 Yue Zeng
Yue Zeng Yilang Li1,2
Yilang Li1,2 Ning Hou
Ning Hou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med. , 25 March 2024
Sec. Cardiovascular Metabolism
Volume 11 - 2024 | https://doi.org/10.3389/fcvm.2024.1375400
Diabetic cardiomyopathy (DCM), one of the most serious complications of diabetes mellitus, has become recognized as a cardiometabolic disease. In normoxic conditions, the majority of the ATP production (>95%) required for heart beating comes from mitochondrial oxidative phosphorylation of fatty acids (FAs) and glucose, with the remaining portion coming from a variety of sources, including fructose, lactate, ketone bodies (KB) and branched chain amino acids (BCAA). Increased FA intake and decreased utilization of glucose and lactic acid were observed in the diabetic hearts of animal models and diabetic patients. Moreover, the polyol pathway is activated, and fructose metabolism is enhanced. The use of ketones as energy sources in human diabetic hearts also increases significantly. Furthermore, elevated BCAA levels and impaired BCAA metabolism were observed in the hearts of diabetic mice and patients. The shift in energy substrate preference in diabetic hearts results in increased oxygen consumption and impaired oxidative phosphorylation, leading to diabetic cardiomyopathy. However, the precise mechanisms by which impaired myocardial metabolic alterations result in diabetes mellitus cardiac disease are not fully understood. Therefore, this review focuses on the molecular mechanisms involved in alterations of myocardial energy metabolism. It not only adds more molecular targets for the diagnosis and treatment, but also provides an experimental foundation for screening novel therapeutic agents for diabetic cardiomyopathy.
Diabetes mellitus incidence has been showing an increasing trend in recent years (1). Diabetic cardiomyopathy (DCM) is characterized by abnormalities in myocardial structure and function that are unrelated to diabetic macrovascular complications such as hypertension, coronary artery disease, and atherosclerosis (2). It consists of cardiac fibrosis, cardiac hypertrophy, diastolic dysfunction, and the progression of systolic dysfunction and heart failure (3, 4). Diabetic patients have a higher rate of heart failure than non-diabetic patients (5).
Prolonged hyperglycemia promotes the accumulation of advanced glycation end products (AGEs) (6). O-GlcNAcylation, which is a post-translational modification of proteins occurring on the hydroxyl group of serine or threonine, is also increased in response to hyperglycemia (7). In diabetic hearts, the utilization and consumption of glucose decrease due to insulin deficiency or insulin resistance (8). Conversely, as fatty acids (FAs) supply and uptake increase, the heart becomes more dependent on FA for energy production (9). Furthermore, the accumulation of lipid metabolism intermediates, namely diacylglycerols and ceramides, results from unbalanced acylcarnitine synthesis and mitochondrial oxidation rate, which additionally contributes to oxidative stress, inflammation, and cardiac dysfunction (10, 11).
Some review articles, such as those written by Ding An and his colleagues, have summarized that dysregulation of glucose and lipid metabolism causes changes in cardiometabolism, which leads to mitochondrial dysfunction and impaired cardiac function (12). In addition to FA and glucose, energy can be generated using other substrates, such as fructose (13, 14), lactate (15), amino acids (16), and ketone bodies (17), in the heart. The metabolic imbalance of these fuels is also linked to the occurrence and development of DCM (Figure 1).
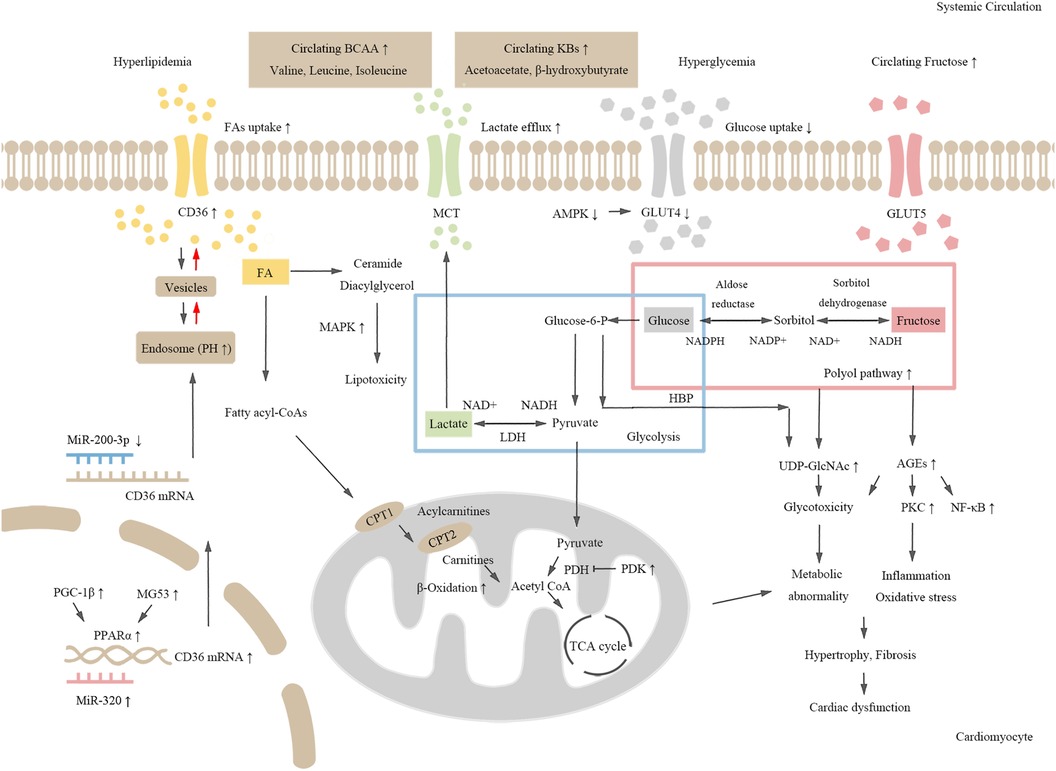
Figure 1. The schematic chart for molecular mechanisms of metabolic dysregulation in DCM. Hyperglycemia and dyslipidemia induce cardiometabolic disorders in diabetes. Both CD36 expression and fatty acid (FA) uptake are increased in diabetic hearts. Activated MAPK signaling pathway induces toxic lipid intermediate accumulation. Increased expression of PGC-1β and MG53, and up-regulation of PPARα and its target genes promote FA oxidation, which further leads to abnormal cardiometabolism and cardiac dysfunction. Decreased glucose and lactate uptake, activation of the polyol pathway, and enhanced fructose metabolism lead to increased production of AGEs and UDP-GlcNAc, which induce inflammation and cardiac dysfunction. Additionally, increased level of amino acids and KBs may also be risk factors for cardiometabolic disorders in diabetes.
This review focuses on changes in myocardial energy metabolism and the relevant signaling pathways connected to those modifications. Table 1 provides a summary of relevant published references on each signaling pathway. Increasing our knowledge of the molecular mechanisms that underlie metabolic disorders in DCM will help us develop effective treatment strategies.
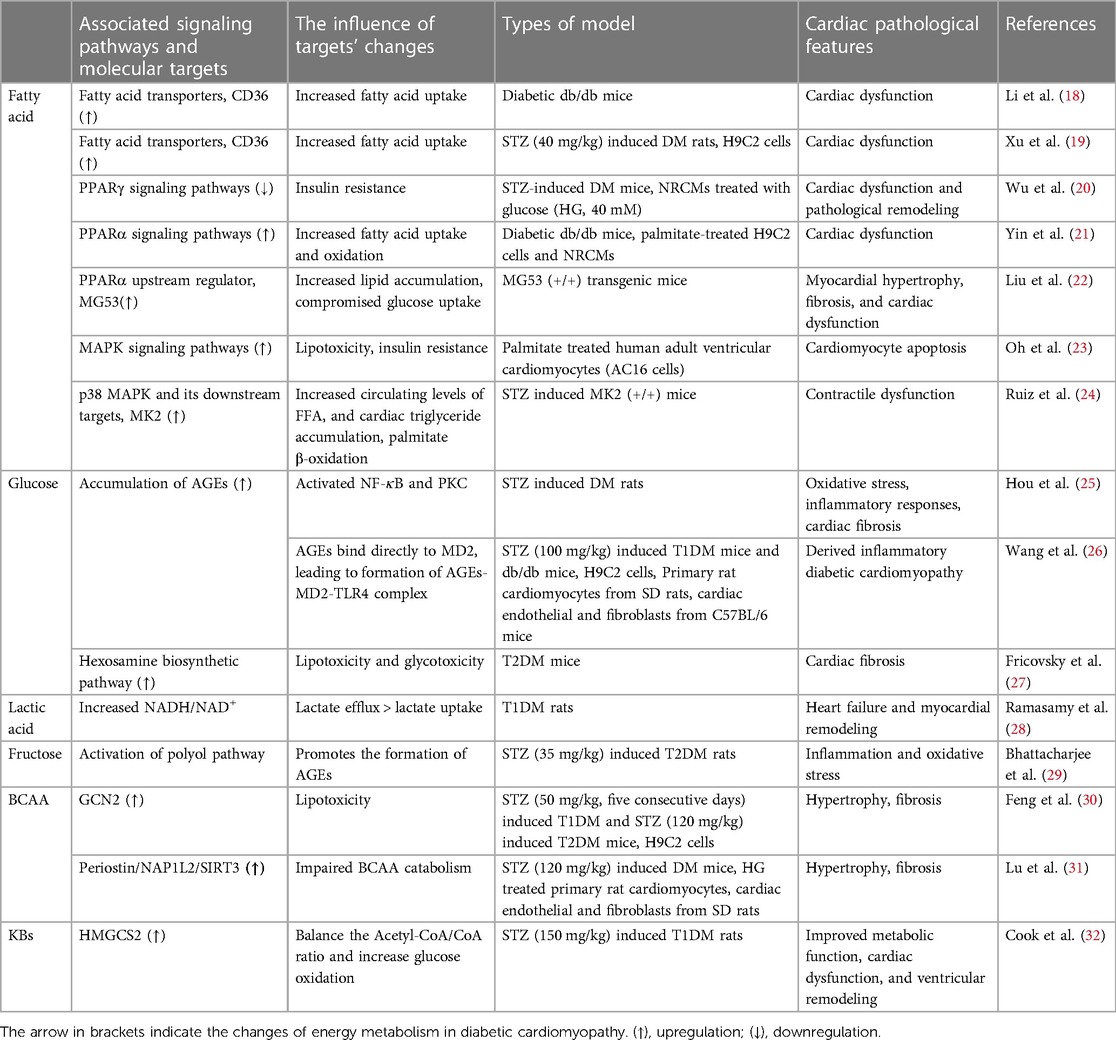
Table 1. Summary of references on signaling pathways relevant to metabolic disorders in DCM.
