Corrigendum: FcγRIIa—dependent platelet activation identified in COVID-19 vaccine-induced immune thrombotic thrombocytopenia-, heparininduced thrombocytopenia, streptokinase- and anisoylated plasminogen-streptokinase activator complex-induced platelet activation
 Mustapha Abdelouahed
Mustapha Abdelouahed Dana Yateem2
Dana Yateem2 Salim Fredericks
Salim Fredericks- 1Department of Medical Sciences and Education, Boston University School of Medicine, Boston, MA, United States
- 2School of Medicine, The Royal College of Surgeons in Ireland, Medical University of Bahrain, Al Sayh, Muharraq Governorate, Bahrain
Coronavirus disease 2019 (COVID-19), which was caused by the coronavirus - severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), was globally responsible for remarkable morbidity and mortality. Several highly effective vaccines for COVID-19 were developed and disseminated worldwide within an unprecedented timescale. Rare but dangerous clotting and thrombocytopenia events, and subsequent coagulation abnormalities, have been reported after massive vaccination against SARS-CoV-2. Soon after their global rollout, reports of a morbid clinical syndrome following vaccination with adenovirus-DNA-based vaccines appeared. In the spring of 2021, reports of a novel, rare and morbid clinical syndrome, with clinically devastating and fatal complication after vaccination with adenovirus-based coronavirus vaccines (Janssen/Johnson & Johnson and Astra-Zeneca vaccines) led to a brief suspension of their use by several countries. Those complications were associated with unusual cerebral and splanchnic venous thrombosis, and circulating autoantibodies directed against anti-platelet factor 4 (PF4), a protein secreted from platelets, leading to the designation: Vaccine-Induced Immune Thrombotic Thrombocytopenia (VITT). The reported VITT incidence remains very low and does not affect the overall benefit of immunization, however, if left untreated, VITT can be debilitating or even fatal. VITT resembled specific adverse drugs' reactions that also involved the production of autoantibodies and subsequent abnormal platelet activation through platelet FcγRIIa. These unusual but well-documented drug reactions were heparin-induced thrombocytopenia (HIT), streptokinase- (SK), and anisoylated plasminogen-streptokinase activator complex- (APSAC) associated with platelet-activating antibodies. There was considerable overlapping of clinical features between VITT, COVID-19 and these adverse drugs' reactions. We review the phenomenon of VITT against the backdrop of shared and common mechanisms that underlie HIT-, SK-, and APSAC-platelet FcγRIIa-dependent platelet activation. An understanding of VITT's pathogenesis may be achieved by comparing and contrasting VITT-, HIT-, SK- and APSAC-induced platelet activation mechanisms, their respective physiopathology and similarities. Discussing these conditions in parallel provides insight into complex immunological disorders and diseases associated with abnormal hemostasis and thrombosis in particular.
Introduction
Coronavirus disease 2019 (COVID-19), which was caused by the coronavirus - severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), was globally responsible for remarkable morbidity and mortality. The SARS-CoV-2 virus deploys its “spike protein” in order to gain entry to cells by chemically associating with angiotensin-converting enzyme 2 (ACE2) located on the outer membrane of the cells of the host (1, 2). The trans-membrane protease serine 2 (TMPRSS2), a serine protease, enzymatically cleaves and proceeds to activate the spike protein to fuse the SARS-CoV-2 virus with the cell membrane. This spike protein, a uniquely recognizable virus component, became the target for vaccine manufacturers. Several highly effective vaccines were developed for COVID-19. These vaccines were designed to trigger the immune system, thus resulting in the production of neutralizing antibodies with the ability to identify the SARS-CoV-2 virus. European and US regulatory authorities approved two technological approaches to producing and administering COVID-19 vaccines. These were lipid nanoparticles encapsulating an mRNA payload acting as a template for the SARS-CoV-2 spike protein (BioNTech/Pfizer and Moderna), and replication-attenuated adenovirus vectors carrying a DNA payload encoding the same spike protein (Ad26.COV.2.S or Janssen/Johnson & Johnson and ChAdOx1 nCoV-19 or Astra-Zeneca). All available vaccines, whether they employed mRNA or DNA, led to the host cells' expression of the SARS-CoV-2 spike protein and elicitation of protective immunity (3). The adenovirus-DNA-based vaccines became caught up in a controversy that led to their temporary suspension in several countries. Midway through 2021, reports of a novel, rare and morbid syndrome with devastating and fatal complications after vaccination were published. It was associated with atypically located cerebral and splanchnic venous thrombosis, abnormal platelet activation, and circulating autoantibodies, which attach themselves to the platelet-secreted protein, platelet factor 4 (PF4). This adverse reaction was designated vaccine-induced immune thrombotic thrombocytopenia (VITT) (4–8).
The proposed pathological mechanism of VITT was the production of these antibodies that were directed against the naturally occurring human protein PF4 as a reaction to the vaccine. This led to massive platelet aggregation. It is also conceivable that the thrombocytopenia following vaccination had multiple causes. Other factors may have been necessary for the development of VITT. The proteins ACE2 and TMPRSS2 are abundantly expressed in platelets. These membrane proteins are responsible for spike protein priming (9, 10). Therefore, it was no surprise that platelets would have been involved in the clinical manifestations of COVID-19 disease. However, the function played by platelets in this post-vaccine adverse reaction was more complex than mere virus-protein to host-protein interactions.
Platelets are crucial conduits conjoining the hemostatic system and immune defenses. This includes those immunological mechanisms and processes protecting against viral disease. Platelets also serve as mediators involved in vital elements of inflammatory processes and are instrumental in modulating responses of the immune system to both “self” and “foreign” molecules (11, 12). The cellular phenomenon of platelet activation is known for its crucial contributions to thrombus formation. Hence platelet-activation's role in preventing bleeding and minimizing vascular injury. However, along with this physiological role comes a pathological role, played out in deleterious thrombotic events and associated cardiovascular sequelae. Not dissimilar to innate immune cells, platelets express an array of immune-associated molecules. These include receptor molecules for: IgG's Fc-region, C-type lectin and various complement and chemokine molecules. They also express toll-like receptors (TLRs), and nucleotide-binding and oligomerization domain–like receptors (13, 14). These receptors recognize pathogens such as dengue, human immunodeficiency, and influenza viruses. When in contact with pathogens, platelets mediate immune responses indirectly through the release of cytokines, anti-microbial peptides and directly through interaction with neutrophils, monocytes, and lymphocytes. The net result of which is the amplification of the immune response (15). Furthermore platelets possess a plethora of coagulation factors and inflammatory factors. These are stowed in their α-granules, only to be secreted upon platelet activation to boost further the cascade of events that comprise coagulation (16, 17). Factors V and XIII function as sites of adhesion for the binding of coagulation factors activation via their cell surface exposure to phosphatidylserine (18).
The descriptions of VITT are recent, and research on its pathophysiology is limited. Pathogenicity of VITT may involve both host and vaccine factors (19). However, several grey areas remain regarding knowledge of VITT and its pathophysiology. This poses a stark standout question. How could a strand of DNA coding for a viral protein delivered in an adenovirus vector generate autoantibodies against a human physiologically functional protein such as PF4? The answer may lay in adverse drug reactions involving totally different chemical compounds. The clinical and laboratory details of this adverse reaction to the COVID-19 vaccines revealed intriguing similarities to previously well-documented adverse drug reactions. Platelet activation and the enhancement of platelet activation by autoantibodies through platelet FcγRIIa is a common feature of VITT with remarkable similarities to heparin-induced thrombocytopenia (HIT), streptokinase (SK), and anisoylated plasminogen-streptokinase activator complex (APSAC) associated platelet-activating antibodies through platelet FcγRIIa. Accounting for commonalities between the various responses to these medications may expand our knowledge of the pathogenesis of VITT. Comparing and contrasting elements of the VITT response to a vaccine with these other abnormal responses to drugs may allow us to better understand VITT and the mechanisms underpinning adverse reactions to heparin, SK and APSAC. In order to comprehensively assess VITT, in light of the responses to these drugs, it is necessary to review elements of the pathology of COVID-19, the vaccines, platelet function and the role of platelets in the above-mentioned adverse drug reactions.
COVID-19 and thrombosis
The putative cause of VITT is anti-PF4 antibodies inducing pathological platelet activation through FcγRIIa, a protein located on the outer membrane of platelets. Circulating immunoglobulin-G antibodies with the ability to induce platelet activation via these FcγRIIa proteins have also been detected in patients with SARS-CoV-2 virus infection. Therefore, this necessitates a brief exploration of some features of COVID-19 itself in relation to VITT.
Thromboembolic complications are prominent clinical features of COVID-19 (20). Thrombosis in atypical locations, such as cerebral veins, has been reported in patients afflicted with COVID-19. A notable overlap exists in the clinical presentation between VITT and COVID-19, i.e., unusual thrombotic complications have been associated with patients with both conditions. Less severe thrombocytopenia occurs frequently with patients with severe COVID-19 with or without it being leagued with hemostasis activation and systemic inflammation, including fibrinolytic shutdown (21, 22). Substantial proinflammatory aspects, indicate a so-called “cytokine storm”, may often occur with COVID-19 patients. Thus being implicated in the pro-thrombotic syndrome (23). Thrombocytopenia occurred more often in patients with the more severe forms of COVID-19 (24). Twenty per cent of patients with COVID-19 had low platelet counts at the time of hospital admission (25). Among survivors of COVID-19, blood platelet counts were inclined to increase within the second week of hospitalization, whereas, among non-survivors, low blood platelet counts were more inclined to remain low or worsen (26). There is a body of evidence that suggests that COVID-19 predisposes individuals to thromboembolic complications, including arterial and venous thrombotic events, as well as thrombocytopenia. These have all been significantly associated with mortality (27). Reports from several independent research groups described platelets as being hyperreactive during COVID-19 disease, this being coupled with abnormalities of gene expression in platelets. This potentially influences interactions between white blood cells and platelets (28). Quiescent platelets isolated from the blood of patients with COVID-19 had abnormally high expression of P-selectin upon activation. These patients also had raised levels of circulating platelet-monocyte, platelet-neutrophil, and platelet-T-cell aggregates (29–31). Other markers of thrombin generation or activation have been associated with COVID-19. These include thrombin-antithrombin complex as well as prothrombin fragments (32).
