
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Cardiovasc. Med. , 19 December 2023
Sec. Cardiac Rhythmology
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1328802
 Hans Ebbinghaus1*
Hans Ebbinghaus1* Laura Ueberham2Daniela Husser-Bollmann1
Laura Ueberham2Daniela Husser-Bollmann1 Andreas Bollmann1Ingo Paetsch1Cosima Jahnke1
Andreas Bollmann1Ingo Paetsch1Cosima Jahnke1 Ulrich Laufs2Borislav Dinov3
Ulrich Laufs2Borislav Dinov3
Cardiac sarcoidosis (CS), a rare condition characterized by non-caseating granulomas, can manifest with symptoms such as atrioventricular block and ventricular tachycardia (VT), as well as mimic inherited cardiomyopathies. A 48-year-old male presented with recurrent VT. The initial 18F-fluorodeoxyglucose positron emission tomography (18FDG-PET) scan showed uptake of the mediastinal lymph node. Cardiovascular magnetic resonance (CMR) demonstrated intramyocardial fibrosis. The follow-up 18FDG-PET scan revealed the presence of tracer uptake in the left ventricular (LV) septum, suggesting the likelihood of CS. Genetic testing identified a pathogenic LMNA variant. A 47-year-old female presented with complaints of palpitations and syncope. An Ajmaline provocation test confirmed Brugada syndrome (BrS). CMR revealed signs of cardiac inflammation. An endomyocardial biopsy (EMB) confirmed the diagnosis of cardiac sarcoidosis. Polymorphic VT was induced during an electrophysiological study, and an implantable cardioverter-defibrillator (ICD) was implanted. A 58-year-old woman presented with sustained VT with a prior diagnosis of hypertrophic cardiomyopathy (HCM). A genetic work-up identified the presence of a heterozygous MYBC3 variant of unknown significance (VUS). CMR revealed late gadolinium enhancement (LGE), while the 18FDG-PET scan demonstrated LV tracer uptake. The immunosuppressive therapy was adjusted, and no further VTs were observed. A 28-year-old male athlete with right ventricular dilatation and syncope experienced a cardiac arrest during training. Genetic testing identified a pathogenic mutation in PKP2. The autopsy has confirmed the presence of ACM and a distinctive extracardiac sarcoidosis. Cardiac sarcoidosis and inherited cardiomyopathies may interact in several different ways, altering the clinical presentation. Overlapping pathologies are frequently overlooked. Delayed or incomplete diagnosis risks inadequate treatment. Thus, genetic testing and endomyocardial biopsies should be recommended to obtain a clear diagnosis.
The prevalence of cardiac sarcoidosis (CS) in patients with pulmonary sarcoidosis has been reported to be approximately 2%–7%, although autopsy studies have revealed myocardial granulomas in 20%–30% of cases, suggesting that the actual prevalence may be underestimated (1). Most probably, sarcoidosis has no single cause, and various exogenous and endogenous factors, including genetic predisposition, may play a role (2). The diagnosis remains challenging due to the non-specific nature of the symptoms that can include atrioventricular (AV) block, ventricular arrhythmias, heart failure, and syncope or even sudden cardiac death (1, 2). Furthermore, CS can present with features of dilated cardiomyopathy (DCM), hypertrophic cardiomyopathy (HCM), or arrhythmogenic cardiomyopathy (ACM), leading to misdiagnosis and delayed treatment (2–4). Potential genetic factors and the possibility of overlap syndromes between CS and inherited cardiomyopathies are not well-studied.
Here, we present four cases of patients diagnosed with CS and who were subsequently diagnosed with another inherited cardiomyopathy. Our purpose is to elucidate the complex interactions between both conditions influencing the symptoms, the course of the disease, and the outcomes.
