 Christina C. Rolling
Christina C. Rolling Tessa J. Barrett
Tessa J. Barrett Jeffrey S. Berger
Jeffrey S. Berger- 1Department of Medicine, New York University Grossman School of Medicine, New York, NY, United States
- 2Department of Oncology and Hematology, University Medical Center Hamburg-Eppendorf, Hamburg, Germany
Platelets, key facilitators of primary hemostasis and thrombosis, have emerged as crucial cellular mediators of innate immunity and inflammation. Exemplified by their ability to alter the phenotype and function of monocytes, activated platelets bind to circulating monocytes to form monocyte-platelet aggregates (MPA). The platelet-monocyte axis has emerged as a key mechanism connecting thrombosis and inflammation. MPA are elevated across the spectrum of inflammatory and autoimmune disorders, including cardiovascular disease, systemic lupus erythematosus (SLE), and COVID-19, and are positively associated with disease severity. These clinical disorders are all characterized by an increased risk of thromboembolic complications. Intriguingly, monocytes in contact with platelets become proinflammatory and procoagulant, highlighting that this interaction is a central element of thromboinflammation.
Introduction
Platelets interact and bind to monocytes by a variety of mechanisms, including attachment of platelets to monocytes via platelet P-selectin and monocyte PSGL1, the release of platelet granules containing chemokines and cytokines, and shedding of platelet-derived microvesicles. These interactions result in the upregulation of monocyte proinflammatory surface markers (e.g., CD40), migration (CD11b/CD18), and procoagulant tissue factor (TF), a principal initiator of coagulation. In addition, monocytes exposed to platelets secrete proinflammatory cytokines (TNF-α, MCP-1, IL-1β) and exhibit a proinflammatory transcriptome. Furthermore, platelets skew monocyte and macrophage differentiation towards a proatherosclerotic phenotype.
Our review covers how platelets affect monocytes in inflammatory diseases, and we present recent findings on potential therapeutic strategies to target the platelet-monocyte proinflammatory axis in thromboinflammation.
Platelets in hemostasis and thrombosis
Derived from megakaryocytes, platelets are small, anucleate cells circulating in the blood for seven to ten days. While crucial to hemostasis and thrombosis, an immunomodulatory effector role for platelets is increasingly apparent (1). Under physiological conditions, circulating platelets become activated when they come in contact with subendothelial collagen following vascular injury. Exposed collagen attaches to von Willebrand factor (VWF) - a large globular multimeric glycoprotein - which then unfolds to a string-like structure. Platelets adhere to tethered VWF via the glycoprotein (GP) Ib-IX-V receptor complex (2) or directly via collagen through the GPVI receptor. Interaction of platelets with collagen and VWF results in platelet activation as shown by changes in the platelet cytoskeleton and release of cytokines, chemokines, and growth factors stored in alpha and dense granules (3). Adenosine diphosphate (ADP) derived from platelet dense granules and thromboxane A2 (TXA2) produced by activated platelets out of arachidonic acid, and other mediators including epinephrine and thrombin generated on the platelet membrane amplify and maintain the initial platelet response by recruiting and activating additional platelets. This ultimately leads to activation of the integrin complex GPIIb/IIIa, a key receptor for platelet adhesion, aggregation, and thrombus stabilization (4). As a result, activated platelets adhere to the vessel wall and form a thrombus, thereby preventing excessive blood loss at the injury site.
In addition to local platelet activation required for thrombosis and wound healing, platelets have effector roles in systemic inflammatory conditions. Platelet activation has been observed in sepsis (5–9), autoimmune disorders (10–14), and chronic proinflammatory conditions, including hyperlipidemia (15–17), atherosclerosis (18–22), and cardiovascular disease (22–26). Resultantly, patients with these diseases express elevated levels of circulating proinflammatory cytokines and chemokines and are also prone to thromboembolic complications (17, 27–29).
The role of monocytes in thromboinflammation
Monocytes originate in the bone marrow and constitute a subpopulation of approximately 10% of all peripheral blood leukocytes. Monocytes are key mediators of innate immunity. They phagocytose and present antigens, secrete chemokines and cytokines, and can terminally differentiate into different macrophage and dendritic cell subtypes that reside in the extravascular tissues (30, 31).
Based on the expression of surface markers CD14 and CD16, circulating monocytes are traditionally classified as either classical (CD14++ CD16−), intermediate (CD14+ CD16+), or nonclassical (CD14+ CD16++) subpopulations that exhibit different functional properties (32). While classical monocytes are phagocytic, CD16+ monocytes are often upregulated in systemic infections, constituting the major cytokine source (33).
The interaction of platelets with monocytes has emerged as a key mechanism connecting thrombosis and inflammation. In support of a proinflammatory and procoagulant platelet effector function, monocyte-platelet aggregates (MPA) are elevated in multiple thromboinflammatory diseases and correlate with disease severity (34). To be able to bind to monocytes, platelets need to become activated (35, 36).
Platelet activation during inflammation
Inflammatory mediators such as complement factors (37), interleukin-6 (IL-6) (38), IL-8 (39), and tumor necrosis factor-alpha (TNF-α) (40) secreted from activated immune and vascular cells contribute to platelet activation by enhancing their adhesion properties to endothelial cells, elevating collagen-induced aggregation, and enforcing release of TXA2.
Under inflammatory conditions, platelet-activating agonists such as TXA2, ADP, and thrombin bind to heterotrimeric G protein-coupled receptors (GPCR) expressed on the platelet surface. GPCR consist of different transmembrane spanning G proteins (GS, Gi, Gq, and G12/13) that are associated with platelet receptors including the ADP receptors P2Y1 (Gq) and P2Y12 (Gi), the thrombin receptors PAR1 (Gq, G12/13) and PAR4 (Gq, G12/13), and the TXA2 receptor (TP; Gq, G12/13) (41). Upon ligand binding, GPCR induce receptor-specific interconnected signaling pathways that lead to platelet alpha and dense granule release and activation of GPIIb/IIIa (inside-out signaling) (42).
Pathways of platelet-monocyte interaction in inflammation
Following platelet activation, P-selectin (CD62P) stored in alpha granules translocates to the platelet surface. Platelet P-selectin binds to glycoprotein ligand 1 (PSGL1) which is constitutively expressed on the surface of monocytes. This initial engagement of platelets with monocytes is strengthened by platelet CD40L binding to monocyte CD40, platelet GPVI binding to extracellular matrix metalloprotease inducer (EMMPRIN, CD147) (43) and platelet GPIb attaching to monocyte CD11b/CD18 (MAC-1) (44). Fibrinogen-mediated binding of platelet GPIIb/IIIa to MAC-1 has also been discussed (45) but appears to play only a minor role in the formation of MPA (46). In addition to platelet and monocyte receptor binding, platelets attract monocytes via chemokines and cytokines released from alpha and dense granules, including soluble CD40L (sCD40L) (47, 48), CXCL4 (platelet factor 4) (49), and CCL5 (50). Platelet-derived extracellular vesicles (PEV) including platelet microparticles, microvesicles, and exosomes are tiny circular fragments shed from the platelet membrane, and have been shown to regulate monocyte properties (51–53).
Interaction of platelets with monocytes by direct cell-to-cell contact and platelet-derived mediators facilitates the transition of monocytes to a proinflammatory and procoagulant phenotype. In the following sections, we will outline the different mechanisms by which platelets modify monocytes, and summarize how these mechanisms contribute to inflammation and thrombosis in the clinical setting (Figure 1).
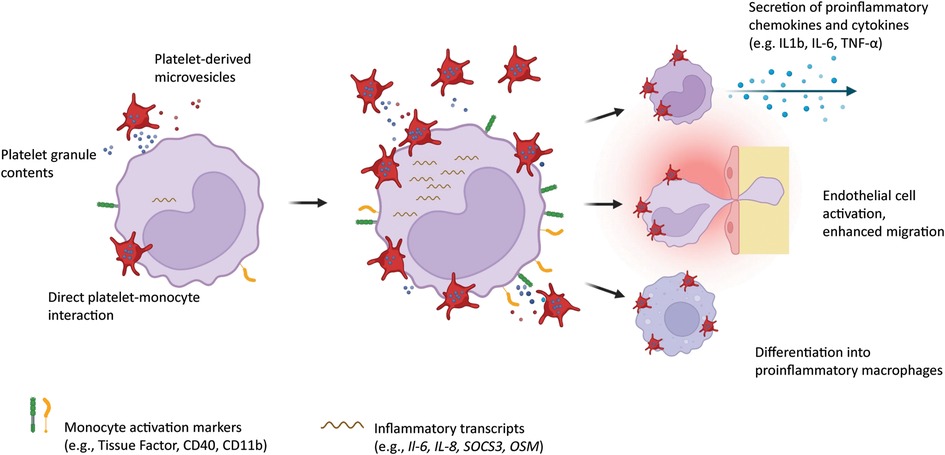
Figure 1. Platelets interact with circulating monocytes by direct attachment onto the monocyte surface, by release of alpha and dense granules containing chemokines and cytokines, and by shedding of microvesicles. Monocytes in turn upregulate inflammation markers on their surface, overexpress proinflammatory transcripts, secrete chemokines and cytokines, show enhanced migration potential, and differentiate into proinflammatory macrophages. Created with BioRender.com
Activated platelets modify the phenotype of circulating monocytes
Monocyte subtypes as defined by their expression of surface markers CD14 and CD16 can be modified by platelet interaction: Platelets appear to preferentially bind to CD16-bearing monocytes (16, 54). When bound to platelets, classical (CD14++CD16−) human monocytes upregulate surface CD16 - likely via platelet-derived transforming growth factor-β (TGF-β) inducing cyclooxygenase 2 (COX-2) upregulation and prostaglandin E2 synthesis in monocytes (55–57). High levels of antigen presenting-related molecules and secretion of proinflammatory cytokines and chemokines characterize these monocyte populations. In support of a proinflammatory platelet effector role, intermediate monocytes are expanded in various inflammatory disorders (34). Several other monocyte activation markers linked to atherosclerosis and inflammation are higher expressed on monocytes attached to platelets than monocytes without adhered platelets, including CD11b/CD18, CD40, PSGL1, HLA-DR, CD86, CD54, and CCR2 (34, 58).
