 Samar A. Tarraf
Samar A. Tarraf Rodrigo Barbosa de Souza
Rodrigo Barbosa de Souza Ashley Herrick1
Ashley Herrick1 Lygia V. Pereira
Lygia V. Pereira Chiara Bellini
Chiara Bellini- 1Department of Bioengineering, Northeastern University, Boston, MA, United States
- 2Department of Genetics and Evolutionary Biology, University of São Paulo, São Paulo, Brazil
Introduction: Ascending thoracic aortic aneurysms arise from pathological tissue remodeling that leads to abnormal wall dilation and increases the risk of fatal dissection/rupture. Large variability in disease manifestations across family members who carry a causative genetic variant for thoracic aortic aneurysms suggests that genetic modifiers may exacerbate clinical outcomes. Decreased perlecan expression in the aorta of mgΔlpn mice with severe Marfan syndrome phenotype advocates for exploring perlecan-encoding Hspg2 as a candidate modifier gene.
Methods: To determine the effect of concurrent Hspg2 and Fbn1 mutations on the progression of thoracic aortopathy, we characterized the microstructure and passive mechanical response of the ascending thoracic aorta in female mice of four genetic backgrounds: wild-type, heterozygous with a mutation in the Fbn1 gene (mgΔlpn), heterozygous with a mutation in the Hspg2 gene (Hspg2+/−), and double mutants carrying both the Fbn1 and Hspg2 variants (dMut).
Results: Elastic fiber fragmentation and medial disarray progress from the internal elastic lamina outward as the ascending thoracic aorta dilates in mgΔlpn and dMut mice. Concurrent increase in total collagen content relative to elastin reduces energy storage capacity and cyclic distensibility of aortic tissues from mice that carry the Fbn1 variant. Inherent circumferential tissue stiffening strongly correlates with the severity of aortic dilatation in mgΔlpn and dMut mice. Perlecan haploinsufficiency superimposed to the mgΔlpn mutation curbs the viability of dMut mice, increases the occurrence of aortic enlargement, and reduces the axial stretch in aortic tissues.
Discussion: Overall, our findings show that dMut mice are more vulnerable than mgΔlpn mice without an Hspg2 mutation, yet later endpoints and additional structural and functional readouts are needed to identify causative mechanisms.
1 Introduction
Advanced DNA sequencing technology and computational tools have been instrumental for the identification of genetic variants that predispose to aortic aneurysms (1). Insights gained from genetic screening aid the clinical management of patients who suffer from an aortic aneurysm and facilitate early diagnosis in family members, thereby preventing premature deaths (2). A broad range of variants in the FBN1 gene encoding for fibrillin-1 have been linked to thoracic aortic disease. The glycoprotein fibrillin-1 assembles into microfibrils to guide the formation of elastic fibers and support their long-term stability (3, 4). Defects or mutations in the FBN1 gene typically give rise to the disarray of elastic fibers in the aorta (4). Since the production of mechanically-competent elastic fibers is limited to the perinatal period, their degradation is often compensated by collagen deposition and reorganization. An increase in the collagen to elastin ratio often leads to tissue stiffening and hinders the aorta from acting as an elastic reservoir to augment diastolic blood flow (5, 6). More than 1,300 mutations in the FBN1 gene cause Marfan syndrome (MFS), a connective tissue disorder characterized by highly penetrant ascending thoracic aortic aneurysms (ATAAs) (7) that progress to life-threatening aortic rupture and/or dissection in the absence of surgical intervention (8). The gold standard for treatment of ATAAs is open repair surgery, whereby the aneurysmal tissue is excised and replaced with a synthetic vascular graft (9). The current clinical guideline for informing surgical intervention is an aortic diameter exceeding 5cm. However, it has long been observed that diameter is a poor predictor of aortic failure in aneurysmal disease (10, 11). For suitable alternative guidelines to be proposed, a better understanding of disease progression as a function of the genetic underpinnings of ATAAs is necessary.
Evolutionary conservation of the genes linked to thoracic aortic aneurysm warranted the development of mouse models that recapitulate human disease (12) and afford longitudinal studies to monitor tissue microstructure and wall mechanics over time. While several mouse models of MFS exist (13–16), we focus here on the mg strain (16). mg mice display classic phenotypic features of patients with MFS, including skeletal kyphosis and ocular defects, as well as enlargement of peripheral air spaces and compromised alveolar wall structures (16–18). Vascular manifestations of MFS in mg mice encompass thickening of the aortic media, disruption of the elastic fiber network, and formation of thoracic aortic aneurysms. Compared to other mouse models of MFS that tend to die prematurely (13, 19), mg mice bred on a C57BL/6 background carry a stable phenotype and live up to 9 months, rendering them a suitable model for tracking aortic remodeling and disease progression (16). Toward this end, we first compared biaxial wall mechanics in the ascending thoracic aorta of mg mice at 4 weeks (pre-aneurysmal), 12 weeks (early aneurysm), and 30 weeks (late-stage aneurysm).
Large intrafamilial variability in the clinical manifestations of MFS (20, 21) suggests that genetic modifiers may exacerbate the phenotype imposed by causative variants. Efforts to identify modifiers genes serve to improve patient screening and recognize individuals at higher risk. It has been shown that mg mice bred on a 129/Sv background mimic the phenotypic range noted across MFS patients (16). These mice have supported recent attempts at recognizing modifier genes (22, 23) that put forth Hspg2 as a candidate modifier for MFS (21). Hspg2 encodes for perlecan, a heparan-sulfate proteoglycan that is present in all basement membranes (24, 25), where it interacts and colocalizes with fibrillin-1 and elastin (26–28). We have recently reported the concurrence of skeletal deformity and ATAA/aortic dissection in mg mice (17). Furthermore, we noted that mg mice presenting a severe phenotype with hyperkyphosis, elastic fiber fragmentation, and aneurysm formation, exhibit lower HSPG2 expression in the aorta than mildly affected mice (21). While perlecan-null mice succumb perinatally (29), perlecan deficiency significantly increases the incidence of aortic dissection in a lethality-rescued transgenic model (Hspg2–Tg) (28). Taken together, these findings suggest a link between HSPG2 deficiency and disease severity that warrants further investigation. To explore the role of perlecan haploinsufficiency on early-stage thoracic aortopathy, we characterized biaxial wall mechanics in the ascending thoracic aorta of 12-week-old Hspg2 (29) and age-matched double-mutant mice carrying mutations in both the Hspg2 and Fbn1 genes.
2 Materials and methods
2.1 Animal models
All animal experiments followed protocols approved by the Institutional Animal Care and Use Committee of the Instituto de Biociências at the University of São Paulo (USP). Mice were bred from heterozygous mg (Fbn1/mg) and Hspg2 mating pairs on a C57BL/6 background, with genotyping performed as described in (16). Mice were kept under controlled conditions of light and temperature in a pathogen-free environment at the Central Animal Facility of the School of Medicine at USP. Female, 12-week-old offspring were organized by genotype in four experimental groups: wild-type (WT, ; Figure 1A), perlecan haploinsufficient (Hspg2, ; Figure 1B), Marfan syndrome (mg, ; Figure 1C), and double-mutant (dMut, ; Figure 1D) mice. In addition, 4-week-old () and 30-week-old () female mg mice were used for monitoring aneurysmal disease over time and compared to age-matched controls (4-week-old WT, ; 30-week-old WT, ). Note, we focused the study on female mice as dMut mice of male sex were not viable, with only a single male dMut mouse born out of 170 offspring over the course of a year (Figure 1E). Moreover, the slower disease progression in female vs. male mg mice permitted tracking progressive aortic dilatation between 4 and 30 weeks-of-age. The 12-week endpoint specifically granted inclusion of mice presenting with different degrees of aortic dilatation. After sacrifice, the thoracic aorta of each mouse was excised from the thoracic cavity, stored in Phosphate-Buffered Saline (PBS) solution, and shipped over dry ice to Northeastern University for mechanical testing. After arriving at Northeastern, samples were kept in a 80C freezer. Prior to testing, samples were thawed overnight in a 4C fridge. Preliminary testing indicated no significant differences in passive mechanics between frozen aortic samples and fresh tissue (30, 31).
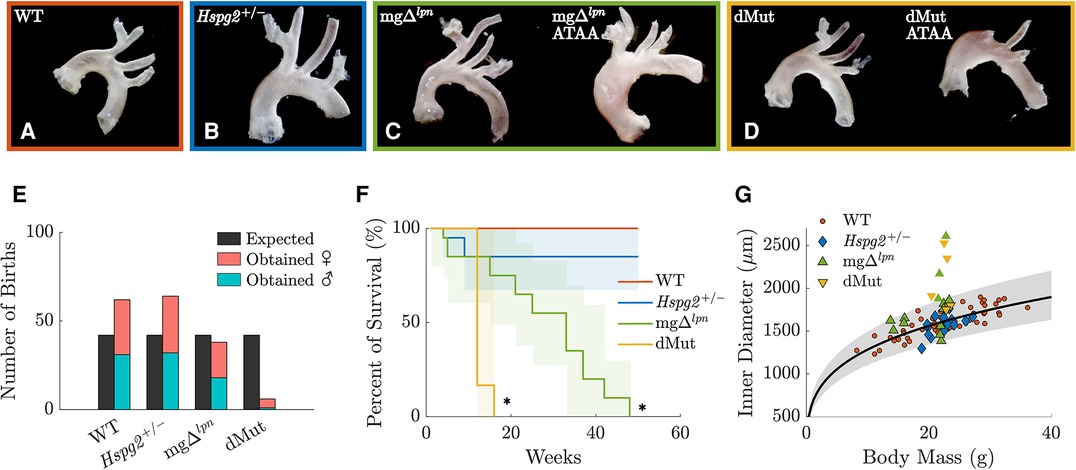
Figure 1. Representative gross anatomy images of the wild-type (WT) control, Hspg2, mg (regular-sized and dilated), and double-mutant (dMut; regular-sized and dilated) ascending thoracic aorta (ATA) following excision from 12-week-old mice (A–D). The yield of dMut mice in the offspring of heterozygous Fbn1/mg and Hspg2 mating pairs falls below expected Mendelian ratio and includes mostly (5/6) female mice (E). Kaplan–Meier curves show normal lifespan in Hspg2 mice and reduced survival in mg mice. Confidence intervals and statistical significance for the Kaplan–Meier curves assessed with the Greenwood’s formula, with * for (F). Allometric scaling relating body mass to luminal diameter for the WT control ATA (black line) supports definition of an expected range of aortic sizes within a 95% confidence interval (gray shaded area). Overall, mg and dMut ATA samples at the 12-week endpoint fall outside normal allometric scaling and are considered dilated. However, only 2 of the dilated ATA samples exceed the current clinical threshold for aneurysmal diagnosis that is set to normalized diameter above 1.5 (G).
