
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 15 July 2024
Sec. Cardiovascular Metabolism
Volume 11 - 2024 | https://doi.org/10.3389/fcvm.2024.1379930
This article is part of the Research Topic New Discoveries on Calcium Handling in Cardiovascular Pathology View all 7 articles
 Theresa Gissibl
Theresa Gissibl Laura Stengel
Laura Stengel Daniel Tarnowski
Daniel Tarnowski Lars S. Maier
Lars S. Maier Stefan WagnerAnna-Lena Feder†
Stefan WagnerAnna-Lena Feder† Can Martin Sag*†
Can Martin Sag*†
Background: Acute stimulation of the late sodium current (INaL) as pharmacologically induced by Anemonia toxin II (ATX-II) results in Na+-dependent Ca2+ overload and enhanced formation of reactive oxygen species (ROS). This is accompanied by an acute increase in the amplitude of the systolic Ca2+ transient. Ca2+ transient amplitude is determined by L-type Ca2+-mediated transsarcolemmal Ca2+ influx (ICa) into the cytosol and by systolic Ca2+ release from the sarcoplasmic reticulum (SR). Type-1 protein kinase A (PKARIα) becomes activated upon increased ROS and is capable of stimulating ICa, thereby sustaining the amplitude of the systolic Ca2+ transient upon oxidative stress.
Objectives: We aimed to investigate whether the increase of the systolic Ca2+ transient as acutely induced by INaL (by ATX-II) may involve stimulation of ICa through oxidized PKARIα.
Methods: We used a transgenic mouse model in which PKARIα was made resistant to oxidative activation by homozygous knock-in replacement of redox-sensitive Cysteine 17 with Serine within the regulatory subunits of PKARIα (KI). ATX-II (at 1 nmol/L) was used to acutely enhance INaL in freshly isolated ventricular myocytes from KI and wild-type (WT) control mice. Epifluorescence and confocal imaging were used to assess intracellular Ca2+ handling and ROS formation. A ruptured-patch whole-cell voltage-clamp was used to measure INaL and ICa. The impact of acutely enhanced INaL on RIα dimer formation and PKA target structures was studied using Western blot analysis.
Results: ATX-II increased INaL to a similar extent in KI and WT cells, which was associated with significant cytosolic and mitochondrial ROS formation in both genotypes. Acutely activated Ca2+ handling in terms of increased Ca2+ transient amplitudes and elevated SR Ca2+ load was equally present in KI and WT cells. Likewise, cellular arrhythmias as approximated by non-triggered Ca2+ elevations during Ca2+ transient decay and by diastolic SR Ca2+-spark frequency occurred in a comparable manner in both genotypes. Most importantly and in contrast to our initial hypothesis, ATX-II did not alter the magnitude or inactivation kinetics of ICa in neither WT nor KI cells and did not result in PKARIα dimerization (i.e., oxidation) despite a clear prooxidant intracellular environment.
Conclusions: The inotropic and arrhythmogenic effects of acutely increased INaL are associated with elevated ROS, but do not involve oxidation of PKARIα.
Impaired intracellular sodium (Na+) and calcium (Ca2+) handling is a key feature of heart failure (HF) contributing to both systolic and diastolic dysfunctions as well as to cardiac arrhythmias (1). An important component of altered Na+ handling in HF is the late sodium current INaL. Most Na+ channels open transiently during the upstroke of the action potential and then close rapidly, forming the peak flow required for action potential generation and conduction. A few Na+ channels, however, fail to inactivate and thus enable the so-called late sodium current (INaL) to persist over hundreds of milliseconds during the plateau phase of the action potential (2). In HF, enhanced INaL (3) contributes to increased Na+ influx into the cell, resulting in prolongation of action potential duration and arrhythmogenesis (4). In addition, intracellular Na+ is closely linked to Ca2+ handling via the Na+-Ca2+ exchanger (NCX). Increased intracellular Na+ concentration [Na+]i leads to a reduced electrochemical gradient for Ca2+ extrusion. Instead, the NCX operates in a “reverse mode” through which Ca2+ is transported into the cell and Na+ out of it (5). The subsequent excessive Ca2+ influx into the cell contributes to diastolic Ca2+ overload in chronic HF (6), as well as to arrhythmias (2) and contractile dysfunction (7). On the cellular level, INaL-related Na+-dependent Ca2+ overload results in pathological overactivation of protein kinases including Ca2+/Calmodulin-dependent protein kinase II (CaMKII) (8) and cAMP-dependent protein kinase A (PKA) (9), which is accompanied by proarrhythmic diastolic Ca2+ loss from the sarcoplasmic reticulum (SR), and mitochondrial reactive oxygen species (ROS) formation (10, 11).
Increased ROS formation results in oxidative stress when ROS levels exceed the antioxidative capacity of myocytes, which represents a well-established pathomechanism of various cardiac diseases including HF (12, 13). ROS serve as signaling molecules under physiological and disease-related conditions and directly influence the function of various channels and transporters in the heart through oxidation, such as those of the ryanodine receptor (RyR2) or SERCA2a (14). Besides, modulation of kinase function through oxidation has been reported for, e.g., CaMKII (15) and type I PKA (16), both of which influence the function of target structures. In that regard, Viatchenko-Karpinski et al. have already demonstrated that increased INaL with subsequent cytosolic Na+ and Ca2+ overload leads to oxidation of CaMKII resulting in dysregulation of Ca2+ handling (17). Moreover, Eiringhaus et al. reported that PKA activity was also increased upon elevated INaL as induced experimentally by Anemonia sulcata toxin II (ATX-II), which was associated with an acute positive inotropic effect that could be blocked pharmacologically using the PKA-inhibitor H89 (9). However, the authors did not assess whether increased PKA activity following ATX-II is solely due to a cAMP-dependent mechanism or may also involve oxidative activation.
PKA is the main effector of beta-adrenergic stimulation in the heart and gets activated in a cAMP-dependent manner. It is composed of two regulatory (R) and two catalytic (C) subunits. Depending on the R subunit, RI or RII, the enzyme is defined as type I or type II PKA (18). Brennan et al. were the first to demonstrate a cAMP-independent activation of type I PKA upon elevated ROS induced by H2O2 exposure. The subunit RIα contains redox-sensitive Cysteine residues forming interprotein disulfide bonds upon oxidation resulting in dimerization of the regulatory subunits and subsequent kinase activation independent of cAMP (16). The pathophysiological significance of oxidative activation of PKARIα in the complex interaction of the various redox-sensitive mechanisms in cardiomyocytes is still a subject of intense research. The clinical relevance of PKARIα has already been shown for tumor-, vascular endothelial growth factor (VEGF)-, and ischemia-induced angiogenesis (19) as well as for platelet-derived growth factor (PDGF) signaling pathways in renal mesangial cells (20). Recent research has focused on elucidating the role of redox-dependent activation of PKARIα in cardiomyocytes. In that regard, Trum et al. found that redox-activated PKARIα inhibits potassium channels and thereby contributes to early after depolarizations suggesting a harmful role for PKARIα in cardiac physiology (21). By contrast, Simon et al. showed that oxidative activation of PKARIα exerts a protective effect in the context of ischemia–reperfusion damage, presumably by inhibiting excessive lysosomal-mediated Ca2+ release (22). In line with this, we have recently found protective effects of oxidized PKARIα with respect to cardiocellular and ventricular contractile functions in the context of acute and chronic oxidative stress through enhanced L-type Ca2+ channel (LTCC) mediated Ca2+ influx (ICa) into ventricular myocytes that results in partially maintained systolic Ca2+ transients despite oxidative stress (23).
However, it is not clear if and to what extent ROS formation may lead to oxidation of PKARIα upon acute ATX-II-mediated Na+/Ca2+ overload and whether oxidized PKARIα is required for the inotropic and arrhythmogenic effects of ATX-II as reported earlier (9). Therefore, we made use of a genetically altered redox-dead mouse model with homozygous knock-in replacement of a redox-sensitive Cysteine 17 with Serine within the regulatory subunits of PKARIα (KI) (19, 22, 23) to test whether acutely induced INaL [by (ATX-II)] may oxidize PKARIα. We acutely exposed KI and wild-type (WT) control ventricular myocytes to ATX-II and comprehensively studied intracellular Ca2+ handling in an in vitro model.
Ventricular myocytes were isolated from a redox-dead transgenic mouse model in which a redox-sensitive Cysteine was made resistant to oxidative activation by knock-in replacement with Serine (PKARIαCys17Ser knock-in mouse model, KI). The point mutation of Cys17Ser was introduced into exon 1 of the Prkar1a gene as described previously (19). Ventricular myocytes from wild-type littermates were used as control. All animal procedures were performed in accordance with the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee.