Disrupted lipid metabolism is an early event in diabetic heart functional abnormalities (33). McGavock et al. discovered myocardial lipid deposition in diabetic patients with normal heart function, implying that metabolic disturbances occur prior to the development of left ventricular dysfunction (33). Diabetic heart lipid metabolism is impacted due to changes in the expression of transporters involved in FA uptake (34), alterations in the PPAR signaling pathway during FA oxidation (35), accumulation of lipotoxicity, and activation of the MAPK signaling pathway (36). Lipid metabolism alterations in type 2 diabetes mellitus (T2DM) have been widely reported, and notably, lipid accumulation in type 1 diabetes mellitus (T1DM) has also been reported by relevant studies (37–39).
Heart FAs are composed of non-esterified FAs that combine with plasma albumin in the bloodstream and esterified FAs in the form of lipoprotein (40). In cardiomyocytes, 70%–90% of cytosolic FA is translocated into mitochondria for β-oxidation, which results in the production of ATP, while the remaining fraction is esterified to triglycerides (TAGs) (11). As the primary metabolic substrate, long-chain FAs (LCFAs) are esterified to fatty acyl-CoAs, which are then converted to acylcarnitines by carnitine palmitoyltransferase-1 (CPT1). Carnitine acylcarnitine translocase then transports acylcarnitines across the inner mitochondrial membrane to convert them into carnitines (41). Finally, mitochondrial FA undergoes β-Oxidation to produce acetyl CoA, which is further metabolized into the tricarboxylic acid cycle to produce ATP (41).
Transporters involved in cardiomyocyte FA uptake include FA translocase (FAT/CD36), FA binding protein (FABPpm), and FA transport protein (FATP) (42). FABPpm works with CD36 to facilitate FA transmembrane transport of FAs (34).
CD36 is known as a scavenger receptor that plays an important role in the uptake of long-chain FAs (34). Abnormal CD36 distribution alters myocardial energy supply, resulting to cardiac dysfunction (18). Reduced lipid uptake, attenuated lipotoxicity, and reduced cardiomyocyte apoptosis are the effects of the downregulation of CD36 in diabetic cardiomyopathy (19).
Hyperglycemia and hyperlipidemia promote CD36 translocation to the cell membrane, leading to increased FA uptake (43). Endosomes store nearly 50% of the CD36 (44). The CD36 recycling occurs in endosomes, intermediate vesicles, and at the membrane (44). The subcellular localization of CD36 is determined by the pH of the endosome, which is kept acidic by the proton pump H+—ATPase (V-ATPase) (45). Increased intracellular LCFA levels cause the V-ATPase subcomplex to disassemble from the intact complex, resulting in endosome alkalinization and increased relocation of CD36 to the cardiomyocyte membrane (45).
Additionally, insulin stimulation promotes CD36 translocation from endosomes to the cell membrane (43). It has been established that the vesicle-associated membrane protein (VAMP) family VAMP2, VAMP3, and VAMP4 mediate the transportation of CD36 to endosomes, intermediate vesicles, and sarcolemma (46). The only member of VAMP family of proteins, VAMP2, which is regulated by the Akt pathway, is responsible for CD36 translocation away from the sarcolemma (46). Hyperinsulinemia activates the insulin receptor substrate 1 (IRS1)/phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB/Akt) pathways, which deactivate Akt substrate 160 (AS160) through Ser/Thr phosphorylation and subsequently reduce the downstream inhibition of Rab GTPase activating proteins (Rabs) (47). The activation of Rabs prevents the transport of CD36, which is carried out by the vesicle-associated membrane protein family (47).
FATP consists of six highly homologous proteins that are encoded by the SLC27A1-6 gene (48). LCFAs can either cross the plasma membrane directly through the FATP complex or accumulate at the plasma membrane first by binding to CD36 and then deliver FAs to FATP (49). FATP has acyl-CoA synthetase activity that stimulates the rate of cellular FA uptake by converting incoming FAs directly into their acyl-CoA thioester (48, 49).
FA uptake and lipid accumulation are increased in mice hearts that specifically overexpress FATP1 (50). Additionally, FATP6, which is primarily expressed in the heart, promotes the uptake of long-chain FAs (51). Because there is no animal model for SLC27A6, the exact role of FATP6 in the heart and other tissues is unkown (51).
A key regulator of FA metabolism is the nuclear hormone receptor superfamily of ligand-activated transcription factors, known as PPAR (peroxisome proliferator activated receptor) (52). Three subtypes of PPAR exist (α, β/δ, γ) (52).
The regulation of lipid biosynthesis and insulin sensitivity is greatly influenced by the PPAR isoform PPARγ (53). The expression of PPARγ is decreased in the hearts of streptozotocin-induced diabetic rats (20). Thiazolidinediones, a type of PPARγ agonist, are used to treat type 2 diabetes mellitus (T2DM) because its effect of promoting insulin sensitization, suggesting that PPARγ activation is beneficial to ameliorate diabetic cardiomyopathy (54).
The PPARγ co-activator-1 (PGC-1) family includes PGC-1α, PGC-1β and PGC-1-related coactivator (PRC). PGC-1α and PGC-1β are involved in mitochondrial biosynthesis (53). Nuclear transcription factor (NRF), which promotes mitochondrial proliferation and regulates cellular energy metabolism, are some of the downstream factors that are stimulated by the activation of PGC-1α signal (55, 56). In the heart of T2DM db/db mice, PGC-1β expression is elevated, enhanced by the transcriptional activity of PPARα (21).
The expression of the genes responsible for FA uptake, mitochondrial FA uptake, and FA oxidation is stimulated by the activation of PPARα and PPARβ/δ (35, 52). PPARα and its target genes are upregulated in DCM, which causes increased FA uptake and decreased glucose utilization, resulting in abnormal cardiac metabolism and cardiac dysfunction (57, 58).
A novel PPARα upstream regulator called Mitsugumin 53 (MG53), also known as TRIM72, controls the expression of PPARα-encoding genes (22). MG53 is protective in cardiac ischemia/reperfusion injury, cardiomyocyte membrane injury, and cardiac fibrosis, but it simultaneously acts as an E3 ligase to promote ubiquitin dependent degradation of the insulin receptor and insulin receptor substrate, leading to insulin resistance and metabolic syndrome (59, 60).
The connection between MG53 and metabolic disorders is still debatable. According to previous studies, mice with heart-specific MG53 overexpression displayed symptoms of diabetic cardiomyopathy (22, 61). By up-regulating PPARα and its downstream targets, MG53 contributes to the pathological development of diabetic cardiomyopathy (61). Recent research, however, also revealed the opposite outcomes. According to Wang and colleagues, when compared to controls, serum MG53 levels in diabetic patients or db/db mice had decreased or remained unchanged on western blot results using a high specificity monoclonal antibody for MG53 (62, 63). The changes in MG53 may play different roles in the heart and serum according to its distribution.
The state of lipids and their intermediates is dynamic. The production and accumulation of two toxic lipids, namely ceramide and diacylglycerol, may be caused by the activation of mitogen-activated protein kinase (MAPK) (36). MAPK has two subfamily members, namely p38 MAPK and c-Jun N-terminal kinase (JNK), which mediate insulin resistance and cardiac dysfunction by promoting or inhibiting the translation of target genes (36).
p38 MAPK regulates lipid and glucose metabolism, mediates insulin resistance, and contributes to diabetic cardiac dysfunction (64). Exposure to high concentrations of palmitate to mimic the lipotoxicity of diabetic hearts increased p38 MAPK phosphorylation and cardiomyocyte apoptosis (23). The activation of p38 MAPK may be associated with reduced IRS1 and IRS2 in insulin resistance. Compared with the control group, IRS1 and IRS2 protein levels and Akt phosphorylation are decreased in the hearts of diabetic mice, whereas p38 MAPK phosphorylation is increased (65).
Downregulation of p38 MAPK is beneficial for diabetic hearts. Atorvastatin improves heart function by reducing inflammation and inhibiting the activation of p38 MAPK in diabetic cardiomyopathy (66). SB203580 and SB202190, which are p38 MAPK inhibitors, reduced cell apoptosis and improved cardiac function in an animal model of STZ-induced diabetes (64).
