreads, for example, reduces the propensity for LDL aggregation (74). Moreover, proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors have been shown to reduce LDL aggregation by altering the lipid composition of LDL particles (75). Therefore, dietary factors that modulate LDL lipid composition and susceptibility to aggregation appear to be key determinants for foam SMC generation. A crucial factor in the diet that determines individual susceptibility to LDL aggregation is the type of fat (76). Camelina sativa oil (CSO), a source of alpha-linolenic acid (ALA), decreases the relative concentrations of saturated and monounsaturated cholesteryl ester species and increases polyunsaturated TGs in LDL (77). Additionally, dietary n-3 polyunsaturated fatty acids (PUFAs) are incorporated into the lipid species of LDL particles, increasing the degree of unsaturation of phospholipids and neutral lipids in the LDLs, thereby altering the tendency of LDL to aggregate. As mentioned above, LDL aggregation seems to be inversely associated with specific unsaturated species of PC and directly associated with those of SM. Therefore, depending on which specific species of phospholipids incorporate the unsaturation and the degree of this unsaturation in the LDL particle, the susceptibility may change (73, 75).
Currently, apolipoprotein-based peptides and their mimetics with the capacity to improve lipid profile and/or lipoprotein functionality have emerged as potential therapeutic tools for atherosclerosis (78). These peptides are usually designed as short amino acid chains based on functional domains of ApoA1 (79) 79 and ApoJ (80, 81) that due to their amphipathic nature have efficacy to rearrange lipids, in particular phospholipids, thereby conferring stability to LDL. Unlike ApoB-based peptides that interact with phospholipids, LRP1-based peptides target a particular sequence of ApoB100, stabilizing its conformation and efficiently preventing LDL aggregation (by more than 85%) (82, 83). These peptidomimetics emerge as potential tools to maintain LDL integrity in the arterial intima.
SMC switching from a contractile to a synthetic phenotype in the arterial intima promotes an increase in the proteoglycan production (84). Since proteoglycans contribute to LDL retention and modification (3, 7), intimal PGs and retained LDL create a vicious cycle driving foam SMC formation. Therefore, active immunization against GAG side chain of PGs has emerged as a promising strategy to target atherosclerosis due to the capacity of GAGs to retain and modify lipoproteins in the arterial intima (85).
Treatment of VSMCs with angiotensin II, the main effector of hypertension, promotes the production of elongated GAG chains, which are associated with enhanced binding to LDL (86). Moreover, the sulfation of GAGs, including both the grade and position of sulfate groups, determines their capacity to bind to LDL (87). chP3R99 monoclonal antibody (mAb), which interacts with sulfated GAGs, efficiently blocks the association of LDL with chondroitin sulfate (CS), thus abrogating LDL oxidation in vitro. In line with this, ChP3R99-immunized rabbits showed reduced aortic arch lesions and decreased macrophage infiltration (88). Additionally, chP3R99 has been shown to be atheroprotective in high-fat, high-cholesterol diet-fed ApoE-deficient (−/−) mice due to the generation of anti-GAG antibodies. These antibodies, as a result of an anti-idiotypic cascade, may interfere with the subendothelial retention of LDL via direct binding to GAGs (89). Besides immunization, there are other therapeutic strategies inhibiting LDL-GAG interactions (90), such as ApoB100 peptidomimetics, ligands of PGs, and growth factor inhibitors (91). LDL and PG interaction involves clusters of amino acids in the ApoB100 that bind to the negatively charged sulfate groups on the galactosaminoglycan chain: site A at residues 3,148–3,158 and site B at residues 3,359–3,369 (92). Blocking this interaction by saturating the GAG chain binding sites with small peptide mimetics successfully prevents atherosclerosis in a mouse model (93).
The cytokine named proliferation-inducing ligand (APRIL) is a PG ligand that inhibits the interaction of LDL with heparan-sulfate proteoglycans (HSPGs) (94). Although the physiology of cytokine/HSPG interaction is largely unknown, APRIL confers atheroprotection by limiting LDL retention, intracellular lipid accumulation in macrophages, and formation of necrotic cores in the arterial intima in experimental in vivo models. Serum levels of a specific form of human APRIL named non-canonical APRIL (nc-APRIL) are associated with long-term cardiovascular mortality in patients with atherosclerosis, independently of well-established cardiovascular risk factors (95). Imatinib, a platelet-derived growth factor receptor inhibitor mainly used in cancer (96), inhibits sulfate incorporation into PGs, elongation of GAG chain, and reduces xyloside binding to LDL in vitro and in vivo experimental models (97). Imatinib reduces lipid deposition in the vessel wall without altering circulating lipid levels in high-fat diet-fed ApoE-deficient (−/−) mice (98).
Extensive reviews have summarized the connections between innate and adaptive immunity in regulating the immune system and the activation of cytokines and chemokines in the vasculature (99). In this manuscript, we will concentrate on new therapies that raise antibodies blocking the capacity of scavenger receptors to interact with modified lipoproteins.
ROS transform LDL retained in the intima extracellular matrix into oxidized LDL, which is then taken up by macrophages through cholesterol-deregulated scavenger receptors such as CD36 and SRA1, among others, leading to foam cell formation (8).
Immunization with oxidized LDL (oxLDL) has been reported to inhibit atherosclerosis progression in HFD-fed mice (91) and rabbits (100, 101). In addition, specific monoclonal antibodies that recognize oxidation-specific epitopes (OSE) in ApoB100 block oxLDL uptake by macrophages in several experimental in vivo models (102–104). Small antibody fragments such as scFv are highly suitable for therapeutic use (105). ASA6 is a new human scFv antibody that interacts with oxLDL and blocks atherosclerosis progression in ApoE−/− mice (106).
In the arterial intima, oxLDL coexists with aggregated LDL, a modified LDL that has a significant impact not only on macrophage function (107, 108), but also on smooth muscle cell (SMC) foam cell formation (109). Low-density lipoprotein receptor-related protein 1 (LRP1) is a key receptor for aggregated LDL-induced foam cell formation (11–13). The inhibition of LRP1 by polyclonal antibodies against the region Gly1127–Cys1140 (P3) located in the CR9 domain of cluster II efficiently blocks aggregated LDL uptake by SMCs (110). Immunotherapy based on the P3 sequence inhibits atherosclerosis in a rabbit model of hypercholesterolemia. Moreover, this immunotherapy, which prevents LRP1 up-regulation, counteracts TNFR1 overexpression involved in VSMC migration into the arterial intima, thus preventing atherosclerosis progression (111).
LRP1 is regulated by the P2Y2 (P2Y purinoreceptor 2) receptor in smooth muscle cells (SMCs) isolated from mice (112). Furthermore, in high-fat diet (HFD)-fed ApoE-deficient (−/−) mice, the P2Y12 receptor increases LRP1-mediated foam cell formation through the upregulation of LRP1 expression (113).