The isolation of ventricular cardiomyocytes was performed as previously reported (23) using adult (12–15 weeks) mice of both sexes in equal shares. Hearts were mounted on a Langendorff perfusion apparatus and were retrogradely perfused for 7–9 min, starting with a calcium-free solution containing (in mmol/L): NaCl 113, KCl 4.7, KH2PO4 0.6, Na2HPO4 0.6, MgSO4 1.2, phenol-red 0.032, NaHCO3 12, KHCO3 10, HEPES 10, taurine 30, BDM (2,3 butanedione monoxime) 10, and glucose 5.5 (at 37°C, pH 7.4). After 4 min, trypsin 0.6% (Thermo Fisher Scientific Inc. of Waltham, MA, USA), 7.5 mg/ml Liberase™ (Roche Diagnostics, Mannheim, Germany), and 0.125 mmol/L CaCl2 were added and the hearts were perfused until they became flaccid. After enzymatic digestion, the ventricular myocardium was dissected from the atrium and mechanical dissociation of the heart was performed in a solution containing 10% bovine calf serum (BCS; Sigma-Aldrich, St. Louis, MO, USA). For immediate measurements, a gradual Ca2+ reintroduction was performed from 0.1 to 0.8 mmol/L. The isolated cardiomyocytes were plated on laminin-coated glass coverslips. Cardiomyocytes were allowed to settle for 15 min at room temperature to allow cell adhesion for the following experiments.
ATX-II, which was used to increase INaL, was purchased from Abcam (ab141870, Cambridge, Great Britain). The sources of the fluorescent dyes and antibodies are given in the following corresponding sections. If not indicated otherwise, Standard Tyrode's solution was used as the experimental solution, containing (in mmol/L) NaCl 140, KCl 4, HEPES 5, MgCl2 1, glucose 10, and CaCl2 1. The pH value was adjusted to 7.40 at 37°C (referred to as “normal Tyrode's solution”, NT).
Ruptured-patch whole-cell voltage-clamp was used to measure INaL and ICa as previously reported (23–25). The myocytes were mounted on the stage of a microscope (Nikon TE2000-U). They were incubated for 15 min with the respective bath solution before measurements were undertaken. Notably, INaL and ICa measurements were performed within the same time limits as the Ca2+ imaging experiments (i.e., following at least 10 min of ATX-II exposure). The experimental groups were additionally treated with 1 nmol/L ATX-II. Cardiomyocytes typically achieved a seal greater than 1 GΩ and a resistance <10 MΩ after rupture. For INaL measurements, the microelectrodes (2–3 MΩ) were filled with (in mmol/L) the following: CsCl 95, Cs-glutamate 40, NaCl 10, MgCl2 0.92, Mg-ATP 5, Li-GTP 0.3, HEPES 5, niflumic acid 0.03, nifedipine 0.02, strophanthidin 0.004, EGTA 1, and CaCl2 0.36 [free (Ca2+)i, 100 nmol/L, pH 7.2, CsOH]. The bath solution contained (in mmol/L) the following: NaCl 135, tetramethylammonium chloride 5, CsCl 4, MgCl2 2, glucose 10, and HEPES 10 (pH 7.4, CsOH). To measure INaL, the cardiomyocytes were held at −120 mV, and INaL was elicited using a train of pulses to −35 mV (1,000 ms duration, 10 pulses, basic cycle length (BCL) 2 s). Recordings were initiated 3–4 min after rupture and were performed at room temperature. The measured current was integrated (between 100 and 500 ms) and normalized to membrane capacitance. For ICa measurements, the microelectrodes (2–3 MΩ) were filled with (in mmol/L) the following: CsCl 86, Cs-glutamate 40, MgCl2 0.92, Mg-ATP 5, Li-GTP 0.3, HEPES 10, EGTA 5, and CaCl2 1.8 [free (Ca2+)i 100 nmol/L, pH 7.2, CsOH]. The bath solution contained (in mmol/L) the following: NaCl 140, CsCl 4, MgCl2 1, glucose 10, HEPES 10, and CaCl2 1 (pH 7.4, CsOH). The signals were filtered with 2.9 and 10 kHz Bessel filters and recorded with an EPC10 amplifier (HEKA Elektronik). The recordings were started 2–3 min after rupture and conducted at room temperature. ICa analysis was performed using LabChart 10 (ADInstruments) and determined by subtracting the steady-state current from the peak ICa current. Subsequently, the amplitude was normalized to cell capacitance. For analysis of ICa inactivation kinetics, time constants τslow (tau slow) and τfast (tau fast) were calculated by biexponential fitting of ICa currents using ClampFit.
For the assessment of cytosolic Ca2+, isolated cardiomyocytes were loaded with 10 µmol/L Fura-2-AM (Thermo Fisher Scientific Inc. of Waltham, MA, USA) in the presence of 0.02% (w/v) pluronic acid (Molecular Probes, Eugene, OR, USA) for 15 min at room temperature in darkness (23, 25). Afterward, the cells were incubated for another 15 min in NT to ensure complete deesterification of intracellular Fura-2. After deesterification, Fura-2-loaded cardiomyocytes were mounted on the stage of a Nikon Eclipse TE200-U inverted epifluorescence microscope. Fura-2 was alternately excited at 340 ± 5 and 380 ± 5 nm (250 pairs per second, HyperSwitch system, IonOptix Corp., Westwood, MA, USA) and emitted fluorescence was collected at 510 ± 20 nm. Excitation light was provided by a 75 W xenon arc lamp (Ushio, Japan). During measurements, the cardiomyocytes were continuously transilluminated with red light (>650 nm), stimulated at a frequency of 0.5 Hz, and superfused with experimental solution (with vehicle control or ATX-II, respectively) at a flow rate of 50 ml/h at 37°C. To estimate SR Ca2+ content, Ca2+ transient amplitude was determined after rapid caffeine application (10 mmol/L, Sigma-Aldrich) during field stimulation pause. After subtraction of background fluorescence, cytosolic Ca2+ levels were assessed as the 340/380 nm fluorescence ratio (F340/F380). Simultaneously with Ca2+ measurements, sarcomere length and fractional shortening of the myocytes were recorded using a sarcomere length detection system (MyoCam, IonOptix Corporation, Westwood, MA, USA). Contractions and Ca2+ transients were averaged over about 10 beats for the analysis of all parameters. Recorded data were analyzed with the software IONWizard (IonOptix Corp.).
Ca2+ spark frequency was analyzed using a laser scanning confocal microscope (Zeiss LSM 700 Pascal; Göttingen, Germany). Isolated cardiomyocytes were loaded with 10 µmol/L Fluo-4-AM (Thermo Fisher Scientific Inc. of Waltham, MA, USA) in the presence of 0.02% (w/v) pluronic acid (Molecular Probes, Eugene, OR, USA) for 15 min at room temperature in darkness (23). Afterward, the solution was replaced by Tyrode's solution. The cardiomyocytes were left to incubate for further 15 min for complete deesterification until the Fluo-4-loaded cardiomyocytes were placed on the stage of a laser scanning confocal microscope. Measurements were conducted in the line-scan mode (10,000 lines per scan). The cardiomyocytes were excited at 488 nm using an argon laser and emission was collected at 505–530 nm through a long-pass emission filter under continuous superfusion with experimental solution (with vehicle or 1 nmol/L ATX-II) and field stimulation at 0.5 Hz at 37°C. Ca2+ sparks were detected and quantified using SparkMaster with manual detection of sparks. Ca2+ spark frequency (CaSpF) was calculated and normalized to scanning interval and cell width. Only cells that showed stimulated Ca2+ transients were included in the evaluation to avoid misinterpretation by an elevated Ca2+ spark frequency in non-viable cells. To assess mitochondrial ROS formation, isolated ventricular cardiomyocytes were loaded with 5 µmol/L MitoSox Red (Thermo Fisher Scientific Inc. of Waltham, MA, USA) in presence of 0.02% (w/v) pluronic acid (Molecular Probes, Eugene, OR, USA) for 15 min at 37°C. The cardiomyocytes were excited at 488 nm using an argon laser and fluorescence emission was collected at 610 nm through a long-pass emission filter. Once every minute, frame scans were acquired and MitoSox Red fluorescence emission was normalized to the initial fluorescence (expressed as F/F0). Likewise, cytosolic ROS formation was assessed by excitation of CellRox Orange-loaded cardiomyocytes at 555 nm while fluorescence emission was collected at 570 nm (Thermo Fisher Scientific Inc. of Waltham, MA, USA, incubation for 30 min with 5 µmol/L at 37°C). Please note that mitochondrial ROS formation as depicted in Figure 1D was assessed in independent sets of experiments in WT vs. KI myocytes with higher initial MitoSox fluorescence in KI cells at baseline (1,665 ± 188 a.u.) as compared with WT (1,004 ± 52 a.u.).