2.2 Passive mechanical testing
All biomechanical testing and data analysis was performed consistent with established methods (12, 32, 33, 34). The thoracic aorta was cleaned of excess fat and perivascular tissues, while preserving lateral branches. Branches were ligated using 9-0 nylon sutures. The ascending thoracic aorta (ATA) was isolated from the aortic root to the left subclavian artery. ATA segments were cannulated at each of the aortic root and brachiocephalic artery ends onto custom-pulled glass micropipettes and secured using double knots of 6-0 silk sutures. Cannulated specimens were then attached to a custom-built, computer-controlled biaxial mechanical testing device (35) and submerged in Hank’s Balanced Salt Solution (HBSS). Luminal pressure was regulated via a hydraulic circuit connected to both cannulae. The outer diameter of each sample was recorded throughout testing using a camera positioned in front of a transparent bath in which the vessel was submerged. Linear actuators adjusted the distance between the two cannulae, thereby modifying the axial stretch, while a load cell coupled to the distal cannula measured the axial force. Luminal pressure and axial length were controlled through a custom LabView (National Instruments, Austin, TX) interface.
The in-vivo axial length of each sample was estimated by gradually stretching the tissue while varying the pressure, to find the energetically optimal stretch at which the axial force remains constant with pressure (36). Vessels were first acclimated via a pulsatile luminal pressurization from 80 to 120 mm Hg while held at their in-vivo axial length, then preconditioned using 4 pressurization cycles between 10 and 140 mm Hg. After re-establishing the traction-free geometry and the in-vivo axial stretch, vessels were subjected to pressure-diameter and force-length tests. In the former protocol set, vessels were held at the estimated in-vivo stretch or 5% of this value, while luminal pressure varied cyclically between 10 and 140 mm Hg. In the latter protocol set, luminal pressure was maintained constant at 10, 60, 100, or 140 mm Hg, while specimens were stretched to vary the axial force between 0.5 and 5 g. Each test is comprised of two cycles, with data collected on the second cycle. Upon completion of all protocols, samples were removed from the biaxial device and a thin ring was cut just distal of the aortic root and imaged with a dissection microscope for estimation of traction-free wall thickness using a custom MATLAB script (MATLAB R2022b; Mathworks, Natick, MA).
2.3 Mechanical analysis
Passive biaxial data were fit via nonlinear regression to the following 8-parameter stored energy function (12, 32)
where and are material parameters with the units of stress, while are dimensionless parameters; is the right Cauchy-Green tensor calculated from the deformation gradient ; is a unit vector in the direction of the fiber family, with the angle defined relative to the axial direction in the reference configuration ( axial, circumferential, and symmetric diagonal); and are invariant measures of the deformation. In cylindrical coordinates, the deformation gradient admits matrix form , with determined by enforcing incompressibility () such that .
Given best fit parameters for the strain energy density function (Eq. 1), the Cauchy stress tensor for any given deformation is
where is a Lagrange multiplier and is the identity tensor.
Components of the linearized stiffness tensor were determined according to the small-on-large deformation approach (37)
where is the Kronecker delta, and the indices and refer to the current and reference configurations, respectively.
Transmural pressures of 80mmHg and 120mmHg were considered representative of in-vivo diastolic and systolic loads, respectively. Geometry, energy storage, biaxial stretch and stress, and tissue stiffness were computed at systolic pressure and individual values of the estimated in-vivo stretch. Cyclic aortic distensibility, an inverse measure of structural stiffness, was calculated as
with and denoting luminal pressure and diameter, respectively, and the subscripts and indicating systole and diastole.
2.4 Allometric scaling
Luminal aortic diameter () scales allometrically with body mass (), according to the relationship (38)
The coefficients and were determined by linear regression on predicted luminal diameters at systole for female C57BL/6 WT mice in this study pooled with prior data by our group (33). Vessels that fell beyond the 95% confidence interval for normal allometric scaling were classified as dilated. Mechanical metrics were compared against aortic dilatation, with normalized diameter defined as the ratio between predicted luminal diameter and expected diameter based on body mass from allometric scaling.
2.5 Histology and immunofluorescence
Following passive mechanical testing, aortic samples were embedded in optimal cutting temperature (OCT) compound (4585, Fisher Scientific), fast-frozen, and stored at 80C. Tissues were cryosectioned to 6 µm in thickness and placed on SuperFrost Plus slides (Thermo Fisher Scientific, Inc., Waltham, MA) with three cross sections per aortic sample. Slides were stained with Verhoeff–Van Gieson (VVG) or picrosirius red (PSR) stains and imaged under bright field or polarized light, respectively, using a 40 objective (DM4 B upright microscope, Leica microsystems). Following stitching (Image Composite Editor, Microsoft 2015), aortic rings were analyzed using a custom MATLAB script (MATLAB R2022b; Mathworks, Natick, MA). Remaining slides were fixed for 10 minutes with 10% neutral buffer, rinsed with 1X PBS, then blocked and permeablized with 5 BSA and 0.1 Tween in 1X PBS for 30 min. Slides were rinsed and then incubated for two hours with the following primary antibodies: anti-collagen I (1:250, Abcam, ab270993), anti-collagen III (1:200, Abcam, ab7778), and anti-collagen IV (1:100, Abcam, ab235296). Following rinsing with PBS, slides were incubated with the appropriate secondary antibody for 1 h. Secondary antibodies consisted of donkey anti-rabbit Alexa Flour 647 (1:500, Abcam, ab150075) and donkey anti-goat Alexa Flour 568 (1:500, Abcam, ab175704). Slides were further stained with DAPI (1:5000, Thermo Scientific, 62,248) for 10 min to allow for visualization of cell nuclei. Two regions per cross section were imaged at 20 using a Zeiss LSM 800 confocal microscope and Zeiss Efficient Navigation (ZEN) software (Carl Zeiss Microscopy, Jena, Germany), using the same acquisition parameters per stain across groups and leveraging the autofluorescence of elastin.
2.6 Statistics
All values are reported as mean standard error of mean (SEM). Since mechanical and histological data were normally distributed as assessed by the Shapiro–Wilk test, differences across groups were determined via one-way ANOVA with post-hoc Tukey test for multiple comparisons. We further relied on the non-parametric Spearman’s rank-order correlation test to probe the relationship between mechanical metrics and aortic dilation, where describes the strength and direction of the correlation. Correlations were deemed mild, moderate, and strong for , , and , respectively.
3 Results
Allometric scaling facilitated segregation of dilated from regular-sized ATAs. Allometric scaling was used to normalize luminal ascending thoracic aortic (ATA) diameter by body mass. A logarithmic transformation followed by linear regression of predicted systolic diameter for ATA samples from female C57BL/6 wild-type mice yielded best-fit values of and (Eq. 5). These estimates are in close agreement with experimental (33, 39, 40) and theoretical (38) values from literature. All 12-week-old Hspg2 specimens fell within the 95% confidence interval for the expected ATA diameter by body mass (Figure 1G). A subset of mice carrying the Fbn1 variant featured ATA dilatation (Figure 1G), including approximately one third (3 out of 9) of the mg and half (3 out of 5) of the dMut mice at the 12-week endpoint. None of the 4-week-old and all of the 30-week-old ATA samples from mg mice were dilated. Note that only 4 out of 8 dilated ATA samples (including vessels from both the 12- and 30-week endpoints) exceeded the current clinical threshold for aneurysmal diagnosis, i.e., they exhibited normalized diameter above 1.5 (41).
Structural stiffening of the ATA progressed with aneurysmal dilatation in mg mice. Despite not yet presenting with an aneurysm (Figure 2A), the ATA of 4-week-old mg mice was structurally stiffer compared to age-matched wild-type (WT) control. The mg mutation abrogated the inflection point that marks the change in convexity in the circumferential pressure vs. diameter behavior of the ATA (Figure 2D), while concurrently reducing axial extensibility in the force vs. stretch response (Figure 2E). Aligned with structural observations, both the circumferential (Figure 2F) and axial (Figure 2G) stress vs. stretch behavior of tissues from the 4-week-old mg ATA remained confined to the left of WT control, delivering comparable stress at lower deformation. Moderate dilatation of the mg ATA at 12 weeks (Figure 2B) coincided with further loss of circumferential distensibility (Figure 2D) and axial extensibility (Figure 2E). By 30 weeks-of-age the mg ATA became aneurysmal (Figure 2C) and featured inherently stiffer tissues that developed higher circumferential stress compared to WT control when extended to the same stretch (Figure 2F). Best fit material parameters at all three endpoints are listed in Table 1, while descriptors of structural and material properties are reported in Supplementary Material Table S1. Consistent with structural and circumferential tissue stiffening, the ratio of collagen (Figure 2H) to elastin (Figure 2I) content within ATA tissues increased as mg mice aged from 4 to 12 weeks, and remained elevated up to the 30-week endpoint (Figure 2K). Meanwhile, the relative content of non-elastin tissue elements, including cell cytoplasm, in VVG-stained tissue cross sections also reached higher values (Figure 2J).