While the interaction between macrophages and smooth muscle cells (SMCs) in atherosclerotic plaques has been proposed to play a pathological role, primarily through the interchange of cholesterol-rich particles (114, 115), macrophages can also exert a beneficial effect on foam cell formation in SMCs. Specifically, macrophages may contribute to decreasing foam cell formation in SMCs by enhancing the cholesterol efflux capacity of these cells. Exogenous lysosomal acid lipase (LAL) secreted by macrophages appears to be capable of hydrolyzing cholesteryl esters accumulated in the lysosomes of foam SMCs, thereby promoting proper cholesterol efflux (116).
High-density lipoproteins (HDLs) play a crucial role in removing excess cholesterol from peripheral tissues and transporting it to the liver for excretion or conversion into bile acids/salts, a process known as reverse cholesterol transport (RCT) (117).
The promotion of free cholesterol (FC) efflux from cholesterol-loaded foam cells situated in the arterial wall is the initial stage of reverse cholesterol transport (RCT). This process is recognized as one of the primary mechanisms responsible for the atheroprotective effects mediated by HDL.
In cholesterol-loaded mouse peritoneal macrophages incubated with diluted human serum, approximately two-thirds of cholesterol efflux occurs through ABCA1 active pathways (118). Moreover, macrophages have been reported to transfer cholesterol to adjacent vascular smooth muscle cells (VSMCs) even in the absence of serum or HDL (119). Interestingly, reverse cholesterol transport (RCT) has been found to be defective in SMCs (120). Both the expression of ATP-binding cassette transporter A1 (ABCA1) and its binding to apolipoprotein A-I (ApoA1) of high-density lipoproteins (HDLs) are reduced in human SMCs, with this impairment appearing to be more pronounced in the advanced stages of atherosclerosis (121). Consistent with these findings, CD45-negative cells (VSMC-derived) from ApoE-deficient (−/−) mice also showed reduced ABCA1 expression and decreased cholesterol efflux to ApoA1 compared to leukocyte-derived foam cells (CD45 positive) (10). However, other studies have reported higher ABCA1 expression in cholesterol-loaded murine SMC (14, 122). Whether SMCs loaded with methyl-β-cyclodextrins-cholesterol reflect mouse SMCs in atherosclerotic lesions is still a matter of debate (121).
Beyond the active efflux through ABCA1 and ABCG1, a recent study showed that HDL-mediated cholesterol efflux is maintained in ABCG1-deficient foam SMCs through the upregulation of SR-BI. This suggests that SR-BI may play a compensatory role in regulating HDL-mediated cholesterol efflux when ABCG1-dependent cholesterol efflux is impaired (123). Lecithin cholesterol acyltransferase (LCAT), a rate-limiting step in reverse cholesterol transport (RCT), plays a crucial role in maintaining free cholesterol (FC) homeostasis between peripheral tissues and HDL. By loading cholesteryl esters (CE) into the core of HDL, LCAT progressively increases particle size, promoting the formation of mature and functional HDL particles (124). New therapeutic approaches based on recombinant human LCAT (rhLCAT), currently in phase II clinical trials (https://www.clinicaltrials.gov/study/NCT04737720), have shown promising results. Preclinical studies indicate that augmenting LCAT levels stimulates reverse cholesterol transport (RCT) and reduces atherosclerosis (125). In familial LCAT deficiency (FLD) patients, intravenous administration of ACP-501, the recombinant human LCAT, improved the abnormal distribution of HDL subfractions and decreased TG levels after meals (126).
In summary, the abundant presence of vascular smooth muscle cells (VSMCs) in atherosclerosis was previously overlooked due to their plasticity and synthetic phenotype. The mechanisms regulating cholesterol influx and efflux in VSMC-derived foam cells are still not fully understood. However, targeting SMC foam cells, alongside traditional leukocyte foam cells, will be crucial for effectively treating cardiovascular disease (CVD), as depicted in Figure 1. A proper crosstalk between macrophages and VSMCs could be vital to inhibit the progression of atherosclerosis.
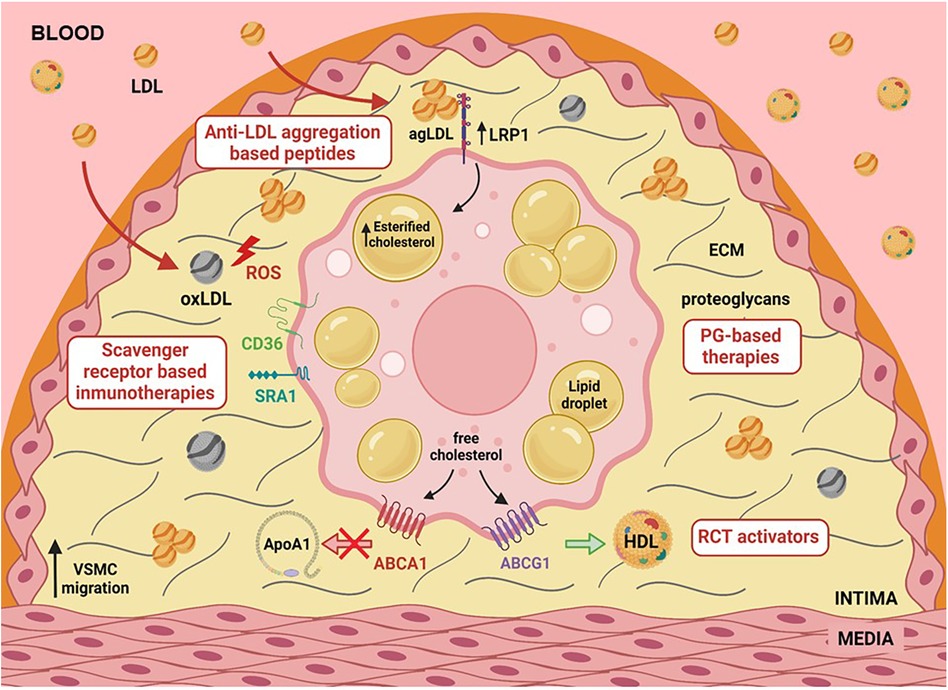
Figure 1 Graphical abstract illustrating various factors such as hypercholesterolemia, hypoxia, and hypertension that upregulate LRP1, a primary lipoprotein receptor involved in human foam SMCs. LRP1 plays a central role in the deregulated uptake of cholesteryl esters (CE) from CE-enriched lipoproteins in SMCs. CE-enriched lipoproteins, predominantly in the form of aggregated LDL, are generated in the extracellular matrix of the arterial intima, interacting with the proteoglycans. The activity of key receptors involved in reverse cholesterol transport (RCT), such as ABCA1 and ABCG1, has been reported to be deficient in foam SMCs. This combination of deregulated CE-enriched lipoprotein uptake and reduced free cholesterol export significantly contributes to the high percentage of foam SMCs in human atherosclerotic plaques.