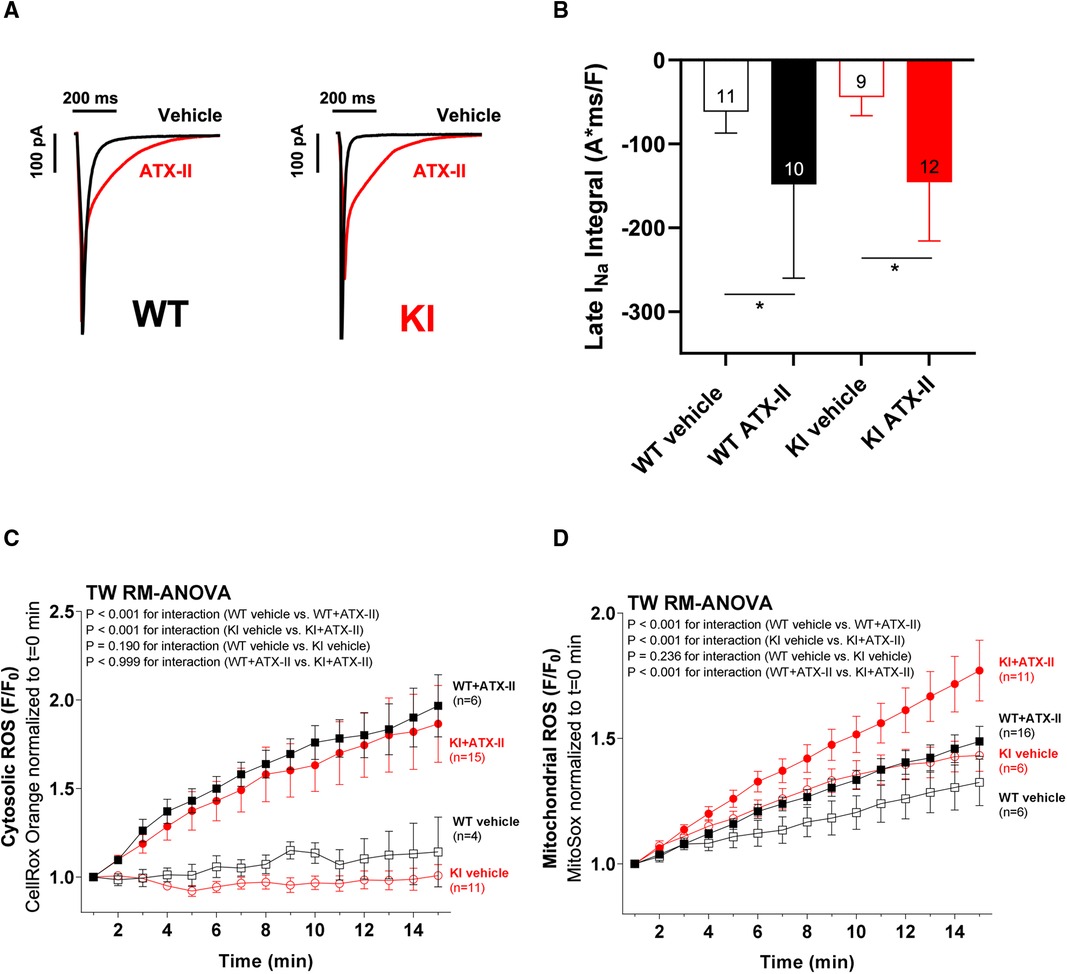
Figure 1 ATX-II increases INaL in WT and KI myocytes and leads to elevated cytosolic and mitochondrial ROS production. Original recordings of INaL (A) and mean data of the INaL integral (B) demonstrate an approximately threefold increase of INaL upon treatment with ATX-II in both WT and KI murine ventricular myocytes. Mean data normalized to fluorescence at t = 0 min of isolated ventricular WT and KI myocytes loaded with ROS sensor CellRox Orange (C) and MitoSox Red (D) show increased cytosolic (C) and mitochondrial (D) ROS formation in cells upon perfusion with ATX-II in comparison with vehicle-treated control myocytes. * indicates significance between groups using the Holm–Sidak post-hoc test.
For Western blot analysis, the cardiomyocytes were treated with vehicle or ATX-II (1 nmol/L) and incubated for 15 min at room temperature. The harvested cardiomyocytes were frozen immediately. The proteins were denatured for 5 min at 95°C under non-reducing conditions for PKARIα dimer and PKARIα monomer. To assess pSer2809 RyR2, pSer2814 RyR2, RyR2, pSer16 phospholamban (PLB), and PLB, the proteins were denatured for 30 min at 37°C in presence of 10% β-mercaptoethanol. They were denatured for 5 min at 95°C in presence of 10% β-mercaptoethanol for assessment of pThr287 CaMKII and CaMKIIδ expression. Following denaturation, the proteins were separated on 5% (RyR2, pSer2809 RyR2, pSer2814 RyR2), 8% (PKARIα dimer, PKARIα monomer, pThr287 CaMKII, and CaMKIIδ), or 12.5% (PLB, pSer16 PLB) SDS-polyacrylamide gels, transferred to a nitrocellulose membrane and incubated with the following primary antibodies: rabbit polyclonal anti-pThr287 CaMKII (1:1,000, PhosphoSolutions, Aurora, CO, USA), rabbit polyclonal anti-CaMKIIδ (1:10.000, Thermo Fisher Scientific Inc. of Waltham, MA, USA), mouse monoclonal anti-PKARIα (1:1,000, BD Biosciences, Heidelberg, Germany), rabbit polyclonal anti-RyR2 antibody (1:10.000, Sigma-Aldrich, St. Louis, MO, USA), rabbit polyclonal anti-pSer2809 RyR2 antibody (1: 1,000, Badrilla, Leeds, United Kingdom), rabbit polyclonal anti-pSer2814 RyR2 antibody (1:1,000, Badrilla, Leeds, United Kingdom), mouse monoclonal anti-PLB (1:10.000, Thermo Fisher Scientific Inc. of Waltham, MA, USA), rabbit polyclonal anti-pSer16 PLB (1:500, Badrilla, Leeds, United Kingdom), and mouse monoclonal anti-GAPDH (1:10.000, Sigma-Aldrich, St. Louis, MO, USA) at 4°C overnight. Secondary antibodies were horseradish peroxidase (HRP)-conjugated sheep anti-mouse and donkey anti-rabbit IgG (1:10.000, GE Healthcare, Chicago, Illinois, USA) and incubated for 1 h at room temperature. For chemiluminescent detection, Immobilon™ Western Chemiluminescent HRP Substrate (Millipore) was used. The values were normalized to GAPDH and WT vehicle values later.
Data are presented as means ± standard error of the mean (SEM). Statistical analyses were performed using unpaired Student's t-test, one-way ANOVA, and two-way ANOVA for repeated measurements (RM) with the Holm–Sidak post-hoc test and Mantel–Cox test for survival. Values of p < 0.05 were considered as statistically significant. The graphs were created using Sigma Plot 12 and Graph Pad Prism 8.0.1.
In a first step, we wanted to exclude the possibility that the knock-in replacement of Cys17 with Serine in the regulatory subunit of PKARIα might lead to a different responsiveness of INaL to ATX-II in KI cells. Therefore, we exposed isolated ventricular cardiomyocytes of both genotypes to 1 nmol/L ATX-II. As depicted in the original measurements in Figure 1A, ATX-II led to an approximately threefold increase of INaL in both WT and KI cells (mean values of the INaL integral are shown in Figure 1B). As previously reported by Viatchenko-Karpinski et al. (17), this increase in INaL was associated with significant cytosolic (Figure 1C) and mitochondrial ROS production (Figure 1D) in both genotypes in our model as well.
In a next step, we aimed to investigate whether oxidatively activated PKARIα is required for the ATX-II-dependent and PKA-mediated activation of intracellular Ca2+ handling that has been previously described by Eiringhaus et al. (9). As shown in Figures 2A,B, ATX-II caused a fairly immediate and time-dependent increase in the amplitude of the systolic Ca2+ transient that reached its maximum about 6–10 min after infusion of ATX-II. However, this pronounced, approximately threefold increase in systolic Ca2+ transient amplitude was largely comparable between WT and KI cells, which indicates that oxidatively activated PKARIα is functionally not required here (p = 0.368 for interaction, see Figure 2B). In line with this, a positive inotropic effect in terms of time-dependently increased fractional myocytes shortening was comparably observed in both WT and KI cells (Figure 2C). Further, rapid caffeine application was used to assess SR Ca2+ content in WT and KI myocytes (Figures 3A,B). Original traces in Figure 3A show that SR Ca2+ content was significantly increased by ATX-II treatment in both WT and KI myocytes without major differences between genotypes. The mean values in Figure 3B illustrate dose-dependency of SR Ca2+ load following ATX-II treatment in both genotypes without significant differences between WT and KI cells (p = 0.873 for interaction). The ATX-II-dependent rise in SR Ca2+ content was further associated with a time-dependent increase in the frequency of diastolic Ca2+ spark events as a measure of diastolic Ca2+ leakage from the SR (see Figure 3C, original confocal line-scans are shown in Figure 3D). Equivalent to Ca2+ transient amplitudes and SR Ca2+ content, this ATX-II-dependent increase in Ca2+ spark frequency did not differ between WT and KI cells (p = 0.765, see Figures 3C,D), further pointing to the fact that activated Ca2+ handling does not require oxidized PKARIα. On the protein level, an unchanged phosphorylation status of Serine 2809 at the RyR2 was observed upon ATX-II exposure that was again not different between genotypes (Ser2809 is the PKA phosphorylation site at the RyR2, see Table 1 and Supplementary Figures S1A and SB). By contrast, increased CaMKII activity in terms of increased autophosphorylation at Threonine 287 was clearly present upon ATX-II, and accompanied by comparable CaMKII-dependent hyperphosphorylation of Serine 2814 at RyR2 in both genotypes (see Supplementary Material Figures S1 and SC–F).