In diabetes, the inactivation or inhibition of p38 MAPK and its downstream targets (for instance, MK2), alleviates lipid metabolism disorders and improves cardiac function (24, 67). FA oxidation and esterification is enhanced in diabetic mice, whereas the levels of free FAs are practically equal in MK2-knockout diabetic mice (MK2−/− mice) and non-diabetic mice (24).
Besides p38 MAPK, decreased JNK signaling also ameliorates diabetic cardiomyopathy (68, 69). Abnormal expression of mammalian sterile 20-like kinase 1 (MST1) is closely related to cardiac diseases (70). In db/db mice, Mst1 down-regulation protects against lipotoxic cardiac injury by inhibiting MEKK1/JNK signaling (69).
Furthermore, mammalian target of rapamycin (mTOR) is an evolutionarily conserved serine/threonine kinase involved in lipid metabolism (71). Canagliflozin (CAN), a sodium-glucose cotransporter 2 inhibitor, binds to mTOR and then inhibits mTOR phosphorylation and the expression of hypoxia inducible factor-1α (HIF-1α), reducing myocardial cellular lipotoxicity and heart injuries in diabetes (72).
In diabetic hearts, lipid oxidation is increased while glucose oxidation is decreased (9). Members of the Sirtuins family overexpression can assist the heart's glucose oxidation efficiency (73). Glycolipid metabolism can be improved by activating AMPK signaling pathway, which promotes glucose uptake and oxidation (74).
Glucose is aerobically oxidized, producing pyruvate, which enters the mitochondria for oxidative decarboxylation. Acetyl CoA and NADH produced by β-oxidation reduce glucose oxidation through activating pyruvate dehydrogenase kinase (PDK) and inhibiting the phosphorylation of the pyruvate dehydrogenase (PDH) enzyme complex (12). This relationship between FA and glucose metabolism is called the glucose-FA cycle or Randle cycle (12, 41).
Seven proteins constitute the Sirtuins (SIRT) are proteins that share a highly conserved NAD+ binding catalytic domain (SIRT1-SIRT7) (73). Sirtuin 3 (SIRT3) is a NAD+ dependent deacetylase, which can improve mitochondrial energy metabolism (75). According to recent research, the SIRT3 pathway promotes the shift of heart energy substrates from FA β-oxidation to glucose oxidation in DCM (75, 76). SIRT1 and SIRT6 up-regulation have also been shown to improve diabetic cardiomyopathy (77, 78).
AMP-activated protein kinase (AMPK) is a myocardial glucose metabolism mediator that regulates energy metabolism exchange under cellular stress. AMPK activation increases GLUT4 expression and promotes GLUT4 redistribution to the muscular membrane, enhancing glucose uptake and improving metabolic disorders in metabolic diseases such as diabetes (74, 79). Carvacrol has previously been shown to restore GLUT4 membrane translocation mediated by PI3K/Akt signaling, lower blood glucose levels, and inhibit cardiac remodeling in both type 1 diabetes mellitus (T2DM) and type 2 diabetes mellitus (T2DM) mice (80).
Changes in the concentrations of ADP, AMP and ATP regulate AMPK. Reduced ATP production caused by hypoxia or increased energy expenditure during muscle contraction can increase cellular AMP concentrations, activating AMPK (81). AMPK activation in this scenario may favor the promotion of catabolic responses (such as FA oxidation and glycolysis) while suppressing anabolic responses (for example, FA, triglyceride, cholesterol and protein synthesis) (82, 83).
Tumor suppressor liver kinase B1 (LKB1), Ca2+/calmodulin-dependent protein kinase kinase β (CaMKKβ, and TGF-β activated kinase 1 (TAK1) are the three upstream kinases of AMPK (84). LKB1 directly phosphorylates AMPK THR-172 to activate its enzyme activity (85). The activation of LKB1-dependent AMPK signaling ameliorates diabetic cardiomyopathy (31, 86, 87).
An increased intracellular Ca2+ concentration promotes CaMKKβ-mediated AMPK activation. Adiponectin induces extracellular Ca2+ influx through adiponectin receptor 1 (AdipoR1), which then activates CaMKKβ and further activates the AMPK signaling pathway, playing a key role in insulin production and secretion (88).
TAK1 was discovered as the third upstream kinase AMPK activator (89). Current studies focus on its role in promoting cytoprotective autophagy through the formation of a complex with its accessory subunit TAK1 binding proteins (TAB1, TAB2, TAB3) (90). TAK1′s mechanisms of action in diabetic with metabolically disrupted hearts are unknown.
AGEs and the occurrence and progression of DCM are closely related phenomena (3, 4). AGEs accumulation is often observed in DCM disease models (91). Reducing the levels of AGEs and Receptor for AGEs (RAGE) is beneficial for structural and functional abnormalities improvement in diabetic heart (91).
AGEs are found in tissues, cells, and blood. They are the collective term for a class of stable end products formed after the free amino groups of substances such as proteins, amino acids, lipids, or nucleic acids undergo a series of reactions involving condensation, rearrangement, cleavage, and oxidative modification with the carbonyl groups of reducing sugars (6, 92).
Blood glucose levels and AGEs levels in the body are closely correlated. Persistently elevated blood sugar levels promote the glycosylation reaction between proteins and glucose, which leads to the production of AGEs in insulin deficient or insulin resistant diabetes (6).
Binding of AGEs to their receptors (RAGEs) activates multiple intracellular signaling pathways. For instance, AGEs can activate nuclear factor kappa A-β (NF-κB) (25) and protein kinase C (PKC) (93, 94) signaling, which can result in the production of reactive oxygen species (ROS), inflammatory responses, and cardiac dysfunction. AGEs induced by high glucose (HG) directly bind to MD2 to form the AGEs-MD2-TLR4 complex, which starts the proinflammatory pathway, leading to inflammatory diabetic cardiomyopathy (26). Fruthermore, AGEs have the ability to alter protein structure, promote collagen cross-linking, and accelerate atherosclerosis development. The activation of AGE/RAGE signaling pathway stimulates the activaion of fibroblast and promotes fibroblast differentiation into myofibroblast, which increases extracellular matrix accumulation and accelerates pathological remodeling of diabetes heart (95).
Future treatments for the chronic diabetic complications may include blocking the AGEs-RAGE system (96). Recent studies have shown that vitamin D reduces NF-κB activity, which decreases RAGE expression (97). Calcitriol has the potential to treat RAGE-mediated cardiovascular complications, because it down-regulates RAGE expression through the proteolysis of RAGE in HL-1 cardiomyocytes, mediated by disintegrin and metalloproteinase 10 (ADAM10) (97). In the DCM disease model, inhibition of protein kinase R (PKR) was found to improve diabetes-induced fibrosis by down-regulating AGEs and ERK 1/2 (98).
The accumulation of O-linked N-acetylglucosamine (O-GlcNAc), a post-translational modification of proteins, in the heart predisposes to glucotoxicity, inducing insulin resistance (4, 99). Diabetic cardiomyopathy can be improved by HBP hyperactive suppression (100, 101).
The primary branch of the glycolysis pathway is HBP. This pathway metabolizes 2%–5% of the glucose (99). O-GlcNAcylation is a dynamic and reversible modification that primarily occurs in the cytoplasm and nucleus, in contrast to advanced glycosylation and other forms of glycosylation in the endoplasmic reticulum and Golgi apparatus (102).
When glucose enters the cell, it is phosphorylated to glucose-6-phosphate and metabolized to fructose-6-phosphate, which feeds into the glucose oxidative metabolism, glycolysis, and gluconeogenesis pathways (7). The first reaction is the rate-limiting conversion of fructose-6-phosphate to glucosamine-6-phosphate by l-glutamine-fructose-6-phosphate amidotransferase (GFAT) with conversion of glucosamine to glutamine. The second reaction involves the use of acetyl CoA as a substrate to convert glucosamine-6-phosphate to n-acetylamino-6-phosphate by glucosamine-6-phosphate acetyltransferase. After that, phosphoglucomutase converts n-acetylglucosamine-6-phosphate to n-acetylglucosamine-1-phosphate. Finally, pyrophosphorylase catalyzes the conjugation of N-acetylglucosamine to uridine nucleotides, resulting in uridine diphosphate N-acetylglucosamine UDP GlcNAc, which acts as the monosaccharide donor for o-glcnacylation. O-GlcNAc transferase (OGT) links O-GlcNAc to protein serine and threonine residues during this process. Conversely, β-n-acetylglucosaminidase (OGA) removes O-GlcNAc (102).
Under physiological conditions, transient activation of O-GlcNAc signals acts as a cellular protective mechanism (103). However, growing evidence suggests that long-term O-Glcnacylation protein elevation in diabetic animals’ hearts is associated with glucose toxicity (103, 104). Hyperglycemia induces glycogen synthase O-Glcnacylation, which reduces its activity and leads to insulin resistance. Increased HBP flux and O-Glcnacylation increased FA oxidation during glucosamine perfusion in hearts in vitro, implying that high O-GlcNAC levels cause both cardiac lipotoxicity and cardiac glycotoxicity (27).
Additionally, the activity of a set of proteins related to metabolic regulation, such as IRS1/2, Akt, AMPK, and GLUT4, decreases through O-GlcNAc modification. Demonstrating that O-GlcNAcylation may be a potential mechanism underlying the typical metabolic dysfunction of hearts (99).