The immune system's responses to PF4 involve separate mechanisms from the immune responses to the spike protein of SARS-CoV-2 virus particle. Further, VITT antibodies have been shown not to bind to the spike protein of SARS-CoV-2. This suggests that the response of the immune system to the SARS-CoV-2 spike protein in the vaccine does not induce VITT (33). When discussing VITT, the intended and non-intended immune responses to the vaccine should not be conflated with the immune response to the viral infection of SAR-CoV-2. However, due to the shared and common elements, discussions of the pathogenesis of VITT are here addressed in parallel with issues related to COVID-19 and the responses to specific drugs.
Vaccine-induced immune thrombotic thrombocytopenia (VITT)
Clinically, VITT is associated with complications related to the hemostatic system (Tables 1, 2). These may be life-threatening thromboses such as deep venous thrombosis, cerebral venous sinus thrombosis, and splanchnic vein thrombosis. These thrombotic events occur paralleled with thrombocytopenia, elevated plasma soluble D-dimer concentrations and raised numbers of activated platelets. Also, there is a detectable amount of anti-PF4-polyanion immunoglobulin antibodies in serum (47, 48). European studies showed an incidence of around 1 per 100,000–250,000 vaccine recipients. More than 90% of these patients were below the age of 60 years and disproportionately female. Fatalities among these VITT patients were reported to be approximately 20%. This relatively low figure may be due to a lag in recognizing and identifying the symptoms and signs by patients and healthcare providers (49).
TABLE 1
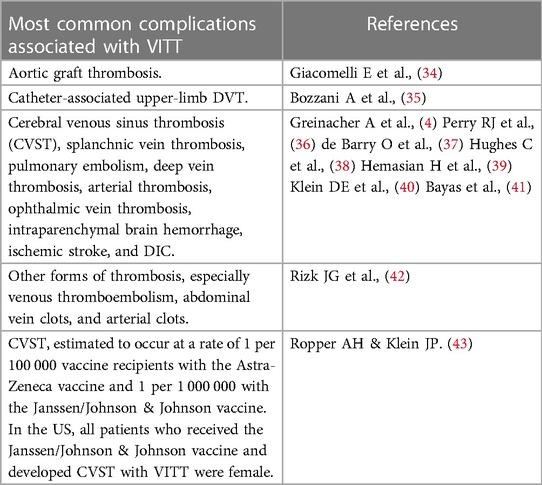
Table 1. Most common complications associated with VITT.
TABLE 2

Table 2. Abnormal laboratory and radiologic abnormalities occurring in VITT.
HIT and the severe form of COVID-19 disease share several common clinical similarities. These two conditions only occur in a small minority of patients infected with SARS-CoV-2 or those patients exposed to heparin. Dissimilar to the related HIT, with an estimated incidence of 1 to 6% of individuals anticoagulated with heparin and a corresponding mortality rate of 25% (50). VITT has been reported as 1:150,000 of Astra-Zeneca recipients and 1:470,000 of Janssen/Johnson & Johnson recipients. These figures carry a mortality rate of 20%–30% for Astra-Zeneca and Janssen/Johnson & Johnson vaccines respectively (51). HIT and VITT are associated with hypercoagulable blood of affected patients and a relatively high occurrence of thrombosis and thrombotic events within atypical blood vessel sites (21). The twain are associated with abnormal full blood cell count values (particularly leukocytes and platelets), coagulation, prothrombin-time, soluble D-dimer plasma concentrations outside the reference intervals, and a potential for disseminated intravascular coagulation. Similar to HIT, VITT has been associated with developing anti-PF4 antibodies. The typical sequela of which may be aggregation of neutrophils and platelets (52).
Antibodies against PF4 characterize VITT
Dissimilar to thrombocytopenia caused by heparin exposure, VITT pathological antibodies are produced independently of exposure to this thrombolytic drug. There are no reports that any of the VITT patients had been previously anticoagulated with heparin prior to the onset of VITT. This suggests that a factor in the vaccine preparation triggered the adverse reaction to the vaccine (45, 53). VITT shares many clinical, laboratory and immunological features with HIT, a pro-thrombotic pathology caused by antibodies that induce platelet-activation by recognizing multimolecular complexes. These structures form between cationic PF4 and anionic heparin (see Figure 1). These features include thrombocytopenia, unusual strokes, anti-PF4 autoantibodies, and platelet activation through platelet FcγRIIa receptor.
FIGURE 1
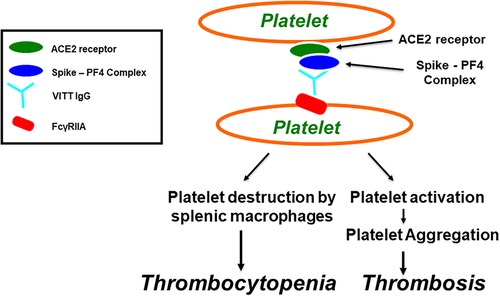
Figure 1. Vaccine-induced immune thrombotic thrombocytopenia (VITT). Antibodies induce platelet aggregation through FcγRIIa. SARS-CoV-2 virions, or its spike protein, produced after COVID-19 vaccination, bind to platelets via ACE2 receptor, leading to activation of platelets and the secretion of platelet factor 4 (PF4). PF4 then biochemically associates with the spike protein, forming PF4-Spike complexes that stimulate VITT anti-PF4 antibody production. VITT pathological IgG antibodies induce platelet aggregation through platelet FcγRIIa and thrombocytopenia through platelet destruction by splenic macrophages.
PF4, a 70-amino-acid protein located in α-granules, is secreted from platelets when activated during the aggregation process and promotes further thrombogenesis. PF4 is cationic in its physical nature and acts as a chemokine (54).. Although the primary function of PF4 is to act as a pro-coagulant factor, it is also plays a role in innate immunological and adaptive immunological processes where thrombocytes are activated. This platelet activation is in response to infections. The positively charged PF4 may readily associate with negatively charged bacterial surfaces, thereby promoting, opsonization and contributing defenses against potentially pathological bacterial infection (55). PF4 also forms molecular associations with polyanions, undergoing a structural change in its conformation, which induces anti-PF4/polyanion antibodies (see Figure 1). These help eliminate pathogens (56) and cause HIT in the absence of heparin molecules. This response is termed “spontaneous HIT” (57).
Because VITT patients were not previously exposed to heparin anticoagulation, it is reasonable to suspect a similar mechanism to “spontaneous HIT”. In vitro experiments demonstrated that proteolysis of plasma proteins leads to sequential release of endogenous glycosaminoglycans, chondroitin sulfate and heparin (43). The dysregulated proteolysis occurring during COVID-19 or VITT triggers a sustained and excessive release of endogenous chondroitin sulfate and/or heparin that may play a fundamental role in their associated and respective pathophysiology (43). Platelets stimulated by endogenous heparin, either directly or with the intermediation of PF4, release additional heparin may lead to the catastrophic thrombotic thrombocytopenia observed in severe COVID-19 or following vaccination (43).
The pathophysiology of VITT is presumably due to the development of antibodies against PF4 that associates with a highly restricted molecular location on the PF4 protein. This may correspond to the PF4-heparin-binding site. The result of which may be platelet consumption and depletion accompanied by thrombus formation (4, 58). Several published studies have described high levels of pathological VITT anti-PF4 antibodies, present in blood specimens from VITT patients, which induce the activation of platelets through platelet FcγRIIa receptors in the presence of PF4 without heparin and potentially other cells, through FcγRIIa receptors (4, 7, 46). The platelet-activating effects of VITT anti-PF4 antibodies can be completely inhibited by anti-FcγRIIa monoclonal antibodies (59–61). The presence of high concentrations of polyclonal immunoglobulins may interfere with the platelet-activating effects of these autoimmune anti-PF4 antibodies. This may be interpreted as a confirmation of the immune system's role in this hyper-activation of platelets working via FcγRIIa (61).
There remains the very real possibility that platelet activation contributing to the pathology of VITT may not be solely attributed to anti-PF4 antibodies. Other factors may underpin the processes resulting in these thrombotic events. The progression of HIT may be ascribed to the many pro-thrombotic mechanisms associated with VITT. These include monocyte activation, over production of tissue factor, and pro-coagulant microparticle generation (58). Restricted to eight amino acid sequences that overlap with the heparin-binding site, VITT antibodies associate with molecular regions similar to PF4 as with heparin (58). Anti VITT antibodies may mimic the physiological function of heparin. Thus, assisting in the clustering of the tetramers of the PF4 protein. These are the building blocks of the immune complexes, which bring together FcγRIIa membrane proteins and induce the activation of platelets (58). It is likely that these VITT pathogenic antibodies associate with and activate other non-platelet cells that also express the mildly ubiquitous FcγRIIa. Noteworthy are endothelial cells as well as the leucocytes (60). Molecular polymorphisms of FcγRIIa may influence the risk of VITT and the development of new thrombotic events. These FcγRIIa polymorphisms impact the affinity of association to the various human IgG subclasses and other molecules involved in FcγRIIa transmembrane signaling. A similar discussion of FcγRIIa polymorphisms applies wholly to HIT (62, 63).
It is now well-established that adenovirus engages with and activate platelets (64, 65). Several viruses can directly lead to platelet hyperactivity (66, 67). Further, constituents of Ad-vector COVID-19 vaccines may play the role of “co-factor” binding to PF4, inducing newly formed immunogenic epitopes that potentially induce the production of the pathological anti-PF4 immunoglobulins. The immunoreactivity observed between the vaccine components and platelets or the PF4 protein may be considered a contributor to the genesis and progression of VITT (4, 7). Platelets may be directly be infected by RNA contained within viruses. Therefore RNA from the virus particles may be translated into spike protein via the protein biosynthesis mechanisms possessed by platelets. Thus, initiating an immune response against the body's own platelets (68). Platelets can translate mRNA and synthesize proteins of viral origin, as is seen with dengue, human immunodeficiency, and influenza viruses (69).