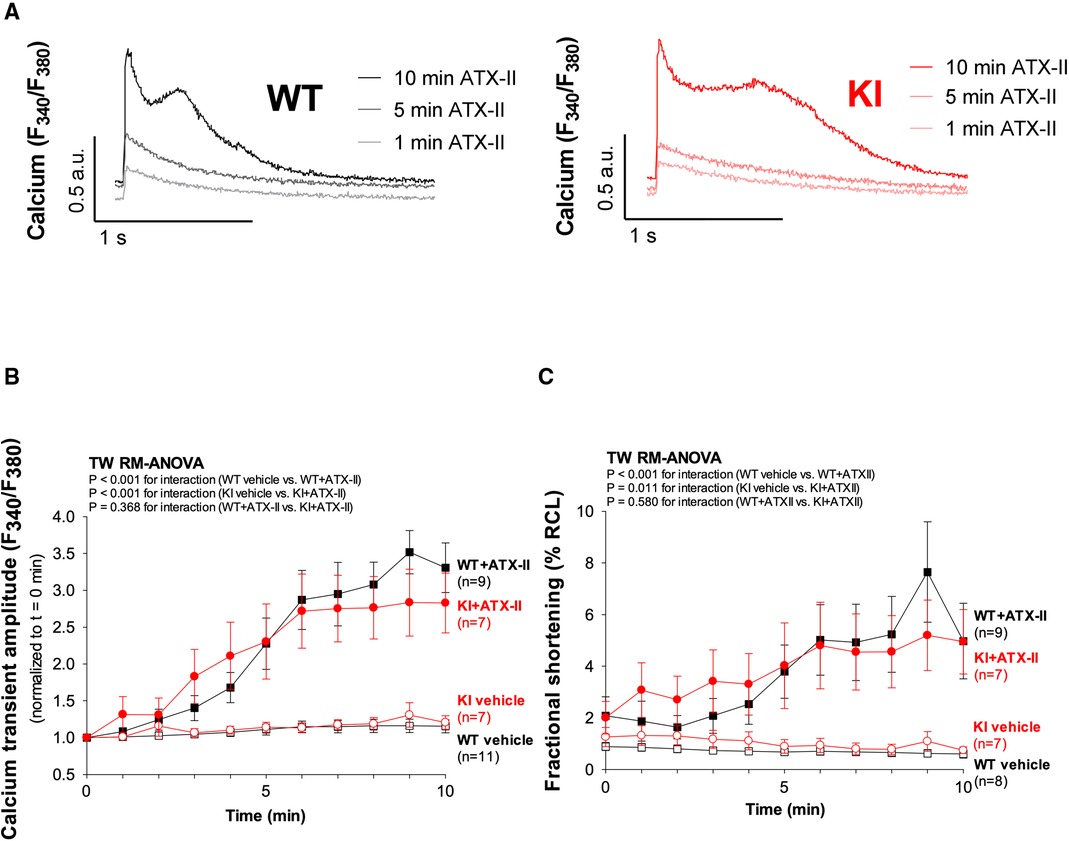
Figure 2 ATX-II activates Ca2+ handling in WT and KI cardiomyocytes independent of oxidized PKARIα. (A) Original traces of intracellular Ca2+ transients measured in Fura-2 AM-loaded ventricular myocytes from WT and PKARIα KI mice at baseline, after 5 min and after 10 min exposure to ATX-II. (B) Mean data for Ca2+-transient amplitudes normalized to t = 0 min during treatment with ATX-II for 10 min show a significant approximately threefold increase in both WT and KI ventricular myocytes to a similar extent. (C) The positive inotropic effect of ATX-II (1 nmol/L) treatment is also represented in the mean data for fractional shortening of myocytes [expressed as % resting cell length (%RCL)].
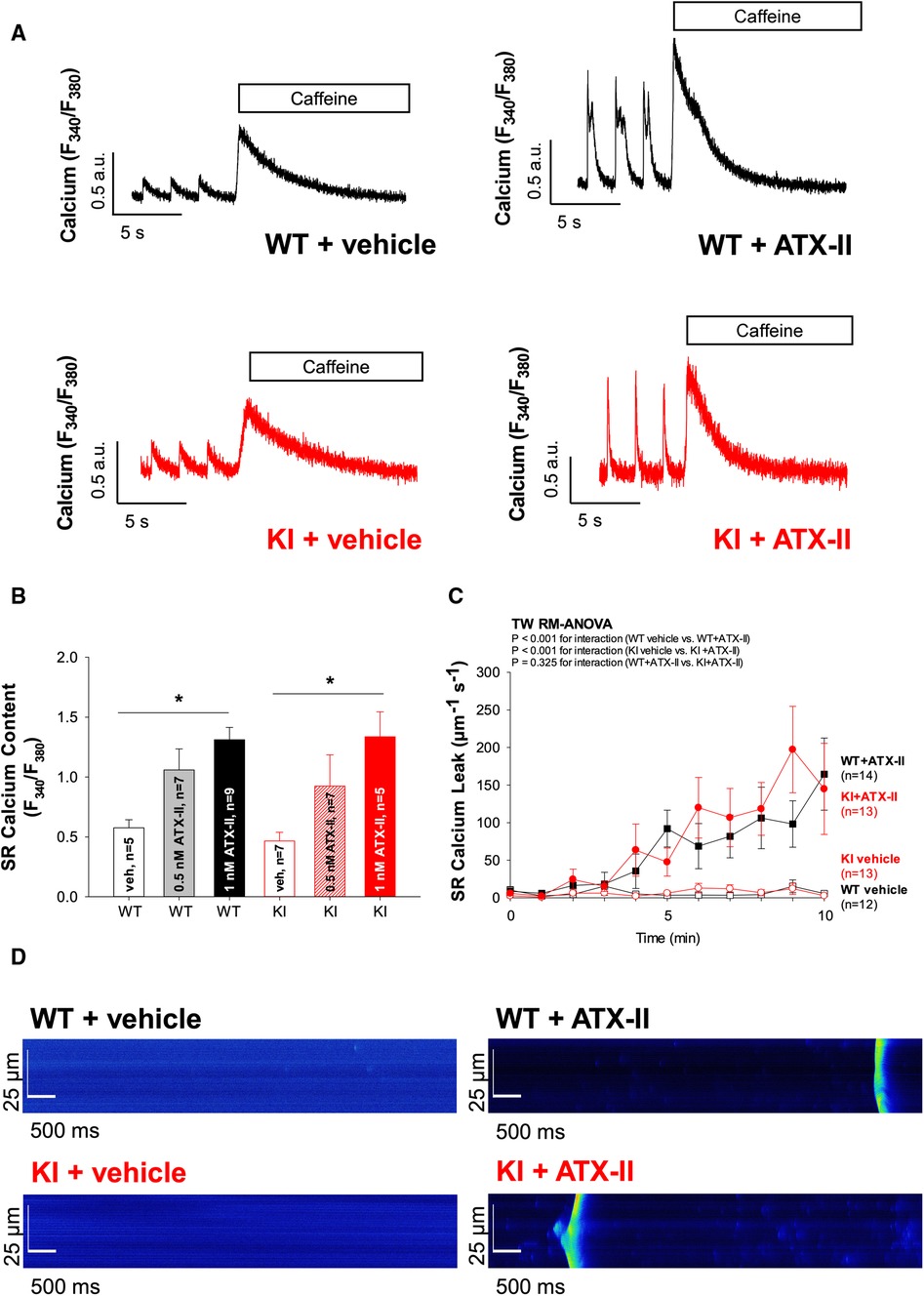
Figure 3 Exposure to ATX-II leads to an increased SR Ca2+ loading associated with increased diastolic SR Ca2+ spark frequency. (A) Original traces of caffeine-induced (10 mmol/L) Ca2+ transients to assess SR Ca2+ content upon exposure to vehicle vs. ATX-II (1 nmol/L) in WT and KI ventricular myocytes. (B) Mean data show that SR Ca2+ content is significantly increased upon treatment with ATX-II (1 nmol/L) in a dose-dependent manner in isolated ventricular myocytes of both WT and KI mice without a significant difference between genotypes. (C) Mean data for diastolic Ca2+ spark frequency illustrate an increased SR Ca2+ leak in WT and in KI cardiomyocytes upon treatment with ATX-II (1 nmol/L). (D) Original confocal line-scan images of isolated ventricular myocytes of WT and KI mice loaded with Fluo-4 at baseline and after 10 min of perfusion with ATX-II (1 nmol/L). * indicates significance between groups using the Holm–Sidak post-hoc test.
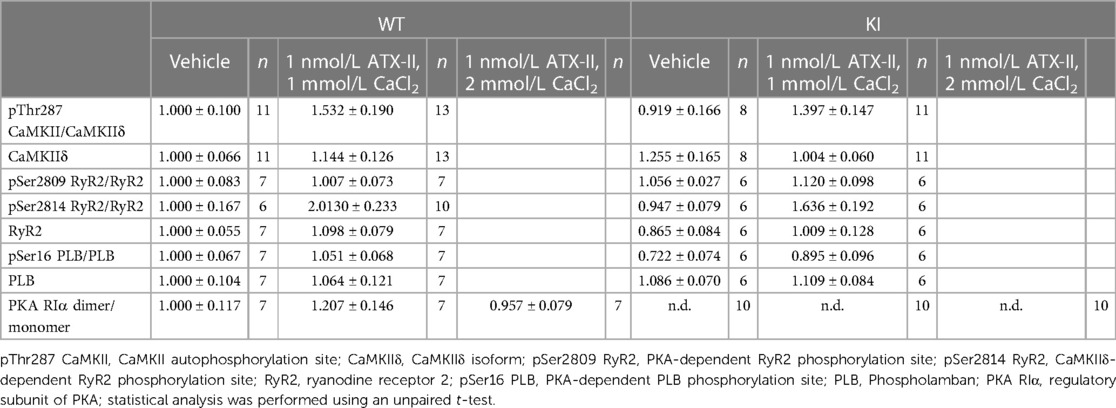
Table 1 Expression and phosphorylation level of Ca2+ handling proteins.
