 Yulong Ma
Yulong Ma Xunjie Zhou
Xunjie Zhou Mingtai Gui
Mingtai Gui Lei Yao
Lei Yao Jianhua Li
Jianhua Li Xiaozhe Chen
Xiaozhe Chen Mingzhu Wang
Mingzhu Wang Bo Lu
Bo Lu Deyu Fu
Deyu Fu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 04 January 2024
Sec. Hypertension
Volume 10 - 2023 | https://doi.org/10.3389/fcvm.2023.1309863
Hypertension constitutes a pervasive chronic ailment on a global scale, frequently inflicting damage upon vital organs, such as the heart, blood vessels, kidneys, brain, and others. And this is a complex clinical dilemma that requires immediate attention. The mitochondria assume a crucial function in the generation of energy, and it is of utmost importance to eliminate any malfunctioning or surplus mitochondria to uphold intracellular homeostasis. Mitophagy is considered a classic example of selective autophagy, an important component of mitochondrial quality control, and is closely associated with many physiological and pathological processes. The ubiquitin-dependent pathway, facilitated by PINK1/Parkin, along with the ubiquitin-independent pathway, orchestrated by receptor proteins such as BNIP3, NIX, and FUNDC1, represent the extensively investigated mechanisms underlying mitophagy. In recent years, research has increasingly shown that mitophagy plays an important role in organ damage associated with hypertension. Exploring the molecular mechanisms of mitophagy in hypertension-mediated organ damage could represent a critical avenue for future research in the development of innovative therapeutic modalities. Therefore, this article provides a comprehensive review of the impact of mitophagy on organ damage due to hypertension.
Hypertension has emerged as the predominant risk factor for mortality in the world (1). Epidemiological surveys indicate that the worldwide incidence of hypertension has surged to encompass 1.3 billion individuals (2). In 2019, hypertension affected 32% of men and 34% of women within the cohort aged 30 to 79 globally (3). Between 2017 and 2020, the National Health and Nutrition Examination Survey revealed a high hypertension prevalence of about 46.7% among adults aged 20 years and older. The rates of hypertension awareness, treatment, and control were 62%, 52.6%, and 25.7% (4, 5). It is estimated that annually, elevated blood pressure results in the deaths of between 7.7 and 10.4 million individuals (6). A prospective study revealed that for each increase of 20 mmHg in systolic blood pressure and 10 mmHg in diastolic blood pressure, the risk of developing ischemic heart disease and stroke doubled. The findings indicate a strong correlation between blood pressure levels and subsequent cardiovascular events (7). Hypertension-mediated organ damage (HMOD) including stroke, ischemic heart disease, kidney disease, and other vascular conditions, is responsible for causing 85 million deaths globally (8, 9). The treatment and management of hypertension pose a significant burden on the healthcare system worldwide. Despite the extensive use of efficient antihypertensive medications, combatting HMOD remains an urgent medical challenge.
Mitochondria are specialized double-membrane organelles that originated from the engulfment of α-amoebae by eukaryotic progenitor cells (10). Mitochondria function as metabolic signaling centers and energy production sites, supplying the necessary biochemical reactions for cellular activity (11). Their role in maintaining cell survival, death, and metabolic homeostasis is paramount (12). Maintaining intracellular mitochondrial homeostasis necessitates a quality control system comprising mitochondrial biogenesis, mitochondrial dynamics, mitophagy, and mitolysosome exocytosis (13, 14). Mitochondria are integral organelles in cardiomyocytes that perform vital functions necessary for maintaining appropriate myocardial activity (15). Mitophagy contributes to intracellular homeostasis by eliminating damaged and excess mitochondria (16). Under typical circumstances, mitophagy supports the preservation of cardiomyocyte energy metabolism homeostasis by eliminating unhealthy mitochondria (17). When mitophagy is either insufficient or excessive, it causes imbalanced mitochondrial homeostasis, which subsequently initiates a range of diseases. Mitophagy can have a positive or negative impact on cardiovascular disease, contingent on the preservation of mitochondrial homeostasis (18). Therefore, this review focus on the correlation between faulty mitophagy, which encompasses inadequate and excessive mitophagy, and multiple HMOD.
Mitochondria, often referred to as the body's “energy production center”, are susceptible to substantial levels of exposure to reactive oxygen species (ROS) due to their unique functions (19). This can easily cause mutations in mitochondrial DNA (mtDNA) and result in structural and functional changes in proteins, leading to mitochondrial harm. Mitophagy (Figure 1), the process responsible for the degradation of dysfunctional mitochondria, is crucial in maintaining intracellular homeostasis (20). The primary modalities that govern mitophagy encompass ubiquitin-dependent, receptor-dependent, and other pathways (21).
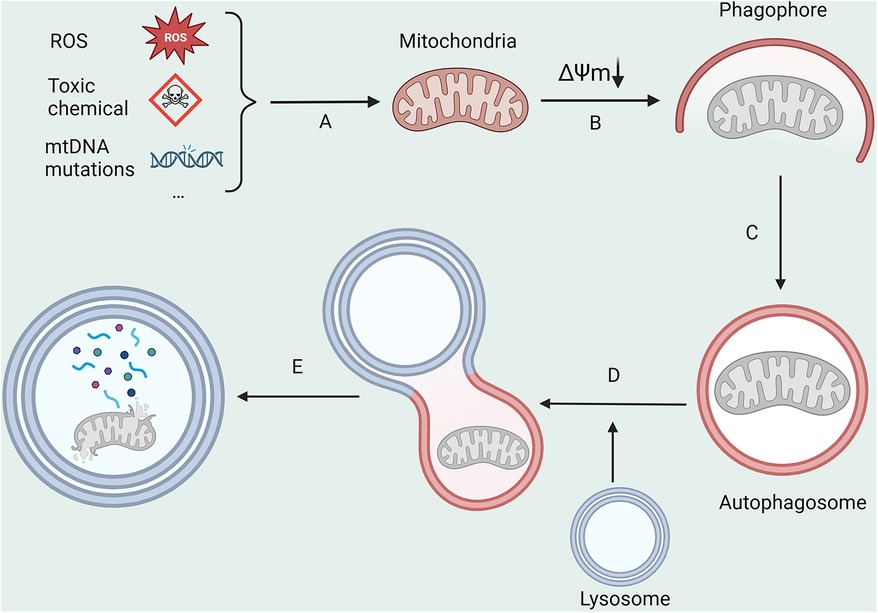
Figure 1. The main processes in mitophagy. (A) ROS, toxic chemicals, and mitochondrial DNA mutations lead to a decrease in mitochondrial membrane potential; (B,C) Damaged mitochondria are encapsulated by autophagosomes; (D) Autophagosomes fuse with lysosomes; (E) Mitochondrial is degraded by lysosomes. All figures were created with BioRender.com and are licensed for use.