Findings from several studies support a platelet-mediated proinflammatory monocyte phenotype (59). For example, activated platelets adhering to monocytes via the P-selectin PSGL1 axis in combination with alpha granule-released, CD40L, CXCL4 and CCL5 induce expression and secretion of monocyte chemoattractant protein-1 (MCP), IL-8, TNF-α, IL-1β, and IL-6 (50). Another proinflammatory mediator secreted by activated platelets is beta (β)-2 microglobulin which has been shown to induce CD16+ monocytes and augment monocyte inflammatory cytokine secretion (60, 61). β-2 microglobulin is also a component of the physiologic plasma compartment. Hence, the composition and interplay of platelet granule-derived and plasma molecules within the close monocyte-platelet interaction may shape the monocyte outcome rather than single molecular mediators. For example, β2 microglobulin and platelet TGF-β exert their effects on the same monocyte receptor but through different downstream signaling cascades, leading to different (proinflammatory vs. pro-reparative) monocyte phenotypes (61).
Intriguingly, monocytes attached to platelets have increased TF expression on their surface (62), resulting in thrombin-mediated fibrin generation and clot formation (63). Under physiologic conditions, TF is expressed on monocytes in an encrypted (inactive) form but can be decrypted (activated) into its procoagulant isoform upon proinflammatory stimuli (64). Activated platelets induced rapid TF upregulation on monocytes (65). This is mediated by platelet-bound P-selectin and does not require de novo protein synthesis (65) albeit incubation of monocytes with soluble P-selectin or platelets over several hours has been shown to induce TF (F3) gene expression (66). Additionally, soluble P-selectin shed from activated platelets contributes to the formation of procoagulant MV (67), emphasizing the contribution of platelets to coagulation activation.
Exposure of circulating monocytes to platelets also affects and modulates their subsequent differentiation into tissue-resident macrophages and dendritic cells: For example, platelets - via direct P-selectin-PSGL1-mediated cell-cell interaction - induced maturation of monocytes into antigen-presenting dendritic cells (68). In a murine sepsis model, activated platelets polarized monocytes toward proinflammatory M1 macrophages (69, 70). This reprogramming was initiated as soon as platelets attached to the monocyte surface and could not be altered later on during the differentiation progress (69). Hence, depending on the underlying pathogenesis, platelets shape the monocyte effector function in circulation and define the fate of macrophage recruitment into extravascular tissues. Platelet-mediated macrophage polarization plays an important role in chronic inflammatory diseases such as atherosclerosis and will be outlined further in the upcoming section.
In summary, activated platelets are required to accelerate monocyte-driven inflammation and thrombosis, two tightly interconnected pathogenic mechanisms that profoundly impact on cardiovascular disease (CVD) and beyond.
Monocyte-platelet interaction in atherosclerosis and cardiovascular disease
Atherosclerosis is the underlying pathology in most cases of CVD including myocardial infarction (MI), stroke, and peripheral artery disease (PAD) (71). Atherosclerosis is a chronic non-resolving inflammatory disease characterized by the formation of lipid-rich calcified plaques within the arterial wall of large and medium-sized vessels due to transmigration and accumulation of activated proinflammatory immune cells consisting of macrophages and T cells. Rupture or erosion of an unstable atherosclerotic plaque results in life-threatening thromboembolic events.
While low MPA levels of around 5%–20% are a normal phenomenon observed in healthy populations (54, 72), their rise is indicative of inflammatory processes and is associated with atherothrombotic complications: Patients with PAD and CVD and/or cardiovascular risk factors, including diabetes mellitus (DM), arterial hypertension and hyperlipidemia exhibit elevated levels of circulating MPA (36, 47, 54, 73–75). Elevated levels of MPA in patients with acute MI were further increased in patients that developed in-hospital adverse events (74).
Circulating monocytes in CVD and other chronic inflammatory disorders are characterized by elevated expression of CD40, a member of the TNF receptor superfamily (76). Proinflammatory cytokines including IFN-γ, IL-1, and TNF-α induce upregulation of monocyte CD40, and monocytes attached to platelets have higher CD40 expression on their surface (34, 77). Importantly, CD40 is a receptor for CD40L which is on the surface of and a marker for activated platelets (48). CD40L-CD40 interaction does not only stabilize MPA but also enhances monocyte migration into the arterial wall (19, 78). CD40-activated macrophages then secrete inflammatory cytokines and matrix metalloproteinases, thereby contributing to plaque destabilization and rupture. In a murine atherosclerosis model, CD40 deficient mice had lower MPA and platelet-mediated leukocyte-endothelium interactions resulting in decreased plaque formation (19). In response to CD40 binding, platelet CD40L is shed from activated platelets into the circulation, which further augments platelet activation (79–81). Notably, platelet-derived CD40L contributes to P-selectin-mediated TF upregulation on monocytes (82, 83). TF expressing macrophages, as well as elevated levels of circulating TF, have been observed in patients with atherosclerosis (84, 85), metabolic cardiovascular risk factors (86–88), and in acute coronary syndrome (89–91).
CXCL4 is one of the most abundant chemokines released from alpha granules upon platelet activation (92). Importantly, CXCL4 has been detected in human carotid atherosclerotic plaques and positively correlates with lesion grade and presence of clinical symptoms (19, 93). The pathogenic role of CXCL4 in accelerating atherosclerosis is mediated, in part, by augmenting the arrest of monocytes on endothelial cells in conjunction with CCL5 (RANTES) (94).
Platelets not only induce attachment and facilitate transmigration of monocytes into the subendothelial space (95) and into atherosclerotic lesions (20) but also shape the subsequent macrophage phenotype: Monocytes exposed to platelet-derived CXCL4 differentiate into proinflammatory M4-type macrophages, which have been suggested as crucial contributors to plaque rupture in coronary artery disease (96). In vitro studies and murine models of atherosclerosis demonstrated that oxidized low-density lipoprotein (oxLDL) generated under chronic inflammatory conditions promoted platelet-monocyte interaction with subsequent monocyte extravasation and foam cell formation (16). Foam cells are lipid-laden macrophages that are a major constituent of atherosclerotic plaques (97).
Findings from our group confirm that platelets drive atherogenesis by inducing proinflammatory monocytes and macrophages: We reported that platelet competent atherogenic Ldlr−/− mice had increased monocyte surface expression of the adhesion receptor CD11b and had higher expression of inflammatory transcripts CCL2, IL6, and CD11b relative to platelet-depleted mice. Furthermore, in the presence of platelets, plaque macrophages were skewed towards a proinflammatory phenotype as defined by upregulation of SOCS3, which promoted proinflammatory cytokine production of IL-6, IL-1b, and TNF-α (20). Consistently, SOCS3 mRNA expression of whole blood correlated with platelet activity and MPA formation in a clinical dataset of women with MI and in patients with symptomatic lower extremity atherosclerosis (20).
Platelets have been shown to also activate dendritic cells (DC) via the P-selectin-PSGL1 axis, thereby contributing to atherosclerosis progression in hyperlipidemic mice (98). This is mediated by toll-like receptor 4 (TLR4) signaling pathways, leading to enhanced secretion of inflammatory cytokines, T cell communication, and adhesion and migration properties of platelet-bearing DC (98).
In addition to physical contact and secretion of granule contents, platelets can modulate monocyte and macrophage responses by shedding of PEV into the circulation. There is evidence that PEV (via P-selectin) adheres to monocytes, thereby inducing cytokine and TF production, and contributing to atherothrombosis in CVD (99). Interestingly, P-selectin-positive and fibrinogen-positive PEV were elevated in patients six months after acute MI (100). Procoagulant PEV may also contribute to monocyte-endothelial interaction, thereby promoting atherosclerosis initiation and progression (101, 102).
Genetic RNA sequencing of platelets from CVD patients allows the identification of new mediators in MPA formation and monocyte reprogramming: Platelets from patients with symptomatic PAD were enriched with myeloid-related protein-14 (MRP-14) mRNA and protein. MRP-14 augmented the expression of P-selectin, thereby enhancing MPA (21). Elevated serum levels of MRP14 were found in PAD patients with incident cardiovascular and limb events, underscoring a clinically important role for this protein (21).
Monocyte-platelet interaction in autoimmune disorders
Patients with autoimmune diseases such as Systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) are at higher risk of developing atherosclerotic and thromboembolic complications (103, 104). Again, elevated levels of MPA in patients with vasculitis, rheumatoid arthritis, and SLE highlight the tight interconnection between inflammation and atherothrombosis (58, 105–108). Monocytes attached to platelets exhibited higher levels of proinflammatory CD54, CD16, CD86, and HLA-DR in SLE patients (58). Additionally, procoagulant microvesicles shed from activated platelets have been implicated in inducing proinflammatory monocytes in RA and SLE (51, 106, 109).
In RA patients, activated platelets attaching to intermediate CD16+ monocytes contributed to elevated MPA formation and inflammatory cytokine secretion via CD147 signaling (106). High platelet activation and platelet-leukocyte aggregation were associated with enhanced TF-dependent global coagulation activation in SLE patients (108).
Monocyte-platelet interaction in infection and inflammation
In addition to a hyperreactive platelet phenotype and a heightened incidence of thromboembolic complications, elevated levels of circulating MPA were a prominent clinical finding in patients with severe COVID-19 (110–113). Platelet-monocyte interaction in COVID-19 was strongly associated with monocyte TF expression and global coagulation activation as shown by elevated fibrinogen and D-dimers (112). Interestingly, upregulation of P-selectin-dependent monocyte TF expression could be reproduced when monocytes from healthy donors were co-cultured with platelets from COVID-19 patients, once more underlining the proinflammatory effector role for platelets in COVID-19 (112).
In dengue virus infection, platelets are activated - evidenced by increased P-selectin and release of CD40L and cytokines- upon direct contact with the virus. As a consequence, elevated MPA can accelerate the monocyte inflammatory response (114–116). In an experimental cerebral malaria model, platelet-derived CXCL4 drove monocyte cytokine production in Plasmodium berghei infected mice (117).
Sepsis is a life-threatening organ dysfunction caused by a dysregulated host response to infection (118) and is characterized by platelet activation and platelet-monocyte interaction (119). Elevated levels of circulating MPA were associated with higher morbidity and mortality and correlated with thromboembolic complications in patients with sepsis (120, 121).
