 Heng Pan1,2,3,†
Heng Pan1,2,3,† Xiyi Lu
Xiyi Lu Di Ye
Di Ye Jun Wan
Jun Wan Jing Ye
Jing Ye
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 07 March 2024
Sec. Atherosclerosis and Vascular Medicine
Volume 11 - 2024 | https://doi.org/10.3389/fcvm.2024.1337586
Cardiovascular diseases have been identified as vital factors in global morbidity and mortality in recent years. The available evidence suggests that various cytokines and pathological proteins participate in these complicated and changeable diseases. The thrombospondin (TSP) family is a series of conserved, multidomain calcium-binding glycoproteins that cause cell-matrix and cell-cell effects via interactions with other extracellular matrix components and cell surface receptors. The TSP family has five members that can be divided into two groups (Group A and Group B) based on their different structures. TSP-1, TSP-2, and TSP-4 are the most studied proteins. Among recent studies and findings, we investigated the functions of several family members, especially TSP-5. We review the basic concepts of TSPs and summarize the relevant molecular mechanisms and cell interactions in the cardiovascular system. Targeting TSPs in CVD and other diseases has a remarkable therapeutic benefit.
Currently, cardiovascular disease (CVD) is a vital cause of human disability and death in both underdeveloped and developed regions (1). In addition to the large cost of treatment during disease onset, the expense of complications and recovery poses a severe burden to families and communities. Although many effective drugs and advanced technologies have been used in clinical practice in recent years, CVD incidence still correlates with poor overall survival and prognosis.
Thrombospondin (TSP) is a matricellular protein that can be secreted by many cell types and is widely distributed in various organs and tissues (2). The TSP family includes five extracellular, conserved matricellular proteins in mammals. The TSP family is divided into two subgroups (Group A and Group B) based on their structure. Group A includes TSP-1 and TSP-2, which are trimers. Group B is composed of TSP-3, TSP-4, and TSP-5, which are pentameric proteins (3). The expression of TSP is low under normal physiological conditions but increases in response to damage, and TSP is subsequently involved in tissue repair or deterioration (4). TSP has a multimeric structure that allows it to bind calcium, cell-surface proteins, bioactive effectors, and other extracellular matrix (ECM) proteins. TSP has many complex and variable functions, such as regulating wound healing, angiogenesis, and tissue remodeling. In the cardiovascular system, TSP participates in regulating vasomotor function, adjusting cell apoptosis and growth, reacting to cardiovascular injuries, and affecting the structural integrity of the heart and blood vessels (5). Many studies have revealed a close link between TSP and CVD. Group A TSPs and TSP-4 from group B are the most thoroughly studied TSPs. We subsequently summarize the functions of TSPs, especially TSP-5, in cardiovascular pathological processes and update the role of group B TSPs in CVD treatment.
CVD and cancer are considered two mutually independent diseases. Along with the increase in cancer survivors and the application of new therapeutic strategies for distinct cancers, patients who suffer from cancer often have a greater risk of cardiovascular complications than individuals in general. Thus, a new discipline called cardio-oncology (6) has gained attention from clinicians and cardiologists. However, the latent links between CVD occurrence and consequential carcinoma have been less investigated. Studies have shown that people with cardiovascular disorders have a greater risk of cancer than does the general public, which is called “reverse cardio-oncology (7, 8)”. This argument is based on the shared risk factors and pathogenic mechanisms involved in these two diseases (9). For example, alcoholism, obesity, and diabetes mellitus are the same causes of CVD and cancer (7, 10). The TSP family also plays a role in shared pathogenic mechanisms and pathways. TSP-4 is involved in CVD and regulates several different cancers, such as colon cancer and prostate cancer, and the effects of TSP-4 on cancer cells are complex and opposite. Overexpression of TSP-4 inhibited the tumor growth of colorectal cancers, but a lack of TSP-4 in prostate cancer cells reduced their invasion and migration (11, 12). In addition, the clinical application of TSPs is closely related to cancer treatment. In this review, we focused on the role of TSPs in the cardiovascular system in two subgroups based on the pathological mechanism, as well as other closely linked diseases and clinical treatment strategies.
The structure of representative TSP polypeptides is shown in Figure 1. TSP subunits have a highly conserved carboxy terminus that binds to many epidermal growth factor (EGF)-like repeats, which are called TSP type 2 and are linked to seven TSP type 3 repeats and a globular C-terminal domain (CTD). The CTD shares common extracellular, calcium-binding, and cell membrane-binding properties (13). In addition to an N-terminal domain (NTD), a procollagen homology domain (PC), and an oligomerization sequence, group A TSPs also have type 1 domains (TSRs), which are composed of three properdin-like repeats called antiangiogenic regions that are involved in modulating antiangiogenic functions and accelerating cell attachment (3). In contrast, group B has no TSRs, and the procollagen homology domain contains four (group A is three) type 2 repeats instead. Inter-subunit disulfide bonds are formed between cysteine residues adjacent to the carboxyl end of the heptad fold repeat sequence in trimer TSP or between the carboxyl end of the pentamer TSP to stabilize the low polymerization of TSP. Because of the variable region, group B has different effects on different tissues (14).
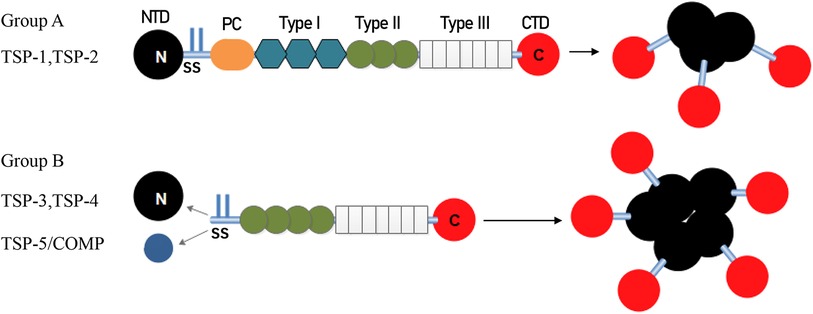
Figure 1. Architecture and oligomerization status of group A and group B TSP. TSP-1 and TSP-2 (Group A) assemble into trimeric structure. TSP-3, 4 and 5 (Group B) assemble into pentamers. TSP, thrombospondin; NTD, N-terminal domain; SS, disulfide bonds; PC, homologous procollagen region, Type I, thrombospondin type 1 domains; Type II, epidermal growth factor-like domains; Type III, thrombospondin type 3 repeats; CTD, C-terminal domain.
Due to the availability of each domain, TSPs can interact with diverse surface receptors and proteins. For example, the C-terminal domain contains a CD47-binding site, EGF-like domains can bind to integrins and Ca2+, TSRs are necessary for binding transforming growth factor (TGF)-β and CD36, and heparin and other integrins bind to the N-terminal domain (15). We analyzed the role of TSP in CVDs according to different receptors, signaling pathways, and immune cells involved.
As stated above, different domains of the TSP can interact with corresponding receptors, including integrins, CD36 and CD47. Integrins are a class of glycosylated, heterodimeric transmembrane receptors that consist of α and β subunits. The interaction of cardiac ECM with specific cell surface integrins is the foundation of cardiomyocyte maturation and remodeling (16).
Moreover, the interaction between TSPs and integrins participates in vascular remodeling and pathology. TSP-1 and TSP-2 are regarded as potent endogenous antiangiogenic proteins that directly affect CD36, CD47, and integrins. Vascular cells express various integrins that bind to TSP-1, including αIibβ3, αvβ3, and α5β1 (17). The N-termini of TSP-1 and TSP-2 can be recognized by α3β1, α4β1, and α6β1 to mediate the adhesion of vascular ECs (18). The binding of integrin α3β1 to TSP-1 promotes EC migration, cell motility, and antiangiogenic effects (19). In another study, TSP-1 was shown to bind to αvβ1 and regulate the nuclear shuttling of Yes-associated protein in response to mechanical stress-induced vascular injury, leading to dynamic remodeling of the aorta in mice (20).
The TGF-β superfamily has a significant role in inducing tissue fibrosis and inflammation. Type I repeats of TSP can interact with latent TGF-β. For instance, TGF-β signaling enables the ERK1/2 pathway to activate fibroblasts and induce interstitial fibrosis in the aging course (21). The TGF-β family is mainly classified into the TGF-β1, -2, and -3 subfamilies. One of the downstream mediators of TGF-β1, connective tissue growth factor (CTGF), also plays an important role in fibrosis.
TSP-1 is a major mediator of TGF-β activation, increasing CTGF and collagen levels and the accumulation of extracellular matrix proteins (22). All of these factors stimulate and are hallmarks of tissue fibrosis. Increased TSP-1 in intermittent hypoxia (IH) patients and mice, which activates the TGF-β pathway via JAK2/STAT3/TSP-1 signaling, has a significant effect on IH-induced fibroblast activation and cardiac fibrosis (23). A TSP-1 antagonist that blocks TGF-β activation can reverse myocardial fibrosis and ensure left ventricular function in hypertensive diabetic rats (24). TSP-1 acted as a protective factor by maintaining fibroblast function and matrix metabolism in pressure-overloaded myocardium. TSP-1 is also downstream of the TGF-β signaling pathway and has been identified as a regulator of microtube formation in glioblastoma (25). In addition to the heart, as a result of TSP-1/TGF-β pathway upregulation, collagen deposition and extensive fibrosis in the arterial wall led to arteriosclerosis, while in TSP-1 knockout (KO) mice or mice treated with TSP-1 antagonist, arterial collagen, CTGF, and arterial stiffness were decreased (26). Ying Xia et al. reported that deletion of TSP-1 disrupted TGF-β signaling, leading to impaired myofibroblast differentiation and decreased collagen expression (27).
TSP-2 can bind to latent TGF-β but cannot activate the TGF-β signal and is regarded as a competitive binding factor to TSP-1 in CVD. However, in the cancer microenvironment, a high level of TSP-2 produced by cancer-associated fibroblasts is activated via the TGF-β1/Smad2/3 pathway, binds to integrin αvβ3/CD36 and activates the MAPK pathway in cancer cells to promote tumor growth and adhesion (28).
Regarding injury to other organs, TSP-1 is highly expressed in patients with sepsis-induced acute kidney injury (AKI). The transcription factor USF2 activates TSP-1 to activate the TGF-β/NLRP3/Caspase-1 signaling pathway, resulting in promotion of the oxidative stress response and stimulation of pyroptosis in sepsis-induced AKI (29). TSP-1 deficiency protects mice against sepsis-induced AKI by decreasing the expression of inflammatory and apoptosis-promoting cytokines, such as the NLRP3 inflammasome, caspase-1, IL-1β, and IL-18, which increase cell viability and partially reverse cell pyroptosis. TSP-1 deletion reduces TGF-β signaling and protects against renal fibrosis in a high-fat diet mouse model (30). In another study, interstitial macrophages secreted TSP-1 after hypoxia exposure, and pathological TSP-1 promoted TGF-β activation and Rho-kinase-mediated vasoconstriction in mice, resulting in pulmonary hypertension (31).