MLCL: Conceptualization, Writing – original draft, Writing – review & editing. AM: Conceptualization, Writing – original draft, Resources, Validation. LC: Writing – original draft, Writing – review & editing. EG: Writing – original draft, Data curation, Software. AB-A: Writing – original draft. AP: Writing – original draft, Validation. JP: Writing – original draft, Investigation, Methodology, Resources, Software, Writing – review & editing. DV: Investigation, Methodology, Resources, Writing – original draft. JG: Methodology, Conceptualization, Software, Writing – original draft. FS: Conceptualization, Investigation, Writing – original draft. NR: Investigation, Data curation, Writing – original draft, Writing – review & editing. JE-G: Writing – original draft, Writing – review & editing, Conceptualization, Supervision. VL-C: Conceptualization, Supervision, Writing – original draft, Writing – review & editing, Funding acquisition, Investigation, Resources.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
The economic support to develop this project was received from FIS PI21/01523 (to VLl-C) from the Instituto de Salud Carlos III (ISCIII) and co-financed with ERDFs, and Fundación BBVA Ayudas a equipos de investigación 2019. Aleyda Benitez Amaro (ABA) is a postdoctoral fellow (FI19/00205) that was granted by the Programme Contratos predoctorales de formación de investigación en salud_from the Instituto de Salud Carlos III (ISCIII) and co-financed with ERDFs. The RIFS methodology required to analyze mitochondrial respiration in frozen cardiac samples was developed thanks to the AYUDAS PARA LA MOVILIDAD DE PERSONAL INVESTIGADOR CONTRATADO EN EL MARCO DE LA AES (M-AES) del ISCIII granted by ABA (MV21/00060). Our group is part of CIBER Enfermedades Cardiovasculares (CIBERCV; CB16/11/00276 to VLl-C), and CIBER de Diabetes y Enfermedades Metabólicas Asociadas (CIBERDEM; CB07/08/016 to JC-EG and NR), projects run by the Instituto de Salud Carlos III. Our group also participates in Redes de investigación (Enfermedades Metabóloicas y Cáncer RED2018-102799-T), a project run by MINECO. We belong to a group recognized by Generalitat de Catalunya (2021 SGR 00834). This work was also funded by Ministerio de Ciencia, Innovación y Universidades (PID2022-137186OB-100), as well as from the Agencia Estatal de Investigación (AEI/10.13039/501100011033) within the Subprograma Ramón y Cajal (RYC-201722879) to NR. The IR SANT PAU is a centre of CERCA Programme/Generalitat de Catalunya
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AgLDL, aggregated LDL; ALA, alpha-linolenic acid; ANGPTL4, angiopoietin-like 4; Apo, apolipoproteins (ApoB100, ApoA1, ApoC1, ApoJ, ApoE); APRIL, A proliferation-inducing ligand; ABC, ATP-binding cassette (ABCG1 and ABCA1); bFGF, basic fibroblast growth factor; CAD, Coronary Artery Disease; cAMP, 3′-5′-cyclic adenosine monophosphate; CAMK2d, calcium/calmodulin dependent protein kinase II delta; CVD, Cardiovascular disease; CE, Cholesteryl Ester; CS, Chondroitin sulfate; Cer, Ceramides; ELK1, ETS Like-1 protein; GAG, Glycosaminoglycan; DDR1, discoidin receptor 1; DIM, Diffuse intimal thickening; ECM, extracellular matrix; FLD, familial LCAT deficiency; FC, Free Cholesterol; HB-EGF, heparin-binding epidermal growth factor-like growth factor; HDL, High Density Lipoprotein; IFNγ, Interferon-γ; IT, intimal thickening; KLF4, Krüppel-like factor 4; LAL, lysosomal acid lipase; LCAT, Lecithin cholesterol acyltransferase; LDL, Low-density lipoprotein; LRP1, Low-density lipoprotein receptor-related protein 1; MRLC, Myosin regulatory light chain; nc- APRIL, Non-canonical APRIL; NOX1, NADPH oxidase 1; oxLDL, oxidized LDL; PAI-1, Plasminogen activator inhibitor-1; PC, Phosphatidylcholine; PL, Phospholipids; PKA protein kinase A; PCSK9, Proprotein convertase subtilisin/kexin type 9; PG, Proteoglycans; PUFAs, Polyunsaturated fatty acids; P2Y2 and P2Y12, P2Y purinoreceptor 2 and 12; RA, Retinoic Acid; RCT, Reverse cholesterol transport; sLRP1, soluble LRP1; SREBP-1 and SREBP-2, Sterol regulatory element-binding protein-1 and 2; SM, Sphingomyelin; SMC, Smooth muscle cell; SSH2, Protein phosphatase Slingshot homolog 2; TF, Tissue Factor; TG, Triglycerides; TNFα, transforming necrosis factor alpha; VSMC, Vascular smooth muscle cell; VLDL, Very low-density lipoprotein.
1. Cardiovascular diseases (CVDs). Available online at: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed February 1, 2024).
2. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Prim. (2019) 5:1–18. doi: 10.1038/s41572-019-0106-z
3. Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. (1995) 15:551–62. doi: 10.1161/01.ATV.15.5.551
4. Olsson U, Camejo G, Hurt-Camejo E, Elfsber K, Wiklund O, Bondjers G. Possible functional interactions of apolipoprotein B-100 segments that associate with cell proteoglycans and the ApoB/E receptor. Arterioscler Thromb Vasc Biol. (1997) 17:149–55. doi: 10.1161/01.ATV.17.1.149
5. Paananen K, Saarinen J, Annila A, Kovanen PT. Proteolysis and fusion of low density lipoprotein particles strengthen their binding to human aortic proteoglycans. J Biol Chem. (1995) 270:12257–62. doi: 10.1074/JBC.270.20.12257
6. Hurt-Camejo E, Rosengren B, Sartipy P, Elfsberg K, Camejo G, Svensson L. CD44, A cell surface chondroitin sulfate proteoglycan, mediates binding of interferon-gamma and some of its biological effects on human vascular smooth muscle cells. J Biol Chem. (1999) 274:18957–64. doi: 10.1074/JBC.274.27.18957
7. Francis GA. The greatly under-represented role of smooth muscle cells in atherosclerosis. Curr Atheroscler Rep. (2023) 25:741–9. doi: 10.1007/S11883-023-01145-8/FIGURES/2
8. Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. (2013) 13:709–21. doi: 10.1038/nri3520
9. Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. (2014) 129:1551–9. doi: 10.1161/CIRCULATIONAHA.113.005015
10. Wang Y, Dubland JA, Allahverdian S, Asonye E, Sahin B, Jaw JE, et al. Smooth muscle cells contribute the majority of foam cells in ApoE (apolipoprotein E)-deficient mouse atherosclerosis. Arterioscler Thromb Vasc Biol. (2019) 39:876–87. doi: 10.1161/ATVBAHA.119.312434
11. Llorente-Cortés V, Martínez-González J, Badimon L. LDL receptor–related protein mediates uptake of aggregated LDL in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. (2000) 20:1572–9. doi: 10.1161/01.ATV.20.6.1572
12. Llorente-Cortés V, Otero-Viñas M, Hurt-Camejo E, Martínez-González J, Badimon L. Human coronary smooth muscle cells internalize versican-modified LDL through LDL receptor–related protein and LDL receptors. Arterioscler Thromb Vasc Biol. (2002) 22:387–93. doi: 10.1161/HQ0302.105367
13. Llorente-Cortés V, Otero-Viñas M, Camino-López S, Costales P, Badimon L. Cholesteryl esters of aggregated LDL are internalized by selective uptake in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. (2006) 26:117–23. doi: 10.1161/01.ATV.0000193618.32611.8B
14. Rong JX, Shapiro M, Trogan E, Fisher EA. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc Natl Acad Sci USA. (2003) 100:13531–6. doi: 10.1073/PNAS.1735526100
15. Bennett MR, Sinha S, Owens GK, Libby P, Bornfeldt KE, Tall AR. Vascular smooth muscle cells in atherosclerosis. Circ Res. (2016) 118:692–702. doi: 10.1161/CIRCRESAHA.115.306361
16. Grootaert MOJ, Bennett MR. Vascular smooth muscle cells in atherosclerosis: time for a re-assessment. Cardiovasc Res. (2021) 117:2326–39. doi: 10.1093/CVR/CVAB046
17. Alencar GF, Owsiany KM, Karnewar S, Sukhavasi K, Mocci G, Nguyen AT, et al. Stem cell pluripotency genes Klf4 and Oct4 regulate complex SMC phenotypic changes critical in late-stage atherosclerotic lesion pathogenesis. Circulation. (2020) 142:2045–59. doi: 10.1161/CIRCULATIONAHA.120.046672
18. Basatemur GL, Jørgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol. (2019) 16:727–44. doi: 10.1038/S41569-019-0227-9
19. Solomon CU, McVey DG, Andreadi C, Gong P, Turner L, Stanczyk PJ, et al. Effects of coronary artery disease-associated variants on vascular smooth muscle cells. Circulation. (2022) 146:917–29. doi: 10.1161/CIRCULATIONAHA.121.058389
20. Aherrahrou R, Lue D, Perry RN, Aberra YT, Khan MD, Soh JY, et al. Genetic regulation of SMC gene expression and splicing predict causal CAD genes. Circ Res. (2023) 132:323–38. doi: 10.1161/CIRCRESAHA.122.321586
21. Davis-Dusenbery BN, Wu C, Hata A. Micromanaging vascular smooth muscle cell differentiation and phenotypic modulation. Arterioscler Thromb Vasc Biol. (2011) 31:2370–7. doi: 10.1161/ATVBAHA.111.226670
22. Feil S, Fehrenbacher B, Lukowski R, Essmann F, Schulze-Osthoff K, Schaller M, et al. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ Res. (2014) 115:662–7. doi: 10.1161/CIRCRESAHA.115.304634/-/DC1
23. Fukumoto Y, Libby P, Rabkin E, Hill CC, Enomoto M, Hirouchi Y, et al. Statins alter smooth muscle cell accumulation and collagen content in established atheroma of watanabe heritable hyperlipidemic rabbits. Circulation. (2001) 103:993–9. doi: 10.1161/01.CIR.103.7.993
24. Son BK, Kozaki K, Iijima K, Eto M, Kojima T, Ota H, et al. Statins protect human aortic smooth muscle cells from inorganic phosphate-induced calcification by restoring Gas6-Axl survival pathway. Circ Res. (2006) 98:1024–31. doi: 10.1161/01.RES.0000218859.90970.8D
25. Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. (2015) 21:628–37. doi: 10.1038/nm.3866
26. Cherepanova OA, Gomez D, Shankman LS, Swiatlowska P, Williams J, Sarmento OF, et al. Activation of the pluripotency factor OCT4 in smooth muscle cells is atheroprotective. Nat Med. (2016) 22:657–65. doi: 10.1038/nm.4109
27. Wirka RC, Wagh D, Paik DT, Pjanic M, Nguyen T, Miller CL, et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med. (2019) 25:1280–9. doi: 10.1038/s41591-019-0512-5
28. Zhou W, Bai Y, Chen J, Li H, Zhang B, Liu H. Revealing the critical regulators of modulated smooth muscle cells in atherosclerosis in mice. Front Genet. (2022) 13:900358. doi: 10.3389/FGENE.2022.900358
29. Cho DI, Ahn MJ, Cho HH, Cho M, Jun JH, Kang BG, et al. ANGPTL4 stabilizes atherosclerotic plaques and modulates the phenotypic transition of vascular smooth muscle cells through KLF4 downregulation. Exp Mol Med. (2023) 55:426–42. doi: 10.1038/S12276-023-00937-X
30. Haskins RM, Nguyen AT, Alencar GF, Billaud M, Kelly-Goss MR, Good ME, et al. Klf4 has an unexpected protective role in perivascular cells within the microvasculature. Am J Physiol Heart Circ Physiol. (2018) 315:H402–14. doi: 10.1152/AJPHEART.00084.2018
31. Hergenreider E, Heydt S, Tréguer K, Boettger T, Horrevoets AJG, Zeiher AM, et al. Atheroprotective communication between endothelial cells and smooth muscle cells through miRNAs. Nat Cell Biol. (2012) 14:249–56. doi: 10.1038/NCB2441
32. Cordes KR, Sheehy NT, White MP, Berry EC, Morton SU, Muth AN, et al. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. (2009) 460:705–10. doi: 10.1038/NATURE08195
33. Xin M, Small EM, Sutherland LB, Qi X, McAnally J, Plato CF, et al. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. (2009) 23:2166–78. doi: 10.1101/GAD.1842409
34. Sala F, Aranda JF, Rotllan N, Ramírez CM, Aryal B, Elia L, et al. MiR-143/145 deficiency protects against progression of atherosclerosis in Ldlr−/− mice. Thromb Haemost. (2014) 112:796. doi: 10.1160/TH13-11-0905
35. Cipollone F, Felicioni L, Sarzani R, Ucchino S, Spigonardo F, Mandolini C, et al. A unique microRNA signature associated with plaque instability in humans. Stroke. (2011) 42:2556–63. doi: 10.1161/STROKEAHA.110.597575