In the realm of ubiquitin-dependent mitophagy, the PTEN-induced putative kinase protein 1 (PINK1)-Parkin pathway stands as an exemplar (20). PINK1 is a serine protease located upstream of Parkin. Normally, PINK1 is transported inside the mitochondria and eventually degraded by the proteasome (22–24). The amount of Parkin in the cytoplasm is minimal. PINK1 stabilizes on the out mitochondrial membrane (OMM) when there is a loss of mitochondrial membrane potential or accumulation of misfolded proteins (25, 26). PINK1 phosphorylates ubiquitin at Ser65, which is a critical step in activating Parkin. This phosphorylation is essential for Parkin to function properly (27–29). However, the phosphorylation of Parkin alone is inadequate to bestow activity. Activated Parkin subsequently forms ubiquitin chains on several proteins in the OMM, ultimately attracting autophagy receptors that bind to ubiquitin (22). A set of receptor proteins, including Sequestosome-1 (SQSTM1 or p62), Optineurin (OPTN), Next to BRCA1 gene 1 protein (NBR1), Calcium-binding and coiled-coil domain-containing protein 2 (NDP52 or CALCOCO2), and Tax1-binding protein 1 (TAX1BP1), interact with microtubule-associated protein 1A/1B-light chain 3 (LC3), thereby initiating the mitophagy process (30, 31). Notably, phosphorylation of ubiquitin induced by PINK1 serves as a signal for mitophagy, which is subsequently amplified by Parkin (26, 32). The deubiquitinase USP30 has demonstrated its ability to remove ubiquitin from depolarized mitochondria and hinder mitophagy (33).
Moreover, multiple signaling pathways influence mitophagy by modulating the expression of PINK1/Parkin. Autophagy and beclin 1 regulator 1 (AMBRA1) plays a important role in mitophagy, regulating this process through its interaction with ATPase family AAA domain containing 3A (ATAD3A) and by strengthening the stability of PINK1 (34). Targeting ATAD3A also overcomes resistance to chemoimmunotherapy by redirecting PD-L1 to mitochondria (35). LncRNA H19 downregulates mitophagy by limiting the translation of Pink1, thereby attenuating cardiac injury induced by obesity (36). Spliced X-box binding protein 1 (XBP1s) regulates mitophagy by interacting with PINK1, an interaction which is transcription- and phosphorylation-dependent (37). In addition to regulating mitophagy, PINK1 plays other roles, including inhibiting tumor growth through and reducing production of acetyl coenzyme A (38).
Mitophagy via the ubiquitin-independent pathway is also commonly referred to as receptor-dependent mitophagy. Unlike mitophagy facilitated by the PINK1/Parkin pathway, the receptor-mediated pathway does not necessitate ubiquitin participation. The OMM contains a range of receptor proteins which have the ability to bind to LC3. For example, BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3), BNIP3-Like (BNIP3l or NIX), and FUN14 domain-containing protein 1 (FUNDC1) can interact directly with LC3, resulting in recognition by the autophagosome. This process is the driving mechanism behind the direction of mitophagy.
BCL2 family members are categorized based on the presence of one or more BCL2 homologous structural domains (39). BNIP3 and BNIP3l, which have structural domain homology to BCL2 in BH3, are proteins produced by the BNIP3 gene located on chromosome 10q26.3. They have a molecular weight of 21.5 kDa and a peptide chain comprising 194 amino acids. These proteins play significant roles in the processes of autophagy (40). BNIP3 protein is expressed at low levels in cells and tissues under normal circumstances, including skeletal muscle, brain, and heart (40, 41). Under hypoxic conditions, the BNIP3 protein undergoes upregulation and becomes anchored to the OMM through its C-terminal transmembrane structural domain. This exposes the N-terminal structural domain to the cytoplasm (42). Like other mitophagy receptor proteins, BNIP3 contains an LC3-interacting region (LIR) motif at its N-terminus. The phosphorylation of the two tandem serine residues, Ser 34 and Ser 35, that are near the LIR motif, stabilizes the NIX-LC3 interaction, leading to the promotion of mitophagy (43). Furthermore, BNIP3 plays a crucial role in regulating ROS production during mitophagy. It has been reported that during hypoxia, hypoxia-inducible factor-1α (HIF1α) promotes BNIP3 expression, which limits ROS production through mitophagy (44, 45). Increased HIF1α expression, in contrast, led to an upregulation of mitochondrial ROS levels in BNIP3-KO tumor cells (46). Furthermore, mitophagy mediated by HIF1α/BNIP3 in renal tubular cells protects against acute kidney injury induced by reperfusion after ischemia by inhibiting apoptosis and ROS generation (47). Similar to BNIP3, the dimerization of NIX, specifically the phosphorylation of its C-terminal region, is crucial for the targeted autophagy in the mitochondria (48). NIX participates in the clearance of mitochondria in the course of reticulocyte maturation (49, 50). The SCF-FBXL4 complex, operating as a ubiquitin E3 ligase in the mitochondria, promotes the degradation of BNIP3 and NIX, resulting in the inhibition of mitophagy (51). However, BNIP3/NIX is not entirely independent of mitophagy mediated by PINK1/Parkin. BNIP3 interacts with PINK1, leading to an accumulation of PINK1 on the OMM. This, in turn, promotes the recruitment of Parkin to mitochondria (52). Inhibition of cyclin dependent kinase 9 (CDK9) inhibits PINK1/Parkin-mediated initiation of mitophagy through regulation of the SIRT1/FOXO3/BNIP3 axis (53). The ubiquitination of NIX by Parkin facilitates the binding of ubiquitin and LC3 through the selective autophagy junction protein NBR1. As a result, autophagosome wrapping around mitochondria is promoted (54).
FUNDC1 is situated within the OMM and is characterized by a canonical LIR motif, in addition to three transmembrane structural domains near its N-terminus (55). This protein serves as a receptor for mitophagy induced by hypoxia. Serine 13 and Tyrosine 18 residues of FUNDC1 can be dephosphorylated under hypoxic stress and in abnormal mitochondrial membrane potential situations. The LIR motif of FUNDC1 interacts hydrophobically with the Y and L pockets of the LC3 protein thereby inducing mitophagy (56, 57). FUNDC1-mediated mitophagy is tightly dependent on the interaction between typical LIR motifs and LC3 (58). In hypoxic conditions, the expression of FUNDC1 was decreased (55). In addition, uULK1 has the ability to phosphorylate FUNDC1, which activates mitophagy. During hypoxia or mitochondrial depolarization, ULK1 expression is induced, and it targets mitochondria to phosphorylate FUNDC1 at Ser17 (near the LIR motif). This leads to an enhanced interaction of FUNDC1 with LC3 (59).
In addition to the mitophagy-mediated mechanisms mentioned earlier, alternative pathways exist for the elimination of dysfunctional mitochondria. Bcl-2-like protein 13 (Bcl2-L-13) is a single-channel membrane protein anchored to the OMM. It binds to LC3 through the LIR motif, recruiting the ULK1 complex to trigger mitophagy in Parkin-deficient cells (60). FK506-binding protein 8 (FKBP8) can induce mitophagy by interacting with LC3A through its LIR motifs (61). minichromosome maintenance complex component 8 (MCM8) initiates mitophagy by binding to LC3 via the LIR motif. Additionally, this type of mitophagy mediated by MCM8 operates independently of previously identified mechanisms (62). While in cells where Bcl2-L-13 is knocked down, there is a reduction in fission and mitophagy due to mitochondrial damage (63). Recent studies have shown that damaged mitochondria are eliminated through extracellular vesicles when lysosomal degradation is inhibited (64). Flunarizine induces the fusion of mitochondria and lysosomes resulting in the generation of a unique structure known as the mitolysosome. This phenomenon occurs through a direct mechanism (65). The mitolysosome is expelled to the extracellular compartment through cytokinesis dependent on VAMP2/ STX4 as vesicles, leading to a reduction in the total number of mitochondria within the cell. This approach works independently of autophagy protein 5 (ATG5) or ras-related protein 9 (RAB9)-mediated mitophagy (65). Migratory cells selectively eliminate impaired mitochondria using migratory bodies to maintain organismal balance, seamlessly linking mitochondrial balance to cell migration (66).