Since ATX-II is known to have proarrhythmogenic effects (26), we also analyzed cellular arrhythmias upon ATX-II treatment in our model [occurring as “non-triggered events” (24, 27, 28) as illustrated in Figure 4A, i.e., as spontaneous Ca2+ elevations during Ca2+ transient decay] to test whether oxidized PKARIα might exert a functional effect in that regard (see Figures 4A,B). As an overview parameter, we assessed the total proportion of arrhythmic cells in each minute at a certain point in time of the measurement (Figure 4B). By doing so, we observed that 78% of WT myocytes and 57% of KI myocytes developed cellular arrhythmias in terms of non-triggered events (as depicted in Figure 4A) within a time frame of 10 min of treatment with ATX-II, which, however, was not different between groups (p = 0.755 for interaction). Likewise, we did not observe a difference with respect to the ATX-II-dependently increased occurrence of non-stimulated Ca2+ waves between genotypes as assessed by confocal microscopy (p = 0.346, data not shown). This lack of change with respect to non-triggered arrhythmogenic events in case of absent oxidative PKARIα activation was mimicked by a similar delay in diastolic Ca2+ elimination that was comparably present in both genotypes upon ATX-II, but not different between WT and KI cells (p = 0.664 for interaction, see Figure 4C). In our in vitro model, PKA-specific phosphorylation of PLB at Serine 16 was not different upon acute ATX-II treatment as compared to the control and did not alter between genotypes (see Table 1).
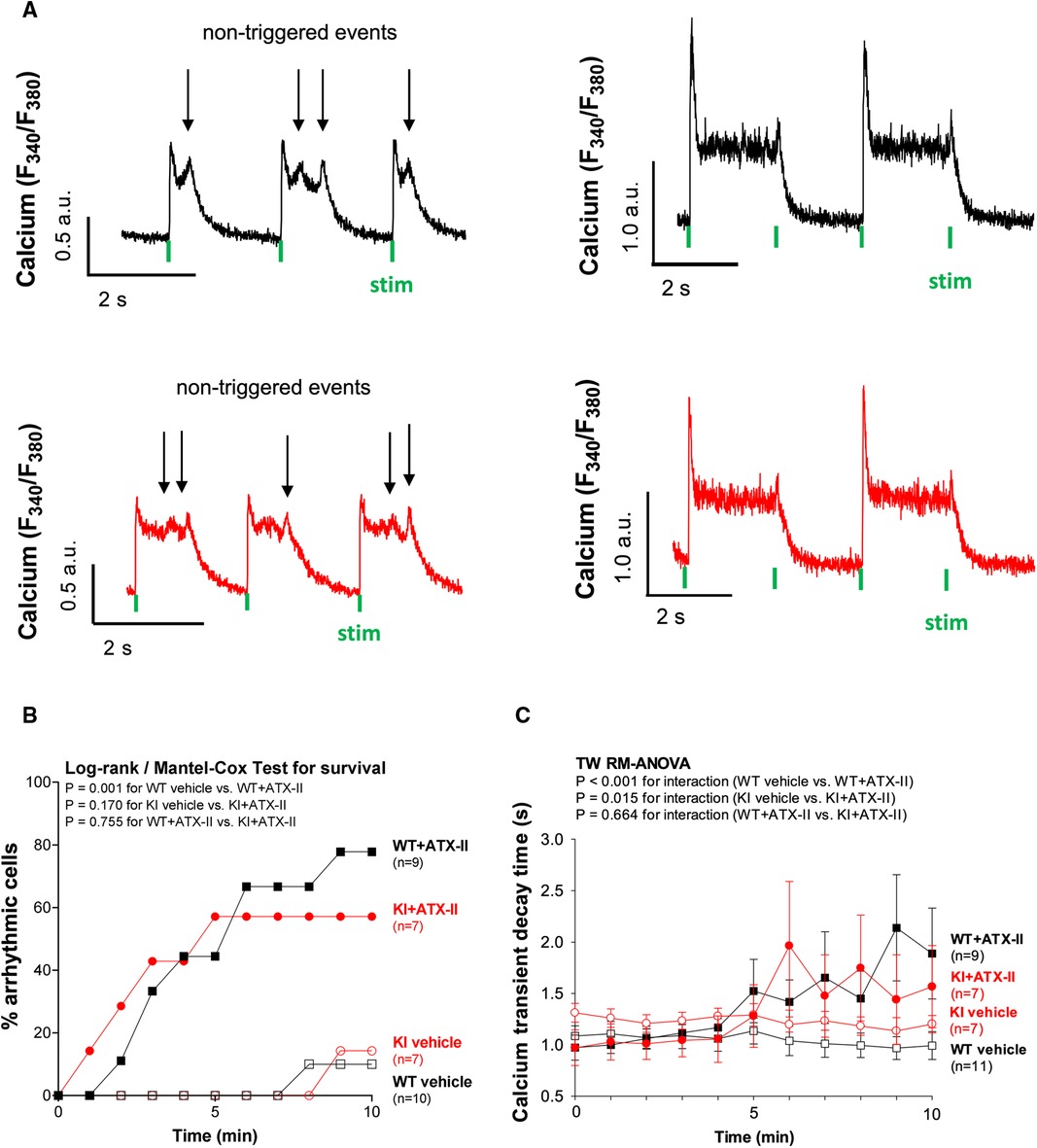
Figure 4 ATX-II causes cellular arrhythmias in WT and KI cells to a similar extent. (A) Original traces of non-triggered arrhythmic events during Ca2+ transient decay (left panel) and impaired relaxation (right panel) in ventricular myocytes of WT and KI mice loaded with Fura-2 AM upon treatment with ATX-II (1 nmol/L). (B) Percentage of ventricular myocytes with arrhythmias during exposure to ATX-II (1 nmol/L) for 10 min show a significant arrhythmogenic effect of ATX-II, which appears to be comparable in both WT and KI myocytes. (C) Decay time (RT90%, relaxation time until 90% decay of the Ca2+ transient) is significantly increased in ventricular myocytes of both WT and KI mice to a similar extent indicating impaired relaxation of cardiomyocytes upon treatment with ATX-II (1 nmol/L).
We have previously reported that oxidized PKARIα exerts a stimulating effect on the L-type-mediated Ca2+ current (ICa) when acutely stimulated with angiotensin II (AngII) as well as in a long-term setting in vivo succeeding transverse aortic constriction (TAC) (23). Since ICa greatly contributes to the amplitude of the systolic Ca2+ transient, the relevance of ICa for the acute activation of Ca2+ handling upon ATX-II was investigated in a next step. As shown in Figures 5A,B and in Supplementary Material Figure S2, ATX-II did not alter the magnitude (Figure 5B) or the inactivation properties (Supplementary Material Figure S2) of ICa in WT myocytes. In contrast to our previous findings, we also failed to observe a functional effect on ICa in KI cells despite clearly increased ROS formation upon ATX-II (compare Figures 1C,D). Notably, we did not observe increased oxidation (i.e., dimerization) of PKARIα upon ATX-II in WT cells at 1 or 2 mmol/L Ca2+ (while no dimerization of PKARIα was observed in KI cells as expected, see Figures 5C,D). Hence, oxidation of PKARIα appears not to be required for the ATX-II-mediated acute activation of Ca2+ handling despite a clearly present pro-oxidant intracellular milieu.
