 Roba Hamed Mostafa1,2,†Ahmed Moustafa1,2,3*†
Roba Hamed Mostafa1,2,†Ahmed Moustafa1,2,3*†
- 1Systems Genomics Laboratory, American University in Cairo, New Cairo, Egypt
- 2Biotechnology Graduate Program, American University in Cairo, New Cairo, Egypt
- 3Department of Biology, American University in Cairo, New Cairo, Egypt
SARS-CoV-2, responsible for the global COVID-19 pandemic, has manifested significant cardiovascular implications for the infected population. These cardiovascular repercussions not only linger beyond the initial phase of illness but have also been observed in individuals who remain asymptomatic. This extended and pervasive impact is often called the post-acute COVID-19 syndrome (PACS) or “Long COVID”. With the number of confirmed global cases approaching an alarming 756 million, the multifaceted challenges of Long COVID are undeniable. These challenges span from individual health complications to considerable burdens on worldwide healthcare systems. Our review comprehensively examines the complications of the persistent cardiovascular complications associated with COVID-19. Furthermore, we shed light on emerging therapeutic strategies that promise to manage and possibly mitigate these complications. We also introduce and discuss the profound concerns regarding the potential transgenerational repercussions of SARS-CoV-2, emphasizing the need for a proactive and informed approach to future research and clinical practice.
Introduction
In December 2019, Wuhan in China experienced a Coronavirus Disease 2019 (COVID-19) outbreak, leading to a global pandemic. This highly infectious virus, the severe acute respiratory syndrome coronavirus (SARS-CoV-2), primarily invades the respiratory system, causing severe pneumonia that can progress to acute respiratory distress syndrome (1). By February 2023, the World Health Organization (WHO) had reported approximately 756 million confirmed cases of COVID-19 globally, along with a staggering 6.8 million cumulative deaths attributable to the virus (2).
The COVID-19 virus contains five structural proteins: spike (S), envelope (E), membrane (M), and nucleocapsid (N). The distinctive crown-like morphology of the coronavirus results from the spike protein, which also facilitates host cell entry and consists of S1 and S2 subunits (3).
SARS-CoV-2, part of the Beta-CoV genus and the Nidovirales order, belongs to the Coronaviridae family. Notably, two previous beta-CoV virus outbreaks—SARS-CoV and Middle East Respiratory Syndrome (MERS) viruses—hit the world in 2002 and 2012, respectively (3).
SARS-CoV-2 has shown higher virulence than SARS-CoV, which is attributed to two key acquired mutations. The receptor binding domain (RBD), a highly variable segment in the SARS-CoV and SARS-CoV-2 genomes, uses six central amino acid residues for adequate recognition of the host angiotensin-converting enzyme 2 (ACE2) receptor. In SARS-CoV-2, mutations in five of the six primary amino acid residues in the RBD allow for a greater binding affinity to the ACE2 receptor. Moreover, an insertion mutation at the S1/S2 fusion site introduces a furin cleavage site, enhancing cellular entry and viral tissue tropism due to S-priming with furin and other proteases (4, 5).
SARS-CoV-2 has led to a significant cardiovascular burden in the infected population, with harmful cardiovascular sequelae persisting even after the acute phase and in asymptomatic patients. This review focuses on the pathophysiology underlying the long-term cardiovascular burden associated with COVID-19 and explores potential new therapeutic targets.
COVID-19 pathogenesis and pathophysiology
Host neuropilin-1 (NRP-1) binding to the NRP-1 binding domain in RBD precedes ACE2 recognition. This action facilitates host ACE2 recognition by the RBD of COVID-19, triggering a conformational change that allows the virus to become closer to the host cells (6).
SARS-CoV-2 can enter cells through one of two pathways: membrane fusion or endocytosis. In membrane fusion, a host protease known as transmembrane protease serine 2 (TMPRSS2) cleaves and primes the spike protein. Subsequently, the fusion peptide within the S2 subunit is inserted into the host cell membrane. The two heptad repeat domains in the S2 subunit, HR1, and HR2, interact, drawing the viral and host membranes together and enabling membrane fusion. This process allows the injection of the viral genome into the host cell. In the endocytosis pathway, the virus-receptor complex is endocytosed into the endosome, where the S protein is cleaved by cathepsin enzymes (7–9).
SARS-CoV-2 exhibits a broad tissue tropism, as its receptor is expressed in various tissues, including the lung, heart, small intestine, oral mucosa, and testis. These tissues have been found to harbor SARS-CoV-2 RNA (10–13). Viral entry triggers multiple immune responses. Toll-like receptor 3 (TLR-3) recognizes the cytosolic viral genome and activates nuclear factor kappa B (NF-κB), which translocates into the nucleus and stimulates the transcription of interferon-gamma and other inflammatory cytokines. Additionally, the NOD-like receptor (NLRP) gene is activated, leading to the production of NLRP3 proteins. NLRP3 initiates the formation of inflammasomes, which induce cell death through pyroptosis and promote inflammation by stimulating the secretion of inflammatory cytokines (14, 15). This immune response activates a wide range of immune cells, including dendritic cells (DCs), T-cells, and B-cells. DCs carry the viral antigen with major histocompatibility complex class II (MHC-II) receptors and migrate to lymph nodes to stimulate further T and B cell responses. T-cells and B-cells are then activated to eliminate the virus through direct cell killing and antibody secretion, respectively (14, 16).
The excessive release of inflammatory cytokines often leads to a cytokine storm, a lethal state of hyperactive immune response associated with systemic inflammation. Persistent systemic inflammation can cause vasculitis, increasing the risk of blood clot formation, hypercoagulability, ischemia, and cell death. Additionally, acute respiratory distress syndrome results in poor perfusion, hypoxia, multiorgan failure, and death (14, 16).
Although COVID-19 was initially considered primarily a respiratory illness, it is now evident that it causes a systemic infection, damaging multiple organs during and after the infection. This damage may arise from a direct viral infection or the detrimental effects of systemic inflammation (17).
COVID-19 and the cardiovascular system
Cardiovascular injury is common among SARS-CoV-2-infected patients, with damage to the myocytes varying from initial injury with elevated troponins to eventual heart failure, indicated by increased levels of the N-terminal-prohormone brain natriuretic peptide BNP (NT-proBNP) (18–20). Among the cardiovascular complications linked to COVID-19 are arrhythmia, myocarditis, acute coronary syndrome, myocardial infarction, and venous thrombosis embolisms, which are detectable through methods such as echocardiography, MRI, electrocardiogram (ECG), coronary angiography, and cardiac autopsies in COVID-19 patients (20–22).
Cardiac involvement in COVID-19 patients predisposes to a higher mortality rate compared to COVID-19 patients without such complications. Higher troponin levels in COVID-19 patients with pre-existing cardiovascular diseases have been associated with doubling the mortality rate (18, 23). Children may also suffer from cardiac dysfunction and coronary abnormalities, such as Kawasaki disease (24).