Recent research has demonstrated that the hypoglycemic drug dapagliflozin, a sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor, reduces cardiac HBP and improves diastolic dysfunction in lipodystrophic T2DM mouse models (101). In diabetic rats with significantly increased O-GlcNAcylation, thiamine may block the biosynthesis of hexosamine and prevent diabetes-induced cardiac fibrosis (100).
Lactic acid is also a vital energy substrate for the myocardium during exercise or myocardial stress (15). Lactic acid is produced by glucose through anaerobic glycolysis (105). The discovery of monocarboxylate transporters (MCTs) laid the foundation for the study of the transmembrane transport of lactate. The MCTs consist of 14 members, and MCTs 1–4 are responsible for transporting monocarboxylates (like L-lactate and pyruvate) and ketone bodies across the plasma membrane (106). Lactate dehydrogenase (LDH) can convert lactic acid into pyruvate in normoxic conditions, providing energy for the tricarboxylic acid cycle (TCA) to produce ATP (106).
Diabetes impairs glucose and lactate metabolism in the myocardium (15). Decreased lactate uptake is associated with an increased cytosolic NADH/NAD+ ratio in the diabetic state (28).
Elevated fructose levels in the hearts of diabetic patients can be divided into extracellular and intracellular sources. Dietary intake is the main extracellular source of fructose, which enters the cardiomyocytes via the systemic circulation. The intracellular source is the polyol pathway, wherein glucose is reduced to sorbitol by aldose reductase and sorbitol is then oxidized to fructose by sorbitol dehydrogenase (SDH) (14).
It has been demonstrated that the proteins required for fructose transport, including GLUT5, GLUT11 and GLUT12, are expressed in the heart (13). The GLUT11 and GLUT12 have little impact on fructose transport in cardiomyocytes, while the glucose transporter GLUT5 is highly specific for fructose and has a low affinity for glucose transport (13).
The formation of fructose-derived AGEs is faster than that of AGEs derived from glucose. Persistent hyperglycemia activates polyol pathways, which cause T2DM to overproduce AGEs (107). Protocatechuic acid, a phenolic from the leaves of Polygonum cuspidatum, significantly suppressed AGEs levels in the serum of T2DM rats by inhibiting the activation of the polyol pathway through reducing the activities of aldose reductase and sorbitol dehydrogenase and increasing glyoxalase I activity (29).
Fructose exposure is associated with metabolic disorders, lipid accumulation, inflammation, and apoptosis (108, 109). Increased cellular fructose metabolism promotes the formation of O-Glcnacylation and AGEs, which are crucial for fructose-mediated cardiomyocyte signaling and dysfunction (13, 14).
In the field of diabetic cardiomyopathy research, the majority of research on metabolic substrates has focused on the changes in FA and glucose metabolism. However, amino acids and ketone bodies (KB) are also used as fuel by cardiomyocytes (17, 110). Branched amino acids (BCAA) and ketone bodies produce acetyl-CoA via branched-chain alpha-ketoate dehydrogenase (BCKD) and beta-hydroxybutyrate dehydrogenase (βDH), respectively, which supplies ATP to the heart (17).
Branched chain amino acids (BCAA) are composed of valine, leucine, and isoleucine (111). In the heart, the first step of BCAA metabolism is the transamination of BCAA into the corresponding branched-chain α-ketoic acid (BCKA) by mitochondrial branched-chain aminotransferase (BCATm). The second step involves oxidative decarboxylation of BCKA by mitochondrial branched-chain alpha-ketoate dehydrogenase (BCKDH). Finally, acetyl-CoA is generated for the TCA cycle (112).
Increased BCAA levels in the blood may be a diabetes risk factor (111). Targeting the gut microbiota to reduce the abnormalities of circulating branched chain amino acids may be a key strategy to improve heart function, according to Yang and his colleagues (112).
General control nonderepressible 2 (GCN2) is an evolutionarily conserved eukaryotic initiation factor 2α (eIF2α) Kinases, which serves as an amino acid sensor (113). When amino acid levels are insufficient, GCN2 can selectively stimulate amino acid biosynthetic gene expression, maintaining amino acid homeostasis (30, 113). GCN2 deficiency in mice improves streptozotocin (STZ) or high-fat diet (HFD) induced diabetic cardiac dysfunction by reducing lipotoxicity and reducing oxidative stress (30).
BCAA also has the function of regulating signaling pathways in the heart. Continuous mTOR signaling, particularly involving leucine, impairs insulin signaling via insulin receptor substrates (IRS) (16, 114). Additionally, impaired BCAA metabolism causes toxic BCAA metabolites accumulation (110). In myocardial fibroblasts, high expression of periosteal protein upregulates nucleosome assembly protein 1-like 2 (NAP1L2) to deacetylate enzymes related to BCAA catabolism, which promotes cardiac fibrosis in diabetic cardiomyopathy (31).
Diabetes can lead to an increase in circulating ketones bodies (KBs) (17, 115). Ketogenesis is the synthesis of KBs through consuming acetyl coenzyme A (acetyl-CoA) produced by lipolysis (116). The two main KBs, acetoacetate and β-hydroxybutyrate (βHB), are essential in maintaining bioenergy homeostasis in diabetic cardiomyopathy (116). It has been established that the expression of hydroxymethylglutaryl-CoA synthase 2 (HMGCS2) is increased in T1DM hearts (32). In comparison with control rats, HMGCS2 protein expression was eight times higher in the hearts of diabetic rats (32). This suggests that the heart opposes “metabolic inflexibility” by transferring excess intramitochondrial Acetyl-CoA of FA oxidation to KBs, thereby releasing free CoA to balance the Acetyl-CoA/CoA ratio in favor of increased glucose oxidation via the pyruvate dehydrogenase complex. Enhanced ketogenesis is likely an adaptive mechanism of cardiac function in diabetic hearts (117).
Increased KBs use in T2DM may help improve cardiac energy efficiency (116). Ketone levels increase in patients with T2DM receiving SGLT-2 inhibitors, which may be associated with a reduced risk of heart failure mortality (118). The increased use of KBs in patients with heart failure or diabetes may be explained by the fact that ketone body breakdown requires less oxygen than FA oxidation to produce the same amount of ATP (119). In contrast with a control group receiving only AGE, Tao and his colleagues found that AGE plus KB treatment inhibited FA oxidation (120). However, KBs leads to DCM cardiac dysfunction and ventricular remodeling despite improvements in metabolic function (120). Therefore, there is still debate regarding how KBs utilization affects cardiac function in diabetes.
The pathological mechanism of diabetic cardiomyopathy is also related to the expression of non-coding RNA. MiRNA binds to the 3′ untranslated region (UTR) of messenger RNA (mRNA) molecule and regulates the expression of cardiac metabolism-related genes at the post-transcriptional level through mRNA translation inhibition or degradation (121). MiR-320 is highly expressed in diabetic cardiomyopathy mice and diabetic patients (18). It translocates to the nucleus and enhances the transcription of the FA metabolism-related gene CD36, which increases the uptake of free FA and induces myocardial lipotoxicity (18). Conversely, the expression of mir-200b-3p was decreased in DCM. Upregulation of mir-200b-3p inhibits CD36 and reduces cardiomyocyte apoptosis (19). Studies have shown that PGC-1β may be a target of miR-30c. MiR-30c reduces transcriptional activity of PPARα regulated by PGC-1β and suppresses the conversion of cardiac metabolism to FA induced by palmitate (21).
LncRNA is a specific transcript comprising over 200 nucleotides that are not translated into proteins. They bind to the miRNA through base pairing and block its regulatory function (122). Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1, also named NEAT2) is a long non-coding RNA with a miR-26a-binding region in the transcriptional sequence, which is significantly upregulated in cardiomyocytes treated with palmitic acid (123). The downregulation of MALAT-1 inhibits the TLR4/NF-κB signaling pathway by regulating HMGB1 expression, which may be the potential mechanism to relieve the inflammatory response and decrease myocardial lipotoxic injury (123).
At present, the research into metabolic disorders in diabetic cardiomyopathy is comprehensive and extensive. The changes in some signaling pathways and molecular targets have been relatively clear, but there are still some limitations that cannot be ignored.
To begin, the signal pathways underlying metabolic disorders in DCM crosstalk with one another, and the relevant molecular mechanisms are complex. For example, CD36 inhibits AMPK activation by forming a molecular complex with Lyn and LKB1 and then participates in the energy regulation process (124). PGC-1α not only regulates FA β-oxidation and excess myocardial lipid accumulation, but it also promotes the AMPK regulated expression of several key players in mitochondrial and glucose metabolism (55, 56).
Second, the multiple pathophysiological mechanisms involved in the development of DCM remain controversial. There is no agreement on whether changes in some signaling pathways and molecular targets (such as HBP and MG53) are beneficial or harmful in the process of disease progression has not yet reached a consensus. More experimental research and theoretical support are required.
In recent years, there has been an increase in the number of studies focusing on mitochondrial dysfunction. The balance between mitochondrial biogenesis and mitophagy is critical for maintaining cellular metabolism in the diabetic heart (125, 126). A comprehensive study of the mitochondrial quality control (MQC) system, which includes mitochondrial fission, fusion, and mitophagy, may shed new light on cardiometabolic disorders in DCM.
Besides, previous research by our group has shown that overexpression of the Hippo pathway effector YAP promotes cardiac remodeling and cardiac fibrosis, leading to cardiac dysfunction (127, 128). Additionally, it may also participate in the glycosylation process and affect diabetic cardiometabolism. However, more investigation into the relationship between these molecular and metabolic disorders in DCM is required. A greater understanding of diabetes and the complications associated with cardiovascular disease will result from an in-depth investigation of these signaling and pathological pathways in myocardial metabolic disorders.