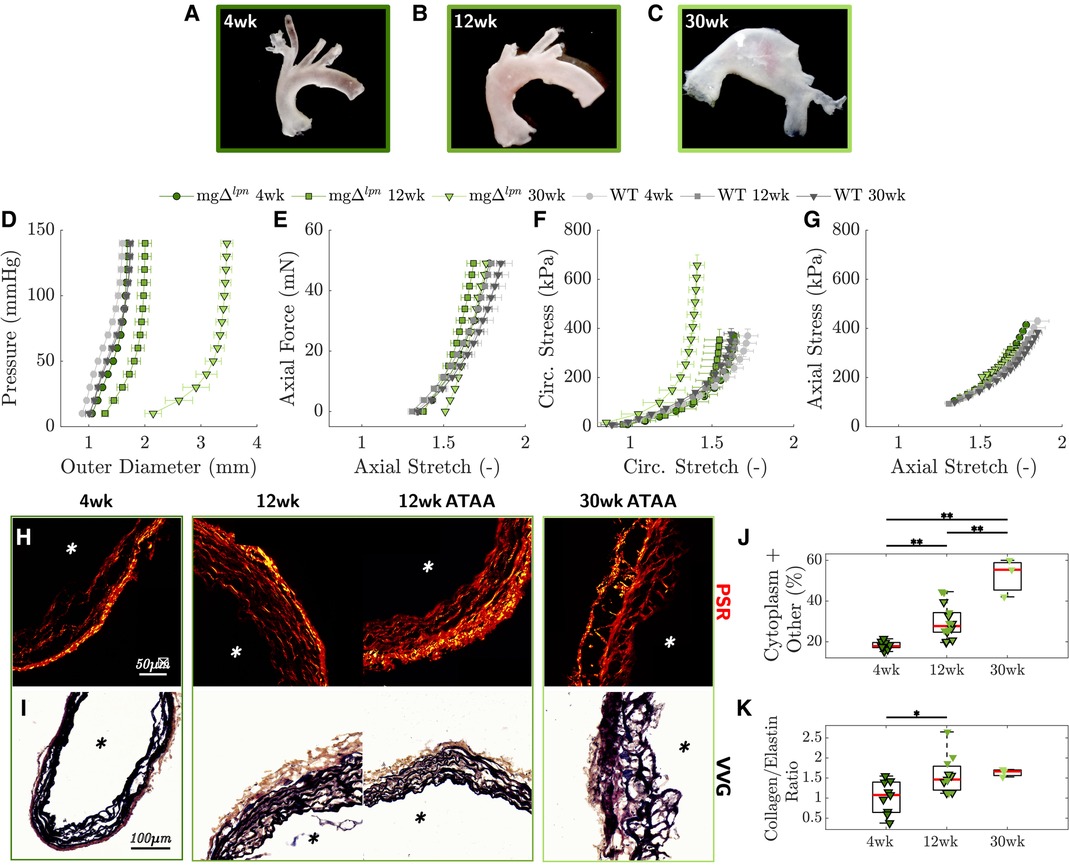
Figure 2. Representative gross anatomy images of the ascending thoracic aorta (ATA) following excision from mg mice show progression of aortic dilatation from 4 to 12 weeks-of-age and development of advanced aneurysm by 30 weeks (A-C). Average pressure vs. diameter (D) and axial force vs. length (E) responses reveal gradual decline of circumferential distensibility and axial extensibility that reflect the leftward shift of biaxial stress vs. stretch responses in the circumferential (F) and axial (G) directions, compared to age-matched wild-type (WT) controls. Anticipation/loss of the inflection point in the pressure vs. diameter behavior is already apparent by the 4-week endpoint and therefore precedes dilatation. Picrosirius red (PSR, H) and Verhoeff–Van Gieson (VVG, I) stains visualize the architecture of collagen and elastic fibers in ATA tissues from mg mice. The progressively higher proportion of cytoplasm and tissues other than elastin in VVG-stained cross sections follows the loss of elastic fibers that occurs with aneurysmal enlargement from 4 to 30 weeks-of-age (J). The ratio of collagen to elastin content likewise increases as mg mice age (K). Asterisks visualize the intimal side in histological cross sections. Markers without a black outline indicate dilated vessels in the boxplot. Statistical significance denoted by overbar, with * for and ** for .
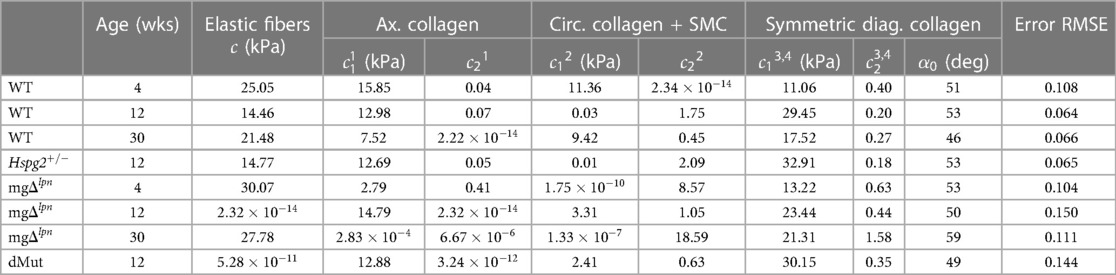
Table 1. Best-fit material parameters for the microstructurally motivated strain energy function in Eq. 1 that describes the representative mechanical response of ascending thoracic aortic (ATA) tissues from wild-type (WT) control, Hspg2, mg, and double-mutant (dMut) mice.
The mg variant dominated the passive mechanical behavior of the dMut ATA. The passive structural (Figures 3A,B) and tissue (Figures 3C,D) responses of the perlecan haploinsufficient (Hspg2) ATA at the 12-week endpoint nearly overlapped with those of age-matched WT controls. Likewise, the global structural (Figures 3A,B) and tissue (Figures 3C,D) behavior of the ATA from mg and dMut mice was comparable at the 12-week endpoint, although the latter extended to larger diameters. Best fit material parameters for all genotypic groups at 12 weeks-of-age are listed in Table 1.
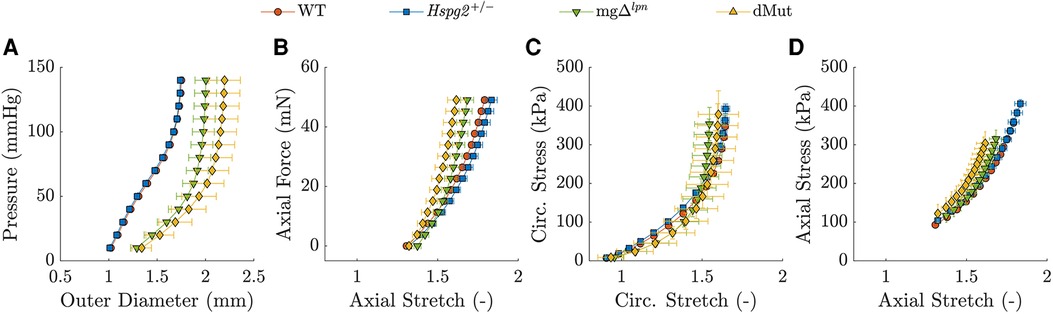
Figure 3. Average pressure vs. diameter (A) and axial force vs. length (B) responses with associated biaxial tissue behavior in the circumferential (C) and axial (D) directions for the wild-type (WT) control, Hspg2, mg (regular-sized and dilated), and double-mutant (dMut; regular-sized and dilated) ascending thoracic aorta (ATA) following excision from 12-week-old mice. The ATA retains normal global mechanical behavior despite perlecan haploinsufficiency and the mg mutation remains a stronger determinant of global mechanical behavior compared to perlecan haploinsufficiency in the dMut ATA by 12 weeks-of-age.
When evaluated at in-vivo loads, ATA tissues from WT and Hspg2 mice experienced comparable biaxial stretch, stress (Eq. 2), and stiffness (Eq. 3), demonstrated similar capacity for energy storage, and exhibited matching wall thickness and cyclic distensibility (Eq. 4), (Figures 4A–I). The Fbn1 variant, either alone or superimposed to perlecan haploinsufficiency, did not affect circumferential stretch levels in ATA tissues (Figure 4A). In contrast, ATA samples from dMut () mice alone extended to lower axial stretch compared to their WT () and Hspg2 () counterparts (Figure 4D; ). The ATA wall was thicker in mg ( µm) and dMut ( µm) than in Hspg2 ( µm) mice (Figure 4G; ). Nevertheless, circumferential stress was remarkably preserved across all groups (Figure 4B), while axial stress was lower in mg ( kPa) and dMut ( kPa) compared to Hspg2 ( kPa) mice. Axial stiffness did not differ significantly across groups (Figure 4F), while ATA tissues from mice carrying the Fbn1 variant exhibited higher circumferential stiffness (mg: MPa, dMut: MPa) compared to both WT ( MPa) and Hspg2 ( MPa) tissues (Figure 4C; ). Importantly, reduced capacity for energy storage (WT: kPa, Hspg2: kPa, mg: kPa, dMut: kPa) and cyclic distensibility (WT: mm Hg, Hspg2: mm Hg, mg: mm Hg, dMut: ) evidenced functional loss in the aorta of mice carrying the Fbn1 variant (Figures 4H,I; ). Descriptors of structural and material properties for all genotypic groups at 12 weeks-of-age are listed in Supplementary Material Table S1.