Mechanisms of COVID-19-associated cardiovascular injury
Although several mechanisms have been proposed, the precise pathophysiological pathway by which SARS-CoV-2 causes cardiac damage has not been entirely established. One such pathway involves SARS-CoV-2-induced hypoxemia secondary to pulmonary dysfunction and respiratory distress syndrome. This results in poor organ perfusion, leading to hypoxic injury in cardiac cells and contributing to myocardial infarction. Another related mechanism involves microvascular injury and thrombosis in the lung microvasculature, which leads to right ventricle heart failure (7, 25).
Systemic inflammation and cytokine storms play a vital role in microvascular injury, endothelial cell activation, venous thrombosis, and the hypercoagulative state associated with COVID-19 (26). This concurs with findings from heart autopsies of COVID-19 patients, where venous thrombosis and immune cell infiltration by CD3+ and CD8+ cytotoxic lymphocytes, CD68+ macrophages, and CD45RO memory cells were identified (27–29). Overexpression of cytokines can cause myocarditis and myocardial injury (27, 30), and cardiac biomarkers have been found to correlate with the levels of inflammatory cytokines (26, 31). Moreover, patients with predisposing cardiovascular risk factors are more susceptible to microvascular injury and endothelial dysfunction since SARS-CoV2 associated inflammation exacerbates the pre-existing endothelial damage aggravating thrombosis which leads to an increased risk of acute coronary syndrome and myocardial injury (32).
Direct virus entry is another proposed mechanism of COVID-19-associated cardiac damage. Inflammation and oxidative stress have also been shown to increase the entry of the SARS-CoV-2 virus into cardiac cells and promote cellular apoptosis (33). Linder et al. documented the presence of SARS-CoV-2 RNA in 61.5% of myocardial autopsies of patients who died of COVID-19, with the viral load exceeding 1,000 copies per μg RNA in 41% of the tissues (27). On the other hand, another study denied the existence of viral RNA and recorded very few RNA copies (34, 35). Similarly, Moustafa et al. found that traces of SARS-CoV-2 RNA are present in peripheral blood mononuclear cells of COVID-19 patients, although in significantly lower quantities compared to bronchoalveolar lavage specimens, suggesting limited viral RNA in the blood (36). Han et al. suggested that COVID-19 could directly infect the heart's pacemaker, causing arrhythmia in hamster models and human embryonic stem cell (hESC)-derived SAN-like pacemaker cells, where the viral RNA was detected inside the pacemaker cells and was capable of inducing ferroptosis (37). In Drosophila and mouse models, non-structural protein 6 (NSP6) of SARS-CoV-2 interacted with host proteins in the heart, potentiating glycolysis, disrupting mitochondrial function, and increasing ROS formation (38).
After viral entry, COVID-19 patients are susceptible to hypertension due to the downregulation of ACE2 receptors in host cells. The ACE2 receptor plays a critical role in the renin-angiotensin-aldosterone (RAAS) system, with its downregulation leading to the upregulation of angiotensin 2, a potent vasoconstrictor (10, 14, 16).
Recent studies using omics-based analyses have provided a more comprehensive understanding of the pathophysiology underlying COVID-19-associated cardiovascular diseases. In the heart tissues of COVID-19 patients, there was an observed upregulation in the transcriptional level of phospholipase C γ2 (PLCG2) in pericytes, fibroblasts, and cardiomyocytes. In contrast, the Afadin level, encoded by the Adherens Junction Formation Factor (AFDN) gene, was predominantly high in endothelial cells (34). Although the mechanism of PLCG2 in the heart is not thoroughly investigated, PLC-γ is known as a regulator of calcium homeostasis within heart tissues and plays a crucial role in cardiac pathogenesis. Its upregulation has been associated with cardiomyocyte apoptosis and mitochondrial dysfunction in patients with myocardial infarction, while its dysregulation can cause cardiac hypertrophy (39, 40). Furthermore, PLC is proposed as an arrhythmogenic factor, exacerbating cardiac fibrosis (41, 42). Whereas Afadin is essential for maintaining endothelial barrier function, the mechanism by which it is linked to SAS-CoV2-associated CVD is not known. Enrichment analysis revealed upregulation in cell differentiation, cell adhesion with elevated immune response, and apoptosis due to oxidative stress in fibroblasts, cardiac cells, and pericytes, respectively (34).
Meanwhile, endocrine senescence pathways in plasma were significantly upregulated in COVID-19 patients with cardiac complications. High FSTL3 and low ADAMTS13 levels were associated with heart failure in COVID-19 patients. The former is an indirect marker of biological aging induced by the TGF-B pathway, and the latter is an antithrombotic agent (19, 43). Another study by Garg et al. found elevated levels of miR-155, miR-499, miR-208a, miR-21, and miR-126 in the blood of critically ill COVID-19 patients. These miRNAs are associated with inflammation, myocardial function, fibroblasts, and endothelial cells (44).
Lipopolysaccharide (LPS)-binding protein (LBP) was upregulated and strongly associated with NT-pro-BNP in critically ill patients with cardiac involvement. LBP levels increase in response to circulating LPS, a major component of the outer membrane of gram-negative bacteria, suggesting gut leakage as an additional mechanism that could exacerbate heart failure in SAR-CoV-2-infected patients (45–48).
Cardiovascular manifestations of long COVID
Recently, it became evident that the adverse effects of COVID-19 are not limited to the acute phase but also extend to a condition known as long COVID. According to the National Institute for Health and Care Excellence (NICE), patients with ongoing symptoms of COVID-19 or newly developed ones that last beyond three months are diagnosed with long-term COVID (49).
In the Netherlands, patients were followed for three months post-infection and showed evidence of elevated inflammatory cytokines and endothelial dysfunction. This is in line with the elevated plasma levels of endothelin-1 (ET-1) that correlate with contact activation factors, hence suggesting endothelial activation and the coagulation process (50). In England, hospitalized COVID-19 patients suffered from an increased rate of major adverse cardiovascular events after discharge. People without any prior cardiovascular events developed cardiovascular diseases at a rate three times higher compared to healthy individuals (51). Another study followed patients for four months post-infection, and the rate of various cardiovascular diseases increased with new onsets of hypercoagulability and cardiomyopathy sequelae post-infection (52). Similarly, in the USA, a substantial excess burden of cardiovascular diseases occurred in a graded fashion according to the severity of the acute phase when patients were followed in two studies for six months and a year (17, 53). The commonly detected long-term cardiovascular sequelae were cerebrovascular disorders, dysrhythmia, pericarditis, myocarditis, ischemic heart diseases, hypertension, heart failure, and thrombotic disorders (17, 52, 53).
Pathophysiology of long COVID cardiovascular complications
Long COVID's clinical burden stresses healthcare systems and demands resources (54), emphasizing the urgency to understand the clinical pathology behind the disease and identify novel clinical targets (Figure 1). However, the absolute mechanism underlying long COVID sequelae has yet to be extensively deciphered.