A 38-year-old male patient presented with persistent shortness of breath on exertion following an upper respiratory tract infection. Transthoracic echocardiography (TTE) revealed a discrete apical hypokinesia. Suspecting myocarditis, a cardiac magnetic resonance imaging (CMR) scan was performed, revealing reduced systolic left ventricular (LV) function (EF 44%) and evidence of focal inferolateral basal edema, consistent with myocardial inflammation. Coronary angiography ruled out coronary artery disease (CAD), and endomyocardial biopsies (EMB) were obtained excluding acute or chronic myocarditis. After innating an angiotensin-converting enzyme (ACE) inhibitor, the patient was subjectively symptom-free, with an improvement in the systolic LV function (EF 60%) during the 1-month follow-up. Three years later, the patient presented again with a third-degree atrioventricular block and a reduced LV function (EF 45%). As a result, a cardiac resynchronization therapy (CRT)-defibrillator was implanted. At 48 years old, he was admitted again due to recurrent ventricular tachycardia (VT). The device interrogation revealed clusters of non-sustained VT, a second-degree AV block, and frequent premature ventricular contractions (PVC). Due to the early onset of AV block and VT, CS was suspected, and the patient underwent 18F-fluorodeoxyglucose positron emission tomography (18FDG-PET), which revealed significant tracer uptake in multiple mediastinal lymph nodes but normal cardiac uptake. In combination with elevated serum levels of angiotensin-converting enzyme (105 U/L, reference range in U/L: 20–70), these findings were indicative for pulmonary sarcoidosis (5). A cardiovascular magnetic resonance (CMR) demonstrated a mildly impaired left ventricular ejection fraction (LVEF 47%) and striated intramyocardial fibrosis areas (Figures 1A,B), which was interpreted as pulmonary sarcoidosis with cardiac involvement in fibrotic, indolent stage. The symptomatic PVC from the LV outflow tract was successfully ablated, followed again by EMB. EMB was negative for cardiotropic viruses, and showed a low-grade lymphocytic inflammation without evidence of non-caseating granulomas consistent with DCM. However, CS could not be ruled out due to a potential sampling error. According to the synopsis of the findings, persistent lymphocytic inflammation, rapid progression, and young age led to the diagnosis of sarcoidosis with cardiac involvement in accordance with the most recent criteria of the Japanese Circulation Society (JCS), and glucocorticoid (GC) therapy was initiated (5). The 18FDG-PET follow-up scan after 6 months revealed tracer uptake in the LV septum (Figure 1C) and mediastinal lymph nodes, indicating active sarcoidosis despite the GC treatment. Thus, the prednisolone dose was increased. The subsequent 18FDG-PET scan showed a decreased lymph node inflammation and no cardiac inflammation (Figure 1D). The positive response to immunosuppressive therapy supported the diagnosis of sarcoidosis. Nonetheless, genetic testing was performed due to a positive family history of DCM (mother and uncle on the maternal side), revealing a heterozygous pathogenic variant (Arg321Ter) in the LMNA gene.
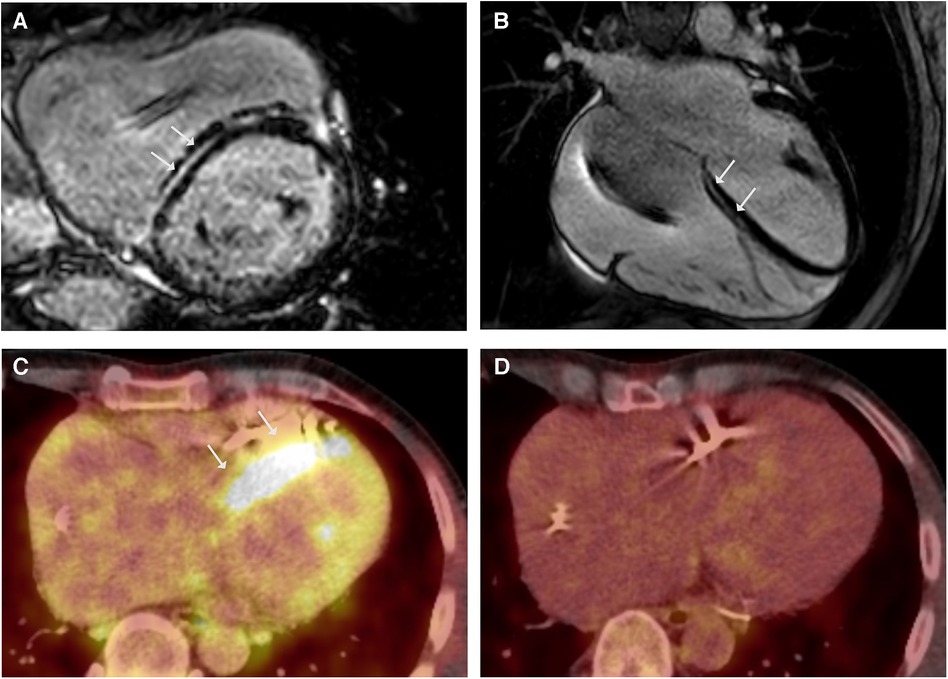
Figure 1. LGE CMR frames in short-axis (A) and 4-chamber (B)-views demonstrating striated midwall fibrosis of the septal LV wall (arrows). Ejection fraction was mildly impaired (LVEF 47%). (C) Axial-view of 18FDG-PET follow-up revealing tracer uptake in the LV Septum (arrows). (D) 18FDG-PET follow-up under immunosuppressive therapy with glucocorticoids (GC) showing no more tracer uptake indicating no cardiac inflammation under GC.