The endoplasmic reticulum (ER) is a large protein processing and transporting region. Mistaken ER protein folding leads to the accumulation of unfolded and misfolded proteins. This disturbance of ER homeostasis initiates the protective stress response and is known as the unfolded protein response (UPR) or ER stress (32). Protein kinase R-like ER kinase (PERK), activating transcription factor 6α (ATF6α), and inositol requiring enzyme 1 alpha (IRE1α) are three primary sensors that strengthen protein folding under ER stress (33). ER stress is a double-edged sword that can restore cell homeostasis and may lead to cell defects (32). In TSP-1-overexpressing transgenic mice, TSP-1 binds to PERK and induces the downstream factor ATF4, thus activating ER stress and inducing autophagy-mediated lethal cardiac atrophy (34).
However, for the other member of group A, TSP-2, further research is needed to reveal the possible links between TSP-2 and ER stress.
Nitric oxide (NO) is a vital physiological regulator of vasomotion and blood flow. TSP-1 affects the proliferation, migration, and angiogenesis of SMCs, ECs, and platelets (PLTs) by regulating NO signaling (22, 35). TSP-1 binds to the cell surface proteins CD47 and CD36 and subsequently participates in the activity of endothelial nitric oxide synthase (eNOS) (36, 37). Moreover, the combination of TSP-1 with CD47 or CD36 can inhibit the SMC cyclic guanosine monophosphate pathway in the presence of a low dose of NO (NO/cGMP) (38), exerting antiangiogenic effects and activating apoptosis in microvascular ECs, which leads to failed endothelial tubule formation and directly affects other antiangiogenic activities. Studies have shown that, compared with CD36, CD47 has a greater affinity for TSP-1 and inhibits NO signaling (39). Through binding to CD47, TSP-1 inhibits eNOS activation and arterial relaxation, manifesting as a blood pressure booster agent (40).
TSP-1 engages the receptor CD47 of ECs to mediate cell phenotypic transformation, and the activation of CD47 inhibits the bioavailability of VEGF (41, 42), as observed in lymphatic endothelial cells, resulting in AKT-eNOS signaling activation and NO reduction (43). Thus, silencing CD47 blocks lymphangiogenesis and atherosclerotic lesion formation (43). Deletion of TSP-1 protects against inflammatory lesion development and vascular smooth muscle cell (VSMC) phenotypic transition in leptin-induced atherogenesis (44). TSP-1 can also suppress the NO signaling pathway by interacting with integrin to regulate platelet aggregation (39).
TSP-2 seems to be an NO target, and suppressing TSP-2 relieves the eNOS-knockout phenotype without restoring NO signaling in mice with injury- and ischemia-induced angiogenesis (45).
Matrix metalloproteinases, such as matrix metallopeptidase (MMP)-2, MMP-3, and MMP-9, participate in tissue remodeling by binding to TSP type I repeats (27).
TSP-1 deficiency increases myocardial MMP-3 and MMP-9 activation under pressure overload, resulting in early cardiac hypertrophy and late dilation. During remodeling of the diabetic heart, TSP-1 deletion increases MMP-2 and MMP-9 activation to degrade collagen and inhibit fibroblast function (46).
TSP-2 acts as a key regulator of cardiac matrix integrity, is required for the myocardium to respond to pressure overload, and plays a role in regulating MMP activity (47). TSP-2 plays a protective role in age-related cardiomyopathy by activating the Src/Akt survival pathway, decreasing inflammation and MMP-2 activity, and maintaining collagen crosslinking (48). High levels of MMP-2 (49) and MMP-9 (50) may cause local disruption of the myocardial matrix, leading to cardiac rupture and dilatation after Ang II infusion in TSP-2-deficient mice (47). Enhanced matrix destruction is shown in the heart of doxorubicin (DOX)-induced TSP-2 KO mice and is accompanied by increased levels of MMP-2 (51). A lack of TSP-2 increases the levels of MMPs in the extracellular matrix and helps with the degradation of fibrillar collagen, thus reducing fibrosis around cardiac cell grafts (52).
MMPs also take part in vascular physiology. TSP-1 can be hydrolyzed into proteolytic fragments, and the different fragments exert pro- or antiangiogenic effects by activating the MAPK pathway to mediate the MMP/tissue inhibitor of metalloproteinase (TIMP) balance (53). TSP-2 inhibits angiogenesis by regulating EC function and modulating MMP-2 and MMP-9 (54).
Reactive oxygen species (ROS) are associated with senility and injury, regulating myocardial metabolism and contributing to vascular dystonia (55). The family of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox) isozymes is the main source of ROS. Nox-4-based NADPH oxidases are the main source of ROS in the vasculature, and downregulation of Nox4 in SMCs inhibits neointimal hyperplasia by decreasing TSP-1 and suppressing SMC proliferation (56). Hypoxia-responsive TSP-1 mediates the critical event in pulmonary hypertension. The level of TSP-1 was increased in hypoxia-induced human pulmonary artery SMCs, and TSP-1-stimulated Nox4 expression was enhanced, causing SMC proliferation and high blood pressure (57). O2− is the first product induced by Nox and rapidly changes into stable H2O2. Moreover, excess accumulation of O2− disrupts the balance of O2−/H2O2, resulting in the impairment of coronary arteriolar vasodilation and heart ischemia (58, 59).
In diabetic conditions, TSP-1 binds to the cell-surface receptor CD47 and significantly increases Nox1-mediated ROS and O2− production, causing endothelial senescence and vascular dysfunction (60–62), while TSP-2 plays an antiangiogenic role. Increasing NADPH oxidase activity and oxidative stress induce the production of TSP-2, which induces bone marrow-derived angiogenic cell dysfunction and vascular impairment (63). The TSP-1/CD47 axis also activates signal-regulatory protein-α, another kind of cell-surface receptor of inflammatory cells, increasing O2− production and promoting renal ischemia‒reperfusion injury (64). H2O2 can induce vasodilation through the release of prostaglandin E2 and calcium-activated potassium channel-related SMC hyperpolarization.
Activation of the plasmin/plasminogen system is necessary for regulating angiogenesis in a variety of diseases (65). A study indicated that plasminogen decreases the expression of TSP-1 and TSP-2, thereby enhancing angiogenesis in damaged brain tissue to help cells resume after ischemic stroke (66). However, no additional studies have evaluated the relationship between TSP-2 and the plasmin/plasminogen system.
The absence of plasminogen suppresses EC migration and decreases cerebrovascular density. Platelet activation releases stored TSP-1 in platelet α-granules; thus, plasma TSP-1 is significantly increased during the acute stage in ST-segment elevation myocardial infarction (STEMI) patients and decreases 1–3 days and 3 months after percutaneous coronary intervention (PCI), which might be associated with major adverse cardiac events (67).
In addition, both plasmin and TSP-1 can activate TGF-β1 signaling in the fibrotic process (68, 69). However, TGF-β1 activation is bistable: the plasmin-mediated mode is characterized by low activation, and the TSP-1-mediated mode is characterized by high activation. Interestingly, when both plasmin and TSP-1 are present, increasing plasmin can disrupt the TSP-1/TGF-β1 feedback loop and thus cause proteolytic cleavage of TSP-1 and inactivation of TGF-β1 signaling (70). This phenomenon corroborates that the activation of plasmin inhibits the expression of TSP-1.
Activation of the TSP-1/CD47 pathway plays a crucial role in the excitation and migration of T regulatory cells (Tregs) to inhibit inflammation in atherosclerosis and abdominal aortic aneurysm (AAA) (71, 72). TSP-1 and TSP-2 also interact with CD47 on the surface of T cells, which triggers T-cell apoptosis to block inflammation. In CD47-knockout mice, TSP-1- or TSP-2-knockout mice, prolonged inflammation occurs along with defects in T-cell apoptosis (73). TSP-2 is also an important regulator of T-lymphocyte migration and differentiation through its interaction with CD47 (71) in humans and mice (74). TSP-2 deficiency results in the generation of fewer Tregs and lower IL-10 and IL-10 receptor levels, thereby leading to an imbalance in the immune response and cardiac damage in Coxsackievirus group B type 3 (CVB3) virus-induced myocarditis (74, 75).
MMPs are necessary for macrophages to persist and migrate into the ECM to induce an inflammatory response in AAA (76). TSP-1 deficiency upregulates the tissue inhibitor MMP-1, which suppresses extracellular gelatinase activity and inhibits MMP activation, thus alleviating AAA development (77). TSP-1 deficiency also enhances atherosclerotic plaque maturation by accelerating inflammation, and macrophage-induced MMP-9 contributes to elastin degradation (78). TSP-1 can bind to CD47 in macrophages to enhance the inflammasome-dependent maturation of IL-1β, which promotes inflammation in response to lipopolysaccharide (LPS) (79). TSP-1 may promote proinflammatory macrophage differentiation to aggravate human aortic dissection (AD) (80). Another study showed that the overexpression of TSP-2 promotes the polarization of macrophages toward an anti-inflammatory phenotype by activating the PI3K pathway in vitro and attenuating LPS-induced pulmonary inflammation (70).
TSP-1 serves as a master regulator of cancer invasion (81, 82). A study showed that the expression of TSP-1 and CD47 is increased in human malignant melanoma tumor tissue, and targeting the TSP-1/CD47 pathway may preserve CD8+ T-cell activation, proliferation, and bioenergetics to alleviate the tumor burden (83). By the way, downregulating TSP-1 levels in skin dendritic cells effectively promoted antitumor reactions through increasing tumor-infiltrating CD4+ and CD8+ T cells. Moreover, TSP-1-knockout bone marrow-derived DCs retarded tumor growth, while targeting TSP-2 did not have the same effect (84). This finding was opposite to the antitumor angiogenesis response of TSP-1/2, indicating that systemic antitumor treatment with TSP-1 could be a double-edged sword.
Group B TSPs binds to integrin to regulate physiological and pathological processes. Overexpression of TSP-3 in the mouse heart significantly damages cardiomyocyte integrity due to reduced sarcolemmal residence of integrins, such as α7β1D and β1 (85). Studies have shown that the expression of TSP-5 is decreased in the hearts of DCM patients and that a lack of TSP-5 in mice spontaneously results in DCM at a young age (86). TSP-5 interacts with cardiomyocyte integrin β1 to maintain cardiomyocyte homeostasis, and TSP-5 or integrin β1 deficiency results in similar F-actin dissolution, connexin-43 defects, and spontaneous apoptosis (86).