Hypertension constitutes a substantial risk factor for various life-threatening diseases (67–69). Consequently, proper mitochondrial function is crucial to cardiovascular health. Alterations in mitochondrial function, structure and homeostasis can be commonly seen in organ tissues of patients with hypertension. Mitochondrial hyperacetylation leads to a cyclic relationship between metabolic disturbances and mitochondrial oxidative stress, which contributes to vascular dysfunction and hypertension (70). A systematic investigation involving 264 hypertensive patients discovered a connection between mitochondrial dysfunction and hypertension. Nevertheless, additional research is necessary to clarify the relationship between mt-DNA mutations and the development of hypertension (71). Abnormalities in mitochondrial function or structure have been discovered in numerous experimental hypertension models (72–74). Overexpression of DNA-damage regulated autophagy modulator 1 (DRAM1) was found to reduce oxidative stress, improve mitochondrial fusion and fission, and increase mitophagy in the placenta of pre-eclamptic mice. These improvements ultimately lead to a reduction in blood pressure (75). Mitophagy serves as a crucial element in HMOD (Figure 2, Table 1), being a component of the mitochondrial quality control system. Studies indicate that there is a correlation between mitophagy activity and hypertension, as genetic variants in the PARK2 gene, which encodes Parkin, have been found to elevate blood pressure level (88).
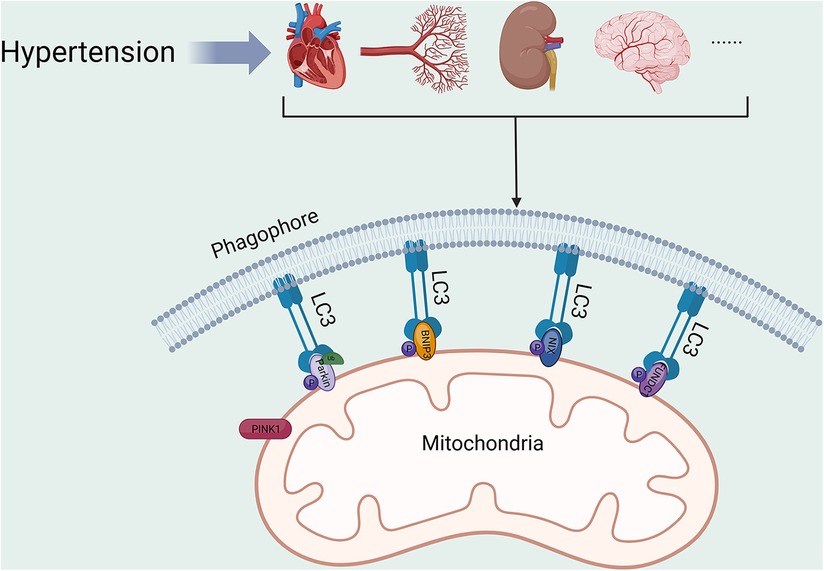
Figure 2. Mitophagy in HMOD. PINK1, PTEN-induced putative kinase protein 1; BNIP3, BCL2/adenovirus E1B 19 kDa protein-interacting protein 3; NIX, BCL2/adenovirus E1B 19 kDa protein-interacting protein 3-like; FUNDC1, FUN14 domain-containing protein 1; LC3, microtubule-associated protein 1A/1B-light chain 3. All figures were created with BioRender.com and are licensed for use.
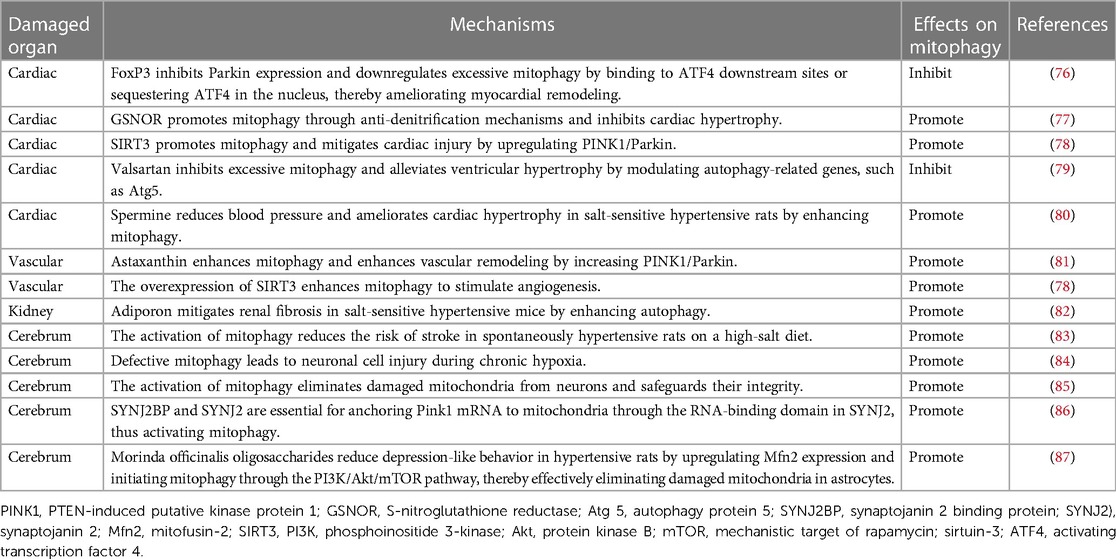
Table 1. The role of mitophagy in HMOD.
Cardiac damage is a common type of organ damage caused by hypertension. A significant quantity of mitophagy is observable in a mouse model experiencing isoprenaline-induced cardiac remodeling. Interestingly, there was also a significant upregulation of mitophagy in the left ventricle of forkhead box protein P3 (FoxP3)-KO mice. Inhibition of FoxP3 expression and down-regulation of nuclear translocation leads to excessive Parkin-mediated mitophagy and more malignant cardiac remodelling. FoxP3 can downregulate activating transcription factor 4 (ATF4), decrease Parkin expression, and inhibit mitophagy, improving cardiac remodeling through two pathways (76). Dr. Tang isolated and cultured neonatal mouse cardiomyocytes from S-nitroglutathione reductase (GSNOR)-cKO mice. Mitophagy markers were detected. The results showed that the expression of PARKIN, PINK1, LC3II/I and p62 was significantly disorganized after Ang Ⅱ treatment, which was significantly ameliorated by overexpression of mtGSNOR, suggesting that mtGSNOR inhibits cardiomyocyte hypertrophy by regulating mitophagy. Furthermore, the researchers illustrated that GSNOR interacts with adenine nucleotide translocator isoform 1 (ANT1) to decrease SNO-ANT1 levels in mitochondria, encourage mitophagy, and improve mitochondrial dysfunction in hypertrophic cardiomyocytes (77). Dr Wei conducted a study on sirtuin-3 (SIRT3)-KO mice whereby they were treated with Ang II. The results of the study showed the presence of hypoxia and microvascular reduction in the cardiac tissue of the mice. In addition, mitochondrial dysfunction and cardiac fibrosis were also observed. They conclude that SIRT3 may promote angiogenesis and thus ameliorate myocardial fibrosis by attenuating mitochondrial dysfunction caused by defective mitophagy (78). Obesity reduces mitochondrial protein and deoxyribonucleic acid content and expands mitochondrial self-degradation in hypertensive hearts. The coexistence of obesity and hypertension also contributes to myocardial fibrosis and left ventricular diastolic dysfunction. Mitochondrial function improves cardiac damage in individuals suffering from hypertension and obesity (89). Valsartan can regulate mitophagy-related genes to inhibit excessive mitophagy, decrease blood pressure, and improve ventricular hypertrophy (79). In addition, spermidine was found to inhibit the progression of heart failure by regulating mitophagy to decrease blood pressure and prevent cardiac hypertrophy and diastolic dysfunction in Dahl salt-sensitive hypertensive rats (80).