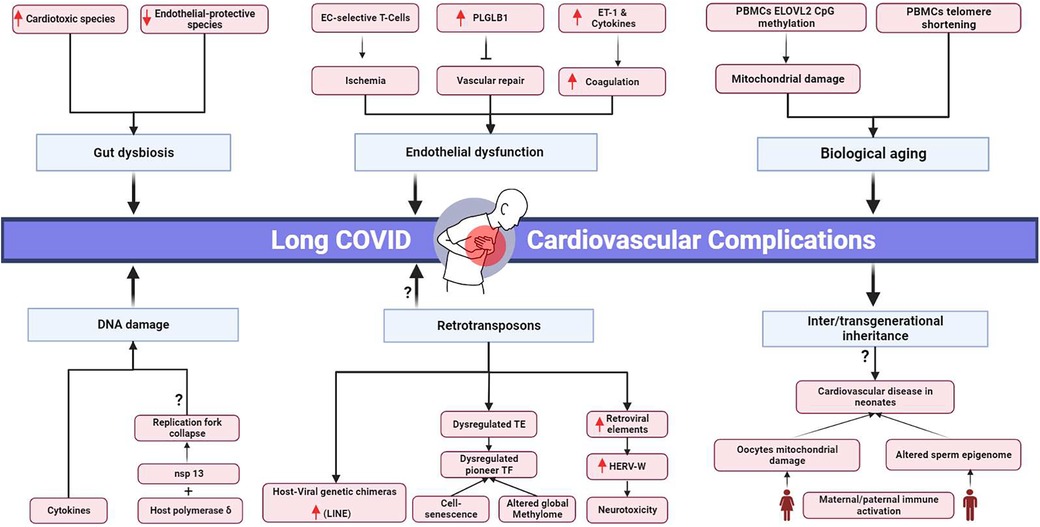
Figure 1. Proposed pathophysiological mechanisms and potential transgenerational cardiovascular impact of long COVID. EC, endothelial cells; PLGLB1, plasminogen-like protein B1; ET-1, Endothelin-1; PBMCs, peripheral blood mononuclear cells; ELOVL2, elongation of very long chain fatty acids-like 2; nsp 13, non-structural protein; HERV-W, human endogenous retrovirus-w. Black arrows represent downstream sequel, bars represent inhibitory downstream sequel, red arrows represent upregulation, question marks represent hypothesized mechanisms requiring further research. Created in BioRender.com.
Cell senescence and mitochondrial dysfunction
The first proposed mechanism is accelerated biological aging. In COVID-19 survivors’ peripheral blood mononuclear cells (PBMCs), the cells were five years above their expected biological age (55). Furthermore, accelerated aging was associated with downregulated ACE2 levels and telomere shortening, which are notable in cardiovascular diseases (56, 57). The senescence-associated secretory phenotype was reported to be correlated with cardiac involvement in COVID-19 patients (19).
Mitochondrial damage is another cornerstone of biological aging (58). In COVID-19 post-acute phase survivors, there was considerable CpG island methylation in the promotor region of elongation of very long chain fatty acids-like 2 (ELOVL2) in PBMCs (55). ELOVL2 is crucial for proper mitochondrial function. Mitochondrial dysfunction in COVID-19 patients’ PBMCs was reported by Ajaz et al. (59). PBMCs with damaged mitochondria are known to aggravate heart failure (60, 61). Another study found downregulated profiles of genes responsible for metabolic and mitochondrial function in SARS-CoV-2-infected cardiac tissues (62). In the iPSC-derived human cardiomyocytes (iPSC-CMs) model, SARS-CoV-2 infection impaired mitochondrial function, cell bioenergetics, and calcium cycling (63).
DNA damage and cell cycle arrest influenced by SARS-CoV-2
DNA damage presented another pathogenic pathway inhibiting the symptoms’ resilience in COVID-19 patients. However, further studies are needed to investigate the longevity of these genetic instabilities and their correlation to long COVID. DNA damage was detected in cultured AC16 cardiomyocyte cells treated with the serum of COVID-19 patients, where markers of DNA double-strand break, γH2Ax, and H3K79me2, were elevated. Persistent inflammatory cytokines were detected in long COVID patients’ serum samples, which are inducers of chromosomal instability (64–66). In agreement, γH2Ax was upregulated in cardiac autopsy tissues from COVID-19 patients (62). SARS-CoV showed an increase in H2AX histone phosphorylation in the infected population due to the interaction between its nonstructural protein nsp13 and the host DNA polymerase δ, leading to replication fork collapse and DNA damage followed by cell cycle arrest. SARS-CoV-induced cycle arrest led to the consumption of host cellular metabolites to allow viral replication (67). Considering the 79% similarity between SARS-CoV and SARS-CoV-2 and the 99.8% similarity between nsp13 in both species (64, 68, 69) and that SARS-CoV-2 can directly infect myocardium and actively replicate inside (27), this could be a potential mechanism by which cardiomyocyte damage occurs during COVID-19 and extends towards long COVID manifestations.
Prolonged immune activation and endothelial damage
Another potential molecular pathway for long COVID is endothelial dysfunction, leading to ischemic injuries and organ damage. Prolonged active CD8+ and CD4+ effector T cells selective for endothelial cells were detected in the blood of COVID-19 convalescent patients (70). In another study, the anti-angiogenic plasminogen-like protein B (PLGLB1) protein was more elevated in the serum of convalescent patients than in the acute phase. PLGLB1 inhibits vascular cell proliferation and prevents vascular repair in damaged vessels due to SARS-CoV-2 infection. Higher PLGLB1 levels in convalescence than in the acute phase indicate endothelial deterioration (71, 72).
Moreover, the immune system remains active in COVID-19 long-haulers, where serum samples showed dominant alterations in type II interferon and NF-κB signaling pathways (65). In PBMCs, miR-155-5p was upregulated during the post-acute phase, which is a factor known to be associated with inflammation and cardiovascular diseases (73, 74).
Retrotransposons and genetic reprogramming in myocytes
SARS-CoV-2 has been implicated in entering heart cells and integrating its reverse-transcribed genome into the host cell genome. In a study where SARS-CoV-2 infection was introduced into human iPSC-derived cardiac cells, chimeric viral-host transcripts were detected, suggesting the possibility of integrating the viral genome into the host’s genetic material. This mechanism is hypothesized to be driven by the long interspersed nuclear elements (LINE-1) retrotransposition mechanism (75). However, more research is required to confirm this hypothesis, which is considered a rare event. Notably, an independent study suggested that these chimeric transcripts might be artifactual and created during the preparation of RNA-seq libraries via RT-switching (76, 77).
An increase in the retrotransposon element (LINE) was observed in the lungs and intestines following the SARS-CoV-2 infection. This surge was attributed to an increase in ten-eleven translocation (TET) enzymes, which alter the methylation profile and activate LINE transcription (78). Transposable elements (TE) were found to be upregulated in bronchoalveolar biopsies but downregulated in PBMCs. The downstream targets for these dysregulated TEs were enriched for pioneer transcription factors (TF) and immune responses. Pioneer TF can alter the global methylome profile of the cells. Compared to other viral infections, TE levels in COVID-19 showed the highest copy number (79). The retroelements are also known to be associated with cell senescence and aging (80, 81). Further studies are needed to analyze their levels in correlation to SARS-CoV-2 long-term cardiovascular complications.
Additionally, the Human Endogenous Retrovirus-W (HERV-W) protein, known to be stimulated in viral infection and to induce immune and neurotoxic deleterious effects, was found elevated in the endothelial cells and pericardial fatty tissues of postmortem cardiac autopsy samples of COVID-19 patients (82–84).