NH: Funding acquisition, Investigation, Supervision, Writing – original draft, Writing – review & editing. YZ: Data curation, Writing – original draft. YL: Formal Analysis, Writing – review & editing. WJ: Methodology, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This work was supported by the Natural Science Foundation of Guangdong Province (grant No. 2023A1515010412), the open research funds from the Sixth Affiliated Hospital of Guangzhou Medical University, Qingyuan People's Hospital (grant No. 202201-203).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: results from the international diabetes federation diabetes atlas, 9(th) edition. Diabetes Res Clin Pract. (2019) 157:107843. doi: 10.1016/j.diabres.2019.107843
2. Dillmann WH. Diabetic cardiomyopathy. Circ Res. (2019) 124:1160–2. doi: 10.1161/CIRCRESAHA.118.314665
3. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. (2018) 122:624–38. doi: 10.1161/CIRCRESAHA.117.311586
4. Ritchie RH, Abel ED. Basic mechanisms of diabetic heart disease. Circ Res. (2020) 126:1501–25. doi: 10.1161/CIRCRESAHA.120.315913
5. Park JJ. Epidemiology, pathophysiology, diagnosis and treatment of heart failure in diabetes. Diabetes Metab J. (2021) 45:146–57. doi: 10.4093/dmj.2020.0282
6. Khalid M, Petroianu G, Adem A. Advanced glycation end products and diabetes Mellitus: mechanisms and perspectives. Biomolecules. (2022) 12. doi: 10.3390/biom12040542
7. Chen Y, Zhao X, Wu H. Metabolic stress and cardiovascular disease in diabetes Mellitus: the role of protein O-GlcNAc modification. Arterioscler Thromb Vasc Biol. (2019) 39:1911–24. doi: 10.1161/ATVBAHA.119.312192
8. Ormazabal V, Nair S, Elfeky O, Aguayo C, Salomon C, Zuñiga FA. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc Diabetol. (2018) 17:122. doi: 10.1186/s12933-018-0762-4
9. de Wit-Verheggen V, van de Weijer T. Changes in cardiac metabolism in prediabetes. Biomolecules. (2021) 11. doi: 10.3390/biom11111680
10. Levelt E, Gulsin G, Neubauer S, McCann GP. Mechanisms in endocrinology: diabetic cardiomyopathy: pathophysiology and potential metabolic interventions state of the art review. Eur J Endocrinol. (2018) 178:R127–39. doi: 10.1530/EJE-17-0724
11. Ho KL, Karwi QG, Connolly D, Pherwani S, Ketema EB, Ussher JR, et al. Metabolic, structural and biochemical changes in diabetes and the development of heart failure. Diabetologia. (2022) 65:411–23. doi: 10.1007/s00125-021-05637-7
12. An D, Rodrigues B. Role of changes in cardiac metabolism in development of diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol. (2006) 291:H1489–506. doi: 10.1152/ajpheart.00278.2006
13. Delbridge LM, Benson VL, Ritchie RH, Mellor KM. Diabetic cardiomyopathy: the case for a role of fructose in disease etiology. Diabetes. (2016) 65:3521–8. doi: 10.2337/db16-0682
14. Annandale M, Daniels LJ, Li X, Neale J, Chau A, Ambalawanar HA, et al. Fructose metabolism and cardiac metabolic stress. Front Pharmacol. (2021) 12:695486. doi: 10.3389/fphar.2021.695486
15. Dong S, Qian L, Cheng Z, Chen C, Wang K, Hu S, et al. Lactate and myocadiac energy metabolism. Front Physiol. (2021) 12:715081. doi: 10.3389/fphys.2021.715081
16. Bloomgarden Z. Diabetes and branched-chain amino acids: what is the link? J Diabetes. (2018) 10:350–2. doi: 10.1111/1753-0407.12645
17. Abdul KA, Clarke K, Evans RD. Cardiac ketone body metabolism. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165739. doi: 10.1016/j.bbadis.2020.165739
18. Li H, Fan J, Zhao Y, Zhang X, Dai B, Zhan J, et al. Nuclear miR-320 mediates diabetes-induced cardiac dysfunction by activating transcription of fatty acid metabolic genes to cause lipotoxicity in the heart. Circ Res. (2019) 125:1106–20. doi: 10.1161/CIRCRESAHA.119.314898
19. Xu L, Chen W, Ma M, Chen A, Tang C, Zhang C, et al. Microarray profiling analysis identifies the mechanism of miR-200b-3p/mRNA-CD36 affecting diabetic cardiomyopathy via peroxisome proliferator activated receptor-γ signaling pathway. J Cell Biochem. (2019) 120:5193–206. doi: 10.1002/jcb.27795
20. Wu X, Zhang T, Lyu P, Chen M, Ni G, Cheng H, et al. Traditional Chinese medication qiliqiangxin attenuates diabetic cardiomyopathy via activating PPAR gamma. Front Cardiovasc Med. (2021) 8. doi: 10.3389/fcvm.2021.698056
21. Yin Z, Zhao Y, He M, Li H, Fan J, Nie X, et al. MiR-30c/PGC-1β protects against diabetic cardiomyopathy via PPARα. Cardiovasc Diabetol. (2019) 18:7. doi: 10.1186/s12933-019-0811-7
22. Liu F, Song R, Feng Y, Guo J, Chen Y, Zhang Y, et al. Upregulation of MG53 induces diabetic cardiomyopathy through transcriptional activation of peroxisome proliferation-activated receptor α. Circulation. (2015) 131:795–804. doi: 10.1161/CIRCULATIONAHA.114.012285
23. Oh CC, Nguy MQ, Schwenke DC, Migrino RQ, Thornburg K, Reaven P. P38α mitogen-activated kinase mediates cardiomyocyte apoptosis induced by palmitate. Biochem Biophys Res Commun. (2014) 450:628–33. doi: 10.1016/j.bbrc.2014.06.023
24. Ruiz M, Coderre L, Lachance D, Houde V, Martel C, Thompson LJ, et al. MK2 deletion in mice prevents diabetes-induced perturbations in lipid metabolism and cardiac dysfunction. Diabetes. (2016) 65:381–92. doi: 10.2337/db15-0238
25. Hou J, Zheng D, Fung G, Deng H, Chen L, Liang J, et al. Mangiferin suppressed advanced glycation end products (AGEs) through NF-κB deactivation and displayed anti-inflammatory effects in streptozotocin and high fat diet-diabetic cardiomyopathy rats. Can J Physiol Pharmacol. (2016) 94:332–40. doi: 10.1139/cjpp-2015-0073
26. Wang Y, Luo W, Han J, Khan ZA, Fang Q, Jin Y, et al. MD2 Activation by direct AGE interaction drives inflammatory diabetic cardiomyopathy. Nat Commun. (2020) 11:2148. doi: 10.1038/s41467-020-15978-3
27. Fricovsky ES, Suarez J, Ihm SH, Scott BT, Suarez-Ramirez JA, Banerjee I, et al. Excess protein O-GlcNAcylation and the progression of diabetic cardiomyopathy. Am J Physiol Regul Integr Comp Physiol. (2012) 303:R689–99. doi: 10.1152/ajpregu.00548.2011
28. Ramasamy R, Oates PJ, Schaefer S. Aldose reductase inhibition protects diabetic and nondiabetic rat hearts from ischemic injury. Diabetes. (1997) 46:292–300. doi: 10.2337/diab.46.2.292
29. Bhattacharjee N, Dua TK, Khanra R, Joardar S, Nandy A, Saha A, et al. Protocatechuic acid, a phenolic from Sansevieria roxburghiana leaves, suppresses diabetic cardiomyopathy via stimulating glucose metabolism, ameliorating oxidative stress, and inhibiting inflammation. Front Pharmacol. (2017) 8:251. doi: 10.3389/fphar.2017.00251
30. Feng W, Lei T, Wang Y, Feng R, Yuan J, Shen X, et al. GCN2 deficiency ameliorates cardiac dysfunction in diabetic mice by reducing lipotoxicity and oxidative stress. Free Radic Biol Med. (2019) 130:128–39. doi: 10.1016/j.freeradbiomed.2018.10.445
31. Lu QB, Fu X, Liu Y, Wang ZC, Liu SY, Li YC, et al. Disrupted cardiac fibroblast BCAA catabolism contributes to diabetic cardiomyopathy via a periostin/NAP1L2/SIRT3 axis. Cell Mol Biol Lett. (2023) 28:93. doi: 10.1186/s11658-023-00510-4
32. Cook GA, Lavrentyev EN, Pham K, Park EA. Streptozotocin diabetes increases mRNA expression of ketogenic enzymes in the rat heart. Biochim Biophys Acta Gen Subj. (2017) 1861:307–12. doi: 10.1016/j.bbagen.2016.11.012
33. McGavock JM, Lingvay I, Zib I, Tillery T, Salas N, Unger R, et al. Cardiac steatosis in diabetes mellitus: a 1H-magnetic resonance spectroscopy study. Circulation. (2007) 116:1170–5. doi: 10.1161/CIRCULATIONAHA.106.645614
34. Zhang X, Fan J, Li H, Chen C, Wang Y. CD36 signaling in diabetic cardiomyopathy. Aging Dis. (2021) 12:826–40. doi: 10.14336/AD.2020.1217