The interaction of TSP-4 and αMβ2 and β3 integrins in macrophages activates p38-MAPK signaling (87). The p38-MAPK pathway plays a pivotal role in the production of proinflammatory mediators and cytokines, as well as in endothelial-leukocyte interactions, resulting in vascular inflammation and arteriosclerosis (87–89). TSP-4 also modulates the proliferation of SMCs and ECs, thereby exerting atherogenic effects (90). Thus, TSP-4 serves as a proangiogenic factor in wound healing. TSP-4 regulates the adhesion, migration, and proliferation of EC cells by interacting with integrin α2 and gabapentin receptor α2δ-1 (91), resulting in a strong proangiogenic effect in vivo and in vitro.
Bone marrow-derived TSP-5 mediates atherosclerotic calcification, and a lack of TSP-5 induces the atherogenic and osteogenic phenotype in macrophages via integrin β3 (92). TSP-5 is involved in the chemotaxis and attachment of VSMCs and inhibits osteochondrogenic phenotypic switching in VSMCs, thereby inhibiting vascular calcification (93, 94). Increased TSP-5 levels during injury or other pathological processes exert protective effects by maintaining the contractile phenotype of VSMCs through interactions with the integrin α7β1 (93, 94). In addition, the C-terminus of TSP-5 binds directly to integrin α5, blocking aberrant activation of ECs in mice and hence reducing vascular inflammation and atherosclerosis (95).
TSP-4 and TSP-5 regulate atherosclerosis, aortic aneurysm, and other vascular tissue remodeling processes through the TGF-β signal pathway (96). Activated TGF-β1 stimulates the Smad3 pathway to upregulate endothelial TSP-4, leading to EC adhesion, migration, and proliferation (97). This strong proangiogenic function can be blocked in TSP-4 KO mice. Smad3 activation in ECs also upregulates TSP-4 expression and angiogenesis via TGF-β during tumor growth (97).
TSP-5, also known as cartilage oligomeric matrix protein (COMP), participates in the differentiation of stem cells and passaged chondrocytes and is dependent on TGF-β signaling (98). TGF-β1 binds to the C-terminal domain of TSP-5, and an additional binding site is the 3-repeat TSP in the presence of manganese. This combination increases TGF-β1-dependent transcription and enhances its bioactivity (98). Along with the activation of the TGF-β signal, the levels of TSP-5 are also elevated, which plays a critical role in skin fibrosis by promoting collagen deposition and modifying fibroblast functions (99, 100). Bone morphogenetic protein (BMP)-2 is a member of the TGF-β superfamily. Yaoyao Du et al. suggested that TSP-5 binds to BMP-2 via the C-terminus to prevent its interaction with the BMP-2 receptor, inhibit the osteochondrogenic transition of VSMCs, and improve vascular calcification (101).
All three members of group B can active ER stress to regulate tissue development. TSP-4 is expressed in the heart and skeletal muscle during injury and damage, and it contributes to sarcolemmal stability and cardioprotection through binding to ATF6α-activated ER stress (102–104). TSP-4 is prominently localized within intracellular vesicles and the ER or its compartments in myocardial cells, with minor accumulation in the extracellular space (102). As a result of ER stress, TSP-4 KO mice infused with Ang II have exaggerated hypertrophic hearts and a high incidence of aneurysm but a protective role in endothelium-dependent relaxation in resistant arteries (105). TSP-3 is highly similar in structure to TSP-4 and is upregulated during cardiac disease. Although TSP-3 also activates ER stress by binding to ATF6α, TSP-3 oppositely enhances cardiac pathological conditions by inhibiting intracellular integrin and destroying myocardial membrane stability (85). The familial mutation in type 3 repeats of TSP-5 induces pseudoachondroplasia and multiple epiphyseal dysplasia resulting from pathological accumulation of mutated TSP-5 in the rough ER and apoptosis of the cells (106). Thus, mutated TSP-5 induces ER stress to regulate cartilage development.
TSP-4 promotes endothelial dysfunction and contributes to the process of hypertension by impairing NO bioavailability and blocking vascular vasodilation in Ang II-infused mice (105). TSP-5 plays a protective role in BP control by improving endothelium-dependent relaxation through CaKMII/eNO signaling. It binds to the C-terminus of Piezo1, which promotes intracellular Ca2+ influx, eNOS activation, and NO generation resulting from activation of endogenous Piezo1 currents (107). TSP-3 has not been found to regulate CVDs through NO signaling.
It is unknown whether TSP-3 acts on immune cells to affect the cardiovascular system. TSP-4 expression directly supports macrophage functions and switching of the proinflammatory phenotype during atherogenesis (108). Inflammation stimulates the expression of TSP-4 in macrophages, while increased TSP-4 promotes the accumulation and proinflammatory phenotypic differentiation of macrophages in LPS-induced peritonitis (108). In vascular inflammation, recombinant TSP-4 contributes to injury-induced restenosis by accelerating macrophage adhesion to VSMCs and increasing VSMC proliferation and migration (109). TSP-5 is also critical for inducing the beneficial phenotype of VSMCs and macrophages to help maintain vascular integrity and function (110, 111). The deletion of TSP-5 promotes VSMC migration and exacerbates VSMC calcification and atherosclerosis (111); moreover, this deletion results in a phenotypic shift of macrophages to atherogenic and osteogenic characteristics (112).
With long-term investigation, we gain a comprehensive understanding of TSP and its effective role in the cardiovascular system. Changes in circulating TSP production also affect the development and prognosis of clinical diseases. For instance, in group A, studies indicate that high circulating TSP-1 is associated with diabetic nephropathy, diabetic cardiovascular disease (113), and pulmonary hypertension (114). Elevated circulating TSP-2 is involved in coronary artery disease (CAD)-induced chronic heart failure (CHF) and increases patient mortality and risk of recurrent hospitalization (115). The group B members also participate in clinical diseases, but the related research is less than group A. Above all, we summarize the role of circulating TSP in clinical cardiovascular diseases (Table 1) and map the relative mechanisms of two TSP subgroups in the physiological and pathological processes of CVD (Figures 2, 3).
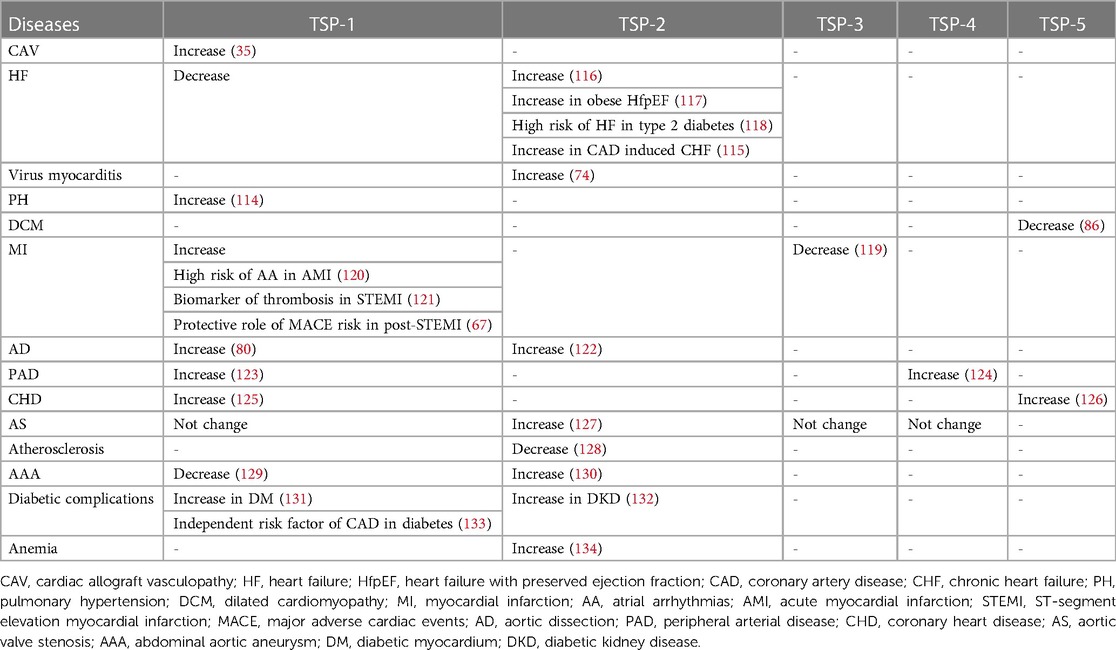
Table 1. Role of the TSP family members in clinical CVDs.
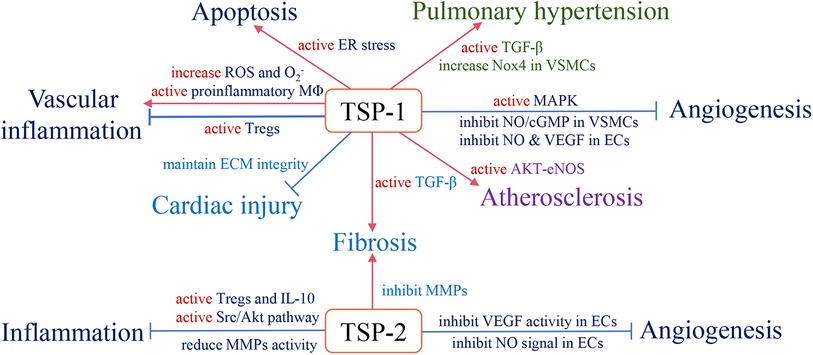
Figure 2. The role of subgroup A of the TSP family and relevant pathological mechanisms in the cardiovascular system. Red arrows define stimulatory effects, and blue arrows define inhibitory effects.
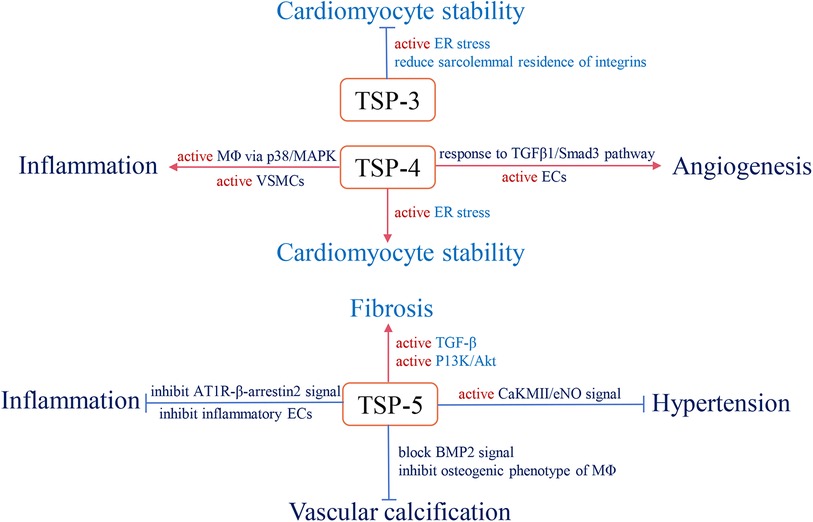
Figure 3. The role and relevant pathological mechanism of subgroup B of the TSP family in cardiovascular pathology. Red arrows define stimulatory effects, and blue arrows define inhibitory effects.