While the majority of studies clearly show a proinflammatory role for MPA, platelets exhibited anti-inflammatory properties on the macrophage phenotype in a murine endotoxemia model of severe septic shock (122). These seemingly divergent findings indicate that the effector role of platelets is complex and depends on the underlying clinical disease and severity. More studies are warranted to delineate and better understand the divergent functions of MPA.
Interaction of platelets with neutrophils and lymphocytes
In addition to monocyte-platelet interaction, formation of neutrophil-platelet aggregates (NPA) represents another intercellular connection linking thrombosis and inflammation. Similar to MPA, elevated circulating NPA have been observed in CVD (123), PAD (124), in COVID-19 (125), and have been linked to an enhanced risk of deep venous thrombosis (126). Platelets – via the P-selectin-PSGL1 axis – activate and induce neutrophils to release neutrophil-extracellular traps (NETs) into circulation (127). NETs consist of DNA, histones, and neutrophile-derived enzymes, most importantly myeloperoxidase, and constitute the most crucial platelet-mediated effect on neutrophils to promote thrombosis in inflammation (128). Since NPA and MPA are found to be simultaneously elevated in proinflammatory conditions (129), activated platelet induce a prothrombogenic state via both leukocyte subpopulations by enhancing their respective inflammatory response in vivo. In contrast, while the magnitude of circulating lymphocyte-platelet aggregates does not change in inflammatory, thrombotic, and atherosclerotic diseases, platelet-lymphocyte interaction has been delineated to exert a specific role in cancer: For example, platelets attenuated T cell activity in cancer patients ex vivo (130), and promoted tumor progression via suppression of CD8 T cells in murine cancer models (131).
In summary, platelets differentially modulate leukocyte activity in response to the underlying disease, with MPA and NPA being important contributors in thromboinflammatory diseases.
Can platelet inhibitors prevent MPA formation?
Targeting MPA has come into focus as an interesting therapeutic strategy. Interrupting the P-selectin-PSGL1-axis offers therapeutic potential in preventing (athero)thrombosis as shown in preclinical models (132–134). Importantly, since P-selectin is also present on activated endothelial cells (135), using an anti-P-selectin antibody allows to attenuate microvascular inflammation in addition to preventing proinflammatory platelet-monocyte interaction.
However, the synthetic P-selectin inhibitor PSI-697 failed to decrease MPA in healthy smokers (136). So far, among P-selectin inhibitors, only crizanlizumab was approved in 2019 (137) and received a conditional marketing authorization by the European Medicines Agency (EMA) in 2020 (138) for the prevention of pain crises in sickle cell disease. In this clinical setting, crizanlizumab is used to prevent sickle erythrocytes to adhere to and activate P-selectin expressing endothelial cells (139). Due to unpublished results of the STAND trial, fate is currently unclear (140).
Recently, a phase 2 clinical trial investigated the effects of crizanlizumab on patients hospitalized with moderate COVID-19 which is associated with endothelial dysfunction and vascular inflammation. Here, crizanlizumab therapy was associated with significantly decreased plasma levels of prothrombin fragments and thrombin-antithrombin complexes, both laboratory markers for coagulation activation (141). Subsequently, the effectiveness of crizanlizumab in preventing adverse clinical outcomes in hospitalized COVID-19 patients is being investigated in a large international phase 4 randomized clinical trial (142).
While PSGL1 and P-selectin inhibitors directly interfere with platelet-monocyte interaction as well as with leukocyte and erythrocyte attachment onto endothelial cells, most therapeutics that are in clinical use block platelet signaling pathways upstream of the release of alpha granule contents like P-selectin. Targeting the ADP-activated P2Y12 pathway effectively reduces MPA in several clinical studies: In patients with acute MI, atherosclerotic vascular disease, and cardiovascular risk factors including diabetes mellitus, intake of P2Y12 inhibitor therapy was associated with MPA reduction in vivo and ex vivo (73, 75, 143, 144). Importantly, the P2Y12 inhibitor ticagrelor resulted in reduced MPA and dampened myocardial inflammation (as shown by increased FGD-uptake and lower cardiac ejection fraction) in acute MI patients (75). Moreover, P2Y12 blockade reduced monocyte TF expression (145, 146) and attenuated circulating levels of IL-8, TNF-α, and CCL2 in an experimental human model of systemic inflammation (147). In patients with acute coronary syndrome, the P2Y12 inhibitor clopidogrel diminished circulating levels of proinflammatory TNF-α and C-reactive protein (144). Of note, pneumonia patients had significantly reduced plasma IL-6 under ticagrelor compared to patients taking placebo and needed less supplemental oxygen (148). In contrast, in non-critically ill patients with COVID-19, P2Y12 blockade in addition to heparin compared to heparin alone did not result in increased odds of improvement in organ support-free days (142) and – equally to COX-1 inhibitors - only had a low likelihood of improving the number of organ support-free days in critically-ill COVID-19 patients when compared to patients without platelet inhibitors (149). Accordingly, depending on the underlying inflammatory condition, the effects of platelet inhibitors on defined clinical outcomes may vary.
COX-1 inhibition with aspirin did not lower MPA assessed in vitro and following 1-week of low-dose aspirin in healthy volunteers ex vivo (54). This might be due to aspirin not directly interfering with GPCR signaling-induced alpha degranulation and P-selectin expression. While aspirin therapy was insufficient to decrease MPA in acute stroke patients, it attenuated MPA ex vivo when whole blood from healthy volunteers was stimulated with thrombin receptor activator peptide 6 (TRAP-6) (150). Different outcomes of MPA under aspirin seem to depend on the measurement methods. Aspirin did not reduce mortality linked to sepsis in healthy elderly patients (151). Importantly, in a murine model of severe bacteremia, aspirin even enhanced inflammation and was associated with higher mortality (122), indicating a potential detrimental effect in severe sepsis.
Clinical data on the effect of PAR1 inhibitors on platelet-monocyte interaction are scarce. PAR1 inhibition attenuated MPA formation in whole blood stimulated with the TXA2 analog U-46619, and consequently, monocyte attached to platelets had less surface expression of CD40 and TF (129). In another in-vitro analysis, incubation of whole blood from healthy donors with PAR1 inhibitors prevented TRAP-6 - but not PAR4 - or collagen-related peptide (CRP)-induced MPA formation, emphasizing that the efficacy of platelet inhibitors is dependent on the activation pathway (152). There is conflicting data on the effect of GPIIb/IIIa inhibitors on MPA with some studies even showing paradoxically elevated MPA levels under eptifibatide (153, 154).
In addition to blocking cell surface receptors, platelet activation can also be targeted by intracellular inhibition of phosphodiesterases (PDE). As a result, intracellular levels of cyclic adenosine monophosphate (cAMP) and/or cyclic guanosine monophosphate (cGMP) accumulate, which eventually disrupts the rearrangement of the actin cytoskeleton required for platelet activation and granule release (155). While the PDE inhibitor dipyridamole did not attenuate thrombin- or collagen-stimulated monocyte-platelet aggregates in vitro (156), patients under combined treatment with aspirin and dipyridamole following a transitory ischemic attack (TIA) had lower MPA when exhibiting dipyridamole responsiveness in a platelet function test in contrast to patients that were identified as dipyridamole non-responders (157).
Platelet GPIb and its ligand MAC-1 on leukocytes are newer potentially therapeutic targets (158). Interestingly, inhibiting this interaction resulted in impaired thrombus formation and delayed thrombosis in murine models (158). In a phase I study on healthy volunteers, GPIb-binding anfibatide, a snaclet (snake C-type lectins) purified from snake venom, showed antithrombotic efficacy without affecting haemostasis (159). Clinical trials are anticipated to investigate the clinical efficacy of GPIb and MAC-1 inhibitors in attenuating thrombosis linked to inflammation.
In conclusion, targeting platelet-monocyte interactions is likely to be beneficial to various inflammatory diseases. Among platelet inhibitors, directly targeting platelet P-selectin or preventing its release from alpha granules by upstream blockade of P2Y12 have been shown to be the most potent strategies to attenuate MPA. Further clinical studies would be welcome to evaluate the effectiveness of antiplatelet therapy-mediated MPA decrease on clinical outcomes within the different inflammatory diseases.
Potential therapies targeting MPA are summarized in (Figure 2).