A 47-year-old female medical assistant was referred to our department due to recurrent palpitations, two prior unexplained presyncope, and suspicion of ventricular septal defect. There was no family history of sudden cardiac death (SCD), cardiomyopathy, or sarcoidosis. The echocardiography showed basal thinning of the interventricular septum but no septal defect. The ECG showed left anterior fascicular block, frequent PVCs, and normal QTc. The Ajmaline provocation test unmasked a coved-type ST segment elevation in V1 with inverted T-waves (Figures 2A,B), suggesting the Brugada syndrome. To exclude the presence of structural heart disease, a CMR was performed 2 days after the Ajmaline provocation test was conducted, revealing an impaired LVEF of 41% with normal-sized heart chambers. Surprisingly, the T2 mapping demonstrated edema at the posterior wall of the right ventricle, as well as at the septal, anterior, and posterior walls of the left ventricle (LV). Furthermore, late gadolinium enhancement (LGE) was observed in the subendocardial to intramyocardial regions of the basal septal and inferolateral LV wall, as well as the entire posterior wall of the right ventricle (Figure 2D). A contrast-enhanced CT showed small, irregular parenchymal foci dispersed in both lungs without evidence of mediastinal lymphadenopathy or fibrosis (Figure 2C). Due to the signs of inflammation shown in the CMR and CT scans, suggesting stage 3 pulmonary sarcoidosis, we performed an EMB, which was positive for epithelioid granuloma with multinucleated cells, confirming the diagnosis of CS. Considering the impaired LVEF, the presence of LGE, and two previous episodes of presyncope, an electrophysiological study for risk stratification was performed that resulted in the induction of polymorphic VT, and a dual chamber implantable cardioverter-defibrillator (ICD) was implanted. During the 6-month follow-up, the device interrogation revealed episodes of polymorphic non-sustained VT and spontaneously terminating ventricular fibrillation (VF).
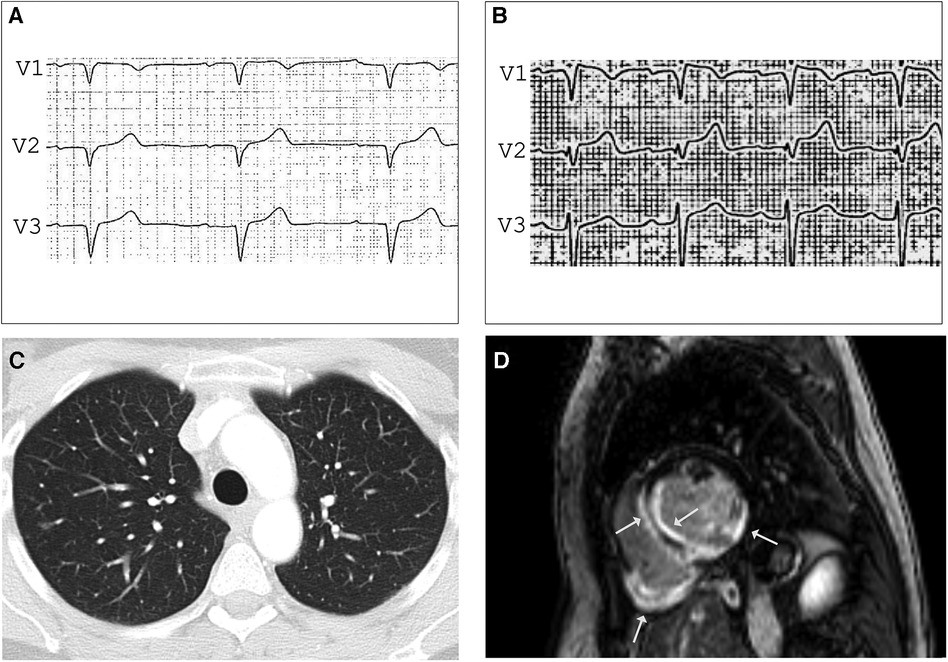
Figure 2. ECG before (A) and after (B) Ajmalin provocation test unmasking coved-type ST segment elevation in V1 suggesting BrS. (C) Contrast-enhanced CT showing small, irregular foci in both lungs compatible with pulmonary sarcoidosis stage 3. LGE CMR frame in short-axis - view (D) revealing subendocardial to intramyocardial LGE in the basal septal and inferolateral LV wall and the entire right ventricular posterior wall (arrows), LVEF was mildly impaired (41%) with normal-sized heart chambers.
A 58-year-old woman was referred to our department due to sustained VT with the diagnosis of HCM in a previous CMR scan. The genetic work-up identified a heterozygous MYBPC3 variant (p.Arg1268Trp) as a variant of unknown significance (VUS). Her history for familial cardiomyopathies or SCD was negative. The ECG showed a first-degree AV block, right bundle-branch block, and left anterior block. Repeated CMR scans confirmed the presence of concentric LV hypertrophy, reduced LVEF (43%), and anterolateral LGE (Figure 3B). Notably, T2 mapping demonstrated regional edema of the LV anterolateral wall (Figure 3A) and raised the suspicion of CS in (sub)acute inflammatory stage. The 18FDG-PET scan showed intense LV septal and lateral FDG uptake (Figures 3C,D), without evidences of extracardiac involvement. The coronary angiography ruled out relevant stenosis, and EMB were taken from the interventricular septum. In the molecular pathological examination, non-caseating granulomas were absent; however, CS was deemed very probable based on the imaging criteria established by Vita et al. (5, 6), and the diagnosis of isolated cardiac sarcoidosis was made according to the JCS criteria. The patient refused ICD implantation and was discharged on GC therapy. Three months later, the 18FDG-PET scan showed increased tracer uptake in the apex as well as decreased enhancement in the septum, and the immunosuppressive therapy was augmented with methotrexate. No further VTs were observed in the patient during the follow-up period.