35. Wang L, Cai Y, Jian L, Cheung CW, Zhang L, Xia Z. Impact of peroxisome proliferator-activated receptor-α on diabetic cardiomyopathy. Cardiovasc Diabetol. (2021) 20:2. doi: 10.1186/s12933-020-01188-0
36. Drosatos K, Schulze PC. Cardiac lipotoxicity: molecular pathways and therapeutic implications. Curr Heart Fail Rep. (2013) 10:109–21. doi: 10.1007/s11897-013-0133-0
37. Ritchie RH, Zerenturk EJ, Prakoso D, Calkin AC. Lipid metabolism and its implications for type 1 diabetes-associated cardiomyopathy. J Mol Endocrinol. (2017) 58:R225–40. doi: 10.1530/JME-16-0249
38. Cao Z, Pan J, Sui X, Fang C, Li N, Huang X, et al. Protective effects of huangqi shengmai yin on type 1 diabetes-induced cardiomyopathy by improving myocardial lipid metabolism. Evid Based Complement Alternat Med. (2021) 2021:5590623. doi: 10.1155/2021/5590623
39. Zhang J, Xiao Y, Hu J, Liu S, Zhou Z, Xie L. Lipid metabolism in type 1 diabetes mellitus: pathogenetic and therapeutic implications. Front Immunol. (2022) 13:999108. doi: 10.3389/fimmu.2022.999108
40. Carpentier AC. Abnormal myocardial dietary fatty acid metabolism and diabetic cardiomyopathy. Can J Cardiol. (2018) 34:605–14. doi: 10.1016/j.cjca.2017.12.029
41. Bayeva M, Sawicki KT, Ardehali H. Taking diabetes to heart–deregulation of myocardial lipid metabolism in diabetic cardiomyopathy. J Am Heart Assoc. (2013) 2:e000433. doi: 10.1161/JAHA.113.000433
42. He Q, Chen Y, Wang Z, He H, Yu P. Cellular uptake, metabolism and sensing of long-chain fatty acids. Front Biosci (Landmark Ed). (2023) 28:10. doi: 10.31083/j.fbl2801010
43. Shu H, Peng Y, Hang W, Nie J, Zhou N, Wang DW. The role of CD36 in cardiovascular disease. Cardiovasc Res. (2020) 118:115–29. doi: 10.1093/cvr/cvaa319
44. Glatz J, Luiken J. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J Lipid Res. (2018) 59:1084–93. doi: 10.1194/jlr.R082933
45. Wang S, Wong LY, Neumann D, Liu Y, Sun A, Antoons G, et al. Augmenting vacuolar H(+)-ATPase function prevents cardiomyocytes from lipid-overload induced dysfunction. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21041520
46. Glatz J, Luiken J, Nabben M. CD36 (SR-B2) as a target to treat lipid overload-induced cardiac dysfunction. J Lipid Atheroscler. (2020) 9:66–78. doi: 10.12997/jla.2020.9.1.66
47. Samovski D, Su X, Xu Y, Abumrad NA, Stahl PD. Insulin and AMPK regulate FA translocase/CD36 plasma membrane recruitment in cardiomyocytes via rab GAP AS160 and Rab8a rab GTPase. J Lipid Res. (2012) 53:709–17. doi: 10.1194/jlr.M023424
48. Anderson CM, Stahl A. SLC27 fatty acid transport proteins. Mol Aspects Med. (2013) 34:516–28. doi: 10.1016/j.mam.2012.07.010
49. Pohl J, Ring A, Ehehalt R, Herrmann T, Stremmel W. New concepts of cellular fatty acid uptake: role of fatty acid transport proteins and of caveolae. Proc Nutr Soc. (2004) 63:259–62. doi: 10.1079/PNS2004341
50. Chiu HC, Kovacs A, Blanton RM, Han X, Courtois M, Weinheimer CJ, et al. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ Res. (2005) 96:225–33. doi: 10.1161/01.RES.0000154079.20681.B9
51. Gimeno RE, Ortegon AM, Patel S, Punreddy S, Ge P, Sun Y, et al. Characterization of a heart-specific fatty acid transport protein. J Biol Chem. (2003) 278:16039–44. doi: 10.1074/jbc.M211412200
52. Montaigne D, Butruille L, Staels B. PPAR control of metabolism and cardiovascular functions. Nat Rev Cardiol. (2021) 18:809–23. doi: 10.1038/s41569-021-00569-6
53. Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med. (2013) 19:557–66. doi: 10.1038/nm.3159
54. Bansal G, Thanikachalam PV, Maurya RK, Chawla P, Ramamurthy S. An overview on medicinal perspective of thiazolidine-2,4-dione: a remarkable scaffold in the treatment of type 2 diabetes. J Adv Res. (2020) 23:163–205. doi: 10.1016/j.jare.2020.01.008
55. Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. (2006) 116:615–22. doi: 10.1172/JCI27794
56. Villena JA. New insights into PGC-1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. FEBS J. (2015) 282:647–72. doi: 10.1111/febs.13175
57. Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. (2002) 109:121–30. doi: 10.1172/JCI14080
58. Finck BN, Han X, Courtois M, Aimond F, Nerbonne JM, Kovacs A, et al. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc Natl Acad Sci U S A. (2003) 100:1226–31. doi: 10.1073/pnas.0336724100
59. Jiang W, Liu M, Gu C, Ma H. The pivotal role of mitsugumin 53 in cardiovascular diseases. Cardiovasc Toxicol. (2021) 21:2–11. doi: 10.1007/s12012-020-09609-y
60. Zhong W, Benissan-Messan DZ, Ma J, Cai C, Lee PHU. Cardiac effects and clinical applications of MG53. Cell Biosci. (2021) 11. doi: 10.1186/s13578-021-00629-x
61. Wu HK, Zhang Y, Cao CM, Hu X, Fang M, Yao Y, et al. Glucose-sensitive myokine/cardiokine MG53 regulates systemic insulin response and metabolic homeostasis. Circulation. (2019) 139:901–14. doi: 10.1161/CIRCULATIONAHA.118.037216
62. Wang Q, Bian Z, Jiang Q, Wang X, Zhou X, Park KH, et al. MG53 does not manifest the development of diabetes in db/db mice. Diabetes. (2020) 69:1052–64. doi: 10.2337/db19-0807
63. Bian Z, Wang Q, Zhou X, Tan T, Park KH, Kramer HF, et al. Sustained elevation of MG53 in the bloodstream increases tissue regenerative capacity without compromising metabolic function. Nat Commun. (2019) 10:4659. doi: 10.1038/s41467-019-12483-0
64. Wang S, Ding L, Ji H, Xu Z, Liu Q, Zheng Y. The role of p38 MAPK in the development of diabetic cardiomyopathy. Int J Mol Sci. (2016) 17. doi: 10.3390/ijms17071037
65. Qi Y, Xu Z, Zhu Q, Thomas C, Kumar R, Feng H, et al. Myocardial loss of IRS1 and IRS2 causes heart failure and is controlled by p38α MAPK during insulin resistance. Diabetes. (2013) 62:3887–900. doi: 10.2337/db13-0095
66. Van Linthout S, Riad A, Dhayat N, Spillmann F, Du J, Dhayat S, et al. Anti-inflammatory effects of atorvastatin improve left ventricular function in experimental diabetic cardiomyopathy. Diabetologia. (2007) 50:1977–86. doi: 10.1007/s00125-007-0719-8
67. Ruiz M, Coderre L, Allen BG, Des Rosiers C. Protecting the heart through MK2 modulation, toward a role in diabetic cardiomyopathy and lipid metabolism. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:1914–22. doi: 10.1016/j.bbadis.2017.07.015
68. Zuo G, Ren X, Qian X, Ye P, Luo J, Gao X, et al. Inhibition of JNK and p38 MAPK-mediated inflammation and apoptosis by ivabradine improves cardiac function in streptozotocin-induced diabetic cardiomyopathy. J Cell Physiol. (2019) 234:1925–36. doi: 10.1002/jcp.27070
69. Xiong Z, Li Y, Zhao Z, Zhang Y, Man W, Lin J, et al. Mst1 knockdown alleviates cardiac lipotoxicity and inhibits the development of diabetic cardiomyopathy in db/db mice. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165806. doi: 10.1016/j.bbadis.2020.165806
70. Shao Y, Wang Y, Sun L, Zhou S, Xu J, Xing D. MST1: a future novel target for cardiac diseases. Int J Biol Macromol. (2023) 239:124296. doi: 10.1016/j.ijbiomac.2023.124296
71. Caron A, Richard D, Laplante M. The roles of mTOR complexes in lipid metabolism. Annu Rev Nutr. (2015) 35:321–48. doi: 10.1146/annurev-nutr-071714-034355
72. Sun P, Wang Y, Ding Y, Luo J, Zhong J, Xu N, et al. Canagliflozin attenuates lipotoxicity in cardiomyocytes and protects diabetic mouse hearts by inhibiting the mTOR/HIF-1α pathway. iScience. (2021) 24:102521. doi: 10.1016/j.isci.2021.102521
73. Palomer X, Aguilar-Recarte D, García R, Nistal JF, Vázquez-Carrera M. Sirtuins: to be or not to be in diabetic cardiomyopathy. Trends Mol Med. (2021) 27:554–71. doi: 10.1016/j.molmed.2021.03.004