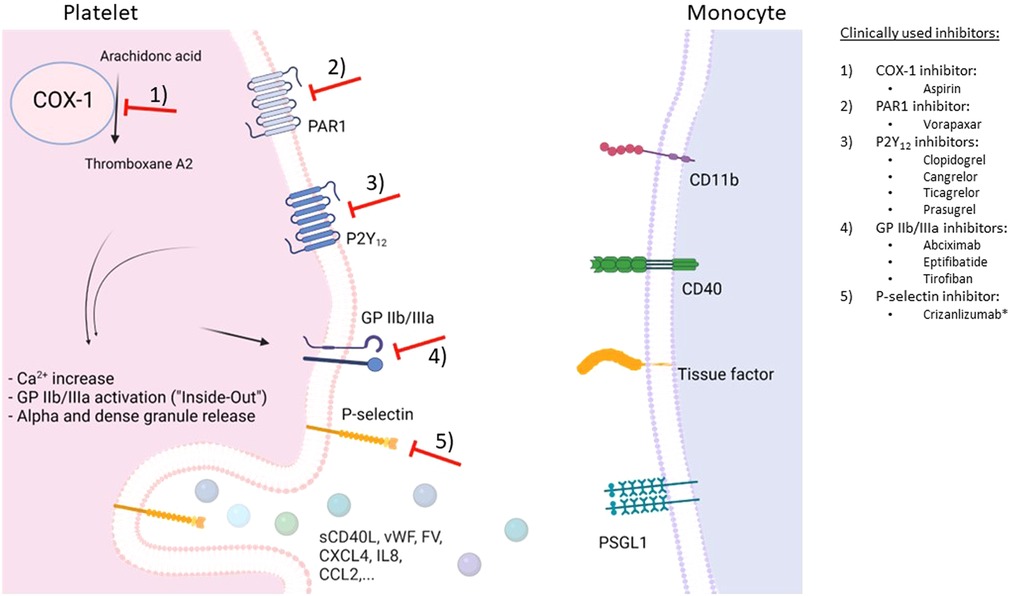
Figure 2. Schematic overview of potential therapeutic targets to prevent monocyte-platelet interactions. Depicted are G protein-coupled receptors (GPCR) associated with platelet receptors P2Y12 and PAR1 as well as GPIIb/IIIa receptors on the platelet surface, and cyclooxygenase-1 (COX-1)-mediated production of platelet-activating thromboxane A2. Following platelet activation, many interconnected pathways result in intracellular calcium increase, activation of GPIIb/IIIa, and release of alpha and dense granule contents. P-selectin (released from alpha granules) translocates to the platelet surface and binds to P-selectin glycoprotein ligand-1 (PSGL1) on the monocyte surface, resulting in monocyte-platelet aggregates (MPA). In turn, monocytes become proinflammatory and procoagulant. *has received a conditional marketing authorization by the European Medicines Agency in 2020. Figure created with BioRender.com
Measurement of monocyte-platelet interactions
Measuring monocyte-platelet aggregates in whole blood by flow cytometry is a straightforward and commonly used method to assess ex vivo markers of thromboinflammation in clinical studies. However, since there is no clear consensus on the best methodological approach, various preanalytical variables such as blood drawing (venous puncture or access via central venous catheter), sample handling (e.g., temperature, transportation, storage), and processing (centrifugation configuration) may lead to incongruent results (72, 160). When interpreting MPA data, it is therefore important to know the experimental set-up and techniques applied for analysis. The following cornerstones need to be considered when measuring MPA by flow cytometry: To avoid any artificial platelet activation, blood collection should always be performed by direct venous puncture without a tourniquet and after an initial discard. The type of anticoagulant (e.g., heparin, EDTA, or sodium citrate) in the collection tube may affect the magnitude of monocyte-platelet aggregate levels (160). Following staining with monocyte and platelet antibodies, blood can be fixed, lysed and stored at 4°C for up to 24 h (160). Monocytes are identified by forward and side scatter properties and by CD14 (and CD16) expression. For platelet labeling, antibodies binding to constitutively expressed platelet surface glycoproteins, e.g., CD41, CD42a, CD42b, or CD61, can be used. MPA are quantified as the percentage of monocytes positive of a platelet marker within the gated monocyte population. Alternatively, multispectral imaging flow cytometry combining flow cytometry and imaging data provides additional information on platelet binding to individual monocytes (161).
Conclusions
Accumulating evidence demonstrates that activated platelets induce a proinflammatory monocyte phenotype, affecting the inflammatory response in acute and chronic inflammation. Mediated by direct platelet-monocyte interactions and platelet-derived cytokines, chemokines, and shedding of procoagulant extracellular vesicles. As a consequence, monocytes release inflammatory cytokines into circulation, become procoagulant, and differentiate into proatherogenic macrophages (Figure 3). Targeting platelet effector cell properties – and hence monocyte-driven inflammation and coagulation – appears to be a promising strategy in inflammatory settings beyond CVD.
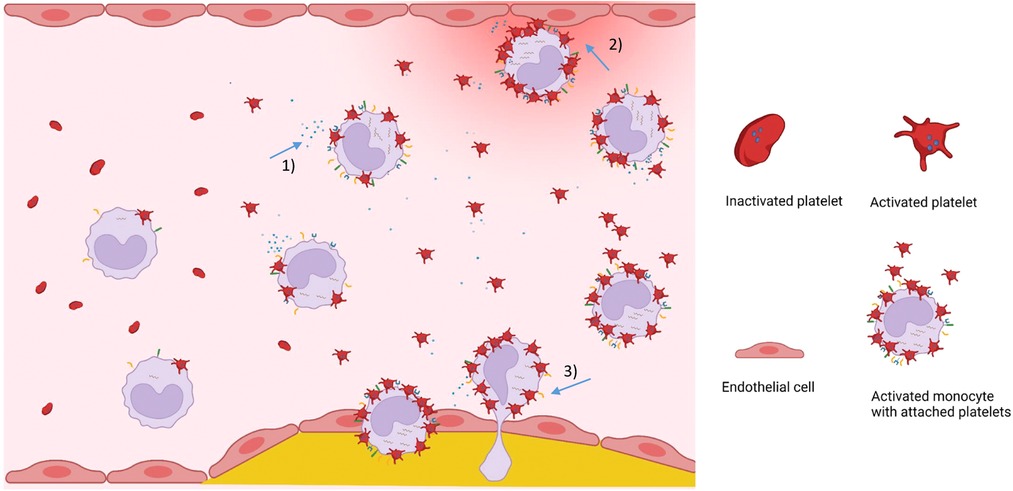
Figure 3. Visual summary on the role of MPA in cardiovascular disease. Plasma levels of circulating monocyte-platelet aggregates are increased in inflammatory diseases. Platelet-induced proinflammatory monocytes secrete inflammatory cytokines and chemokines (1), attach to and activate endothelial cells (2), show enhanced transendothelial migration potential (3), and differentiate into proinflammatory macrophages that contribute to the progression of atherosclerosis. Figure created with BioRender.com
Author contributions
CR: drafted the first version of the manuscript. TB and JB: critically revised and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This review was supported by Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) grant no. RO 6121 1/1 to C.C.R. and the National Heart, Lung and Blood Institute of the National Institutes of Health (R35HL144993 to JB).
Conflict of interest
Jeffrey Berger is PI for the NIH-funded ACTIV4a trial investigating P2Y12 inhibitors in patients hospitalized with COVID-19.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets as immune and inflammatory cells. Blood. (2014) 123(18):2759–67. doi: 10.1182/blood-2013-11-462432
2. Peyvandi F, Garagiola I, Baronciani L. Role of von Willebrand factor in the haemostasis. Blood Transfus. (2011) 9(Suppl 2):S3–S8. doi: 10.2450/2011.002S
3. Coppinger JA, Maguire PB. Insights into the platelet releasate. Curr Pharm Des. (2007) 13(26):2640–6. doi: 10.2174/138161207781662885
4. Lordan R, Tsoupras A, Zabetakis I. Platelet activation and prothrombotic mediators at the nexus of inflammation and atherosclerosis: potential role of antiplatelet agents. Blood Rev. (2021) 45:100694. doi: 10.1016/j.blre.2020.100694
5. Semple JW. Platelets play a direct role in sepsis-associated endothelial cell death. Thromb Haemost. (2008) 99(2):249. doi: 10.1160/TH08-01-0010
6. Middleton EA, Rowley JW, Campbell RA, Grissom CK, Brown SM, Beesley SJ, et al. Sepsis alters the transcriptional and translational landscape of human and murine platelets. Blood. (2019) 134(12):911–23. doi: 10.1182/blood.2019000067
7. Szilagyi B, Fejes Z, Poliska S, Pocsi M, Czimmerer Z, Patsalos A, et al. Reduced miR-26b expression in megakaryocytes and platelets contributes to elevated level of platelet activation Status in sepsis. Int J Mol Sci. (2020) 21(3):1–22. doi: 10.3390/ijms21030866
8. Poli V, Di Gioia M, Sola-Visner M, Granucci F, Frelinger AL 3rd, Michelson AD, et al. Inhibition of transcription factor NFAT activity in activated platelets enhances their aggregation and exacerbates gram-negative bacterial septicemia. Immunity. (2022) 55(2):224–36 e225. doi: 10.1016/j.immuni.2021.12.002
9. Cornelius DC, Baik CH, Travis OK, White DL, Young CM, Austin Pierce W, et al. NLRP3 Inflammasome activation in platelets in response to sepsis. Physiol Rep. (2019) 7(9):e14073. doi: 10.14814/phy2.14073
10. Habets KL, Huizinga TW, Toes RE. Platelets and autoimmunity. Eur J Clin Invest. (2013) 43(7):746–57. doi: 10.1111/eci.12101
11. Berlacher MD, Vieth JA, Heflin BC, Gay SR, Antczak AJ, Tasma BE, et al. FcgammaRIIa ligation induces platelet hypersensitivity to thrombotic stimuli. Am J Pathol. (2013) 182(1):244–54. doi: 10.1016/j.ajpath.2012.09.005
12. Cloutier N, Allaeys I, Marcoux G, Machlus KR, Mailhot B, Zufferey A, et al. Platelets release pathogenic serotonin and return to circulation after immune complex-mediated sequestration. Proc Natl Acad Sci U S A. (2018) 115(7):E1550–9. doi: 10.1073/pnas.1720553115
13. Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME, et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. (2010) 327(5965):580–3. doi: 10.1126/science.1181928
14. Garshick MS, Tawil M, Barrett TJ, Salud-Gnilo CM, Eppler M, Lee A, et al. Activated platelets induce endothelial cell inflammatory response in psoriasis via COX-1. Arterioscler Thromb Vasc Biol. (2020) 40(5):1340–51. doi: 10.1161/ATVBAHA.119.314008
15. Daub K, Seizer P, Stellos K, Kramer BF, Bigalke B, Schaller M, et al. Oxidized LDL-activated platelets induce vascular inflammation. Semin Thromb Hemost. (2010) 36(2):146–56. doi: 10.1055/s-0030-1251498
16. Badrnya S, Schrottmaier WC, Kral JB, Yaiw KC, Volf I, Schabbauer G, et al. Platelets mediate oxidized low-density lipoprotein-induced monocyte extravasation and foam cell formation. Arterioscler Thromb Vasc Biol. (2014) 34(3):571–80. doi: 10.1161/ATVBAHA.113.302919
17. Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. (2007) 13(9):1086–95. doi: 10.1038/nm1626
18. Momi S, Falcinelli E, Petito E, Ciarrocca Taranta G, Ossoli A, Gresele P. Matrix metalloproteinase-2 on activated platelets triggers endothelial PAR-1 initiating atherosclerosis. Eur Heart J. (2022) 43(6):504–14. doi: 10.1093/eurheartj/ehab631
19. Gerdes N, Seijkens T, Lievens D, Kuijpers MJ, Winkels H, Projahn D, et al. Platelet CD40 exacerbates atherosclerosis by transcellular activation of endothelial cells and leukocytes. Arterioscler Thromb Vasc Biol. (2016) 36(3):482–90. doi: 10.1161/ATVBAHA.115.307074
20. Barrett TJ, Schlegel M, Zhou F, Gorenchtein M, Bolstorff J, Moore KJ, et al. Platelet regulation of myeloid suppressor of cytokine signaling 3 accelerates atherosclerosis. Sci Transl Med. (2019) 11(517):1–14. doi: 10.1126/scitranslmed.aax0481