36. Rykaczewska U, Zhao Q, Saliba-Gustafsson P, Lengquist M, Kronqvist M, Bergman O, et al. Plaque evaluation by ultrasound and transcriptomics reveals BCLAF1 as a regulator of smooth muscle cell lipid transdifferentiation in atherosclerosis. Arterioscler Thromb Vasc Biol. (2022) 42:659–76. doi: 10.1161/ATVBAHA.121.317018
37. Neuville P, Yan ZQ, Gidlöf A, Pepper MS, Hansson GK, Gabbiani G, et al. Retinoic acid regulates arterial smooth muscle cell proliferation and phenotypic features in vivo and in vitro through an RARalpha-dependent signaling pathway. Arterioscler Thromb Vasc Biol. (1999) 19:1430–6. doi: 10.1161/01.ATV.19.6.1430
38. Watanabe A, Kurabayashi M, Arai M, Sekiguchi K, Nagai R. Combined effect of retinoic acid and basic FGF on PAI-1 gene expression in vascular smooth muscle cells. Cardiovasc Res. (2001) 51:151–9. doi: 10.1016/S0008-6363(01)00274-7
39. Wang J, Xie SA, Li N, Zhang T, Yao W, Zhao H, et al. Matrix stiffness exacerbates the proinflammatory responses of vascular smooth muscle cell through the DDR1-DNMT1 mechanotransduction axis. Bioact Mater. (2022) 17:406–24. doi: 10.1016/J.BIOACTMAT.2022.01.012
40. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. (2004) 84:767–801. doi: 10.1152/PHYSREV.00041.2003
41. Peoples GE, Blotnick S, Takahashii K, Freeman MR, Klagsbrun M, Eberlein TJ. T lymphocytes that infiltrate tumors and atherosclerotic plaques produce heparin-binding epidermal growth factor-like growth factor and basic fibroblast growth factor: a potential pathologic role. Proc Natl Acad Sci USA. (1995) 92:6547–51. doi: 10.1073/PNAS.92.14.6547
42. Chen P, Qin L, Li G, Tellides G, Simons M. Smooth muscle FGF/TGFβ cross talk regulates atherosclerosis progression. EMBO Mol Med. (2016) 8:712–28. doi: 10.15252/EMMM.201506181
43. Tang Y, Urs S, Boucher J, Bernaiche T, Venkatesh D, Spicer DB, et al. Notch and transforming growth factor-β (TGFβ) signaling pathways cooperatively regulate vascular smooth muscle cell differentiation. J Biol Chem. (2010) 285:17556. doi: 10.1074/JBC.M109.076414
44. Yu XH, Zhang J, Zheng XL, Yang YH, Tang CK. Interferon-γ in foam cell formation and progression of atherosclerosis. Clin Chim Acta. (2015) 441:33–43. doi: 10.1016/J.CCA.2014.12.007
45. Michael DR, Ashlin TG, Davies CS, Gallagher H, Stoneman TW, Buckley ML, et al. Differential regulation of macropinocytosis in macrophages by cytokines: implications for foam cell formation and atherosclerosis⋆. Cytokine. (2013) 64:357–61. doi: 10.1016/J.CYTO.2013.05.016
46. Persson J, Nilsson J, Lindholm MW. Interleukin-1beta and tumour necrosis factor-alpha impede neutral lipid turnover in macrophage-derived foam cells. BMC Immunol. (2008) 9:70–81. doi: 10.1186/1471-2172-9-70
47. Cagnina A, Chabot O, Davin L, Lempereur M, Maréchal P, Oury C, et al. Atherosclerosis — an inflammatory disease. Rev Med Liege. (1999) 77:302–9. doi: 10.1056/NEJM199901143400207
48. Taniyama Y, Griendling KK. Reactive oxygen species in the vasculature. Hypertension. (2003) 42:1075–81. doi: 10.1161/01.HYP.0000100443.09293.4F
49. Alexander RW. Hypertension and the pathogenesis of atherosclerosis. Hypertension. (1995) 25:155–61. doi: 10.1161/01.HYP.25.2.155
50. Rensen SSM, Doevendans PAFM, Van Eys GJJM. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Netherlands Hear J. (2007) 15:100–8. doi: 10.1007/BF03085963/METRICS
51. Nishio E, Watanabe Y. The involvement of reactive oxygen species and arachidonic acid in alpha 1-adrenoceptor-induced smooth muscle cell proliferation and migration. Br J Pharmacol. (1997) 121:665–70. doi: 10.1038/SJ.BJP.0701171
52. Wang Z, Castresana MR, Newman WH. Reactive oxygen and NF-κB in VEGF-induced migration of human vascular smooth muscle cells. Biochem Biophys Res Commun. (2001) 285:669–74. doi: 10.1006/bbrc.2001.5232
53. Ammarguellat F, Llovera M, Kelly PA, Goffin V. Low doses of EPO activate MAP kinases but not JAK2-STAT5 in rat vascular smooth muscle cells. Biochem Biophys Res Commun. (2001) 284:1031–8. doi: 10.1006/BBRC.2001.5085
54. Grote K, Flach I, Luchtefeld M, Akin E, Holland SM, Drexler H, et al. Mechanical stretch enhances mRNA expression and proenzyme release of matrix metalloproteinase-2 (MMP-2) via NAD(P)H oxidase–derived reactive oxygen species. Circ Res. (2003) 92:e80–6. doi: 10.1161/01.RES.0000077044.60138.7C
55. Southgate KM, Davies M, Booth RFG, Newby AC. Involvement of extracellular-matrix-degrading metalloproteinases in rabbit aortic smooth-muscle cell proliferation. Biochem J. (1992) 288(Pt 1):93–9. doi: 10.1042/BJ2880093
56. Mason DP, Kenagy RD, Hasenstab D, Bowen-Pope DF, Seifert RA, Coats S, et al. Matrix metalloproteinase-9 overexpression enhances vascular smooth muscle cell migration and alters remodeling in the injured rat carotid artery. Circ Res. (1999) 85:1179–85. doi: 10.1161/01.RES.85.12.1179
57. Lassègue B, Clempus RE. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am J Physiol—Regul Integr Comp Physiol. (2003) 285:R277–97. doi: 10.1152/ajpregu.00758.2002
58. Konior A, Schramm A, Czesnikiewicz-Guzik M, Guzik TJ. NADPH Oxidases in vascular pathology. Antioxidants Redox Signal. (2014) 20:2794–814. doi: 10.1089/ars.2013.5607
59. Lee MY, Martin AS, Mehta PK, Dikalova AE, Garrido AM, Datla SR, et al. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler Thromb Vasc Biol. (2009) 29:480–7. doi: 10.1161/ATVBAHA.108.181925
60. Vendrov AE, Madamanchi NR, Hakim ZS, Rojas M, Runge MS. Thrombin and NAD(P)H oxidase-mediated regulation of CD44 and BMP4-Id pathway in VSMC, restenosis, and atherosclerosis. Circ Res. (2006) 98:1254–63. doi: 10.1161/01.RES.0000221214.37803.79
61. Vendrov AE, Madamanchi NR, Niu XL, Molnar KC, Runge M, Szyndralewiez C, et al. NADPH oxidases regulate CD44 and hyaluronic acid expression in thrombin-treated vascular smooth muscle cells and in atherosclerosis. J Biol Chem. (2010) 285:26545–57. doi: 10.1074/JBC.M110.143917