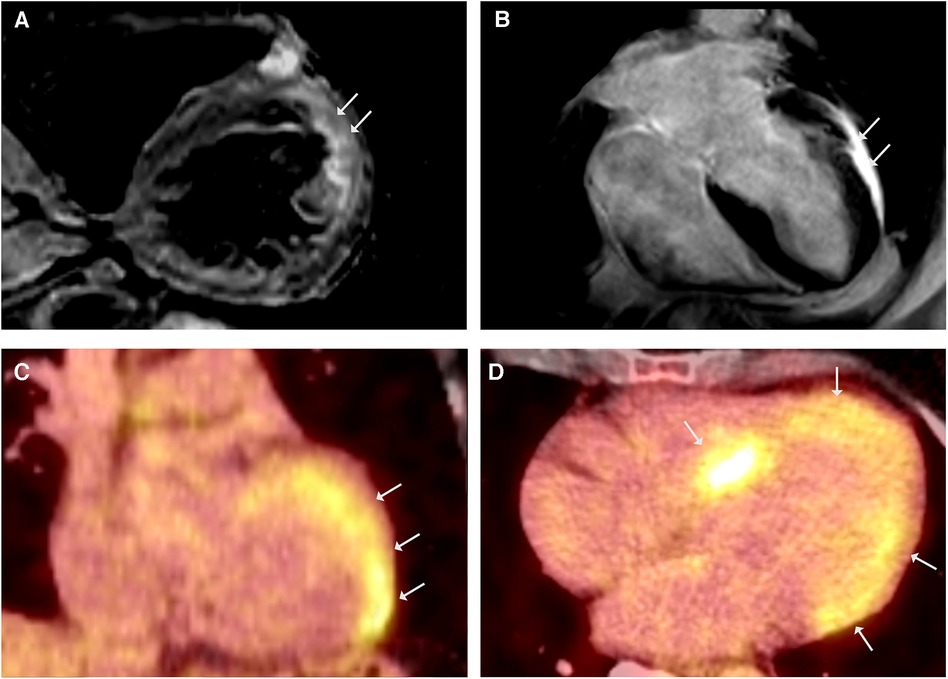
Figure 3. CMR showed simultaneously occurring edematous and fibrosing processes. (A) CMR, T2-weighted black blood imaging in short - axis view demonstrating regional edema of the LV anterolateral wall. (B) LGE CMR frame in 4-chamber view showing LV hypertrophy and anterolateral LGE, LVEF was mildly reduced (43%). 18FDG-PET coronal (C) and axial (D) views revealing intense FDG uptake in septal and lateral LV.
A 28-year-old male athlete with a history of exercise-induced syncope presented at our clinic. He reported palpitations and loss of consciousness during intensive physical activity as an American football player. His family history was negative for sudden cardiac death. The clinical examination, blood testing, and 12-lead ECG were unremarkable. The transthoracic echocardiogram showed right ventricular enlargement without dyskinesia. Exercise testing and signal-averaged ECG were normal. The CMR revealed a dilated right ventricle with regional dyssynchrony and moderately decreased right ventricular ejection fraction of 42% (Figures 4A–D). The RV end-diastolic volume index was measured at 130 ml/m2. Despite the strong recommendation against competitive sports, he suffered a cardiac arrest during training, with ECG showing ventricular fibrillation. Immediate defibrillation failed, and he was transferred to our institution under ongoing cardiopulmonary resuscitation. Sinus rhythm was restored post-ECMO, but the patient died due to cerebral edema. The postmortem genetic testing identified a novel pathogenic mutation in plakophilin-2 (PKP2), leading to premature truncation (NM_004572.3 c.1540_1543delAAAC). The autopsy confirmed ACM, and extracardiac sarcoidosis was found in multiple organs including the lungs, pleura, spleen, liver, lymph nodes, and pelvis. Notably, there was no evidence of cardiac involvement in the samples examined.