21. Dann R, Hadi T, Montenont E, Boytard L, Alebrahim D, Feinstein J, et al. Platelet-derived MRP-14 induces monocyte activation in patients with symptomatic peripheral artery disease. J Am Coll Cardiol. (2018) 71(1):53–65. doi: 10.1016/j.jacc.2017.10.072
22. Eicher JD, Wakabayashi Y, Vitseva O, Esa N, Yang Y, Zhu J, et al. Characterization of the platelet transcriptome by RNA sequencing in patients with acute myocardial infarction. Platelets. (2016) 27(3):230–9. doi: 10.3109/09537104.2015.1083543
23. Stokes KY, Granger DN. Platelets: a critical link between inflammation and microvascular dysfunction. J Physiol. (2012) 590(5):1023–34. doi: 10.1113/jphysiol.2011.225417
24. Schmidt RA, Morrell CN, Ling FS, Simlote P, Fernandez G, Rich DQ, et al. The platelet phenotype in patients with ST-segment elevation myocardial infarction is different from non-ST-segment elevation myocardial infarction. Transl Res. (2018) 195:1–12. doi: 10.1016/j.trsl.2017.11.006
25. Gidlof O, van der Brug M, Ohman J, Gilje P, Olde B, Wahlestedt C, et al. Platelets activated during myocardial infarction release functional miRNA, which can be taken up by endothelial cells and regulate ICAM1 expression. Blood. (2013) 121(19):3908–17. S3901–3926. doi: 10.1182/blood-2012-10-461798
26. Kleinschnitz C, Pozgajova M, Pham M, Bendszus M, Nieswandt B, Stoll G. Targeting platelets in acute experimental stroke: impact of glycoprotein Ib, VI, and IIb/IIIa blockade on infarct size, functional outcome, and intracranial bleeding. Circulation. (2007) 115(17):2323–30. doi: 10.1161/CIRCULATIONAHA.107.691279
27. Stark K, Massberg S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat Rev Cardiol. (2021) 18(9):666–82. doi: 10.1038/s41569-021-00552-1
28. Yafasova A, Fosbol EL, Schou M, Baslund B, Faurschou M, Docherty KF, et al. Long-term cardiovascular outcomes in systemic lupus erythematosus. J Am Coll Cardiol. (2021) 77(14):1717–27. doi: 10.1016/j.jacc.2021.02.029
29. Levine RL, LeClerc JR, Bailey JE, Monberg MJ, Sarwat S. Venous and arterial thromboembolism in severe sepsis. Thromb Haemost. (2008) 99(5):892–8. doi: 10.1160/TH08-01-0004
30. Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. (2006) 177(10):7303–11. doi: 10.4049/jimmunol.177.10.7303
31. Saeed S, Quintin J, Kerstens HH, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science. (2014) 345(6204):1251086. doi: 10.1126/science.1251086
32. Kapellos TS, Bonaguro L, Gemund I, Reusch N, Saglam A, Hinkley ER, et al. Human monocyte subsets and phenotypes in major chronic inflammatory diseases. Front Immunol. (2019) 10:2035. doi: 10.3389/fimmu.2019.02035
33. Zawada AM, Rogacev KS, Rotter B, Winter P, Marell RR, Fliser D, et al. SuperSAGE evidence for CD14++CD16+ monocytes as a third monocyte subset. Blood. (2011) 118(12):e50–e61. doi: 10.1182/blood-2011-01-326827
34. Kral JB, Schrottmaier WC, Salzmann M, Assinger A. Platelet interaction with innate immune cells. Transfus Med Hemother. (2016) 43(2):78–88. doi: 10.1159/000444807
35. Barnard MR, Linden MD, Frelinger AL 3rd, et al.. Effects of platelet binding on whole blood flow cytometry assays of monocyte and neutrophil procoagulant activity. J Thromb Haemost. (2005) 3(11):2563–70. doi: 10.1111/j.1538-7836.2005.01603.x
36. Furman MI, Benoit SE, Barnard MR, Valeri CR, Borbone ML, Becker RC, et al. Increased platelet reactivity and circulating monocyte-platelet aggregates in patients with stable coronary artery disease. J Am Coll Cardiol. (1998) 31(2):352–8. doi: 10.1016/S0735-1097(97)00510-X
37. Speth C, Rambach G, Wurzner R, Lass-Florl C, Kozarcanin H, Hamad OA, et al. Complement and platelets: mutual interference in the immune network. Mol Immunol. (2015) 67(1):108–18. doi: 10.1016/j.molimm.2015.03.244
38. Oleksowicz L, Mrowiec Z, Zuckerman D, Isaacs R, Dutcher J, Puszkin E. Platelet activation induced by interleukin-6: evidence for a mechanism involving arachidonic acid metabolism. Thromb Haemost. (1994) 72(2):302–8. doi: 10.1055/s-0038-1648857
39. Regnault V, de Maistre E, Carteaux JP, Gruel Y, Nguyen P, Tardy B, et al. Platelet activation induced by human antibodies to interleukin-8. Blood. (2003) 101(4):1419–21. doi: 10.1182/blood-2002-02-0620
40. Pignatelli P, De Biase L, Lenti L, Tocci G, Brunelli A, Cangemi R, et al. Tumor necrosis factor-alpha as trigger of platelet activation in patients with heart failure. Blood. (2005) 106(6):1992–4. doi: 10.1182/blood-2005-03-1247
41. Woulfe DS. Platelet G protein-coupled receptors in hemostasis and thrombosis. J Thromb Haemost. (2005) 3(10):2193–200. doi: 10.1111/j.1538-7836.2005.01338.x
42. Offermanns S. Activation of platelet function through G protein-coupled receptors. Circ Res. (2006) 99(12):1293–304. doi: 10.1161/01.RES.0000251742.71301.16
43. Schrottmaier WC, Kral JB, Badrnya S, Assinger A. Aspirin and P2Y12 inhibitors in platelet-mediated activation of neutrophils and monocytes. Thromb Haemost. (2015) 114(3):478–89. doi: 10.1160/TH14-11-0943
44. Ehlers R, Ustinov V, Chen Z, Zhang X, Rao R, Luscinskas FW, et al. Targeting platelet-leukocyte interactions: identification of the integrin mac-1 binding site for the platelet counter receptor glycoprotein Ibalpha. J Exp Med. (2003) 198(7):1077–88. doi: 10.1084/jem.20022181
45. Patko Z, Csaszar A, Acsady G, Peter K, Schwarz M. Roles of mac-1 and glycoprotein IIb/IIIa integrins in leukocyte-platelet aggregate formation: stabilization by mac-1 and inhibition by GpIIb/IIIa blockers. Platelets. (2012) 23(5):368–75. doi: 10.3109/09537104.2011.625098
46. Ostrovsky L, King AJ, Bond S, Mitchell D, Lorant DE, Zimmerman GA, et al. A juxtacrine mechanism for neutrophil adhesion on platelets involves platelet-activating factor and a selectin-dependent activation process. Blood. (1998) 91(8):3028–36. doi: 10.1182/blood.V91.8.3028.3028_3028_3036
47. Harding SA, Sommerfield AJ, Sarma J, Twomey PJ, Newby DE, Frier BM, et al. Increased CD40 ligand and platelet-monocyte aggregates in patients with type 1 diabetes mellitus. Atherosclerosis. (2004) 176(2):321–5. doi: 10.1016/j.atherosclerosis.2004.05.008
48. Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. (1998) 391(6667):591–4. doi: 10.1038/35393
49. Woller G, Brandt E, Mittelstadt J, Rybakowski C, Petersen F. Platelet factor 4/CXCL4-stimulated human monocytes induce apoptosis in endothelial cells by the release of oxygen radicals. J Leukoc Biol. (2008) 83(4):936–45. doi: 10.1189/jlb.0907592
50. Suzuki J, Hamada E, Shodai T, Kamoshida G, Kudo S, Itoh S, et al. Cytokine secretion from human monocytes potentiated by P-selectin-mediated cell adhesion. Int Arch Allergy Immunol. (2013) 160(2):152–60. doi: 10.1159/000339857
51. Villar-Vesga J, Grajales C, Burbano C, Vanegas-Garcia A, Munoz-Vahos CH, Vasquez G, et al. Platelet-derived microparticles generated in vitro resemble circulating vesicles of patients with rheumatoid arthritis and activate monocytes. Cell Immunol. (2019) 336:1–11. doi: 10.1016/j.cellimm.2018.12.002
52. Chimen M, Evryviadou A, Box CL, Harrison MJ, Hazeldine J, Dib LH, et al. Appropriation of GPIbalpha from platelet-derived extracellular vesicles supports monocyte recruitment in systemic inflammation. Haematologica. (2020) 105(5):1248–61. doi: 10.3324/haematol.2018.215145
53. Poon KS, Palanisamy K, Chang SS, Sun KT, Chen KB, Li PC, et al. Plasma exosomal miR-223 expression regulates inflammatory responses during cardiac surgery with cardiopulmonary bypass. Sci Rep. (2017) 7(1):10807. doi: 10.1038/s41598-017-09709-w
54. Allen N, Barrett TJ, Guo Y, Nardi M, Ramkhelawon B, Rockman CB, et al. Circulating monocyte-platelet aggregates are a robust marker of platelet activity in cardiovascular disease. Atherosclerosis. (2019) 282:11–8. doi: 10.1016/j.atherosclerosis.2018.12.029
55. Lee SJ, Yoon BR, Kim HY, Yoo SJ, Kang SW, Lee WW. Activated platelets convert CD14(+)CD16(-) into CD14(+)CD16(+) monocytes with enhanced FcgammaR-mediated phagocytosis and Skewed M2 polarization. Front Immunol. (2020) 11:611133. doi: 10.3389/fimmu.2020.611133
56. Phillips JH, Chang CW, Lanier LL. Platelet-induced expression of Fc gamma RIII (CD16) on human monocytes. Eur J Immunol. (1991) 21(4):895–9. doi: 10.1002/eji.1830210406
57. Passacquale G, Vamadevan P, Pereira L, Hamid C, Corrigall V, Ferro A. Monocyte-platelet interaction induces a pro-inflammatory phenotype in circulating monocytes. PLoS One. (2011) 6(10):e25595. doi: 10.1371/journal.pone.0025595
58. Mariscal A, Zamora C, Magallares B, Salman-Monte TC, Ortiz MA, Diaz-Torne C, et al. Phenotypic and functional consequences of PLT binding to monocytes and its association with clinical features in SLE. Int J Mol Sci. (2021) 22(9):1–17. doi: 10.3390/ijms22094719
59. Schrottmaier WC, Mussbacher M, Salzmann M, Assinger A. Platelet-leukocyte interplay during vascular disease. Atherosclerosis. (2020) 307:109–20. doi: 10.1016/j.atherosclerosis.2020.04.018