Vascular damage is prevalent among patients with hypertension, and it constitutes a significant contributing factor to the development of hypertension. Defective mitochondrial function promotes autophagy in endothelial cells, leading to reduced migratory and vasculogenic capacity, vascular endothelial dysfunction, and ultimately resulting in hypertension (90). Oxidative stress enhances mitochondrial damage, which in turn leads to vascular dysfunction, a process that is an important mechanism in the pathogenesis of hypertension. Astaxanthin promotes mitophagy and biosynthesis by upregulating the expression of Pink, Parkin, and mtDNA, resulting in restored mitochondrial function and improved vascular remodeling (81). It is likely that the decrease in angiogenic capacity of late EPCs, mediated by mitochondrial dysfunction due to defective CXCR4/JAK2/SIRT5 signal pathway, is responsible for capillary rarefaction in hypertension (91). Significant mitochondrial dysfunction was observed in rat aortic vascular smooth muscle cells treated with angiotensin II. This included increased oxygen consumption, elevated levels of ROS, reduced ATP production, decreased mtDNA levels, upregulated expression of Parkin and dynamin 1-like protein 1 (Drp1), and increased expression of peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) and transcription factor A (TFAM). Astragaloside IV has the potential to reverse the described phenomena. Based on the results, it can be inferred that Astragaloside IV is able to ameliorate the injury to vascular smooth muscle cells caused by Ang Ⅱ-induced mitochondrial dysfunction in rat aortic cells by promoting mitophagy and biosynthesis (92). Another study has shown that overexpression of SIRT3 enhances mitophagy for angiogenesis and ameliorates cardiac remodeling in hypertension (78). Increased division of endothelial mitochondria induced by Drp1 upregulation may be an important mechanism mediating vascular dysfunction in hypertension. Physical exercise or Drp1 inhibition effectively reduces mitochondrial division in endothelial cells, leading to the improvement of vascular endothelial function and the reduction of blood pressure (93). In mice injected with Ang II, Mito2HOBA enhanced Sirt3 activity and production of endothelial nitric oxide. It also reduced vascular superoxide levels, thereby improving endothelium-dependent relaxation, and decreased blood pressure. Furthermore, Mito2HOBA exhibited the ability to preserve mitochondrial respiration, protect ATP production, and reduce the opening of mitochondrial permeability pores in these Ang Ⅱ-injected mice (94).
Hypertension elevates the risk of developing tubulointerstitial fibrosis, tubular atrophy, and glomerulosclerosis (95). The SIRT2/Septin4 deacetylase pathway mitigates hypertensive kidney injury by improving oxidative stress and preventing apoptosis in renal peduncle cells (96). Stimulator of interferon genes (STING) regulates renal inflammatory response and fibrosis induced by hypertension through acyl-CoA synthetase long chain family member 4 (ACSL4)-mediated fibroblasts. The inhibitors of ACSL4, rosiglitazone, and fibroblasts inhibitor, Fer-1, downregulated mtDNA/STING dependent renal inflammation induced by Ang Ⅱ (97). Adiporon, a lipocalin receptor agonist, improves renal fibrosis by promoting autophagy in a mouse model of hypertension induced by a high salt diet (82). Alfonso Eirin's team analyzed biological samples from 25 patients diagnosed with primary hypertension, 34 patients diagnosed with secondary hypertension caused by renal vascular disease, and 22 healthy volunteers. The findings indicate that blood pressure, urinary neutrophil gelatinase protein, and kidney injury molecule-1 levels were elevated in patients with both essential hypertension and secondary hypertension caused by renal vascular disease. The researchers concluded that heightened urinary mitochondrial DNA copy number may serve as an indicator for hypertensive patients and those with renal impairment and insufficiency. This indicates that there is mitochondrial impairment in hypertensive kidney disease in human (98).
Hypertension represents a substantial risk factor for various cerebrovascular conditions, such as stroke and vascular dementia. Mitochondrial quality is crucial for the survival of neurons following ischemic insults (99). One study discovered that mitochondria located in the brain tissue of spontaneously hypertensive rats had a decreased ability to produce ATP (100). Another study indicated that losartan enhanced the metabolic of mitochondria in the brain tissue of spontaneously hypertensive rats (101). Mitophagy deficiency has been associated with various neurological disorders (102). Dr. Maurizio found that the brains of spontaneously hypertensive rats on a high-salt diet display impaired mitochondrial function. Mitophagy activation was found to reduce stroke risk (83). Chronic cerebral hypoperfusion is thought to severely affect cognitive function. Impaired autophagy or BNIP3-mediated mitophagy may serve as a mechanism for neuronal cell injury during chronic hypoxia (84). Interestingly, the process of mitophagy in ischemic neurons does not occur directly in the axon but instead takes place during the transit back to the neuronal cytosol. By activating mitophagy, damaged mitochondria are removed from neurons, ultimately providing protection (85). Interestingly, another study has indicated that synaptojanin 2 binding protein (SYNJ2BP) and synaptojanin 2 (SYNJ2) are required to tether Pink1 mRNA to mitochondria via the RNA-binding domain in SYNJ2 to activate mitophagy (86). More than half of individuals with hypertension suffer from depression (103). A recent study discovered that morinda officinalis oligosaccharides decrease depression-like behavior in hypertensive rats by increasing Mfn2 expression and initiating mitophagy through the PI3K/Akt/mTOR pathway, effectively removing damaged mitochondria in astrocytes (87). Moreover, research on hypertension induced by stress has indicated that inhibiting the HMGB1/RAGE axis increases stress-induced mitochondrial autophagic flux and reduces microglia-mediated neuroinflammation, which reduces sympathetic vasoconstriction in the ventral lateral aspect of the medulla cephaladis and decreases blood pressure (104).
The projected prevalence of hypertension suggests that it will affect 1/3 of the global population until 2025 (105). Organ damage resulting from hypertension is a significant issue that requires attention. Hypertension is tightly linked to energy metabolism, and maintaining mitochondrial homeostasis is essential for the prevention and treatment of cardiovascular disease. A significant amount of evidence indicates that mitochondrial dysfunction is strongly related to HMOD. Mitophagy, an indispensable pathway for maintaining mitochondrial quality control, plays a significant role in hypertension and HMOD, such as improving cardiac remodelling, renal fibrosis, vascular remodelling, and nerve cell damage due to hypertension. Regulation of mitophagy has a positive impact on hypertension treatment. The PINK1/Parkin-mediated pathway for mitophagy is presently one of the most comprehensively researched pathways. PINK1 kinase activity and its localization to mitochondria are necessary for inducing Parkin translocation to depolarized mitochondria. In addition, the process by which p62 is recruited to the OMM is essential for mitophagy (106). It is apparent that PINK1/Parkin plays an extremely important role in mitophagy. Furthermore, PINK1 has the ability to regulate mitophagy-independent mitochondrial dynamics through phosphorylates Drp1 S616 (107). In the absence of PINK1/Parkin, a variety of receptor proteins exist to mediate mitophagy, such as BNIP3, NIX, FUNDC1, and FKBP8.