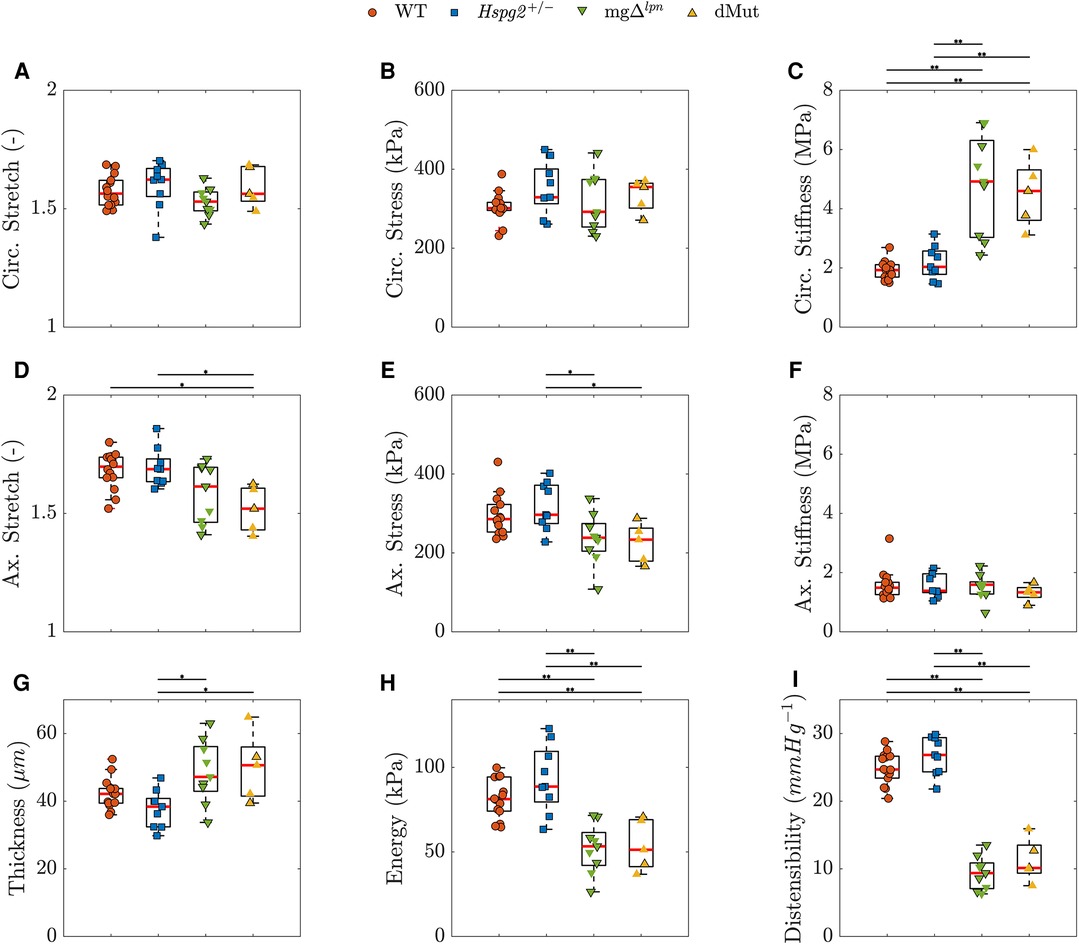
Figure 4. Predicted in vivo material, geometrical, and structural properties of the ascending thoracic aorta (ATA) from 12-week-old wild-type (WT) control, Hspg2, mg (regular-sized and dilated), and double-mutant (dMut; regular-sized and dilated) mice grouped by genotype. Perlecan haploinsufficiency preserves inferred mechanical metrics in the Hspg2 ATA. The Fbn1 variant overall supports tissue circumferential stiffening (C) and radial thickening (G), lowers the axial stress (E), limits the capacity for energy storage (H), and reduces the cyclic distensiblity (I) of the ATA from both mg and dMut mice. However, the dMut ATA alone exhibits reduced axial stretch (D). Circumferential stretch (A), circumferential stress (B), and axial stiffness (F) are remarkably consistent across genotypic groups. Markers without a black outline indicate dilated vessels in the boxplots. Statistical significance denoted by overbar, with * for and ** for .
Intrinsic circumferential tissue stiffness positively correlated with aortic dilatation. Consistent with previous findings (12), circumferential tissue stiffening strongly correlated with aortic dilatation (, ; Figure 5C). Widening of the lumen moderately correlated with the rise in circumferential stress (, ; Figure 5B) as well as the decline in axial stretch (, ; Figure 5E). A mild negative correlation further emerged between aortic dilatation and axial stress (, , ; Figure 5F). Functional metrics of cyclic distensibility (, ; Figure 5H) and energy storage (, ; Figure 5D) exhibited negative correlations with aortic dilatation. No meaningful correlation was noted between circumferential stretch (Figure 5A) or axial stiffness (Figure 5G) and aortic diameter.
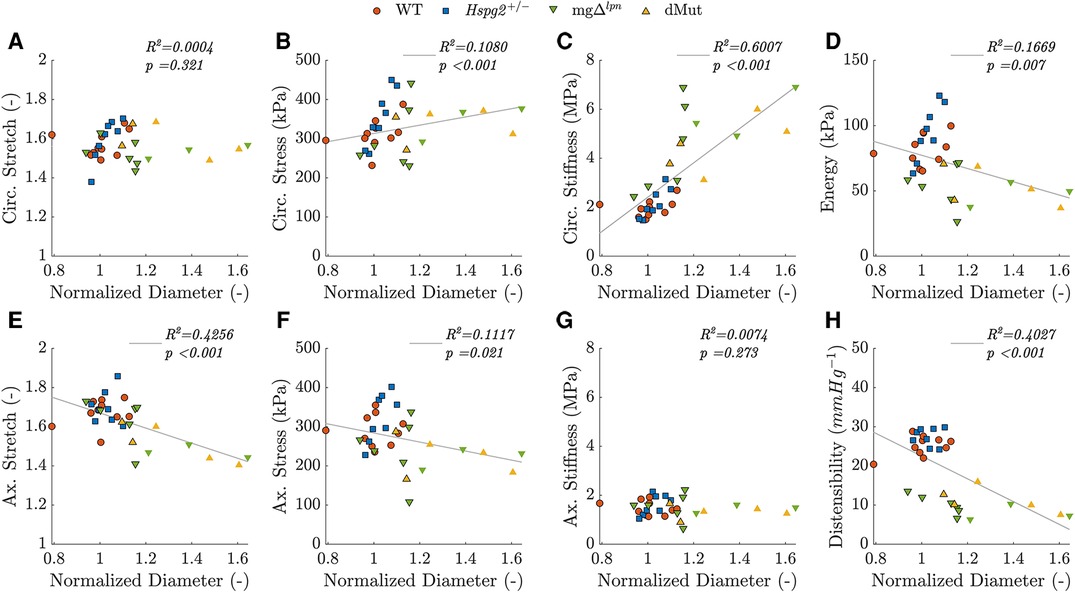
Figure 5. Predicted in vivo material, geometrical, and structural properties of the ascending thoracic aorta (ATA) from 12-week-old (WT) control, Hspg2, mg (regular-sized and dilated), and double-mutant (dMut; regular-sized and dilated) mice visualized as a function of normalized ATA diameter, i.e., the ratio between current diameter and the expected diameter for a WT mouse with the same body mass. Aortic diameter is strongly and positively correlated with circumferential tissue stiffness (, ; C), moderately and negatively correlated with distensibility (, ; H) and axial stretch (, ; E), moderately and positively correlated with circumferential stress (, ; B), and mildly and negatively correlated with energy storage capacity (, ; D) and axial stress (, ; F). Aortic diameter is not significantly correlated with either circumferential stretch (A) or axial stiffness (G). Markers without a black outline indicate a dilated vessel. Note, although Spearman’s rank is a non-parametric measure of correlation that does not assume linearity, best fit lines are added to aid trend visualization for each mechanical metric.
Increased tissue accumulation of collagen III compensated for elastic fiber degradation in the dMut ATA. Supporting mechanical findings, ATA tissues collected from WT and Hspg2 mice exhibited similar microstructure at the 12-week endpoint (Figures 6A–D), with preserved collagen to elastin ratio (Figure 6E), competent elastic fiber network (Figure 6F), and comparable collagen I, III, and IV accumulation (Figures 6G,I,J). ATA tissues from 12-week-old mg and dMut mice followed consistent patterns of microstructural disarray that worsened with the severity of aortic dilatation (Figures 6A–D). The ratio of collagen to elastin content was higher in mg and dMut mice compared to WT control and Hspg2 mice, respectively (Figure 6E). Similarly, the relative content of cytoplasm and other tissues in VVG-stained cross sections was larger in mice carrying the Fbn1 variant (Figure 6H) contrasted to both WT control and Hspg2 mice. Qualitative interrogation of the ATA wall microstructure via immunofluorescence staining revealed gradual loss of elastin integrity that began at the inner elastic lamina (IEL) and expanded outward in mg and dMut mice. Disarray of elastic fibers in the proximity of the IEL became apparent in regular-sized ATA tissues from 12-week-old mice, while fiber fragmentation extended toward the adventitial interface in mice of the same age presenting with ATA dilatation (Figure 6C). This coincided with a decrease in mean elastin fluorescence intensity in both genotypes carrying the Fbn1 variant compared to WT controls (Figure 6F). Unlike collagen I and IV (Figures 6G,J), collagen III accumulation was enhanced in the aortic media of ATA samples from mg and dMut mice, compared to both WT control and Hspg2 mice (Figure 6I).