TSP-1 and TSP-2 have significant antiangiogenic effects because of type I repeats (135). ABT-510 is a nonpeptide analog of the type I repeat and is constructed with a single D-amino acid substitution that results in 1,000-fold antiangiogenic activity (136). ABT-510 inhibited VEGF-induced microvascular EC migration and exerted strong antiangiogenic effects to prevent graft arteriosclerosis in rats (137); moreover, ABT-510 induced inflammation in mice with inflammatory bowel disease (138). In humans, the use of ABT-510 alone or in combination with other therapies for the treatment of advanced parenchymal or epithelial carcinoma has been tested in phase I and II clinical trials (139–141). Several studies have shown that ABT-510 is safe and efficient at restraining tumor growth and inhibiting tumor neovascularization, and combination therapy increases the efficiency of anticancer therapy (142–144).
The TSP/CD47 axis plays a vital role in many pathological processes in the cardiovascular system. Preclinical trials of blocking CD47 with monoclonal antibodies in animal ischemia models have shown improved angiogenesis and a significant increase in tissue survival (39, 145). The CD47 antibody (CC-90002) has been tested for biosafety/tolerability in phase I clinical trials and beneficial effects for neoplastic hematologic disorders, while another humanized anti-CD47 monoclonal antibody (Hu5F9-G4) may be beneficial for acute myeloid leukemia and advanced solid malignancy (146). The activated TGF-β pathway has strong profibrotic activity, the activation sequence (LSKL) in TSP-1 was mapped, and an LSKL peptide was developed for competitive binding (22). The LSKL peptide blocks TGF-β release and inhibits fibrosis in various animal models, such as diabetic nephropathy (147), liver fibrosis (148), and skin scarring (149). However, in Ang II-infused apolipoprotein E-deficient mice, decreased activity of the TGF-β pathway promotes AAA and atherosclerosis (150). Thus, antifibrotic therapy has dual effects on different pathological conditions, concentrating on preventing advanced parenchymal organ fibrosis or local treatment, which may be potential therapeutic directions.
Numerous studies have shown that patients with CVD have a greater cancer risk than do healthy individuals. There are connections between cancer and atherosclerotic cardiovascular disease (ASCVD) (151), myocardial infarction (152) and heart failure (153). Thus, a new discipline called “reverse cardio-oncology” was established, and studies need to identify the shared mechanisms and pathways between these 2 diseases (9). We summarize the shared mechanisms and pathways of the TSP proteins in cardiovascular disease and cancer in Table 2. Thrombospondins strongly regulate angiogenesis, tissue fibrosis and inflammation via effects on the cardiovascular system and tumor microenvironment. TSP-1 is involved in myocardial fibrosis and tumor growth by activating the TGF-β pathway (22, 160). TSP-4 mediates inflammatory macrophage infiltration, which not only exacerbates atherogenesis in the vasculature but also induces breast cancer cell growth in mice (89, 161). ABT-510, a type I repeat of TSP-1/TSP-2, can prevent graft arteriosclerosis in animal models (137) while inhibiting tumor neovascularization as an antineoplastic drug (139). Although there is no direct evidence that TSPs participate in the crosstalk between CVD and cancer, we reasonably hypothesize that this occurs.
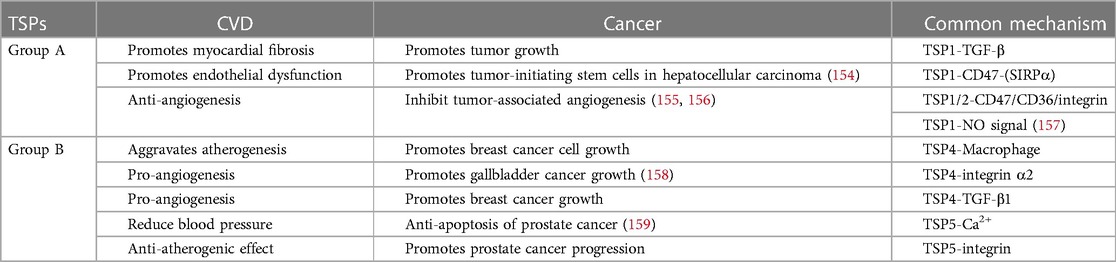
Table 2. Common mechanisms of TSP in CVD and cancer.
TSP-5 has been shown to play a protective role in the cardiovascular system and affects conditions, including hypertension, atherosclerosis, and AAA. TSP-5 helps sustain the contractile phenotypes of VSMCs. It has been demonstrated that TSP-5 deficiency induces VSMC migration while aggravating VSMC calcification and atherosclerosis (111); moreover, the angiotensin II type 1 receptor/β-arrestin-2 signaling pathway is also activated by the absence of TSP-5, resulting in a high risk of AAA (162). TSP-5 supplementation is a potentially effective therapy for CVD treatment. However, TSP-5 also plays a critical role in the migration and invasion of various cancer cells and is a potential target for cancer treatment (163). Cancer patients may benefit from systematic administration of anti-TSP-5 therapy while under the threat of CVD. In conclusion, local targeted therapy or treatment that focuses on interactions with TSPs and signaling pathways may be meaningful (112).
HP: Writing – original draft. XL: Writing – original draft. DY: Writing – review & editing. YF: Writing – review & editing. JW: Writing – review & editing. JY: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This work was supported by the National Natural Science Foundation of China (82070436).
The author gratefully acknowledged Victor Cheng for his support and appreciation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Leong DP, Joseph PG, McKee M, Anand SS, Teo KK, Schwalm JD, et al. Reducing the global burden of cardiovascular disease, part 2: prevention and treatment of cardiovascular disease. Circ Res. (2017) 121(6):695–710. doi: 10.1161/CIRCRESAHA.117.311849
2. Adams JC, Lawler J. The thrombospondins. Int J Biochem Cell Biol. (2004) 36(6):961–8. doi: 10.1016/j.biocel.2004.01.004
3. Frangogiannis NG. Matricellular proteins in cardiac adaptation and disease. Physiol Rev. (2012) 92(2):635–88. doi: 10.1152/physrev.00008.2011
4. Stenina-Adognravi O. Thrombospondins: old players, new games. Curr Opin Lipidol. (2013) 24(5):401–9. doi: 10.1097/MOL.0b013e3283642912
5. Chistiakov DA, Melnichenko AA, Myasoedova VA, Grechko AV, Orekhov AN. Thrombospondins: a role in cardiovascular disease. Int J Mol Sci. (2017) 18(7):1540. doi: 10.3390/ijms18071540
6. Tan LL, Lyon AR. Cardio-oncology for the general cardiologist. Heart (Br Cardiac Soc). (2021) 107(15):1254–66. doi: 10.1136/heartjnl-2020-317871
7. de Boer RA, Meijers WC, van der Meer P, van Veldhuisen DJ. Cancer and heart disease: associations and relations. Eur J Heart Fail. (2019) 21(12):1515–25. doi: 10.1002/ejhf.1539
8. Liang Z, He Y, Hu X. Cardio-oncology: mechanisms, drug combinations, and reverse cardio-oncology. Int J Mol Sci. (2022) 23(18):10617. doi: 10.3390/ijms231810617
9. Aboumsallem JP, Moslehi J, de Boer RA. Reverse cardio-oncology: cancer development in patients with cardiovascular disease. J Am Heart Assoc. (2020) 9(2):e13754. doi: 10.1161/JAHA.119.013754
10. Koene RJ, Prizment AE, Blaes A, Konety SH. Shared risk factors in cardiovascular disease and cancer. Circulation. (2016) 133(11):1104–14. doi: 10.1161/CIRCULATIONAHA.115.020406
11. Greco SA, Chia J, Inglis KJ, Cozzi SJ, Ramsnes I, Buttenshaw RL, et al. Thrombospondin-4 is a putative tumour-suppressor gene in colorectal cancer that exhibits age-related methylation. BMC Cancer. (2010) 10:494. doi: 10.1186/1471-2407-10-494
12. Liu J, Cheng G, Yang H, Deng X, Qin C, Hua L, et al. Reciprocal regulation of long noncoding rnas thbs4-003 and thbs4 control migration and invasion in prostate cancer cell lines. Mol Med Rep. (2016) 14(2):1451–8. doi: 10.3892/mmr.2016.5443
13. Adams JC, Lawler J. The thrombospondins. Cold Spring Harb Perspect Biol. (2011) 3(10):a9712. doi: 10.1101/cshperspect.a009712
14. Murphy-Ullrich JE, Iozzo RV. Thrombospondins in physiology and disease: new tricks for old dogs. Matrix Biol J Int Soc Matrix Biol. (2012) 31(3):152–4. doi: 10.1016/j.matbio.2012.01.002
15. Mustonen E, Ruskoaho H, Rysä J. Thrombospondins, potential drug targets for cardiovascular diseases. Basic Clin Pharmacol Toxicol. (2013) 112(1):4–12. doi: 10.1111/bcpt.12026
16. Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev. (2007) 87(4):1285–342. doi: 10.1152/physrev.00012.2007
17. Chen H, Herndon ME, Lawler J. The cell biology of thrombospondin-1. Matrix Biol J Int Soc Matrix Biol. (2000) 19(7):597–614. doi: 10.1016/S0945-053X(00)00107-4
18. Calzada MJ, Sipes JM, Krutzsch HC, Yurchenco PD, Annis DS, Mosher DF, et al. Recognition of the n-terminal modules of thrombospondin-1 and thrombospondin-2 by alpha6beta1 integrin. J Biol Chem. (2003) 278(42):40679–87. doi: 10.1074/jbc.M302014200
19. Furrer J, Luy B, Basrur V, Roberts DD, Barchi JJ. Conformational analysis of an alpha3beta1 integrin-binding peptide from thrombospondin-1: implications for antiangiogenic drug design. J Med Chem. (2006) 49(21):6324–33. doi: 10.1021/jm060833l
20. Yamashiro Y, Thang BQ, Ramirez K, Shin SJ, Kohata T, Ohata S, et al. Matrix mechanotransduction mediated by thrombospondin-1/integrin/yap in the vascular remodeling. Proc Natl Acad Sci USA. (2020) 117(18):9896–905. doi: 10.1073/pnas.1919702117