SARS-CoV-2 spike protein chemically associates directly and increases the activation of platelets
The virus SARS-CoV-2 uses receptors in order to infect lung epithelial cells. COVID-19 patients in a critical condition expressing ACE2 as well as TMPRSS2, have hyperactive platelets, mean platelet volume above their reference interval, increased activation of αIIbβ3 and P-selectin expression on thrombocytes (10, 30). MAPK pathway, downstream of ACE2 mediates the potentiating role of SARS-CoV-2 on platelet activation (29). Neutrophils isolated from severe COVID-19 patients demonstrate a clear association with IgG immune complexes. Patients' sera with severe COVID-19 contain high amounts of immune complexes. These complexes potentially activate neutrophils via a mechanism that is partially independent of FcγRIIa platelets (70).
The Spike protein of the SARS-CoV-2 virus particle binds directly to ACE2 on the surface of platelets, enhancing platelet activation and causing the release of coagulation factors and inflammatory cytokines. In addition, this causes the formation of leukocyte–platelet aggregates and induces thrombus formation (10, 28, 30). Like SARS-CoV-2, through this chemical association with ACE2, the spike protein of SARS-CoV-2 stimulates the secretion of PF4 from platelets and activates cells of the immune system to initiate the inflammatory reaction, thus triggering the production of anti-PF4 antibodies (71).
Structural similarities have been evidenced between platelet factor 4 and SARS-CoV-2′s spike protein (4, 72). The chains are similar in their molecular structural organization, including anti-parallel β-pleated sheets uncased within a pair of α-helices. They both possess common amino acid sequences corresponding to 323–335 residues in RBD and 15–27 amino acidic residues in PF4 (72). Additional molecular similarities between the spike and PF4 have been reported upon, either in the RBD or the other spike protein domains (4). The anti-Spike IgG antibodies do not recognize PF4; the anti-PF4 antibodies demonstrate immunoreactivity with “Spike-RBD” (72). In a similar fashion to heparin-PF4 complexes, since RBD and PF4 can interact with a significant affinity, they form PF4-spike complexes that bind to platelet ACE2 receptors that could have an involvement in the immune production of anti-PF4 antibodies, leading to platelets aggregation through platelet FcγRIIa (72).
There also remains the possibility that other receptor proteins mediate SARS-CoV-2 spike-platelet interactions. Heparin sulphate, located on the cell surface, is implicated as a binding site for viruses (73). The molecular association of the SARS-CoV-2′s spike protein to cell membrane-bound heparin sulphate was attenuated in the presence of heparin. Thus hindering the passage of viral particles into host cells (74). A substitution mutation within the spike protein at residue 614 (aspartic acid to glycine) has been identified and suggested to be a clear “fitness advantage” in increasing SARS-CoV-2′s transmissibility in The Americas and Europe (75). Platelets interact with the spike protein of SARS-CoV-2 through various C-type lectin receptor proteins and proteoglycans. Variants generated through alternative-splicing of the spike protein exist, as well as for CD147. This being a transmembrane protein highly expressed in blood cells, including platelets. Further, adenovirus vectors also interact with the CD46 receptor.
VITT: laboratory assessment, prevention, management, and treatment
The reported laboratory aspects associated with VITT point to a consumptive coagulopathy. This involved thrombocytopenia, low plasma fibrinogen concentrations, and elevated plasma concentrations of D-dimer. This was reported with the severest of HIT cases (76). Most reports strongly correlate VITT with the strong presence of anti-platelet factor 4 in serum and platelet activation as assessed by function laboratory assays (4, 7).
Initial testing should be performed for recently vaccinated patients (i.e., between 4 and 30 days post-vaccination) presenting clinically and with new laboratory findings, including a full blood count, soluble D-dimer measures, and plasma fibrinogen measures. Deviations from reference intervals would identify patients with severe manifestations of VITT. New clinical symptoms may include nausea, vomiting, headaches, vision problems, pain in the abdomen and chest, as well as dyspnea (49). If lab findings are within the reference intervals, then there is a low likelihood of VITT. However, these VITT patients should be continually monitored, and laboratory investigations should be repeated if symptoms reoccur or persist. Suppose there is solely a single isolated incident of thrombocytopenia, with plasma soluble D-dimer and plasma fibrinogen within their reference intervals and an absence of thrombosis. In that case, vaccine-induced ITP should be considered a probable diagnosis, particularly when the index patient had received one of the vaccines containing mRNA, supposing there is suggestive evidence of consumptive coagulopathy (e.g., thrombocytopenia and plasma soluble D-dimer) (49). In that case, diagnostic imaging investigations and anti-PF4 IgG immunoassays should be performed to document VITT. If diagnostic imaging investigations reveal thrombosis, hospitalization and the initiation of clinical interventions involving non-heparin anticoagulant drugs are recommended without the transfusion of platelets (49). For complications with hemorrhaging, intravenous immunoglobulin (IVIG) and the steroid prednisone are suggested to resolve the low platelet counts, cryoprecipitate and fresh frozen plasma should be administered to manage coagulopathy (49).
There is sufficient evidence of common clinical and laboratory characteristics observed between VITT and HIT to adopt similar management approaches to the two syndromes (Table 3). Although the pathogenesis of VITT is not yet clear, recently published findings were consistent in describing that the three phenomena of thromboembolic events, low platelet count, and the detection of anti-PF4 antibodies appear as the hallmarks of VITT (9). Society of Thrombosis and Haemostasis Research suggested screening for anti-PF4 antibodies if thromboembolic events or low platelet counts occurs within two weeks post-vaccination (38). Treatment recommendations issued by The American Society of Hematology for VITT are similar to those for severe HIT in patients with low platelet count, thrombosis, and the positive detection PF4-heparin IgG as assessed by immunoassay (38). The regimens for successful management of HIT of autoimmune origin can also be similarly applied to VITT. Interventions may include IVIG and glucocorticoids given in high doses (42).
TABLE 3
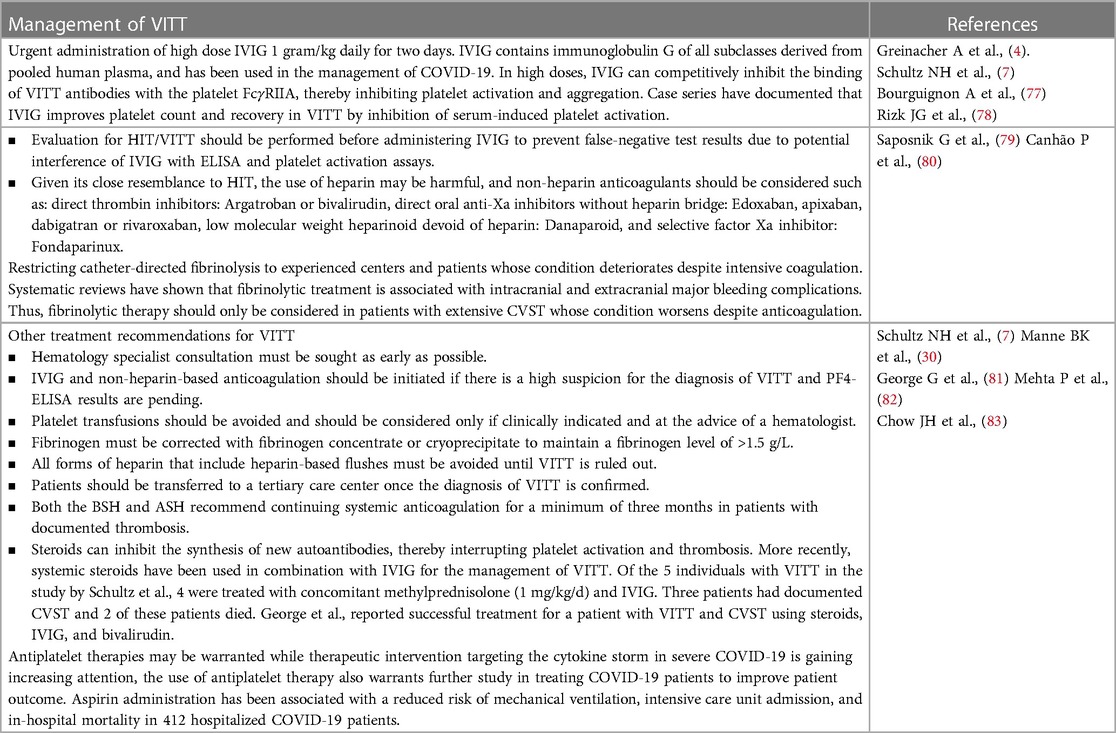
Table 3. VITT: Management and treatment.
Platelet FcγRIIa activation associated with HIT
Both VITT and HIT disorders are associated with thrombocytopenia, thrombosis, the presence of autoantibodies to PF4, and platelet activation through platelet FcγRIIa. The key adverse drug reaction to the anticoagulant heparin is HIT. It is a pharmacologically induced immunoglobulin-mediated thrombocytopenic disorder associated with both arterial and venous thrombosis resulting from the generation of platelet thrombi (84, 85). Approximately 5% of patients who receive heparin anticoagulation therapy develop HIT. Out of those patients, about 5%–80% develop thrombosis, which can involve arterial vessels, venous vessels, or both vessel types (86–88). HIT is characterized by the presence of platelet-activating antibodies that recognize multimolecular complexes between the cationic PF4 and the anionic heparin, leading to platelet activation through FcγRIIa receptors, resulting in thrombosis, endothelial cell injury, and the release of procoagulant-rich microparticles (see Figure 2) (89–93). The removal of platelets via phagocytosis by splenic macrophages, in a FcγRIIa-dependent mechanism, or platelet consumption caused by thrombi formation, can explain thrombocytopenia (4, 94).
FIGURE 2
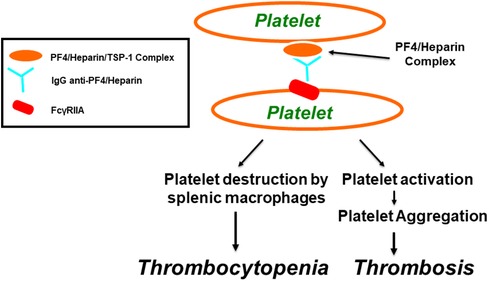
Figure 2. Pathogenesis of heparin-induced thrombocytopenia (HIT) as well as thrombosis through FcγRIIa. Within α-granules of platelets, PF4 is associated with a proteoglycan carrier that contains chondroitin-4-sulfate. Platelet factor 4 biochemically associates with physiologic concentrations of heparin, resulting in the molecular formation of the PF4/heparin combined complexes that leads to generation of anti-PF4/heparin antibodies that lead to the generation of anti-PF4/heparin antibodies. Multimolecular complexes of PF4-heparin and antibodies lead to cross-linking of FcγRIIA, inducing platelet aggregation and thrombocytopenia, through platelet destruction by splenic macrophages.