Altered gut microbiome
The gut microbiota has been implicated in the pathophysiology of various diseases, including COVID-19. During the acute phase of COVID-19, the gut leakage marker Lipopolysaccharide Binding Protein (LBP) was found to be elevated in plasma samples and was associated with cardiovascular complications via the activation of inflammasomes (46, 48). Persistent gut dysbiosis was found in hospitalized COVID-19 patients, and this imbalance between opportunistic pathogens and beneficial symbionts prolonged after the acute phase (85–87).
In a 6-month period of COVID-19, a significant difference in gut microbiota composition was observed between patients with long COVID symptoms and those without symptoms or healthy individuals (88). Ruminococcus gnavus was found to be elevated in post-acute COVID-19 syndrome (PACS), known for its association with atherosclerosis and coronary artery disease. Faecalibacterium prausnitzii, a protective species against atherosclerosis, was depleted in PACS patients. Collinsella aerofaciens level was low in PACS, and its decreased level was associated with CAD and cardiac valve calcification (88–94). Furthermore, Bifidobacterium pseudocatenulatum, known to ameliorate TNF inflammatory signals and protect against endothelial damage, had an inverse relationship with PACS symptoms. On the other hand, Bacteroides vulgatus, an atherosclerosis attenuator species, was found to be abundant in PACS patients (88, 95–98).
Intergenerational and transgenerational inheritance
Long-term cardiovascular effects of COVID-19 have been observed in the infected population. However, whether these effects extend to future generations is still unknown (17, 53). Systematic reviews suggest that 20%–30% of neonates born to SARS-CoV-2-positive mothers are infected due to vertical transmission, exhibiting clinical manifestations such as hypotension and tachycardia (99, 100).
Neonates born to positive mothers or preconception-positive parents are still at high risk of developing adverse events, even if they are not infected. Growing evidence shows that parental environmental factors, stress, and infection before or during conception can influence the offspring's phenotype by modulating their epigenetics (101–103). These epigenetic markers can be inherited by the offspring or grandoffspring, suggesting a potential role for the intergenerational and transgenerational inheritance of SARS-CoV-2 adverse events due to parental exposure to SARS-CoV-2 (102).
In the context of paternal contributions, infection and immune activation can alter the sperm epigenome and affect future generations. The sperm transfers its epigenetic information into the embryos, impacting embryonic development (104–106). Moreover, cardiovascular disease inheritance by sperm non-coding RNA was suggested by Wagner et al. (107). There is mounting evidence that SARS-CoV-2 negatively affects sperm, altering the semen proteomics of convalescent patients, which is associated with multiple pathways, including inflammatory cytokines, sperm differentiation, and ROS formation. DNA fragmentation has also been reported in the sperm of convalescent COVID-19 patients (108–110).
Furthermore, maternal immune activation during pregnancy can have adverse effects on the cardiovascular system of the offspring and may potentiate the activation of inflammatory pathways in neonates and ROS formation (102). SARS-CoV-2 can influence mitochondrial damage in peripheral blood cells and cardiac cells, which can potentially affect oocyte mitochondria (59, 62, 63). However, the quality of oocytes in recovered patients has been reported as normal (111). Since maternal mitochondria are transferred to the zygote during fertilization and mitochondrial DNA mutations can predispose individuals to cardiovascular diseases, there may be risks associated with mitochondrial damage due to COVID-19 (112, 113).
Conclusion
SARS-CoV-2's impact extends beyond the acute respiratory distress often spotlighted in medical narratives. Its ability to affect multiple organs and lead to serious cardiovascular complications provides a broader perspective on the virus's toll. As highlighted in our review, while the mechanisms underlying these complications are not fully defined, they likely involve hypoxia, endothelial dysfunction, and an exacerbated immune response. Notably, persistent cardiovascular ramifications have emerged not just in symptomatic COVID-19 survivors but also in those who remained asymptomatic.
With an astounding 756 million confirmed COVID-19 cases worldwide, the repercussions of long COVID, especially its cardiovascular sequelae, present both a challenge and a burden to global healthcare systems. We have reviewed several proposed pathophysiological mechanisms for these complications, including systemic biological aging, gut microbiome disruptions, mitochondrial and direct cardiac damage, sustained systemic inflammation, and epigenomic alterations.
Nevertheless, a significant gap remains in our comprehensive understanding of these mechanisms, with many still hypothesized and awaiting rigorous scientific validation. The urgency of the current situation underscores the need for continued research into the pathophysiology of long COVID. Only by elucidating these pathways can we hope to identify effective therapeutic targets and address the extensive cardiovascular consequences of this pandemic.
Author contributions
RM: Investigation, Visualization, Writing – original draft, Writing – review & editing. AM: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wu D, Wu T, Liu Q, Yang Z. The SARS-CoV-2 outbreak: what we know. Int J Infect Dis. (2020) 94:44–8. doi: 10.1016/j.ijid.2020.03.004