21. Trial J, Cieslik KA. Changes in cardiac resident fibroblast physiology and phenotype in aging. Am J Physiol Heart Circ Physiol. (2018) 315(4):H745–55. doi: 10.1152/ajpheart.00237.2018
22. Murphy-Ullrich JE, Suto MJ. Thrombospondin-1 regulation of latent tgf-β activation: a therapeutic target for fibrotic disease. Matrix Biol. (2018) 68-69:28–43. doi: 10.1016/j.matbio.2017.12.009
23. Bao Q, Zhang B, Suo Y, Liu C, Yang Q, Zhang K, et al. Intermittent hypoxia mediated by tsp1 dependent on stat3 induces cardiac fibroblast activation and cardiac fibrosis. Elife. (2020) 9:e49923. doi: 10.7554/eLife.49923
24. Belmadani S, Bernal J, Wei CC, Pallero MA, Dell'italia L, Murphy-Ullrich JE, et al. A thrombospondin-1 antagonist of transforming growth factor-beta activation blocks cardiomyopathy in rats with diabetes and elevated angiotensin ii. Am J Pathol. (2007) 171(3):777–89. doi: 10.2353/ajpath.2007.070056
25. Joseph JV, Magaut CR, Storevik S, Geraldo LH, Mathivet T, Latif MA, et al. Tgf-β promotes microtube formation in glioblastoma through thrombospondin 1. Neuro Oncol. (2022) 24(4):541–53. doi: 10.1093/neuonc/noab212
26. Kim CW, Pokutta-Paskaleva A, Kumar S, Timmins LH, Morris AD, Kang DW, et al. Disturbed flow promotes arterial stiffening through thrombospondin-1. Circulation. (2017) 136(13):1217–32. doi: 10.1161/CIRCULATIONAHA.116.026361
27. Xia Y, Dobaczewski M, Gonzalez-Quesada C, Chen W, Biernacka A, Li N, et al. Endogenous thrombospondin 1 protects the pressure-overloaded myocardium by modulating fibroblast phenotype and matrix metabolism. Hypertension. (2011) 58(5):902–11. doi: 10.1161/HYPERTENSIONAHA.111.175323
28. Nan P, Dong X, Bai X, Lu H, Liu F, Sun Y, et al. Tumor-stroma tgf-β1-thbs2 feedback circuit drives pancreatic ductal adenocarcinoma progression via integrin αβ/cd36-mediated activation of the mapk pathway. Cancer Lett. (2022) 528:59–75. doi: 10.1016/j.canlet.2021.12.025
29. Sun J, Ge X, Wang Y, Niu L, Tang L, Pan S. Usf2 knockdown downregulates thbs1 to inhibit the tgf-β signaling pathway and reduce pyroptosis in sepsis-induced acute kidney injury. Pharmacol Res. (2022) 176:105962. doi: 10.1016/j.phrs.2021.105962
30. Cui W, Maimaitiyiming H, Qi X, Norman H, Wang S. Thrombospondin 1 mediates renal dysfunction in a mouse model of high-fat diet-induced obesity. Am J Physiol Renal Physiol. (2013) 305(6):F871–80. doi: 10.1152/ajprenal.00209.2013
31. Kumar R, Mickael C, Kassa B, Sanders L, Hernandez-Saavedra D, Koyanagi DE, et al. Interstitial macrophage-derived thrombospondin-1 contributes to hypoxia-induced pulmonary hypertension. Cardiovasc Res. (2020) 116(12):2021–30. doi: 10.1093/cvr/cvz304
32. Ren J, Bi Y, Sowers JR, Hetz C, Zhang Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat Rev Cardiol. (2021) 18(7):499–521. doi: 10.1038/s41569-021-00511-w
33. Hetz C, Zhang K, Kaufman RJ. Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol. (2020) 21(8):421–38. doi: 10.1038/s41580-020-0250-z
34. Vanhoutte D, Schips TG, Vo A, Grimes KM, Baldwin TA, Brody MJ, et al. Thbs1 induces lethal cardiac atrophy through perk-atf4 regulated autophagy. Nat Commun. (2021) 12(1):3928. doi: 10.1038/s41467-021-24215-4
35. Zhao XM, Hu Y, Miller GG, Mitchell RN, Libby P. Association of thrombospondin-1 and cardiac allograft vasculopathy in human cardiac allografts. Circulation. (2001) 103(4):525–31. doi: 10.1161/01.CIR.103.4.525
36. Isenberg JS, Ridnour LA, Dimitry J, Frazier WA, Wink DA, Roberts DD. Cd47 is necessary for inhibition of nitric oxide-stimulated vascular cell responses by thrombospondin-1. J Biol Chem. (2006) 281(36):26069–80. doi: 10.1074/jbc.M605040200
37. Isenberg JS, Jia Y, Fukuyama J, Switzer CH, Wink DA, Roberts DD. Thrombospondin-1 inhibits nitric oxide signaling via cd36 by inhibiting myristic acid uptake. J Biol Chem. (2007) 282(21):15404–15. doi: 10.1074/jbc.M701638200
38. Yao M, Roberts DD, Isenberg JS. Thrombospondin-1 inhibition of vascular smooth muscle cell responses occurs via modulation of both camp and cgmp. Pharmacol Res. (2011) 63(1):13–22. doi: 10.1016/j.phrs.2010.10.014
39. Rogers NM, Sharifi-Sanjani M, Csányi G, Pagano PJ, Isenberg JS. Thrombospondin-1 and cd47 regulation of cardiac, pulmonary and vascular responses in health and disease. Matrix Biol J Int Soc Matrix Biol. (2014) 37:92–101. doi: 10.1016/j.matbio.2014.01.002
40. Bauer EM, Qin Y, Miller TW, Bandle RW, Csanyi G, Pagano PJ, et al. Thrombospondin-1 supports blood pressure by limiting enos activation and endothelial-dependent vasorelaxation. Cardiovasc Res. (2010) 88(3):471–81. doi: 10.1093/cvr/cvq218
41. Bazzazi H, Zhang Y, Jafarnejad M, Isenberg JS, Annex BH, Popel AS. Computer simulation of tsp1 inhibition of vegf-akt-enos: an angiogenesis triple threat. Front Physiol. (2018) 9:644. doi: 10.3389/fphys.2018.00644
42. Kaur S, Martin-Manso G, Pendrak ML, Garfield SH, Isenberg JS, Roberts DD. Thrombospondin-1 inhibits vegf receptor-2 signaling by disrupting its association with cd47. J Biol Chem. (2010) 285(50):38923–32. doi: 10.1074/jbc.M110.172304
43. Singla B, Aithbathula RV, Pervaiz N, Kathuria I, Swanson M, Ekuban FA, et al. Cd47 activation by thrombospondin-1 in lymphatic endothelial cells suppresses lymphangiogenesis and promotes atherosclerosis. Arterioscler Thromb Vasc Biol. (2023) 43(7):1234–50. doi: 10.1161/ATVBAHA.122.318904
44. Ganguly R, Khanal S, Mathias A, Gupta S, Lallo J, Sahu S, et al. Tsp-1 (thrombospondin-1) deficiency protects apoe mice against leptin-induced atherosclerosis. Arterioscler, Thromb, Vasc Biol. (2021) 41(2):e112–27. doi: 10.1161/ATVBAHA.120.314962
45. MacLauchlan S, Yu J, Parrish M, Asoulin TA, Schleicher M, Krady MM, et al. Endothelial nitric oxide synthase controls the expression of the angiogenesis inhibitor thrombospondin 2. Proc Natl Acad Sci USA. (2011) 108(46):E1137–45. doi: 10.1073/pnas.1104357108
46. Ma Y, Yabluchanskiy A, Lindsey ML. Thrombospondin-1: the good, the bad, and the complicated. Circ Res. (2013) 113(12):1272–4. doi: 10.1161/CIRCRESAHA.113.302749
47. Schroen B, Heymans S, Sharma U, Blankesteijn WM, Pokharel S, Cleutjens JP, et al. Thrombospondin-2 is essential for myocardial matrix integrity: increased expression identifies failure-prone cardiac hypertrophy. Circ Res. (2004) 95(5):515–22. doi: 10.1161/01.RES.0000141019.20332.3e
48. Swinnen M, Vanhoutte D, Van Almen GC, Hamdani N, Schellings MW, D'hooge J, et al. Absence of thrombospondin-2 causes age-related dilated cardiomyopathy. Circulation. (2009) 120(16):1585–97. doi: 10.1161/CIRCULATIONAHA.109.863266
49. Hayashidani S, Tsutsui H, Ikeuchi M, Shiomi T, Matsusaka H, Kubota T, et al. Targeted deletion of mmp-2 attenuates early lv rupture and late remodeling after experimental myocardial infarction. Am J Physiol Heart Circ Physiol (2003) 285(3):H1229–35. doi: 10.1152/ajpheart.00207.2003
50. Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. (2000) 106(1):55–62. doi: 10.1172/JCI8768
51. van Almen GC, Swinnen M, Carai P, Verhesen W, Cleutjens JP, D'hooge J, et al. Absence of thrombospondin-2 increases cardiomyocyte damage and matrix disruption in doxorubicin-induced cardiomyopathy. J Mol Cell Cardiol. (2011) 51(3):318–28. doi: 10.1016/j.yjmcc.2011.05.010
52. Reinecke H, Robey TE, Mignone JL, Muskheli V, Bornstein P, Murry CE. Lack of thrombospondin-2 reduces fibrosis and increases vascularity around cardiac cell grafts. Cardiovasc Pathol. (2013) 22(1):91–5. doi: 10.1016/j.carpath.2012.03.005
53. Donnini S, Morbidelli L, Taraboletti G, Ziche M. Erk1-2 and p38 mapk regulate mmp/timp balance and function in response to thrombospondin-1 fragments in the microvascular endothelium. Life Sci. (2004) 74(24):2975–85. doi: 10.1016/j.lfs.2003.09.075
54. Krady MM, Zeng J, Yu J, MacLauchlan S, Skokos EA, Tian W, et al. Thrombospondin-2 modulates extracellular matrix remodeling during physiological angiogenesis. Am J Pathol. (2008) 173(3):879–91. doi: 10.2353/ajpath.2008.080128
55. Mourmoura E, Couturier K, Hininger-Favier I, Malpuech-Brugère C, Azarnoush K, Richardson M, et al. Functional changes of the coronary microvasculature with aging regarding glucose tolerance, energy metabolism, and oxidative stress. Age. (2014) 36(4):9670. doi: 10.1007/s11357-014-9670-z
56. Tong X, Khandelwal AR, Qin Z, Wu X, Chen L, Ago T, et al. Role of smooth muscle nox4-based nadph oxidase in neointimal hyperplasia. J Mol Cell Cardiol. (2015) 89:185–94. doi: 10.1016/j.yjmcc.2015.11.013
57. Green DE, Kang BY, Murphy TC, Hart CM. Peroxisome proliferator-activated receptor gamma (pparγ) regulates thrombospondin-1 and nox4 expression in hypoxia-induced human pulmonary artery smooth muscle cell proliferation. Pulm Circ. (2012) 2(4):483–91. doi: 10.4103/2045-8932.105037
58. Csiszar A, Ungvari Z, Edwards JG, Kaminski P, Wolin MS, Koller A, et al. Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ Res. (2002) 90(11):1159–66. doi: 10.1161/01.RES.0000020401.61826.EA