Typically, mitophagy is regarded as a safeguard or adaptive mechanism in pathological settings. Nevertheless, in certain instances, mitophagy can have negative effects. Maintaining cellular homeostasis relies on a delicate balance in mitophagy. Swift and effective removal of damaged mitochondria is imperative to prevent the buildup of dysfunctional organelles that may disrupt cellular function. Therefore, it is of paramount importance to comprehend the regulatory mechanisms and factors influencing mitophagy activity. In summary, two potential scenarios of mitophagy exist in hypertensive states: one displaying deficient mitophagy and the other exhibiting excessive mitophagy. Both scenarios prove detrimental to organismal homeostasis. Deficient mitophagy leads to excessive accumulation of mtROS and reduction of mtDNA, which in turn impairs mitochondrial and intracellular homeostasis. When excessive mitophagy occurs, the number of normal mitochondria in the cell decreases, resulting in a shortage in the cellular energy supply chain. Most of the current evidence suggests that promoting mitophagy reduces organ or tissue damage caused by hypertension. Nonetheless, other studies have found that inhibiting excessive mitophagy may have a protective effect. These two completely contrasting phenomena may be correlated with the disease type and stage. Therefore, it is imperative to solve this enigma as soon as possible. And that is also the direction of our research. The regulation of mitophagy is incredibly complicated, and its significance in disease is of utmost importance. The investigation of molecular mechanisms that underlie mitophagy in disease progression presents an auspicious approach for creating innovative therapeutic methods. This necessitates understanding the process involved, including its underlying biological principles, to combine with such mechanisms. Moreover, the clinical implementation of these findings demands meticulous assessment of their applicability in a clinical milieu.
Despite the large number of meaningful studies on mitophagy conducted by scientists, there are still many limitations. Firstly, mitophagy is a dynamic process, and most of the current studies only observe the changes in mitophagy at a certain point in time or over a period of time, which does not fully show the development of mitophagy in an organism. This is also related to the current detection technology. Secondly, mitophagy may have opposite effects in different diseases or at different stages of the same disease, which makes the study more difficult. Finally, most of the current studies are still at the stage of basic research, and translating basic research into clinical therapeutic strategies requires a more in-depth understanding of the mechanism of mitophagy in HMOD. Although many effective antihypertensive medications are widely available, the rate of control of hypertension remains unsatisfactory. Uncontrolled hypertension results in damage to multiple target organs. The development of artificial intelligence has greatly facilitated our work. Perhaps we can use AI to discover more gene targets that play a role in mitophagy, and then screen for mitochondria-targeted drugs to treat HMOD, or develop novel nanomaterials to deliver drugs to mitochondria. In addition, searching for reliable biomarkers of mitophagy in HMOD patients and developing non-invasive methods to assess mitochondrial autophagic activity could be used for early diagnosis and monitoring of treatment effects. All these are yet to be explored and investigated.
YM: Writing – original draft. XZ: Writing – original draft. MG: Data curation, Investigation, Writing – review & editing. LY: Data curation, Investigation, Writing – review & editing. JL: Data curation, Investigation, Writing – review & editing. XC: Data curation, Investigation, Writing – review & editing. MW: Writing – review & editing. BL: Writing – review & editing. DF: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article.
This study was funded by the National Natural Science Foundation of China (82174130 and 82274262).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Collaborators GBDRF. Global burden of 87 risk factors in 204 countries and territories, 1990–2019: a systematic analysis for the global burden of disease study 2019. Lancet. (2020) 396:1223–49. doi: 10.1016/S0140-6736(20)30752-2
2. Mills KT, Stefanescu A, He J. The global epidemiology of hypertension. Nat Rev Nephrol. (2020) 16:223–37. doi: 10.1038/s41581-019-0244-2
3. Collaboration NCDRF. Worldwide trends in hypertension prevalence and progress in treatment and control from 1990 to 2019: a pooled analysis of 1201 population-representative studies with 104 million participants. Lancet. (2021) 398:957–80. doi: 10.1016/S0140-6736(21)01330-1
4. Whelton PK, Carey RM, Aronow WS, Casey DE Jr., Collins KJ, Dennison Himmelfarb C, et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American college of cardiology/American heart association task force on clinical practice guidelines. J Am Coll Cardiol. (2018) 71:e127–248. doi: 10.1016/j.jacc.2017.11.006
5. Tsao CW, Aday AW, Almarzooq ZI, Anderson CAM, Arora P, Avery CL, et al. Heart disease and stroke statistics-2023 update: a report from the American heart association. Circulation. (2023) 147:e93–e621. doi: 10.1161/CIR.0000000000001123
6. Global Burden of Metabolic Risk Factors for Chronic Diseases C. Cardiovascular disease, chronic kidney disease, and diabetes mortality burden of cardiometabolic risk factors from 1980 to 2010: a comparative risk assessment. Lancet Diabetes Endocrinol. (2014) 2:634–47. doi: 10.1016/S2213-8587(14)70102-0
7. Lewington S, Clarke R, Qizilbash N, Peto R, Collins R, Prospective Studies C. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet. (2002) 360:1903–13. doi: 10.1016/s0140-6736(02)11911-8
8. Collaborators GBDRF. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990-2015: a systematic analysis for the global burden of disease study 2015. Lancet. (2016) 388:1659–724. doi: 10.1016/S0140-6736(16)31679-8
9. Olsen MH, Angell SY, Asma S, Boutouyrie P, Burger D, Chirinos JA, et al. A call to action and a lifecourse strategy to address the global burden of raised blood pressure on current and future generations: the lancet commission on hypertension. Lancet. (2016) 388:2665–712. doi: 10.1016/S0140-6736(16)31134-5
10. Lane N, Martin W. The energetics of genome complexity. Nature. (2010) 467:929–34. doi: 10.1038/nature09486
11. Nunnari J, Suomalainen A. Mitochondria: in sickness and in health. Cell. (2012) 148:1145–59. doi: 10.1016/j.cell.2012.02.035