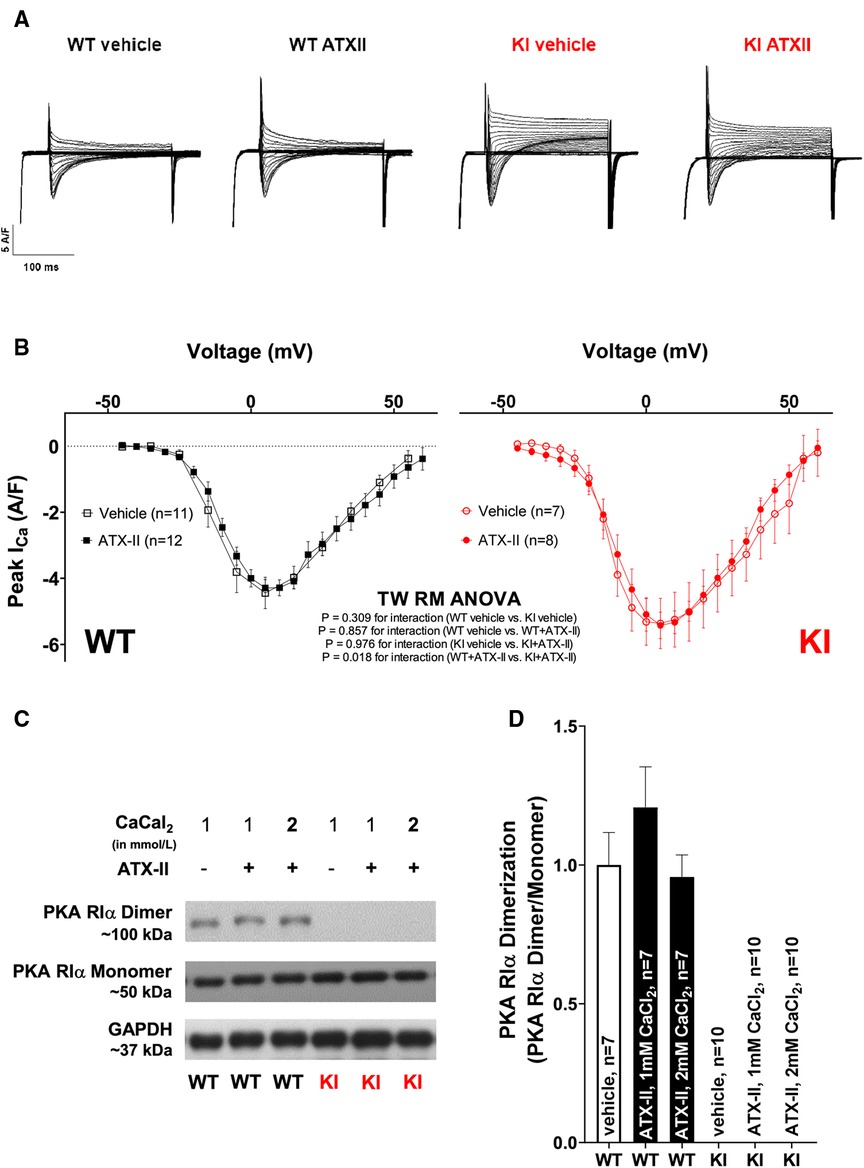
Figure 5 Unchanged ICa and absent PKARIα dimerization upon acute ATX-II treatment. (A) Original traces of ICa measured by whole-cell rupture-patch clamp technique in WT and KI ventricular myocytes upon treatment with ATX-II (1 nmol/L) vs. vehicle-treated control cells. (B) Mean data for peak ICa-voltage relationship in isolated murine ventricular myocytes demonstrate unchanged ICa upon treatment with ATX-II (1 nmol/L) with no functional effect of absent PKARIα oxidation in KI cells. (C,D) Original Western blots and mean data for PKARIα dimer/monomer ratio show PKARIα dimer formation in WT murine ventricular myocytes upon vehicle treatment that is not significantly increased upon ATX-II treatment (1 nmol/L) upon 1 or 2 mmol/L CaCl2. Dimerization is completely absent in KI samples.
In the present study, the role of oxidatively activated PKARIα in ATX-II-mediated Na+/Ca2+-mishandling in an in vitro model of acutely increased INaL was investigated for the first time. The main finding of our study is that oxidatively activated PKARIα does not contribute to the acute disturbance of Na+ and Ca2+ handling as induced by INaL (at least within a time frame of 15 min), which is accompanied by the unexpected fact that PKARIα does not become oxidized despite a clear pro-oxidant intracellular environment. We, therefore, conclude that oxidative activation of PKARIα is not crucially involved in mediating the positive inotropic and arrhythmogenic effects as induced by acute ATX-II exposure.
To induce an increase of INaL, which typically occurs in heart failure, we treated mouse cardiomyocytes from WT and PKARIα KI mice with ATX-II. An increased INaL in heart failure is known to be associated with arrhythmias (29) and impaired Na + -Ca2+ handling (4, 6). However, Na+ channels in ventricular myocytes are influenced by numerous factors. Among others, INaL is enhanced by CaMKII (30) and can be progressively induced by ROS (31, 32). Most importantly in regard to our study, protein kinase A is also involved in the regulation of Na+ channels (33, 34) and the modulation of INaL (35). Hence, PKA activation may potentially induce a positive feedback mechanism and further enhance INaL. To exclude the possibility that treatment with equal concentrations of ATX-II affects ion currents in genotypes differently, we assessed the INaL in cardiomyocytes of both genotypes under control conditions and upon ATX-II treatment. Treatment with 1 nmol/L ATX-II resulted in an approximately threefold increase in INaL without significant differences between genotypes. Hence, it could be assumed that initial conditions were equal in both genotypes and that INaL is not subject to oxidatively-activated PKARIα (oxPKA)-dependent regulation upon acute ATX-II exposure.
We next tested whether ATX-II-dependently increased Na+ influx into the cell led to increased ROS formation in our setting as it was proposed previously (10). Various studies have shown that altered Na+ and Ca2+ handling in cardiomyocytes can lead to increased mitochondrial ROS production. Interestingly, there are several mechanisms leading to increased mitochondrial ROS production that may be relevant in our model, as follows: Increased cytosolic Na+ levels have been shown to reduce mitochondrial Ca2+ accumulation by promoting Ca2+ efflux through the mitochondrial Na+-Ca2+-exchanger (mNCE) (10, 11). Kohlhaas et al. demonstrated that decreased mitochondrial Ca2+ levels were associated with increased mitochondrial ROS production (10). In addition, it has also been suggested that enhanced oxidative phosphorylation during beta-adrenergic stimulation (that is needed to meet the increased energy demand) leads to increased electron leakage and thus mitochondrial ROS formation (36). In our present study, we found significantly increased mitochondrial as well as cytosolic ROS formation in both WT and KI myocytes following acute ATX-II exposure in the face of a comparable positive inotropic effect in both groups. We therefore interpret increased ROS formation in our model as the result of at least two mechanisms, namely, (i) cytosolic Na+/Ca2+ overload leading to mitochondrial ROS formation and (ii) enhanced oxidative phosphorylation in the mitochondria as a consequence of positive inotropy. Regardless of the exact mechanism, we clearly observed increased ROS production in WT and KI cells following ATX-II treatment so that we could largely exclude the possibility that ATX-II might have failed to increase ROS formation in one of the genotypes.
We have previously found that oxidized PKARIα is an important factor for the maintenance of functional Ca2+ handling in the setting of enhanced oxidative stress by sustaining ICa (23). Hence, we now sought to determine the impact of oxidatively activated PKARIα on Ca2+ handling in the setting of acutely increased INaL with consecutively enhanced oxidative stress. In line with previous studies (9), we observed a positive inotropic effect upon ATX-II in terms of significantly increased Ca2+ transients and increased fractional shortening. In addition, we found an impeded diastolic decay of the Ca2+ transient that was paralleled by a time-dependent increase in arrhythmogenic non-triggered Ca2+ elevation events during Ca2+ transient decay, which altogether resulted in a delay of diastolic Ca2+ elimination. However, these effects were equally present in both genotypes, which speaks against a crucial involvement of oxidized PKARIα.
Likewise, ATX-II treatment induced diastolic SR Ca2+ leakage in both genotypes to a similar extent. An increase in SR Ca2+ leakage following enhanced INaL is generally in accordance with previous studies. Various factors such as activation of CaMKII (24) and direct ROS-dependent regulation of Ca2+ spark frequency (37) have been shown to modify this effect. Fischer et al. investigated the effect of INaL on the induction of SR Ca2+ leakage in atrial murine myocytes and uncovered that it was due to the activation of both CaMKII and PKA (38). By contrast, Wagner et al. ruled out a Nox2-dependent (and therefore redox-related) influence of PKA on SR Ca2+ leak and concluded that the leak was CaMKII-mediated (39), which is in accordance with another study from our group in which we have observed that acutely increased oxidative stress induces SR Ca2+ leakage independent of RIα dimer formation (23). Since we did not observe increased phosphorylation of Ser-2809 (which is the PKA-dependent phosphorylation site at the RyR2) here, it can be assumed that the increase in Ca2+ spark frequency upon ATX-II-related ROS generation as observed in our model is likely also not PKA dependent. Moreover, this finding indicates again that redox-activated PKA is not a major driver of SR Ca2+ leak in cardiac myocytes. Instead, in our experimental setting, ATX-II-activated CaMKII in WT and KI cells as well resulted in subsequently hyperphosphorylated RyR2 at the CaMKII-dependent phosphorylation site 2814 in both genotypes, which points to the fact that the induction of the ATX-II/ROS-mediated SR Ca2+ leak is primarily driven by increased CaMKII activity. In addition, ATX-II-dependent SR Ca2+ leakage may be a consequence of a sensitization of the RyR2 to the increased Ca2+ load of the SR (which increases the driving force for Ca2+ release for a given Ca2+ load of the SR that was also present as increased systolic fractional Ca2+ release).