2. WHO Coronavirus (COVID-19) Dashboard. (2023). Available online at: https://covid19.who.int/ (accessed July 25, 2023).
3. Li H, Liu S-M, Yu X-H, Tang S-L, Tang C-K. Coronavirus disease 2019 (COVID-19): current status and future perspectives. Int J Antimicrob Agents. (2020) 55(5):105951. doi: 10.1016/j.ijantimicag.2020.105951
4. Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat Med. (2020) 26(4):450–2. doi: 10.1038/s41591-020-0820-9
5. Nishiga M, Wang DW, Han Y, Lewis DB, Wu JC. COVID-19 and cardiovascular disease: from basic mechanisms to clinical perspectives. Nat Rev Cardiol. (2020) 17(9):543–58. doi: 10.1038/s41569-020-0413-9
6. Daly JL, Simonetti B, Klein K, Chen KE, Williamson MK, Antón-Plágaro C, et al. Neuropilin-1 is a host factor for SARS-CoV-2 infection. Science. (2020) 370(6518): 861–5. doi: 10.1126/science.abd3072
7. Chung MK, Zidar DA, Bristow MR, Cameron SJ, Chan T, Harding CV III, et al. COVID-19 and cardiovascular disease. Circ Res. (2021) 128:1214–36. doi: 10.1161/CIRCRESAHA.121.317997
8. Kwan JYY, Lin L-T, Bell R, Bruce JP, Richardson C, Pugh TJ, et al. Elevation in viral entry genes and innate immunity compromise underlying increased infectivity and severity of COVID-19 in cancer patients. Sci Rep. (2021) 11(1):4533. doi: 10.1038/s41598-021-83366-y
9. Liu S, Xiao G, Chen Y, He Y, Niu J, Escalante CR, et al. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet. (2004) 363(9413):938–47. doi: 10.1016/S0140-6736(04)15788-7
10. Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. (2002) 417(6891):822–8. doi: 10.1038/nature00786
11. Hamming I, Timens W, Bulthuis MLC, Lely AT, Navis GJ, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. (2004) 203(2):631–7. doi: 10.1002/path.1570
12. Qi J, Zhou Y, Hua J, Zhang L, Bian J, Liu B, et al. The scRNA-Seq expression profiling of the receptor ACE2 and the cellular protease TMPRSS2 reveals human organs susceptible to SARS-CoV-2 infection. Int J Environ Res Public Health. (2021) 18(1):284. doi: 10.3390/ijerph18010284
13. Salamanna F, Maglio M, Landini MP, Fini M. Body localization of ACE-2: on the trail of the keyhole of SARS-CoV-2. Front Med. (2020) 7:594495. doi: 10.3389/fmed.2020.594495
14. Brodin P. Immune determinants of COVID-19 disease presentation and severity. Nat Med. (2021) 27(1):28–33. doi: 10.1038/s41591-020-01202-8
15. Wang M, Yu F, Chang W, Zhang Y, Zhang L, Li P. Inflammasomes: a rising star on the horizon of COVID-19 pathophysiology. Front Immunol. (2023) 14:1185233. doi: 10.3389/fimmu.2023.1185233
16. Jackson LA, Anderson EJ, Rouphael NG, Roberts PC, Makhene M, Coler RN, et al. An mRNA vaccine against SARS-CoV-2 — preliminary report. N Engl J Med. (2020) 383:1920–31. doi: 10.1056/NEJMoa2022483
17. Al-Aly Z, Xie Y, Bowe B. High-dimensional characterization of post-acute sequelae of COVID-19. Nature. (2021) 594(7862):259–64. doi: 10.1038/s41586-021-03553-9
18. Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, et al. Cardiovascular implications of fatal outcomes of patients with coronavirus disease 2019 (COVID-19). JAMA Cardiology. (2020) 5(7):811–8. doi: 10.1001/jamacardio.2020.1017
19. Roh JD, Kitchen RR, Guseh JS, McNeill JN, Aid M, Martinot AJ, et al. Plasma proteomics of COVID-19-associated cardiovascular complications: implications for pathophysiology and therapeutics. JACC Basic Transl Sci. (2022) 7(5):425–41. doi: 10.1016/j.jacbts.2022.01.013
20. Shi S, Qin M, Shen B, Cai Y, Liu T, Yang F, et al. Association of cardiac injury with mortality in hospitalized patients with COVID-19 in Wuhan, China. JAMA Cardiol. (2020) 5(7):802–10. doi: 10.1001/jamacardio.2020.0950
21. Lala A, Johnson KW, Januzzi JL, Russak AJ, Paranjpe I, Richter F, et al. Prevalence and impact of myocardial injury in patients hospitalized with COVID-19 infection. J Am Coll Cardiol. (2020) 76(5):533–46. doi: 10.1016/j.jacc.2020.06.007
22. Long B, Brady WJ, Koyfman A, Gottlieb M. Cardiovascular complications in COVID-19. Am J Emerg Med. (2020) 38(7):1504–7. doi: 10.1016/j.ajem.2020.04.048
23. Razavi AC, Kelly TN, He J, Fernandez C, Whelton PK, Krousel-Wood M, et al. Cardiovascular disease prevention and implications of coronavirus disease 2019: an evolving case study in the crescent city. J Am Heart Assoc. (2020) 9(13):e016997. doi: 10.1161/JAHA.120.016997
24. Riphagen S, Gomez X, Gonzalez-Martinez C, Wilkinson N, Theocharis P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet. (2020) 395(10237):1607–8. doi: 10.1016/S0140-6736(20)31094-1
25. Han MK, McLaughlin VV, Criner GJ, Martinez FJ. Pulmonary diseases and the heart. Circulation. (2007) 116(25):2992–3005. doi: 10.1161/CIRCULATIONAHA.106.685206
26. Arhontoulis DC, Kerr CM, Richards D, Tjen K, Hyams N, Jones JA, et al. Human cardiac organoids to model COVID-19 cytokine storm induced cardiac injuries. J Tissue Eng Regen Med. (2022) 16(9):799–811. doi: 10.1002/term.3327
27. Lindner D, Fitzek A, Bräuninger H, Aleshcheva G, Edler C, Meissner K, et al. Association of cardiac infection with SARS-CoV-2 in confirmed COVID-19 autopsy cases. JAMA Cardiol. (2020) 5(11):1281–5. doi: 10.1001/jamacardio.2020.3551
28. Maiese A, Frati P, Del Duca F, Santoro P, Manetti AC, La Russa R, et al. Myocardial pathology in COVID-19-associated cardiac injury: a systematic review. Diagnostics (Basel, Switzerland). (2021) 11(9):1647. doi: 10.3390/diagnostics11091647
29. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. (2020) 8(4):420–2. doi: 10.1016/S2213-2600(20)30076-X
30. Jarrah AA, Schwarskopf M, Wang ER, LaRocca T, Dhume A, Zhang S, et al. SDF-1 induces TNF-mediated apoptosis in cardiac myocytes. Apoptosis. (2018) 23(1):79–91. doi: 10.1007/s10495-017-1438-3
31. Yang L, Gou J, Gao J, Huang L, Zhu Z, Ji S, et al. Immune characteristics of severe and critical COVID-19 patients. Signal Transduct Target Ther. (2020) 5(1):179. doi: 10.1038/s41392-020-00296-3
32. Kovanen PT, Vuorio A. SARS-CoV-2 reinfection: adding insult to dysfunctional endothelium in patients with atherosclerotic cardiovascular disease. Atherosclerosis Plus. (2023) 53:1–5. doi: 10.1016/j.athplu.2023.06.002