74. Haye A, Ansari MA, Rahman SO, Shamsi Y, Ahmed D, Sharma M. Role of AMP-activated protein kinase on cardio-metabolic abnormalities in the development of diabetic cardiomyopathy: a molecular landscape. Eur J Pharmacol. (2020) 888. doi: 10.1016/j.ejphar.2020.173376
75. Li L, Zeng H, He X, Chen JX. Sirtuin 3 alleviates diabetic cardiomyopathy by regulating TIGAR and cardiomyocyte metabolism. J Am Heart Assoc. (2021) 10:e018913. doi: 10.1161/JAHA.120.018913
76. Sun Y, Tian Z, Liu N, Zhang L, Gao Z, Sun X, et al. Exogenous H(2)S switches cardiac energy substrate metabolism by regulating SIRT3 expression in db/db mice. J Mol Med (Berl). (2018) 96:281–99. doi: 10.1007/s00109-017-1616-3
77. D'Onofrio N, Servillo L, Balestrieri ML. SIRT1 and SIRT6 signaling pathways in cardiovascular disease protection. Antioxid Redox Signal. (2018) 28:711–32. doi: 10.1089/ars.2017.7178
78. Karbasforooshan H, Karimi G. The role of SIRT1 in diabetic cardiomyopathy. Biomed Pharmacother. (2017) 90:386–92. doi: 10.1016/j.biopha.2017.03.056
79. Horman S, Beauloye C, Vanoverschelde JL, Bertrand L. AMP-activated protein kinase in the control of cardiac metabolism and remodeling. Curr Heart Fail Rep. (2012) 9:164–73. doi: 10.1007/s11897-012-0102-z
80. Hou N, Mai Y, Qiu X, Yuan W, Li Y, Luo C, et al. Carvacrol attenuates diabetic cardiomyopathy by modulating the PI3K/AKT/GLUT4 pathway in diabetic mice. Front Pharmacol. (2019) 10:998. doi: 10.3389/fphar.2019.00998
81. Wu S, Zou MH. AMPK, mitochondrial function, and cardiovascular disease. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21144987
82. Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol. (2018) 19:121–35. doi: 10.1038/nrm.2017.95
83. Lin SC, Hardie DG. AMPK: sensing glucose as well as cellular energy Status. Cell Metab. (2018) 27:299–313. doi: 10.1016/j.cmet.2017.10.009
84. Entezari M, Hashemi D, Taheriazam A, Zabolian A, Mohammadi S, Fakhri F, et al. AMPK signaling in diabetes mellitus, insulin resistance and diabetic complications: a pre-clinical and clinical investigation. Biomed Pharmacother. (2022) 146:112563. doi: 10.1016/j.biopha.2021.112563
85. Molaei A, Molaei E, Sadeghnia H, Hayes AW, Karimi G. LKB1: an emerging therapeutic target for cardiovascular diseases. Life Sci. (2022) 306:120844. doi: 10.1016/j.lfs.2022.120844
86. Chai D, Lin X, Zheng Q, Xu C, Xie H, Ruan Q, et al. Retinoid X receptor agonists attenuates cardiomyopathy in streptozotocin-induced type 1 diabetes through LKB1-dependent anti-fibrosis effects. Clin Sci (Lond). (2020) 134:609–28. doi: 10.1042/CS20190985
87. Lu QB, Ding Y, Liu Y, Wang ZC, Wu YJ, Niu KM, et al. Metrnl ameliorates diabetic cardiomyopathy via inactivation of cGAS/STING signaling dependent on LKB1/AMPK/ULK1-mediated autophagy. J Adv Res. (2023) 51:161–79. doi: 10.1016/j.jare.2022.10.014
88. Hegyi B, Bers DM, Bossuyt J. CaMKII signaling in heart diseases: emerging role in diabetic cardiomyopathy. J Mol Cell Cardiol. (2019) 127:246–59. doi: 10.1016/j.yjmcc.2019.01.001
89. Xie M, Zhang D, Dyck JR, Li Y, Zhang H, Morishima M, et al. A pivotal role for endogenous TGF-beta-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proc Natl Acad Sci U S A. (2006) 103:17378–83. doi: 10.1073/pnas.0604708103
90. Neumann D. Is TAK1 a direct upstream kinase of AMPK? Int J Mol Sci. (2018) 19. doi: 10.3390/ijms19082412
91. Pei Z, Deng Q, Babcock SA, He EY, Ren J, Zhang Y. Inhibition of advanced glycation endproduct (AGE) rescues against streptozotocin-induced diabetic cardiomyopathy: role of autophagy and ER stress. Toxicol Lett. (2018) 284:10–20. doi: 10.1016/j.toxlet.2017.11.018
92. Montagnani M. Diabetic cardiomyopathy: how much does it depend on AGE? Br J Pharmacol. (2008) 154:725–6. doi: 10.1038/bjp.2008.121
93. Hsieh DJ, Ng SC, Zeng RY, Padma VV, Huang CY, Kuo WW. Diallyl trisulfide (DATS) suppresses AGE-induced cardiomyocyte apoptosis by targeting ROS-mediated PKCδ activation. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21072608
94. Preetha RM, Anupama N, Sreelekshmi M, Raghu KG. Chlorogenic acid attenuates glucotoxicity in H9c2 cells via inhibition of glycation and PKC α upregulation and safeguarding innate antioxidant status. Biomed Pharmacother. (2018) 100:467–77. doi: 10.1016/j.biopha.2018.02.027
95. Cheng Y., Wang Y., Yin R., Xu Y., Zhang L., Zhang Y., Yang L., and Zhao D. (2023). Central role of cardiac fibroblasts in myocardial fibrosis of diabetic cardiomyopathy. Front Endocrinol (Lausanne) 14, 1162754. doi: 10.3389/fendo.2023.1162754
96. Chandra KP, Shiwalkar A, Kotecha J, Thakkar P, Srivastava A, Chauthaiwale V, et al. Phase I clinical studies of the advanced glycation end-product (AGE)-breaker TRC4186: safety, tolerability and pharmacokinetics in healthy subjects. Clin Drug Investig. (2009) 29:559–75. doi: 10.2165/11315260-000000000-00000
97. Lee TW, Kao YH, Chen YJ, Chao TF, Lee TI. Therapeutic potential of vitamin D in AGE/RAGE-related cardiovascular diseases. Cell Mol Life Sci. (2019) 76:4103–15. doi: 10.1007/s00018-019-03204-3
98. Mangali S, Bhat A, Jadhav K, Kalra J, Sriram D, Vamsi KVV, et al. Upregulation of PKR pathway mediates glucolipotoxicity induced diabetic cardiomyopathy in vivo in wistar rats and in vitro in cultured cardiomyocytes. Biochem Pharmacol. (2020) 177:113948. doi: 10.1016/j.bcp.2020.113948
99. Ma J, Hart GW. Protein O-GlcNAcylation in diabetes and diabetic complications. Expert Rev Proteomics. (2013) 10:365–80. doi: 10.1586/14789450.2013.820536
100. Kohda Y, Shirakawa H, Yamane K, Otsuka K, Kono T, Terasaki F, et al. Prevention of incipient diabetic cardiomyopathy by high-dose thiamine. J Toxicol Sci. (2008) 33:459–72. doi: 10.2131/jts.33.459
101. Joubert M, Jagu B, Montaigne D, Marechal X, Tesse A, Ayer A, et al. The sodium-glucose cotransporter 2 inhibitor dapagliflozin prevents cardiomyopathy in a diabetic lipodystrophic mouse model. Diabetes. (2017) 66:1030–40. doi: 10.2337/db16-0733
102. Qin CX, Sleaby R, Davidoff AJ, Bell JR, De Blasio MJ, Delbridge LM, et al. Insights into the role of maladaptive hexosamine biosynthesis and O-GlcNAcylation in development of diabetic cardiac complications. Pharmacol Res. (2017) 116:45–56. doi: 10.1016/j.phrs.2016.12.016
103. Peterson SB, Hart GW. New insights: a role for O-GlcNAcylation in diabetic complications. Crit Rev Biochem Mol Biol. (2016) 51:150–61. doi: 10.3109/10409238.2015.1135102
104. Ducheix S., Magré J., Cariou B., and Prieur X. (2018). Chronic O-GlcNAcylation and diabetic cardiomyopathy: the bitterness of glucose. Front Endocrinol (Lausanne) 9, 642. doi: 10.3389/fendo.2018.00642
105. Rabinowitz JD, Enerbäck S. Lactate: the ugly duckling of energy metabolism. Nat Metab. (2020) 2:566–71. doi: 10.1038/s42255-020-0243-4
106. Cluntun AA, Badolia R, Lettlova S, Parnell KM, Shankar TS, Diakos NA, et al. The pyruvate-lactate axis modulates cardiac hypertrophy and heart failure. Cell Metab. (2021) 33:629–648.10. doi: 10.1016/j.cmet.2020.12.003
107. Singh VP, Bali A, Singh N, Jaggi AS. Advanced glycation end products and diabetic complications. Korean J Physiol Pharmacol. (2014) 18:1–14. doi: 10.4196/kjpp.2014.18.1.1
108. Zhao C, Zhang Y, Liu H, Li P, Zhang H, Cheng G. Fortunellin protects against high fructose-induced diabetic heart injury in mice by suppressing inflammation and oxidative stress via AMPK/nrf-2 pathway regulation. Biochem Biophys Res Commun. (2017) 490:552–9. doi: 10.1016/j.bbrc.2017.06.076
109. Fan XD, Wan LL, Duan M, Lu S. HDAC11 deletion reduces fructose-induced cardiac dyslipidemia, apoptosis and inflammation by attenuating oxidative stress injury. Biochem Biophys Res Commun. (2018) 503:444–51. doi: 10.1016/j.bbrc.2018.04.090