Thrombotic risk in HIT is strongly correlated with high levels of circulating anti-PF4 antibodies, as detected by immunoassays and functional platelet activation assays (95–97). Platelet activation induced by antibody-PF4/heparin complexes is accompanied by other cascading reactions that result in thrombotic complications (98). HIT immune complexes also activate other cells, such as monocytes (99), neutrophils (100), and endothelium (89, 101). HIT antibodies cross-link FcγRIIa on platelets, monocytes, and neutrophils, initiating pro-coagulant cellular responses that generate a profound hypercoagulable state (102). Although HIT is considered a platelet activation disorder, neutrophilia is common, particularly in thrombosis patients (103).
Platelet FcγRIIa activation associated with SK and APSAC
Platelet activation and the enhancement of platelet activation by autoantibodies binding to platelet membrane FcγRIIa is a common feature of VITT (47), HIT (4), SK, and APSAC (59, 104–107). Both SK and APSAC thrombolytic drugs act predominantly by bringing about the biomolecular transformation of inactive plasminogen to physiologically and biochemically active plasmin. This, in turn, breaks down the physicochemical structures of both fibrin molecules and fibrinogen molecules, thus achieving thrombolysis.
Streptokinase, an exogenous plasminogen activator derived from bacteria, has the latent potential to trigger an innate immune response, subsequently producing antibodies against SK. These anti-SK antibodies inhibit the breakdown of fibrin whilst simultaneously inducing platelet activation. These antibodies are highly prevalent among particular populations, such as individuals exposed to β-hemolytic streptococci infection or those with previous pharmacological exposure to SK (108). Following SK exposure, there is a progressive increase in detectable plasma levels of anti-SK antibodies, reaching a maximum at around two weeks post-SK thrombolytic therapy (109).
Both SK and APSAC modify the in vitro platelet aggregation predominantly via two main mechanisms. These being fibrinogen breakdown (107) and immune responses (see Figure 3) (59, 104–107). When SK or APSAC are added to normal subjects' platelet-rich plasma (PRP), there is an inhibition of “platelet aggregation” by way of response to platelet-activating pharmacological agonists [e.g., adenosine diphosphate (ADP) and collagen] (104, 106, 110). The SK or APSAC inhibition effect is mediated by plasmin generation, as it is attenuated in the presence of aprotinin (110). Platelet aggregation inhibition by SK is caused by the fibrinogen fragment E (110). Using PRP from healthy blood donors, the prevalence of SK- (or APSAC) reducing the aggregation of platelets by ADP was found to be 47% (104).
FIGURE 3
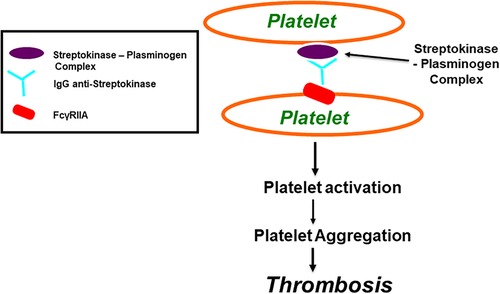
Figure 3. Streptokinase (SK) and Anisoylated Plasminogen-Streptokinase Activator Complex (APSAC) induced platelet aggregation through FcγRIIa. Both Streptokinase and APSAC modify in vitro platelet aggregation by two mechanisms; reduced aggregation due to fibrinogenolysis, and enhanced aggregation via an immunological reaction. The reduced aggregation by SK (or APSAC) is mediated by plasmin generation and the fibrinogen degradation product, fragment E. As shown in this figure, SK (or APSAC) may also trigger platelet aggregation by a mechanism involving specific IgG anti-SK. Both SK- (or APSAC) induced platelet aggregation and SK- (or APSAC) enhanced ADP-induced platelet aggregation require the interaction of the Fc domain of the anti-SK antibodies with the platelet FcγRIIA.
SK or APSAC also potentially induce platelet aggregation via specific circulating anti-SK IgG antibodies (104, 107) (see Figure 3). A previous infection with streptococcus or a history of treatment with SK (or APSAC) may influence the presence or absence of a patient's circulating anti-SK antibodies. Awareness of the broader deleterious effects of SK and APSAC may prove paramount when selecting from the array of available thrombolytic therapies so as to minimize the risk of excessive blood loss or thrombosis (104).
Using the PRP collected from healthy donors, we previously reported 5.8% and 53% prevalences for thrombolytic-associated “platelet aggregation” for streptokinase and APSAC, respectively (104). This thrombolytic drug-associated “platelet aggregation” reaction was accompanied by the release of a relatively large concentration of 4.5 µmol/L ATP. However, without streptokinase-induced “platelet aggregation”, activation of platelets occurred to some extent with SK-induced platelet shape change only with the release of a mere 0.05 µmol/L ATP (104). SK and APSAC-inducing platelet aggregation or enhancing of platelet aggregation are unaffected by the presence of aprotinin, indicating that the observed effect is specifically associated with SK and works independently of the generation of plasmin, a product from its plasminogen precursor (104). In PRP, SK- or APSAC-induced platelet aggregation showed no discernable correlation with the amount of healthy subject plasma anti-SK antibodies (104). A higher titre of anti-streptokinase antibodies was bereft of any such association with platelet aggregation in response to SK or APSAC exposure, using in vitro experimental settings, suggesting that anti-SK antibodies- activating platelets constitute a clearly defined sub-group of whole anti-SK antibodies in plasma (104).
Platelets perform a pivotal part in stimulating the response of the immune system in bringing about the said thrombolytic drug-induced “platelet aggregation” in the SK- (or APSAC) “enhanced ADP-induced platelet aggregation” as demonstrated by our in vitro experiments (59, 107). This was evidenced by mixing IgG anti-SK, isolated from patients recently diagnosed with an acute myocardial infarction who received SK for thrombolysis, and washed platelets from apparently healthy volunteer donors (59, 107). The combination of platelets from healthy subjects, “responders” to platelet-activating monoclonal antibody PL2-49, with SK (or APSAC) and purified IgG-induced platelets aggregation whilst simultaneously enhancing the SK platelet aggregation response to ADP. This response is mediated via platelet FcγRIIa and accompanied by the generation of inositol triphosphate, the subsequent cellular influx of Ca2+, and TXA2 release (59, 107). Further, compounds known to inhibit the catalytic activities of protein tyrosine kinase (PTK), as well as the enzyme protein kinase-C (PKC) and the biological catalyst phospholipase-C (PLC), have been employed to investigate “platelet aggregation” as induced by “anti-SK antibodies” and additional APSAC in isolated washed platelets. These compounds being: erbstatin - a PTK inhibitor; GF 109203X - a PCK inhibitor; and neomycin – an inhibitor of PLC. The presence of these enzyme inhibitors prevented the aggregation of platelets induced by “anti-SK antibodies” in addition to APSAC, suggesting that PTK, PCK and PCL pathways are fundamental to activating platelets via the immune system (59). The TXA2 receptor antagonist – SQ 29548 and the cyclooxygenase inhibitor – aspirin significantly attenuated the activation of platelets in this in vitro experimental setting (59). The release of ADP also contributed considerably to the “platelet aggregation” response, as confirmed by employing apyrase, a compound known to attenuate the immunoglobulin-G anti-streptokinase with the addition of APSAC-induced aggregation of platelets (59).
Two distinct pathways and their dynamic interactions enable “anti-SK antibodies” to elicit streptokinase-induced “platelet aggregation” and “streptokinase-enhanced platelet aggregation”. These pathways are: (i) the activation of platelets by the immune system via membrane-bound FcγRIIa, and (ii) the involvement of a demarcated subgroup of thrombocytes eliciting antibodies directed against SK (59, 104–107).
FcγRIIa structural polymorphisms correlate with platelet's functional response following stimulation and activation by antibodies of mouse origin or aggregated in response to antibodies of human origin (111). Several monoclonal antibodies induce the activation of platelets via interactions between their Fc-domains and membrane FcγRIIa. Platelets from apparently healthy donors demonstrate a vast variation in response to activating monoclonal antibodies (112–116). Although the mechanisms involved in these response disparities have not been definitively elucidated, the majority of reports addressing the platelet FcγRIIa functional polymorphism were performed in PRP (112, 113, 116, 117).
In our previous PRP studies, all platelets from subjects with SK- (or APSAC) elicited aggregation of platelets and SK- (or APSAC) enhanced “platelet aggregation” induced were assigned as “responders” in response to the monoclonal activating antibody PL-249 (88, 89). However, all subjects with only streptokinase (or APSAC) enhanced platelet aggregation were considered “intermediate-responders” and as an alternative group, “non-responders” in the biological reaction of these cohorts to the PL-249 (88, 89). However, in washed platelets, SK- (or APSAC) induced platelet aggregation and SK- (or APSAC-) “enhanced ADP-induced platelet aggregation”, observed using platelets from “responders”, “intermediate-responders” or “non-responders” to activating monoclonal antibody PL-249, indicating that the functional platelet FcγRIIa polymorphism is observed only when platelets are activated in PRP and not observed when using washed isolated platelets (104, 105). Because the platelet FcγRIIa functional polymorphism was observed only in PRP and not in washed platelets, we suggested that other important plasma factor(s) and mechanisms are involved in platelet FcγRIIa functional polymorphism (105).
The risk of developing thrombosis in VITT may depend on platelet FcγRIIa polymorphism; this has been reported to be a risk factor for HIT (62, 63). Further, in vitro studies and investigations may be needed to determine the role of platelet FcγRIIa polymorphism in VITT by comparing the platelet aggregation elicited in vitro by purified VITT IgG anti-PF4 in PRP vs. washed platelets, including platelets from different apparently healthy subjects defined as “responders”, “intermediate-responders” or “non-responders” to mouse activating platelet antibodies; such as the PL-249 monoclonal antibody.