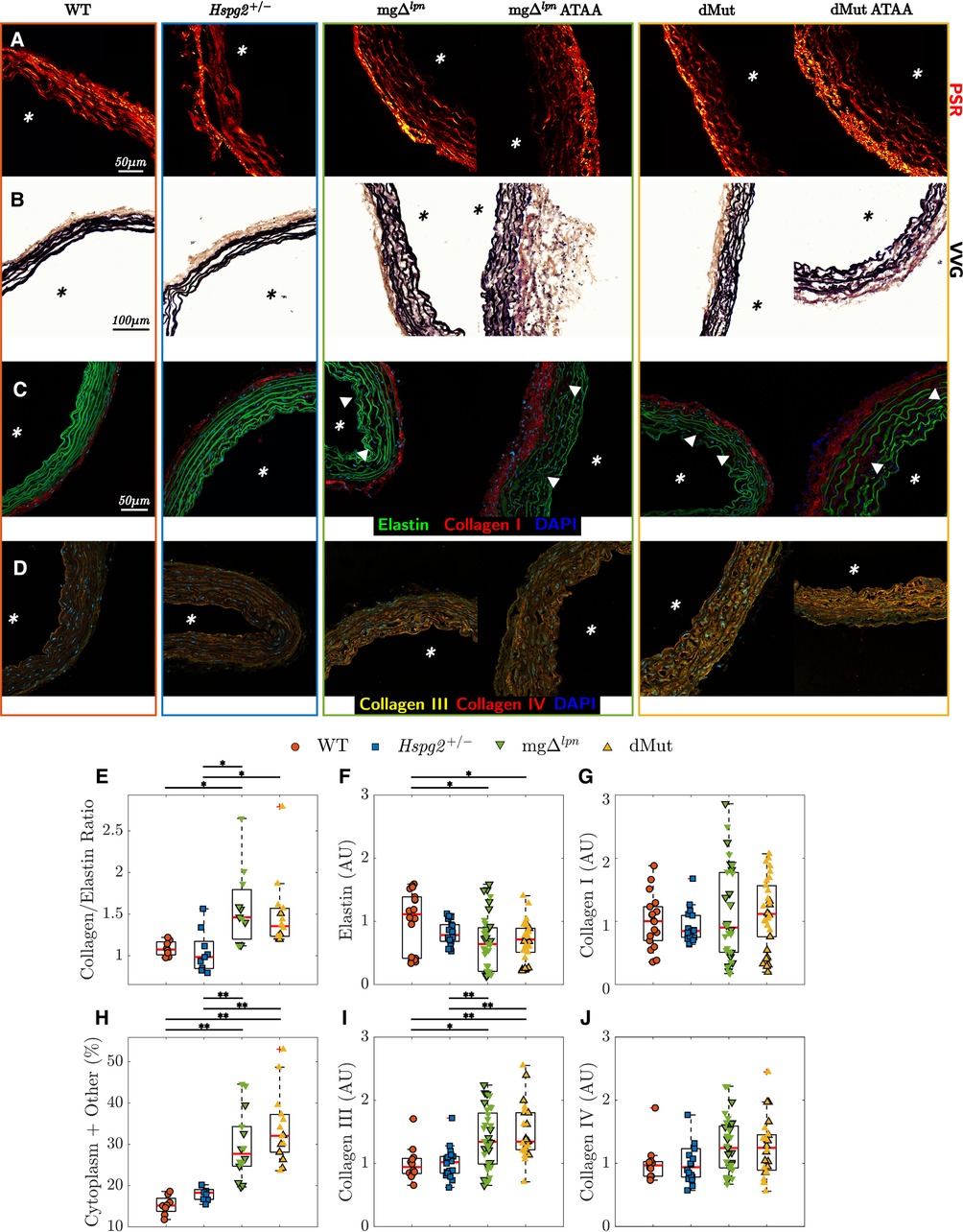
Figure 6. Elastic and collagen fiber architecture in tissues from the ascending thoracic aortic (ATA) of 12-week-old wild-type (WT) control, Hspg2, mg (regular-sized and dilated), and double-mutant (dMut; regular-sized and dilated) mice visualized by picrosirius red (PSR, A), Verhoeff–Van Gieson (VVG, B), and immunofluorescence staining (C,D). Perlecan haploinsufficiency alone maintains normal organization of elastic and collagen fibers in Hspg2 ATA tissues. Both mg and dMut mice present ATA cross sections with larger collagen to elastin content ratio (E) and higher relative content of cytoplasm and other non-elastin tissues (H), compared to WT and Hspg2 mice. Consistently, the mean fluorescence intensity of elastin by area (F) decreases, yet the mean fluorescence intensity of collagen III by area (I) increases in the ATA of mice carrying the Fbn1 variant. The mean fluorescence intensity of collagen I (G) and collagen IV (J) by area is comparable across all genotypes. Asterisks visualize the intimal side in histological cross sections, and white triangles show fragmentation, gaps, or disarray in the elastic fiber network. Markers without a black outline indicate dilated vessels in all boxplots. Statistical significance denoted by overbar, with * for and ** for .
4 Discussion
Surgical intervention remains the most effective treatment option for patients suffering from an aortic aneurysm, yet resolving when best to operate poses numerous challenges (42). Appreciation of the diffuse mechanical and structural remodeling that accompanies progressive focal aortic dilatations could support clinical decision-making. However, ex-vivo mechanical characterization of human tissues often relies on aortic specimens excised from patients during surgery for advanced-stage aortopathy. To overcome this obstacle, animal models offer the unique opportunity to track mechanical and structural features of aortic aneurysms across all stages of the disease (43). We submit here that the mg mouse model of Marfan syndrome (MFS) is a viable candidate for this purpose. Many snapshot studies on aneurysmal tissue mechanics leverage Fbn1 mice (13) that exhibit a severe MFS phenotype and die spontaneously of aortic rupture by 9 weeks-of-age (44). Noting that the murine aorta reaches mechanical maturity around 8 weeks (45) deters from performing longitudinal studies in Fbn1 mice. As an alternative, the Fbn1 mouse model of thoracic disease (13, 14) enjoys a longer life-span at the expense of a mild phenotype that does not manifest until after 9 months (46, 47). On account of the stable phenotype (16), the early presentation of thoracic aortopathy (12 weeks), and the long life-span, mg mice therefore embody a suitable option for evaluating the progression of aneurysmal disease.
While most literature probes the mechanical response of murine aneurysmal tissues at 9 weeks due to the low survival of Fbn1 mice beyond this age, we opted to incorporate three endpoints with the purpose of capturing pre- (4 weeks), early- (12 weeks), and advanced- (30 weeks) aortic dilatation. Since male dMut mice are not viable (1 male vs. 5 female mice born out of 170 offspring; Figure 1E), we restricted our study to females. Strong sexual dimorphism in aortic aneurysm development has been previously reported in MFS, with male mice exhibiting a worse phenotype that is more vulnerable to environmental triggers (47). Since male sex enhances the susceptibility to thoracic aortopathy (46, 48), we acknowledge that female mg mice may experience milder aortic disease yielding lower likelihood to encounter premature death than their male litter mates (49). As such, there remains a need for elucidating sex differences in mg mice, which we were unable to capture in this work. Regardless, the progressive shift toward a collagen-driven pressure vs. diameter response (Figure 2D) and the gradual loss of axial extensibility (Figure 2E) in the mg ascending thoracic aorta are consistent with qualitative snapshot observations in male Fbn1 and Fbn1 mice (12, 44, 47, 50–52). The inherent biaxial stiffening that ascending aortic tissues experience between 4 and 30 weeks-of-age (Figures 2F,G, Supplementary Material Table S1) further reflects structural findings and aligns with clinical observations (53–55). Importantly, circumferential stiffening of 4 week-old mg tissues beyond control levels (Figure 2F) despite preserved ascending aortic caliber (Figure 2A) advocates for structural and functional remodeling to precede aneurysmal dilatation and increase the propensity thereof. Weiss et al. similarly proposed that tissue stiffening between 8 and 12 weeks-of-age may prime the Fbn1 aorta for later aneurysm formation (47).
Expected heterogeneity in the time of aortic disease onset (50) and low viability of dMut mice prompted limiting the comparison across genotypes (WT, Hspg2, mg, and dMut) and aortic phenotypes to the 12-week endpoint. By then, 3 out of 9 mg and 3 out of 5 dMut mice departed from the 95% confidence interval range for expected ascending aortic diameter by body mass (Figure 1G) (50, 51). To explore the effect of lumen widening on aortic function we further examined non-parametric correlations between mechanical behavior and luminal diameter. While energy storage and cyclic distensibility decline in mice carrying the mg mutation (Figures 4H,I), these metrics only moderately or mildly correlate with ascending aortic size (Figures 5D,H). The functional impairment of the aorta as imposed by the Fbn1 variant does not therefore reflect aneurysm progression. Note that the structural stiffening of the aorta nonetheless portends to worse clinical outcomes in MFS patients (56). Echoing our earlier inferences (12), circumferential tissue stiffness is elevated in mg and dMut mice (Figure 4C) and most strongly correlates with ascending aortic diameter (Figure 5C), thereby alluding to severe microstructural remodeling concurrent with aneurysm formation.
Consistent with previous findings (57–62), collagen deposition and progressive loss of elastic fiber integrity contribute to medial deterioration as the ascending aorta dilates in aging mg mice (Figure 2, Supplementary Material Figure S1). Furthermore, patterns of microstructural remodeling are comparable in all 12-week-old mice carrying the Fbn1 variant and mostly depend on the severity of aneurysmal disease. In support of wall thickening and lumen widening, the total area of collagen compared to elastin and the relative content of cytoplasm and other non-elastin tissue elements are both larger in the aorta of mg and dMut mice (Figures 6E,H). Noting that mechanical competence in addition to mere content supports normal aortic function (63, 64), we analyzed elastic fiber architecture in the aorta of mice carrying the mg mutation. While normal-sized vessels exhibit a radial gradient in medial degeneration that advances from severe fiber fragmentation at the inner elastic lamina to preserved fiber integrity at the adventitial interface, the microstructural disarray encompasses the entire medial compartment in aneurysmal aortic samples (Figure 6C). Interestingly, Ferruzzi et al. noted a reverse radial gradient in elastic fiber fragmentation when characterizing aortic microstructure in the Fbln5 mouse model of impaired elastogenesis (65). Given that Fbln5 mice develop severe arterial elastopathy in the absence of aneurysmal dilatation or dissection (66), preserving the integrity of the elastic laminae adjacent to the lumen appears to be protective against ascending aortic dilatation. It has been suggested that an early breach of the internal elastic lamina may facilitate immune cell infiltration and perpetuate pathological remodeling that promotes aneurysm formation (67).