110. Neinast M, Murashige D, Arany Z. Branched chain amino acids. Annu Rev Physiol. (2019) 81:139–64. doi: 10.1146/annurev-physiol-020518-114455
111. Ramzan I, Ardavani A, Vanweert F, Mellett A, Atherton PJ, Idris I. The association between circulating branched chain amino acids and the temporal risk of developing type 2 diabetes Mellitus: a systematic review & meta-analysis. Nutrients. (2022) 14. doi: 10.3390/nu14204411
112. Yang Y, Zhao M, He X, Wu Q, Li DL, Zang WJ. Pyridostigmine protects against diabetic cardiomyopathy by regulating vagal activity, gut Microbiota, and branched-chain amino acid catabolism in diabetic mice. Front Pharmacol. (2021) 12:647481. doi: 10.3389/fphar.2021.647481
113. Wang Y, Lei T, Yuan J, Wu Y, Shen X, Gao J, et al. GCN2 deficiency ameliorates doxorubicin-induced cardiotoxicity by decreasing cardiomyocyte apoptosis and myocardial oxidative stress. Redox Biol. (2018) 17:25–34. doi: 10.1016/j.redox.2018.04.009
114. Zhang L, Zhang H, Xie X, Tie R, Shang X, Zhao Q, et al. Empagliflozin ameliorates diabetic cardiomyopathy via regulated branched-chain amino acid metabolism and mTOR/p-ULK1 signaling pathway-mediated autophagy. Diabetol Metab Syndr. (2023) 15:93. doi: 10.1186/s13098-023-01061-6
115. Mizuno Y, Harada E, Nakagawa H, Morikawa Y, Shono M, Kugimiya F, et al. The diabetic heart utilizes ketone bodies as an energy source. Metab Clin Exp. (2017) 77:65–72. doi: 10.1016/j.metabol.2017.08.005
116. Mishra PK. Why the diabetic heart is energy inefficient: a ketogenesis and ketolysis perspective. Am J Physiol Heart Circ Physiol. (2021) 321:H751–5. doi: 10.1152/ajpheart.00260.2021
117. Luong TV, Abild CB, Bangshaab M, Gormsen LC, Søndergaard E. Ketogenic diet and cardiac substrate metabolism. Nutrients. (2022) 14. doi: 10.3390/nu14071322
118. Nirengi S, Peres VDSC, Stanford KI. Disruption of energy utilization in diabetic cardiomyopathy; a mini review. Curr Opin Pharmacol. (2020) 54:82–90. doi: 10.1016/j.coph.2020.08.015
119. Trang NN, Lee TW, Kao YH, Chao TF, Lee TI, Chen YJ. Ketogenic diet modulates cardiac metabolic dysregulation in streptozocin-induced diabetic rats. J Nutr Biochem. (2023) 111:109161. doi: 10.1016/j.jnutbio.2022.109161
120. Tao J, Chen H, Wang YJ, Qiu JX, Meng QQ, Zou RJ, et al. Ketogenic diet suppressed T-regulatory cells and promoted cardiac fibrosis via reducing mitochondria-associated membranes and inhibiting mitochondrial function. Oxid Med Cell Longev. (2021) 2021:5512322. doi: 10.1155/2021/5512322
121. Sumaiya K, Ponnusamy T, Natarajaseenivasan K, Shanmughapriya S. Cardiac metabolism and MiRNA interference. Int J Mol Sci. (2022) 24. doi: 10.3390/ijms24010050
122. Jakubik D, Fitas A, Eyileten C, Jarosz-Popek J, Nowak A, Czajka P, et al. MicroRNAs and long non-coding RNAs in the pathophysiological processes of diabetic cardiomyopathy: emerging biomarkers and potential therapeutics. Cardiovasc Diabetol. (2021) 20:55. doi: 10.1186/s12933-021-01245-2
123. Jia P, Wu N, Jia D, Sun Y. Downregulation of MALAT1 alleviates saturated fatty acid-induced myocardial inflammatory injury via the miR-26a/HMGB1/TLR4/NF-κB axis. Diabetes Metab Syndr Obes. (2019) 12:655–65. doi: 10.2147/DMSO.S203151
124. Samovski D, Sun J, Pietka T, Gross RW, Eckel RH, Su X, et al. Regulation of AMPK activation by CD36 links fatty acid uptake to β-oxidation. Diabetes. (2015) 64:353–9. doi: 10.2337/db14-0582
125. Cai C, Wu F, He J, Zhang Y, Shi N, Peng X, et al. Mitochondrial quality control in diabetic cardiomyopathy: from molecular mechanisms to therapeutic strategies. Int J Biol Sci. (2022) 18:5276–90. doi: 10.7150/ijbs.75402
126. Peng C, Zhang Y, Lang X, Zhang Y. Role of mitochondrial metabolic disorder and immune infiltration in diabetic cardiomyopathy: new insights from bioinformatics analysis. J Transl Med. (2023) 21:66. doi: 10.1186/s12967-023-03928-8
127. Chen X, Li Y, Luo J, Hou N. Molecular mechanism of hippo-YAP1/TAZ pathway in heart development, disease, and regeneration. Front Physiol. (2020) 11:389. doi: 10.3389/fphys.2020.00389
Keywords: diabetic cardiomyopathy, metabolism, fatty acid oxidation, glucotoxicity, heart failure, cardiac function
Citation: Zeng Y, Li Y, Jiang W and Hou N (2024) Molecular mechanisms of metabolic dysregulation in diabetic cardiomyopathy. Front. Cardiovasc. Med. 11:1375400. doi: 10.3389/fcvm.2024.1375400
Received: 23 January 2024; Accepted: 8 March 2024;
Published: 25 March 2024.
Edited by:
Wang Wang, University of Washington, United StatesReviewed by:
Fuyang Zhang, Air Force Medical University, China© 2024 Zeng, Li, Jiang and Hou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ning Hou aG91bmluZ0BnemhtdS5lZHUuY24=
Abbreviations ADAM10, Disintegrin and metalloproteinase 10; AdipoR1, adiponectin receptor 1; ADP, adenosine diphosphate; AGEs, advanced glycation end products; AMP, adenosine monophosphate; AMPK, AMP-activated protein kinase; AS160, Akt substrate 160; ATP, adenosine triphosphate; BAT, brown adipose tissue; BCAA, branched chain amino acids; BCATm, mitochondrial branched-chain aminotransferase; BCKA, branched-chain α-ketoic acid; BCKDH, branched-chain alpha-ketoate dehydrogenase; CaMKKβ, calcium/calmodulin-dependent kinase kinase 2; CAN, canagliflozin; CPT1, carnitine palmitoyltransferase-1; DCM, diabetic cardiomyopathy; eIF2α, eukaryotic initiation factor 2α; ERK 1/2, extracellular signal-regulated kinase-1/2; FA, fatty acids; FABPpm, plasma membrane-associated fatty acid-binding protein; FAT, fatty acid translocase; FATP, fatty acid transport protein; FoxO3, forkhead box O3; GAPs, GTPase activating proteins; GFAT, L-glutamine-fructose-6-phosphate amidotransferase; GLUT4, GLUCOSE transporter type 4; HBP, hexosamine biosynthetic pathway; HDAC4, histone deacetylase 4; HFD, high-fat diet; HIF-1α, hypoxia inducible factor-1α; HMGB1, high mobility group box 1; HMGCS2, hydroxymethylglutaryl-CoA synthase 2 ; IRS1, insulin receptor substrate 1; JNK, c-Jun N-terminal kinase; KB, ketone bodies; LCFA, long-chain fatty acids ; LDH, lactate dehydrogenase; LKB1, liver kinase B1; MALAT1, metastasis-associated lung adenocarcinoma transcript 1; MAPK, mitogen-activated protein kinase; MCTs, monocarboxylate transporters; MD2, myeloid differentiation 2; MEKK1, mitogen-activated protein kinase kinase kinase 1; MG53, mitsugumin 53; miRNA, microrna; MK2, mapk-activated protein kinase 2; MQC, mitochondrial quality control; mRNA, messenger RNA; Mst1, macrophage stimulating 1; mTOR, mammalian target of rapamycin; NADH, nicotinamide adenine dinucleotide; NAP1L2, nucleosome assembly protein 1-like 2; NF-κB, nuclear factor-kappa B; OGA, O-GlcNAcase; O-GlcNAc, O-linked N-acetylglucosamine; OGT, O-GlcNAc transferase; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; PGC-1, peroxisome proliferator-activated receptor γ coactivator-1; PGE2, prostaglandin E2; PI3K, phosphatidylinositol 3-kinase; PKB, protein kinase B; PKC, protein kinase C; PKR, protein kinase R; PPARs, peroxisome proliferator-activated receptors; PRC, Peroxisome proliferator-activated receptor γ coactivator-1-related coactivator; RAGE, receptor for advanced glycation end-products; ROS, REACTIVE oxygen species; SGLT2, sodium-dependent glucose cotransporter 2; SIRT, sirtuins; SLC27A1-6, solute Carrier Family 27 Member 1–6; STZ, streptozotocin; TAK1, TGF-β activated kinase 1; TCA cycle, tricarboxylic acid cycle; TGs, triglycerides; TLR4, TOLL-like receptor 4; T1DM, type 1 diabetes mellitus; T2DM, type 2 diabetes mellitus; VAMP, vesicle-associated membrane protein; WAT, white adipose tissue; βDH1, β-hydroxybutyrate dehydrogenase; βHB, β-hydroxybutyrate.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.