VITT incidence rates: COVID-19 Oxford/AstraZeneca and Johnson & Johnson/Janssen vaccines and their relation to VITT
VITT is very rare, however, it can be life threatening, especially if the diagnosis and treatment are delayed. It remains highly relevant in countries that afforded and can only afford adenoviral vector-based vaccines for COVID-19 vaccination campaigns. The vaccination campaign, combined with an educational program informing the public and especially physicians about VITT symptoms and appropriate treatment, allowed a reduction in mortality from 50% to 5% - 6% in Australia (118). In Table 4, we focus on the COVID-19 Oxford/AstraZeneca and Johnson & Johnson/Janssen vaccines, used in the Western world with most reported VITT cases. The VITT incidence caused by COVID-19 Oxford/AstraZeneca and Johnson & Johnson/Janssen vaccines varies by the reporting country.
TABLE 4
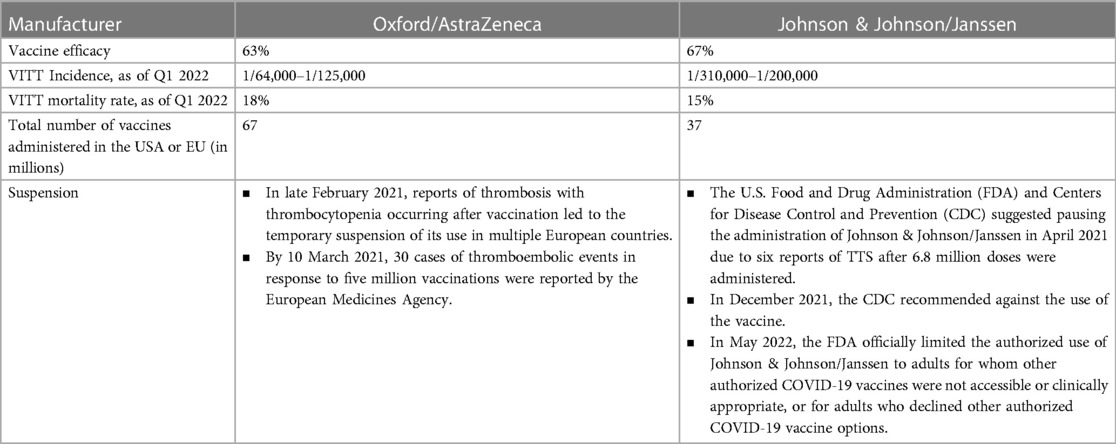
Table 4. Incidence of VITT in relation to COVID-19 Oxford/AstraZeneca and Johnson & Johnson/Janssen vaccines (119).
Worldwide population and demographics reaction to COVID-19 vaccines
Vaccination is critical for controlling the COVID-19 pandemic. As of 30 June 2022, 66.4% of the world's population had received at least one dose of a COVID-19 vaccine, however only 17.4% of people in low-income countries had received a first dose, underlying unequal access, availability and delivery (120–122). Despite the reductions in COVID-19 disease severity, hospitalizations and deaths, since the introduction of multiple safe and effective COVID-19 vaccines, vaccine hesitancy and refusal remains substantial, caused in part by misinformation, lower education, mistrust in science and governments, and elusive broad public support (123). The study of COVID-19 vaccine hesitancy among 23,000 respondents in 23 countries during 2022, representing almost 60% of the world's population, showed willingness to accept vaccination at 79.1%, however one out of eight (12.1%) vaccinated respondents are hesitant about booster doses (123). Twice-yearly COVID-19 booster vaccinations are recommended in some countries based on eligibility and availability. The same factors that influence hesitancy to accept an initial COVID-19 dose also drive booster hesitancy (124). COVID-19 vaccine hesitancy persists for vaccine boosters, which may present a serious challenge to anticipated routine COVID-19 immunization programs. Worldwide, policymakers and public health officials must address COVID-19 vaccine hesitancy and resistance as a component of their overall prevention and vaccination strategy.
Discussion & conclusion
Vaccines are vital and invaluable to control pandemics and to build up herd immunity. They have considerably reduced the incidence of childhood illnesses, notably measles, mumps, and rubella, and have facilitated the eradication of many infectious diseases, such as poliomyelitis, diphtheria, and smallpox (9). From the onset of COVID-19 pandemic to November 2021, more than 771 million cases and 6,960,7835 million deaths were documented worldwide. Vaccination for prevention against COVID-19 is crucial in containing the spread of SARS-CoV-2 and controlling this pandemic. For the Covid-19 pandemic, the use of diverse technologies allowed the development and production of highly effective vaccines, all produced with unprecedented timelines. The two most common COVID-19 vaccine platforms currently in use are the messenger RNA (mRNA) and adenovirus vector vaccines. The development of antibodies directed against the SARS-CoV-2 spike protein is the strategy chosen by most vaccine developers.
The overwhelming inflammatory response in patients with SARS-CoV-2 infection can lead to VITT associated with a hypercoagulable state, thrombosis, large vessel thrombosis, and, ultimately, death. VITT's overwhelming inflammatory bodily reaction to COVID-19 vaccination was observed in a small minority of vaccine recipients. The risk of VITT associated with thrombosis and mortality should be considered considerably subordinate to the relative risk of loss of life and morbidity attributable to the SARS-CoV-2 infection. VITT recognition raised concerns regarding the safety of COVID-19 vaccines and led to the reconsideration of vaccination strategies in many countries. Prior to vaccination, individuals should be educated about the clinical symptoms associated with VITT syndrome and should be advised to seek urgent medical treatment. VITT appears to be similar to HIT-, SK-, and APSC - induced platelet activation through platelet FcγRIIa, all showed strong, dose-dependent respective antibodies-mediated platelet activation, which is completely inhibited by IV.3, an FcγRIIa-blocking monoclonal antibody. In the event of major thrombotic events 4–30 days after COVID-19 vaccination, VITT should be considered; its treatment consists of therapeutic anticoagulation with nonheparin anticoagulants, and the infusion of high-dose IVIG to neutralize the anti - PF4 antibody-FcγRIIa binding. Routine platelet transfusions, aspirin, and warfarin should be avoided because of the possibility of worsening thrombosis and magnifying bleeding risk. There are no well-known risk factors for VITT with CVST post vaccination.
VITT was observed in both males and females across a wide age group, thus, there may not be a gender or age restriction in regard to suspected VITT. Therapeutic anticoagulation with nonheparin anticoagulants is the primary treatment for VITT with or without CVST. Given the concern of VITT, healthcare providers should be familiar with the VITT clinical presentations, pathophysiology, diagnostic criteria, and management consideration. Furthermore, they should maintain a high index of suspicion in patients presenting with symptoms on laboratory abnormalities suggestive of VITT as early recognition and management of this syndrome can prevent catastrophic complications. With increased VITT awareness and recognition by providers and the public alike, future cases should help refine VITT clinical understanding and improve its clinical outcomes. The better understanding of the mechanisms behind the VITT thrombotic complications is needed to aid in better disease management and improve the prognosis. Vigilance should be maintained regarding large-scale injections of vaccines, which contain adenoviruses and genetic material from the SARS-CoV-2 spike protein.
In immune complex disorders, platelets interact with IgG-containing immune complex through platelet FcγRIIa. The similarities of the mechanisms of VITT-, HIT-, SK, and APSAC-induced platelet activation could be relevant for a better understanding of immune diseases associated with abnormal hemostasis and thrombosis in general, and to VITT in particular. The exact VITT pathogenic mechanism is yet to be fully elucidated, further, in vitro as well as ex vivo experiments, investigations and specific lines of inquiry are warranted to reveal the true relationship that lies between COVID-19 vaccinations with VITT, and VITT possible risk factors in order to implement risk minimization strategies.