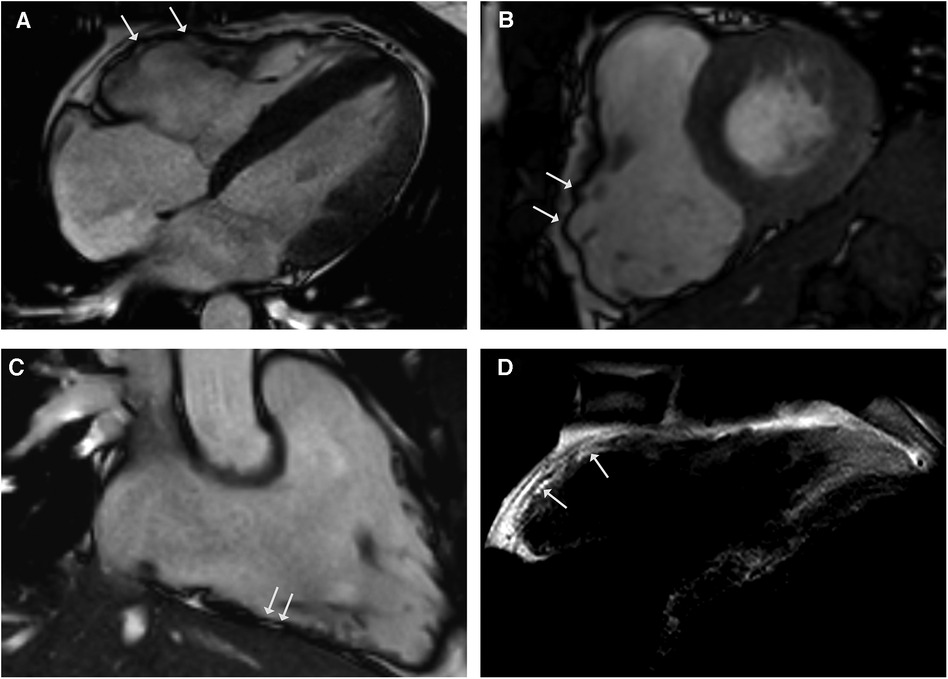
Figure 4. CMR cine images in 4-chamber (A) and short-axis - views (B) revealing right ventricular dilatation with bulging (arrows) and regional dyssynchrony in end-systolic phase. Right ventricular systolic function was mildly reduced (RV EF 42%). CMR 3-chamber view of the RV (C) demonstrating fatty spots. (D) T1-weighted black blood spin CMR showing fatty replacement of the RV free wall.
This case series reveals the complex interactions between CS and inherited cardiomyopathies ranging from simple coexistence or phenocopying to overlap cardiomyopathy with distinct clinical presentation and outcomes.
The diagnosis of cardiac sarcoidosis was made according to the most recent guidelines by the JCS for diagnosing cardiac sarcoidosis (5). We employed these criteria due to the diagnostic challenges associated with isolated cardiac sarcoidosis, primarily arising from potential sampling errors in endomyocardial biopsy. Notably, Kandolin et al. (7) found that at least two-thirds of patients remain undiagnosed after a single endomyocardial biopsy session. In contrast to the criteria set by the Heart Rhythm Society (HRS), the criteria established by the Japanese Circulation Society allow for diagnosis even in the absence of histological evidence of cardiac or extracardiac non-caseating granulomas (5, 8). Sato et al. (9) have substantiated the reliability of the updated JCS diagnostic criteria, using a multimodal imaging approach. However, the lack of histological evidence remains a limitation in the context of overlap cardiomyopathies.
LMNA/C-related DCM and CS can conceivably coexist without influencing each other. Due to the similar clinical presentation, they cannot be reliably distinguished from one another. Both CS and LMNA/C-associated DCM usually present at early to mid-adulthood with conduction system disease, LV dilatation, and VT (10). Moreover, the typical CMR finding in LMNA/C-DCM is the presence of linear mid-wall fibrosis in the basal interventricular septum, which is also a predilection site of CS and limits the diagnostic utility of CMR to differentiate between CS and LMNA/C-DCM (6, 11). The interaction between both diseases is conceivable, wherein the pathogenic mutation renders the myocardium susceptible to secondary damage. In particular, the LMNA/C variant presented in the first case is suspected to cause a nonsense-mediated mRNA decay (NMD), which prevents the expression of the truncated mutant protein, and has additional trans effects on the levels of lamin A protein expressed by the wild-type allele, decreasing its levels and causing a shift in the ratio of lamin A to lamin C (12). The release of proinflammatory cytokines such as TNF-α, IL-2, and Interferon-γ may accelerate disease deterioration in a susceptible mutation carrier. Interestingly, abnormal myocardial 18FDG uptake was previously observed in a patient with LMNA/C DCM, providing more evidences that inflammation may be a key factor in the development of LMNA/C-associated DCM (13).