60. Hilt ZT, Pariser DN, Ture SK, Mohan A, Quijada P, Asante AA, et al. Platelet-derived beta2M regulates monocyte inflammatory responses. JCI Insight. (2019) 4(5):1–21. doi: 10.1172/jci.insight.122943
61. Hilt ZT, Maurya P, Tesoro L, Pariser DN, Ture SK, Cleary SJ, et al. Beta2m signals monocytes through non-canonical TGFbeta receptor signal transduction. Circ Res. (2021) 128(5):655–69. doi: 10.1161/CIRCRESAHA.120.317119
62. Chu AJ. Tissue factor, blood coagulation, and beyond: an overview. Int J Inflam. (2011) 2011:367284. doi: 10.4061/2011/367284
63. Mackman N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler Thromb Vasc Biol. (2004) 24(6):1015–22. doi: 10.1161/01.ATV.0000130465.23430.74
64. Langer F, Ruf W. Synergies of phosphatidylserine and protein disulfide isomerase in tissue factor activation. Thromb Haemost. (2014) 111(4):590–7. doi: 10.1160/TH13-09-0802
65. Ivanov II, Apta BHR, Bonna AM, Harper MT. Platelet P-selectin triggers rapid surface exposure of tissue factor in monocytes. Sci Rep. (2019) 9(1):13397. doi: 10.1038/s41598-019-49635-7
66. Celi A, Pellegrini G, Lorenzet R, De Blasi A, Ready N, Furie BC, et al. P-selectin induces the expression of tissue factor on monocytes. Proc Natl Acad Sci U S A. (1994) 91(19):8767–71. doi: 10.1073/pnas.91.19.8767
67. Andre P, Hartwell D, Hrachovinova I, Saffaripour S, Wagner DD. Pro-coagulant state resulting from high levels of soluble P-selectin in blood. Proc Natl Acad Sci U S A. (2000) 97(25):13835–40. doi: 10.1073/pnas.250475997
68. Han P, Hanlon D, Arshad N, Lee JS, Tatsuno K, Robinson E, et al. Platelet P-selectin initiates cross-presentation and dendritic cell differentiation in blood monocytes. Sci Adv. (2020) 6(11):eaaz1580. doi: 10.1126/sciadv.aaz1580
69. Carestia A, Mena HA, Olexen CM, Ortiz Wilczynski JM, Negrotto S, Errasti AE, et al. Platelets promote macrophage polarization toward pro-inflammatory phenotype and increase survival of septic mice. Cell Rep. (2019) 28(4):896–908 e895. doi: 10.1016/j.celrep.2019.06.062
70. Singhal R, Chawla S, Batra H, Gupta S, Ojha A, Rathore DK, et al. Engulfment of Hb-activated platelets differentiates monocytes into pro-inflammatory macrophages in PNH patients. Eur J Immunol. (2018) 48(8):1285–94. doi: 10.1002/eji.201747449
71. Engel D, Seijkens T, Poggi M, Sanati M, Thevissen L, Beckers L, et al. The immunobiology of CD154-CD40-TRAF interactions in atherosclerosis. Semin Immunol. (2009) 21(5):308–12. doi: 10.1016/j.smim.2009.06.004
72. Li N, Goodall AH, Hjemdahl P. A sensitive flow cytometric assay for circulating platelet-leucocyte aggregates. Br J Haematol. (1997) 99(4):808–16. doi: 10.1046/j.1365-2141.1997.4993305.x
73. Klinkhardt U, Bauersachs R, Adams J, Graff J, Lindhoff-Last E, Harder S. Clopidogrel but not aspirin reduces P-selectin expression and formation of platelet-leukocyte aggregates in patients with atherosclerotic vascular disease. Clin Pharmacol Ther. (2003) 73(3):232–41. doi: 10.1067/mcp.2003.13
74. Loguinova M, Pinegina N, Kogan V, Vagida M, Arakelyan A, Shpektor A, et al. Monocytes of different subsets in complexes with platelets in patients with myocardial infarction. Thromb Haemost. (2018) 118(11):1969–81. doi: 10.1055/s-0038-1673342
75. Kossmann H, Rischpler C, Hanus F, Nekolla SG, Kunze KP, Gotze K, et al. Monocyte-platelet aggregates affect local inflammation in patients with acute myocardial infarction. Int J Cardiol. (2019) 287:7–12. doi: 10.1016/j.ijcard.2019.04.009
76. Jansen MF, Hollander MR, van Royen N, Horrevoets AJ, Lutgens E. CD40 In coronary artery disease: a matter of macrophages? Basic Res Cardiol. (2016) 111(4):38. doi: 10.1007/s00395-016-0554-5
77. Schonbeck U, Libby P. The CD40/CD154 receptor/ligand dyad. Cell Mol Life Sci. (2001) 58(1):4–43. doi: 10.1007/PL00000776
78. Kotowicz K, Dixon GL, Klein NJ, Peters MJ, Callard RE. Biological function of CD40 on human endothelial cells: costimulation with CD40 ligand and interleukin-4 selectively induces expression of vascular cell adhesion molecule-1 and P-selectin resulting in preferential adhesion of lymphocytes. Immunology. (2000) 100(4):441–8. doi: 10.1046/j.1365-2567.2000.00061.x
79. Henn V, Steinbach S, Buchner K, Presek P, Kroczek RA. The inflammatory action of CD40 ligand (CD154) expressed on activated human platelets is temporally limited by coexpressed CD40. Blood. (2001) 98(4):1047–54. doi: 10.1182/blood.V98.4.1047
80. Aukrust P, Muller F, Ueland T, Berget T, Aaser E, Brunsvig A, et al. Enhanced levels of soluble and membrane-bound CD40 ligand in patients with unstable angina. Possible reflection of T lymphocyte and platelet involvement in the pathogenesis of acute coronary syndromes. Circulation. (1999) 100(6):614–20. doi: 10.1161/01.CIR.100.6.614
81. Prasad KS, Andre P, Yan Y, Phillips DR. The platelet CD40l/GP IIb-IIIa axis in atherothrombotic disease. Curr Opin Hematol. (2003) 10(5):356–61. doi: 10.1097/00062752-200309000-00006
82. Lindmark E, Tenno T, Siegbahn A. Role of platelet P-selectin and CD40 ligand in the induction of monocytic tissue factor expression. Arterioscler Thromb Vasc Biol. (2000) 20(10):2322–8. doi: 10.1161/01.ATV.20.10.2322
83. Mach F, Schonbeck U, Bonnefoy JY, Pober JS, Libby P. Activation of monocyte/macrophage functions related to acute atheroma complication by ligation of CD40: induction of collagenase, stromelysin, and tissue factor. Circulation. (1997) 96(2):396–9. doi: 10.1161/01.CIR.96.2.396
84. Wilcox JN, Smith KM, Schwartz SM, Gordon D. Localization of tissue factor in the normal vessel wall and in the atherosclerotic plaque. Proc Natl Acad Sci U S A. (1989) 86(8):2839–43. doi: 10.1073/pnas.86.8.2839
85. Annex BH, Denning SM, Channon KM, Sketch MH Jr, Stack RS, Morrissey JH, et al. Differential expression of tissue factor protein in directional atherectomy specimens from patients with stable and unstable coronary syndromes. Circulation. (1995) 91(3):619–22. doi: 10.1161/01.CIR.91.3.619
86. Singh A, Boden G, Homko C, Gunawardana J, Rao AK. Whole-blood tissue factor procoagulant activity is elevated in type 1 diabetes: effects of hyperglycemia and hyperinsulinemia. Diabetes Care. (2012) 35(6):1322–7. doi: 10.2337/dc11-2114
87. Ayer JG, Song C, Steinbeck K, Celermajer DS, Ben Freedman S. Increased tissue factor activity in monocytes from obese young adults. Clin Exp Pharmacol Physiol. (2010) 37(11):1049–54. doi: 10.1111/j.1440-1681.2010.05430.x
88. Ferro D, Basili S, Alessandri C, Mantovani B, Cordova C, Violi F. Simvastatin reduces monocyte-tissue-factor expression type IIa hypercholesterolaemia. Lancet. (1997) 350(9086):1222. doi: 10.1016/S0140-6736(05)63452-6
89. Steffel J, Luscher TF, Tanner FC. Tissue factor in cardiovascular diseases: molecular mechanisms and clinical implications. Circulation. (2006) 113(5):722–31. doi: 10.1161/CIRCULATIONAHA.105.567297
90. Suefuji H, Ogawa H, Yasue H, Kaikita K, Soejima H, Motoyama T, et al. Increased plasma tissue factor levels in acute myocardial infarction. Am Heart J. (1997) 134(2 Pt 1):253–9. doi: 10.1016/S0002-8703(97)70132-7
91. Chiva-Blanch G, Laake K, Myhre P, Bratseth V, Arnesen H, Solheim S, et al. Platelet-, monocyte-derived and tissue factor-carrying circulating microparticles are related to acute myocardial infarction severity. PLoS One. (2017) 12(2):e0172558. doi: 10.1371/journal.pone.0172558
92. Brandt E, Petersen F, Ludwig A, Ehlert JE, Bock L, Flad HD. The beta-thromboglobulins and platelet factor 4: blood platelet-derived CXC chemokines with divergent roles in early neutrophil regulation. J Leukoc Biol. (2000) 67(4):471–8. doi: 10.1002/jlb.67.4.471
93. Pitsilos S, Hunt J, Mohler ER, Prabhakar AM, Poncz M, Dawicki J, et al. Platelet factor 4 localization in carotid atherosclerotic plaques: correlation with clinical parameters. Thromb Haemost. (2003) 90(6):1112–20. doi: 10.1160/TH03-02-0069
94. von Hundelshausen P, Koenen RR, Sack M, Mause SF, Adriaens W, Proudfoot AE, et al. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood. (2005) 105(3):924–30. doi: 10.1182/blood-2004-06-2475
95. van Gils JM, da Costa Martins PA, Mol A, Hordijk PL, Zwaginga JJ. Transendothelial migration drives dissociation of plateletmonocyte complexes. Thromb Haemost. (2008) 100(2):271–9. doi: 10.1160/TH08-03-0165
96. Gleissner CA. Macrophage phenotype modulation by CXCL4 in atherosclerosis. Front Physiol. (2012) 3:1. doi: 10.3389/fphys.2012.00001
97. Choudhury RP, Lee JM, Greaves DR. Mechanisms of disease: macrophage-derived foam cells emerging as therapeutic targets in atherosclerosis. Nat Clin Pract Cardiovasc Med. (2005) 2(6):309–15. doi: 10.1038/ncpcardio0195
98. Ye Z, Zhong L, Zhu S, Wang Y, Zheng J, Wang S, et al. The P-selectin and PSGL-1 axis accelerates atherosclerosis via activation of dendritic cells by the TLR4 signaling pathway. Cell Death Dis. (2019) 10(7):507. doi: 10.1038/s41419-019-1736-5