Author contributions
MA: Writing – original draft, Writing – review & editing. DY: Writing – original draft, Writing – review & editing. SF: Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflicts of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. (2020) 367(6483):1260–3. doi: 10.1126/science.abb2507
2. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. (2020) 181(2):281–92.e6. doi: 10.1016/j.cell.2020.02.058
3. Parker EPK, Shrotri M, Kampmann B. Keeping track of the SARS-CoV-2 vaccine pipeline. Nat Rev Immunol. (2020) 20(11):650. doi: 10.1038/s41577-020-00455-1
4. Greinacher A, Thiele T, Warkentin TE, Weisser K, Kyrle PA, Eichinger S. Thrombotic thrombocytopenia after ChAdOx1 nCov-19 vaccination. N Engl J Med. (2021) 384(22):2092–101. doi: 10.1056/NEJMoa2104840
5. Sadoff J, Gray G, Vandebosch A, Cardenas V, Shukarev G, Grinsztejn B, et al. Safety and efficacy of single-dose Ad26.COV2.S vaccine against COVID-19. N Engl J Med. (2021) 384(23):2187–201. doi: 10.1056/NEJMoa2101544
6. Muir KL, Kallam A, Koepsell SA, Gundabolu K. Thrombotic thrombocytopenia after Ad26.COV2.S vaccination. N Engl J Med. (2021) 384(20):1964–5. doi: 10.1056/NEJMc2105869
7. Schultz NH, Sørvoll IH, Michelsen AE, Munthe LA, Lund-Johansen F, Ahlen MT, et al. Thrombosis and thrombocytopenia after ChAdOx1 nCoV-19 vaccination. N Engl J Med. (2021) 384(22):2124–30. doi: 10.1056/NEJMoa2104882
8. See I, Su JR, Lale A, Woo EJ, Guh AY, Shimabukuro TT, et al. US case reports of cerebral venous sinus thrombosis with thrombocytopenia after Ad26.COV2.S vaccination, March 2 to April 21, 2021. JAMA. (2021) 325(24):2448–56. doi: 10.1001/jama.2021.7517
9. Cines DB, Bussel JB. SARS-CoV-2 vaccine-induced immune thrombotic thrombocytopenia. N Engl J Med. (2021) 384(23):2254–6. doi: 10.1056/NEJMe2106315
10. Zhang S, Liu Y, Wang X, Yang L, Li H, Wang Y, et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J Hematol Oncol. (2020) 13(1):120. doi: 10.1186/s13045-020-00954-7
11. Guo L, Rondina MT. The era of thromboinflammation: platelets are dynamic sensors and effector cells during infectious diseases. Front Immunol. (2019) 10:2204. doi: 10.3389/fimmu.2019.02204
12. Ribeiro LS, Migliari Branco L, Franklin BS. Regulation of innate immune responses by platelets. Front Immunol. (2019) 10:1320. doi: 10.3389/fimmu.2019.01320
13. Allaoui A, Khawaja AA, Badad O, Naciri M, Lordkipanidzé M, Guessous F, et al. Platelet function in viral immunity and SARS-CoV-2 infection. Semin Thromb Hemost. (2021) 47(4):419–26. doi: 10.1055/s-0041-1726033
14. Semple JW, Italiano JE Jr, Freedman J. Platelets and the immune continuum. Nat Rev Immunol. (2011) 11(4):264–74. doi: 10.1038/nri2956
15. Tang YQ, Yeaman MR, Selsted ME. Antimicrobial peptides from human platelets. Infect Immun. (2002) 70(12):6524–33. doi: 10.1128/IAI.70.12.6524-6533.2002
16. Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev. (2013) 93(1):327–58. doi: 10.1152/physrev.00016.2011
17. Heemskerk JW, Mattheij NJ, Cosemans JM. Platelet-based coagulation: different populations, different functions. J Thromb Haemost. (2013) 11(1):2–16. doi: 10.1111/jth.12045
18. Golebiewska EM, Poole AW. Platelet secretion: from haemostasis to wound healing and beyond. Blood Rev. (2015) 29(3):153–62. doi: 10.1016/j.blre.2014.10.003
19. Elrashdy F, Tambuwala MM, Hassan SS, Adadi P, Seyran M, Abd El-Aziz TM, et al. Autoimmunity roots of the thrombotic events after COVID-19 vaccination. Autoimmun Rev. (2021) 20(11):102941. doi: 10.1016/j.autrev.2021.102941
20. Shibeeb S, Ahmad MN. Thrombotic and hypercoagulability complications of COVID-19: an update. J Blood Med. (2021) 12:785–93. doi: 10.2147/JBM.S316014
21. Warkentin TE, Kaatz S. COVID-19 versus HIT hypercoagulability. Thromb Res. (2020) 196:38–51. doi: 10.1016/j.thromres.2020.08.017
22. Whyte CS, Morrow GB, Mitchell JL, Chowdary P, Mutch NJ. Fibrinolytic abnormalities in acute respiratory distress syndrome (ARDS) and versatility of thrombolytic drugs to treat COVID-19. J Thromb Haemost. (2020) 18(7):1548–55. doi: 10.1111/jth.14872
23. McGonagle D, Sharif K, O'Regan A, Bridgewood C. The role of cytokines including interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun Rev. (2020) 19(6):102537. doi: 10.1016/j.autrev.2020.102537
24. Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. (2020) 382(18):1708–20. doi: 10.1056/NEJMoa2002032
25. Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. (2020) 180(7):934–43. doi: 10.1001/jamainternmed.2020.0994
26. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus-infected pneumonia in Wuhan, China. JAMA. (2020) 323(11):1061–9. doi: 10.1001/jama.2020.1585
27. Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. (2020) 191:145–7. doi: 10.1016/j.thromres.2020.04.013
28. Campbell RA, Boilard E, Rondina MT. Is there a role for the ACE2 receptor in SARS-CoV-2 interactions with platelets? J Thromb Haemost. (2021) 19(1):46–50. doi: 10.1111/jth.15156
29. Elalamy I, Gerotziafas G, Alamowitch S, Laroche JP, Van Dreden P, Ageno W, et al. SARS-CoV-2 vaccine and thrombosis: an expert consensus on vaccine-induced immune thrombotic thrombocytopenia. J Thromb Haemostasis. (2021) 121(8):982–91. doi: 10.1055/a-1499-0119
30. Manne BK, Denorme F, Middleton EA, Portier I, Rowley JW, Stubben C, et al. Platelet gene expression and function in patients with COVID-19. Blood. (2020) 136(11):1317–29. doi: 10.1182/blood.2020007214
31. Barrett TJ, Lee AH, Xia Y, Lin LH, Black M, Cotzia P, et al. Platelet and vascular biomarkers associate with thrombosis and death in coronavirus disease. Circ Res. (2020) 127(7):945–7. doi: 10.1161/CIRCRESAHA.120.317803
32. Moosavi M, Wooten M, Goodman A, Nahab FB, Duncan A, Maier C, et al. Retrospective analyses associate hemostasis activation biomarkers with poor outcomes in patients with COVID-19. Am J Clin Pathol. (2021) 155(4):498–505. doi: 10.1093/ajcp/aqaa266
33. Greinacher A, Selleng K, Mayerle J, Palankar R, Wesche J, Reiche S, et al. Anti-platelet factor 4 antibodies causing VITT do not cross-react with SARS-CoV-2 spike protein. Blood. (2021) 138(14):1269–77. doi: 10.1182/blood.2021012938
34. Giacomelli E, Dorigo W, Fargion A, Calugi G, Cianchi G, Pratesi C. Acute thrombosis of an aortic prosthetic graft in a patient with severe COVID-19-related pneumonia. Ann Vasc Surg. (2020) 66:8–10. doi: 10.1016/j.avsg.2020.04.040
35. Bozzani A, Arici V, Franciscone MM, Danesino V, Cascina A, Ticozzelli G, et al. Severe acute respiratory syndrome coronavirus 2 infection and the upper limb deep vein thrombosis risk. Ann Vasc Surg. (2020) 66:11–3. doi: 10.1016/j.avsg.2020.04.037
36. Perry RJ, Tamborska A, Singh B, Craven B, Marigold R, Arthur-Farraj P, et al. Cerebral venous thrombosis after vaccination against COVID-19 in the UK: a multicentre cohort study. Lancet. (2021) 398(10306):1147–56. doi: 10.1016/S0140-6736(21)01608-1
37. de Barry O, Cabral D, Kahn JE, Vidal F, Carlier RY, El Hajjam M. 18-FDG pseudotumoral lesion with quick flowering to a typical lung CT COVID-19. Radiol Case Rep. (2020) 15(10):1813–6. doi: 10.1016/j.radcr.2020.07.035
38. Hughes C, Nichols T, Pike M, Subbe C, Elghenzai S. Cerebral venous sinus thrombosis as a presentation of COVID-19. Eur J Case Rep Intern Med. (2020) 7(5):001691. PMID: 32399457; PMCID: 7213833. doi: 10.12890/2020_001691
39. Hemasian H, Ansari B. First case of COVID-19 presented with cerebral venous thrombosis: a rare and dreaded case. Rev Neurol. (2020) 176(6):521–3. doi: 10.1016/j.neurol.2020.04.013
40. Klein DE, Libman R, Kirsch C, Arora R. Cerebral venous thrombosis: a typical presentation of COVID-19 in the young. J Stroke Cerebrovasc Dis. (2020) 29(8):104989. doi: 10.1016/j.jstrokecerebrovasdis.2020.104989
41. Bayas A, Menacher M, Christ M, Behrens L, Rank A, Naumann M. Bilateral superior ophthalmic vein thrombosis, ischaemic stroke, and immune thrombocytopenia after ChAdOx1 nCoV-19 vaccination. Lancet. (2021) 397(10285):e11. doi: 10.1016/S0140-6736(21)00872-2
42. Rizk JG, Gupta A, Sardar P, Henry BM, Lewin JC, Lippi G, et al. Clinical characteristics and pharmacological management of COVID-19 vaccine-induced immune thrombotic thrombocytopenia with cerebral venous sinus thrombosis: a review. JAMA cardiology. (2021) 6(12):1451–60. doi: 10.1001/jamacardio.2021.3444
43. Ropper AH, Klein JP. Cerebral venous thrombosis. N Engl J Med. (2021) 385(1):59–64. doi: 10.1056/NEJMra2106545
44. Sadoff J, Davis K, Douoguih M. Thrombotic thrombocytopenia after Ad26.COV2.S vaccination - response from the manufacturer. N Engl J Med. (2021) 384(20):1965–6. doi: 10.1056/NEJMc2106075
45. Oldenburg J, Klamroth R, Langer F, Albisetti M, von Auer C, Ay C, et al. Diagnosis and management of vaccine-related thrombosis following AstraZeneca COVID-19 vaccination: guidance statement from the GTH. Hamostaseologie. (2021) 41(3):184–9. doi: 10.1055/a-1469-7481