Large variability in phenotypic severity and intrafamilial disease manifestations across MFS patients (20, 68) rationalizes efforts to identify modifier genes and quantify their contribution to clinical outcomes. Perlecan haploinsufficiency due to heterozygous mutation in the candidate modifier gene Hspg2 reduces the viability of mg mice (Figures 1E,F), thereby suggesting that concurrence of the Hspg2 and Fbn1 variants may compromise the long term survival of patients with MFS. Interestingly, necropsies on dMut mice revealed cardiac abnormalities that were reminiscent of congestive heart failure with left atrial dilatation. This is consistent with the syndromic nature of MFS, which affects multiple organ systems and presents diverse cardiovascular complications including enlargement of the left atrium (69). Furthermore, similar to our observations in dMut mice, congestive heart failure rather than aortic enlargement is the leading cause of early mortality in neonatal MFS patients within the first 2 years of life (70). Since perlecan has been shown to play an important role in heart stability (71), concurrent Hspg2 and Fbn1 mutations may escalate cardiac symptoms beyond threshold for clinical relevance.
Although Nonaka et al. reported spontaneous aortic dissection in lethality-rescued Hspg2 mice by 10 weeks-of-age (28), the aorta of Hspg2 mice exhibits normal mechanical behavior at the 12-week endpoint (Figures 3 and 4). The mg variant likewise imposes an overall stronger imprinting than perlecan haploinsufficiency on the global mechanical response of the dMut aorta at 12 weeks (Figures 3A–D). Nevertheless, the dMut but not the mg aorta endures a decline in axial stretch (Figure 4D), which is a common early sign of maladaptive arterial wall remodeling (72). Consistent with mechanical readouts, 12 week-old dMut mice experience comparable incidence of fatal aortic dissection/rupture but nonetheless suffer from aortic dilatation at a moderately higher frequency than mg mice. As an important note, perlecan-null mice surviving to birth display normal basement membranes that quickly deteriorate when exposed to mechanical loads, leading to premature death (73). While our Hspg2 and dMut mice express significantly lower though not negligible levels of perlecan compared to their respective controls (Supplementary Material Figure S2), it is possible that overexpression of other extracellular matrix elements may strengthen elastic fibers and protect them from fragmentation, at least early in the mouse life (74).
Mounting evidence advocates for evaluating the effect of perlecan haploinsufficiency on the mechanical response of the ascending thoracic aorta within the context of MFS (21, 28). As a first step, we performed passive inflation/extension tests to interrogate elastic and collagenous fibers as load-bearing components of the aortic wall, but neglected vasoactive responses involving vascular smooth muscle cells (vSMC). Phenotypic switching of vSMC from contractile to synthetic occurs at a higher rate in aneurysmal disease and leads to increased production of proteolytic enzymes that degrade the extracellular matrix and weaken the arterial wall (75, 76). Disruption of endothelial nitric oxide signaling is yet another common feature of thoracic aortopathies (77) that prevents vSMC relaxation and hinders flow-mediated vasodilatation (78). Perlecan notably localizes in the basement membrane of the aorta (79) where it acts as a shear flow sensor for endothelial cells (73), and perlecan deficiency reduces the expression of endothelial nitric oxide synthase (80). Further evidence suggests that perlecan and other heparan sulfate proteoglycans may influence smooth muscle cell activation and proliferation following vascular injury (81). Future efforts should therefore probe adrenergic vasoconstriction as well as endothelium-dependent and independent vasodilatation of the Hspg2 and dMut aorta, which may provide further insight on the lower viability of mice with concurrent Fbn1 and Hspg2 mutations. Since the effect of modifier genes often becomes apparent under stress conditions (82), it will also be important to characterize the structure and function of ascending thoracic aortic tissues from dMut mice that have been rendered hypertensive, a known risk factor for aneurysmal disease (7, 83, 84).
4.1 Conclusions
The mg mouse strain carries a Fbn1 variant that replicates in mice the classic phenotypic traits of patients with Marfan syndrome (MFS). The longer life span of mg mice compared to alternative mouse models of MFS enabled us to examine the effect of a causative Fbn1 mutation on ascending thoracic aorta (ATA) dilatation, tissue microstructure, and mechanical function. We then explored the effect of the candidate modifier gene Hspg2 on the thoracic aortopathy associated with MFS by cross-breeding Hspg2 with mg mice to create dMut mice. We show that elastic fiber fragmentation in mice carrying the mg mutation first occurs in the proximity of the internal elastic lamina and precedes aortic dilatation. Loss of elastic fiber integrity then expands radially toward the adventitial side as the aneurysm forms and enlarges. Throughout this process, the ratio of total collagen to elastin area increases, tissues become stiffer and lose capacity for energy storage, while the ATA as a structure experiences reduced cyclic distensibility. Tissue stiffening strongly correlates with dilatation while the functional decline only moderately or mildly depends on ATA size. Perlecan haploinsufficiency reduces the overall viability of dMut mice, increases the occurrence of aortic dilatation by 12 weeks-of-age, and reduces the axial stretch, yet it does not aggravate the microstructural disarray nor the stiffening of ATA tissues at the 12-week endpoint. Further effort is therefore warranted to evaluate the effect of Hspg2 and other modifier genes on later-stage thoracic aortopathy in MFS, with the hope of assisting the risk stratification of patients suffering from the disease.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The animal study was approved by Institutional Animal Care and Use Committee of the Instituto de Biociências at the University of São Paulo (USP). The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
SAT: Formal Analysis, Investigation, Visualization, Writing – original draft, Writing – review & editing; RdeS: Formal Analysis, Investigation, Writing – review & editing; AH: Formal Analysis, Investigation, Visualization, Writing - review & editing; LVP: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – original draft, Writing – review & editing; CB: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – original draft, Writing – review & editing.
Funding
We acknowledge financial support from the National Institute of Health (R01HL168473 to CB), the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brazil (CAPES - 001 to LVP and RBS), and the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP - 2016/16077-0 to LVP and RBS).
Acknowledgments
We thank the Institute for Chemical Imaging of Living Systems at Northeastern University for consultation and imaging support. We express our gratitude to Mr. Nathan Li from the Animal Histology Core at Tufts University for performing VVG and PSR staining of aortic tissues.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2024.1319164/full#supplementary-material
References
1. Renard M, Francis C, Ghosh R, Scott AF, Witmer PD, Adès LC, et al. Clinical validity of genes for heritable thoracic aortic aneurysm, dissection. J Am Coll Cardiol. (2018) 72:605–15. doi: 10.1016/j.jacc.2018.04.089
2. Ostberg NP, Zafar MA, Ziganshin BA, Elefteriades JA. The genetics of thoracic aortic aneurysms, dissection: a clinical perspective. Biomolecules. (2020) 10:182. doi: 10.3390/biom10020182
3. Ramirez F, Dietz HC. Fibrillin-rich microfibrils: structural determinants of morphogenetic, homeostatic events. J Cell Physiol. (2007) 213:326–30. doi: 10.1002/jcp.21189
4. Karimi A, Milewicz DM. Structure of the elastin-contractile units in the thoracic aorta, how genes that cause thoracic aortic aneurysms and dissections disrupt this structure. Can J Cardiol. (2016) 32:26–34. doi: 10.1016/j.cjca.2015.11.004
5. Wagenseil JE, Mecham RP. Elastin in large artery stiffness and hypertension. J Cardiovasc Transl Res. (2012) 5:264–73. doi: 10.1007/s12265-012-9349-8
6. Cocciolone AJ, Hawes JZ, Staiculescu MC, Johnson EO, Murshed M, Wagenseil JE. Elastin, arterial mechanics, and cardiovascular disease. Am J Physiol Heart Circ Physiol. (2018) 315:H189–H205. doi: 10.1152/ajpheart.00087.2018
7. Pinard A, Jones GT, Milewicz DM. Genetics of thoracic and abdominal aortic diseases. Circ Res. (2019) 124:588–606. doi: 10.1161/CIRCRESAHA.118.312436
8. Keane MG, Pyeritz RE. Medical management of marfan syndrome. Circulation. (2008) 117:2802–13. doi: 10.1161/CIRCULATIONAHA.107.693523