Despite increased diastolic SR Ca2+ loss, however, SR Ca2+ content was still significantly elevated after treatment with ATX-II as compared with untreated control cardiomyocytes in both groups, which points to the fact that SR Ca2+ loading prevailed over SR Ca2+ loss in our model. Enhanced SR Ca2+ loading may be due to increased SERCA2a activity. In that regard, Eiringhaus et al. observed maintained, and upon CaMKII inhibition even increased, SR Ca2+ load despite INaL-related diastolic SR Ca2+ loss, which they attributed to increased SERCA2a activity due to PKA-mediated PLB phosphorylation (9). However, in our model, we did not find a functional acceleration of SR reuptake [that can be approximated by Ca2+ transient decay kinetics in mouse myocytes (40)] nor evidence for PKA-dependent hyperphosphorylation of PLB (at Ser-16). This lack in ATX-II-dependent and PKA-mediated activation of central target structures of intracellular Ca2+ handling in our model may be explained by slightly different incubation strategies with ATX-II resulting in different exposure times to ATX-II (e.g., we did not add ATX-II to the fluorescent dye during incubation here). By contrast, we do not believe that this difference is due to different experimental Ca2+ concentrations, because we still failed to observe increased RIα dimerization following ATX-II even upon elevated Ca2+ concentrations as used by Eiringhaus et al. (compare Figures 5C,D). Conversely, since we observed clearly activated Ca2+ handling, we believe that PKA-dependent phosphorylation of PLB and RyR2 are at least not crucially involved in the very acute effects of ATX-II-dependent Ca2+ activation as observed here. Instead, in our murine model, diastolic Ca2+ overload and increased Ca2+ transient amplitudes are presumably a consequence of increased NCX activity in its reverse mode leading to severe cytosolic Ca2+ overload, since ATX-II does not enhance transsarcolemmal Ca2+ influx via the LTCC (which would be an alternative mechanism of cytosolic Ca2+ overload, see the following). While this effect may be different in larger animals and humans (41), our observation would be in line with previous reports that have demonstrated that inhibition of NCX significantly attenuates intracellular Ca2+ overload and also reduces arrhythmias upon increased INaL (30, 42). In contrast to our previous study (23), in which oxidized PKARIα maintained ICa upon oxidative stress, we failed to detect any functional effect of ATX-II treatment on the amplitude or the inactivation properties of ICa in both WT and KI cells here (while ICa was slightly but significantly enhanced in KI vs. WT upon ATX-II treatment in the face of a generally lower ICa amplitude in WT cells). Our observation that ATX-II-related ROS formation did not result in PKARIα oxidation in WT cells further suggests that this unexpected lack of functional effect on ICa is rather a result of absent oxidative PKARIα activation in comparison with a potentially differential regulation of ICa by oxidized PKARIα in the context of ATX-II.
We acknowledge that our present study raises many new questions. Most importantly, it remains unclear why elevated cytosolic and mitochondrial ROS as acutely induced by ATX-II do not oxidize PKARIα, yet we believe that this aspect is beyond the scope of this study. Nevertheless, there is no question that this finding is in contrast to earlier studies in which an increased dimerization of the RIα subunit was clearly induced by various forms of oxidative stress such as by chronic pressure overload (23) or by pharmacologically induced oxidative stress using 1-nitrosocyclohexalycetate (NCA) (43). Interestingly, preliminary and as yet unpublished data by Simon et al. suggest that endogenous ROS indeed do not necessarily oxidize PKARIα in the heart, which appears to be correct for ROS as induced by NADPH oxidase 2 and 4 (NOX2 and NOX4), xanthine oxidase (XO), and the mitochondria (44). Instead, nitric oxide might turn out to be the relevant source for PKARIα dimer formation in the heart. Hence, our results may be at least hypothesis-generating in a way that it supports the notion that different stimuli and sources of ROS (or even NO) might differentially facilitate the dimerization of the PKARIα subunit. Conversely, we cannot rule out that chronically elevated INaL (such as in HF or in experimental models leading to HF such as TAC or myocardial infarction) would lead to PKARIα oxidation with consecutive functional consequences in intracellular Ca2+ handling and should therefore be subsequently investigated.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethical approval was not required for the study involving animals in accordance with the local legislation and institutional requirements because no experiments using living animals are presented in this manuscript.
TG: Validation, Software, Methodology, Data curation, Writing – review & editing, Writing – original draft, Visualization, Investigation, Formal Analysis, Conceptualization. LS: Writing – review & editing, Writing – original draft, Visualization, Validation, Software, Methodology, Investigation, Formal Analysis, Data curation. DT: Writing – review & editing, Writing – original draft, Validation, Methodology. LSM: Writing – review & editing, Writing – original draft, Validation, Supervision, Resources, Project administration. SW: Validation, Supervision, Methodology, Investigation, Formal Analysis, Writing – review & editing. ALF: Visualization, Supervision, Software, Investigation, Formal Analysis, Data curation, Conceptualization, Writing – review & editing, Writing – original draft, Validation, Methodology. CMS: Resources, Project administration, Writing – review & editing, Writing – original draft, Visualization, Supervision, Investigation, Formal Analysis, Conceptualization.
The authors declare financial support was received for the research, authorship, and/or publication of this article.
This work was supported by grants from Deutsche Forschungsgemeinschaft (AZ 3282/1-1 and 3282/1-2) to CMS.
We thank Thomas Sowa for his technical expertise.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2024.1379930/full#supplementary-material
Supplementary Figure S1
ATX-II activates CaMKII and leads to hyperphosphorylated RyR2 at the CaMKII-dependent phosphorylation site Serine 2814 in KI and WT cells (A,B) Original Western blots and mean data for PKA-dependent phosphorylation of Serine 2809 at RyR2 and for (C,D) CaMKII-dependent phosphorylation of Serine 2814 at RyR2 in the absence and presence of ATX-II. (E,F) depict increased CaMKII autophosphorylation at Threonine 287 (normalized to CaMKIIδ expression) following ATX-II exposure in WT and KI cells. * indicates significance between groups using unpaired Student’s t-test.
Supplementary Figure S2
Inactivation kinetics of ICa are unchanged upon acute ATX-II treatment. (A) Original traces of ICa inactivation at 0 mV as measured by whole-cell rupture-patch clamp technique in WT and KI ventricular myocytes upon treatment with ATX-II (1 nmol/L) vs. vehicle-treated control cells. (B,C) Voltage-dependent ICa inactivation kinetics displayed as mean values of τslow and τfast (double exponential fit) in isolated ventricular myocytes demonstrate no differences upon treatment with ATX-II (1 nmol/L) and no functional effect in case of absent oxidative PKARIα activation.
1. Sag CM, Wagner S, Maier LS. Role of oxidants on calcium and sodium movement in healthy and diseased cardiac myocytes. Free Radic Biol Med. (2013) 63:338–49. doi: 10.1016/j.freeradbiomed.2013.05.035
2. Noble D, Noble PJ. Late sodium current in the pathophysiology of cardiovascular disease: consequences of sodium-calcium overload. Heart. (2006) 92(Suppl 4):iv1–5. doi: 10.1136/hrt.2005.078782
3. Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, et al. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. (2005) 38(3):475–83. doi: 10.1016/j.yjmcc.2004.12.012
4. Maltsev VA, Silverman N, Sabbah HN, Undrovinas AI. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: implications for repolarization variability. Eur J Heart Fail. (2007) 9(3):219–27. doi: 10.1016/j.ejheart.2006.08.007
5. Baartscheer A. [Na+]i and the driving force of the Na+/Ca2+-exchanger in heart failure. Cardiovasc Res. (2003) 57(4):986–95. doi: 10.1016/S0008-6363(02)00848-9
6. Undrovinas NA, Maltsev VA, Belardinelli L, Sabbah HN, Undrovinas A. Late sodium current contributes to diastolic cell Ca2+ accumulation in chronic heart failure. J Physiol Sci. (2010) 60(4):245–57. doi: 10.1007/s12576-010-0092-0
7. Vassalle M, Lin C-I. Calcium overload and cardiac function. J Biomed Sci. (2004) 11(5):542–65. doi: 10.1007/BF02256119
8. Hegyi B, Pölönen R-P, Hellgren KT, Ko CY, Ginsburg KS, Bossuyt J, et al. Cardiomyocyte Na+ and Ca2+ mishandling drives vicious cycle involving CaMKII, ROS, and ryanodine receptors. Basic Res Cardiol. (2021) 116(1):58. doi: 10.1007/s00395-021-00900-9