Recurrent presyncope of unclear mechanism, ventricular fibrillation in ICD interrogation, and induced Brugada type-1 ECG were indicating a probable/definite diagnosis of BrS according to the Shanghai Score System (14). The VF episode is more suggestive of BrS, while a monomorphic VT resulting from scar-related re-entry is a typical finding in cardiac sarcoidosis (15, 16). Rarely, ventricular fibrillation and polymorphic VT are observed in CS, which can be explained by triggered activity resulting from early or delayed afterdepolarizations caused by increased intracellular calcium release (16). However, CS can imitate BrS due to extensive right ventricular involvement and inflammation. One theory regarding BrS suggests that the characteristic ECG pattern and the occurrence of ventricular arrhythmias may be attributed to the conduction slowing in the right ventricular outflow tract alongside with a delay in epicardial activation (17). Coronel et al. (18) demonstrated that conduction slowing in epicardial RVOT is associated with extensive regional fatty infiltration and fibrosis without evidence of associated mutations. A similar mechanism was suggested in ACM and could be the missing link between CS, ACM, and BrS phenocopies (19). The pronounced inflammatory involvement in this case, with evidence of edema and LGE in both the RV and LV, along with potential alterations in electrical properties, could potentially have led to a phenotypic overlap during the Ajmaline provocation test. On the other hand, the presence of CS could potentially impact the penetrance of an oligogenic BrS within the context of an overlap syndrome, stemming from an epigenetic effect induced by inflammation. BrS is considered an autosomal dominant disease, although alternative mechanisms of oligogenic inheritance have been proposed. Genetic studies have revealed that 30% of diagnosed BrS are associated with >350 rare variants in SCN5A, although almost 60% of these cases have negative results in genetic testing (20). The same SCN5A variant (p.C335R) has been linked to BrS, familial atrial fibrillation, and DCM (21). Since the same SCN5A variant can result in different phenotypes, it is reasonable to assume that additional factors contribute to the development of the corresponding phenotype, e.g., oligogenic, polygenic, epigenetic, or inflammatory mechanisms.
In the third case of suspected HCM, the CMR and 18FDG-PET imaging raised suspicion of CS due to the unexpected evidence of acute myocardial inflammation. A heterozygous MYBPC3 variant (p.Arg1268Trp) of unclear significance and an allele count higher than expected for a pathogenic variant (ExAC 0.03%) were found, which has not previously been reported in MYBPC3-related cardiomyopathy (22). However, MYBPC3 mutations are frequently associated with autosomal dominant DCM, NCCM, and RCM and are the most common cause of familial HCM (23). Notably, incomplete penetrance of the MYBPC3 mutations is suggested (24). In this case, no pathological genetic variant was identified; instead, a VUS was found, leading us to hypothesize that CS might be associated with an enhancement of the penetrance and expression of a VUS within the context of an overlap syndrome, thereby leading to the HCM phenotype. As in the previous case, this could be attributed to an epigenetic effect induced by inflammation.
The overlap between cardiac sarcoidosis and ACM, as well as phenocopies of ACM caused by cardiac sarcoidosis, has been previously described, including a case from our group (25). CS represents one of the most common phenocopies of ARVC (26). Vasaiwala et al. (27) reported that 15% of initially diagnosed ACM cases were reclassified as CS based on invasive findings. The revised international Task Force Criteria (TFC) of 2010 lack reliability in differentiating between CS and ACM. Gasperetti et al. (26) distinguished ACM by noting larger right RVOT dimensions (RVOT dilation ≥ 35 mm) and T-wave inversions, while CS exhibited greater LV involvement, lower LVEF, and high-grade AVB. The positive 18FDG-PET results were exclusive to CS in this study. However, ACM may also exhibit a positive 18FDG-PET result during inflammatory phases, marking the transition from subclinical to overt disease phenotype (28–30). Both Protonotarios et al. and Smith et al. (29, 31) observed elevated myocardial FDG uptake in ACM patients, particularly in those with mutations in the desmoplakin gene. As a result, ACM may be misdiagnosed as CS, warranting a myocardial biopsy to clarify the diagnosis. In our case, it remains unclear whether CS was incidentally discovered or if the early death of the patient resulted from the combined unfavorable effects of CS and ACM due to the inability to perform 18FDG-PET scans, owing to the rapid and severe progression of the disease. However, based on the negative autopsy for sarcoid granulomas, we rather assume a coexistence in this case.
Cardiac sarcoidosis and all types of inherited cardiomyopathies may interact in several different ways, changing the clinical presentation and likely the course of the disease. Overlapping pathologies are frequently overlooked and associated with an unfavorable clinical course. Delayed or incomplete diagnosis bears the risk of suboptimal treatment. 18FDG-PET, endomyocardial biopsies, and genetic evaluation should be considered to all patients with suspected inherited cardiomyopathy and/or cardiac sarcoidosis to obtain a clear diagnosis.
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
HE: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Visualization, Writing – original draft. LU: Writing – review & editing. DH-B: Writing – review & editing. AB: Writing – review & editing. UL: Writing – review & editing. BD: Conceptualization, Writing – review & editing, Supervision.
The authors declare financial support was received for the research, authorship, and/or publication of this article.