9. Erbel R, Aboyans V, Boileau C, Bossone E, Bartolomeo RD, Eggebrecht H, et al. 2014 ESC Guidelines on the diagnosis and treatment of aortic diseases: document covering acute and chronic aortic diseases of the thoracic and abdominal aorta of the adult. The task force for the diagnosis and treatment of Aortic Diseases of the European. Eur Heart J. (2014) 35:2873–926. doi: 10.1093/eurheartj/ehu281
10. Pape LA, Tsai TT, Isselbacher EM, Oh JK, O’gara PT, Evangelista A, et al. Aortic diameter >or = 5.5 cm is not a good predictor of type A aortic dissection: observations from the International Registry of Acute Aortic Dissection (IRAD). Circulation. (2007) 116:1120–7. doi: 10.1161/CIRCULATIONAHA.107.702720
11. Vianna E, Kramer B, Tarraf S, Gillespie C, Colbrunn R, Bellini C, et al. Aortic diameter is a poor predictor of aortic tissue failure metrics in patients with ascending aneurysms. J Thorac Cardiovasc Surg. (2022). doi: 10.1016/j.jtcvs.2022.10.021. [Epub ahead of print].36528437
12. Bellini C, Bersi MR, Caulk AW, Ferruzzi J, Milewicz DM, Ramirez F, et al. Comparison of 10 murine models reveals a distinct biomechanical phenotype in thoracic aortic aneurysms. J R Soc Interface. (2017) 14:20161036. doi: 10.1098/rsif.2016.1036
13. Pereira L, Lee SY, Gayraud B, Andrikopoulos K, Shapiro SD, Bunton T, et al. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc Natl Acad Sci USA. (1999) 96:3819–23. doi: 10.1073/pnas.96.7.3819
14. Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL, et al. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest. (2004) 114:172–81. doi: 10.1172/JCI20641
15. Huang J, Davis EC, Chapman SL, Budatha M, Marmorstein LY, Word RA, et al. Fibulin-4 deficiency results in ascending aortic aneurysms. Circ Res. (2010) 106:583–92. doi: 10.1161/CIRCRESAHA.109.207852
16. Lima BL, Santos EJC, Fernandes GR, Merkel C, Mello MRB, Gomes JPA, et al. A new mouse model for Marfan syndrome presents phenotypic variability associated with the genetic background and overall levels of Fbn1 expression. PLoS ONE. (2010) 5:e14136. doi: 10.1371/journal.pone.0014136
17. de Souza RB, Farinha-Arcieri LE, Catroxo MHB, Martins AMCRPF, Tedesco RC, Alonso LG, et al. Association of thoracic spine deformity and cardiovascular disease in a mouse model for Marfan syndrome. PLoS ONE. (2019) 14:e0224581. doi: 10.1371/journal.pone.0224581
18. Souza RB, Gyuricza IG, Cassiano LL, Farinha-Arcieri LE, Alvim Liberatore AM, Schuindt do Carmo S, et al. The mglpn mouse model for Marfan syndrome recapitulates the ocular phenotypes of the disease. Exp Eye Res. (2021) 204:108461. doi: 10.1016/j.exer.2021.108461
19. Cook JR, Clayton NP, Carta L, Galatioto J, Chiu E, Smaldone S, et al. Dimorphic effects of transforming growth factor- signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arterioscler Thromb Vasc Biol. (2015) 35:911–7. doi: 10.1161/ATVBAHA.114.305150
20. Díaz de Bustamante A, Ruiz-Casares E, Darnaude MT, Perucho T, Martínez-Quesada G. Variabilidad fenotípica del síndrome de Marfan en una familia con una nueva mutación en el gen FBN1. Rev Esp Cardiol. (2012) 65:380–1. doi: 10.1016/j.recesp.2011.05.027
21. Gerdes Gyuricza I, Barbosa de Souza R, Farinha-Arcieri LE, Ribeiro Fernandes G, Veiga Pereira L. Is HSPG2 a modifier gene for Marfan syndrome? Eur J Hum Genet. (2020) 28:1292–6. doi: 10.1038/s41431-020-0666-0
22. Hutchinson S, Furger A, Halliday D, Judge DP, Jefferson A, Dietz HC, et al. Allelic variation in normal human FBN1 expression in a family with Marfan syndrome: a potential modifier of phenotype? Hum Mol Genet. (2003) 12:2269–76. doi: 10.1093/hmg/ddg241
23. Aubart M, Gazal S, Arnaud P, Benarroch L, Gross MS, Buratti J, et al. Association of modifiers and other genetic factors explain Marfan syndrome clinical variability. Eur J Hum Genet. (2018) 26:1759–72. doi: 10.1038/s41431-018-0164-9
24. Hassell JR, Robey PG, Barrach HJ, Wilczek J, Rennard SI, Martin GR. Isolation of a heparan sulfate-containing proteoglycan from basement membrane. Proc Natl Acad Sci USA. (1980) 77:4494–8. doi: 10.1073/pnas.77.8.4494
25. Arikawa-Hirasawa E, Watanabe H, Takami H, Hassell JR, Yamada Y. Perlecan is essential for cartilage and cephalic development. Nat Genet. (1999) 23:354–8. doi: 10.1038/15537
26. Tiedemann K, Sasaki T, Gustafsson E, Göhring W, Bätge B, Notbohm H, et al. Microfibrils at basement membrane zones interact with perlecan via fibrillin-1. J Biol Chem. (2005) 280:11404–12. doi: 10.1074/jbc.M409882200
27. Hayes AJ, Lord MS, Smith SM, Smith MM, Whitelock JM, Weiss AS, et al. Colocalization in vivo and association in vitro of perlecan and elastin. Histochem Cell Biol. (2011) 136:437–54. doi: 10.1007/s00418-011-0854-7
28. Nonaka R, Iesaki T, Kerever A, Arikawa-Hirasawa E. Increased risk of aortic dissection with perlecan deficiency. Int J Mol Sci. (2021) 23:315. doi: 10.3390/ijms23010315
29. Costell M, Gustafsson E, Aszódi A, Mörgelin M, Bloch W, Hunziker E, et al. Perlecan maintains the integrity of cartilage and some basement membranes. J Cell Biol. (1999) 147:1109–22. doi: 10.1083/jcb.147.5.1109
30. Stemper BD, Yoganandan N, Stineman MR, Gennarelli TA, Baisden JL, Pintar FA. Mechanics of fresh, refrigerated, and frozen arterial tissue. J Surg Res. (2007) 139:236–42. doi: 10.1016/j.jss.2006.09.001
31. Salinas SD, Clark MM, Amini R. The effects of 80C short-term storage on the mechanical response of tricuspid valve leaflets. J Biomech. (2020) 98:109462. doi: 10.1016/j.jbiomech.2019.109462
32. Ferruzzi J, Bersi MR, Humphrey JD. Biomechanical phenotyping of central arteries in health and disease: advantages of and methods for murine models. Ann Biomed Eng. (2013) 41:1311–30. doi: 10.1007/s10439-013-0799-1
33. Farra YM, Rivera C, Silvestro M, Matz J, Pratama Y, Kumari P, et al. Mapping the unicellular transcriptome of the ascending thoracic aorta to changes in mechanosensing and mechanoadaptation during aging. bioRxiv (2022). doi:10.1101/2022.07.21.501037
34. Vargas AI, Tarraf SA, Fitzgibbons TP, Bellini C, Amini R. Biomechanical remodeling of the murine descending thoracic aorta during late-gestation pregnancy. Curr Res Physiol. (2023) 6:100102. doi: 10.1016/j.crphys.2023.100102
35. Farra YM, Matz J, Ramkhelawon B, Oakes JM, Bellini C. Structural and functional remodeling of the female Apoe mouse aorta due to chronic cigarette smoke exposure. Am J Physiol Heart Circ Physiol. (2021) 320:H2270–82. doi: 10.1152/ajpheart.00893.2020
36. Weizsäcker HW, Lambert H, Pascale K, Weizsacker HW, Lambert H, Pascale K. Analysis of the passive mechanical properties of rat carotid arteries. J Biomech. (1983) 16:703–15. doi: 10.1016/0021-9290(83)90080-5
37. Baek S, Gleason RL, Rajagopal KR, Humphrey JD. Theory of small on large: potential utility in computations of fluid–solid interactions in arteries. Comput Methods Appl Mech Eng. (2007) 196:3070–8. doi: 10.1016/j.cma.2006.06.018
38. West GB, Brown JH, Enquist BJ. A general model for the origin of allometric scaling laws in biology. Science. (1997) 276:122–6. doi: 10.1126/science.276.5309.122
39. Goergen CJ, Johnson BL, Greve JM, Taylor CA, Zarins CK. Increased anterior abdominal aortic wall motion: possible role in aneurysm pathogenesis and design of endovascular devices. J Endovasc Ther. (2007) 14:574–84. doi: 10.1177/152660280701400421
40. Prim DA, Mohamed MA, Lane BA, Poblete K, Wierzbicki MA, Lessner SM, et al. Comparative mechanics of diverse mammalian carotid arteries. PLoS ONE. (2018) 13:e0202123. doi: 10.1371/journal.pone.0202123
41. Hiratzka LF, Bakris GL, Beckman JA, Bersin RM, Carr VF, Casey DEJ, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with Thoracic Aortic Disease: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. (2010) 121:266–369. doi: 10.1161/CIR.0b013e3181d4739e
42. Palumbo MC, Rong LQ, Kim J, Navid P, Sultana R, Butcher J, et al. Prosthetic aortic graft replacement of the ascending thoracic aorta alters biomechanics of the native descending aorta as assessed by transthoracic echocardiography. PLoS ONE. (2020) 15:e0230208. doi: 10.1371/journal.pone.0230208
43. Milewicz DM, Braverman AC, De Backer J, Morris SA, Boileau C, Maumenee IH, et al. Marfan syndrome. Nat Rev Dis Primers. (2021) 7:64. doi: 10.1038/s41572-021-00298-7
44. Cavinato C, Chen M, Weiss D, Ruiz-Rodríguez MJ, Schwartz MA, Humphrey JD. Progressive microstructural deterioration dictates evolving biomechanical dysfunction in the Marfan aorta. Front Cardiovasc Med. (2021) 8:800730. doi: 10.3389/fcvm.2021.800730
45. Murtada SI, Kawamura Y, Li G, Schwartz MA, Tellides G, Humphrey JD. Developmental origins of mechanical homeostasis in the aorta. Dev Dyn. (2021) 250:629–39. doi: 10.1002/dvdy.283
46. Gharraee N, Sun Y, Swisher JA, Lessner SM. Age and sex dependency of thoracic aortopathy in a mouse model of Marfan syndrome. Am J Physiol Heart Circ Physiol. (2022) 322:H44–H56. doi: 10.1152/ajpheart.00255.2021
47. Weiss D, Rego BV, Cavinato C, Li DS, Kawamura Y, Emuna N, et al. Compensatory aortic remodeling in Marfan syndrome protects against sexually dimorphic rupture during a BAPN challenge. bioRxiv (2022). doi:10.1101/2022.06.21.497029