62. Sheehan AL, Carrell S, Johnson B, Stanic B, Banfi B, Miller FJ. Role for Nox1 NADPH oxidase in atherosclerosis. Atherosclerosis. (2011) 216:321–6. doi: 10.1016/J.ATHEROSCLEROSIS.2011.02.028
63. Ambasta RK, Schreiber JG, Janiszewski M, Busse R, Brandes RP. Noxa1 is a central component of the smooth muscle NADPH oxidase in mice. Free Radic Biol Med. (2006) 41:193–201. doi: 10.1016/J.FREERADBIOMED.2005.12.035
64. Niu XL, Madamanchi NR, Vendrov AE, Tchivilev I, Rojas M, Madamanchi C, et al. Nox activator 1. Circulation. (2010) 121:549–59. doi: 10.1161/CIRCULATIONAHA.109.908319
65. Vendrov AE, Sumida A, Canugovi C, Lozhkin A, Hayami T, Madamanchi NR, et al. NOXA1-dependent NADPH oxidase regulates redox signaling and phenotype of vascular smooth muscle cell during atherogenesis. Redox Biol. (2019) 21:101063. doi: 10.1016/j.redox.2018.11.021
66. Clempus RE, Sorescu D, Dikalova AE, Pounkova L, Jo P, Sorescu GP, et al. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol. (2007) 27:42–8. doi: 10.1161/01.ATV.0000251500.94478.18
67. Craige SM, Kant S, Reif M, Chen K, Pei Y, Angoff R, et al. Endothelial NADPH oxidase 4 protects ApoE−/− mice from atherosclerotic lesions. Free Radic Biol Med. (2015) 89:1–7. doi: 10.1016/j.freeradbiomed.2015.07.004
68. Di Marco E, Gray SP, Kennedy K, Szyndralewiez C, Lyle AN, Lassègue B, et al. NOX4-derived reactive oxygen species limit fibrosis and inhibit proliferation of vascular smooth muscle cells in diabetic atherosclerosis. Free Radic Biol Med. (2016) 97:556–67. doi: 10.1016/J.FREERADBIOMED.2016.07.013
69. Schürmann C, Rezende F, Kruse C, Yasar Y, Löwe O, Fork C, et al. The NADPH oxidase Nox4 has anti-atherosclerotic functions. Eur Heart J. (2015) 36:3447–56. doi: 10.1093/EURHEARTJ/EHV460
70. Tertov VV, Orekhov AN, Sobenin IA, Gabbasov ZA, Popov EG, Yaroslavov AA, et al. Three types of naturally occurring modified lipoproteins induce intracellular lipid accumulation due to lipoprotein aggregation. Circ Res. (1992) 71:218–28. doi: 10.1161/01.RES.71.1.218
71. Tertov VV, Sobenin IA, Gabbasov ZA, Popov EG, Orekhov AN. Lipoprotein aggregation as an essential condition of intracellular lipid accumulation caused by modified low density lipoproteins. Biochem Biophys Res Commun. (1989) 163:489–94. doi: 10.1016/0006-291X(89)92163-3
72. Ehsan Ismail NA, Alavi MZ, Moore S. Lipoprotein-proteoglycan complexes from injured rabbit aortas accelerate lipoprotein uptake by arterial smooth muscle cells. Atherosclerosis. (1994) 105:79–87. doi: 10.1016/0021-9150(94)90010-8
73. Ruuth M, Nguyen SD, Vihervaara T, Hilvo M, Laajala TD, Kondadi PK, et al. Susceptibility of low-density lipoprotein particles to aggregate depends on particle lipidome, is modifiable, and associates with future cardiovascular deaths. Eur Heart J. (2018) 39:2562–73. doi: 10.1093/EURHEARTJ/EHY319
74. Ruuth M, Äikäs L, Tigistu-Sahle F, Käkelä R, Lindholm H, Simonen P, et al. Plant lipoprotein) stanol aggregation esters reduce by LDL altering (low-density LDL surface lipids. Arterioscler Thromb Vasc Biol. (2020) 40:2310–21. doi: 10.1161/ATVBAHA.120.314329
75. Hilvo M, Simolin H, Metso J, Ruuth M, Öörni K, Jauhiainen M, et al. PCSK9 inhibition alters the lipidome of plasma and lipoprotein fractions. Atherosclerosis. (2018) 269:159–65. doi: 10.1016/J.ATHEROSCLEROSIS.2018.01.004
76. Ruuth M, Lahelma M, Luukkonen PK, Lorey MB, Qadri S, Sädevirta S, et al. Overfeeding saturated fat increases LDL (low-density lipoprotein) aggregation susceptibility while overfeeding unsaturated fat decreases proteoglycan-binding of lipoproteins. Arterioscler Thromb Vasc Biol. (2021) 41:2823–36. doi: 10.1161/ATVBAHA.120.315766
77. Erkkilä AT, Manninen S, Fredrikson L, Bhalke M, Holopainen M, Ruuth M, et al. Lipidomic changes of LDL after consumption of camelina sativa oil, fatty fish and lean fish in subjects with impaired glucose metabolism—a randomized controlled trial. J Clin Lipidol. (2021) 15:743–51. doi: 10.1016/J.JACL.2021.08.060
78. Benitez-Amaro A, Solanelles Curco S, Garcia E, Julve J, Rives J, Benitez S, et al. Apolipoprotein and LRP1-based peptides as new therapeutic tools in atherosclerosis. J Clin Med. (2021) 10:3571–90. doi: 10.3390/JCM10163571
79. Nguyen SD, Javanainen M, Rissanen S, Zhao H, Huusko J, Kivelä AM, et al. Apolipoprotein A-I mimetic peptide 4F blocks sphingomyelinase-induced LDL aggregation. J Lipid Res. (2015) 56:1206–21. doi: 10.1194/JLR.M059485
80. Martínez-Bujidos M, Rull A, González-Cura B, Pérez-Cuéllar M, Montoliu-Gaya L, Villegas S, et al. Clusterin/apolipoprotein J binds to aggregated LDL in human plasma and plays a protective role against LDL aggregation. FASEB J. (2015) 29:1688–700. doi: 10.1096/FJ.14-264036
81. Rivas-Urbina A, Rull A, Montoliu-Gaya L, Pérez-Cuellar M, Ordóñez-Llanos J, Villegas S, et al. Low-density lipoprotein aggregation is inhibited by apolipoprotein J-derived mimetic peptide D-[113–122]apoJ. Biochim Biophys Acta Mol Cell Biol Lipids. (2020) 1865:158541. doi: 10.1016/J.BBALIP.2019.158541
82. Benitez-Amaro A, Pallara C, Nasarre L, Rivas-Urbina A, Benitez S, Vea A, et al. Molecular basis for the protective effects of low-density lipoprotein receptor-related protein 1 (LRP1)-derived peptides against LDL aggregation. Biochim Biophys Acta Biomembr. (2019) 1861:1302–16. doi: 10.1016/J.BBAMEM.2019.05.003
83. Benitez-Amaro A, Pallara C, Nasarre L, Ferreira R, de Gonzalo-Calvo D, Prades R, et al. Development of innovative antiatherosclerotic peptides through the combination of molecular modeling and a dual (biochemical-cellular) screening system. Adv Ther. (2020) 3:2000037. doi: 10.1002/ADTP.202000037
84. Allahverdian S, Ortega C, Francis GA. Smooth muscle cell-proteoglycan-lipoprotein interactions as drivers of atherosclerosis. Handb Exp Pharmacol. (2022) 270:335–58. doi: 10.1007/164_2020_364