99. van Es N, Bleker S, Sturk A, Nieuwland R. Clinical significance of tissue factor-exposing microparticles in arterial and venous thrombosis. Semin Thromb Hemost. (2015) 41(7):718–27. doi: 10.1055/s-0035-1556047
100. Gasecka A, Rogula S, Eyileten C, Postula M, Jaguszewski MJ, Kochman J, et al. Role of P2Y receptors in platelet extracellular vesicle release. Int J Mol Sci. (2020) 21(17):1–14. doi: 10.3390/ijms21176065
101. Boulanger CM, Loyer X, Rautou PE, Amabile N. Extracellular vesicles in coronary artery disease. Nat Rev Cardiol. (2017) 14(5):259–72. doi: 10.1038/nrcardio.2017.7
102. Tan KT, Lip GY. The potential role of platelet microparticles in atherosclerosis. Thromb Haemost. (2005) 94(3):488–92. doi: 10.1160/TH05-03-0201
103. Katz G, Smilowitz NR, Blazer A, Clancy R, Buyon JP, Berger JS. Systemic lupus erythematosus and increased prevalence of atherosclerotic cardiovascular disease in hospitalized patients. Mayo Clin Proc. (2019) 94(8):1436–43. doi: 10.1016/j.mayocp.2019.01.044
104. Crowson CS, Liao KP, Davis JM 3rd, Solomon DH, Matteson EL, Knutson KL, et al.. Rheumatoid arthritis and cardiovascular disease. Am Heart J. (2013) 166(4):622–8 e621. doi: 10.1016/j.ahj.2013.07.010
105. Miao D, Li DY, Chen M, Zhao MH. Platelets are activated in ANCA-associated vasculitis via thrombin-PARs pathway and can activate the alternative complement pathway. Arthritis Res Ther. (2017) 19(1):252. doi: 10.1186/s13075-017-1458-y
106. Rong MY, Wang CH, Wu ZB, Zeng W, Zheng ZH, Han Q, et al. Platelets induce a proinflammatory phenotype in monocytes via the CD147 pathway in rheumatoid arthritis. Arthritis Res Ther. (2014) 16(6):478. doi: 10.1186/s13075-014-0478-0
107. Joseph JE, Harrison P, Mackie IJ, Isenberg DA, Machin SJ. Increased circulating platelet-leucocyte complexes and platelet activation in patients with antiphospholipid syndrome, systemic lupus erythematosus and rheumatoid arthritis. Br J Haematol. (2001) 115(2):451–9. doi: 10.1046/j.1365-2141.2001.03101.x
108. Monzon Manzano E, Fernandez-Bello I, Justo Sanz R, Robles Marhuenda A, Lopez-Longo FJ, Acuna P, et al. Insights into the procoagulant profile of patients with systemic lupus erythematosus without antiphospholipid antibodies. J Clin Med. (2020) 9(10):1–17. doi: 10.3390/jcm9103297
109. Burbano C, Villar-Vesga J, Orejuela J, Munoz C, Vanegas A, Vasquez G, et al. Potential involvement of platelet-derived microparticles and microparticles forming immune complexes during monocyte activation in patients with systemic lupus erythematosus. Front Immunol. (2018) 9:322. doi: 10.3389/fimmu.2018.00322
110. Barrett TJ, Cornwell M, Myndzar K, Rolling CC, Xia Y, Drenkova K, et al. Platelets amplify endotheliopathy in COVID-19. Sci Adv. (2021) 7(37):eabh2434. doi: 10.1126/sciadv.abh2434
111. Barrett TJ, Bilaloglu S, Cornwell M, Burgess HM, Virginio VW, Drenkova K, et al. Platelets contribute to disease severity in COVID-19. J Thromb Haemost. (2021) 19(12):3139–53. doi: 10.1111/jth.15534
112. Hottz ED, Azevedo-Quintanilha IG, Palhinha L, Teixeira L, Barreto EA, Pao CRR, et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood. (2020) 136(11):1330–41. doi: 10.1182/blood.2020007252
113. Chao Y, Rebetz J, Blackberg A, Hovold G, Sunnerhagen T, Rasmussen M, et al. Distinct phenotypes of platelet, monocyte, and neutrophil activation occur during the acute and convalescent phase of COVID-19. Platelets. (2021) 32(8):1092–102. doi: 10.1080/09537104.2021.1921721
114. Chao CH, Wu WC, Lai YC, Tsai PJ, Perng GC, Lin YS, et al. Dengue virus nonstructural protein 1 activates platelets via toll-like receptor 4, leading to thrombocytopenia and hemorrhage. PLoS Pathog. (2019) 15(4):e1007625. doi: 10.1371/journal.ppat.1007625
115. Nunez-Avellaneda D, Mosso-Pani MA, Sanchez-Torres LE, Castro-Mussot ME, Corona-de la Pena NA, Salazar MI. Dengue virus induces the release of sCD40l and changes in levels of membranal CD42b and CD40l molecules in human platelets. Viruses. (2018) 10(7):1–13. doi: 10.3390/v10070357
116. Hottz ED, Medeiros-de-Moraes IM, Vieira-de-Abreu A, de Assis EF, Vals-de-Souza R, Castro-Faria-Neto HC, et al. Platelet activation and apoptosis modulate monocyte inflammatory responses in dengue. J Immunol. (2014) 193(4):1864–72. doi: 10.4049/jimmunol.1400091
117. Srivastava K, Field DJ, Aggrey A, Yamakuchi M, Morrell CN. Platelet factor 4 regulation of monocyte KLF4 in experimental cerebral malaria. PLoS One. (2010) 5(5):e10413. doi: 10.1371/journal.pone.0010413
118. Fernando SM, Rochwerg B, Seely AJE. Clinical implications of the third international consensus definitions for sepsis and septic shock (sepsis-3). CMAJ. (2018) 190(36):E1058–9. doi: 10.1503/cmaj.170149
119. Margraf A, Zarbock A. Platelets in inflammation and resolution. J Immunol. (2019) 203(9):2357–67. doi: 10.4049/jimmunol.1900899
120. Vardon-Bounes F, Ruiz S, Gratacap MP, Garcia C, Payrastre B, Minville V. Platelets are critical key players in sepsis. Int J Mol Sci. (2019) 20(14):1–13. doi: 10.3390/ijms20143494
121. Fu G, Deng M, Neal MD, Billiar TR, Scott MJ. Platelet-monocyte aggregates: understanding mechanisms and functions in sepsis. Shock. (2021) 55(2):156–66. doi: 10.1097/SHK.0000000000001619
122. Xiang B, Zhang G, Guo L, Li XA, Morris AJ, Daugherty A, et al. Platelets protect from septic shock by inhibiting macrophage-dependent inflammation via the cyclooxygenase 1 signalling pathway. Nat Commun. (2013) 4:2657. doi: 10.1038/ncomms3657
123. Nijm J, Wikby A, Tompa A, Olsson AG, Jonasson L. Circulating levels of proinflammatory cytokines and neutrophil-platelet aggregates in patients with coronary artery disease. Am J Cardiol. (2005) 95(4):452–6. doi: 10.1016/j.amjcard.2004.10.009
124. Dopheide JF, Rubrech J, Trumpp A, Geissler P, Zeller GC, Bock K, et al. Leukocyte-platelet aggregates-a phenotypic characterization of different stages of peripheral arterial disease. Platelets. (2016) 27(7):658–67. doi: 10.3109/09537104.2016.1153619
125. Le Joncour A, Biard L, Vautier M, Bugaut H, Mekinian A, Maalouf G, et al. Neutrophil-platelet and monocyte-platelet aggregates in COVID-19 patients. Thromb Haemost. (2020) 120(12):1733–5. doi: 10.1055/s-0040-1718732
126. Zhou J, Xu E, Shao K, Shen W, Gu Y, Li M, et al. Circulating platelet-neutrophil aggregates as risk factor for deep venous thrombosis. Clin Chem Lab Med. (2019) 57(5):707–15. doi: 10.1515/cclm-2018-0909
127. Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood. (2015) 126(2):242–6. doi: 10.1182/blood-2015-01-624023
128. Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos DC, Metallidis S, Rafailidis P, et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Invest. (2020) 130(11):6151–7. doi: 10.1172/JCI141374
129. Rolling CC, Sowa MA, Wang TT, Cornwell M, Myndzar K, Schwartz T, et al. P2y12 inhibition suppresses proinflammatory platelet-monocyte interactions. Thromb Haemost. (2023) 123(2):231–44. doi: 10.1055/s-0042-1758655
130. Lutz MS, Klimovich B, Maurer S, Heitmann JS, Marklin M, Zekri L, et al. Platelets subvert antitumor efficacy of T cell-recruiting bispecific antibodies. J Immunother Cancer. (2022) 10(2):1–8. doi: 10.1136/jitc-2021-003655
131. Tyagi T, Jain K, Yarovinsky TO, Chiorazzi M, Du J, Castro C, et al. Platelet-derived TLT-1 promotes tumor progression by suppressing CD8+ T cells. J Exp Med. (2023) 220(1):1–20. doi: 10.1084/jem.20212218
132. Myers DD Jr, Wrobleski SK, Longo C, Bedard PW, Kaila N, Shaw GD, et al. Resolution of venous thrombosis using a novel oral small-molecule inhibitor of P-selectin (PSI-697) without anticoagulation. Thromb Haemost. (2007) 97(3):400–7. doi: 10.1160/TH06-11-0658
133. Myers DD Jr, Henke PK, Bedard PW, Wrobleski SK, Kaila N, Shaw G, et al. Treatment with an oral small molecule inhibitor of P-selectin (PSI-697) decreases vein wall injury in a rat stenosis model of venous thrombosis. J Vasc Surg. (2006) 44(3):625–32. doi: 10.1016/j.jvs.2006.05.021
134. Wong DJ, Park DD, Park S, Haller CA, Chen J, Dai E, et al. A PSGL-1 glycomimetic reduces thrombus burden without affecting hemostasis. Blood. (2021) 138(13):1182–93. doi: 10.1182/blood.2020009428
135. Gotsch U, Jager U, Dominis M, Vestweber D. Expression of P-selectin on endothelial cells is upregulated by LPS and TNF-alpha in vivo. Cell Adhes Commun. (1994) 2(1):7–14. doi: 10.3109/15419069409014198
136. Japp AG, Chelliah R, Tattersall L, Lang NN, Meng X, Weisel K, et al. Effect of PSI-697, a novel P-selectin inhibitor, on platelet-monocyte aggregate formation in humans. J Am Heart Assoc. (2013) 2(1):e006007. doi: 10.1161/JAHA.112.006007
137. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-crizanlizumab-tmca-sickle-cell-disease; (2019).