33. Tangos M, Budde H, Kolijn D, Sieme M, Zhazykbayeva S, Lódi M, et al. SARS-CoV-2 infects human cardiomyocytes promoted by inflammation and oxidative stress. Int J Cardiol. (2022) 362:196–205. doi: 10.1016/j.ijcard.2022.05.055
34. Delorey TM, Ziegler CGK, Heimberg G, Normand R, Yang Y, Segerstolpe Å, et al. COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature. (2021) 595(7865):107–13. doi: 10.1038/s41586-021-03570-8
35. Puelles VG, Lütgehetmann M, Lindenmeyer MT, Sperhake JP, Wong MN, Allweiss L, et al. Multiorgan and renal tropism of SARS-CoV-2. N Engl J Med. (2020) 383(6):590–2. doi: 10.1056/NEJMc2011400
36. Moustafa A, Khalel RS, Aziz RK. Traces of SARS-CoV-2 RNA in peripheral blood cells of patients with COVID-19. Omics: A Journal of Integrative Biology. (2021) 25(8):475–83. doi: 10.1089/omi.2021.0068
37. Han Y, Zhu J, Yang L, Nilsson-Payant BE, Hurtado R, Lacko LA, et al. SARS-CoV-2 infection induces ferroptosis of sinoatrial node pacemaker cells. Circ Res. (2022) 130(7):963–77. doi: 10.1161/CIRCRESAHA.121.320518
38. Zhu J-Y, Wang G, Huang X, Lee H, Lee J-G, Yang P, et al. SARS-CoV-2 Nsp6 damages drosophila heart and mouse cardiomyocytes through MGA/MAX complex-mediated increased glycolysis. Commun Biol. (2022) 5(1):1039. doi: 10.1038/s42003-022-03986-6
39. Faul C, Amaral AP, Oskouei B, Hu M-C, Sloan A, Isakova T, et al. FGF23 induces left ventricular hypertrophy. J Clin Invest. (2011) 121(11):4393–408. doi: 10.1172/JCI46122
40. Gao T, Yang P, Fu D, Liu M, Deng X, Shao M, et al. The protective effect of allicin on myocardial ischemia-reperfusion by inhibition of ca overload-induced cardiomyocyte apoptosis via the PI3K/GRK2/PLC-γ/IP3R signaling pathway. Aging. (2021) 13(15):19643–56. doi: 10.18632/aging.203375
41. Kao Y-H, Chen Y-C, Lin Y-K, Shiu R-J, Chao T-F, Chen S-A, et al. FGF-23 dysregulates calcium homeostasis and electrophysiological properties in HL-1 atrial cells. Eur J Clin Investig. (2014) 44(8):795–801. doi: 10.1111/eci.12296
42. Lee TW, Chung CC, Lee TI, Lin YK, Kao YH, Chen YJ. Fibroblast growth factor 23 stimulates cardiac fibroblast activity through phospholipase C-mediated calcium signaling. Int J Mol Sci. (2021) 23(1):166. doi: 10.3390/ijms23010166
43. Tominaga K, Suzuki HI. TGF-β signaling in cellular senescence and aging-related pathology. Int J Mol Sci. (2019) 20(20):5002. doi: 10.3390/ijms20205002
44. Garg A, Seeliger B, Derda AA, Xiao K, Gietz A, Scherf K, et al. Circulating cardiovascular microRNAs in critically ill COVID-19 patients. Eur J Heart Fail. (2021) 23(3):468–75. doi: 10.1002/ejhf.2096
45. Alexander C, Rietschel ET. Bacterial lipopolysaccharides and innate immunity. J Endotoxin Res. (2001) 7(3):167–202.11581570
46. Hoel H, Heggelund L, Reikvam DH, Stiksrud B, Ueland T, Michelsen AE, et al. Elevated markers of gut leakage and inflammasome activation in COVID-19 patients with cardiac involvement. J Intern Med. (2021) 289(4):523–31. doi: 10.1111/joim.13178
47. Kitchens RL, Thompson PA. Modulatory effects of sCD14 and LBP on LPS-host cell interactions. J Endotoxin Res. (2005) 11(4):225–9. doi: 10.1179/096805105X46565
48. Roberts LM, Buford TW. Lipopolysaccharide binding protein is associated with CVD risk in older adults. Aging Clin Exp Res. (2021) 33(6):1651. doi: 10.1007/s40520-020-01684-z
49. Venkatesan P. NICE guideline on long COVID. Lancet Respir Med. (2021) 9(2):129. doi: 10.1016/S2213-2600(21)00031-X
50. Willems LH, Nagy M, Ten Cate H, Spronk HMH, Groh LA, Leentjens J, et al. Sustained inflammation, coagulation activation and elevated endothelin-1 levels without macrovascular dysfunction at 3 months after COVID-19. Thromb Res. (2022) 209:106–14. doi: 10.1016/j.thromres.2021.11.027
51. Ayoubkhani D, Khunti K, Nafilyan V, Maddox T, Humberstone B, Diamond I, et al. Post-COVID syndrome in individuals admitted to hospital with COVID-19: retrospective cohort study. Br Med J. (2021) 372:n693. doi: 10.1136/bmj.n693
52. Daugherty SE, Guo Y, Heath K, Dasmariñas MC, Jubilo KG, Samranvedhya J, et al. Risk of clinical sequelae after the acute phase of SARS-CoV-2 infection: retrospective cohort study. Br Med J. (2021) 373:n1098. doi: 10.1136/bmj.n1098
53. Xie Y, Xu E, Bowe B, Al-Aly Z. Long-term cardiovascular outcomes of COVID-19. Nat Med. (2022) 28(3):583–90. doi: 10.1038/s41591-022-01689-3
54. Menges D, Ballouz T, Anagnostopoulos A, Aschmann HE, Domenghino A, Fehr JS, et al. Burden of post-COVID-19 syndrome and implications for healthcare service planning: a population-based cohort study. PloS One. (2021) 16(7):e0254523. doi: 10.1371/journal.pone.0254523
55. Mongelli A, Barbi V, Gottardi Zamperla M, Atlante S, Forleo L, Nesta M, et al. Evidence for biological age acceleration and telomere shortening in COVID-19 survivors. Int J Mol Sci. (2021) 22:6151. doi: 10.3390/ijms22116151
56. Bekaert S, De Meyer T, Rietzschel ER, De Buyzere ML, De Bacquer D, Langlois M, et al. Telomere length and cardiovascular risk factors in a middle-aged population free of overt cardiovascular disease. Aging Cell. (2007) 6(5):639–47. doi: 10.1111/j.1474-9726.2007.00321.x
57. Cheng F, Luk AO, Tam CHT, Fan B, Wu H, Yang A, et al. Shortened relative leukocyte telomere length is associated with prevalent and incident cardiovascular complications in type 2 diabetes: analysis from the Hong Kong diabetes register. Diabetes Care. (2020) 43(9):2257–65. doi: 10.2337/dc20-0028
58. Lima T, Li TY, Mottis A, Auwerx J. Pleiotropic effects of mitochondria in aging. Nat Aging. (2022) 2(3):199–213. doi: 10.1038/s43587-022-00191-2
59. Ajaz S, McPhail MJ, Singh KK, Mujib S, Trovato FM, Napoli S, et al. Mitochondrial metabolic manipulation by SARS-CoV-2 in peripheral blood mononuclear cells of patients with COVID-19. Am J Physiol Cell Physiol. (2021) 320(1):C57–65. doi: 10.1152/ajpcell.00426.2020
60. Alfatni A, Riou M, Charles A-L, Meyer A, Barnig C, Andres E, et al. Peripheral blood mononuclear cells and platelets mitochondrial dysfunction, oxidative stress, and circulating mtDNA in cardiovascular diseases. J Clin Med Res. (2020) 9(2):311. doi: 10.3390/jcm9020311
61. Sauer F, Riou M, Charles A-L, Meyer A, Andres E, Geny B, et al. Pathophysiology of heart failure: a role for peripheral blood mononuclear cells mitochondrial dysfunction? J Clin Med Res. (2022) 11(3):741. doi: 10.3390/jcm11030741