12. Ni HM, Williams JA, Ding WX. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. (2015) 4:6–13. doi: 10.1016/j.redox.2014.11.006
13. Sygitowicz G, Sitkiewicz D. Mitochondrial quality control: the role in cardiac injury. Front Biosci. (2022) 27:96. doi: 10.31083/j.fbl2703096
14. Bao F, Zhou L, Xiao J, Liu X. Mitolysosome exocytosis: a novel mitochondrial quality control pathway linked with parkinsonism-like symptoms. Biochem Soc Trans. (2022) 50:1773–83. doi: 10.1042/BST20220726
15. Abate M, Festa A, Falco M, Lombardi A, Luce A, Grimaldi A, et al. Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin Cell Dev Biol. (2020) 98:139–53. doi: 10.1016/j.semcdb.2019.05.022
16. Wu NN, Zhang Y, Ren J. Mitophagy, mitochondrial dynamics, and homeostasis in cardiovascular aging. Oxid Med Cell Longev. (2019) 2019:9825061. doi: 10.1155/2019/9825061
17. Morales PE, Arias-Duran C, Avalos-Guajardo Y, Aedo G, Verdejo HE, Parra V, et al. Emerging role of mitophagy in cardiovascular physiology and pathology. Mol Aspects Med. (2020) 71:100822. doi: 10.1016/j.mam.2019.09.006
18. Zhou H, He L, Xu G, Chen L. Mitophagy in cardiovascular disease. Clin Chim Acta. (2020) 507:210–8. doi: 10.1016/j.cca.2020.04.033
19. Annesley SJ, Fisher PR. Mitochondria in health and disease. Cells. (2019) 8:680. doi: 10.3390/cells8070680
20. Pickles S, Vigie P, Youle RJ. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr Biol. (2018) 28:R170–R85. doi: 10.1016/j.cub.2018.01.004
21. Khaminets A, Behl C, Dikic I. Ubiquitin-dependent and independent signals in selective autophagy. Trends Cell Biol. (2016) 26:6–16. doi: 10.1016/j.tcb.2015.08.010
22. Harper JW, Ordureau A, Heo JM. Building and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol. (2018) 19:93–108. doi: 10.1038/nrm.2017.129
23. Sekine S, Youle RJ. PINK1 Import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. (2018) 16:2. doi: 10.1186/s12915-017-0470-7
24. Hasson SA, Kane LA, Yamano K, Huang CH, Sliter DA, Buehler E, et al. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature. (2013) 504:291–5. doi: 10.1038/nature12748
25. Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, et al. PINK1 Is selectively stabilized on impaired mitochondria to activate parkin. PLoS Biol. (2010) 8:e1000298. doi: 10.1371/journal.pbio.1000298
26. Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. (2015) 524:309–14. doi: 10.1038/nature14893
27. Kondo-Okamoto N, Noda NN, Suzuki SW, Nakatogawa H, Takahashi I, Matsunami M, et al. Autophagy-related protein 32 acts as autophagic degron and directly initiates mitophagy. J Biol Chem. (2012) 287:10631–8. doi: 10.1074/jbc.M111.299917
28. Iguchi M, Kujuro Y, Okatsu K, Koyano F, Kosako H, Kimura M, et al. Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J Biol Chem. (2013) 288:22019–32. doi: 10.1074/jbc.M113.467530
29. Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, et al. PINK1 Phosphorylates ubiquitin to activate parkin E3 ubiquitin ligase activity. J Cell Biol. (2014) 205:143–53. doi: 10.1083/jcb.201402104
30. Palikaras K, Lionaki E, Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat Cell Biol. (2018) 20:1013–22. doi: 10.1038/s41556-018-0176-2
31. Lamark T, Johansen T. Mechanisms of selective autophagy. Annu Rev Cell Dev Biol. (2021) 37:143–69. doi: 10.1146/annurev-cellbio-120219-035530
32. Okatsu K, Koyano F, Kimura M, Kosako H, Saeki Y, Tanaka K, et al. Phosphorylated ubiquitin chain is the genuine parkin receptor. J Cell Biol. (2015) 209:111–28. doi: 10.1083/jcb.201410050
33. Bingol B, Sheng M. Mechanisms of mitophagy: PINK1, parkin, USP30 and beyond. Free Radic Biol Med. (2016) 100:210–22. doi: 10.1016/j.freeradbiomed.2016.04.015
34. Di Rienzo M, Romagnoli A, Ciccosanti F, Refolo G, Consalvi V, Arena G, et al. AMBRA1 regulates mitophagy by interacting with ATAD3A and promoting PINK1 stability. Autophagy. (2022) 18:1752–62. doi: 10.1080/15548627.2021.1997052
35. Xie XQ, Yang Y, Wang Q, Liu HF, Fang XY, Li CL, et al. Targeting ATAD3A-PINK1-mitophagy axis overcomes chemoimmunotherapy resistance by redirecting PD-L1 to mitochondria. Cell Res. (2023) 33:215–28. doi: 10.1038/s41422-022-00766-z
36. Wang SH, Zhu XL, Wang F, Chen SX, Chen ZT, Qiu Q, et al. LncRNA H19 governs mitophagy and restores mitochondrial respiration in the heart through Pink1/parkin signaling during obesity. Cell Death Dis. (2021) 12:557. doi: 10.1038/s41419-021-03821-6
37. Manaa WE, Duplan E, Goiran T, Lauritzen I, Vaillant Beuchot L, Lacas-Gervais S, et al. Transcription- and phosphorylation-dependent control of a functional interplay between XBP1s and PINK1 governs mitophagy and potentially impacts Parkinson disease pathophysiology. Autophagy. (2021) 17:4363–85. doi: 10.1080/15548627.2021.1917129
38. Yin K, Lee J, Liu Z, Kim H, Martin DR, Wu D, et al. Mitophagy protein PINK1 suppresses colon tumor growth by metabolic reprogramming via p53 activation and reducing acetyl-CoA production. Cell Death Differ. (2021) 28:2421–35. doi: 10.1038/s41418-021-00760-9
39. Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. (2004) 116:205–19. doi: 10.1016/s0092-8674(04)00046-7
40. Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. (2009) 16:939–46. doi: 10.1038/cdd.2009.16
41. Lee H, Paik SG. Regulation of BNIP3 in normal and cancer cells. Mol Cells. (2006) 21:1–6. doi: 10.1016/j.molcel.2005.12.001
42. Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. (2012) 287:19094–104. doi: 10.1074/jbc.M111.322933
43. Rogov VV, Suzuki H, Marinkovic M, Lang V, Kato R, Kawasaki M, et al. Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci Rep. (2017) 7:1131. doi: 10.1038/s41598-017-01258-6
44. Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. (2001) 61:6669–73.11559532
45. Tracy K, Dibling BC, Spike BT, Knabb JR, Schumacker P, Macleod KF. BNIP3 Is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol. (2007) 27:6229–42. doi: 10.1128/MCB.02246-06
46. Chourasia AH, Tracy K, Frankenberger C, Boland ML, Sharifi MN, Drake LE, et al. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. (2015) 16:1145–63. doi: 10.15252/embr.201540759
47. Fu ZJ, Wang ZY, Xu L, Chen XH, Li XX, Liao WT, et al. HIF-1alpha-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol. (2020) 36:101671. doi: 10.1016/j.redox.2020.101671
48. Marinkovic M, Sprung M, Novak I. Dimerization of mitophagy receptor BNIP3l/NIX is essential for recruitment of autophagic machinery. Autophagy. (2021) 17:1232–43. doi: 10.1080/15548627.2020.1755120
49. Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, et al. NIX Is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci U S A. (2007) 104:19500–5. doi: 10.1073/pnas.0708818104