85. Delgado-Roche L, Brito V, Acosta E, Pérez A, Fernández JR, Hernández-Matos Y, et al. Arresting progressive atherosclerosis by immunization with an anti-glycosaminoglycan monoclonal antibody in apolipoprotein E-deficient mice. Free Radic Biol Med. (2015) 89:557–66. doi: 10.1016/J.FREERADBIOMED.2015.08.027
86. Figueroa JE, Vijayagopal P. Angiotensin II stimulates synthesis of vascular smooth muscle cell proteoglycans with enhanced low density lipoprotein binding properties. Atherosclerosis. (2002) 162:261–8. doi: 10.1016/S0021-9150(01)00714-6
87. Ballinger ML, Nigro J, Frontanilla KV, Dart AM, Little PJ. Regulation of glycosaminoglycan structure and atherogenesis. Cell Mol Life Sci. (2004) 61:1296–306. doi: 10.1007/S00018-004-3389-4
88. Soto Y, Acosta E, Delgado L, Pérez A, Falcón V, Bécquer MA, et al. Antiatherosclerotic effect of an antibody that binds to extracellular matrix glycosaminoglycans. Arterioscler Thromb Vasc Biol. (2012) 32:595–604. doi: 10.1161/ATVBAHA.111.238659
89. Brito V, Mellal K, Portelance SG, Pérez A, Soto Y, Deblois D, et al. Induction of anti-anti-idiotype antibodies against sulfated glycosaminoglycans reduces atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. (2012) 32:2847–54. doi: 10.1161/ATVBAHA.112.300444
90. Afroz R, Cao Y, Rostam MA, Ta H, Xu S, Zheng W, et al. Signalling pathways regulating galactosaminoglycan synthesis and structure in vascular smooth muscle: implications for lipoprotein binding and atherosclerosis. Pharmacol Ther. (2018) 187:88–97. doi: 10.1016/J.PHARMTHERA.2018.02.005
91. Fredrikson GN, Söderberg I, Lindholm M, Dimayuga P, Chyu KY, Shah PK, et al. Inhibition of atherosclerosis in apoE-null mice by immunization with apoB-100 peptide sequences. Arterioscler Thromb Vasc Biol. (2003) 23:879–84. doi: 10.1161/01.ATV.0000067937.93716.DB
92. Borén J, Olin K, Lee I, Chait A, Wight TN, Innerarity TL. Identification of the principal proteoglycan-binding site in LDL. A single-point mutation in apo-B100 severely affects proteoglycan interaction without affecting LDL receptor binding. J Clin Invest. (1998) 101:2658–64. doi: 10.1172/JCI2265
93. Skålén K, Gustafsson M, Knutsen Rydberg E, Hultén LM, Wiklund O, Innerarity TL, et al. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. (2002) 417:750–4. doi: 10.1038/NATURE00804
94. Ingold K, Zumsteg A, Tardivel A, Huard B, Steiner QG, Cachero TG, et al. Identification of proteoglycans as the APRIL-specific binding partners. J Exp Med. (2005) 201:1375–83. doi: 10.1084/JEM.20042309
95. Tsiantoulas D, Eslami M, Obermayer G, Clement M, Smeets D, Mayer FJ, et al. APRIL limits atherosclerosis by binding to heparan sulfate proteoglycans. Nature. (2021) 597:92–6. doi: 10.1038/S41586-021-03818-3
96. Cohen P, Cross D, Jänne PA. Kinase drug discovery 20 years after imatinib: progress and future directions. Nat Rev Drug Discov. (2021) 20:551–69. doi: 10.1038/S41573-021-00195-4
97. Getachew R, Ballinger ML, Burch ML, Little PJ, Osman N. Characterisation of Ki11502 as a potent inhibitor of PDGF beta receptor-mediated proteoglycan synthesis in vascular smooth muscle cells. Eur J Pharmacol. (2010) 626:186–92. doi: 10.1016/J.EJPHAR.2009.09.066
98. Ballinger ML, Osman N, Hashimura K, Haan JB, Jandeleit-Dahm K, Allen T, et al. Imatinib inhibits vascular smooth muscle proteoglycan synthesis and reduces LDL binding in vitro and aortic lipid deposition in vivo. J Cell Mol Med. (2010) 14:1408–18. doi: 10.1111/J.1582-4934.2009.00902.X
99. Williams JW, Huang Lh, Randolph GJ. Cytokine circuits in cardiovascular disease. Immunity. (2019) 50:941–54. doi: 10.1016/J.IMMUNI.2019.03.007
100. Palinski W, Miller E, Witztum JL. Immunization of low density lipoprotein (LDL) receptor-deficient rabbits with homologous malondialdehyde-modified LDL reduces atherogenesis. Proc Natl Acad Sci USA. (1995) 92:821–5. doi: 10.1073/PNAS.92.3.821
101. Ameli S, Hultgårdh-Nilsson A, Regnström J, Calara F, Yano J, Cercek B, et al. Effect of immunization with homologous LDL and oxidized LDL on early atherosclerosis in hypercholesterolemic rabbits. Arterioscler Thromb Vasc Biol. (1996) 16:1074–9. doi: 10.1161/01.ATV.16.8.1074
102. Schiopu A, Bengtsson J, Söderberg I, Janciauskiene S, Lindgren S, Mikko AP, et al. Recombinant human antibodies against aldehyde-modified apolipoprotein B-100 peptide sequences inhibit atherosclerosis. Circulation. (2004) 110:2047–52. doi: 10.1161/01.CIR.0000143162.56057.B5
103. Schiopu A, Frendéus B, Jansson B, Söderberg I, Ljungcrantz I, Araya Z, et al. Recombinant antibodies to an oxidized low-density lipoprotein epitope induce rapid regression of atherosclerosis in apobec-1(−/−)/low-density lipoprotein receptor(−/−) mice. J Am Coll Cardiol. (2007) 50:2313–8. doi: 10.1016/J.JACC.2007.07.081
104. Tsimikas S, Miyanohara A, Hartvigsen K, Merki E, Shaw PX, Chou MY, et al. Human oxidation-specific antibodies reduce foam cell formation and atherosclerosis progression. J Am Coll Cardiol. (2011) 58:1715–27. doi: 10.1016/J.JACC.2011.07.017
105. Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. (2005) 23:1126–36. doi: 10.1038/NBT1142
106. Zhang L, Xue S, Ren F, Huang S, Zhou R, Wang Y, et al. An atherosclerotic plaque-targeted single-chain antibody for MR/NIR-II imaging of atherosclerosis and anti-atherosclerosis therapy. J Nanobiotechnology. (2021) 19:296–315. doi: 10.1186/S12951-021-01047-4
107. Haberland ME, Mottino G, Le M, Frank JS. Sequestration of aggregated LDL by macrophages studied with freeze-etch electron microscopy. J Lipid Res. (2001) 42:605–19. doi: 10.1016/S0022-2275(20)31170-6
108. Grosheva I, Haka AS, Qin C, Pierini LM, Maxfield FR. Aggregated LDL in contact with macrophages induces local increases in free cholesterol levels that regulate local actin polymerization. Arterioscler Thromb Vasc Biol. (2009) 29:1615–21. doi: 10.1161/ATVBAHA.109.191882