59. Kang LS, Reyes RA, Muller-Delp JM. Aging impairs flow-induced dilation in coronary arterioles: role of no and h(2)o(2). Am J Physiol Heart Circ Physiol. (2009) 297(3):H1087–95. doi: 10.1152/ajpheart.00356.2009
60. Meijles DN, Sahoo S, Al Ghouleh I, Amaral JH, Bienes-Martinez R, Knupp HE, et al. The matricellular protein tsp1 promotes human and mouse endothelial cell senescence through cd47 and nox1. Sci Signal. (2017) 10(501):eaaj1784. doi: 10.1126/scisignal.aaj1784
61. Csányi G, Yao M, Rodríguez AI, Al Ghouleh I, Sharifi-Sanjani M, Frazziano G, et al. Thrombospondin-1 regulates blood flow via cd47 receptor-mediated activation of nadph oxidase 1. Arterioscler Thromb Vasc Biol. (2012) 32(12):2966–73. doi: 10.1161/ATVBAHA.112.300031
62. Bitar MS. Diabetes impairs angiogenesis and induces endothelial cell senescence by up-regulating thrombospondin-cd47-dependent signaling. Int J Mol Sci. (2019) 20(3):673. doi: 10.3390/ijms20030673
63. Bae ON, Wang JM, Baek SH, Wang Q, Yuan H, Chen AF. Oxidative stress-mediated thrombospondin-2 upregulation impairs bone marrow-derived angiogenic cell function in diabetes mellitus. Arterioscler Thromb Vasc Biol. (2013) 33(8):1920–7. doi: 10.1161/ATVBAHA.113.301609
64. Yao M, Rogers NM, Csányi G, Rodriguez AI, Ross MA, St Croix C, et al. Thrombospondin-1 activation of signal-regulatory protein-α stimulates reactive oxygen species production and promotes renal ischemia reperfusion injury. J Am Soc Nephrol. (2014) 25(6):1171–86. doi: 10.1681/ASN.2013040433
65. Ismail AA, Shaker BT, Bajou K. The plasminogen-activator plasmin system in physiological and pathophysiological angiogenesis. Int J Mol Sci. (2021) 23(1):337. doi: 10.3390/ijms23010337
66. Fang J, Chopp M, Xin H, Zhang L, Wang F, Golembieski W, et al. Plasminogen deficiency causes reduced angiogenesis and behavioral recovery after stroke in mice. J Cereb Blood Flow Metab. (2021) 41(10):2583–92. doi: 10.1177/0271678X211007958
67. Kaiser R, Grotemeyer K, Kälsch T, Gräber S, Wilkens H, Elmas E. Decreased tsp-1 following percutaneous coronary intervention is associated with major adverse cardiac events in st-elevation myocardial infarction. Clin Hemorheol Microcirc. (2013) 54(1):59–73. doi: 10.3233/CH-2012-1565
68. Hugo C. The thrombospondin 1-tgf-beta axis in fibrotic renal disease. Nephrol Dial Transplant. (2003) 18(7):1241–5. doi: 10.1093/ndt/gfg159
69. Blakytny R, Ludlow A, Martin GE, Ireland G, Lund LR, Ferguson MW, et al. Latent tgf-beta1 activation by platelets. J Cell Physiol. (2004) 199(1):67–76. doi: 10.1002/jcp.10454
70. Venkatraman L, Chia SM, Narmada BC, White JK, Bhowmick SS, Forbes Dewey C, et al. Plasmin triggers a switch-like decrease in thrombospondin-dependent activation of tgf-β1. Biophys J. (2012) 103(5):1060–8. doi: 10.1016/j.bpj.2012.06.050
71. Grimbert P, Bouguermouh S, Baba N, Nakajima T, Allakhverdi Z, Braun D, et al. Thrombospondin/cd47 interaction: a pathway to generate regulatory t cells from human cd4 + cd25- t cells in response to inflammation. J Immunol. (2006) 177(6):3534–41. doi: 10.4049/jimmunol.177.6.3534
72. Futagami Y, Sugita S, Vega J, Ishida K, Takase H, Maruyama K, et al. Role of thrombospondin-1 in t cell response to ocular pigment epithelial cells. J Immunol. (2007) 178(11):6994–7005. doi: 10.4049/jimmunol.178.11.6994
73. Lamy L, Foussat A, Brown EJ, Bornstein P, Ticchioni M, Bernard A. Interactions between cd47 and thrombospondin reduce inflammation. J Immunol. (2007) 178(9):5930–9. doi: 10.4049/jimmunol.178.9.5930
74. Papageorgiou AP, Swinnen M, Vanhoutte D, VandenDriessche T, Chuah M, Lindner D, et al. Thrombospondin-2 prevents cardiac injury and dysfunction in viral myocarditis through the activation of regulatory t-cells. Cardiovasc Res. (2012) 94(1):115–24. doi: 10.1093/cvr/cvs077
75. Shi Y, Fukuoka M, Li G, Liu Y, Chen M, Konviser M, et al. Regulatory t cells protect mice against coxsackievirus-induced myocarditis through the transforming growth factor beta-coxsackie-adenovirus receptor pathway. Circulation. (2010) 121(24):2624–34. doi: 10.1161/CIRCULATIONAHA.109.893248
76. Raffort J, Lareyre F, Clément M, Hassen-Khodja R, Chinetti G, Mallat Z. Monocytes and macrophages in abdominal aortic aneurysm. Nat Rev Cardiol. (2017) 14(8):457–71. doi: 10.1038/nrcardio.2017.52
77. Yang H, Zhou T, Sorenson CM, Sheibani N, Liu B. Myeloid-derived tsp1 (thrombospondin-1) contributes to abdominal aortic aneurysm through suppressing tissue inhibitor of metalloproteinases-1. Arterioscler Thromb Vasc Biol. (2020) 40(12):e350–66. doi: 10.1161/ATVBAHA.120.314913
78. Moura R, Tjwa M, Vandervoort P, Van Kerckhoven S, Holvoet P, Hoylaerts MF. Thrombospondin-1 deficiency accelerates atherosclerotic plaque maturation in apoe-/- mice. Circ Res. (2008) 103(10):1181–9. doi: 10.1161/CIRCRESAHA.108.185645
79. Stein EV, Miller TW, Ivins-O'Keefe K, Kaur S, Roberts DD. Secreted thrombospondin-1 regulates macrophage interleukin-1β production and activation through cd47. Sci Rep. (2016) 6:19684. doi: 10.1038/srep19684
80. Zeng T, Yuan J, Gan J, Liu Y, Shi L, Lu Z, et al. Thrombospondin 1 is increased in the aorta and plasma of patients with acute aortic dissection. Can J Cardiol. (2019) 35(1):42–50. doi: 10.1016/j.cjca.2018.11.008
81. Kitami K, Yoshihara M, Tamauchi S, Sugiyama M, Koya Y, Yamakita Y, et al. Peritoneal restoration by repurposing vitamin d inhibits ovarian cancer dissemination via blockade of the tgf-β1/thrombospondin-1 axis. Matrix Biol J Int Soc Matrix Biol. (2022) 109:70–90. doi: 10.1016/j.matbio.2022.03.003
82. Patwardhan S, Mahadik P, Shetty O, Sen S. Ecm stiffness-tuned exosomes drive breast cancer motility through thrombospondin-1. Biomaterials. (2021) 279:121185. doi: 10.1016/j.biomaterials.2021.121185
83. Stirling ER, Terabe M, Wilson AS, Kooshki M, Yamaleyeva LM, Alexander-Miller MA, et al. Targeting the cd47/thrombospondin-1 signaling axis regulates immune cell bioenergetics in the tumor microenvironment to potentiate antitumor immune response. J Immunother Cancer. (2022) 10(11):e004712. doi: 10.1136/jitc-2022-004712
84. Weng TY, Huang SS, Yen MC, Lin CC, Chen YL, Lin CM, et al. A novel cancer therapeutic using thrombospondin 1 in dendritic cells. Mol Therapy. (2014) 22(2):292–302. doi: 10.1038/mt.2013.236
85. Schips TG, Vanhoutte D, Vo A, Correll RN, Brody MJ, Khalil H, et al. Thrombospondin-3 augments injury-induced cardiomyopathy by intracellular integrin inhibition and sarcolemmal instability. Nat Commun. (2019) 10(1):76. doi: 10.1038/s41467-018-08026-8
86. Huang Y, Xia J, Zheng J, Geng B, Liu P, Yu F, et al. Deficiency of cartilage oligomeric matrix protein causes dilated cardiomyopathy. Basic Res Cardiol. (2013) 108(5):374. doi: 10.1007/s00395-013-0374-9
87. Cuadrado A, Nebreda AR. Mechanisms and functions of p38 mapk signalling. Biochem J. (2010) 429(3):403–17. doi: 10.1042/BJ20100323
88. Wang C, Hockerman S, Jacobsen EJ, Alippe Y, Selness SR, Hope HR, et al. Selective inhibition of the p38α mapk-mk2 axis inhibits inflammatory cues including inflammasome priming signals. J Exp Med. (2018) 215(5):1315–25. doi: 10.1084/jem.20172063
89. Frolova EG, Pluskota E, Krukovets I, Burke T, Drumm C, Smith JD, et al. Thrombospondin-4 regulates vascular inflammation and atherogenesis. Circ Res. (2010) 107(11):1313–25. doi: 10.1161/CIRCRESAHA.110.232371
90. Stenina OI, Desai SY, Krukovets I, Kight K, Janigro D, Topol EJ, et al. Thrombospondin-4 and its variants: expression and differential effects on endothelial cells. Circulation. (2003) 108(12):1514–9. doi: 10.1161/01.CIR.0000089085.76320.4E
91. Muppala S, Frolova E, Xiao R, Krukovets I, Yoon S, Hoppe G, et al. Proangiogenic properties of thrombospondin-4. Arterioscler Thromb Vasc Biol. (2015) 35(9):1975–86. doi: 10.1161/ATVBAHA.115.305912
92. Jia Y, Wang M, Mao C, Yu F, Wang Y, Xiao R, et al. Comp-prohibitin 2 interaction maintains mitochondrial homeostasis and controls smooth muscle cell identity. Cell Death Dis. (2018) 9(6):676. doi: 10.1038/s41419-018-0703-x
93. Wang L, Zheng J, Du Y, Huang Y, Li J, Liu B, et al. Cartilage oligomeric matrix protein maintains the contractile phenotype of vascular smooth muscle cells by interacting with alpha(7)beta(1) integrin. Circ Res. (2010) 106(3):514–25. doi: 10.1161/CIRCRESAHA.109.202762
94. Riessen R, Fenchel M, Chen H, Axel DI, Karsch KR, Lawler J. Cartilage oligomeric matrix protein (thrombospondin-5) is expressed by human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. (2001) 21(1):47–54. doi: 10.1161/01.ATV.21.1.47
95. Lv H, Wang H, Quan M, Zhang C, Fu Y, Zhang L, et al. Cartilage oligomeric matrix protein fine-tunes disturbed flow-induced endothelial activation and atherogenesis. Matrix Biol J Int Soc Matrix Biol. (2021) 95:32–51. doi: 10.1016/j.matbio.2020.10.003