50. Zhang J, Ney PA. NIX Induces mitochondrial autophagy in reticulocytes. Autophagy. (2008) 4:354–6. doi: 10.4161/auto.5552
51. Cao Y, Zheng J, Wan H, Sun Y, Fu S, Liu S, et al. A mitochondrial SCF-FBXL4 ubiquitin E3 ligase complex degrades BNIP3 and NIX to restrain mitophagy and prevent mitochondrial disease. EMBO J. (2023) 42:e113033. doi: 10.15252/embj.2022113033
52. Salminen A, Kaarniranta K, Kauppinen A, Ojala J, Haapasalo A, Soininen H, et al. Impaired autophagy and APP processing in Alzheimer’s disease: the potential role of Beclin 1 interactome. Prog Neurobiol. (2013) 106–107:33–54. doi: 10.1016/j.pneurobio.2013.06.002
53. Yao J, Wang J, Xu Y, Guo Q, Sun Y, Liu J, et al. CDK9 Inhibition blocks the initiation of PINK1-PRKN-mediated mitophagy by regulating the SIRT1-FOXO3-BNIP3 axis and enhances the therapeutic effects involving mitochondrial dysfunction in hepatocellular carcinoma. Autophagy. (2022) 18:1879–97. doi: 10.1080/15548627.2021.2007027
54. Gao F, Chen D, Si J, Hu Q, Qin Z, Fang M, et al. The mitochondrial protein BNIP3l is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum Mol Genet. (2015) 24:2528–38. doi: 10.1093/hmg/ddv017
55. Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. (2012) 14:177–85. doi: 10.1038/ncb2422
56. Zhang W. The mitophagy receptor FUN14 domain-containing 1 (FUNDC1): a promising biomarker and potential therapeutic target of human diseases. Genes Dis. (2021) 8:640–54. doi: 10.1016/j.gendis.2020.08.011
57. Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell. (2014) 54:362–77. doi: 10.1016/j.molcel.2014.02.034
58. Zhang W, Siraj S, Zhang R, Chen Q. Mitophagy receptor FUNDC1 regulates mitochondrial homeostasis and protects the heart from I/R injury. Autophagy. (2017) 13:1080–1. doi: 10.1080/15548627.2017.1300224
59. Wu W, Tian W, Hu Z, Chen G, Huang L, Li W, et al. ULK1 Translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep. (2014) 15:566–75. doi: 10.1002/embr.201438501
60. Murakawa T, Okamoto K, Omiya S, Taneike M, Yamaguchi O, Otsu K. A mammalian mitophagy receptor, Bcl2-L-13, recruits the ULK1 complex to induce mitophagy. Cell Rep. (2019) 26:338–45.e6. doi: 10.1016/j.celrep.2018.12.050
61. Lim GG, Lim KL. Parkin-independent mitophagy-FKBP8 takes the stage. EMBO Rep. (2017) 18:864–5. doi: 10.15252/embr.201744313
62. Lin M, Xian H, Chen Z, Wang S, Liu M, Liang W, et al. MCM8-mediated mitophagy protects vascular health in response to nitric oxide signaling in a mouse model of kawasaki disease. Nat Cardiovasc Res. (2023) 2:778–92. doi: 10.1038/s44161-023-00314-x
63. Murakawa T, Yamaguchi O, Hashimoto A, Hikoso S, Takeda T, Oka T, et al. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun. (2015) 6:7527. doi: 10.1038/ncomms8527
64. Liang W, Sagar S, Ravindran R, Najor RH, Quiles JM, Chi L, et al. Mitochondria are secreted in extracellular vesicles when lysosomal function is impaired. Nat Commun. (2023) 14:5031. doi: 10.1038/s41467-023-40680-5
65. Bao F, Zhou L, Zhou R, Huang Q, Chen J, Zeng S, et al. Mitolysosome exocytosis, a mitophagy-independent mitochondrial quality control in flunarizine-induced parkinsonism-like symptoms. Sci Adv. (2022) 8:eabk2376. doi: 10.1126/sciadv.abk2376
66. Jiao H, Jiang D, Hu X, Du W, Ji L, Yang Y, et al. Mitocytosis, a migrasome-mediated mitochondrial quality-control process. Cell. (2021) 184:2896–910.e13. doi: 10.1016/j.cell.2021.04.027
67. Xie X, Atkins E, Lv J, Bennett A, Neal B, Ninomiya T, et al. Effects of intensive blood pressure lowering on cardiovascular and renal outcomes: updated systematic review and meta-analysis. Lancet. (2016) 387:435–43. doi: 10.1016/S0140-6736(15)00805-3
68. Kennelly SP, Lawlor BA, Kenny RA. Blood pressure and dementia—a comprehensive review. Ther Adv Neurol Disord. (2009) 2:241–60. doi: 10.1177/1756285609103483
69. Sharp SI, Aarsland D, Day S, Sonnesyn H, Alzheimer’s Society Vascular Dementia Systematic Review G, Ballard C. Hypertension is a potential risk factor for vascular dementia: systematic review. Int J Geriatr Psychiatry. (2011) 26:661–9. doi: 10.1002/gps.2572
70. Dikalov SI, Dikalova AE. Crosstalk between mitochondrial hyperacetylation and oxidative stress in vascular dysfunction and hypertension. Antioxid Redox Signal. (2019) 31:710–21. doi: 10.1089/ars.2018.7632
71. Liu Y, Li Y, Wang X, Ma Q, Zhu C, Li Z, et al. Mitochondrial tRNA mutations in Chinese hypertensive individuals. Mitochondrion. (2016) 28:1–7. doi: 10.1016/j.mito.2016.02.007
72. Eirin A, Williams BJ, Ebrahimi B, Zhang X, Crane JA, Lerman A, et al. Mitochondrial targeted peptides attenuate residual myocardial damage after reversal of experimental renovascular hypertension. J Hypertens. (2014) 32:154–65. doi: 10.1097/HJH.0b013e3283658a53
73. Tang Y, Mi C, Liu J, Gao F, Long J. Compromised mitochondrial remodeling in compensatory hypertrophied myocardium of spontaneously hypertensive rat. Cardiovasc Pathol. (2014) 23:101–6. doi: 10.1016/j.carpath.2013.11.002
74. Rimbaud S, Ruiz M, Piquereau J, Mateo P, Fortin D, Veksler V, et al. Resveratrol improves survival, hemodynamics and energetics in a rat model of hypertension leading to heart failure. PLoS One. (2011) 6:e26391. doi: 10.1371/journal.pone.0026391
75. Chen G, Lin Y, Chen L, Zeng F, Zhang L, Huang Y, et al. Role of DRAM1 in mitophagy contributes to preeclampsia regulation in mice. Mol Med Rep. (2020) 22:1847–58. doi: 10.3892/mmr.2020.11269
76. Pan XC, Xiong YL, Hong JH, Liu Y, Cen YY, Liu T, et al. Cardiomyocytic FoxP3 is involved in Parkin-mediated mitophagy during cardiac remodeling and the regulatory role of triptolide. Theranostics. (2022) 12:2483–501. doi: 10.7150/thno.71102
77. Tang X, Zhao S, Liu J, Liu X, Sha X, Huang C, et al. Mitochondrial GSNOR alleviates cardiac dysfunction via ANT1 denitrosylation. Circ Res. (2023) 133:220–36. doi: 10.1161/CIRCRESAHA.123.322654
78. Wei T, Huang G, Gao J, Huang C, Sun M, Wu J, et al. Sirtuin 3 deficiency accelerates hypertensive cardiac remodeling by impairing angiogenesis. J Am Heart Assoc. (2017) 6:e006114. doi: 10.1161/JAHA.117.006114
79. Zhang X, Li ZL, Crane JA, Jordan KL, Pawar AS, Textor SC, et al. Valsartan regulates myocardial autophagy and mitochondrial turnover in experimental hypertension. Hypertension. (2014) 64:87–93. doi: 10.1161/HYPERTENSIONAHA.113.02151
80. Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. (2016) 22:1428–38. doi: 10.1038/nm.4222