HE is thankful for the support of the German Heart Foundation (DSHF, F 28 / 22). Supported by the Open Access Publishing Fund of Leipzig University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Caforio ALP, Adler Y, Agostini C, Allanore Y, Anastasakis A, Arad M, et al. Diagnosis and management of myocardial involvement in systemic immune-mediated diseases: a position statement of the European Society of Cardiology working group on myocardial and pericardial disease. Eur Heart J. (2017) 38:2649–62. doi: 10.1093/eurheartj/ehx321
2. Ueberham L, Hagendorff A, Klingel K, Paetsch I, Jahnke C, Kluge T, et al. Pathophysiological gaps, diagnostic challenges, and uncertainties in cardiac sarcoidosis. J Am Heart Assoc. (2023) 12(6):e027971. doi: 10.1161/JAHA.122.027971
3. Adlan AMA, Prasad SK, Varnava AM. Sarcoidosis presenting as dilated cardiomyopathy. Heart. (2011) 97:1896. doi: 10.1136/heartjnl-2011-300379
4. Waki H, Eguchi K, Toriumi S, Ikemoto T, Suzuki T, Fukushima N, et al. Isolated cardiac sarcoidosis mimicking arrhythmogenic right ventricular cardiomyopathy. Intern Med Tokyo Jpn. (2018) 57:835–9. doi: 10.2169/internalmedicine.9395-17
5. Terasaki F, Azuma A, Anzai T, Ishizaka N, Ishida Y, Isobe M, et al. JCS 2016 guideline on diagnosis and treatment of cardiac sarcoidosis – digest version –. Circ J. (2019) 83:2329–88. doi: 10.1253/circj.CJ-19-0508
6. Vita T, Okada DR, Veillet-Chowdhury M, Bravo PE, Mullins E, Hulten E, et al. Complementary value of cardiac magnetic resonance imaging and positron emission tomography/computed tomography in the assessment of cardiac sarcoidosis. Circ Cardiovasc Imaging. (2018) 11:e007030. doi: 10.1161/CIRCIMAGING.117.007030
7. Kandolin R, Lehtonen J, Graner M, Schildt J, Salmenkivi K, Kivistö SM, et al. Diagnosing isolated cardiac sarcoidosis. J Intern Med. (2011) 270:461–8. doi: 10.1111/j.1365-2796.2011.02396.x
8. Birnie DH, Sauer WH, Bogun F, Cooper JM, Culver DA, Duvernoy CS, et al. HRS expert consensus statement on the diagnosis and management of arrhythmias associated with cardiac sarcoidosis. Heart Rhythm. (2014) 11:1304–23. doi: 10.1016/j.hrthm.2014.03.043
9. Sato K, Kawamatsu N, Yamamoto M, Machino-Ohtsuka T, Ishizu T, Ieda M. Utility of updated Japanese Circulation Society guidelines to diagnose isolated cardiac sarcoidosis. J Am Heart Assoc. (2022) 11:e025565. doi: 10.1161/JAHA.122.025565
10. Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. (1999) 341:1715–24. doi: 10.1056/NEJM199912023412302
11. Holmström M, Kivistö S, Heliö T, Jurkko R, Kaartinen M, Antila M, et al. Late gadolinium enhanced cardiovascular magnetic resonance of lamin A/C gene mutation related dilated cardiomyopathy. J Cardiovasc Magn Reson. (2011) 13:30. doi: 10.1186/2F1532-429X-13-30
12. Al-Saaidi R, Rasmussen TB, Palmfeldt J, Nissen PH, Beqqali A, Hansen J, et al. The LMNA mutation p.Arg321Ter associated with dilated cardiomyopathy leads to reduced expression and a skewed ratio of lamin A and lamin C proteins. Exp Cell Res. (2013) 319:3010–9. doi: 10.1016/j.yexcr.2013.08.024
13. Rosse C, Reddy A, Patel K, Brien TO. A deceptive case of lamin A/C-associated cardiomyopathy. J Am Coll Cardiol. (2021) 77:2255. doi: 10.1016/S0735-1097(21)03610-X
14. Antzelevitch C, Yan G-X, Ackerman MJ, Borggrefe M, Corrado D, Guo J, et al. J-Wave syndromes expert consensus conference report: emerging concepts and gaps in knowledge. Heart Rhythm. (2016) 13:e295–324. doi: 10.1016/j.hrthm.2016.05.024
15. Kumar S, Barbhaiya C, Nagashima K, Choi E-K, Epstein LM, John RM, et al. Ventricular tachycardia in cardiac sarcoidosis: characterization of ventricular substrate and outcomes of catheter ablation. Circ Arrhythm Electrophysiol. (2015) 8:87–93. doi: 10.1161/CIRCEP.114.002145
16. Hamzeh N, Steckman DA, Sauer WH, Judson MA. Pathophysiology and clinical management of cardiac sarcoidosis. Nat Rev Cardiol. (2015) 12:278–88. doi: 10.1038/nrcardio.2015.22
17. Blok M, Boukens BJ. Mechanisms of arrhythmias in the Brugada syndrome. Int J Mol Sci. (2020) 21:7051. doi: 10.3390/ijms21197051
18. Coronel R, Casini S, Koopmann TT, Wilms-Schopman FJG, Verkerk AO, de Groot JR, et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: a combined electrophysiological, genetic, histopathologic, and computational study. Circulation. (2005) 112:2769–77. doi: 10.1161/CIRCULATIONAHA.105.532614