46. Scully M, Singh D, Lown R, Poles A, Solomon T, Levi M, et al. Pathologic antibodies to platelet factor 4 after ChAdOx1 nCoV-19 vaccination. N Engl J Med. (2021) 384(23):2202–11. doi: 10.1056/NEJMoa2105385
47. McFadyen JD, Sharma P, Moon MJ, Noonan J, Goodall E, Tran HA, et al. Activation of circulating platelets in vaccine-induced thrombotic thrombocytopenia and its reversal by intravenous immunoglobulin. Br J Haematol. (2022) 196(1):234–7. doi: 10.1111/bjh.17750
48. Carli G, Nichele I, Ruggeri M, Barra S, Tosetto A. Deep vein thrombosis (DVT) occurring shortly after the second dose of mRNA SARS-CoV-2 vaccine. Intern Emerg Med. (2021) 16(3):803–4. doi: 10.1007/s11739-021-02685-0
49. Arepally GM, Ortel TL. Vaccine-induced immune thrombotic thrombocytopenia: what we know and do not know. Blood. (2021) 138(4):293–8. doi: 10.1182/blood.2021012152
50. Liu Z, Ji S, Sheng J, Wang F. Pharmacological effects and clinical applications of ultra low molecular weight heparins. Drug Discov Ther. (2014) 8(1):1–10. doi: 10.5582/ddt.8.1
51. Siegler JE, Klein P, Yaghi S, Vigilante N, Abdalkader M, Coutinho JM, et al. Cerebral vein thrombosis with vaccine-induced immune thrombotic thrombocytopenia. Stroke. (2021) 52(9):3045–53. doi: 10.1161/STROKEAHA.121.035613
52. Kashir J, Ambia AR, Shafqat A, Sajid MR, AlKattan K, Yaqinuddin A. Scientific premise for the involvement of neutrophil extracellular traps (NETs) in vaccine-induced thrombotic thrombocytopenia (VITT). J Leukocyte Biol. (2022) 111(3):725–34. doi: 10.1002/JLB.5COVR0621-320RR
53. Dotan A, Shoenfeld Y. Perspectives on vaccine induced thrombotic thrombocytopenia. J Autoimmun. (2021) 121:102663. doi: 10.1016/j.jaut.2021.102663
54. Slungaard A. Platelet factor 4: a chemokine enigma. Int J Biochem Cell Biol. (2005) 37(6):1162–7. doi: 10.1016/j.biocel.2004.12.003
55. Comer SP, Cullivan S, Szklanna PB, Weiss L, Cullen S, Kelliher S, et al. COVID-19 induces a hyperactive phenotype in circulating platelets. PLoS Biol. (2021) 19(2):e3001109. doi: 10.1371/journal.pbio.3001109
56. Hoylaerts MF, Vanassche T, Verhamme P. Bacterial killing by platelets: making sense of (H)IT. J Thromb Haemostasis. (2018) 16(6):1182–6. doi: 10.1111/jth.14012
57. Warkentin TE. High-dose intravenous immunoglobulin for the treatment and prevention of heparin-induced thrombocytopenia: a review. Expert Rev Hematol. (2019) 12(8):685–98. doi: 10.1080/17474086.2019.1636645
58. Huynh A, Kelton JG, Arnold DM, Daka M, Nazy I. Antibody epitopes in vaccine-induced immune thrombotic thrombocytopaenia. Nature. (2021) 596(7873):565–9. doi: 10.1038/s41586-021-03744-4
59. Abdelouahed M, Hatmi M, Helft G, Emadi S, Elalamy I, Samama MM. Comparative effects of recombinant staphylokinase and streptokinase on platelet aggregation. Thromb Haemostasis. (1997) 77(5):815–7. doi: 10.1055/s-0038-1656058
60. McFadyen JD, Peter K. Platelet CXCL14: introducing a new player and potential therapeutic target in thromboinflammation. Cardiovasc Res. (2021) 117(3):645–7. doi: 10.1093/cvr/cvaa351
61. Padmanabhan A, Jones CG, Pechauer SM, Curtis BR, Bougie DW, Irani MS, et al. IVIg for treatment of severe refractory heparin-induced thrombocytopenia. Chest. (2017) 152(3):478–85. doi: 10.1016/j.chest.2017.03.050
62. Rollin J, Pouplard C, Gruel Y. Risk factors for heparin-induced thrombocytopenia: focus on fcγ receptors. Thromb Haemostasis. (2016) 116(5):799–805. doi: 10.1160/TH16-02-0109
63. Rollin J, Pouplard C, Sung HC, Leroux D, Saada A, Gouilleux-Gruart V, et al. Increased risk of thrombosis in FcγRIIA 131RR patients with HIT due to defective control of platelet activation by plasma IgG2. Blood. (2015) 125(15):2397–404. doi: 10.1182/blood-2014-09-594515
64. Stone D, Liu Y, Shayakhmetov D, Li ZY, Ni S, Lieber A. Adenovirus-platelet interaction in blood causes virus sequestration to the reticuloendothelial system of the liver. J Virol. (2007) 81(9):4866–71. doi: 10.1128/JVI.02819-06
65. Othman M, Labelle A, Mazzetti I, Elbatarny HS, Lillicrap D. Adenovirus-induced thrombocytopenia: the role of von Willebrand factor and P-selectin in mediating accelerated platelet clearance. Blood. (2007) 109(7):2832–9. doi: 10.1182/blood-2006-06-032524
66. Simon AY, Sutherland MR, Pryzdial EL. Dengue virus binding and replication by platelets. Blood. (2015) 126(3):378–85. doi: 10.1182/blood-2014-09-598029
67. Assinger A, Kral JB, Yaiw KC, Schrottmaier WC, Kurzejamska E, Wang Y, et al. Human cytomegalovirus-platelet interaction triggers toll-like receptor 2-dependent proinflammatory and proangiogenic responses. Arterioscler, Thromb, Vasc Biol. (2014) 34(4):801–9. doi: 10.1161/ATVBAHA.114.303287
68. Wise J. COVID-19: European countries suspend use of Oxford-AstraZeneca vaccine after reports of blood clots. BMJ. (2021) 372:n699. PMID: 33707182. doi: 10.1136/bmj.n699
69. Weyrich AS, Schwertz H, Kraiss LW, Zimmerman GA. Protein synthesis by platelets: historical and new perspectives. J Thromb Haemost. (2009) 7(2):241–6. doi: 10.1111/j.1538-7836.2008.03211.x
70. Mazzitelli I, Bleichmar L, Ludueña MG, Pisarevsky A, Labato M, Chiaradia V, et al. Immunoglobulin G immune complexes may contribute to neutrophil activation in the course of severe coronavirus disease 2019. J Infect Dis. (2021) 224(4):575–85. doi: 10.1093/infdis/jiab174
71. Angeli F, Spanevello A, Reboldi G, Visca D, Verdecchia P. SARS-CoV-2 vaccines: lights and shadows. Eur J Intern Med. (2021) 88:1–8. doi: 10.1016/j.ejim.2021.04.019
72. Passariello M, Vetrei C, Amato F, De Lorenzo C. Interactions of spike-RBD of SARS-CoV-2 and platelet factor 4: new insights in the etiopathogenesis of thrombosis. Int J Mol Sci. (2021) 22(16):1–9. doi: 10.3390/ijms22168562
73. Liu J, Thorp SC. Cell surface heparan sulfate and its roles in assisting viral infections. Med Res Rev. (2002) 22(1):1–25. doi: 10.1002/med.1026
74. Mycroft-West CJ, Su D, Pagani I, Rudd TR, Elli S, Gandhi NS, et al. Heparin inhibits cellular invasion by SARS-CoV-2: structural dependence of the interaction of the spike S1 receptor-binding domain with heparin. Thromb Haemostasis. (2020) 120(12):1700–15. doi: 10.1055/s-0040-1721319
75. Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell. (2020) 182(4):812–27.e19. doi: 10.1016/j.cell.2020.06.043
76. Gruel Y, Pouplard C, Nguyen P, Borg JY, Derlon A, Juhan-Vague I, et al. Biological and clinical features of low-molecular-weight heparin-induced thrombocytopenia. Br J Haematol. (2003) 121(5):786–92. doi: 10.1046/j.1365-2141.2003.04363.x
77. Bourguignon A, Arnold DM, Warkentin TE, Smith JW, Pannu T, Shrum JM, et al. Adjunct immune globulin for vaccine-induced immune thrombotic thrombocytopenia. N Engl J Med. (2021) 385(8):720–8. doi: 10.1056/NEJMoa2107051
78. Rizk JG, Kalantar-Zadeh K, Mehra MR, Lavie CJ, Rizk Y, Forthal DN. Pharmaco-immunomodulatory therapy in COVID-19. Drugs. (2020) 80:1267–92. doi: 10.1007/s40265-020-01367-z
79. Saposnik G, Barinagarrementeria F, Brown RD Jr, Bushnell CD, Cucchiara B, Cushman M, et al. Diagnosis and management of cerebral venous thrombosis: a statement for healthcare professionals from the American heart association/American stroke association. Stroke. (2011) 42(4):1158–92. doi: 10.1161/STR.0b013e31820a8364
80. Canhão P, Falcão F, Ferro JM. Thrombolytics for cerebral sinus thrombosis: a systematic review. Cerebrovascular Diseases. (2003) 15(3):159–66. doi: 10.1159/000068833
81. George G, Friedman KD, Curtis BR, Lind SE. Successful treatment of thrombotic thrombocytopenia with cerebral sinus venous thrombosis following Ad26.COV2.S vaccination. Am J Hematol. (2021) 96(8):E301–3. doi: 10.1002/ajh.26237
82. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. (2020) 395(10229):1033–4. doi: 10.1016/S0140-6736(20)30628-0
83. Chow JH, Khanna AK, Kethireddy S, Yamane D, Levine A, Jackson AM, et al. Aspirin use is associated with decreased mechanical ventilation, intensive care unit admission, and in-hospital mortality in hospitalized patients with coronavirus disease 2019. Anesth Analg. (2021) 132(4):930–41. doi: 10.1213/ANE.0000000000005292
84. Cines DB, Tomaski A, Tannenbaum S. Immune endothelial-cell injury in heparin-associated thrombocytopenia. N Engl J Med. (1987) 316(10):581–9. doi: 10.1056/NEJM198703053161004
85. Celoria GM, Steingart RH, Banson B, Friedmann P, Rhee SW, Berman JA. Coumarin skin necrosis in a patient with heparin-induced thrombocytopenia–a case report. Angiology. (1988) 39(10):915–20. doi: 10.1177/000331978803901009
86. Gupta AK, Kovacs MJ, Sauder DN. Heparin-induced thrombocytopenia. Ann Pharmacother. (1998) 32(1):55–9. doi: 10.1345/aph.16388
87. Warkentin TE, Kelton JG. A 14-year study of heparin-induced thrombocytopenia. Am J Med. (1996) 101(5):502–7. doi: 10.1016/S0002-9343(96)00258-6
88. Chong BH. Heparin-induced thrombocytopenia. Br J Haematol. (1995) 89(3):431–9. doi: 10.1111/j.1365-2141.1995.tb08346.x