109. Llorente-Cortés V, Royo T, Juan-Babot O, Badimon L. Adipocyte differentiation-related protein is induced by LRP1-mediated aggregated LDL internalization in human vascular smooth muscle cells and macrophages. J Lipid Res. (2007) 48:2133–40. doi: 10.1194/JLR.M700039-JLR200
110. Costales P, Fuentes-Prior P, Castellano J, Revuelta-Lopez E, Corral-Rodríguez MÁ, Nasarre L, et al. K domain CR9 of low density lipoprotein (LDL) receptor-related protein 1 (LRP1) is critical for aggregated LDL-induced foam cell formation from human vascular smooth muscle cells. J Biol Chem. (2015) 290:14852–65. doi: 10.1074/JBC.M115.638361
111. Bornachea O, Benitez-Amaro A, Vea A, Nasarre L, de Gonzalo-Calvo D, Escola-Gil JC, et al. Immunization with the Gly1127-Cys1140 amino acid sequence of the LRP1 receptor reduces atherosclerosis in rabbits. Molecular, immunohistochemical and nuclear imaging studies. Theranostics. (2020) 10:3263–80. doi: 10.7150/THNO.37305
112. Dissmore T, Seye CI, Medeiros DM, Weisman GA, Bardford B, Mamedova L. The P2Y2 receptor mediates uptake of matrix-retained and aggregated low density lipoprotein in primary vascular smooth muscle cells. Atherosclerosis. (2016) 252:128–35. doi: 10.1016/J.ATHEROSCLEROSIS.2016.07.927
113. Chen J, Pi S, Yu C, Shi H, Liu Y, Guo X, et al. SLRP1 (soluble low-density lipoprotein receptor-related protein 1): a novel biomarker for P2Y12 (P2Y purinoceptor 12) receptor expression in atherosclerotic plaques. Arterioscler Thromb Vasc Biol. (2020) 40:E166–79. doi: 10.1161/ATVBAHA.120.314350
114. He C, Hu X, Weston TA, Jung RS, Sandhu J, Huang S, et al. Macrophages release plasma membrane-derived particles rich in accessible cholesterol. Proc Natl Acad Sci USA. (2018) 115:E8499–508. doi: 10.1073/PNAS.1810724115/SUPPL_FILE/PNAS.1810724115.SAPP.PDF
115. Hu X, Weston TA, He C, Jung RS, Heizer PJ, Young BD, et al. Release of cholesterol-rich particles from the macrophage plasma membrane during movement of filopodia and lamellipodia. Elife. (2019) 8:e50231. doi: 10.7554/ELIFE.50231
116. Dubland JA, Allahverdian S, Besler KJ, Ortega C, Wang Y, Pryma CS, et al. Low LAL (lysosomal acid lipase) expression by smooth muscle cells relative to macrophages as a mechanism for arterial foam cell formation. Arterioscler Thromb Vasc Biol. (2021) 41:E354–68. doi: 10.1161/ATVBAHA.120.316063
117. Phillips MC. Molecular mechanisms of cellular cholesterol efflux. J Biol Chem. (2014) 289:24020–9. doi: 10.1074/JBC.R114.583658
118. Adorni MP, Zimetti F, Billheimer JT, Wang N, Rader DJ, Phillips MC, et al. The roles of different pathways in the release of cholesterol from macrophages. J Lipid Res. (2007) 48:2453–62. doi: 10.1194/JLR.M700274-JLR200
119. He C, Jiang H, Song W, Riezman H, Tontonoz P, Weston TA, et al. Cultured macrophages transfer surplus cholesterol into adjacent cells in the absence of serum or high-density lipoproteins. Proc Natl Acad Sci USA. (2020) 117:10476–83. doi: 10.1073/PNAS.1922879117
120. Choi HY, Rahmani M, Wong BW, Allahverdian S, McManus BM, Pickering JG, et al. ATP-binding cassette transporter A1 expression and apolipoprotein A-I binding are impaired in intima-type arterial smooth muscle cells. Circulation. (2009) 119:3223–31. doi: 10.1161/CIRCULATIONAHA.108.841130
121. Conklin AC, Nishi H, Schlamp F, Ord T, Ounap K, Kaikkonen MU, et al. Meta-analysis of smooth muscle lineage transcriptomes in atherosclerosis and their relationships to in vitro models. Immunometabolism. (2021) 3:e210022–53. doi: 10.20900/IMMUNOMETAB20210022
122. Vengrenyuk Y, Nishi H, Long X, Ouimet M, Savji N, Martinez FO, et al. Cholesterol loading reprograms the microRNA-143/145-myocardin axis to convert aortic smooth muscle cells to a dysfunctional macrophage-like phenotype. Arterioscler Thromb Vasc Biol. (2015) 35:535–46. doi: 10.1161/ATVBAHA.114.304029/-/DC1
123. Oladosu O, Esobi IC, Powell RR, Bruce T, Stamatikos A. Dissecting the impact of vascular smooth muscle cell ABCA1 versus ABCG1 expression on cholesterol efflux and macrophage-like cell transdifferentiation: the role of SR-BI. J Cardiovasc Dev Dis. (2023) 10:416–28. doi: 10.3390/JCDD10100416
124. Ossoli A, Simonelli S, Vitali C, Franceschini G, Calabresi L. Role of LCAT in atherosclerosis. J Atheroscler Thromb. (2016) 23:119–27. doi: 10.5551/JAT.32854
125. Rousset X, Vaisman B, Auerbach B, Krause BR, Homan R, Stonik J, et al. Effect of recombinant human lecithin-cholesterol:acyltransferase infusion on lipoprotein metabolism in mice. J Pharmacol Exp Ther. (2010) 335:140–8. doi: 10.1124/JPET.110.169540
Keywords: apolipoproteins (ApoB100, apoA1, apoC1, apoJ, apoE), foam-like SMC, atherosclerosis, cardiovascular diseases, peptidomimetics, lipoproteins, transcription factors, reverse cholesterol transport
Citation: La Chica Lhoëst MT, Martinez A, Claudi L, Garcia E, Benitez-Amaro A, Polishchuk A, Piñero J, Vilades D, Guerra JM, Sanz F, Rotllan N, Escolà-Gil JC and Llorente-Cortés V (2024) Mechanisms modulating foam cell formation in the arterial intima: exploring new therapeutic opportunities in atherosclerosis. Front. Cardiovasc. Med. 11:1381520. doi: 10.3389/fcvm.2024.1381520
Received: 20 February 2024; Accepted: 28 May 2024;
Published: 17 June 2024.
Edited by:
Hong Jin, Karolinska Institutet (KI), SwedenReviewed by:
Robert C. Bauer, Columbia University, United States© 2024 La Chica Lhoëst, Martinez, Claudi, Garcia, Benitez-Amaro, Polishchuk, Piñero, Vilades, Guerra, Sanz, Rotllan, Escolà-Gil and Llorente-Cortés. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: V. Llorente-Cortés, dmljZW50YS5sbG9yZW50ZUBpaWJiLmNzaWMuZXM=; Y2xsb3JlbnRlQHNhbnRwYXUuY2F0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.