9. Eiringhaus J, Herting J, Schatter F, Nikolaev VO, Sprenger J, Wang Y, et al. Protein kinase/phosphatase balance mediates the effects of increased late sodium current on ventricular calcium cycling. Basic Res Cardiol. (2019) 114(2):13. doi: 10.1007/s00395-019-0720-7
10. Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Böhm M, et al. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation. (2010) 121(14):1606–13. doi: 10.1161/CIRCULATIONAHA.109.914911
11. Maack C, Cortassa S, Aon MA, Ganesan AN, Liu T, O’Rourke B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ Res. (2006) 99(2):172–82. doi: 10.1161/01.RES.0000232546.92777.05
12. Ide T, Tsutsui H, Kinugawa S, Suematsu N, Hayashidani S, Ichikawa K, et al. Direct evidence for increased hydroxyl radicals originating from superoxide in the failing myocardium. Circ Res. (2000) 86(2):152–7. doi: 10.1161/01.RES.86.2.152
13. Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. (2011) 301(6):H2181–90. doi: 10.1152/ajpheart.00554.2011
14. Wagner S, Rokita AG, Anderson ME, Maier LS. Redox regulation of sodium and calcium handling. Antioxid Redox Signal. (2013) 18(9):1063–77. doi: 10.1089/ars.2012.4818
15. Erickson JR, Joiner MA, Guan X, Kutschke W, Yang J, Oddis CV, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. (2008) 133(3):462–74. doi: 10.1016/j.cell.2008.02.048
16. Brennan JP, Bardswell SC, Burgoyne JR, Fuller W, Schröder E, Wait R, et al. Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J Biol Chem. (2006) 281(31):21827–36. doi: 10.1074/jbc.M603952200
17. Viatchenko-Karpinski S, Kornyeyev D, El-Bizri N, Budas G, Fan P, Jiang Z, et al. Intracellular Na+ overload causes oxidation of CaMKII and leads to Ca2+ mishandling in isolated ventricular myocytes. J Mol Cell Cardiol. (2014) 76:247–56. doi: 10.1016/j.yjmcc.2014.09.009
18. Liu Y, Chen J, Fontes SK, Bautista EN, Cheng Z. Physiological and pathological roles of protein kinase A in the heart. Cardiovasc Res. (2022) 118(2):386–98. doi: 10.1093/cvr/cvab008
19. Burgoyne JR, Rudyk O, Cho H, Prysyazhna O, Hathaway N, Weeks A, et al. Deficient angiogenesis in redox-dead Cys17Ser PKARIα knock-in mice. Nat Commun. (2015) 6:7920. doi: 10.1038/ncomms8920
20. Eisel F, Boosen M, Beck M, Heide H, Wittig I, Beck K-F, et al. Platelet-derived growth factor triggers PKA-mediated signalling by a redox-dependent mechanism in rat renal mesangial cells. Biochem Pharmacol. (2013) 85(1):101–8. doi: 10.1016/j.bcp.2012.10.017
21. Trum M, Islam MMT, Lebek S, Baier M, Hegner P, Eaton P, et al. Inhibition of cardiac potassium currents by oxidation-activated protein kinase A contributes to early afterdepolarizations in the heart. Am J Physiol Heart Circ Physiol. (2020) 319(6):H1347–57. doi: 10.1152/ajpheart.00182.2020
22. Simon JN, Vrellaku B, Monterisi S, Chu SM, Rawlings N, Lomas O, et al. Oxidation of protein kinase A regulatory subunit PKARIα protects against myocardial ischemia-reperfusion injury by inhibiting lysosomal-triggered calcium release. Circulation. (2021) 143(5):449–65. doi: 10.1161/CIRCULATIONAHA.120.046761
23. Islam MMT, Tarnowski D, Zhang M, Trum M, Lebek S, Mustroph J, et al. Enhanced heart failure in redox-dead Cys17Ser PKARIα knock-in mice. J Am Heart Assoc. (2021) 10(19):e021985. doi: 10.1161/JAHA.121.021985
24. Sag CM, Mallwitz A, Wagner S, Hartmann N, Schotola H, Fischer TH, et al. Enhanced late INa induces proarrhythmogenic SR Ca leak in a CaMKII-dependent manner. J Mol Cell Cardiol. (2014) 76:94–105. doi: 10.1016/j.yjmcc.2014.08.016
25. Tarnowski D, Feder A-L, Trum M, Kreitmeier K-G, Stengel L, Maier LS, et al. Ibrutinib impairs IGF-1-dependent activation of intracellular Ca handling in isolated mouse ventricular myocytes. Front Cardiovasc Med. (2023) 10:1190099. doi: 10.3389/fcvm.2023.1190099
26. el-Sherif N, Fozzard HA, Hanck DA. Dose-dependent modulation of the cardiac sodium channel by sea anemone toxin ATXII. Circ Res. (1992) 70(2):285–301. doi: 10.1161/01.RES.70.2.285
27. Sag CM, Wadsack DP, Khabbazzadeh S, Abesser M, Grefe C, Neumann K, et al. Calcium/calmodulin-dependent protein kinase II contributes to cardiac arrhythmogenesis in heart failure. Circ Heart Fail. (2009) 2(6):664–75. doi: 10.1161/CIRCHEARTFAILURE.109.865279
28. Kreitmeier KG, Tarnowski D, Nanadikar MS, Baier MJ, Wagner S, Katschinski DM, et al. CaMKIIδ Met281/282 oxidation is not required for recovery of calcium transients during acidosis. Am J Physiol Heart Circ Physiol. (2021) 320(3):H1199–212. doi: 10.1152/ajpheart.00040.2020
29. Shryock JC, Song Y, Rajamani S, Antzelevitch C, Belardinelli L. The arrhythmogenic consequences of increasing late INa in the cardiomyocyte. Cardiovasc Res. (2013) 99(4):600–11. doi: 10.1093/cvr/cvt145
30. Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, et al. Reactive oxygen species-activated Ca/calmodulin kinase IIδ is required for late I(Na) augmentation leading to cellular Na and Ca overload. Circ Res. (2011) 108(5):555–65. doi: 10.1161/CIRCRESAHA.110.221911
31. Song Y, Shryock JC, Wagner S, Maier LS, Belardinelli L. Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J Pharmacol Exp Ther. (2006) 318(1):214–22. doi: 10.1124/jpet.106.101832
32. Kassmann M, Hansel A, Leipold E, Birkenbeil J, Lu S-Q, Hoshi T, et al. Oxidation of multiple methionine residues impairs rapid sodium channel inactivation. Pflugers Arch. (2008) 456(6):1085–95. doi: 10.1007/s00424-008-0477-6
33. Frohnwieser B, Chen L-Q, Schreibmayer W, Kallen RG. Modulation of the human cardiac sodium channel ac-subunit by cAMP-dependent protein kinase and the responsible sequence domain. J Physiol. (1997) 498(2):309–18. doi: 10.1113/jphysiol.1997.sp021859
34. Zhou J, Yi J, Hu N, George AL, Murrax KT. Activation of protein kinase A modulates trafficking of the human cardiac sodium channel in Xenopus oocytes. Circ Res. (2000) 87(1):33–8. doi: 10.1161/01.RES.87.1.33
35. Hegyi B, Bányász T, Izu LT, Belardinelli L, Bers DM, Chen-Izu Y. β-adrenergic regulation of late Na+ current during cardiac action potential is mediated by both PKA and CaMKII. J Mol Cell Cardiol. (2018) 123:168–79. doi: 10.1016/j.yjmcc.2018.09.006
36. Hamilton S, Terentyeva R, Martin B, Perger F, Li J, Stepanov A, et al. Increased RyR2 activity is exacerbated by calcium leak-induced mitochondrial ROS. Basic Res Cardiol. (2020) 115(4):38. doi: 10.1007/s00395-020-0797-z
37. Yan Y, Liu J, Wei C, Li K, Xie W, Wang Y, et al. Bidirectional regulation of Ca2+ sparks by mitochondria-derived reactive oxygen species in cardiac myocytes. Cardiovasc Res. (2008) 77(2):432–41. doi: 10.1093/cvr/cvm047
38. Fischer TH, Herting J, Mason FE, Hartmann N, Watanabe S, Nikolaev VO, et al. Late INa increases diastolic SR-Ca2+-leak in atrial myocardium by activating PKA and CaMKII. Cardiovasc Res. (2015) 107(1):184–96. doi: 10.1093/cvr/cvv153
39. Wagner SD, Christian F, Hannah AA, Sag CM, Engels SE, Möllencamp J, et al. NADPH oxidase 2 mediates angiotensin II-dependent cellular arrhythmias via PKA and CaMKII. J Mol Cell Cardiol. (2014) 75:206–15. doi: 10.1016/j.yjmcc.2014.07.011
40. Maier LS, Zhang T, Chen L, DeSantiago J, Brown JH, Bers DM. Transgenic CaMKIIdeltaC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res. (2003) 92(8):904–11. doi: 10.1161/01.RES.0000069685.20258.F1
41. Nagy N, Tóth N, Nánási PP. Antiarrhythmic and inotropic effects of selective Na+/Ca2+ exchanger inhibition: what can we learn from the pharmacological studies? Int J Mol Sci. (2022) 23(23):14651. doi: 10.3390/ijms232314651
42. Nagy N, Kormos A, Kohajda Z, Szebeni Á, Szepesi J, Pollesello P, et al. Selective Na(+)/Ca(2+) exchanger inhibition prevents Ca(2+) overload-induced triggered arrhythmias. Br J Pharmacol. (2014) 171(24):5665–81. doi: 10.1111/bph.12867
43. Diering S, Stathopoulou K, Goetz M, Rathjens L, Harder S, Piasecki A, et al. Receptor-independent modulation of cAMP-dependent protein kinase and protein phosphatase signaling in cardiac myocytes by oxidizing agents. J Biol Chem. (2020) 295(45):15342–65. doi: 10.1074/jbc.RA120.014467
Keywords: PKARIα, excitation contraction coupling (ECC), CaMKII, oxidative stress, L-type Ca2+ current (ICa)
Citation: Gissibl T, Stengel L, Tarnowski D, Maier LS, Wagner S, Feder A-L and Sag CM (2024) The inotropic and arrhythmogenic effects of acutely increased late INa are associated with elevated ROS but not oxidation of PKARIα. Front. Cardiovasc. Med. 11: 1379930. doi: 10.3389/fcvm.2024.1379930
Received: 31 January 2024; Accepted: 10 June 2024;
Published: 15 July 2024.
Edited by:
Andreas Rinne, University of Medicine and Pharmacy “Carol Davila”, RomaniaReviewed by:
Dmitry Terentyev, The Ohio State University, United States© 2024 Gissibl, Stengel, Tarnowski, Maier, Wagner, Feder and Sag. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Can Martin Sag, Y2FuLW1hcnRpbi5zYWdAdWtyLmRl
†These authors have contributed equally to this work and share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.