89. Warkentin TE, Hayward CP, Boshkov LK, Santos AV, Sheppard JA, Bode AP, et al. Sera from patients with heparin-induced thrombocytopenia generate platelet-derived microparticles with procoagulant activity: an explanation for the thrombotic complications of heparin-induced thrombocytopenia. Blood. (1994) 84(11):3691–9. doi: 10.1182/blood.V84.11.3691.bloodjournal84113691
90. Amiral J, Bridey F, Wolf M, Boyer-Neumann C, Fressinaud E, Vissac AM, et al. Antibodies to macromolecular platelet factor 4-heparin complexes in heparin-induced thrombocytopenia: a study of 44 cases. Thromb Haemostasis. (1995) 73(1):21–8. doi: 10.1055/s-0038-1651670
91. Cai Z, Yarovoi SV, Zhu Z, Rauova L, Hayes V, Lebedeva T, et al. Atomic description of the immune complex involved in heparin-induced thrombocytopenia. Nat Commun. (2015) 6:8277. doi: 10.1038/ncomms9277
92. Cancio LC, Cohen DJ. Heparin-induced thrombocytopenia and thrombosis. J Am Coll Surg. (1998) 186(1):76–91. doi: 10.1016/S1072-7515(97)00134-8
93. Warkentin TE, Chong BH, Greinacher A. Heparin-induced thrombocytopenia: towards consensus. Thromb Haemostasis. (1998) 79(1):1–7. doi: 10.1055/s-0037-1614206
94. Kuwana M, Okazaki Y, Ikeda Y. Splenic macrophages maintain the anti-platelet autoimmune response via uptake of opsonized platelets in patients with immune thrombocytopenic purpura. J Thromb Haemost. (2009) 7(2):322–9. doi: 10.1111/j.1538-7836.2008.03161.x
95. Baroletti S, Hurwitz S, Conti NA, Fanikos J, Piazza G, Goldhaber SZ. Thrombosis in suspected heparin-induced thrombocytopenia occurs more often with high antibody levels. Am J Med. (2012) 125(1):44–9. doi: 10.1016/j.amjmed.2011.06.025
96. Warkentin TE, Sheppard JI, Moore JC, Sigouin CS, Kelton JG. Quantitative interpretation of optical density measurements using PF4-dependent enzyme-immunoassays. J Thromb Haemost. (2008) 6(8):1304–12. doi: 10.1111/j.1538-7836.2008.03025.x
97. Zwicker JI, Uhl L, Huang WY, Shaz BH, Bauer KA. Thrombosis and ELISA optical density values in hospitalized patients with heparin-induced thrombocytopenia. J Thromb Haemost. (2004) 2(12):2133–7. doi: 10.1111/j.1538-7836.2004.01039.x
98. Datta P, Zhang F, Dordick JS, Linhardt RJ. Platelet factor 4 polyanion immune complexes: heparin induced thrombocytopenia and vaccine-induced immune thrombotic thrombocytopenia. Thromb J. (2021) 19(1):66. doi: 10.1186/s12959-021-00318-2
99. Rauova L, Hirsch JD, Greene TK, Zhai L, Hayes VM, Kowalska MA, et al. Monocyte-bound PF4 in the pathogenesis of heparin-induced thrombocytopenia. Blood. (2010) 116(23):5021–31. doi: 10.1182/blood-2010-03-276964
100. Perdomo J, Leung HHL, Ahmadi Z, Yan F, Chong JJH, Passam FH, et al. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat Commun. (2019) 10(1):1322. doi: 10.1038/s41467-019-09160-7
101. Kelton JG, Sheridan D, Santos A, Smith J, Steeves K, Smith C, et al. Heparin-induced thrombocytopenia: laboratory studies. Blood. (1988) 72(3):925–30. doi: 10.1182/blood.V72.3.925.bloodjournal723925
102. Arepally GM, Padmanabhan A. Heparin-induced thrombocytopenia: a focus on thrombosis. Arterioscler, Thromb, Vasc Biol. (2021) 41(1):141–52. PMID: 33267665; PMCID: 7769912. doi: 10.1161/ATVBAHA.120.315445
103. Hui M, Sheppard JI, Li N, Warkentin TE. Neutrophil and monocyte counts in heparin-induced thrombocytopenia. Thromb Haemostasis. (2019) 119(6):941–51. doi: 10.1055/s-0039-1683913
104. Abdelouahed M, Elalamy I, Lebrazi J, Helft G, Mirshahi M, Lecrubier C, et al. Original article: streptokinase modifies in vitro platelet aggregation by two mechanisms: reduced aggregation due to fibrinogenolysis and enhanced aggregation via an immunological reaction. Platelets. (1995) 6(6):317–25. doi: 10.3109/09537109509078465
105. Abdelouahed M, Elalamy I, Samama MM, Hatmi M. Platelet aggregation by IgG anti-streptokinase and anisoylated plasminogen-streptokinase activator complex: heterogenous responses in platelet-rich plasma but not in washed platelets. Thromb Res. (1997) 86(3):255–62. doi: 10.1016/S0049-3848(97)00068-6
106. Abdelouahed M, Emadi S, Elalamy I, Samama MM, Hatmi M. Signal transduction in the platelet activation induced by IgG anti-streptokinase and anisoylated plasminogen-streptokinase activator complex. Platelets. (1997) 8(2-3):135–41. doi: 10.1080/09537109709169328
107. Lebrazi J, Helft G, Abdelouahed M, Elalamy I, Mirshahi M, Samama MM, et al. Human anti-streptokinase antibodies induce platelet aggregation in an fc receptor (CD32) dependent manner. Thromb Haemostasis. (1995) 74(3):938–42. doi: 10.1055/s-0038-1649851
108. Bachmann F. Development of antibodies against perorally and rectally administered streptokinase in man. J Lab Clin Med. (1968) 72(2):228–38.5671199
109. Lynch M, Littler WA, Pentecost BL, Stockley RA. Immunoglobulin response to intravenous streptokinase in acute myocardial infarction. Br Heart J. (1991) 66(2):139–42. doi: 10.1136/hrt.66.2.139
110. Lebrazi J, Abdelouahed M, Mirshahi M, Samama MM, Lecompte T. Streptokinase and APSAC inhibit platelet aggregation in vitro by fibrinogenolysis: effect of plasma fibrinogen degradation products X and E. Fibrinolysis. (1995) 9(2):113–9. doi: 10.1016/S0268-9499(95)80073-5
111. Looney RJ, Anderson CL, Ryan DH, Rosenfeld SI. Structural polymorphism of the human platelet Fc gamma receptor. J Immunol (Baltimore, Md: 1950). (1988) 141(8):2680–3. doi: 10.4049/jimmunol.141.8.2680
112. Brandt JT, Isenhart CE, Osborne JM, Ahmed A, Anderson CL. On the role of platelet Fc gamma RIIa phenotype in heparin-induced thrombocytopenia. Thromb Haemostasis. (1995) 74(6):1564–72. doi: 10.1055/s-0038-1649983
113. Bachelot C, Saffroy R, Gandrille S, Aiach M, Rendu F. Role of Fc gamma RIIA gene polymorphism in human platelet activation by monoclonal antibodies. Thromb Haemostasis. (1995) 74(6):1557–63. doi: 10.1055/s-0038-1649982
114. Mazurov AV, Vinogradov DV, Vlasik TN, Burns GF, Berndt MC. Heterogeneity of platelet Fc-receptor-dependent response to activating monoclonal antibodies. Platelets. (1992) 3(4):181–8. doi: 10.3109/09537109209013181
115. Tomiyama Y, Kunicki TJ, Zipf TF, Ford SB, Aster RH. Response of human platelets to activating monoclonal antibodies: importance of Fc gamma RII (CD32) phenotype and level of expression. Blood. (1992) 80(9):2261–8. doi: 10.1182/blood.V80.9.2261.2261
116. Horsewood P, Hayward CP, Warkentin TE, Kelton JG. Investigation of the mechanisms of monoclonal antibody-induced platelet activation. Blood. (1991) 78(4):1019–26. doi: 10.1182/blood.V78.4.1019.1019
117. Rubinstein E, Kouns WC, Jennings LK, Boucheix C, Carroll RC. Interaction of two GPIIb/IIIa monoclonal antibodies with platelet Fc receptor (Fc gamma RII). Br J Haematol. (1991) 78(1):80–6. doi: 10.1111/j.1365-2141.1991.tb04386.x
118. The Department of Health and Aged Care A. COVID-19 vaccine weekly safety report - 23-09-2021. Available at: https://www.tga.gov.au/news/covid-19-vaccine-safety-reports/covid-19-vaccine-weekly-safety-report-23-09-2021: The Department of Health and Aged Care, Australia 2021.
119. Roytenberg R, García-Sastre A, Li W. Vaccine-induced immune thrombotic thrombocytopenia: what do we know hitherto? Front Med). (2023) 10:1155727. doi: 10.3389/fmed.2023.1155727
120. Singh JA, Kochhar S, Wolff J, Atuire C, Bhan A, Emanuel E, et al. WHO Guidance on COVID-19 vaccine trial designs in the context of authorized COVID-19 vaccines and expanding global access: ethical considerations. Vaccine. (2022) 40(14):2140–9. doi: 10.1016/j.vaccine.2022.02.038
121. Hassan MA, Aliyu S. Delayed access to COVID-19 vaccines: a perspective on low-income countries in Africa. Int J Health Serv. (2022) 52(3):323–9. doi: 10.1177/00207314221096365
122. Mathieu E, Ritchie H, Rodés-Guirao L, Appel C, Giattino C, Hasell J, et al. Coronavirus Pandemic (COVID-19). OurWorldInDataorg. 2020.
123. Lazarus JV, Wyka K, White TM, Picchio CA, Gostin LO, Larson HJ, et al. A survey of COVID-19 vaccine acceptance across 23 countries in 2022. Nat Med. (2023) 29(2):366–75. doi: 10.1038/s41591-022-02185-4
Keywords: vaccine-induced immune thrombotic thrombocytopenia, COVID-19, platelet activation, FcγRIIa, heparin-induced thrombocytopenia, vaccination
Citation: Abdelouahed M, Yateem D and Fredericks S (2023) FcγRIIa - dependent platelet activation identified in COVID-19 vaccine-induced immune thrombotic thrombocytopenia-, heparin-induced thrombocytopenia, streptokinase- and anisoylated plasminogen-streptokinase activator complex-induced platelet activation. Front. Cardiovasc. Med. 10:1282637. doi: 10.3389/fcvm.2023.1282637
Received: 24 August 2023; Accepted: 30 October 2023;
Published: 15 November 2023.
Edited by:
Cristina Tudoran, Victor Babes University of Medicine and Pharmacy, RomaniaReviewed by:
Omer Iqbal, Loyola University Chicago, United StatesLarisa Anghel, Institute of Cardiovascular Diseases, Romania
© 2023 Abdelouahed, Yateem and Fredericks. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Salim Fredericks c2ZyZWRlcmlja3NAcmNzaS1tdWIuY29t