48. Takeda N, Inuzuka R, Maemura S, Morita H, Nawata K, Fujita D, et al. Impact of pathogenic FBN1 variant types on the progression of aortic disease in patients with Marfan syndrome. Circulation. (2018) 11:e002058. doi: 10.1161/CIRCGEN.117.002058
49. Souza RB, Kawahara EI, Farinha-Arcieri LE, Gyuricza IG, Neofiti-Papi B, Miranda-Rodrigues M, et al. Hyperkyphosis is not dependent on bone mass and quality in the mouse model of Marfan syndrome. Bone. (2021) 152:116073. doi: 10.1016/j.bone.2021.116073
50. Bellini C, Korneva A, Zilberberg L, Ramirez F, Rifkin DB, Humphrey JD. Differential ascending and descending aortic mechanics parallel aneurysmal propensity in a mouse model of Marfan syndrome. J Biomech. (2016) 49:2383–9. doi: 10.1016/j.jbiomech.2015.11.059
51. Korneva A, Zilberberg L, Rifkin DB, Humphrey JD, Bellini C. Absence of LTBP-3 attenuates the aneurysmal phenotype but not spinal effects on the aorta in Marfan syndrome. Biomech Model Mechanobiol. (2019) 18:261–73. doi: 10.1007/s10237-018-1080-1
52. Chen M, Cavinato C, Hansen J, Tanaka K, Ren P, Hassab A, et al. FN (fibronectin)-integrin 5 signaling promotes thoracic aortic aneurysm in a mouse model of Marfan syndrome. Arterioscler Thromb Vasc Biol. (2023) 43:e132–50. doi: 10.1161/ATVBAHA.123.319120
53. Groenink M, de Roos A, Mulder BJ, Spaan JA, van der Wall EE. Changes in aortic distensibility and pulse wave velocity assessed with magnetic resonance imaging following beta-blocker therapy in the marfan syndrome. Am J Cardiol. (1998) 82:203–8. doi: 10.1016/S0002-9149(98)00315-4
54. Andrea G, Jose RP, Lydia DS, Gisela TT, Giuliana M, Laura G, et al. Influence of aortic dilation on the regional aortic stiffness of bicuspid aortic valve assessed by 4-dimensional flow cardiac magnetic resonance. JACC: Cardiovasc Imaging. (2019) 12:1020–9. doi: 10.1016/j.jcmg.2018.03.017
55. Tarraf SA, Kramer B, Vianna E, Gillespie C, Germano E, Emerton KB, et al. Lengthwise regional mechanics of the human aneurysmal ascending thoracic aorta. Acta Biomater. (2023) 162:266–77. doi: 10.1016/j.actbio.2023.03.023
56. Selamet Tierney ES, Levine JC, Sleeper LA, Roman MJ, Bradley TJ, Colan SD, et al. Influence of aortic stiffness on aortic-root growth rate and outcome in patients with the Marfan syndrome. Am J Cardiol. (2018) 121:1094–101. doi: 10.1016/j.amjcard.2018.01.016
57. Schlatmann TJ, Becker AE. Pathogenesis of dissecting aneurysm of aorta. Am J Cardiol. (1977) 39:21–6. doi: 10.1016/S0002-9149(77)80005-2
58. Bode-Jänisch S, Schmidt A, Günther D, Stuhrmann M, Fieguth A. Aortic dissecting aneurysms—histopathological findings. Forensic Sci Int. (2012) 214:13–7. doi: 10.1016/j.forsciint.2011.07.006
59. Wu D, Shen YH, Russell L, Coselli JS, LeMaire SA. Molecular mechanisms of thoracic aortic dissection. J Surg Res. (2013) 184:907–24. doi: 10.1016/j.jss.2013.06.007
60. Halushka MK, Angelini A, Bartoloni G, Basso C, Batoroeva L, Bruneval P, et al. Consensus statement on surgical pathology of the aorta from the Society for Cardiovascular Pathology and the Association For European Cardiovascular Pathology: II. Noninflammatory degenerative diseases—nomenclature and diagnostic criteria. Cardiovasc Pathol. (2016) 25:247–57. doi: 10.1016/j.carpath.2016.03.002
61. Sherifova S, Holzapfel GA. Biomechanics of aortic wall failure with a focus on dissection and aneurysm: a review. Acta Biomater. (2019) 99:1–17. doi: 10.1016/j.actbio.2019.08.017
62. Yousef S, Matsumoto N, Dabe I, Mori M, Landry AB, Lee SR, et al. Quantitative not qualitative histology differentiates aneurysmal from nondilated ascending aortas and reveals a net gain of medial components. Sci Rep. (2021) 11:13185. doi: 10.1038/s41598-021-92659-1
63. Ferruzzi J, Bersi MR, Mecham RP, Ramirez F, Yanagisawa H, Tellides G, et al. Loss of elastic fiber integrity compromises common carotid artery function: implications for vascular aging. Artery Res. (2016) 14:41–52. doi: 10.1016/j.artres.2016.04.001
64. Weiss D, Latorre M, Rego B, Cavinato C, Tanski B, Berman A, et al. Biomechanical consequences of compromised elastic fiber integrity and matrix cross-linking on abdominal aortic aneurysmal enlargement. Acta Biomater. (2021) 134:422–34. doi: 10.1016/j.actbio.2021.07.059
65. Ferruzzi J, Bersi MR, Uman S, Yanagisawa H, Humphrey JD. Decreased elastic energy storage, not increased material stiffness, characterizes central artery dysfunction in fibulin-5 deficiency independent of sex. J Biomech Eng. (2015) 137:031007. doi: 10.1115/1.4029431
66. Yanagisawa H, Davis EC, Starcher BC, Ouchi T, Yanagisawa M, Richardson JA, et al. Fibulin-5 is an elastin-binding protein essential for elastic fibre development in vivo. Nature. (2002) 415:168–71. doi: 10.1038/415168a
67. Seeburun S, Wu S, Hemani D, Pham L, Ju D, Xie Y, et al. Insights into elastic fiber fragmentation: mechanisms and treatment of aortic aneurysm in Marfan syndrome. Vascul Pharmacol. (2023) 153:107215. doi: 10.1016/j.vph.2023.107215
68. Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. (2010) 47:476–85. doi: 10.1136/jmg.2009.072785
69. Ernst L, Zhang K, Pieske-Kraigher E, Pieske B, Heinzel FR, Gehle P. Morphological and secretory dysfunction of left atrium in Marfan syndrome. Eur Heart J. (2021) 42:ehab724.1863. doi: 10.1093/eurheartj/ehab724.1863
70. Hennekam RC. Severe infantile Marfan syndrome versus neonatal Marfan syndrome. Am J Med Genet A. (2005) 139:1– . doi: 10.1002/ajmg.a.30979
71. Sasse P, Malan D, Fleischmann M, Roell W, Gustafsson E, Bostani T, et al. Perlecan is critical for heart stability. Cardiovasc Res. (2008) 80:435–44. doi: 10.1093/cvr/cvn225
72. Jackson ZS, Dajnowiec D, Gotlieb AI, Langille BL. Partial off-loading of longitudinal tension induces arterial tortuosity. Arterioscler Thromb Vasc Biol. (2005) 25:957–62. doi: 10.1161/01.ATV.0000161277.46464.11
73. Hayes AJ, Farrugia BL, Biose IJ, Bix GJ, Melrose J. Perlecan, a multi-functional, cell-instructive, matrix-stabilizing proteoglycan with roles in tissue development has relevance to connective tissue repair and regeneration. Front Cell Dev Biol. (2022) 10:105849. doi: 10.3389/fcell.2022.856261
74. Singh P, Carraher C, Schwarzbauer JE. Assembly of fibronectin extracellular matrix. Annu Rev Cell Dev Biol. (2010) 26:397–419. doi: 10.1146/annurev-cellbio-100109-104020
75. Malashicheva A, Kostina D, Kostina A, Irtyuga O, Voronkina I, Smagina L, et al. Phenotypic and functional changes of endothelial and smooth muscle cells in thoracic aortic aneurysms. Int J Vasc Med. (2016) 2016:3107879. doi: 10.1155/2016/3107879
76. Rombouts KB, van Merrienboer TAR, Ket JCF, Bogunovic N, van der Velden J, Yeung KK. The role of vascular smooth muscle cells in the development of aortic aneurysms and dissections. Eur J Clin Invest. (2022) 52:e13697. doi: 10.1111/eci.13697
77. van de Pol V, Kurakula K, DeRuiter MC, Goumans MJ. Thoracic aortic aneurysm development in patients with bicuspid aortic valve: what is the role of endothelial cells? Front Physiol. (2017) 8:938. doi: 10.3389/fphys.2017.00938
78. Van Hove CE, Van der Donckt C, Herman AG, Bult H, Fransen P. Vasodilator efficacy of nitric oxide depends on mechanisms of intracellular calcium mobilization in mouse aortic smooth muscle cells. Br J Pharmacol. (2009) 158:920–30. doi: 10.1111/j.1476-5381.2009.00396.x
79. Bix G, Iozzo RV. Novel interactions of perlecan: unraveling perlecan’s role in angiogenesis. Microsc Res Tech. (2008) 71:339–48. doi: 10.1002/jemt.20562
80. Nonaka R, Iesaki T, de Vega S, Daida H, Okada T, Sasaki T, et al. Perlecan deficiency causes endothelial dysfunction by reducing the expression of endothelial nitric oxide synthase. Physiol Rep. (2015) 3:e12272. doi: 10.14814/phy2.12272
81. Lundmark K, Tran PK, Kinsella MG, Clowes AW, Wight TN, Hedin U. Perlecan inhibits smooth muscle cell adhesion to fibronectin: role of heparan sulfate. J Cell Physiol. (2001) 188:67–74. doi: 10.1002/jcp.1094
82. Riordan JD, Nadeau JH. From peas to disease: modifier genes, network resilience, and the genetics of health. Am J Hum Genet. (2017) 101:177–91. doi: 10.1016/j.ajhg.2017.06.004
83. Ince H, Nienaber CA. Etiology, pathogenesis and management of thoracic aortic aneurysm. Nat Clin Pract Cardiovasc Med. (2007) 4:418–27. doi: 10.1038/ncpcardio0937
Keywords: ascending aneurysm, biomechanics, Marfan syndrome, fibrillin-1, perlecan
Citation: Tarraf SA, de Souza RB, Herrick A, Pereira LV and Bellini C (2024) The Fbn1 gene variant governs passive ascending aortic mechanics in the mgΔlpn mouse model of Marfan syndrome when superimposed to perlecan haploinsufficiency. Front. Cardiovasc. Med. 11:1319164. doi: 10.3389/fcvm.2024.1319164
Received: 11 October 2023; Accepted: 12 February 2024;
Published: 13 March 2024.
Edited by:
Xuerui Tan, First Affiliated Hospital of Shantou University Medical College, ChinaReviewed by:
Yanming Li, Baylor College of Medicine, United StatesCallie Kwartler, University of Texas Health Science Center at Houston, United States
© 2024 Tarraf, de Souza, Herrick, Pereira and Bellini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chiara Bellini Yy5iZWxsaW5pQG5vcnRoZWFzdGVybi5lZHU= Lygia V. Pereira bHBlcmVpcmFAdXNwLmJy
†These authors have contributed equally to this work