96. Goumans MJ, Liu Z, Ten Dijke P. Tgf-beta signaling in vascular biology and dysfunction. Cell Res. (2009) 19(1):116–27. doi: 10.1038/cr.2008.326
97. Muppala S, Xiao R, Krukovets I, Verbovetsky D, Yendamuri R, Habib N, et al. Thrombospondin-4 mediates tgf-β-induced angiogenesis. Oncogene. (2017) 36(36):5189–98. doi: 10.1038/onc.2017.140
98. Haudenschild DR, Hong E, Yik JH, Chromy B, Mörgelin M, Snow KD, et al. Enhanced activity of transforming growth factor β1 (tgf-β1) bound to cartilage oligomeric matrix protein. J Biol Chem. (2011) 286(50):43250–8. doi: 10.1074/jbc.M111.234716
99. Farina G, Lemaire R, Pancari P, Bayle J, Widom RL, Lafyatis R. Cartilage oligomeric matrix protein expression in systemic sclerosis reveals heterogeneity of dermal fibroblast responses to transforming growth factor beta. Ann Rheum Dis. (2009) 68(3):435–41. doi: 10.1136/ard.2007.086850
100. Schulz JN, Plomann M, Sengle G, Gullberg D, Krieg T, Eckes B. New developments on skin fibrosis—essential signals emanating from the extracellular matrix for the control of myofibroblasts. Matrix Biol J Int Soc Matrix Biol. (2018):522–32. doi: 10.1016/j.matbio.2018.01.025
101. Du Y, Wang Y, Wang L, Liu B, Tian Q, Liu CJ, et al. Cartilage oligomeric matrix protein inhibits vascular smooth muscle calcification by interacting with bone morphogenetic protein-2. Circ Res. (2011) 108(8):917–28. doi: 10.1161/CIRCRESAHA.110.234328
102. Lynch JM, Maillet M, Vanhoutte D, Schloemer A, Sargent MA, Blair NS, et al. A thrombospondin-dependent pathway for a protective er stress response. Cell. (2012) 149(6):1257–68. doi: 10.1016/j.cell.2012.03.050
103. Brody MJ, Vanhoutte D, Schips TG, Boyer JG, Bakshi CV, Sargent MA, et al. Defective flux of thrombospondin-4 through the secretory pathway impairs cardiomyocyte membrane stability and causes cardiomyopathy. Mol Cell Biol. (2018) 38(14):e00114–8. doi: 10.1128/MCB.00114-18
104. Doroudgar S, Glembotski CC. Atf6 [corrected] and thrombospondin 4: the dynamic duo of the adaptive endoplasmic reticulum stress response. Circ Res. (2013) 112(1):9–12. doi: 10.1161/CIRCRESAHA.112.280560
105. Palao T, Rippe C, van Veen H, VanBavel E, Swärd K, Bakker EN. Thrombospondin-4 knockout in hypertension protects small-artery endothelial function but induces aortic aneurysms. Am J Physiol Heart Circ Physiol. (2016) 310(11):H1486–93. doi: 10.1152/ajpheart.00046.2016
106. Hashimoto Y, Tomiyama T, Yamano Y, Mori H. Mutation (d472y) in the type 3 repeat domain of cartilage oligomeric matrix protein affects its early vesicle trafficking in endoplasmic reticulum and induces apoptosis. Am J Pathol. (2003) 163(1):101–10. doi: 10.1016/S0002-9440(10)63634-6
107. Wang H, Yuan Z, Wang B, Li B, Lv H, He J, et al. Comp (cartilage oligomeric matrix protein), a novel piezo1 regulator that controls blood pressure. Hypertension. (2022) 79(3):549–61. doi: 10.1161/HYPERTENSIONAHA.121.17972
108. Rahman MT, Muppala S, Wu J, Krukovets I, Solovjev D, Verbovetskiy D, et al. Effects of thrombospondin-4 on pro-inflammatory phenotype differentiation and apoptosis in macrophages. Cell Death Dis. (2020) 11(1):53. doi: 10.1038/s41419-020-2237-2
109. Lv L, Liang W, Ye M, Zhang J, Zhang H, Xue G, et al. Thrombospondin-4 ablation reduces macrophage recruitment in adipose tissue and neointima and suppresses injury-induced restenosis in mice. Atherosclerosis. (2016) 247:70–7. doi: 10.1016/j.atherosclerosis.2016.02.005
110. Wang M, Fu Y, Gao C, Jia Y, Huang Y, Liu L, et al. Cartilage oligomeric matrix protein prevents vascular aging and vascular smooth muscle cells senescence. Biochem Biophys Res Commun. (2016) 478(2):1006–13. doi: 10.1016/j.bbrc.2016.08.004
111. Fu Y, Kong W. Cartilage oligomeric matrix protein: matricellular and matricrine signaling in cardiovascular homeostasis and disease. Curr Vasc Pharmacol. (2017) 15(3):186–96. doi: 10.2174/1570161115666170201121232
112. Fu Y, Gao C, Liang Y, Wang M, Huang Y, Ma W, et al. Shift of macrophage phenotype due to cartilage oligomeric matrix protein deficiency drives atherosclerotic calcification. Circ Res. (2016) 119(2):261–76. doi: 10.1161/CIRCRESAHA.115.308021
113. Guo N, Yang L, Wan X, Qiu D, Sun W, Ma H. Relationship between elevated circulating thrombospondin-1 levels and vascular complications in diabetes mellitus. J Diabetes Investig. (2024) 15(2):197–207. doi: 10.1111/jdi.14095
114. Kaiser R, Frantz C, Bals R, Wilkens H. The role of circulating thrombospondin-1 in patients with precapillary pulmonary hypertension. Respir Res. (2016) 17(1):96. doi: 10.1186/s12931-016-0412-x
115. Berezin AE, Kremzer AA, Samura TA. Circulating thrombospondine-2 in patients with moderate-to-severe chronic heart failure due to coronary artery disease. J Biomed Res. (2015) 2:30. doi: 10.7555/JBR.29.20140025
116. Egerstedt A, Berntsson J, Smith ML, Gidlöf O, Nilsson R, Benson M, et al. Profiling of the plasma proteome across different stages of human heart failure. Nat Commun. (2019) 10(1):5830. doi: 10.1038/s41467-019-13306-y
117. Kresoja KP, Rommel KP, Wachter R, Henger S, Besler C, Klöting N, et al. Proteomics to improve phenotyping in obese patients with heart failure with preserved ejection fraction. Eur J Heart Fail. (2021) 23(10):1633–44. doi: 10.1002/ejhf.2291
118. Lee CH, Wu MZ, Lui D, Fong C, Ren QW, Yu SY, et al. Prospective associations of circulating thrombospondin-2 level with heart failure hospitalization, left ventricular remodeling and diastolic function in type 2 diabetes. Cardiovasc Diabetol. (2022) 21(1):231. doi: 10.1186/s12933-022-01646-x
119. Chen Y, Meng H, Meng X, Yan Z, Wang J, Meng F. Correlation between low thbs3 expression in peripheral blood and acute myocardial infarction. Front Biosci. (2022) 27(10):291. doi: 10.31083/j.fbl2710291
120. Liao W, Xu L, Pan Y, Wei J, Wang P, Yang X, et al. Association of atrial arrhythmias with thrombospondin-1 in patients with acute myocardial infarction. Bmc Cardiovasc Disord. (2021) 21(1):507. doi: 10.1186/s12872-021-02322-w
121. Alonso-Orgaz S, Moreno-Luna R, López JA, Gil-Dones F, Padial LR, Moreu J, et al. Proteomic characterization of human coronary thrombus in patients with st-segment elevation acute myocardial infarction. J Proteomics. (2014) 109:368–81. doi: 10.1016/j.jprot.2014.07.016
122. Qi L, Wu K, Shi S, Ji Q, Miao H, Bin Q. Thrombospondin-2 is upregulated in patients with aortic dissection and enhances angiotensin ii-induced smooth muscle cell apoptosis. Exp Ther Med. (2020) 20(6):150. doi: 10.3892/etm.2020.9279
123. Smadja DM, D'Audigier C, Bièche I, Evrard S, Mauge L, Dias JV, et al. Thrombospondin-1 is a plasmatic marker of peripheral arterial disease that modulates endothelial progenitor cell angiogenic properties. Arterioscler, Thromb, Vasc Biol. (2011) 31(3):551–9. doi: 10.1161/ATVBAHA.110.220624
124. Yazman S, Depboylu BC, Saruhan E, Cenikli U, Harmandar B, Arslan K, et al. New biomarkers (endocan, interleukin-17, and thrombospondin-4) for the diagnosis, assessment of severity, and follow-up of peripheral arterial disease. Angiology. (2023) 74(7):631–9. doi: 10.1177/00033197231166492
125. Prentice RL, Zhao S, Johnson M, Aragaki A, Hsia J, Jackson RD, et al. Proteomic risk markers for coronary heart disease and stroke: validation and mediation of randomized trial hormone therapy effects on these diseases. Genome Med. (2013) 5(12):112. doi: 10.1186/gm517
126. Wang FF, Ha L, Yu HY, Mi L, Han JL, Gao W. Altered serum level of cartilage oligomeric matrix protein and its association with coronary calcification in patients with coronary heart disease. J Geriatr Cardiol. (2017) 14(2):87–92. doi: 10.11909/j.issn.1671-5411.2017.02.002
127. Pohjolainen V, Mustonen E, Taskinen P, Näpänkangas J, Leskinen H, Ohukainen P, et al. Increased thrombospondin-2 in human fibrosclerotic and stenotic aortic valves. Atherosclerosis. (2012) 220(1):66–71. doi: 10.1016/j.atherosclerosis.2011.10.003
128. Sonmez FC, Yildiz P, Akhtar MS, Aydin C, Sonmez O, Ay N, et al. New markers in atherosclerosis: thrombospondin-2 (thbs-2) and leukocyte cell-derived chemotaxin-2 (lect-2); an immunohistochemical study. Med Sci Monit. (2016) 22:5234–9. doi: 10.12659/MSM.898889
129. Moxon JV, Padula MP, Clancy P, Emeto TI, Herbert BR, Norman PE, et al. Proteomic analysis of intra-arterial thrombus secretions reveals a negative association of clusterin and thrombospondin-1 with abdominal aortic aneurysm. Atherosclerosis. (2011) 219(2):432–9. doi: 10.1016/j.atherosclerosis.2011.08.013
130. Golledge J, Clancy P, Hankey GJ, Norman PE. Relation between serum thrombospondin-2 and cardiovascular mortality in older men screened for abdominal aortic aneurysm. Am J Cardiol. (2013) 111(12):1800–4. doi: 10.1016/j.amjcard.2013.02.038