19. Hoffmayer KS, Scheinman MM. Electrocardiographic patterns of ventricular arrhythmias in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Front Physiol. (2012) 3:23. doi: 10.3389/fphys.2012.00023
20. Campuzano O, Sarquella-Brugada G, Cesar S, Arbelo E, Brugada J, Brugada R. Update on genetic basis of Brugada syndrome: monogenic, polygenic or oligogenic? Int J Mol Sci. (2020) 21:7155. doi: 10.3390/ijms21197155
21. Sedaghat-Hamedani F, Rebs S, El-Battrawy I, Chasan S, Krause T, Haas J, et al. Identification of SCN5a p.C335R variant in a large family with dilated cardiomyopathy and conduction disease. Int J Mol Sci. (2021) 22:12990. doi: 10.3390/ijms222312990
22. Kobayashi Y, Yang S, Nykamp K, Garcia J, Lincoln SE, Topper SE. Pathogenic variant burden in the ExAC database: an empirical approach to evaluating population data for clinical variant interpretation. Genome Med. (2017) 9:13. doi: 10.1186/s13073-017-0403-7
23. Brodehl A, Ebbinghaus H, Deutsch M-A, Gummert J, Gärtner A, Ratnavadivel S, et al. Human induced pluripotent stem-cell-derived cardiomyocytes as models for genetic cardiomyopathies. Int J Mol Sci. (2019) 20:3–8. doi: 10.3390/ijms20184381
24. Ho CY, Day SM, Ashley EA, Michels M, Pereira AC, Jacoby D, et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: insights from the sarcomeric human cardiomyopathy registry (SHaRe). Circulation. (2018) 138:1387–98. doi: 10.1161/CIRCULATIONAHA.117.033200
25. Müssigbrodt A, Knopp H, Czimbalmos C, Jahnke C, Richter S, Husser D, et al. Exercise-related sudden cardiac death of an American football player with arrhythmogenic right ventricular dysplasia/cardiomyopathy AND sarcoidosis. Clin Case Rep. (2019) 7:686–8. doi: 10.1002/ccr3.2071
26. Gasperetti A, Rossi VA, Chiodini A, Casella M, Costa S, Akdis D, et al. Differentiating hereditary arrhythmogenic right ventricular cardiomyopathy from cardiac sarcoidosis fulfilling 2010 ARVC task force criteria. Heart Rhythm. (2021) 18:231–8. doi: 10.1016/j.hrthm.2020.09.015
27. Vasaiwala SC, Finn C, Delpriore J, Leya F, Gagermeier J, Akar JG, et al. Prospective study of cardiac sarcoid mimicking arrhythmogenic right ventricular dysplasia. J Cardiovasc Electrophysiol. (2009) 20:473–6. doi: 10.1111/j.1540-8167.2008.01351.x
28. Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M. Arrhythmogenic right ventricular cardiomyopathy: dysplasia, dystrophy, or myocarditis? Circulation. (1996) 94:983–91. doi: 10.1161/01.CIR.94.5.983
29. Protonotarios A, Wicks E, Ashworth M, Stephenson E, Guttmann O, Savvatis K, et al. Prevalence of 18F-fluorodeoxyglucose positron emission tomography abnormalities in patients with arrhythmogenic right ventricular cardiomyopathy. Int J Cardiol. (2019) 284:99–104. doi: 10.1016/j.ijcard.2018.10.083
30. Mavrogeni S, Protonotarios N, Tsatsopoulou A, Papachristou P, Sfendouraki E, Papadopoulos G. Naxos disease evolution mimicking acute myocarditis: the role of cardiovascular magnetic resonance imaging. Int J Cardiol. (2013) 166:e14–5. doi: 10.1016/j.ijcard.2012.12.078
31. Smith ED, Lakdawala NK, Papoutsidakis N, Aubert G, Mazzanti A, McCanta AC, et al. Desmoplakin cardiomyopathy, a fibrotic and inflammatory form of cardiomyopathy distinct from typical dilated or arrhythmogenic right ventricular cardiomyopathy. Circulation. (2020) 141:1872–84. doi: 10.1161/CIRCULATIONAHA.119.044934
Keywords: cardiac sarcoidosis, non-ischemic cardiomyopathy, familial cardiomyopathy, ventricular tachyarrhythmias, conduction disease
Citation: Ebbinghaus H, Ueberham L, Husser-Bollmann D, Bollmann A, Paetsch I, Jahnke C, Laufs U and Dinov B (2023) Case Report: Four cases of cardiac sarcoidosis in patients with inherited cardiomyopathy—a phenotypic overlap, co-existence of two rare cardiomyopathies or a second-hit disease. Front. Cardiovasc. Med. 10:1328802. doi: 10.3389/fcvm.2023.1328802
Received: 27 October 2023; Accepted: 4 December 2023;
Published: 19 December 2023.
Edited by:
Marina Cerrone, Grossman School of Medicine, New York University, United StatesReviewed by:
Francesca Romana Gentile, University of Pavia, Italy© 2023 Ebbinghaus, Ueberham, Husser-Bollmann, Bollmann, Laufs and Dinov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hans Ebbinghaus aGFucy5lYmJpbmdoYXVzQG1lZGl6aW4udW5pLWxlaXB6aWcuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.