62. Kulasinghe A, Liu N, Tan CW, Monkman J, Sinclair JE, Bhuva DD, et al. Transcriptomic profiling of cardiac tissues from SARS-CoV-2 patients identifies DNA damage. Immunology. (2023) 168(3):403–19. doi: 10.1111/imm.13577
63. Ramachandran K, Maity S, Muthukumar AR, Kandala S, Tomar D, Abd El-Aziz TM, et al. SARS-CoV-2 infection enhances mitochondrial PTP complex activity to perturb cardiac energetics. iScience. (2022) 25(1):103722. doi: 10.1016/j.isci.2021.103722
64. Pánico P, Ostrosky-Wegman P, Salazar AM. The potential role of COVID-19 in the induction of DNA damage. Mutat Res Rev Mutat Res. (2022) 789:108411. doi: 10.1016/j.mrrev.2022.108411
65. Talla A, Vasaikar SV, Szeto GL, Lemos MP, Czartoski JL, MacMillan H, et al. Persistent serum protein signatures define an inflammatory subset of long COVID. Nat Commun. (2023) 14:3417. doi: 10.1038/s41467-023-38682-4
66. Zhou H, Ren X, Yang Y, Xu B, Li Y, Feng Y, et al. An alternative way of SARS-COV-2 to induce cell stress and elevated DNA damage risk in cardiomyocytes without direct infection. Immun Inflamm Dis. (2022) 10(7):e638. doi: 10.1002/iid3.638
67. Xu LH, Huang M, Fang SG, Liu DX. Coronavirus infection induces DNA replication stress partly through interaction of its nonstructural protein 13 with the p125 subunit of DNA polymerase δ. J Biol Chem. (2011) 286(45):39546–59. doi: 10.1074/jbc.M111.242206
68. Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. (2020) 395(10224):565–74. doi: 10.1016/S0140-6736(20)30251-8
69. Yoshimoto FK. The proteins of severe acute respiratory syndrome coronavirus-2 (SARS CoV-2 or n-COV19), the cause of COVID-19. Protein J. (2020) 39(3):198–216. doi: 10.1007/s10930-020-09901-4
70. Chioh FW, Fong S-W, Young BE, Wu K-X, Siau A, Krishnan S, et al. Convalescent COVID-19 patients are susceptible to endothelial dysfunction due to persistent immune activation. eLife. (2021) 10:e64909. doi: 10.7554/eLife.64909
71. Morioka H, Morii T, Vogel T, Hornicek FJ, Weissbach L. Interaction of plasminogen-related protein B with endothelial and smooth muscle cells in vitro. Exp Cell Res. (2003) 287(1):166–77. doi: 10.1016/S0014-4827(03)00137-X
72. Wu S, Xu Y, Zhang J, Ran X, Jia X, Wang J, et al. Longitudinal serum proteome characterization of COVID-19 patients with different severities revealed potential therapeutic strategies. Front Immunol. (2022) 13:893943. doi: 10.3389/fimmu.2022.893943
73. Abbasi-Kolli M, Sadri Nahand J, Kiani SJ, Khanaliha K, Khatami A, Taghizadieh M, et al. The expression patterns of MALAT-1, NEAT-1, THRIL, and miR-155-5p in the acute to the post-acute phase of COVID-19 disease. Braz J Infect Dis. (2022) 26(3):102354. doi: 10.1016/j.bjid.2022.102354
74. Cao RY, Li Q, Miao Y, Zhang Y, Yuan W, Fan L, et al. The emerging role of MicroRNA-155 in cardiovascular diseases. BioMed Res Int. (2016) 2016:9869208. doi: 10.1155/2016/9869208
75. Zhang L, Richards A, Barrasa MI, Hughes SH, Young RA, Jaenisch R. Reverse-transcribed SARS-CoV-2 RNA can integrate into the genome of cultured human cells and can be expressed in patient-derived tissues. Proc Natl Acad Sci U S A. (2021) 118(21):e2105968118. doi: 10.1073/pnas.2105968118
76. Kazachenka A, Kassiotis G. SARS-CoV-2-host chimeric RNA-Sequencing reads do not necessarily signify virus integration into the host DNA. Front Microbiol. (2021) 12:676693. doi: 10.3389/fmicb.2021.676693
77. Yan B, Chakravorty S, Mirabelli C, Wang L, Trujillo-Ochoa JL, Chauss D, et al. Host-virus chimeric events in SARS-CoV2 infected cells are infrequent and artifactual. J Virol. (2021) 95:e0029421. doi: 10.1128/JVI.00294-21
78. Yin Y, Liu X-Z, He X, Zhou L-Q. Exogenous coronavirus interacts with endogenous retrotransposon in human cells. Front Cell Infect Microbiol. (2021) 11:609160. doi: 10.3389/fcimb.2021.609160
79. Marston JL, Greenig M, Singh M, Bendall ML, Duarte RRR, Feschotte C, et al. SARS-CoV-2 infection mediates differential expression of human endogenous retroviruses and long interspersed nuclear elements. JCI Insight. (2021) 6(24):e147170. doi: 10.1172/jci.insight.147170
80. Cardelli M, Giacconi R, Malavolta M, Provinciali M. Endogenous retroelements in cellular senescence and related pathogenic processes: promising drug targets in age-related diseases. Curr Drug Targets. (2016) 17(4):416–27. doi: 10.2174/1389450116666150518095829
81. Mustafin RN, Khusnutdinova EK. COVID-19, retroelements, and aging. Adv Gerontol. (2021) 11(1):83–92. doi: 10.1134/S2079057021010458
82. Charvet B, Reynaud JM, Gourru-Lesimple G, Perron H, Marche PN, Horvat B. Induction of proinflammatory multiple sclerosis-associated retrovirus envelope protein by human herpesvirus-6A and CD46 receptor engagement. Front Immunol. (2018) 9:2803. doi: 10.3389/fimmu.2018.02803
83. Charvet B, Brunel J, Pierquin J, Iampietro M, Decimo D, Queruel N, et al. SARS-CoV-2 induces human endogenous retrovirus type W envelope protein expression in blood lymphocytes and in tissues of COVID-19 patients. iScience. (2023) 26:106604. doi: 10.1016/j.isci.2023.106604
84. Duperray A, Barbe D, Raguenez G, Weksler BB, Romero IA, Couraud P-O, et al. Inflammatory response of endothelial cells to a human endogenous retrovirus associated with multiple sclerosis is mediated by TLR4. Int Immunol. (2015) 27(11):545–53. doi: 10.1093/intimm/dxv025
85. Liu Q, Su Q, Zhang F, Tun HM, Mak JWY, Lui GC-Y, et al. Multi-kingdom gut microbiota analyses define COVID-19 severity and post-acute COVID-19 syndrome. Nat Commun. (2022) 13(1):6806. doi: 10.1038/s41467-022-34535-8
86. Yeoh YK, Zuo T, Lui GC-Y, Zhang F, Liu Q, Li AY, et al. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut. (2021) 70(4):698–706. doi: 10.1136/gutjnl-2020-323020
87. Zuo T, Zhang F, Lui GCY, Yeoh YK, Li AYL, Zhan H, et al. Alterations in gut microbiota of patients with COVID-19 during time of hospitalization. Gastroenterology. (2020) 159(3):944–955.e8. doi: 10.1053/j.gastro.2020.05.048
88. Liu Q, Mak JWY, Su Q, Yeoh YK, Lui GC-Y, Ng SSS, et al. Gut microbiota dynamics in a prospective cohort of patients with post-acute COVID-19 syndrome. Gut. (2022) 71(3):544–52. doi: 10.1136/gutjnl-2021-325989