131. Xu L, Zhang Y, Chen J, Xu Y. Thrombospondin-1: a key protein that induces fibrosis in diabetic complications. J Diabetes Res. (2020) 2020:8043135. doi: 10.1155/2020/8043135
132. Lin Z, Zhang D, Zhang X, Guo W, Wang W, Zhang Y, et al. Extracellular status of thrombospondin-2 in type 2 diabetes mellitus and utility as a biomarker in the determination of early diabetic kidney disease. Bmc Nephrol. (2023) 24(1):154. doi: 10.1186/s12882-023-03216-z
133. Choi KY, Kim DB, Kim MJ, Kwon BJ, Chang SY, Jang SW, et al. Higher plasma thrombospondin-1 levels in patients with coronary artery disease and diabetes mellitus. Korean Circ J. (2012) 42(2):100–6. doi: 10.4070/kcj.2012.42.2.100
134. Lopes FC, Traina F, Almeida CB, Leonardo FC, Franco-Penteado CF, Garrido VT, et al. Key endothelial cell angiogenic mechanisms are stimulated by the circulating milieu in sickle cell disease and attenuated by hydroxyurea. Haematologica. (2015) 100(6):730–9. doi: 10.3324/haematol.2014.119727
135. Carlson CB, Lawler J, Mosher DF. Structures of thrombospondins. Cell Mol Life Sci. (2008) 65(5):672–86. doi: 10.1007/s00018-007-7484-1
136. Dawson DW, Volpert OV, Pearce SF, Schneider AJ, Silverstein RL, Henkin J, et al. Three distinct d-amino acid substitutions confer potent antiangiogenic activity on an inactive peptide derived from a thrombospondin-1 type 1 repeat. Mol Pharmacol. (1999) 55(2):332–8. doi: 10.1124/mol.55.2.332
137. Thaunat O, Louedec L, Graff-Dubois S, Dai J, Groyer E, Yacoub-Youssef H, et al. Antiangiogenic treatment prevents adventitial constrictive remodeling in graft arteriosclerosis. Transplantation. (2008) 85(2):281–9. doi: 10.1097/TP.0b013e318160500a
138. Punekar S, Zak S, Kalter VG, Dobransky L, Punekar I, Lawler JW, et al. Thrombospondin 1 and its mimetic peptide abt-510 decrease angiogenesis and inflammation in a murine model of inflammatory bowel disease. Pathobiol J Immunopathol Mol Cell Biol. (2008) 75(1):9–21. doi: 10.1159/000113790
139. Gietema JA, Hoekstra R, de Vos FY, Uges DR, van der Gaast A, Groen HJ, et al. A phase i study assessing the safety and pharmacokinetics of the thrombospondin-1-mimetic angiogenesis inhibitor abt-510 with gemcitabine and cisplatin in patients with solid tumors. Ann Oncol. (2006) 17(8):1320–7. doi: 10.1093/annonc/mdl102
140. Baker LH, Rowinsky EK, Mendelson D, Humerickhouse RA, Knight RA, Qian J, et al. Randomized, phase ii study of the thrombospondin-1-mimetic angiogenesis inhibitor abt-510 in patients with advanced soft tissue sarcoma. J Clin Oncol. (2008) 26(34):5583–8. doi: 10.1200/JCO.2008.17.4706
141. Campbell NE, Greenaway J, Henkin J, Moorehead RA, Petrik J. The thrombospondin-1 mimetic abt-510 increases the uptake and effectiveness of cisplatin and paclitaxel in a mouse model of epithelial ovarian cancer. Neoplasia. (2010) 12(3):275–83. doi: 10.1593/neo.91880
142. Uronis HE, Cushman SM, Bendell JC, Blobe GC, Morse MA, Nixon AB, et al. A phase I study of abt-510 plus bevacizumab in advanced solid tumors. Cancer Med. (2013) 2(3):316–24. doi: 10.1002/cam4.65
143. Hoekstra R, de Vos FY, Eskens FA, de Vries EG, Uges DR, Knight R, et al. Phase I study of the thrombospondin-1-mimetic angiogenesis inhibitor abt-510 with 5-fluorouracil and leucovorin: a safe combination. Eur J Cancer. (2006) 42(4):467–72. doi: 10.1016/j.ejca.2005.08.040
144. Ebbinghaus S, Hussain M, Tannir N, Gordon M, Desai AA, Knight RA, et al. Phase 2 study of abt-510 in patients with previously untreated advanced renal cell carcinoma. Clin Cancer Res. (2007) 13:6689–95. doi: 10.1158/1078-0432.CCR-07-1477
145. Isenberg JS, Romeo MJ, Abu-Asab M, Tsokos M, Oldenborg A, Pappan L, et al. Increasing survival of ischemic tissue by targeting cd47. Circ Res. (2007) 100(5):712–20. doi: 10.1161/01.RES.0000259579.35787.4e
146. Liu J, Wang L, Zhao F, Tseng S, Narayanan C, Shura L, et al. Pre-clinical development of a humanized anti-cd47 antibody with anti-cancer therapeutic potential. Plos One. (2015) 10(9):e137345. doi: 10.1016/j.ajpath.2011.02.039
147. Lu A, Miao M, Schoeb TR, Agarwal A, Murphy-Ullrich JE. Blockade of tsp1-dependent tgf-β activity reduces renal injury and proteinuria in a murine model of diabetic nephropathy. Am J Pathol. (2011) 178(6):2573–86. doi: 10.1016/j.ajpath.2011.02.039
148. Kuroki H, Hayashi H, Nakagawa S, Sakamoto K, Higashi T, Nitta H, et al. Effect of lskl peptide on thrombospondin 1-mediated transforming growth factor β signal activation and liver regeneration after hepatectomy in an experimental model. Br J Surg. (2015) 102(7):813–25. doi: 10.1002/bjs.9765
149. Xu X, Khoong YM, Gu S, Huang X, Ren JY, Gu YH, et al. Investigating the potential of lskl peptide as a novel hypertrophic scar treatment. Biomed Pharmacother. (2020) 124:109824. doi: 10.1016/j.biopha.2020.109824
150. Krishna SM, Seto SW, Jose RJ, Biros E, Moran CS, Wang Y, et al. A peptide antagonist of thrombospondin-1 promotes abdominal aortic aneurysm progression in the angiotensin ii-infused apolipoprotein-e-deficient mouse. Arterioscler, Thromb, Vasc Biol. (2015) 35(2):389–98. doi: 10.1161/ATVBAHA.114.304732
151. Jain V, Rifai MA, Brinzevich D, Taj M, Saleh M, Krittanawong C, et al. Association of premature atherosclerotic cardiovascular disease with higher risk of cancer: a behavioral risk factor surveillance system study. Eur J Prev Cardiol. (2022) 29(3):493–501. doi: 10.1093/eurjpc/zwab084
152. Malmborg M, Christiansen CB, Schmiegelow MD, Torp-Pedersen C, Gislason G, Schou M. Incidence of new onset cancer in patients with a myocardial infarction—a nationwide cohort study. Bmc Cardiovasc Disord. (2018) 18(1):198. doi: 10.1186/s12872-018-0932-z
153. Meijers WC, de Boer RA. Common risk factors for heart failure and cancer. Cardiovasc Res. (2019) 115(5):844–53. doi: 10.1093/cvr/cvz035
154. Zheng Y, Zou F, Wang J, Yin G, Le V, Fei Z, et al. Photodynamic therapy-mediated cancer vaccination enhances stem-like phenotype and immune escape, which can be blocked by thrombospondin-1 signaling through cd47 receptor protein. J Biol Chem. (2015) 290(14):8975–86. doi: 10.1074/jbc.M114.624965
155. Liao X, Yan S, Li J, Jiang C, Huang S, Liu S, et al. Cd36 and its role in regulating the tumor microenvironment. Curr Oncol. (2022) 29(11):8133–45. doi: 10.3390/curroncol29110642
156. Saumet A, Slimane MB, Lanotte M, Lawler J, Dubernard V. Type 3 repeat/c-terminal domain of thrombospondin-1 triggers caspase-independent cell death through cd47/alphavbeta3 in promyelocytic leukemia nb4 cells. Blood. (2005) 106(2):658–67. doi: 10.1182/blood-2004-09-3585
157. Roberts DD, Isenberg JS, Ridnour LA, Wink DA. Nitric oxide and its gatekeeper thrombospondin-1 in tumor angiogenesis. Clin Cancer Res. (2007) 13(3):795–8. doi: 10.1158/1078-0432.CCR-06-1758
158. Shi Y, Sun L, Zhang R, Hu Y, Wu Y, Dong X, et al. Thrombospondin 4/integrin α2/hsf1 axis promotes proliferation and cancer stem-like traits of gallbladder cancer by enhancing reciprocal crosstalk between cancer-associated fibroblasts and tumor cells. J Exp Clin Cancer Res. (2021) 40(1):14. doi: 10.1186/s13046-020-01812-7
159. Englund E, Canesin G, Papadakos KS, Vishnu N, Persson E, Reitsma B, et al. Cartilage oligomeric matrix protein promotes prostate cancer progression by enhancing invasion and disrupting intracellular calcium homeostasis. Oncotarget. (2017) 8(58):98298–311. doi: 10.18632/oncotarget.21176
160. Kaur S, Bronson SM, Pal-Nath D, Miller TW, Soto-Pantoja DR, Roberts DD. Functions of thrombospondin-1 in the tumor microenvironment. Int J Mol Sci. (2021) 22(9):4570. doi: 10.3390/ijms22094570
161. Muppala S, Xiao R, Gajeton J, Krukovets I, Verbovetskiy D, Stenina-Adognravi O. Thrombospondin-4 mediates hyperglycemia- and tgf-beta-induced inflammation in breast cancer. Int J Cancer. (2021) 148(8):2010–22. doi: 10.1002/ijc.33439
162. Fu Y, Huang Y, Yang Z, Chen Y, Zheng J, Mao C, et al. Cartilage oligomeric matrix protein is an endogenous β-arrestin-2-selective allosteric modulator of at1 receptor counteracting vascular injury. Cell Res. (2021) 31(7):773–90. doi: 10.1038/s41422-020-00464-8
Keywords: cardiovascular disease, thrombospondin, cartilage oligomeric matrix protein, myocardial remodeling, vascular remodeling
Citation: Pan H, Lu X, Ye D, Feng Y, Wan J and Ye J (2024) The molecular mechanism of thrombospondin family members in cardiovascular diseases. Front. Cardiovasc. Med. 11:1337586. doi: 10.3389/fcvm.2024.1337586
Received: 20 November 2023; Accepted: 14 February 2024;
Published: 7 March 2024.
Edited by:
Sasha A. Singh, Harvard Medical School, United StatesReviewed by:
Priya Raman, Northeast Ohio Medical University, United States© 2024 Pan, Lu, Ye, Feng, Wan and Ye. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Wan d2FuanVuQHdodS5lZHUuY24= Jing Ye d2h1eWVqaW5nQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.