138. Delgado J, Voltz C, Stain M, Lapvetelainen T, Urach S, Lahteenvuo J, et al. The European medicines agency review of crizanlizumab for the prevention of recurrent vaso-occlusive crises in patients with sickle cell disease. Hemasphere. (2021) 5(7):e604. doi: 10.1097/HS9.0000000000000604
139. Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. (2017) 376(5):429–39. doi: 10.1056/NEJMoa1611770
140. https://www.novartis.com/news/novartis-provides-update-phase-iii-stand-trial-assessing-crizanlizumab; (2023).
141. Leucker TM, Osburn WO, Reventun P, et al. Effect of crizanlizumab, a P-selectin inhibitor, in COVID-19: a placebo-controlled. Randomized Trial. JACC Basic Transl Sci. (2021) 6(12):935–45. doi: 10.1016/j.jacbts.2021.09.013
142. Berger JS, Kornblith LZ, Gong MN, Reynolds HR, Cushman M, Cheng Y, et al. Effect of P2Y12 inhibitors on survival free of organ support among non-critically Ill hospitalized patients with COVID-19: a randomized clinical trial. JAMA. (2022) 327(3):227–36. doi: 10.1001/jama.2021.23605
143. Layne K, Di Giosia P, Ferro A, Passacquale G. Anti-platelet drugs attenuate the expansion of circulating CD14highCD16+ monocytes under pro-inflammatory conditions. Cardiovasc Res. (2016) 111(1):26–33. doi: 10.1093/cvr/cvw089
144. Thomas MR, Storey RF. Effect of P2Y12 inhibitors on inflammation and immunity. Thromb Haemost. (2015) 114(3):490–7. doi: 10.1160/TH14-12-1068
145. Evangelista V, Manarini S, Dell'Elba G, et al. Clopidogrel inhibits platelet-leukocyte adhesion and platelet-dependent leukocyte activation. Thromb Haemost. (2005) 94(3):568–77. doi: 10.1160/TH05-01-0020
146. Frelinger AL 3rd, Jakubowski JA, Li Y, Barnard MR, Linden MD, Tarnow I, et al. The active metabolite of prasugrel inhibits adenosine diphosphate- and collagen-stimulated platelet procoagulant activities. J Thromb Haemost. (2008) 6(2):359–65. doi: 10.1111/j.1538-7836.2007.02838.x
147. Thomas MR, Outteridge SN, Ajjan RA, Phoenix F, Sangha GK, Faulkner RE, et al. Platelet P2Y12 inhibitors reduce systemic inflammation and its prothrombotic effects in an experimental human model. Arterioscler Thromb Vasc Biol. (2015) 35(12):2562–70. doi: 10.1161/ATVBAHA.115.306528
148. Sexton TR, Zhang G, Macaulay TE, Callahan LA, Charnigo R, Vsevolozhskaya OA, et al. Ticagrelor reduces thromboinflammatory markers in patients with pneumonia. JACC Basic Transl Sci. (2018) 3(4):435–49. doi: 10.1016/j.jacbts.2018.05.005
149. Investigators R-CWCftR-C, Bradbury CA, Lawler PR, Stanworth SJ, McVerry BJ, McQuilten Z, et al. Effect of antiplatelet therapy on survival and organ support-free days in critically ill patients with COVID-19: a randomized clinical trial. JAMA. (2022) 327(13):1247–59. doi: 10.1001/jama.2022.2910
150. Lukasik M, Dworacki G, Michalak S, Kufel-Grabowska J, Golanski J, Watala C, et al. Aspirin treatment influences platelet-related inflammatory biomarkers in healthy individuals but not in acute stroke patients. Thromb Res. (2011) 128(5):e73–e80. doi: 10.1016/j.thromres.2011.06.016
151. Eisen DP, Leder K, Woods RL, Lockery JE, McGuinness SL, Wolfe R, et al. Effect of aspirin on deaths associated with sepsis in healthy older people (ANTISEPSIS): a randomised, double-blind, placebo-controlled primary prevention trial. Lancet Respir Med. (2021) 9(2):186–95. doi: 10.1016/S2213-2600(20)30411-2
152. Rigg RA, Healy LD, Chu TT, Ngo ATP, Mitrugno A, Zilberman-Rudenko J, et al. Protease-activated receptor 4 activity promotes platelet granule release and platelet-leukocyte interactions. Platelets. (2019) 30(1):126–35. doi: 10.1080/09537104.2017.1406076
153. Schneider DJ, Taatjes DJ, Sobel BE. Paradoxical inhibition of fibrinogen binding and potentiation of alpha-granule release by specific types of inhibitors of glycoprotein IIb-IIIa. Cardiovasc Res. (2000) 45(2):437–46. doi: 10.1016/S0008-6363(99)00253-9
154. Holmes MB, Sobel BE, Schneider DJ. Variable responses to inhibition of fibrinogen binding induced by tirofiban and eptifibatide in blood from healthy subjects. Am J Cardiol. (1999) 84(2):203–7. doi: 10.1016/S0002-9149(99)00235-0
155. Rondina MT, Weyrich AS. Targeting phosphodiesterases in anti-platelet therapy. Handb Exp Pharmacol. (2012) 210:225–38. doi: 10.1007/978-3-642-29423-5_9
156. Weyrich AS, Denis MM, Kuhlmann-Eyre JR, et al. Dipyridamole selectively inhibits inflammatory gene expression in platelet-monocyte aggregates. Circulation. (2005) 111(5):633–42. doi: 10.1161/01.CIR.0000154607.90506.45
157. Tobin WO, Kinsella JA, Collins DR, Coughlan T, O'Neill D, Egan B, et al. Enhanced ex vivo inhibition of platelet function following addition of dipyridamole to aspirin after transient ischaemic attack or ischaemic stroke: first results from the TRinity AntiPlatelet responsiveness (TrAP) study. Br J Haematol. (2011) 152(5):640–7. doi: 10.1111/j.1365-2141.2010.08539.x
158. Wang Y, Gao H, Shi C, Erhardt PW, Pavlovsky A, D AS, et al. Leukocyte integrin Mac-1 regulates thrombosis via interaction with platelet GPIbalpha. Nat Commun. (2017) 8:15559. doi: 10.1038/ncomms15559
159. Li BX, Dai X, Xu XR, Adili R, Neves MAD, Lei X, et al. In vitro assessment and phase I randomized clinical trial of anfibatide a snake venom derived anti-thrombotic agent targeting human platelet GPIbalpha. Sci Rep. (2021) 11(1):11663. doi: 10.1038/s41598-021-91165-8
160. Harding SA, Din JN, Sarma J, Jessop A, Weatherall M, Fox KA, et al. Flow cytometric analysis of circulating platelet-monocyte aggregates in whole blood: methodological considerations. Thromb Haemost. (2007) 98(2):451–6. doi: 10.1160/TH06-11-0654
Keywords: monocyte-platelet aggregates, thromboinflammation, antiplatelet therapy, P2Y12 inhibitor, inflammatory diseases, atherosclerosis
Citation: Rolling CC, Barrett TJ and Berger JS (2023) Platelet-monocyte aggregates: molecular mediators of thromboinflammation. Front. Cardiovasc. Med. 10:960398. doi: 10.3389/fcvm.2023.960398
Received: 2 June 2022; Accepted: 24 April 2023;
Published: 15 May 2023.
Edited by:
Masanori Aikawa, Harvard Medical School, United StatesReviewed by:
Matthew Dean Linden, University of Western Australia, AustraliaKatsuya Hirano, Kagawa University, Japan
Ole Weis Bjerrum, Odense University Hospital, Denmark
James David McFadyen, Baker Heart and Diabetes Institute, Australia
© 2023 Rolling, Barrett and Berger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christina C. Rolling Yy5yb2xsaW5nQHVrZS5kZQ== Jeffrey S. Berger SmVmZnJleS5CZXJnZXJAbnl1bGFuZ29uZS5vcmc=