89. Gang J, Wang H, Xue X, Zhang S. Microbiota and COVID-19: long-term and complex influencing factors. Front Microbiol. (2022) 13:963488. doi: 10.3389/fmicb.2022.963488
90. Jie Z, Xia H, Zhong S-L, Feng Q, Li S, Liang S, et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat Commun. (2017) 8(1):845. doi: 10.1038/s41467-017-00900-1
91. Kim M, Huda MN, Bennett BJ. Sequence meets function-microbiota and cardiovascular disease. Cardiovasc Res. (2022) 118(2):399–412. doi: 10.1093/cvr/cvab030
92. Liu Z, Li J, Liu H, Tang Y, Zhan Q, Lai W, et al. The intestinal microbiota associated with cardiac valve calcification differs from that of coronary artery disease. Atherosclerosis. (2019) 284:121–8. doi: 10.1016/j.atherosclerosis.2018.11.038
93. Toya T, Corban MT, Marrietta E, Horwath IE, Lerman LO, Murray JA, et al. Coronary artery disease is associated with an altered gut microbiome composition. PloS One. (2020) 15(1):e0227147. doi: 10.1371/journal.pone.0227147
94. Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, et al. Non-lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell. (2015) 163(7):1585–95. doi: 10.1016/j.cell.2015.11.055
95. Giménez-Bastida JA, González-Sarrías A, Larrosa M, Tomás-Barberán F, Espín JC, García-Conesa M-T. Ellagitannin metabolites, urolithin A glucuronide and its aglycone urolithin A, ameliorate TNF-α-induced inflammation and associated molecular markers in human aortic endothelial cells. Mol Nutr Food Res. (2012) 56(5):784–96. doi: 10.1002/mnfr.201100677
96. Spigoni V, Mena P, Cito M, Fantuzzi F, Bonadonna RC, Brighenti F, et al. Effects on nitric oxide production of urolithins, gut-derived ellagitannin metabolites, in human aortic endothelial cells. Molecules. (2016) 21(8):1009. doi: 10.3390/molecules21081009
97. Yoshida N, Emoto T, Yamashita T, Watanabe H, Hayashi T, Tabata T, et al. Bacteroides vulgatus and Bacteroides dorei reduce gut microbial lipopolysaccharide production and inhibit atherosclerosis. Circulation. (2018) 138(22):2486–98. doi: 10.1161/CIRCULATIONAHA.118.033714
98. Zhao Y, Wang Z. Gut microbiome and cardiovascular disease. Curr Opin Cardiol. (2020) 35(3):207–18. doi: 10.1097/HCO.0000000000000720
99. Musa SS, Bello UM, Zhao S, Abdullahi ZU, Lawan MA, He D. Vertical transmission of SARS-CoV-2: a systematic review of systematic reviews. Viruses. (2021) 13(9):1877. doi: 10.3390/v13091877
100. Raschetti R, Vivanti AJ, Vauloup-Fellous C, Loi B, Benachi A, De Luca D. Synthesis and systematic review of reported neonatal SARS-CoV-2 infections. Nat Commun. (2020) 11(1):5164. doi: 10.1038/s41467-020-18982-9
101. Kaati G, Bygren LO, Edvinsson S. Cardiovascular and diabetes mortality determined by nutrition during parents’ and grandparents’ slow growth period. Eur J Human Genet. (2002) 10(11):682–8. doi: 10.1038/sj.ejhg.5200859
102. Kleeman EA, Gubert C, Hannan AJ. Transgenerational epigenetic impacts of parental infection on offspring health and disease susceptibility. Trends Genet. (2022) 38(7):662–75. doi: 10.1016/j.tig.2022.03.006
103. Patti M-E. Intergenerational programming of metabolic disease: evidence from human populations and experimental animal models. Cell Mol Life Sci. (2013) 70(9):1597–608. doi: 10.1007/s00018-013-1298-0
104. Conine CC, Sun F, Song L, Rivera-Pérez JA, Rando OJ. Small RNAs gained during epididymal transit of sperm are essential for embryonic development in mice. Dev Cell. (2018) 46(4):470–480.e3. doi: 10.1016/j.devcel.2018.06.024
105. Sharma U. Paternal contributions to offspring health: role of sperm small RNAs in intergenerational transmission of epigenetic information. Front Cell Dev Biol. (2019) 7:215. doi: 10.3389/fcell.2019.00215
106. Tyebji S, Hannan AJ, Tonkin CJ. Pathogenic infection in male mice changes sperm small RNA profiles and transgenerationally alters offspring behavior. Cell Rep. (2020) 31(4):107573. doi: 10.1016/j.celrep.2020.107573
107. Wagner KD, Wagner N, Ghanbarian H, Grandjean V, Gounon P, Cuzin F, et al. RNA induction and inheritance of epigenetic cardiac hypertrophy in the mouse. Dev Cell. (2008) 14(6):962–9. doi: 10.1016/j.devcel.2008.03.009
108. Ghosh S, Parikh S, Nissa MU, Acharjee A, Singh A, Patwa D, et al. Semen proteomics of COVID-19 convalescent men reveals disruption of key biological pathways relevant to male reproductive function. ACS Omega. (2022) 7:8601–12. doi: 10.1021/acsomega.1c06551
109. Hajizadeh Maleki B, Tartibian B. COVID-19 and male reproductive function: a prospective, longitudinal cohort study. Reproduction. (2021) 161(3):319–31. doi: 10.1530/REP-20-0382
110. Shcherbitskaia AD, Komarova EM, Milyutina YP, Ishchuk MA, Sagurova YM, Safaryan GK, et al. Oxidative stress markers and sperm DNA fragmentation in men recovered from COVID-19. Int J Mol Sci. (2022) 23(17):10060. doi: 10.3390/ijms231710060
111. Castiglione Morelli MA, Iuliano A, Schettini SCA, Ferri A, Colucci P, Viggiani L, et al. Are the follicular fluid characteristics of recovered coronavirus disease 2019 patients different from those of vaccinated women approaching fertilization? Front Physiol. (2022) 13:840109. doi: 10.3389/fphys.2022.840109
112. Chiaratti MR, Macabelli CH, Augusto Neto JD, Grejo MP, Pandey AK, Perecin F, et al. Maternal transmission of mitochondrial diseases. Genet Mol Biol. (2020) 43(1 suppl. 1):e20190095. doi: 10.1590/1678-4685-gmb-2019-0095
Keywords: SARS-CoV-2, cardiovascular, long COVID, post-acute COVID-19 syndrome (PACS), transgenerational inheritance
Citation: Mostafa RH and Moustafa A (2024) Beyond acute infection: molecular mechanisms underpinning cardiovascular complications in long COVID. Front. Cardiovasc. Med. 11:1268571. doi: 10.3389/fcvm.2024.1268571
Received: 28 July 2023; Accepted: 29 January 2024;
Published: 14 February 2024.
Edited by:
Patricia Bozza, Oswaldo Cruz Institute, BrazilReviewed by:
Petri T. Kovanen, Wihuri Research Institute, Finland© 2024 Mostafa and Moustafa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ahmed Moustafa amoustafa@aucegypt.edu
†ORCID Roba Hamed Mostafa orcid.org/0000-0001-5367-5691 Ahmed Moustafa orcid.org/0000-0002-0111-3555