81. Chen Y, Li S, Guo Y, Yu H, Bao Y, Xin X, et al. Astaxanthin attenuates hypertensive vascular remodeling by protecting vascular smooth muscle cells from oxidative stress-induced mitochondrial dysfunction. Oxid Med Cell Longev. (2020) 2020:4629189. doi: 10.1155/2020/4629189
82. Li Y, Song B, Ruan C, Xue W, Zhao J. Adiporon attenuates hypertension-induced epithelial-mesenchymal transition and renal fibrosis via promoting epithelial autophagy. J Cardiovasc Transl Res. (2021) 14:538–45. doi: 10.1007/s12265-020-10075-8
83. Forte M, Bianchi F, Cotugno M, Marchitti S, De Falco E, Raffa S, et al. Pharmacological restoration of autophagy reduces hypertension-related stroke occurrence. Autophagy. (2020) 16:1468–81. doi: 10.1080/15548627.2019.1687215
84. Song Y, Du Y, Zou W, Luo Y, Zhang X, Fu J. Involvement of impaired autophagy and mitophagy in neuro-2a cell damage under hypoxic and/or high-glucose conditions. Sci Rep. (2018) 8:3301. doi: 10.1038/s41598-018-20162-1
85. Zheng Y, Zhang X, Wu X, Jiang L, Ahsan A, Ma S, et al. Somatic autophagy of axonal mitochondria in ischemic neurons. J Cell Biol. (2019) 218:1891–907. doi: 10.1083/jcb.201804101
86. Harbauer AB, Hees JT, Wanderoy S, Segura I, Gibbs W, Cheng Y, et al. Neuronal mitochondria transport Pink1 mRNA via synaptojanin 2 to support local mitophagy. Neuron. (2022) 110:1516–31.e9. doi: 10.1016/j.neuron.2022.01.035
87. Yang L, Ao Y, Li Y, Dai B, Li J, Duan W, et al. Morinda officinalis oligosaccharides mitigate depression-like behaviors in hypertension rats by regulating Mfn2-mediated mitophagy. J Neuroinflammation. (2023) 20:31. doi: 10.1186/s12974-023-02715-y
88. Jin HS, Hong KW, Kim BY, Kim J, Yoo YH, Oh B, et al. Replicated association between genetic variation in the PARK2 gene and blood pressure. Clin Chim Acta. (2011) 412:1673–7. doi: 10.1016/j.cca.2011.05.026
89. Badimon L, Onate B, Vilahur G. Adipose-derived mesenchymal stem cells and their reparative potential in ischemic heart disease. Rev Esp Cardiol (Engl Ed). (2015) 68:599–611. doi: 10.1016/j.rec.2015.02.025
90. Zhao X, Cui L, Xiao Y, Mao Q, Aishanjiang M, Kong W, et al. Hypertension-associated mitochondrial DNA 4401A >G mutation caused the aberrant processing of tRNAMet, all 8 tRNAs and ND6 mRNA in the light-strand transcript. Nucleic Acids Res. (2019) 47:10340–56. doi: 10.1093/nar/gkz742
91. Yu BB, Zhi H, Zhang XY, Liang JW, He J, Su C, et al. Mitochondrial dysfunction-mediated decline in angiogenic capacity of endothelial progenitor cells is associated with capillary rarefaction in patients with hypertension via downregulation of CXCR4/JAK2/SIRT5 signaling. EBioMedicine. (2019) 42:64–75. doi: 10.1016/j.ebiom.2019.03.031
92. Lu Y, Li S, Wu H, Bian Z, Xu J, Gu C, et al. Beneficial effects of astragaloside IV against angiotensin II-induced mitochondrial dysfunction in rat vascular smooth muscle cells. Int J Mol Med. (2015) 36:1223–32. doi: 10.3892/ijmm.2015.2345
93. Li G, Xu K, Xing W, Yang H, Li Y, Wang X, et al. Swimming exercise alleviates endothelial mitochondrial fragmentation via inhibiting dynamin-related protein-1 to improve vascular function in hypertension. Hypertension. (2022) 79:e116–e28. doi: 10.1161/HYPERTENSIONAHA.122.19126
94. Dikalova A, Mayorov V, Xiao L, Panov A, Amarnath V, Zagol-Ikapitte I, et al. Mitochondrial isolevuglandins contribute to vascular oxidative stress and mitochondria-targeted scavenger of isolevuglandins reduces mitochondrial dysfunction and hypertension. Hypertension. (2020) 76:1980–91. doi: 10.1161/HYPERTENSIONAHA.120.15236
95. Seccia TM, Caroccia B, Calo LA. Hypertensive nephropathy. Moving from classic to emerging pathogenetic mechanisms. J Hypertens. (2017) 35:205–12. doi: 10.1097/HJH.0000000000001170
96. Zhang Y, Zhang N, Zou Y, Song C, Cao K, Wu B, et al. Deacetylation of Septin4 by SIRT2 (silent mating type information regulation 2 homolog-2) mitigates damaging of hypertensive nephropathy. Circ Res. (2023) 132:601–24. doi: 10.1161/CIRCRESAHA.122.321591
97. Gao L, Zhang J, Yang T, Jiang L, Liu X, Wang S, et al. STING/ACSL4 axis-dependent ferroptosis and inflammation promote hypertension-associated chronic kidney disease. Mol Ther. (2023) 31:3084–103. doi: 10.1016/j.ymthe.2023.07.026
98. Eirin A, Saad A, Tang H, Herrmann SM, Woollard JR, Lerman A, et al. Urinary mitochondrial DNA copy number identifies chronic renal injury in hypertensive patients. Hypertension. (2016) 68:401–10. doi: 10.1161/HYPERTENSIONAHA.116.07849
99. An H, Zhou B, Ji X. Mitochondrial quality control in acute ischemic stroke. J Cereb Blood Flow Metab. (2021) 41:3157–70. doi: 10.1177/0271678X211046992
100. Doroshchuk AD, Postnov A, Afanas’eva GV, Budnikov E, Postnov Iu V. Decreased ATP-synthesis ability of brain mitochondria in spontaneously hypertensive rats. Kardiologiia. (2004) 44:64–5.15489849
101. Sumbalova Z, Kucharska J, Kristek F. Losartan improved respiratory function and coenzyme Q content in brain mitochondria of young spontaneously hypertensive rats. Cell Mol Neurobiol. (2010) 30:751–8. doi: 10.1007/s10571-010-9501-4
102. Karbowski M, Neutzner A. Neurodegeneration as a consequence of failed mitochondrial maintenance. Acta Neuropathol. (2012) 123:157–71. doi: 10.1007/s00401-011-0921-0
103. Guan S, Fang X, Gu X, Zhang Z, Tang Z, Wu X, et al. The link of depression, untreated hypertension, and diabetes with mortality in postmenopausal women: a cohort study. Clin Exp Hypertens. (2021) 43:1–6. doi: 10.1080/10641963.2020.1790584
104. Zhang S, Hu L, Jiang J, Li H, Wu Q, Ooi K, et al. HMGB1/RAGE axis mediates stress-induced RVLM neuroinflammation in mice via impairing mitophagy flux in microglia. J Neuroinflammation. (2020) 17:15. doi: 10.1186/s12974-019-1673-3
105. Oliveros E, Patel H, Kyung S, Fugar S, Goldberg A, Madan N, et al. Hypertension in older adults: assessment, management, and challenges. Clin Cardiol. (2020) 43:99–107. doi: 10.1002/clc.23303
106. Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. (2010) 12:119–31. doi: 10.1038/ncb2012
Keywords: hypertension, organ damage, mitochondria, mitochondrial quality control, mitophagy
Citation: Ma Y, Zhou X, Gui M, Yao L, Li J, Chen X, Wang M, Lu B and Fu D (2024) Mitophagy in hypertension-mediated organ damage. Front. Cardiovasc. Med. 10:1309863. doi: 10.3389/fcvm.2023.1309863
Received: 8 October 2023; Accepted: 14 December 2023;
Published: 4 January 2024.
Edited by:
Gaurav Kumar, Medical College of Wisconsin, United StatesReviewed by:
Aravind Parthasarathy, Medical College of Wisconsin, United States© 2024 Ma, Zhou, Gui, Yao, Li, Chen, Wang, Lu and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deyu Fu ZmR5NjVAMTI2LmNvbQ== Bo Lu bHVibzIwMDYwOUAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.